A new Merluccius polli reference genome to investigate the effects of global change in West African waters
 Juan L. Mateo
Juan L. Mateo Carmen Blanco-Fernandez
Carmen Blanco-Fernandez Eva Garcia-Vazquez
Eva Garcia-Vazquez Gonzalo Machado-Schiaffino
Gonzalo Machado-Schiaffino- 1Department of Informatics, University of Oviedo, Oviedo, Spain
- 2Department of Functional Biology, University of Oviedo, Oviedo, Spain
Genome resources have become crucial to assess genome-wide level of variation as well as to detect adaptive variation. This is particularly important for studying diversity in marine species inhabiting regions highly affected by accelerated climate warming and pollution, also known as global change. A greater awareness of the impacts of global change is urgently needed to ensure sustainable marine fisheries. Despite recent efforts, there are still many gaps in fish reference genomes, both geographical and taxonomic. Here, we sequence, assemble and annotate the genome of Merluccius polli. The total length of this new assembly (~582 Kb, N50 = 168Kb) is approximately 40% longer and much less fragmented than a previous version. Even though it might not be intrinsic of this species, low level of heterozygosity (1.16 SNPs/Kb) and low proportion of repeat content (9.21%) was found in this genome. This hake species has a wide latitudinal distribution; therefore, it is exposed to a changing temperature gradient and to a variety of contaminants in part of its distribution along West African coast. Special emphasis was laid on the identification and characterization of candidate genes known to respond to different stressors (depth, temperature, hypoxia, and heavy metals) happening along its geographical distribution. A total of 68 of the selected candidate genes known to be associated with responses to these stressors were found in the current assembly of the genome, and their predicted sequence can be considered as full-length. Therefore, it is expected that this genome would serve as a tool to further investigations of global change in one of the most stressed marine regions in the planet.
1 Introduction
Genome projects are an essential tool for multiple disciplines. In evolutionary sciences, having whole genomes sequenced helps to understand the relationships between different species and taxonomic groups (e.g. Kautt et al., 2020), to identify episodes of hybridization (Harrison et al., 2017) or selection processes (Fuentes-Pardo and Ruzzante, 2017), for example. The genome-wide variation at SNP is used to determine the number of populations within a species (Supple & Shapiro, 2018), or to estimate the connectivity between populations and subpopulations (Grummer et al., 2019) amongst other applications of importance for species and population management and conservation. This is crucial in marine species that are very difficult to monitor using conventional sighting or tagging methods (Yan et al., 2021); for example, the population units of yellowfin tuna in African waters were discovered only when whole-genome variation was analyzed (Mullins et al., 2018). Overfishing of target and by-catch species is affecting many stocks (Cochrane, 2021; Yan et al., 2021). Thus the identification of recent bottlenecks and robust estimates of effective population size based on whole-genome variation (Morin et al., 2021; Atmore et al., 2022) are necessary.
A new application of genomic analysis could be the prediction of the effects of global change, which is essential to design timely measures to ensure a species is managed in a way compatible with its resilience in a changing world (Sumaila & Tai, 2020). A greater awareness of the impacts of global change is urgently needed to ensure sustainable marine fisheries (Cochrane, 2020). Global change encompasses accelerated climate warming and pollution by different substances that can be very intense in some regions. For this, knowing the genes that respond to different environmental challenges, and their variation at a population level, is very important (Hansen et al., 2012). The expression of these genes, their modulation, and signals of selection at a DNA level (e.g. genome scans), will indicate how the species is responding to the environmental changes in a region (Stillman & Armstrong, 2015; Cline et al., 2020; Beemelmanns et al., 2021).
Being marine diversity particularly vulnerable to global change (Cheung et al., 2009), having the complete sequence of genomes of key species inhabiting marine regions most affected by global change is especially important. The west coast of Africa is an example. The Gulf of Guinea, located in the Equator, is a contamination hotspot that include leaks from oil pipelines and catastrophic oil spills (Najoui et al., 2022), heavy metals from waste dumping (Steinhausen et al., 2022) and mining (Garcia-Vazquez et al., 2021), and emerging pollutants like microplastics (Masiá et al., 2022). Fishing resources are very important there; the local populations rely on fish for protein supply, and will suffer from stock declines predicted under global change (Golden et al., 2016). The availability of reference genomes of marine fish exploited in that region seems especially important to understand both the effects of global change on marine species and the resource value. Accessible reference genomes allow the detection of signals of evolutionary selection, and of regions of reduced genetic diversity that are signals of high risk of species extinction (Zoonomia Consortium, 2020).
Despite recent efforts such as 10K Genome consortium (Rhie et al., 2021) and the 10K fish genomes (Fan et al., 2020), there are many gaps in fish reference genomes, both geographical and taxonomic. We will focus on a group of high economic interest that comprises many species exploited by fisheries, like cods and hakes: the Gadids. For reference genomes, within this group some families are quite well represented (Figure 1), like the Gadidae family which is in the upper right part of the plot. This family contains the Atlantic cod Gadus morhua. There is a noticeable direct relationship between the economic importance of each family and the number of species with an assembled genome. However, there are two clear outliers, the Melanonidae and Merlucciidae families. The regression line in Figure 1 shows this trend without the two outliers (R2 = 0.75, p-value=0.025). The Merlucciidae family, which is second in importance for fisheries for the abundant catch of Merluccius species, is underrepresented with only three species that have been sequenced to date whose assemblies are highly fragmented.
