 Peng-Fei Zhang1*†
Peng-Fei Zhang1*† Dan Xie2,3†
Dan Xie2,3†- 1Gastric Cancer Center, Division of Medical Oncology, Cancer Center, West China Hospital, Sichuan University, Chengdu, China
- 2Department of Medical Genetics, West China Second University Hospital, Sichuan University, Chengdu, China
- 3Key Laboratory of Birth Defects and Related Diseases of Women and Children (Sichuan University), Ministry of Education, Chengdu, China
Chimeric antigen receptor (CAR) -T cell therapy has achieved tremendous efficacy in the treatment of hematologic malignancies and represents a promising treatment regimen for cancer. Despite the striking response in patients with hematologic malignancies, most patients with solid tumors treated with CAR-T cells have a low response rate and experience major adverse effects, which indicates the need for biomarkers that can predict and improve clinical outcomes with future CAR-T cell treatments. Recently, the role of the gut microbiota in cancer therapy has been established, and growing evidence has suggested that gut microbiota signatures may be harnessed to personally predict therapeutic response or adverse effects in optimizing CAR-T cell therapy. In this review, we discuss current understanding of CAR-T cell therapy and the gut microbiota, and the interplay between the gut microbiota and CAR-T cell therapy. Above all, we highlight potential strategies and challenges in harnessing the gut microbiota as a predictor and modifier of CAR-T cell therapy efficacy while attenuating toxicity.
Introduction
Immunotherapy, which includes a variety of approaches, such as tumor vaccines, immune checkpoint inhibitors (ICIs), and adoptive cell therapy (ACT), has made tremendous advances in cancer treatment in recent decades (1–3). Chimeric antigen receptor T (CAR-T) cell therapy, as the most concerned treatment in immunotherapy, has impacted the history of cancer therapy and attracted increasing amount of attention worldwide (4). CARs are versatile synthetic receptors that are genetically engineered to express in T cells. CARs comprise an ectodomain with an antigen-binding module and a hinge, a transmembrane region, and costimulatory/activation domains (such as the signaling domains of CD3ζ, CD28 and/or CD137) (5, 6). Currently, CAR-T cell therapy is being increasingly studied in a variety of tumors, especially in patients with recurrent/refractory (R/R) B cell leukemia or lymphoma (7–10). To date, a series of CAR-T cell products, such as Kymriah, Yescarta, Tecartus and Breyanzi, have been approved for the treatment of R/R B cell leukemia or lymphoma (11–14). The unprecedented success of CAR-T cell therapy in treating hematological malignancies has sparked interest in broadening the application of CAR-T cell therapy in solid tumors. Currently, more than 200 clinical trials investigating CAR-T cells for the treatment of solid tumors have been launched around the world; however, the results of applying CAR-T cells to solid tumors have been disappointing. In most studies, only a few patients achieved partial response (PR) (15, 16). In addition to poor efficacy in solid tumors, several other disadvantages also limit the wide application of CAR-T cells. CAR-T cells therapy is often accompanied by self-reactivity mediated by on-target, off-tumor antigen expression, increased systemic inflammation leading to cytokine release syndrome (CRS), neurotoxicity, and suppression of humoral immunity due to B-cell aplasia (17–19). These disadvantages limit CAR-T cells from reaching their full therapeutic potential and therefore highlight the critical need for biomarkers that can predict and improve clinical outcomes with future CAR-T cell treatments.
The gut microbiota refers to the vast collection of microbes living in the gastrointestinal tract (20). With the development and wide application of novel molecular technologies (such as 16S ribosomal RNA sequencing, metagenomics and metabolomics), a multitude of preclinical and clinical studies have been performed to explore intricate host-microbiota interactions over the last decade (21). First, the gut microbiota plays a key role in several physiological processes, including immunity, metabolism, and the inflammatory response (22, 23). Second, gastrointestinal tract dysbiosis promotes the occurrence and progression of a variety of malignant tumors (24). Third, the gut microbiota has been demonstrated to affect the response to several anticancer therapeutics, such as chemotherapy, radiotherapy, and immune checkpoint blockade (ICB) (25, 26). In light of these findings, there is emerging interest in investigating the role of the gut microbiota in CAR-T cell therapy. For example, in a recent study, Smith et al. investigated the influence of the gut microbiota on the response and toxicity of hematologic malignancies to CD19 CAR-T cell therapy. A total of 228 patients treated with CD19 CAR-T cells were recruited and stratified based on exposure to broad-spectrum antibiotics, including piperacillin, imipenem or meropenem (PIM), within four weeks prior to the first treatment with CAR-T cells. Strikingly, patients exposed to broad-spectrum antibiotics had significantly shorter overall survival (OS), an increased incidence of immune effector cell-associated neurotoxicity syndrome (ICANS) and a trend toward an increased incidence of CRS compared to patients without broad-spectrum antibiotic exposure (27). In this review, we summarize the general aspects of CAR-T cell therapy and the gut microbiota, focus on the current understanding of the interplay between the gut microbiota and CAR-T cell therapy, and highlight the potential of microbiota-based prognostic prediction and gut microbiota-based biotherapy to potentiate CAR-T cell efficacy while attenuating toxicity.
CAR-T cell therapy
In recent years, CAR-T cell therapy has shown unprecedented antitumor efficacy and stands at the novel forefront of current cancer therapy (28). CAR-T cell therapy involves a series of complex processes, including genetically modified T cells to express CAR toward a tumor-specific antigen (TSA) or tumor-associated antigen (TAA) via viral and non-viral transfection methods. After in vitro T-cell amplification, CAR-T cells will be reinfused back into patients to eradicate tumors (29). CARs are synthetic receptors, and the basic structure of CARs contains an extracellular antigen-binding region that binds to the antigen (usually a single-chain variable fragment (scFv) of an antibody), a hinge region providing flexibility, a transmembrane region and an intracellular signal transduction region that activates the cytotoxic functions of CAR-T cells upon antigen recognition. Over the past decade, the design of CARs has progressed rapidly. To date, fourth generation CARs have been developed based on different intracellular signals (30). First-generation CARs contain only one CD3ζ signaling domain to stimulate cytotoxic CAR-T cell activity, which mimics the process of natural biologic T-cell activation. However, the clinical efficacy of first-generation CAR-T cells has been disappointing in the majority of early trials because of the short persistence and low expansion ability of CAR-T cells in vivo (31, 32). To solve this problem, second-generation CARs, which contain an additional costimulatory molecule, such as CD28, OX40 or 4-1BB, have been developed. The addition of a costimulatory signal domain can recapitulate the natural costimulation of T cells and significantly improve the antitumor ability of CAR-T cells (33, 34). To further enhance the antitumor effect of CAR-T cells, third-generation CARs that combine multiple costimulatory molecules have also been developed (35, 36). Fourth-generation CARs are designed to express more powerful weapons, such as cytokines, receptors for chemokines, and a controlled suicide gene. Compared with second- and third-generation CAR-T cells, fourth-generation CAR-T cells exhibit rapid expansion, high tumor killing activity, and obvious advantages in terms of safety and persistence (37–39). In the past decade, CAR-T cell therapy has shown remarkable efficacy in the treatment of patients with R/R hematological malignancies (7–10). Thus, several CAR-T cell medicines, such as CD19-targeted Tisagenlecleucel (Kymriah®), Axicabtagene ciloleucel (Yescarta®), brexucabtagene autoleucel (Tecartus®), lisocabtagene maraleucel (Breyanzi®), B cell maturation antigen (BCMA)-targeted idecabtagene vicleucel (Abecma®), and ciltacabtagene autoleucel (Carvykti®), have been approved by the U.S. Food and Drug Administration (FDA) and European Medicines Agency (EMA) for the treatment of hematological malignancies, including lymphomas, some forms of leukemia, and most recently for the treatment of multiple myeloma (MM) (11–14, 40, 41).
Inspired by the tremendous success of CAR-T cell therapy in hematological malignancies, researchers are gradually expanding the application of CAR-T cell therapy to the treatment of solid tumors. Over the years, a series of CAR-T cells targeting antigens in solid tumor cells, including carcinoembryonic antigen (CEA), epidermal growth factor receptor (EGFR), human epidermal growth factor receptor 2 (HER2), and prostate-specific membrane antigen (PSMA), have been investigated in preclinical and clinical settings (42–48). However, the antitumor efficacy of CAR-T cell therapy in these solid tumor settings has been disappointing. Several factors have impeded the utility of CAR-T cell therapy for solid tumors, including barriers that inhibit the trafficking and infiltration of CAR-T cells into the tumor, lack of specific tumor antigens that are highly and uniformly expressed in tumor cells, and hostile tumor microenvironment (TME) characterized by oxidative stress, nutritional depletion, acidic pH, and hypoxia (15, 16, 49, 50). Therefore, biomarkers for identifying the favorable prognostic patients receiving CAR-T cells are urgently needed.
CAR-T cell therapy-related toxicity
In addition to the low response or high relapse rate in patients treated with CAR-T cells, treatment with CAR-T cells also shows severe adverse events (AEs), which include CRS, ICANS, cardiotoxicity, hypersensitivity reactions, fatal macrophage activation syndrome (MAS), and uveitis (17). These fatal AEs result in doctors walking on thin ice when prescribing CAR-T cells. CRS, which is defined as an excessive release of cytokines, such as interleukin (IL)-1, IL-6, interferon (IFN)-γ, and IL-10, following the activation of immune cells during immunotherapy, represents the most common AE associated with CAR-T cell therapy and occurs in 40–93% of patients receiving CAR-T cell therapy (51). CRS usually occurs within 1-2 weeks after CAR-T cells administration (52, 53). Common symptoms of CRS include fever, exhaustion, anorexia, myalgia, and arthralgia. CRS remains a major hurdle for the widespread use of CAR-T cells, if not properly identified and managed, CRS can be fatal and further progresses to more severe forms of the syndrome, including cardiac arrhythmia, tachycardia, and respiratory or multi-organ conditions (54, 55). Another severe AE in patients treated with CAR-T cells is ICANS, which occurs in up to 67% of leukemia patients and 62% of lymphoma patients (17). Impaired cognition and overall confusion, such as aphagia, lethargy, and delirium, represent the initial symptoms of ICANS (56, 57). With the progression of the disease, symptoms, including hallucinations, encephalopathy, seizures, and cerebral edema, can be observed in patients treated with CAR-T cells (58). Although the exact causes of ICANS are not fully understood, blood brain barrier disruptions, the influx of cytokines into the central nervous system (CNS), and microglial and myeloid activation within the CNS are regarded as key factors in the development of ICANS (59–61). Although a series of treatment options, including intravenous hydration corticosteroids, monoclonal antibodies against the IL-6 receptor, IL-1 receptor antagonist, or targeting granulocyte-macrophage colony-stimulating factor (GM-CSF), have been approved to relieve these AEs, toxicity is still a major challenge for the wide application of CAR-T cells. Therefore, methods for assessing and predicting potential toxicities before administration are also urgently needed (62).
