 Simran Venkatraman1
Simran Venkatraman1 Brinda Balasubramanian2
Brinda Balasubramanian2 Chanitra Thuwajit3
Chanitra Thuwajit3 Jaroslaw Meller4,5,6
Jaroslaw Meller4,5,6 Rutaiwan Tohtong1*
Rutaiwan Tohtong1* Somchai Chutipongtanate4,7*
Somchai Chutipongtanate4,7*- 1Department of Biochemistry, Faculty of Science, Mahidol University, Bangkok, Thailand
- 2Division of Cancer and Stem Cells, Biodiscovery Institute, School of Medicine, University of Nottingham, Nottingham, United Kingdom
- 3Department of Immunology, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand
- 4Department of Environmental and Public Health Sciences, University of Cincinnati College of Medicine, Cincinnati, OH, United States
- 5Department of Biomedical Informatics, University of Cincinnati College of Medicine, Cincinnati, OH, United States
- 6Division of Biomedical Informatics, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, United States
- 7Milk, microbiome, Immunity and Lactation research for Child Health (MILCH) and Novel Therapeutics Lab, Division of Epidemiology, Department of Environmental and Public Health Sciences, University of Cincinnati College of Medicine, Cincinnati, OH, United States
MYC activation is a known hallmark of cancer as it governs the gene targets involved in various facets of cancer progression. Of interest, MYC governs oncometabolism through the interactions with its partners and cofactors, as well as cancer immunity via its gene targets. Recent investigations have taken interest in characterizing these interactions through multi-Omic approaches, to better understand the vastness of the MYC network. Of the several gene targets of MYC involved in either oncometabolism or oncoimmunology, few of them overlap in function. Prominent interactions have been observed with MYC and HIF-1α, in promoting glucose and glutamine metabolism and activation of antigen presentation on regulatory T cells, and its subsequent metabolic reprogramming. This review explores existing knowledge of the role of MYC in oncometabolism and oncoimmunology. It also unravels how MYC governs transcription and influences cellular metabolism to facilitate the induction of pro- or anti-tumoral immunity. Moreover, considering the significant roles MYC holds in cancer development, the present study discusses effective direct or indirect therapeutic strategies to combat MYC-driven cancer progression.
1 Introduction
MYC is a proto-oncogenic transcription factor that governs a myriad of cellular processes including cell proliferation, survival, DNA damage repairs, histone modifications, and cellular metabolism (1). MYC is a family of transcription factors, i.e., MYC(c-MYC), MYCN (N-Myc) and MYCL (L-Myc), all of these contain a basic helix-loop-helix structure (bHLH) and leucine zipper (LZ) structural motifs with 6 conserved regions known as the MYC homology boxes (2). MYC family shares similar functions but has distinct tissue specificity; c-MYC is ubiquitously expressed in a broad variety of tissue development, n-MYC in neural and hematopoietic tissues, and L-MYC in lungs. The bHLH structure allows the interaction of MYC with DNA, while the LZ structure allows interaction with its partner transcription factor MAX. This MYC-MAX heterodimer interacts with numerous elements to either promote or repress transcription of gene targets (3).
Dysregulation of MYC implicates a wide array of diseases including neurodegenerative diseases (4), immune disorders (5), and cancers (6). Of the known hallmarks of cancer, MYC dysregulation has been reported to result in angiogenesis (7), cell replicative immortality (8), cell invasion and migration (8), alterations in cellular energetics (9), insensitivity to growth signals (10), and evading immune recognition and programmed cell death (6, 11). Because of its multifaceted dysregulation, MYC-driven cancers are often associated with poor prognosis (12–14). The involvement of MYC in both metabolism and immune evasion is highly concerning, especially in the context of malignant transformation. MYC promotes cell proliferation under conditions that would typically prove fatal for normal cells by manipulating glucose metabolism and eluding immunosurveillance by releasing metabolites within the tumor microenvironment (TME) (15, 16). While this facet has great implications for tumor progression, it also poses a particular threat in both tumorigenesis and potential tumor recurrence (17, 18).