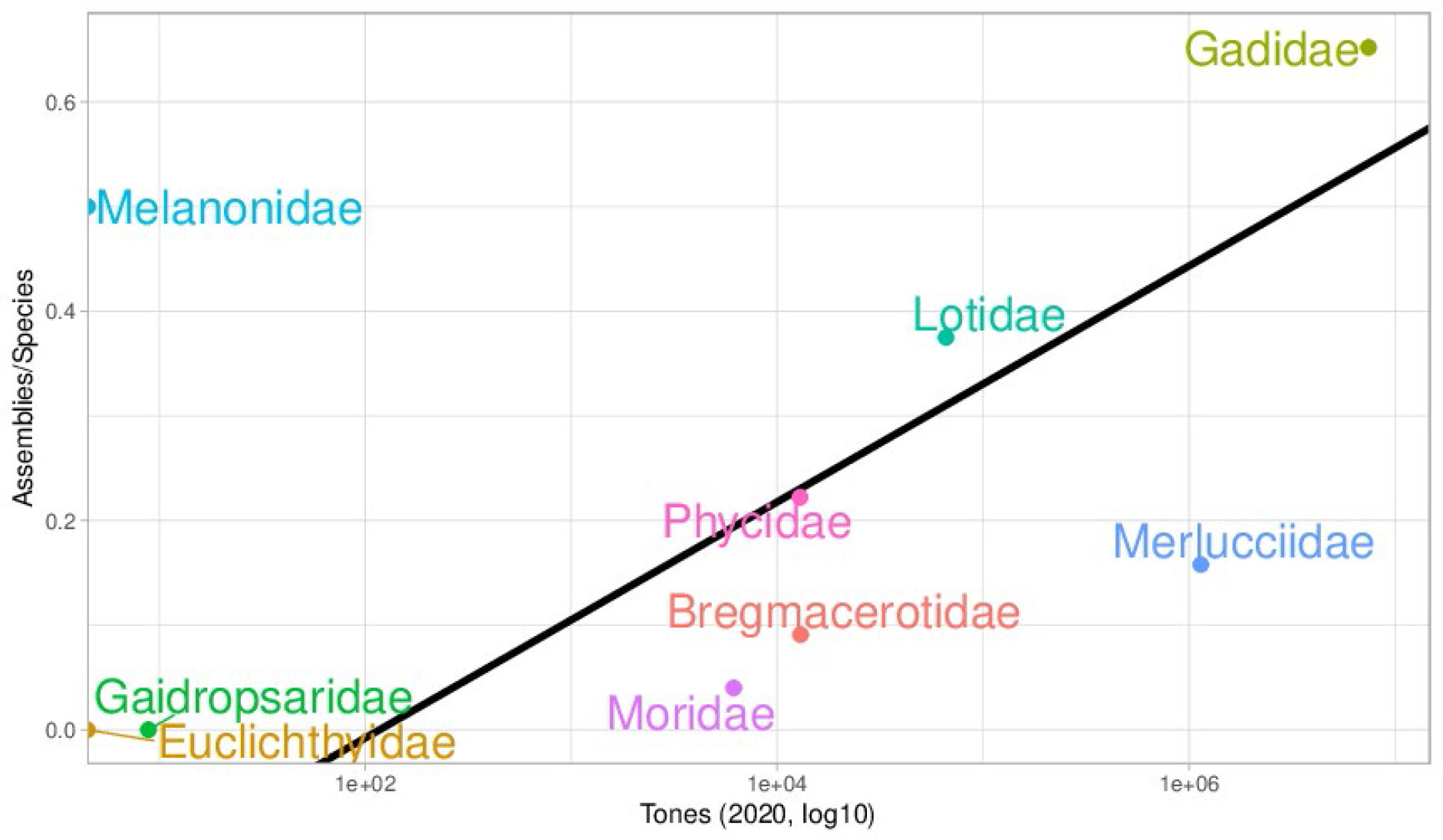
Figure 1 Chart showing the relationship between the global production in 2020 (https://www.fao.org/fishery/statistics-query/en/capture) and the number of assemblies per species of each family (https://www.ncbi.nlm.nih.gov/assembly/). The black line depicts the linear regression model.
When completely assembled genomes are not available, which is frequent in non-model marine fish, genome scans on candidate genes/genomic regions can be employed as a proxy for understanding responses to global change. Merluccius polli presents a wide latitudinal distribution, thus, being exposed to a changing temperature gradient and to a variety of contaminants – at least in part of its distribution, with the Gulf of Guinea at the centre of its distribution. Furthermore, at the southernmost end of its range, Benguela Niño causes anoxic periods that have been associated with spawning alterations of other African Merluccius species (Miralles et al., 2014). Due to these characteristics, Merluccius polli can be a great asset for the study of adaptive responses to global change. Therefore, the aim of the present study is to sequence, assemble and annotate the genome of Merluccius polli. Additionally, we aim to identify a set of candidate genes known to respond to different stressors happening in its distribution, for this genome to serve as a tool to further investigations of global change in one of the most stressed marine regions in the planet. Before this study, only three genomes were available for species of the genus Merluccius: M. capensis, M. merluccius and M. polli, all with relatively short assembled sequences, being the longest shorter than 70,000 base pairs long.
2 Materials and methods
2.1 DNA extraction and library preparation
Samples of Merluccius polli were collected from the waters of Mauritania (18°18’N, 16°40’W) and stored in absolute ethanol at 4°C. The morphological identification by experts was later validated by PCR amplification and Sanger sequencing of one mitochondrial (Control Region) and one nuclear (5s rDNA) marker. Later, 1µg of genomic DNA was extracted from fin tissue of a sole individual of Merluccius polli using a phenol:chloroform based protocol. The resulting DNA extraction was purified by Mag- Bind® Total Pure NGS magnetic beads. The library was then submitted to a quality control check: DNA concentration was measured with Qubit 2.0 Fluorometer, and the integrity of the sample was checked in Bioanalyzer DNA 12000 chip. (DIN value = 8.3).
2.2 Sequencing strategy, annotation and bioinformatic pipeline
Sequencing was carried out combining Illumina NovaSeq and PacBio Sequel II sequencing in Macrogen.Inc, South Korea. SMRT libraries were prepared and subsequently sequenced with PacBio Sequel II, generating long subreads of a targeted insert size between 10 and 20 kb. PacBio subreads were assembled using Hierarchical Genome Assembly Process (HGAP 4) and default settings (Chin et al., 2013) for an estimated genome size of 534 Mb. A second library of paired-end 150 bp Illumina short reads was sequenced to correct consensus sequence generated from the PacBio data. Illumina reads with a phred score below 30 were filtered. The assemblage was corrected using Pilon v1.21 (Walker et al., 2014). K-mer analysis was employed to estimate the genome size. CDS from close species (i.e., Gadus morhua, Myripristis murdjan, Acanthopagrus latus, Archocentrus centrarchus and Anabas testudineus) were downloaded from GenBank (GCF_902167405.1, GCF_902150065.1, GCF_904848185.1, GCF_007364275.1, GCF_900324465.2) and used to make a training model using SNAP v2.31.8. Then, gene model prediction was carried out by Maker (Holt & Yandell, 2011) v2.31.8 and predictions = 1 were removed to avoid false positives. Protein prediction sets were also tested using InterProScan v5.30-69.0 (Jones et al., 2014) and psiblast v2.4.0 (Camacho et al., 2009) with EggNOG DB v4.5 (Huerta-Cepas et al., 2016). Finally, the level of completion of the genome was assessed using Benchmarking Universal Single-Copy Orthologs software (BUSCO) version 5.1.2 (Manni et al., 2021).
2.3 Candidate gene selection
We conducted a bibliographic search using various search engines (i.e. Google Scholar, Web of Science) to select a set of candidate genes that, according to current scientific literature, are associated with changes in environmental factors linked to global change occurring along the distribution of Merluccius polli. Such association may be evidenced from correlational studies based on SNP, experiments for gene functions and/or other signals. The environmental factors considered were: temperature, heavy metal pollution, hypoxia, depth, and heavy metals concentration. Temperature is indeed the key factor in current climate change, and many studies describe genes involved in fish adaptation to changes in temperature (e.g. Hori et al., 2010; Dietrich et al., 2018; Lou et al., 2022). Heavy metals are main pollutants in West African waters, as explained above. The genomic mechanisms of defense of Teleostean fish against these pollutants have been described in Gadids (Olsvik et al., 2011) as well as in other species (Kim et al., 2016; Eide et al., 2021). Hypoxia is known to happen associated with the Benguela regime shift (Hutchings et al., 2009) and Benguela Niño (Monteiro et al., 2008) at the south of M. polli distribution. The genes involved in responses to hypoxia have been also described in marine fish (Tiedke et al., 2014; Xin et al., 2022). Finally, fish species change depth as a response to escape adverse conditions caused by climate change (Thresher et al., 2007; Dulvy et al., 2008), and those changes encompass a variety of physiological and genomic responses (Brown & Thatje, 2014).