The gut microbiome and cancer
The gut microbiota, which refers to the vast collection of microbes inhabiting in the gastrointestinal tract, is involved in several key processes of human health, including providing nutrients and vitamins, protecting against pathogens, helping the development of the immune system and maintaining epithelial mucosa homeostasis (22, 23, 63). The balance between human cells and the gut microbiota is critical for maintaining the host’s health status. The gut microbiota can be modulated through several methods, including consuming dietary fibers, taking probiotics, or performing physical activity (64–68). In contrast, antibiotics, proton pump inhibitors (PPIs), smoking, and chronic stress may impair bacterial variability (69–72). Recently, several studies have suggested that the gut microbiota of cancer patients is very different from that of healthy individuals, and the role of the gut microbiota as a contributor to carcinogenesis has been well studied (73, 74). To date, the exact mechanisms through which the gut microbiota contributes to oncogenesis are not fully understood; however, several possible mechanisms can account for the process: (1) direct oncogenic effects of microorganisms and their products; (2) increased circulating pro-carcinogenic metabolites; and (3) disruption of cancer immunosurveillance through pro-inflammatory and immunosuppressive pathways (75).
In addition to its pro-carcinogenic properties, recent preclinical and few clinical studies suggest that the gut microbiota can modulate the response and susceptibility to side effects of different therapeutic strategies, including surgery, chemotherapy, radiotherapy and immunotherapy (76, 77) (Figure 1). Although the underlying mechanisms are not well understood, some of them have been described as TIMER framework mechanisms, including translocation, immunomodulation, metabolism, enzymatic degradation and reduced diversity (78, 79). In this section, we summarize the role of the gut microbiota in modulating the efficacy, resistance and toxicity of cancer therapies.
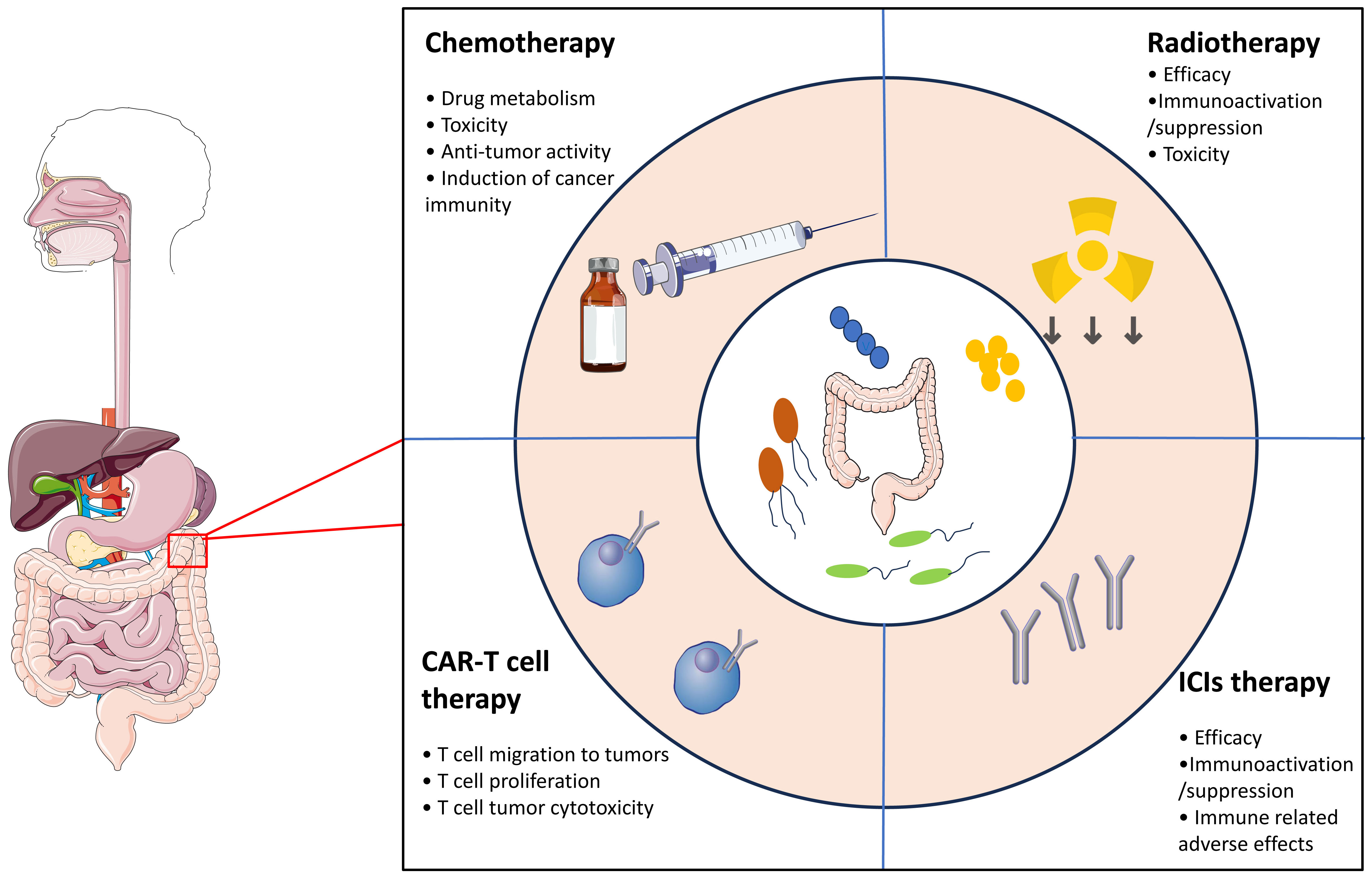
Figure 1 The gut microbiota modulates the response and susceptibility to side effects in response to different therapeutic strategies.
Chemotherapy
Recently, the gut microbiota has been demonstrated to be a key factor affecting the efficacy, pharmacokinetics, and toxicity of chemotherapy (80, 81). To date, a series of mechanisms, including xenometabolism, immune interactions, and altered community structure, have been proposed to account for the impact of the gut microbiota on chemotherapy efficacy (79). The gut microbiota can directly modify the metabolism of antitumor drugs, which is linked to an increase in toxicity and a decrease in treatment efficacy (82, 83). For example, the toxicity of irinotecan, a topoisomerase I inhibitor widely used to treat colorectal cancer, is dependent on glucuronosyltransferases in the liver. The active form of irinotecan, SN-38, can be detoxified into the inactive form SN-38-G by glucuronosyltransferases (84). However, in a in vivo rat model, Bacteroides species and other bacteria that express β-glucuronidase (such as Faecalibacterium prausnitzii and Clostridum spp.), can convert SN-38-G back into SN-38 (85). This process is associated with the accumulation of SN-38 in the gut, which leads to diarrhea. In addition, toxicity is preventable with broad-spectrum antibiotics or inhibitors of β-glucuronidase in in vivo studies (86). On the other hand, dysbiosis of the gut microbiota have also been suggested to be correlated with severe side effects, such as intestinal mucositis induced by doxorubicin, 5-FU or irinotecan (87, 88). For example, several studies in mice have shown that 5-FU therapy induces intestinal dysbiosis by increasing the abundance of Staphylococcus and Clostridium species and decreasing the abundance of Enterobacteriaceae, Lactobacillus and Bacteroides, thus exacerbating severe side effects in murine cancer models and in cancer patients (89). In addition to its role in reducing the toxicity of chemotherapy, the efficacy of chemotherapy can also be modulated by the gut microbiota (90, 91). Possible mechanisms include remote immune modulations by the gut microbiota and/or bacterial translocation to lymphoid organs. Several studies have also suggested that the gut microbiota provides a tumor microenvironment that can favor the toxic effect of drugs on cancer cells and sustain anticancer adaptive immunity following drug-induced immunogenic cell death (84). In a pioneer in vivo study, Iida et al. reported that oxaliplatin and cisplatin had reduced antitumor efficacy and chemoresistance in germ-free or antibiotic-treated mice, compared to specific-pathogen-free (SPF) mice (92). The gut microbiota primes myeloid cells in tumors to produce reactive oxygen species (ROS) via NADPH oxidase 2 (NOX2), which are important for DNA damage and apoptosis in response to platinum compounds. In addition, several in vivo studies have demonstrated that cyclophosphamide (CTX) can modulate the immune microenvironment of tumors by reducing regulatory T cells (Tregs) and increasing Th1 and Th17 cells in a manner dependent on the gut microbiota (90, 91, 93). The translocation of Enterococcus hirae to lymph nodes and the accumulation of Barnesiella intestinihominis in the colon promoted cancer immunity following CTX treatment. Although further clinical studies are needed to evaluate these innovative findings, the gut microbiota has been demonstrated to be an essential biomarker for cancer chemotherapy efficacy.
Radiotherapy
Radiotherapy, which directly induces DNA damage through the production of ROS or reactive nitrogen species (RNS), is another important treatment option for cancer patients (80, 94). Moreover, radiotherapy can also induce local immunogenic effects and stimulate the innate immune system (95, 96). However, the tumor response after radiotherapy is heterogeneous, and the causes of this heterogeneity remain unclear. Recently, growing evidence has suggested that the gut microbiota may contribute to the interpatient heterogeneity of radiotherapy. In a in vivo mouse study, Cui et al. investigated the role of the circadian rhythm in the effect of radiotherapy and reported that mice with a normal 12-h dark/12-h light cycle had significantly better survival than did those with different cycles (8-h dark/16-h light or 16-h light/8-h dark) (97). In this process, alteration of the gut microbiota was suggested to account for the radio-resistance. Moreover, several other studies have suggested that depletion of the gut microbiota by broad-spectrum antibiotics results in an expansion of the Saccharomycetes class of fungi, which decreases radiotherapy efficacy by inhibiting tumor immunity by signaling through the β-glucan receptor Dectin 1 (98). Another hypothesis concerns the link between radio-resistance and gut microbiota-involved autophagy regulation. In a cohort study, Digomann et al. reported that several autophagy-related proteins were correlated with the clinical prognosis of patients with head and neck squamous cell carcinoma treated with radiotherapy (99).