Estimating up to 70% of cancers are affected by MYC aberration (19, 20), MYC therefore has been perceived as one of the most valuable targets for cancer therapy. However, direct pharmacological inhibition of MYC has remained challenging due to its lack of enzymatic activity or binding sites. Hence, this has raised interest in exploring the interactome of MYC to identify druggable targets, thereby modulating MYC-dependent transcriptome. A prototype of this approach is Omomyc, a MAX-interfering peptide. Omomyc was found to halt breast cancer progression, and regressed lung cancer in preclinical models (21). Currently, clinical trials are underway to determine the safety and efficacy of this drug in non-small cell lung cancer and colorectal cancer (ClinicalTrials.gov identifier NCT04808362). The success of this proof-of-concept inhibition of protein-protein interactions of MYC encourages the development of many such small molecules in therapeutically targeting MYC.
In this review, we enumerate the recent studies that characterize the targets and partners of MYC involved in cancer metabolism and immunology. Further, we discuss current evidence of the overlap between cellular functions governed by MYC and how one function may influence another. This guides us to further unravel how MYC orchestrates cancer growth by mediating metabolism and oncoimmunology. Lastly, in the growing interest of mitigating the ‘undruggable’ nature of MYC, we discuss currently available therapeutic strategies to combat MYC, a central target in the grand scheme of cancer.
2 Key MYC partners and targets
MYC structure consists of several domains that allow binding interactions of coactivators, heterodimers, or ligases. Each of these interactors facilitates the function of MYC in carrying out various biological processes. Its organization begins with a transcription activation site, which is a conserved region known as the MYC homology box (MBI and II), followed by a proline, glutamine, threonine-rich region, two more MYC homology boxes (MBIII and IV), and lastly, a basic HLH-LZ, at the carboxy-terminal (22). Because of the various regions available for interactions, and the implication of MYC in various cellular processes and molecular functions, there is a growing interest in unraveling the vast network of MYC and its interactome. In Figure 1, we summarized the text-mined sources of MYC protein-protein interactions with key partners.
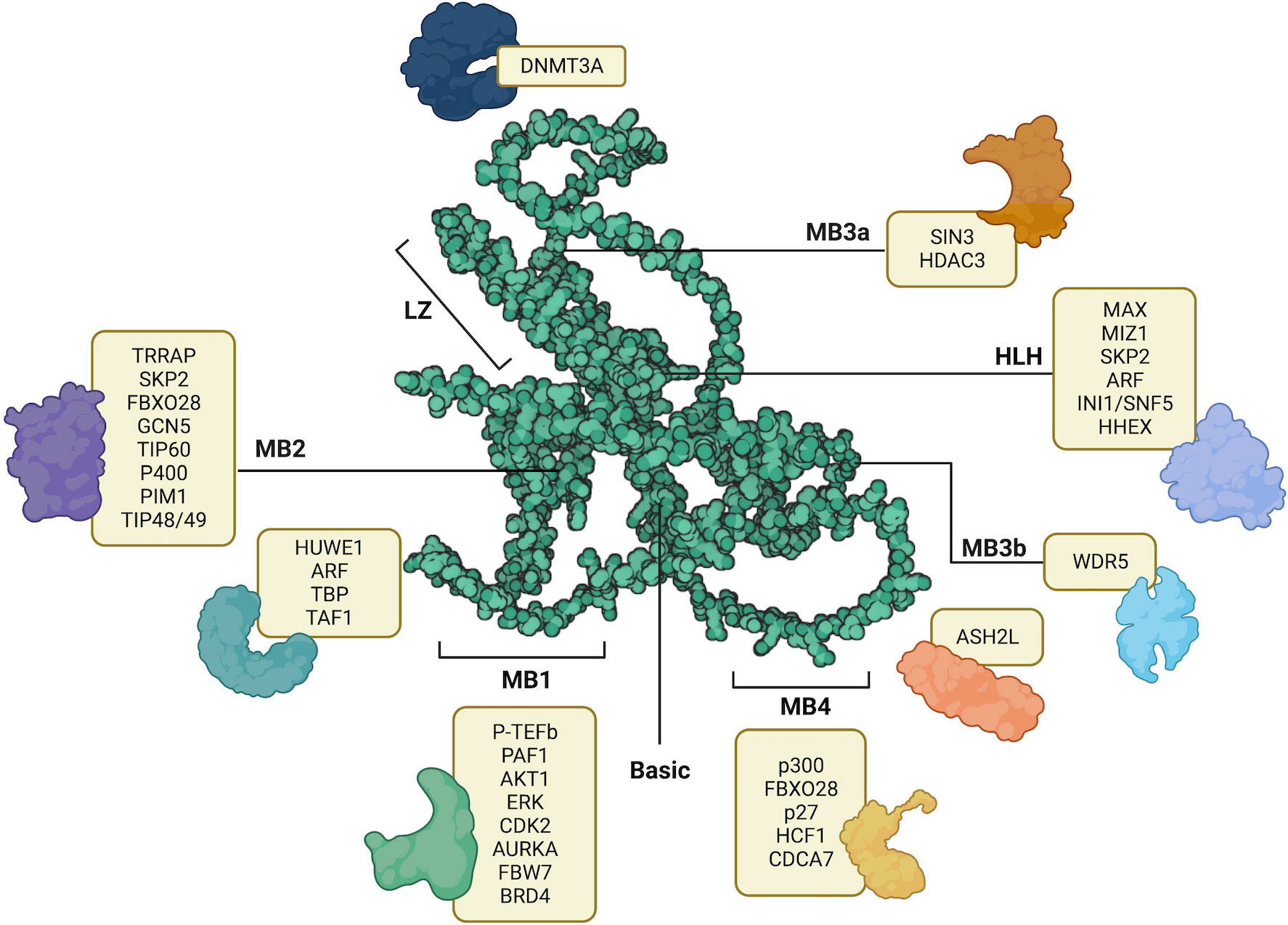