A candidate gene was selected when a relation between such gene and any of the aforementioned environmental factors had been previously established on Teleosts in the literature. Genes found in phylogenetically close species (i.e. Gadiformes) were prioritized, but any Teleostean was considered a valuable example. Data were collected on the name of the gene, gene family, target factor/s to which the gene was associated, species where it was found, NCBI accession number and bibliographic reference.
To identify these candidate genes on the M. polli genome, first they were searched in the reference genes from Gadus morhua or, in case that one gene could not be found, from Danio rerio, in both cases from Ensembl version 107 (Cunningham et al., 2022). In the case that the candidate gene was identified in the source publication with a valid accession code or the proper sequence, the corresponding gene in Gadus morhua or Danio rerio was assigned by the best alignment result (the highest bit-score and lowest e-value) against the annotated protein sequences. The alignment was performed using the tool glsearch from the FASTA suite version 36 (Pearson and Lipman, 1988) taking as subject the sequence of the candidate gene using as a threshold an e-value of 1e-15.
For the genes that had obsolete accession codes or were not found in the original publication, the corresponding gene in Gadus morhua or Danio rerio was searched by gene symbol. Next, to identify the orthologous gene in M. polli, OrthoFinder (Emms and Kelly, 2019) version 2.5. was run with default settings, specifying the parameter –f with the folder containing the protein sequences of the following species retrieved from Ensembl version 107: Danio rerio, Gadus morhua, Oreochromis niloticus, Oryzias latipes, Salmo salar and M. polli.
2.4 Assessment of candidate genes and genome completeness
The candidate genes found in M. polli genome were classified by their response to environmental factors (according to literature as explained above) in four categories: responsive to temperature, depth, hypoxia, and exposure to heavy metals. Some of them may be classed in more than one category because, as part of the fish defensome, they may respond to more than one stressor (Eide et al., 2021), like regulatory transcription factors, antioxidant proteins and others.
To compute the proportion of target protein covered by its orthologous in Gadus morhua or Danio rerio, a global alignment using the Needleman-Wunsch algorithm was performed, and the proportion was defined as one minus the ratio between the number of deletions and the length of the target.
Additionally, we selected five GO functions expected to be involved in responses to the selected factors (GO:0003774, cytoskeletal motor activity; GO:0051015, actin filament binding; GO:0019825, oxygen binding; GO:0046872, metal ion binding; GO:0001666, response to hypoxia; GO:0009408, response to heat) to assess completion of our annotation in relation to functions that may potentially be linked to responses to global change. For this purpose, we searched genes associated to these GO terms in Danio rerio and Gadus morhua and matched those to our annotation of M. polli genome.
2.5 Heterozygosity
The Illumina reads generated in this study were mapped to the M. polli assembly with bwa version 0.7.17 (Li, 2013) with default parameters. The SNP calling was done using bcftools version 1.11 (Danecek et al., 2021) and default settings. The resulting SNPs were filtered so that SNPs with at least 15 reads and no more than 90 reads were considered. These values correspond to one third and twice the average coverage of the Illumina reads in the assembly.
2.6 Repeat content
RepeatModeler version 2.0.3 (Flynn et al., 2020) was used to build the repeat library of the M. polli assembly with the option –LTRStruct. Then, RepeatMasker version 4.1.3 was used with the Dfam database version 3.6 to scan the assembly.
3 Results
3.1 New Merluccius polli reference genome
This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession JAOPHQ000000000. The version described in this paper is version JAOPHQ010000000; organism Merluccius polli C29. This new assembly was built from a combination of PacBio and Illumina sequencing data. A total of 7,349,474 subreads were obtained using PacBio Sequel sequencing accounting for 81,293,939,445 bases with a subread N50 of 16,960 (SRR21859253). From Illumina NovaSeq sequencing 157,168,555 pairs-end reads were obtained initially (SRR21859252), and after filtering the final number were 86,964,178. The details of the assembly produced are shown in Table 1, in comparison to available assemblies to date of the genus Merluccius.
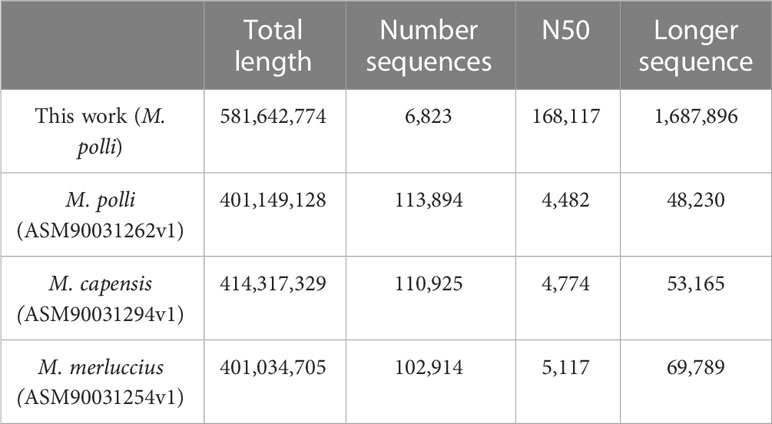
Table 1 Description of the assembly presented in this work, together with available to date of the genus Merluccius.
The total length of this new assembly is approximately 40% longer than previous ones (Table 1). However, the most significant improvement is the level of fragmentation, which is highly reduced. The number of total sequences is down to 6,823 from more than one hundred thousand, and the statistic N50 clearly shows that the majority of the new scaffolds are longer than 160kb while in the previous assemblies, even the longer sequence was not beyond 70kb.
In order to assess the completeness of the assembly, the tool BUSCO (Manni et al., 2021) was used against the Actinopterygii lineage. The result (Table 2) shows that more than 78% of the gene groups searched could be found in the assembly, with most of them complete.
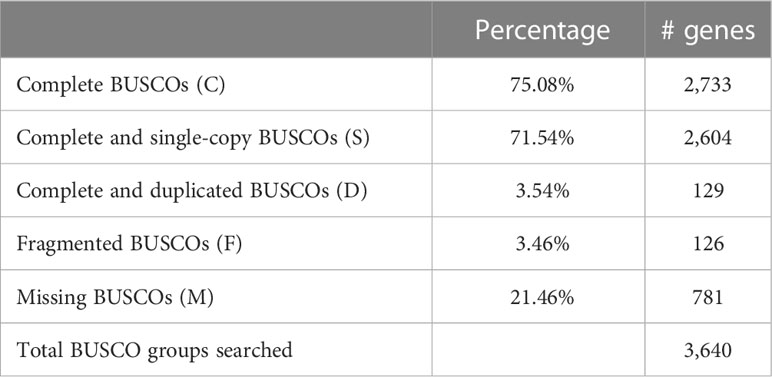
Table 2 Analysis of assembly completeness against Actinopterygii.
The annotation carried out with Maker (Holt & Yandell, 2011) revealed a total of 26,143 genes (Supplementary Table 1).
3.2 Candidate genes potentially affected by global change
The list of candidate genes is available in Supplementary Table 2. We identified 42, 28, 37 and 28 candidates related with the response of Teleostean fish to depth changes, exposure to heavy metals, temperature changes and hypoxia, respectively. It is worth noting that some of the candidate genes were associated with more than one factors. A total of 68 of those candidate genes were located in the new reference genome of M. polli (Supplementary Table 2): 26 for depth (62% of the potential candidates), 19 for heavy metals (67.8%), 23 for temperature (62.2%) and 18 for hypoxia (64.3%).