On the other hand, radiotherapy side effects alter quality of life and are integral parts of treatment decisions. The gut microbiota plays a key role in radio-induced toxicity. In a study, Ferreira et al. reported a close correlation between the gut microbiota abundances and radiation enteropathy (100). In patients with radiation enteropathy, the composition of Clostridium, Roseburia and Phascolarctobacterium were significantly increased. In another study, a significant alteration in the Firmicutes/Bacteroidetes ratio was observed in patients with pelvic radiation-induced diarrhea (101). In a mouse model, fecal transplantation improved gastrointestinal tract function in irradiated mice and protected against radiation-induced death (102). Taken together, these data suggested that the gut microbiota plays a key role in the modulation of radiosensitivity and radiation-induced toxicity. However, further preclinical and clinical studies are needed to determine the detailed underlying mechanisms involved.
Immunotherapy
Immunotherapy, which includes ICB therapy that inhibits programmed cell death protein 1/programmed cell death ligand 1 (PD-1/PD-L1) and cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) signaling, tumor vaccines, and ACT, represents a hotspot of cancer therapy (103). Despite the remarkable effectiveness of immunotherapy in a subset of patients, most patients treated with immunotherapy will experience primary or acquired resistance (104, 105). Furthermore, immune-related adverse events (irAEs) are another challenge that limits the wide application of immunotherapy (106). Recently, accumulating evidence has pinpointed the indispensable roles of the gut microbiota in cancer immunotherapy. In a murine melanoma model generated by Paulos et al., the antitumor efficacy of CD8+ T cells was strongly increased after total body irradiation through the translocation of gut bacteria into mesenteric lymph nodes (107). This effect could be attributed to the fact that irradiation induces the release of microbial lipopolysaccharide (LPS), which activates the innate immune response and enhances the efficacy of antitumor CD8+ T cells. Antibiotic treatment or LPS neutralization can significantly decrease the antitumor response. In another in vivo study, Iida et al. reported that the efficacy of anti-IL-10/CpG oligodeoxynucleotide (ODN) immunotherapy was impaired by antibiotic treatment in both murine models of MC38 colon carcinoma and B16 melanoma (92). Antibiotic treatment can decrease the gut microbiota load and the number of proinflammatory cytokine-producing monocytes in tumors, leading to failure of the immunotherapy response. All these studies highlighted the important role of the gut microbiota in the antitumor efficacy of immunotherapy.
After the relationship between the gut microbiota and immunotherapy was recognized, an increasing number of studies have been conducted to explore the impact of the gut microbiota on the efficacy and toxicity of ICI therapy. Although the underlying mechanisms are not well understood, growing evidence confirms the central role of remote lymphoid and myeloid cell modulation by the gut microbiota. In a study, GF mice or antibiotic-treated mice were not capable of responding to the CTLA-4 antibody compared to SPF mice (108). The gut microbiota, which includes Bacteroides thetaiotaomicron, Bacteroides fragilis and Burkholderia cepacia, was suggested to be associated with the antitumor efficacy and toxicity of anti-CTLA-4 inhibitors. After oral administration of Bacteroides spp., the antitumor efficacy of the anti-CTLA-4 inhibitor was restored, with an increase in intratumoral mature DCs and an increase in the Th1 response in tumor-draining lymph nodes. Moreover, the microbiota composition could predict the status of solid tumor responders and nonresponders to anti-PD-1/PD-L1 therapy. In a study conducted by Gopalakrishnan et al., enrichment of Faecalibacterium species in the gut microbiota of melanoma patients was associated with a high response to anti-PD-L1 therapy, while enrichment of Bacteroides thetaiotaomicron, Escherichia coli, and Anaerotruncus coli hominis was found mainly in nonresponders (109). Fecal microbiota transplantation (FMT) via responder patients into GF mice improved ICI efficacy, which was associated with increased numbers of intratumoral mature DCs, IFN-γ+CD8+ and/or CD4+ antitumor T cells and decreased numbers of intratumoral CD4+FoxP3+ Tregs (109–111). In total, these promising results strongly support the use of microbial targeting in antitumor immunotherapy to enhance tumor efficacy.
Influence of the gut microbiota on the effectiveness of CAR-T cell therapy
Currently, CAR-T-cell therapy is one of the most promising cancer therapies and has led to unprecedented responses in patients with R/R hematologic malignancies, including lymphoma, leukemia, and MM. In the past decade, the correlation between the gut microbiota and the antitumor efficacy of CAR-T cells has been explored in a series of preclinical and clinical studies, and it is not unexpected that the gut microbiota represents a key factor in predicting and determining the outcomes of CAR-T-cell therapy. Smith et al. conducted the first human study to investigate the influence of the gut microbiota on the response and toxicity of CD19 CAR-T-cell-based therapies (27). A retrospective cohort of 228 patients from the Memorial Sloane Kettering Cancer Center (MSKCC) and University of Pennsylvania who were treated with second-generation CD19 CAR-T cells was established; 137 of these patients were non-Hodgkin lymphoma (NHL) patients, 91 of whom were acute lymphocytic leukemia (ALL) patients. Then, they analyzed the fecal microbiota composition of patients receiving CD19 CAR-T-cell therapy and hypothesized that the microbiota is associated with antitumor efficacy and toxicity. The baseline stool samples obtained prior to CAR-T-cell therapy were heterogeneous for bacteria at the phylum level, as indicated by a decreased Shannon index for alpha diversity compared to that of healthy controls. The authors stratified patients based on exposure to antibiotics, given that antibiotics are commonly used to treat secondary infections in these patients. Indeed, 60% of the patients received antibiotics, and 20.6% of them specifically received broad-spectrum antibiotics such as piperacillin/tazobactam, imipenem/cilastatin, and meropenem (PIM), which target anaerobic gut commensal bacteria. Using OS and progression-free survival (PFS) as indices of treatment response, the authors found that PIM exposure prior to CAR-T-cell therapy was correlated with worse OS and PFS. To further validate this hypothesis, the authors established a prospective cohort of 48 NHL or ALL patients with matched baseline gut microbiota profiles and found that response (measured as complete response (CR) on day 100 post treatment) to CAR-T-cell therapy was positively correlated with baseline microbiota diversity and enrichment of specific bacterial taxa, such as Ruminococcus, Faecalibacterium, and Bacteroides. In another recent study by Stein-Thoeringer et al., a large cohort of lymphoma patients receiving second-generation CD19 CAR-T cells (122 were treated with axicabtagene ciloleucel (axi-cel), 49 received tisagenlecleucel (tisa-cel) and 1 received lisocabtagene maraleucel (liso-cel)) in Germany and the U.S. were established (112). Consistent with the findings of previous studies, an association between exposure to antibiotics prior to CAR-T-cell infusion and increased incidences of cancer relapse/disease progression and a decrease in OS was also observed. Moreover, based on machine learning methods used to explore microbiome-based predictions of treatment outcomes, Bacteroides, Ruminococcus, Eubacteria, and Akkermansia spp. were identified as major potential drivers of therapy responsiveness. Similarly, Hu and colleagues also revealed significant differences in the enrichment of Bifidobacterium, Prevotella, Sutterella, and Collinsella between MM patients treated with second-generation BCMA CAR-T cells in complete remission and those in partial remission (113). However, in a study investigating how vancomycin-induced gut microbiota dysbiosis affects CAR-T-cell therapy, the results were different (114). Vancomycin is a branched tricyclic peptide antibiotic that targets mostly gram-positive bacteria. When administrated orally, vancomycin is poorly absorbed and does not reach an effective dose systemically due to its large size. In this study directed by Uribe-Herranz et al., mice receiving vancomycin in combination with murine CD19bbz CAR-T-cell therapy showed an increased tumor response and tumor-associated antigen (TAA) cross-presentation compared with those of mice receiving CD19 CAR-T-cell therapy alone, both in lymphoma and melanoma murine models. FMT from healthy human donors to preconditioned mice recapitulated the results obtained in naive gut microbiota mice. 16s Metagenomic analysis demonstrated that oral vancomycin affected the composition of human gut microbiome of engrafted avatar mice, with a significant decrease of alpha diversity and significant expansion of vancomycin-resistant bacteria (such as Enterobacteriaceae and Sutterellaceae). The decrease or depletion of gram-positive bacteria included several families, genera, and species, including Ruminococcaceae and Lachnospiraceae, which are known to impair antigen presentation. Similarly, under clinical conditions, compared with unexposed patients, B-ALL patients treated with CD19 CAR-T cells and exposed to oral vancomycin had greater CAR-T-cell peak expansion. Taken together, although research on the role of the gut microbiota in CAR-T-cell therapy is still in its infancy, these findings suggest the tremendous potential of the gut microbiota as a noninvasive prognostic marker for CAR-T-cell therapy.
Interactions between the gut microbiota and toxicities related to CAR-T cell therapy
In addition to the effect of the gut microbiota on the antitumor efficacy of CAR-T cells, the gut microbiota was also demonstrated to alter the development of toxicity in patients treated with CAR-T cells. Smith and colleagues investigated the role of the gut microbiota in the toxicity of CAR-T cells in a multicenter study of patients with B-cell lymphoma and leukemia (27). In a retrospective cohort (n= 228), they found that exposure to antibiotics, particularly PIM, was associated with shorter survival and increased neurotoxicity. Multiple bacterial species were associated with the absence of toxicity, but microbes associated with toxicity were unidentifiable according to the linear discriminant analysis effect size. The nonoxidative branch of the pentose phosphate pathway was upregulated in patients with reported toxicities, indicating that metabolites from these bacterial taxa may serve as biomarkers for side effects after CD19 CAR-T-cell therapy (112). In another recent study by Hu et al., researchers investigated second-generation BCMA CAR-T-cell toxicity in patients with relapsed/refractory MM, NHL, or ALL (113). Microbiota changes were longitudinally monitored throughout CAR-T-cell therapy. Stool samples were collected prior to CAR-T-cell infusion, during CAR-T-cell infusion but prior to the development of CRS, during active CRS, and up to fourteen days after CAR-T-cell infusion. Severe CRS was associated with changes in the abundance of several gut microbiota species. As indicated by the Shannon index, there was a significantly decreased abundance of Bifidobacteria. Alpha diversity was observed after CAR-T-cell infusion. Furthermore, an increase in the abundance of the Actinomyces and Enterococcus genera was also suggested to be associated with an increased incidence of CRS. Overall, antibiotic exposure and subsequent alteration of the gut microbiota are associated with increased toxicity, including CRS and ICANS, and with worsened CAR-T-cell responses.