One of the main genes involved in heavy metal responses, the metallothionein, was not found according to the annotation tool used. However, taking as a template the nucleotide sequence of this gene from Gadus morhua and using the tool blat (Kent, 2002), it is possible to identify this gene in the new M. polli in contig3709 (see the sequence in Supplementary Table 3). Other metal-responsive genes associated with heavy metals were indeed found, as well as genes of response to oxidative stress that are expressed in response to heavy metal pollution in other fish (Supplementary Table 2).
Furthermore, the comparison of presence of genes from GO functions GO:0003774, GO:0051015, GO:0019825, GO:0046872, GO:0001666, and GO:0009408 between Danio rerio and Gadus morhua and our genome showed different percentage of representativeness among the different functions (Supplementary Figure 1). GO function representativeness was overall lower for the comparison between Danio rerio and M. polli (averaging at 53.1%) than against Gadus morhua (averaging at 76.8%). This is to be expected due to the phylogenetic distance between species, as well as for the higher completion of Danio rerio genome annotation, as this species is widely used as a model organism.
From the list of candidate genes not only the majority (68 out of 109) of them can be found in this assembly, but also the predicted sequence can be considered as full-length (Figure 2), taking as reference the protein sequence of G. morhua or Danio rerio used as target. The proportion of the target sequence that is covered by the identified sequence in M. polli is greater than 0.90 in 70% of them. This shows that, although the annotation was based on predictions without experimental data, the genes found are generally not fragmented.
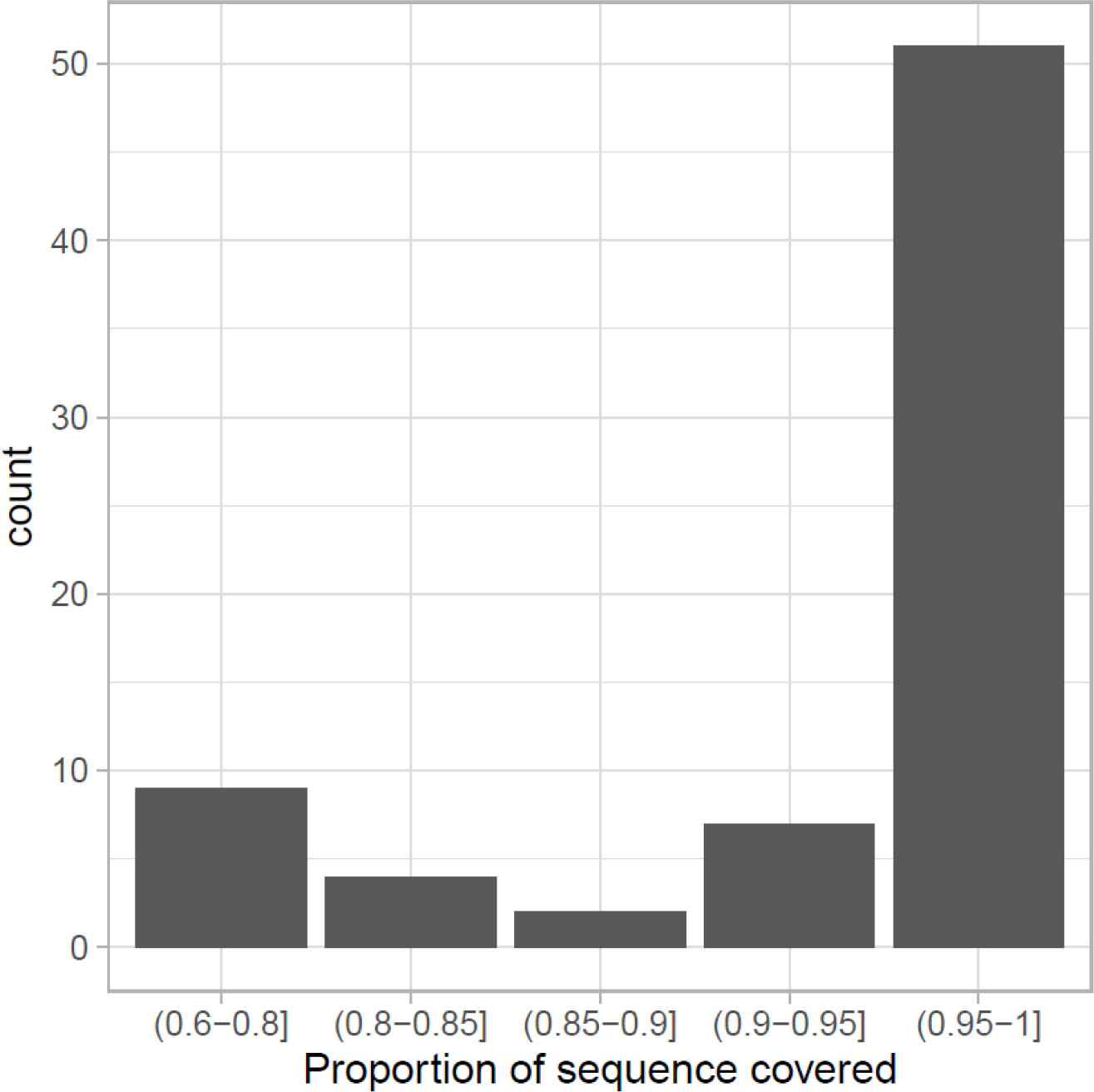
Figure 2 Candidate genes that can be considered as full-length in the newly assembled Merluccius polli genome or that cover most of the sequence of the corresponding genes in Gadus morhua or Danio rerio.
3.3 Repeat content
The repeat content of this assembly is 9.24%. The amount of each type of repeats is shown in Table 3. Of those, simple repeats are the most represented with over 7% of the genome. From interspersed repeats, that account 1% of the genome, LINEs and LTRs cover 0.5% and 0.26%, respectively. These values are much lower than related species. For example, in the Atlantic cod, gadMor2 assembly (Tørresen et al., 2017), the percentage of interspersed repeats is 23%. However, the difference is lower when looking at LINEs and LTRs where the proportion of the cod genome is 2.86% and 3.47%, respectively.
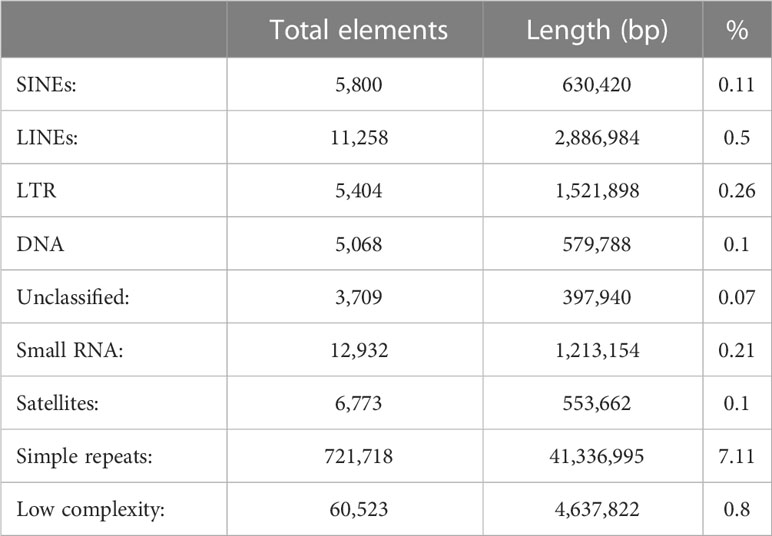
Table 3 Repeat content of the assembly.