Potential strategies for modifying the gut microbiota to enhance the therapeutic efficacy of CAR-T cells
As mentioned above, the gut microbiota has been demonstrated to be an extrinsic factor that can predict and prospectively dictate the efficacy and toxicity of CAR-T-cell therapy, providing a novel target for improving the efficacy of CAR-T-cell therapy. Currently, several potential strategies, including FMT, administration of defined taxa and diet, have been demonstrated to improve the treatment efficacy while decreasing the toxicity of ICIs therapy through modulation of the gut microbiota. In this section, we focused on the development and future application of these potential strategies to optimize the microbiota composition to improve the efficacy of CAR-T-cell therapy and decrease the incidence of AEs (Figure 2).
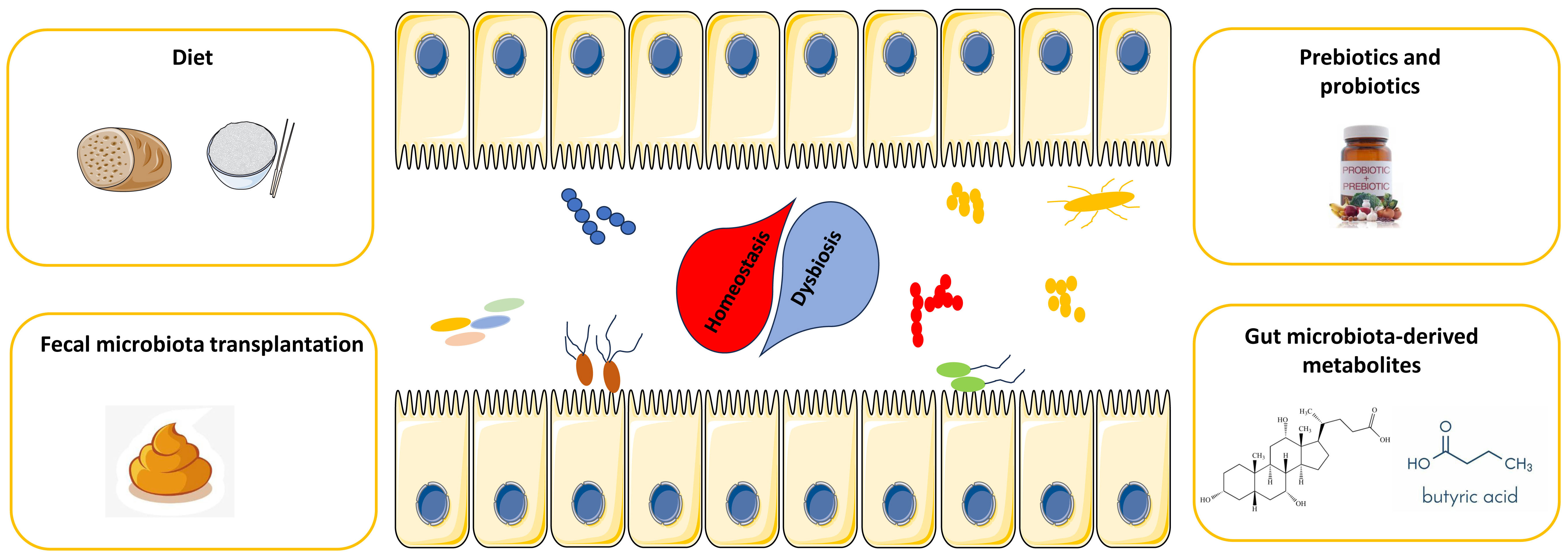
Figure 2 Potential strategies for harnessing the gut microbiota to potentiate CAR-T-cell efficacy while attenuating toxicity.
FMT
FMT represents the transfer of fecal microbial content from a healthy donor to the intestine of a recipient and has been widely tested in the treatment of resistant Clostridium difficile infection, other opportunistic infections and inflammatory bowel diseases (115, 116). Moreover, FMT has also demonstrated to improve the efficacy of a series of cancer therapies. For example, Gopalakrishnan et al. established melanoma-bearing GF mice and orally transferred the fecal microbiota of responder or nonresponder patients to anti-PD-1 therapy into these mice. Ultimately, mice treated with FMT from responder patients had significantly reduced tumor size (median tumor volume: 403.7 mm3 vs. 2301 mm3) and improved responses compared to those treated with FMT from nonresponder patients (109). Recently, the results of several clinical trials incorporating FMT to improve cancer therapy have been published. In a clinical trial led by Dr. Davar (NCT03341143), investigators assessed whether resistance to anti-PD-1 therapy can be overcome by changing the gut microbiota in patients with advanced melanoma (117). The patients received FMT from seven donors (including four with complete response (CR) and three with partial response (PR)) in combination with pembrolizumab treatment. The objective response rate (ORR) was 20% (3 PRs out of 15 patients), whereas the percentage of patients with durable SDs lasting >12 months was 20% (3 out of 15 patients). Another phase I clinical trial (NCT03353402) showed similar results, and the ORR in patients receiving FMT and PD-1 inhibitors was 30% (3 out of 10, including two PRs and one CR) (118). In addition to overcoming resistance to ICIs in patients with refractory cancer, FMT also showed promising efficacy in enhancing the antitumor efficacy of ICIs in first-line treatment settings in a recent study of previously untreated patients with advanced melanoma. (NCT03772988). The ORR was 65% (13 of 20, including 4 (20%) CRs), and only five patients (25%) experienced grade 3 irAEs from FMT combined with the PD-1 inhibitor. Responders experienced an enrichment of immunogenic bacteria and a loss of deleterious bacteria following FMT (119). As mentioned above, significant correlation of gut microbiome with treatment outcome and CRS grade of CAR-T therapy has been observed. Thus, it is reasonable that FMT can also help to rescued the diversity and function of gut microbiota in cancer patients. Although additional evidence is needed, combination with FMT provides a promising way to enhance the efficacy and safety of immunotherapy and may also constitute a potential strategy for enhancing the antitumor efficacy of CAR-T-cell therapy in the near future.
Diet, prebiotics and probiotics
In addition to antibiotics and FMT, which can directly modulate the gut microbiota, diet is a critical means of regulating microbial composition and function. In recent decades, several different dietary strategies, including long-term caloric restriction, intermittent fasting, short-term starvation, high-fiber diets, ketogenic diets and fermented food, have been widely studied in the context of cancer and have been demonstrated to improve the antitumor efficacy of cancer therapy (120). Despite several limitations, dietary intervention represents a tractable strategy for modulating the function of the gut microbiota (121–125). For example, a high-fiber diet has been demonstrated to be associated with a better response and survival rate when treated with ICIs in preclinical models and in clinical cohorts and was considered to be correlated with enhanced T-cell activation and monocytic reprogramming within the TME (126). In addition, caloric restriction can promote memory T cell accumulation in the bone marrow and enhance T cell immunity to bacterial infection and tumors in T cell adoptive transfer models (127). The ketogenic diet has represented another popular diet in recent years. Ketone body 3-hydroxybutyrate can induce T cell immunity, thereby enhancing the antitumor effect of anti-PD-1 therapy in in vivo mice model (128). In addition to diet modulation, several naturally occurring soluble fibers, such as inulin and pectin, can be found in many vegetables and fruits. These fibers are called prebiotics and cannot be digested by gastrointestinal enzymes but can be fermented by bacteria (129, 130). Recently, prebiotics have been shown to affect the functional status of the gut microbiota in preclinical models. For example, oral inulin-gel treatments increase the abundance of short-chain fatty acid (SCFA)-producing microorganisms and amplify the antitumor efficacy of anti-PD-1 therapy (131). Pectin feeding can also enhance the ability of mice transplanted with the fecal microbiota of a patient with cancer to respond to anti-PD-1 therapy by expanding the fiber-fermenting and SCFA-producing microbiota (132). All these data suggested that traditional diet modulation or dietary components, such as prebiotics, may represent a promising way to improve immunotherapy responsiveness by targeting the microbiota.
The concept of probiotics, which refers to the specific transplantation of single microbial species and/or designer microbial consortia to enhance the response to ICIs and other forms of cancer treatment, has also emerged as an important research field (133). In 1993, a multicenter, randomized controlled study of 223 patients with uterine cervix carcinoma treated with a combination of heat-killed Lactobacillus casei strains (LC9018) and radiation therapy revealed that tumor regression was enhanced through the induction of an immune response against cancer cells (134). Since then, the potential applications of combination probiotics/cancer treatment have sparked interest in future studies. For example, in a small, open-label trial, 58% of patients with metastatic renal cell carcinoma (RCC) treated with CBM588 (a formulation that includes a strain of Clostridium butyricum) in combination with ICIs responded to treatment, whereas 20% of patients received only ICIs (135). Additionally, PFS was significantly prolonged in CBM588-treated patients to 12.7 months compared with 2.5 months in patients receiving ICB alone, thus highlighting that the addition of bifidogenic bacterial products can improve the clinical outcome of patients with RCC. Despite several limitations, there are tremendous opportunities to develop informed, ‘next-generation’ probiotics, as recent studies suggest that specific microorganisms in the gut may enhance antitumor immune responses in part through the induction of highly therapeutic TLSs in the TME, which have been favorably associated with patients’ response to ICIs across cancer types (136, 137). Together, these lines of investigation open up new possibilities for transplanting and targeting specific therapeutic microorganisms and can also be applied to enhance the antitumor efficacy of CAR-T cells in the future.
Targeting gut microbiota-derived metabolites
Gut microbiota-derived metabolites represent a variety of small molecules produced or transformed by intestinal microorganisms that not only exert direct effects in the intestine but also modulate the function of cells in remote organs. Unlike gut bacteria, which are predominantly located in the luminal compartment of the intestine, gut microbiota-derived small molecules can easily cross the epithelial layer and diffuse through the entire circulation (138). It is estimated that 5 and 10% of all plasma metabolites are derived from the gut microbiota; however, these products generated by the gut microbiota were considered merely dead-end byproducts of their metabolic pathways for a long time (139, 140). In the past decade, gut microbiota-derived metabolites have received increasing attention in cancer research (141–143). For example, short-chain fatty acids (SCFAs), which are synthesized by the bacterial fermentation of dietary fiber, are the most abundant class of microbial metabolites and are composed of carboxylic acids with aliphatic tails of 1-5 carbons (144). Bacterial SCFAs, such as acetate (C2), propionate (C3), butyrate (C4) and valerate (C5), play key roles in regulating anticancer immunity and cancer immune surveillance (145). Butyrate reportedly enhances the therapeutic efficacy of anti-PD-1 agents by increasing CD4+ and CD8+ T-cell infiltration in the TME in tumor-bearing mice humanized with intestinal microbes from colorectal cancer (CRC) patients (132). Moreover, replenishing butyrate prior to anti-PD-1 treatment was sufficient to improve the therapeutic efficacy in nonresponders. Similarly, He and colleagues indicated that the SCFA butyrate could directly potentiate the antitumor CD8+ T-cell response via ID2-dependent IL-12 signaling, suggesting the potential beneficial role of butyrate supplementation in anticancer immunity therapy (146). In a study directed by Luu et al, microbiota-modulated SCFA, may also enhance the antitumor action of cytotoxic lymphocytes and CAR-T cells through metabolic and epigenetic remodeling of CAR-T cells. Mechanically, they found that in vitro treatment of CTLs and CAR-T cells with pentanoate and butyrate increases the function of mTOR as a central cellular metabolic sensor, and inhibits class I histone deacetylase activity, which increased expression of effector molecules such as CD25, IFNγ, and TNFα in syngeneic murine melanoma and pancreatic cancer models (147, 148). In addition to SCFA, Other microbial-derived metabolites, signaling through aryl hydrocarbon receptors (AhR), may also play a role in contributing to CD8+ T cell exhaustion by upregulating inhibitory receptors and downregulating cytokine production, thereby altering the ability of T cells to kill tumor cells. Supplementation or inhibition of such microbially secreted bioactive metabolites may potentially be used to reinvigorate the immune response (149, 150). Taken together, gut microbiota-derived metabolites represent another promising target to enhance the efficacy and safety of CAR-T-cell therapy in the future.