3.4 Heterozygosity
Taking advantage of the Illumina short reads generated in this study, the level of heterozygosity of the M. polli assembly was assessed. Using the standard pipeline of bcftools (Danecek et al., 2021) 680,445 heterozygous SNPs were identified, what means a heterozygosity rate of 1.16 SNPs/Kb. These rates of heterozygosity, while slightly lower than what was found in other marine fishes fall within the expected range. Other species, namely Gadus morhua (Star et al., 2011) or Larimichthys crocea (Wu et al., 2014) have higher heterozygosity rates (2.09 SNPs/Kb and 3.58 SNPs/Kb respectively). Lower heterozygosities are found in some species of sharks: both the great hammerhead and the whale shark present very low heterozygosity rates (0.53 SNPs/Kb and 0.65 SNPs/Kb respectively) (Stanhope et al., 2023). Although these heterozygosity estimates come from a single individual, this represents the first estimate of genomic variation in a species from the genus Merluccius.
4 Discussion
The new genome of M. polli assembled in this study is a promising tool for the investigation of the effects of global change in the stressed West African waters. Genomic resources for this purpose are much needed because the region is subjected to many stresses intrinsic to global change. With at least 20 candidate genes to investigate the effects of each stressor considered, the species could be a good model to understand how marine species are responding to current global change.
Another effect of climate change is the increase of interspecific hybridization as long as species move to higher latitudes (Hoffmann and Sgrò, 2011; Potts et al., 2014; Muhlfeld et al., 2017). The fact that M. polli overlaps with other species of the same genus: M. capensis at the south and M. senegalensis at the north of its distribution (Figure 3), together with the fact that hybridization has been found between sympatric Merluccius species (Machado-Schiaffino et al., 2010), it seems possible that the likely expansion of M. polli to higher latitudes encompasses interspecific hybridization. The new reference assembly is much less fragmented, therefore, the longer scaffolds provides the necessary flanking sequences around the genes for robust analyses such as genome scans, allowing also to infer the potential role of hybridization, followed by adaptive introgression, as a response to global change.
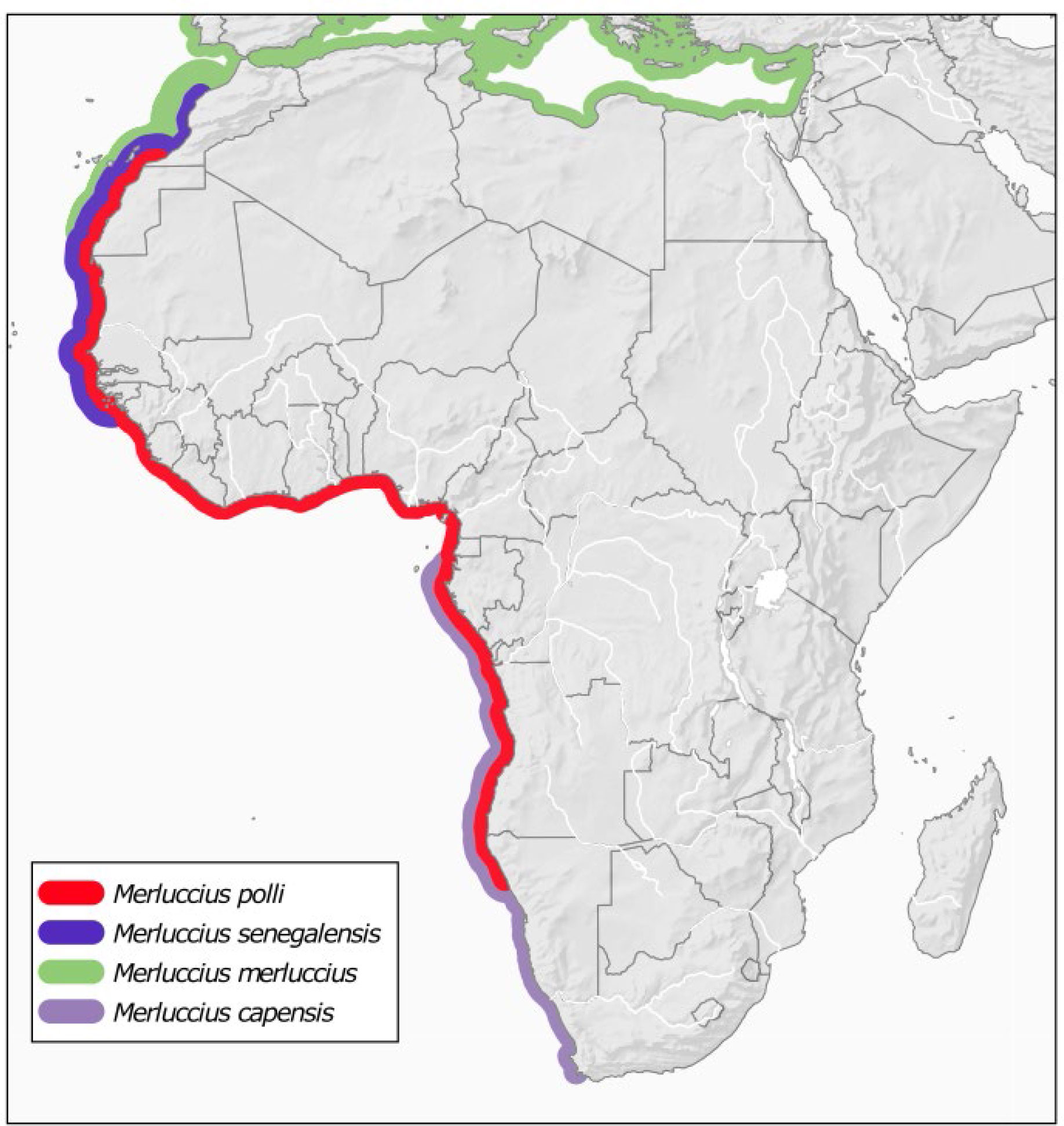
Figure 3 Distribution of Benguela hake M. polli and overlapping species of the genus Merluccius. Species distributions were taken from Pitcher and Alheit (1995). M. polli and M. senegalensis northern limits are updated as reported in Manchih et al. (2018).
Regarding the low proportion of repeat content in this genome, no evidence seems to indicate that this is due to an intrinsic feature of M. polli genome, and could be attributed to an artefact caused by the sequencing. Unfortunately, the available genomes from the other Merluccius species are not complete enough to compare with. In any case, it seems to exist a positive relation between the size of genome and the proportion of repeat contents in fishes (Yuan et al., 2018). Therefore, the proportion of repeated elements in M. polli hake, much lower than those found for other teleosts such as medaka (17.5% of 700 Mb; Kasahara et al., 2007) or Atlantic cod (25.4% of 830 Mb; Star et al., 2011), might be one of the reasons why Benguela hake has a smaller-size genome (~584Mb) than those species.
We have observed low levels of heterozygosity in Merluccius polli as compared to other fish. While low heterozygosity may sometimes be associated with endangered or declining species (Stanhope et al., 2023), this does not seem to be the case for M. polli in particular, since it has been reported to be growing at least in the northernmost part of its distribution (Manchih et al., 2018), where this particular individual was taken from.