Current challenges and future perspectives
In the past decade, immunotherapy has made significant progress and has gradually become the most important treatment for tumors. CAR-T-cell therapy represents the most promising treatment option for cancer, and a series of CAR-T-cell products have been approved for the treatment of R/R B-cell leukemia or lymphoma. Although promising in terms of efficacy, several limitations still exist, including poor efficacy in solid tumors and toxicity, such as CRS and ICAN. In contrast to the stability of the human genome, the modifiable nature of the gut microbiota renders it a promising opportunity for CAR-T-cell therapy. Recently, the role of the gut microbiota in cancer therapy has been established, and growing evidence has suggested that targeting the gut microbiota is a promising method for enhancing the antitumor efficacy of CAR-T-cell therapy. As mentioned above, FMT, microbiota-derived metabolites, diet, prebiotics and probiotics have been explored in several studies and are regarded as effective strategies for enhancing the antitumor efficacy of CAR-T cells through modifying the composition and function of the gut microbiota.
Although it is believed that modulating the gut microflora will likely be a promising method for managing CAR-T-cell therapy, several potential challenges should be mentioned. First, most of the evidence supporting the relationship between the microbiota and immunotherapy has been obtained from mouse models. As the gut microbiota in mice is not identical to that in humans and the innate and adaptive immune systems of mice are also different from those of humans, a deeper comprehension of the underlying molecular mechanisms and additional humanized animal models or clinical trials are needed to further explore the underlying correlation. Second, the exact mechanisms underlying the correlation between the gut microbiota and cancer genesis/therapy have not been fully characterized, and evidence of causation between them is still lacking. However, further in vitro and in vivo studies are needed to determine the causal mechanisms involved. In the context of CAR-T-cell therapy, transferring defined microbiota constituents from responders and nonresponders into GF cancer mice would enable us to elucidate the causal contribution of the gut microbiota and its bioactive metabolites. Interindividual microbiota variability represents another formidable challenge in identifying the impacts of CAR-T-cell therapy and AEs on the gut microbiota and their bioactive metabolites. Indeed, the microbiota populations of CAR-T-cell-treated patients reported by Smith et al. and Hu et al. are quite different (27, 113). In different trials, varying taxa have been associated with impaired immunotherapy responsiveness, indicating that defining a generalizable CAR-T-cell therapy optimizing the microbial signature will be difficult. Thus, multicentric clinical trials with high-quality training and validation sets for identifying CAR-T-cell therapy-related gut microbiota are urgently needed. Moreover, artificial intelligence technologies can be used in this process. Fecal microbiota transfer into CAR-T-cell-resistant individuals may optimize responsiveness and even lead to the conversion of nonresponders to responders. However, as a living body, such treatment can pose risks of introducing potentially harmful bacteria into patients who are already severely immunocompromised, and it is difficult to guarantee its safety. Thus, identification and isolation of defined microbiota that mediate such favorable effects may offer a safer, more reproducible, and universal treatment option. Another challenge is the complex physiological conditions (such as gastric acid and diverse enzymes) that might digest or deactivate microbial agents before they reach the action site. Thus, the appropriate delivery route and dose should also be optimized.
Conclusions
CAR-T-cell therapy has revolutionized the history of cancer treatment. However, several disadvantages, such as low efficacy and high toxicity, limit the widespread application of CAR-T cells in solid tumors. Recently, an increasing number of studies have highlighted the key role of the gut microbiota in affecting the efficacy of cancer therapies and their side effects. Similarly, overwhelming preclinical and clinical evidence also supports a critical role of the gut microbiota in the response to and toxicity of CAR-T-cell therapy, which indicates that modulating the gut microbiota is a promising therapeutic strategy for enhancing the antitumor efficacy and attenuating the toxicity of CAR-T-cell therapy. Currently, research on microbial therapy for cancer is still in its infancy, and further mechanistic dissection via cellular and animal studies as well as validation with larger longitudinal clinical cohorts are needed. Despite these challenges, targeting the gut microbiota remains a promising strategy for improving CAR-T-cell therapy in the future.
Author contributions
P-FZ: Conceptualization, Funding acquisition, Investigation, Methodology, Writing – original draft, Writing – review & editing. DX: Conceptualization, Investigation, Methodology, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was supported by Science & Technology Department of Sichuan Province Funding Project (2022NSFSC1348).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Igarashi Y, Sasada T. Cancer vaccines: toward the next breakthrough in cancer immunotherapy. J Immunol Res. (2020) 2020:5825401. doi: 10.1155/2020/5825401
2. Bagchi S, Yuan R, Engleman EG. Immune checkpoint inhibitors for the treatment of cancer: clinical impact and mechanisms of response and resistance. Annu Rev Pathol: Mech Disease. (2021) 16:223–49. doi: 10.1146/annurev-pathol-042020-042741
3. Hong M, Clubb JD, Chen YY. Engineering CAR-T cells for next-generation cancer therapy. Cancer Cell. (2020) 38:473–88. doi: 10.1016/j.ccell.2020.07.005
4. Jogalekar MP, Rajendran RL, Khan F, Dmello C, Gangadaran P, Ahn B-C. CAR T-cell-based gene therapy for cancers: new perspectives, challenges, and clinical developments. Front Immunol. (2022) 13:925985. doi: 10.3389/fimmu.2022.925985
5. Huang R, Li X, He Y, Zhu W, Gao L, Liu Y, et al. Recent advances in CAR-T cell engineering. J Hematol Oncol. (2020) 13:1–19. doi: 10.1186/s13045-020-00910-5
6. Jayaraman J, Mellody MP, Hou AJ, Desai RP, Fung AW, Pham AHT, et al. CAR-T design: Elements and their synergistic function. EBioMedicine. (2020) 58:102931. doi: 10.1016/j.ebiom.2020.102931
7. Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. New Engl J Med. (2019) 380:1726–37. doi: 10.1056/NEJMoa1817226
8. Jacobson CA, Chavez JC, Sehgal AR, William BM, Munoz J, Salles G, et al. Axicabtagene ciloleucel in relapsed or refractory indolent non-Hodgkin lymphoma (ZUMA-5): a single-arm, multicentre, phase 2 trial. Lancet Oncol. (2022) 23:91–103. doi: 10.1016/S1470-2045(21)00591-X
9. Locke FL, Miklos DB, Jacobson CA, Perales M-A, Kersten M-J, Oluwole OO, et al. Axicabtagene ciloleucel as second-line therapy for large B-cell lymphoma. New Engl J Med. (2022) 386:640–54. doi: 10.1056/NEJMoa2116133
10. Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood J Am Soc Hematol. (2011) 118:4817–28. doi: 10.1182/blood-2011-04-348540
11. Mullard A. FDA approves first CAR T therapy. Nat Rev Drug Discovery. (2017) 16:669–70. doi: 10.1038/nrd.2017.196
12. FDA okays second CAR-T for Kite. Nat Biotechnol. (2020) 38:1012–. doi: 10.1038/s41587-020-0676-z
13. Frey NV. Approval of brexucabtagene autoleucel for adults with relapsed and refractory acute lymphocytic leukemia. Blood J Am Soc Hematol. (2022) 140:11–5. doi: 10.1182/blood.2021014892
14. Mullard A. FDA approves fourth CAR-T cell therapy. Nat Rev Drug Discovery. (2021) 20:166–7. doi: 10.1038/d41573-021-00031-9
15. Wagner J, Wickman E, DeRenzo C, Gottschalk S. CAR T cell therapy for solid tumors: bright future or dark reality? Mol Ther. (2020) 28:2320–39. doi: 10.1016/j.ymthe.2020.09.015
16. Maalej KM, Merhi M, Inchakalody VP, Mestiri S, Alam M, Maccalli C, et al. CAR-cell therapy in the era of solid tumor treatment: current challenges and emerging therapeutic advances. Mol Cancer. (2023) 22:20. doi: 10.1186/s12943-023-01723-z
17. Brudno JN, Kochenderfer JN. Recent advances in CAR T-cell toxicity: mechanisms, manifestations and management. Blood Rev. (2019) 34:45–55. doi: 10.1016/j.blre.2018.11.002
18. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. (2021) 11:69. doi: 10.1038/s41408-021-00459-7
19. Flugel CL, Majzner RG, Krenciute G, Dotti G, Riddell SR, Wagner DL, et al. Overcoming on-target, off-tumour toxicity of CAR T cell therapy for solid tumours. Nat Rev Clin Oncol. (2023) 20:49–62. doi: 10.1038/s41571-022-00704-3
20. Aggarwal N, Kitano S, Puah GRY, Kittelmann S, Hwang IY, Chang MW. Microbiome and human health: Current understanding, engineering, and enabling technologies. Chem Rev. (2022) 123:31–72. doi: 10.1021/acs.chemrev.2c00431
21. de Vos WM, Tilg H, Van Hul M, Cani PD. Gut microbiome and health: mechanistic insights. Gut. (2022) 71:1020–32. doi: 10.1136/gutjnl-2021-326789
22. Scott AJ, Alexander JL, Merrifield CA, Cunningham D, Jobin C, Brown R, et al. International Cancer Microbiome Consortium consensus statement on the role of the human microbiome in carcinogenesis. Gut. (2019) 68:1624–32. doi: 10.1136/gutjnl-2019-318556
23. Shi Z, Zhang M. Emerging roles for the gut microbiome in lymphoid neoplasms. Clin Med Insights: Oncol. (2021) 15:11795549211024197. doi: 10.1177/11795549211024197
24. Cullin N, Antunes CA, Straussman R, Stein-Thoeringer CK, Elinav E. Microbiome and cancer. Cancer Cell. (2021) 39:1317–41. doi: 10.1016/j.ccell.2021.08.006
25. Bhatt AP, Redinbo MR, Bultman SJ. The role of the microbiome in cancer development and therapy. CA: Cancer J Clin. (2017) 67:326–44. doi: 10.3322/caac.21398
26. Villéger R, Lopès A, Carrier G, Veziant J, Billard E, Barnich N, et al. Intestinal microbiota: a novel target to improve anti-tumor treatment? Int J Mol Sci. (2019) 20:4584. doi: 10.3390/ijms20184584
27. Smith M, Dai A, Ghilardi G, Amelsberg KV, Devlin SM, Pajarillo R, et al. Gut microbiome correlates of response and toxicity following anti-CD19 CAR T cell therapy. Nat Med. (2022) 28:713–23. doi: 10.1038/s41591-022-01702-9
28. Bourbon E, Ghesquières H, Bachy E. CAR-T cells, from principle to clinical applications. Bull du Cancer. (2021) 108:S4–S17. doi: 10.1016/j.bulcan.2021.02.017
29. Labanieh L, Mackall CL. CAR immune cells: design principles, resistance and the next generation. Nature. (2023) 614:635–48. doi: 10.1038/s41586-023-05707-3
30. Lee Y-H, Kim CH. Evolution of chimeric antigen receptor (CAR) T cell therapy: current status and future perspectives. Arch pharmacal Res. (2019) 42:607–16. doi: 10.1007/s12272-019-01136-x
31. Brocker T, Karjalainen K. Signals through T cell receptor-zeta chain alone are insufficient to prime resting T lymphocytes. J Exp Med. (1995) 181:1653–9. doi: 10.1084/jem.181.5.1653
32. Thistlethwaite FC, Gilham DE, Guest RD, Rothwell DG, Pillai M, Burt DJ, et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol Immunother. (2017) 66:1425–36. doi: 10.1007/s00262-017-2034-7
33. Katsarou A, Sjöstrand M, Naik J, Mansilla-Soto J, Kefala D, Kladis G, et al. Combining a CAR and a chimeric costimulatory receptor enhances T cell sensitivity to low antigen density and promotes persistence. Sci Trans Med. (2021) 13:eabh1962. doi: 10.1126/scitranslmed.abh1962
34. Ye L, Lou Y, Lu L, Fan X. Mesothelin−targeted second generation CAR-T cells inhibit growth of mesothelin-expressing tumors in vivo. Exp Ther Med. (2019) 17:739–47. doi: 10.3892/etm.2018.7015
35. Guedan S, Posey AD Jr., Shaw C, Wing A, Da T, Patel PR, et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight. (2018) 3(1):e96976. doi: 10.1172/jci.insight.96976
36. Roselli E, Boucher JC, Li G, Kotani H, Spitler K, Reid K, et al. 4-1BB and optimized CD28 co-stimulation enhances function of human mono-specific and bi-specific third-generation CAR T cells. J Immunother Cancer. (2021) 9(10):e003354. doi: 10.1136/jitc-2021-003354
37. Tang L, Pan S, Wei X, Xu X, Wei Q. Arming CAR-T cells with cytokines and more: Innovations in the fourth-generation CAR-T development. Mol Ther. (2023) 31:3146–62. doi: 10.1016/j.ymthe.2023.09.021
38. Duan D, Wang K, Wei C, Feng D, Liu Y, He Q, et al. The BCMA-targeted fourth-generation CAR-T cells secreting IL-7 and CCL19 for therapy of refractory/recurrent multiple myeloma. Front Immunol. (2021) 12:609421. doi: 10.3389/fimmu.2021.609421
39. Dimitri A, Herbst F, Fraietta JA. Engineering the next-generation of CAR T-cells with CRISPR-Cas9 gene editing. Mol Cancer. (2022) 21:78. doi: 10.1186/s12943-022-01559-z
40. Anderson LD Jr. Idecabtagene vicleucel (ide-cel) CAR T-cell therapy for relapsed and refractory multiple myeloma. Future Oncol. (2021) 18:277–89. doi: 10.2217/fon-2021-1090
41. Ciltacabtagene autoleucel (Carvykti) for multiple myeloma. Med Lett Drugs Ther. (2022) 64(1663):e188–e189.
42. Zhang Y, Zhang Z, Ding Y, Fang Y, Wang P, Chu W, et al. Phase I clinical trial of EGFR-specific CAR-T cells generated by the piggyBac transposon system in advanced relapsed/refractory non-small cell lung cancer patients. J Cancer Res Clin Oncol. (2021) 147:3725–34. doi: 10.1007/s00432-021-03613-7
43. Zhang C, Wang Z, Yang Z, Wang M, Li S, Li Y, et al. Phase I escalating-dose trial of CAR-T therapy targeting CEA+ metastatic colorectal cancers. Mol Ther. (2017) 25:1248–58. doi: 10.1016/j.ymthe.2017.03.010
44. Li H, Huang Y, Jiang D-Q, Cui L-Z, He Z, Wang C, et al. Antitumor activity of EGFR-specific CAR T cells against non-small-cell lung cancer cells in vitro and in mice. Cell Death Disease. (2018) 9:177. doi: 10.1038/s41419-017-0238-6
45. Bughda R, Dimou P, D’Souza RR, Klampatsa A. Fibroblast activation protein (FAP)-targeted CAR-T cells: launching an attack on tumor stroma. ImmunoTargets Ther. (2021) 10:313–23. doi: 10.2147/ITT.S291767
46. Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human epidermal growth factor receptor 2 (HER2)–specific chimeric antigen receptor–modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol. (2015) 33:1688. doi: 10.1200/JCO.2014.58.0225
47. Forsberg EM, Lindberg MF, Jespersen H, Alsén S, Bagge RO, Donia M, et al. HER2 CAR-T cells eradicate uveal melanoma and T-cell therapy–resistant human melanoma in IL2 transgenic NOD/SCID IL2 receptor knockout mice. Cancer Res. (2019) 79:899–904. doi: 10.1158/0008-5472.CAN-18-3158
48. Junghans RP, Ma Q, Rathore R, Gomes EM, Bais AJ, Lo AS, et al. Phase I trial of anti-PSMA designer CAR-T cells in prostate cancer: possible role for interacting interleukin 2-T cell pharmacodynamics as a determinant of clinical response. Prostate. (2016) 76:1257–70. doi: 10.1002/pros.23214
49. Martinez M, Moon EK. CAR T cells for solid tumors: new strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front Immunol. (2019) 10:128. doi: 10.3389/fimmu.2019.00128
50. Liu G, Rui W, Zhao X, Lin X. Enhancing CAR-T cell efficacy in solid tumors by targeting the tumor microenvironment. Cell Mol Immunol. (2021) 18:1085–95. doi: 10.1038/s41423-021-00655-2
51. Morris EC, Neelapu SS, Giavridis T, Sadelain M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat Rev Immunol. (2022) 22:85–96. doi: 10.1038/s41577-021-00547-6
52. Hay KA, Hanafi L-A, Li D, Gust J, Liles WC, Wurfel MM, et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor–modified T-cell therapy. Blood J Am Soc Hematol. (2017) 130:2295–306. doi: 10.1182/blood-2017-06-793141
53. Teachey DT, Lacey SF, Shaw PA, Melenhorst JJ, Maude SL, Frey N, et al. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Cancer Discovery. (2016) 6:664–79. doi: 10.1158/2159-8290.CD-16-0040
54. Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, et al. Chimeric antigen receptor T-cell therapy—assessment and management of toxicities. Nat Rev Clin Oncol. (2018) 15:47–62. doi: 10.1038/nrclinonc.2017.148
55. Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood J Am Soc Hematol. (2016) 127:3321–30. doi: 10.1182/blood-2016-04-703751
56. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. New Engl J Med. (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
57. Hunter BD, Jacobson CA. CAR T-cell associated neurotoxicity: mechanisms, clinicopathologic correlates, and future directions. JNCI: J Natl Cancer Institute. (2019) 111:646–54. doi: 10.1093/jnci/djz017
58. Nastoupil LJ, Jain MD, Feng L, Spiegel JY, Ghobadi A, Lin Y, et al. Standard-of-care axicabtagene ciloleucel for relapsed or refractory large B-cell lymphoma: results from the US Lymphoma CAR T Consortium. J Clin Oncol. (2020) 38:3119. doi: 10.1200/JCO.19.02104
59. Gust J, Hay KA, Hanafi L-A, Li D, Myerson D, Gonzalez-Cuyar LF, et al. Endothelial activation and blood–brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discovery. (2017) 7:1404–19. doi: 10.1158/2159-8290.CD-17-0698
60. Gust J, Finney OC, Li D, Brakke HM, Hicks RM, Futrell RB, et al. Glial injury in neurotoxicity after pediatric CD19-directed chimeric antigen receptor T cell therapy. Ann Neurol. (2019) 86:42–54. doi: 10.1002/ana.25502
61. Santomasso BD, Park JH, Salloum D, Riviere I, Flynn J, Mead E, et al. Clinical and biological correlates of neurotoxicity associated with CAR T-cell therapy in patients with B-cell acute lymphoblastic leukemia. Cancer Discovery. (2018) 8:958–71. doi: 10.1158/2159-8290.CD-17-1319
62. Schubert M-L, Schmitt M, Wang L, Ramos C, Jordan K, Müller-Tidow C, et al. Side-effect management of chimeric antigen receptor (CAR) T-cell therapy. Ann Oncol. (2021) 32:34–48. doi: 10.1016/j.annonc.2020.10.478
63. Fan Y, Pedersen O. Gut microbiota in human metabolic health and disease. Nat Rev Microbiol. (2021) 19:55–71. doi: 10.1038/s41579-020-0433-9
64. Feng W, Liu J, Cheng H, Zhang D, Tan Y, Peng C. Dietary compounds in modulation of gut microbiota-derived metabolites. Front Nutr. (2022) 9:939571. doi: 10.3389/fnut.2022.939571
65. Chung WSF, Walker AW, Louis P, Parkhill J, Vermeiren J, Bosscher D, et al. Modulation of the human gut microbiota by dietary fibres occurs at the species level. BMC Biol. (2016) 14:1–13. doi: 10.1186/s12915-015-0224-3