The majority of candidate genes found in the genome of M. polli seem to be related with only one of the factors considered; for example, the gene prdx3 coding for peroxiredoxin 3 (putative locus 026202-RA in M. polli genome) responds to warmer temperatures in the Antarctic emerald rockcod Trematomus bernacchii (Tolomeo et al., 2019), but we found no references of this gene to be involved in fish responses to depth, hypoxia or heavy metals in our search (Supplementary Table 1). However, 14 of them (corresponding to 18 loci since four were duplicated) were reported to respond to several of the considered stressors in fish, being therefore good candidates to assess responses to global change from a faster and more economical methods, targeting a reduced number of SNPs or a small number of genes, if needed (Supplementary Table 1). As an example, the gene HbB2 (putative loci 009158-RA and 029449-RA in M. polli genome) coding for the subunit beta-2 of the hemoglobin is involved in the response to depth, temperature, and hypoxia in other Gadids (Bradbury et al., 2010; Baalsrud et al., 2017; Pan et al., 2017; Hahn et al., 2017). While the number of sequences from candidate genes retrieved directly from the literature is relatively low (40.6% of them found in M. polli genome), this is to be expected due to many of the studies dealing with the response to the four environmental factors considered are carried out in species where reference genomes are not available, therefore, only data about relative expression (e.g. qPCR, microarrays) are reported for several candidate genes and no sequences to compare are provided, therefore these genes had to be searched by name instead.
An added value of this new reference genome is that M. polli is an African species. The species inhabiting African regions are scarcely represented in genome projects. This is partially due to the unequal contribution of the different continents to the generation of genomics resources, which are still a challenge for many African countries (Adebamowo et al., 2018). For example, in 2021 it was only one land plant assembly generated in Africa in contrast to 235 assemblies from China, 2012 from the USA or 168 from Europe (Marks et al., 2021). Although the genome presented here was generated in Europe, following recommended good practices for marine genetics resources (Saeedi et al., 2019), it is publicly available for all the researchers worldwide.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
JM, originated the idea, conducted data analysis and write-up. CB-F, originated the idea, conducted lab duties, data analysis and write-up. EG-V, originated the idea, funding acquisition, lead the research design, analysis and write-up. GM-S, originated the idea, funding acquisition, led the research design, supervised the project, contributed to the analysis and write-up. All authors contributed to interpreting the results. All authors contributed to the article and approved the submitted version.
Funding
This study has been supported by the Spanish project GLOBALHAKE, reference PID2019-108347RB-I00, and the Government of Asturias Principality, Grant AYUD/2021/50967.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1111107/full#supplementary-material
References
Adebamowo S. N., Francis V., Tambo E., Diallo S. H., Landouré G., Nembaware V., et al. (2018). Implementation of genomics research in Africa: challenges and recommendations. Global Health Action 11 (1), 1419033. doi: 10.1080/16549716.2017.1419033
Atmore L. M., Martínez-García L., Makowiecki D., André C., Lõugas L., Barrett J. H., et al. (2022). Population dynamics of Baltic herring since the Viking age revealed by ancient DNA and genomics. Proc. Natl. Acad. Sci. 119 (45), e2208703119. doi: 10.1073/pnas.2208703119
Baalsrud H. T., Voje K. L., Tørresen O. K., Solbakken M. H., Matschiner M., Malmstrøm M., et al. (2017). Evolution of hemoglobin genes in codfishes influenced by ocean depth. Sci. Rep. 7, 7956. doi: 10.1038/s41598-017-08286-2
Beemelmanns A., Zanuzzo F. S., Xue X., Sandrelli R. M., Rise M. L., Gamperl A. K. (2021). The transcriptomic responses of Atlantic salmon (Salmo salar) to high temperature stress alone, and in combination with moderate hypoxia. BMC Genomics 22 (1), 1–33. doi: 10.1186/s12864-021-07464-x
Bradbury I. R., Hubert S., Higgins B., Borza T., Bowman S., Paterson I. G., et al. (2010). Parallel adaptive evolution of Atlantic cod on both sides of the Atlantic ocean in response to temperature. Proc. R. Soc. B 277, 3725–3734. doi: 10.1098/rspb.2010.0985
Brown A., Thatje S. (2014). Explaining bathymetric diversity patterns in marine benthic invertebrates and demersal fishes: physiological contributions to adaptation of life at depth. Biol. Rev. 89, 406–426. doi: 10.1111/brv.12061
Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., et al. (2009). BLAST+: architecture and applications. BMC Bioinf. 10, 1–9. doi: 10.1186/1471-2105-10-421
Cheung W. W., Lam V. W., Sarmiento J. L., Kearney K., Watson R., Pauly D. (2009). Projecting global marine biodiversity impacts under climate change scenarios. Fish Fisheries 10, 235–251. doi: 10.1111/j.1467-2979.2008.00315.x
Chin C. S., Alexander D. H., Marks P., Klammer A. A., Drake J., Heiner C., et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569. doi: 10.1038/nmeth.2474
Cline A. J., Hamilton S. L., Logan C. A. (2020). Effects of multiple climate change stressors on gene expression in blue rockfish (Sebastes mystinus). Comp. Biochem. Physiol. Part A: Mol. Integr. Physiol. 239, 110580. doi: 10.1016/j.cbpa.2019.110580
Cochrane K. L. (2021). Reconciling sustainability, economic efficiency and equity in marine fisheries: has there been progress in the last 20 years? Fish Fisheries 22, 298–323. doi: 10.1111/faf.12521
Cunningham F., Allen J. E., Allen J., Alvarez-Jarreta J., Amode M. R., Armean I. M., et al. (2022). Ensembl 2022. Nucleic Acids Res. 50 (D1), D988–D995. doi: 10.1093/nar/gkab1049
Danecek P., Bonfield J. K., Liddle J., Marshall J., Ohan V., Pollard M. O., et al. (2021). Twelve years of SAMtools and BCFtools. GigaScience 10 (2), giab008. doi: 10.1093/gigascience/giab008
Dietrich M. A., Hliwa P., Adamek M., Steinhagen D., Karol H., Ciereszko A. (2018). Acclimation to cold and warm temperatures is associated with differential expression of male carp blood proteins involved in acute phase and stress responses, and lipid metabolism. Fish Shellfish Immunol. 76, 305–315. doi: 10.1016/j.fsi.2018.03.018
Dulvy N. K., Rogers S. I., Jennings S., Stelzenmüller V., Dye S. R., Skjoldal H. R. (2008). Climate change and deepening of the north Sea fish assemblage: a biotic indicator of warming seas. J. Appl. Ecol. 45, 1029–1039. doi: 10.1111/j.1365-2664.2008.01488.x
Eide M., Zhang X., Karlsen O. A., Goldstone J. V., Stegeman J., Jonassen I., et al. (2021). The chemical defensome of five model teleost fish. Sci. Rep. 11, 10546. doi: 10.1038/s41598-021-89948-0
Emms D. M., Kelly S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238. doi: 10.1186/s13059-019-1832-y
Fan G., Song Y., Yang L., Huang X., Zhang S., Zhang M., et al. (2020). Initial data release and announcement of the 10,000 fish genomes project (Fish10K). GigaScience 9 (8), giaa080. doi: 10.1093/gigascience/giaa080
Flynn J. M., Hubley R., Goubert C., Rosen J., Clark A. G., Feschotte C., et al. (2020). RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. U.S.A. 117 (17), 9451–9457. doi: 10.1073/pnas.1921046117