66. Zhang L, Liu Y, Sun Y, Zhang X. Combined physical exercise and diet: regulation of gut microbiota to prevent and treat of metabolic disease: a review. Nutrients. (2022) 14:4774. doi: 10.3390/nu14224774
67. Campaniello D, Corbo MR, Sinigaglia M, Speranza B, Racioppo A, Altieri C, et al. How diet and physical activity modulate gut microbiota: Evidence, and perspectives. Nutrients. (2022) 14:2456. doi: 10.3390/nu14122456
68. Li J, Sung CYJ, Lee N, Ni Y, Pihlajamäki J, Panagiotou G, et al. Probiotics modulated gut microbiota suppresses hepatocellular carcinoma growth in mice. Proc Natl Acad Sci. (2016) 113:E1306–E15. doi: 10.1073/pnas.1518189113
69. Mu C, Zhu W. Antibiotic effects on gut microbiota, metabolism, and beyond. Appl Microbiol Biotechnol. (2019) 103:9277–85. doi: 10.1007/s00253-019-10165-x
70. Kiecka A, Szczepanik M. Proton pump inhibitor-induced gut dysbiosis and immunomodulation: current knowledge and potential restoration by probiotics. Pharmacol Rep. (2023) 75:791–804. doi: 10.1007/s43440-023-00489-x
71. Gui X, Yang Z, Li MD. Effect of cigarette smoke on gut microbiota: state of knowledge. Front Physiol. (2021) 12:673341. doi: 10.3389/fphys.2021.673341
72. Westfall S, Caracci F, Estill M, Frolinger T, Shen L, Pasinetti GM. Chronic stress-induced depression and anxiety priming modulated by gut-brain-axis immunity. Front Immunol. (2021) 12:670500. doi: 10.3389/fimmu.2021.670500
73. Schwabe RF, Jobin C. The microbiome and cancer. Nat Rev Cancer. (2013) 13:800–12. doi: 10.1038/nrc3610
74. Whisner CM, Athena Aktipis C. The role of the microbiome in cancer initiation and progression: how microbes and cancer cells utilize excess energy and promote one another’s growth. Curr Nutr Rep. (2019) 8:42–51. doi: 10.1007/s13668-019-0257-2
75. Zitvogel L, Daillère R, Roberti MP, Routy B, Kroemer G. Anticancer effects of the microbiome and its products. Nat Rev Microbiol. (2017) 15:465–78. doi: 10.1038/nrmicro.2017.44
76. Scott AJ, Merrifield CA, Younes JA, Pekelharing EP. Pre-, pro-and synbiotics in cancer prevention and treatment—a review of basic and clinical research. Ecancermedicalscience. (2018) 12:869. doi: 10.3332/ecancer.2018.869
77. Bashiardes S, Tuganbaev T, Federici S, Elinav E. The microbiome in anti-cancer therapy. Semin Immunol. (2017) 32:74–81. doi: 10.1016/j.smim.2017.04.001
78. Ma W, Mao Q, Xia W, Dong G, Yu C, Jiang F. Gut microbiota shapes the efficiency of cancer therapy. Front Microbiol. (2019) 10:1050. doi: 10.3389/fmicb.2019.01050
79. Alexander JL, Wilson ID, Teare J, Marchesi JR, Nicholson JK, Kinross JM. Gut microbiota modulation of chemotherapy efficacy and toxicity. Nat Rev Gastroenterol Hepatol. (2017) 14:356–65. doi: 10.1038/nrgastro.2017.20
80. Roy S, Trinchieri G. Microbiota: a key orchestrator of cancer therapy. Nat Rev Cancer. (2017) 17:271–85. doi: 10.1038/nrc.2017.13
81. Fujita K-i, Sparreboom A. Pharmacogenetics of irinotecan disposition and toxicity: a review. Curr Clin Pharmacol. (2010) 5:209–17. doi: 10.2174/157488410791498806
82. Guthrie L, Gupta S, Daily J, Kelly L. Human microbiome signatures of differential colorectal cancer drug metabolism. NPJ Biofilms Microbiomes. (2017) 3:27. doi: 10.1038/s41522-017-0034-1
83. Wang J, Feng W, Zhang S, Chen L, Tang F, Sheng Y, et al. Gut microbial modulation in the treatment of chemotherapy-induced diarrhea with Shenzhu Capsule. BMC Complementary Altern Med. (2019) 19:1–12. doi: 10.1186/s12906-019-2548-y
84. Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell. (2015) 28:690–714. doi: 10.1016/j.ccell.2015.10.012
85. Stringer AM, Gibson RJ, Logan RM, Bowen JM, Yeoh AS, Keefe DM. Faecal microflora and β-glucuronidase expression are altered in an irinotecan-induced diarrhea model in rats. Cancer Biol Ther. (2008) 7:1919–25. doi: 10.4161/cbt.7.12.6940
86. Wallace BD, Wang H, Lane KT, Scott JE, Orans J, Koo JS, et al. Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science. (2010) 330:831–5. doi: 10.1126/science.1191175
87. Fijlstra M, Ferdous M, Koning AM, Rings EH, Harmsen HJ, Tissing WJ. Substantial decreases in the number and diversity of microbiota during chemotherapy-induced gastrointestinal mucositis in a rat model. Supportive Care Cancer. (2015) 23:1513–22. doi: 10.1007/s00520-014-2487-6
88. Hong B-Y, Sobue T, Choquette L, Dupuy AK, Thompson A, Burleson JA, et al. Chemotherapy-induced oral mucositis is associated with detrimental bacterial dysbiosis. Microbiome. (2019) 7:1–18. doi: 10.1186/s40168-019-0679-5
89. Stringer AM, Gibson RJ, Logan RM, Bowen JM, Yeoh AS, Hamilton J, et al. Gastrointestinal microflora and mucins may play a critical role in the development of 5-fluorouracil-induced gastrointestinal mucositis. Exp Biol Med. (2009) 234:430–41. doi: 10.3181/0810-RM-301
90. Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillère R, Hannani D, et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science. (2013) 342:971–6. doi: 10.1126/science.1240537
91. Daillère R, Vétizou M, Waldschmitt N, Yamazaki T, Isnard C, Poirier-Colame V, et al. Enterococcus hirae and Barnesiella intestinihominis facilitate cyclophosphamide-induced therapeutic immunomodulatory effects. Immunity. (2016) 45:931–43. doi: 10.1016/j.immuni.2016.09.009
92. Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science. (2013) 342:967–70. doi: 10.1126/science.1240527
93. Xu X, Zhang X. Effects of cyclophosphamide on immune system and gut microbiota in mice. Microbiological Res. (2015) 171:97–106. doi: 10.1016/j.micres.2014.11.002
94. Baskar R, Dai J, Wenlong N, Yeo R, Yeoh K-W. Biological response of cancer cells to radiation treatment. Front Mol Biosci. (2014) 1:24. doi: 10.3389/fmolb.2014.00024
95. Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. (2013) 31:51–72. doi: 10.1146/annurev-immunol-032712-100008
96. Barker HE, Paget JT, Khan AA, Harrington KJ. The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat Rev Cancer. (2015) 15:409–25. doi: 10.1038/nrc3958
97. Cui M, Xiao H, Luo D, Zhang X, Zhao S, Zheng Q, et al. Circadian rhythm shapes the gut microbiota affecting host radiosensitivity. Int J Mol Sci. (2016) 17:1786. doi: 10.3390/ijms17111786
98. Shiao SL, Kershaw KM, Limon JJ, You S, Yoon J, Ko EY, et al. Commensal bacteria and fungi differentially regulate tumor responses to radiation therapy. Cancer Cell. (2021) 39:1202–13.e6. doi: 10.1016/j.ccell.2021.07.002
99. Digomann D, Kurth I, Tyutyunnykova A, Chen O, Löck S, Gorodetska I, et al. The CD98 heavy chain is a marker and regulator of head and neck squamous cell carcinoma radiosensitivity. Clin Cancer Res. (2019) 25:3152–63. doi: 10.1158/1078-0432.CCR-18-2951
100. Ferreira MR, Muls A, Dearnaley DP, Andreyev HJN. Microbiota and radiation-induced bowel toxicity: lessons from inflammatory bowel disease for the radiation oncologist. Lancet Oncol. (2014) 15:e139–e47. doi: 10.1016/S1470-2045(13)70504-7
101. Wang A, Ling Z, Yang Z, Kiela PR, Wang T, Wang C, et al. Gut microbial dysbiosis may predict diarrhea and fatigue in patients undergoing pelvic cancer radiotherapy: a pilot study. PloS One. (2015) 10:e0126312. doi: 10.1371/journal.pone.0126312
102. Cui M, Xiao H, Li Y, Zhou L, Zhao S, Luo D, et al. Faecal microbiota transplantation protects against radiation-induced toxicity. EMBO Mol Med. (2017) 9:448–61. doi: 10.15252/emmm.201606932
103. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. (2018) 359:1350–5. doi: 10.1126/science.aar4060
104. Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, et al. Pembrolizumab versus chemotherapy for PD-L1–positive non–small-cell lung cancer. New Engl J Med. (2016) 375:1823–33. doi: 10.1056/NEJMoa1606774
105. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. New Engl J Med. (2015) 372:2509–20. doi: 10.1056/NEJMoa1500596
106. Morad G, Helmink BA, Sharma P, Wargo JA. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell. (2021) 184:5309–37. doi: 10.1016/j.cell.2021.09.020
107. Paulos CM, Wrzesinski C, Kaiser A, Hinrichs CS, Chieppa M, Cassard L, et al. Microbial translocation augments the function of adoptively transferred self/tumor-specific CD8+ T cells via TLR4 signaling. J Clin Invest. (2007) 117:2197–204. doi: 10.1172/JCI32205
108. Vétizou M, Pitt JM, Daillère R, Lepage P, Waldschmitt N, Flament C, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. (2015) 350:1079–84. doi: 10.1126/science.aad1329
109. Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews M, Karpinets T, et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science. (2018) 359:97–103. doi: 10.1126/science.aan4236
110. Routy B, Le Chatelier E, Derosa L, Duong CP, Alou MT, Daillère R, et al. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science. (2018) 359:91–7. doi: 10.1126/science.aan3706
111. Tanoue T, Morita S, Plichta DR, Skelly AN, Suda W, Sugiura Y, et al. A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature. (2019) 565:600–5. doi: 10.1038/s41586-019-0878-z
112. Stein-Thoeringer CK, Saini NY, Zamir E, Blumenberg V, Schubert M-L, Mor U, et al. A non-antibiotic-disrupted gut microbiome is associated with clinical responses to CD19-CAR-T cell cancer immunotherapy. Nat Med. (2023) 29:906–16. doi: 10.1038/s41591-023-02234-6
113. Hu Y, Li J, Ni F, Yang Z, Gui X, Bao Z, et al. CAR-T cell therapy-related cytokine release syndrome and therapeutic response is modulated by the gut microbiome in hematologic Malignancies. Nat Commun. (2022) 13:5313. doi: 10.1038/s41467-022-32960-3
114. Elkrief A, Derosa L, Kroemer G, Zitvogel L, Routy B. The negative impact of antibiotics on outcomes in cancer patients treated with immunotherapy: a new independent prognostic factor? Ann Oncol. (2019) 30:1572–9. doi: 10.1093/annonc/mdz206
115. Gough E, Shaikh H, Manges AR. Systematic review of intestinal microbiota transplantation (fecal bacteriotherapy) for recurrent Clostridium difficile infection. Clin Infect Diseases. (2011) 53:994–1002. doi: 10.1093/cid/cir632
116. Merrick B, Allen L, Zain NMM, Forbes B, Shawcross DL, Goldenberg SD. Regulation, risk and safety of faecal microbiota transplant. Infect Prev Practice. (2020) 2:100069. doi: 10.1016/j.infpip.2020.100069
117. Davar D, Dzutsev AK, McCulloch JA, Rodrigues RR, Chauvin J-M, Morrison RM, et al. Fecal microbiota transplant overcomes resistance to anti–PD-1 therapy in melanoma patients. Science. (2021) 371:595–602. doi: 10.1126/science.abf3363
118. Baruch EN, Youngster I, Ben-Betzalel G, Ortenberg R, Lahat A, Katz L, et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science. (2021) 371:602–9. doi: 10.1126/science.abb5920
119. Routy B, Lenehan JG, Miller WH Jr., Jamal R, Messaoudene M, Daisley BA, et al. Fecal microbiota transplantation plus anti-PD-1 immunotherapy in advanced melanoma: a phase I trial. Nat Med. (2023) 29:2121–32. doi: 10.1038/s41591-023-02453-x
120. Wastyk HC, Fragiadakis GK, Perelman D, Dahan D, Merrill BD, Feiqiao BY, et al. Gut-microbiota-targeted diets modulate human immune status. Cell. (2021) 184:4137–53.e14. doi: 10.1016/j.cell.2021.06.019
121. Weber DD, Aminazdeh-Gohari S, Kofler B. Ketogenic diet in cancer therapy. Aging (Albany NY). (2018) 10:164. doi: 10.18632/aging.101382
122. Chen J, Su Z, Wang G. Dietary reprogram of macrophages for cell competition: a promising strategy for Malignant cancer control. Signal Transduction Targeted Ther. (2023) 8:411. doi: 10.1038/s41392-023-01664-5
123. Talib WH, Mahmod AI, Kamal A, Rashid HM, Alashqar AM, Khater S, et al. Ketogenic diet in cancer prevention and therapy: molecular targets and therapeutic opportunities. Curr Issues Mol Biol. (2021) 43:558–89. doi: 10.3390/cimb43020042
124. Plotti F, Terranova C, Luvero D, Bartolone M, Messina G, Feole L, et al. Diet and chemotherapy: the effects of fasting and ketogenic diet on cancer treatment. Chemotherapy. (2020) 65:77–84. doi: 10.1159/000510839
125. Kanarek N, Petrova B, Sabatini DM. Dietary modifications for enhanced cancer therapy. Nature. (2020) 579:507–17. doi: 10.1038/s41586-020-2124-0
126. Spencer CN, McQuade JL, Gopalakrishnan V, McCulloch JA, Vetizou M, Cogdill AP, et al. Dietary fiber and probiotics influence the gut microbiome and melanoma immunotherapy response. Science. (2021) 374:1632–40. doi: 10.1126/science.aaz7015
127. Eriau E, Paillet J, Kroemer G, Pol JG. Metabolic reprogramming by reduced calorie intake or pharmacological caloric restriction mimetics for improved cancer immunotherapy. Cancers. (2021) 13:1260. doi: 10.3390/cancers13061260
128. Ferrere G, Alou MT, Liu P, Goubet A-G, Fidelle M, Kepp O, et al. Ketogenic diet and ketone bodies enhance the anticancer effects of PD-1 blockade. JCI Insight. (2021) 6(2):e145207. doi: 10.1172/jci.insight.145207
129. Beukema M, Faas MM, de Vos P. The effects of different dietary fiber pectin structures on the gastrointestinal immune barrier: impact via gut microbiota and direct effects on immune cells. Exp Mol Med. (2020) 52:1364–76. doi: 10.1038/s12276-020-0449-2
130. Le Bastard Q, Chapelet G, Javaudin F, Lepelletier D, Batard E, Montassier E. The effects of inulin on gut microbial composition: a systematic review of evidence from human studies. Eur J Clin Microbiol Infect Diseases. (2020) 39:403–13. doi: 10.1007/s10096-019-03721-w
131. Han K, Nam J, Xu J, Sun X, Huang X, Animasahun O, et al. Generation of systemic antitumour immunity via the in situ modulation of the gut microbiome by an orally administered inulin gel. Nat Biomed Eng. (2021) 5:1377–88. doi: 10.1038/s41551-021-00749-2
132. Zhang S-L, Mao Y-Q, Zhang Z-Y, Li Z-M, Kong C-Y, Chen H-L, et al. Pectin supplement significantly enhanced the anti-PD-1 efficacy in tumor-bearing mice humanized with gut microbiota from patients with colorectal cancer. Theranostics. (2021) 11:4155. doi: 10.7150/thno.54476
133. Kristensen NB, Bryrup T, Allin KH, Nielsen T, Hansen TH, Pedersen O. Alterations in fecal microbiota composition by probiotic supplementation in healthy adults: a systematic review of randomized controlled trials. Genome Med. (2016) 8:1–11. doi: 10.1186/s13073-016-0300-5
134. Okawa T, Niibe H, Arai T, Sekiba K, Noda K, Takeuchi S, et al. Effect of LC9018 combined with radiation therapy on carcinoma of the uterine cervix. A phase III, multicenter, randomized, controlled study. Cancer. (1993) 72:1949–54. doi: 10.1002/1097-0142(19930915)72:6<1949::AID-CNCR2820720626>3.0.CO;2-W
135. Dizman N, Meza L, Bergerot P, Alcantara M, Dorff T, Lyou Y, et al. Nivolumab plus ipilimumab with or without live bacterial supplementation in metastatic renal cell carcinoma: a randomized phase 1 trial. Nat Med. (2022) 28:704–12. doi: 10.1038/s41591-022-01694-6
136. Bernard NJ. Probiotics boost immunotherapy. Nat Immunol. (2023) 24(5):732. doi: 10.1038/s41590-023-01512-2
137. Badgeley A, Anwar H, Modi K, Murphy P, Lakshmikuttyamma A. Effect of probiotics and gut microbiota on anti-cancer drugs: Mechanistic perspectives. Biochim Biophys Acta (BBA)-Reviews Cancer. (2021) 1875:188494. doi: 10.1016/j.bbcan.2020.188494
138. Agus A, Clément K, Sokol H. Gut microbiota-derived metabolites as central regulators in metabolic disorders. Gut. (2021) 70:1174–82. doi: 10.1136/gutjnl-2020-323071
139. Chen L, Zhernakova DV, Kurilshikov A, Andreu-Sánchez S, Wang D, Augustijn HE, et al. Influence of the microbiome, diet and genetics on inter-individual variation in the human plasma metabolome. Nat Med. (2022) 28:2333–43. doi: 10.1038/s41591-022-02014-8
140. Den Besten G, Van Eunen K, Groen AK, Venema K, Reijngoud D-J, Bakker BM. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res. (2013) 54:2325–40. doi: 10.1194/jlr.R036012
141. Yang Q, Wang B, Zheng Q, Li H, Meng X, Zhou F, et al. A review of gut microbiota-derived metabolites in tumor progression and cancer therapy. Advanced Science. (2023) 10:2207366. doi: 10.1002/advs.202207366
142. Cai J, Sun L, Gonzalez FJ. Gut microbiota-derived bile acids in intestinal immunity, inflammation, and tumorigenesis. Cell Host Microbe. (2022) 30:289–300. doi: 10.1016/j.chom.2022.02.004
143. Hou H, Chen D, Zhang K, Zhang W, Liu T, Wang S, et al. Gut microbiota-derived short-chain fatty acids and colorectal cancer: Ready for clinical translation? Cancer Lett. (2022) 526:225–35. doi: 10.1016/j.canlet.2021.11.027
144. Koh A, De Vadder F, Kovatcheva-Datchary P, Bäckhed F. From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell. (2016) 165:1332–45. doi: 10.1016/j.cell.2016.05.041
145. Kim M, Qie Y, Park J, Kim CH. Gut microbial metabolites fuel host antibody responses. Cell Host Microbe. (2016) 20:202–14.
146. He Y, Fu L, Li Y, Wang W, Gong M, Zhang J, et al. Gut microbial metabolites facilitate anticancer therapy efficacy by modulating cytotoxic CD8+ T cell immunity. Cell Metab. (2021) 33:988–1000.e7. doi: 10.1016/j.cmet.2021.03.002
147. Rangan P, Mondino A. Microbial short-chain fatty acids: a strategy to tune adoptive T cell therapy. J ImmunoTherapy Cancer. (2022) 10(7):e004147. doi: 10.1136/jitc-2021-004147
148. Luu M, Riester Z, Baldrich A, Reichardt N, Yuille S, Busetti A, et al. Microbial short-chain fatty acids modulate CD8+ T cell responses and improve adoptive immunotherapy for cancer. Nat Commun. (2021) 12:4077. doi: 10.1038/s41467-021-24331-1
149. Liu Y, Zhou N, Zhou L, Wang J, Zhou Y, Zhang T, et al. IL-2 regulates tumor-reactive CD8+ T cell exhaustion by activating the aryl hydrocarbon receptor. Nat Immunol. (2021) 22:358–69. doi: 10.1038/s41590-020-00850-9
Keywords: gut microbiota, CAR-T cell therapy, immunotherapy, antitumor efficacy, toxicity
Citation: Zhang P-F and Xie D (2024) Targeting the gut microbiota to enhance the antitumor efficacy and attenuate the toxicity of CAR-T cell therapy: a new hope? Front. Immunol. 15:1362133. doi: 10.3389/fimmu.2024.1362133
Received: 27 December 2023; Accepted: 05 March 2024;
Published: 15 March 2024.
Edited by:
Sonia G. Sáyago-Ayerdi, Instituto Tecnológico de Tepic, MexicoReviewed by:
Océane C. B. Martin, Université de Bordeaux, FranceYuho Najima, Tokyo Metropolitan Komagome Hospital, Japan
Copyright © 2024 Zhang and Xie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peng-Fei Zhang, fly_121988@126.com
†These authors have contributed equally to this work