Fuentes-Pardo A. P., Ruzzante D. E. (2017). Whole-genome sequencing approaches for conservation biology: advantages, limitations and practical recommendations. Mol. Ecol. 26, 5369–5406. doi: 10.1111/mec.14264
Garcia-Vazquez E., Geslin V., Turrero P., Rodriguez N., Machado-Schiaffino G., Ardura A. (2021). Oceanic karma? eco-ethical gaps in African EEE metal cycle may hit back through seafood contamination. Sci. Total Environ. 762, 143098. doi: 10.1016/j.scitotenv.2020.143098
Golden C. D., Allison E. H., Cheung W. W. L., Dey M. M., Halpern B. S., McCauley D. J., et al. (2016). Fall in fish catch threatens human health. Nature 534, 317–320. doi: 10.1038/534317a
Grummer J. A., Beheregaray L. B., Bernatchez L., Hand B. K., Luikart G., Narum S. R., et al. (2019). Aquatic landscape genomics and environmental effects on genetic variation. Trends Ecol. Evol. 34 (7), 2019641–654. doi: 10.1016/j.tree.2019.02.013
Hahn C., Genner M. J., Turner G. F., Joyce D. A. (2017). The genomic basis of cichlid fish adaptation within the deepwater “twilight zone” of lake Malawi. Evol. Lett. 1, 184–198. doi: 10.1002/evl3.20
Hansen M. M., Olivieri I., Waller D. M., Nielsen E. E., The GeM Working Group (2012). Monitoring adaptive genetic responses to environmental change. Mol. Ecol. 21, 1311–1329. doi: 10.1111/j.1365-294X.2011.05463.x
Harrison H., Berumen M., Saenz-Agudelo P., Salas E., Williamson D., Jones G. (2017). Widespread hybridization and bidirectional introgression in sympatric species of coral reef fish. Mol. Ecol. 26, 5692–5704. doi: 10.1111/mec.14279
Hoffmann A. A., Sgrò C. M. (2011). Climate change and evolutionary adaptation. Nature 470 (7335), 479–485. doi: 10.1038/nature09670
Holt C., Yandell M. (2011). MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinf. 12, 491. doi: 10.1186/1471-2105-12-491
Hori T. S., Gamperl A. K., Afonso L. O. B., Johnson S. C., Hubert S., Kimball J., et al. (2010). Heat-shock responsive genes identified and validated in Atlantic cod (Gadus morhua) liver, head kidney and skeletal muscle using genomic techniques. BMC Genomics 11, 1–22. doi: 10.1186/1471-2164-11-72
Huerta-Cepas J., Szklarczyk D., Forslund K., Cook H., Heller D., Walter M. C., et al. (2016). eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 44 (D1), D286–D293. doi: 10.1093/nar/gkv1248
Hutchings L., van der Lingen C. D., Shannon L. J., Crawford R. J. M., Verheye H. M. S., Bartholomae C. H., et al. (2009). The benguela current: an ecosystem of four components. Prog. Oceanogr. 83, 15–32. doi: 10.1016/j.pocean.2009.07.046
Jones P., Binns D., Chang H. Y., Fraser M., Li W., McAnulla C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Kasahara M., Naruse K., Sasaki S., Nakatani Y., Qu W., Ahsan B., et al. (2007). The medaka draft genome and insights into vertebrate genome evolution. Nature 447, 714. doi: 10.1038/nature05846
Kautt A. F., Kratochwil C. F., Nater A., Machado-Schiaffino G., Olave M., Henning F., et al. (2020). Contrasting signatures of genomic divergence during sympatric speciation. Nature 588 (7836), 106–111. doi: 10.1038/s41586-020-2845-0
Kent W. J. (2002). BLAT —the BLAST -like alignment tool. Genome Res. 12 (4), 656–664. doi: 10.1101/gr.229202
Kim Y. J., Lee N., Woo S., Ryu J. C., Yum S. (2016). Transcriptomic change as evidence for cadmium-induced endocrine disruption in marine fish model of medaka, oryzias javanicus. Mol. Cell. Toxicol. 12, 409–420. doi: 10.1007/s13273-016-0045-7
Li H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:1303.3997
Lou F., Liu M., Han Z., Gao T. (2022). Comparative transcriptome reveals the thermal stress response differences between heilongjiang population and xinjiang population of Lota lota. Comp. Biochem. Physiol. Part D: Genomics Proteomics 42, 100960. doi: 10.1016/j.cbd.2022.100960
Machado-Schiaffino G., Juanes F., Garcia-Vazquez E. (2010). Introgressive hybridization in north American hakes after secondary contact. Mol. Phylogenet. Evol. 55, 552–558. doi: 10.1016/j.ympev.2010.01.034
Manchih K., Peralta L. F., Bensbai J., Najd A., Bekkali M. (2018). Distribution of black hakes merluccius senegalensis and merluccius polli along the Moroccan Atlantic coast. AACL Bioflux 11, 245–258.
Manni M., Berkeley M. R., Seppey M., Simão F. A., Zdobnov E. M. (2021). BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38 (10), 4647–4654. doi: 10.1093/molbev/msab199
Marks R. A., Hotaling S., Frandsen P. B., VanBuren R. (2021). Representation and participation across 20 years of plant genome sequencing. Nat. Plants 7, 1571–1578. doi: 10.1038/s41477-021-01031-8
Masiá P., Mateo J. L., Arias A., Bartolomé M., Blanco C., Erzini K., et al. (2022). Potential microplastics impacts on African fishing resources. Sci. Total Environ. 806 (2), 150671. doi: 10.1016/j.scitotenv.2021.150671
Miralles L., Machado-Schiaffino G., Garcia-Vazquez E. (2014). Genetic markers reveal a gradient of hybridization between cape hakes (Merluccius capensis and Merluccius paradoxus) in their sympatric geographic distribution. J. Sea Res. 86, 69–75. doi: 10.1016/j.seares.2013.11.009
Monteiro P. M. S., van der Plas A. K., Melice J. L., Florenchi P. (2008). Interannual hypoxia variability in a coastal upwelling system: ocean–shelf exchange, climate and ecosystem-state implications. Deep Sea Res. 55, 435–450. doi: 10.1016/j.dsr.2007.12.010
Morin P. A., Archer F. I., Avila C. D., Balacco J. R., Bukhman Y. V., Chow W., et al. (2021). Reference genome and demographic history of the most endangered marine mammal, the vaquita. Mol. Ecol. Resour. 21, 1008–1020. doi: 10.1111/1755-0998.13284
Muhlfeld C. C., Kovach R. P., Al-Chokhachy R., Amish S. J., Kershner J. L., Leary R. F., et al. (2017). Legacy introductions and climatic variation explain spatiotemporal patterns of invasive hybridization in a native trout. Global Change Biol. 23, 4663–4674. doi: 10.1111/gcb.13681
Mullins R. B., McKeown N. J., Sauer W. H. H., Shaw P. W. (2018). Genomic analysis reveals multiple mismatches between biological and management units in yellowfin tuna (Thunnus albacares). ICES J. Mar. Sci. 75 (6), 2145–2152. doi: 10.1093/icesjms/fsy102
Najoui Z., Amoussou N., Riazanoff S., Aurel G., Frappart F. (2022). Oil slicks in the gulf of Guinea – 10 years of envisat advanced synthetic aperture radar observations. Earth System Sci. Data 14, 4569–4588. doi: 10.5194/essd-14-4569-2022
Olsvik P. A., Brattås M., Lie K. K., Goksøyr A. (2011). Transcriptional responses in juvenile Atlantic cod (Gadus morhua) after exposure to mercury-contaminated sediments obtained near the wreck of the German WW2 submarine U-864, and from Bergen harbor, Western Norway. Chemosphere 83, 552–563. doi: 10.1016/j.chemosphere.2010.12.019
Pan Y. K., Ern R., Morrison P. R., Brauner C. J., Esbaugh A. J. (2017). Acclimation to prolonged hypoxia alters hemoglobin isoform expression and increases hemoglobin oxygen affinity and aerobic performance in a marine fish. Sci. Rep. 7, 7834. doi: 10.1038/s41598-017-07696-6
Pearson W. R, Lipman D. J. (1988). Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. U.S.A 85 (8), 2444–2448. doi: 10.1073/pnas.85.8.2444
Pitcher T. J., Alheit J. (1995). “What makes a hake? a review of the critical biological features that sustain global hake fisheries,” in Hake: biology, fisheries and markets. Eds. Alheit J., Pitcher T. J. (London: Chapman & Hall), 1–14.
Potts W., Henriques R., Santos C., Munnik K., Ansorge I., Dufois F., et al. (2014). Ocean warming, a rapid distributional shift and the hybridization of a coastal fish species. Global Change Biol. 20, 2765–2777. doi: 10.1111/gcb.12612
Rhie A., McCarthy S. A., Fedrigo O., Damas J., Formenti G., Koren S., et al. (2021). Towards complete and error-free genome assemblies of all vertebrate species. Nature 592, 737–746. doi: 10.1038/s41586-021-03451-0
Saeedi H., Reimer J. D., Brandt M. I., Dumais P., Jażdżewska A. M., Jeffery N. W., et al. (2019). Global marine biodiversity in the context of achieving the aichi targets: ways forward and addressing data gaps. PeerJ 7, e7221. doi: 10.7717/peerj.7221
Stanhope M. J., Ceres K. M., Sun Q., Wang M., Zehr J. D., Marra N. J., et al. (2023). Genomes of endangered great hammerhead and shortfin mako sharks reveal historic population declines and high levels of inbreeding in great hammerhead. Iscience 26 (1), 105815. doi: 10.1016/j.isci.2022.105815
Star B., Nederbragt A. J., Jentoft S., Grimholt U., Malmstrøm M., Gregers T. F., et al. (2011). The genome sequence of Atlantic cod reveals a unique immune system. Nature 477, 207–210. doi: 10.1038/nature10342
Steinhausen S. L., Agyeman N., Ardura A., Turrero P., Garcia-Vazquez E. (2022). Heavy metals in fish nearby WEEE may threaten consumer’s health. examples from Accra, Ghana. Mar. Pollut. Bull. 175, 113162. doi: 10.1016/j.marpolbul.2021.113162
Stillman J. H., Armstrong E. (2015). Genomics are transforming our understanding of responses to climate change. BioScience 65 (3), 237–246. doi: 10.1093/biosci/biu219
Sumaila U. R., Tai T. C. (2020). End overfishing and increase the resilience of the ocean to climate change. Front. Mar. Sci. 7. doi: 10.3389/fmars.2020.00523
Supple M. A., Shapiro B. (2018). Conservation of biodiversity in the genomics era. Genome Biol. 19 (1), 1–12. doi: 10.1186/s13059-018-1520-3
Tørresen O. K., Star B., Jentoft S., Reinar W. B., Grove H., Miller J. R., et al. (2017). An improved genome assembly uncovers prolific tandem repeats in Atlantic cod. BMC Genomics 18, 95. doi: 10.1186/s12864-016-3448-x
Thresher R. E., Koslow J. A., Morison A. K., Smith D. C. (2007). Depth-mediated reversal of the effects of climate change on long-term growth rates of exploited marine fish. Proc. Natl. Acad. Sci. 104 (18), 7461–7465. doi: 10.1073/pnas.0610546104
Tiedke J., Thiel R., Burmester T. (2014). Molecular response of estuarine fish to hypoxia: a comparative study with ruffe and flounder from field and laboratory. PloS One 9 (3), e90778. doi: 10.1371/journal.pone.0090778
Tolomeo A. M., Carraro A., Bakiu R., Toppo S., Garofalo F., Pellegrino D., et al. (2019). Molecular characterization of novel mitochondrial peroxiredoxins from the Antarctic emerald rockcod and their gene expression in response to environmental warming. Comp. Biochem. Physiol. Part C: Toxicol. Pharmacol. 225, 108580. doi: 10.1016/j.cbpc.2019.108580
Walker B. J., Abeel T., Shea T., Priest M., Abouelliel A., Sakthikumar S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS One 9 (11), e112963. doi: 10.1371/journal.pone.0112963
Wu C., Zhang D., Kan M., Lv Z., Zhu A., Su Y., et al. (2014). The draft genome of the large yellow croaker reveals well-developed innate immunity. Nat. Commun. 5, 5227. doi: 10.1038/ncomms622
Xin Y., Yang Z., Zhu Y., Li Y., Yu J., Zhong W., et al. (2022). Hypoxia induces oxidative injury and apoptosis via mediating the nrf-2/Hippo pathway in blood cells of largemouth bass (Micropterus salmoides). Front. Ecol. Evol. 10. doi: 10.3389/fevo.2022.841318
Yan H. F., Kyne P. M., Jabado R. W., Leeney R. H., Davidson L. N. K., Derrick D. H., et al. (2021). Overfishing and habitat loss drive range contraction of iconic marine fishes to near extinction. Sci. Adv. 7 (7), eabb6026. doi: 10.1126/sciadv.abb6026
Yuan Z., Liu S., Zhou T., Tian C., Bao L., Dunham R., et al. (2018). Comparative genome analysis of 52 fish species suggests differential associations of repetitive elements with their living aquatic environments. BMC Genomics 19, 141. doi: 10.1186/s12864-018-4516-1
Keywords: genome, global change, candidate genes, environmental challenges, Benguela hake
Citation: Mateo JL, Blanco-Fernandez C, Garcia-Vazquez E and Machado-Schiaffino G (2023) A new Merluccius polli reference genome to investigate the effects of global change in West African waters. Front. Mar. Sci. 10:1111107. doi: 10.3389/fmars.2023.1111107
Received: 29 November 2022; Accepted: 10 April 2023;
Published: 24 April 2023.
Edited by:
Fran Saborido-Rey, Spanish National Research Council (CSIC), SpainReviewed by:
Natalia Petit-Marty, Institute of Marine Research (CSIC), SpainQiuming Yao, University of Nebraska-Lincoln, United States
Copyright © 2023 Mateo, Blanco-Fernandez, Garcia-Vazquez and Machado-Schiaffino. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gonzalo Machado-Schiaffino, machadogonzalo@uniovi.es
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share senior authorship