Figure 1 AlphaFold predicted structure of MYC (AF- P01106-F1) its annotated structural domains and their respective interactors. LZ – Leucine Zipper, HLH – Helix-Loop-Helix, MB1-MB4 – MYC binding boxes. Created with BioRender.com.
Investigating the mechanisms of action revealed crucial insights into MYC functions; MYC utilizes its transcription activation domain to recruit cofactors containing chromatin modifiers, specifically histone acetyltransferases (HATs). One such cofactor, p300 (EP300) HAT, was identified as having a novel functional interaction with MYC (3). Moreover, p300 was also found to interact with N-MYC in regulating cell proliferation in MYCN-amplified neuroblastoma cell lines (23). Conversely, MYC transcriptionally represses gene expression of its targets by interacting with transcription factors such as MIZ-1 and NFY-B, which facilitates the recruitment of histone deacetylases (HDACs) (24). This finding highlights the multifaceted role of MYC, whereby it acts as a regulator by binding to the promoter region of target genes and modulates DNA methylation through the recruitment of HATs and HDACs.
MYC is considered a systemic regulator of diverse functions, because of the multidomain structure and the requirement of chromatin-modulating cofactors. MYC functions as a molecular switch of activating and/or repressing the transcription of its gene targets, depending on the position at which specific cofactors bind. The transactivation domain spans the MB1 and MB2 regions (22). Within this domain, cofactors that are shown to bind and activate gene transcription include FBW7 (25, 26), TAF1 (27), TBP (27), p-TEFb (28) TRRAP (3, McMahon et al., 1998), GCN5 (29), TIP60 (30), TIP48 (31), p400 (32), and SKP2 (33). These transactivating cofactors promote the transcription of target genes related to cell proliferation and survival, including CDK4 (34), CDC25A (35), and E2F1. Moreover, beyond sustained proliferative signaling, MYC has roles in various other hallmarks of cancer mediated by its gene targets. For instance, in promoting angiogenesis, MYC binds to the promoter region of VEGFA, thereby increasing its production (36). Moreover, MYC regulates invasion and migration by inducing the transcriptional activation of LGALS1 (37).
Conversely, the repression of MYC gene targets is triggered by cofactors binding in and between the regions of MB2 and MB3, and the bHLHLZ region. Cofactors that contribute to transrepression of MYC gene targets include TIP48/49, DNMT3a (38), PRC2 (39), HDAC1 (40), HDAC3 (41), KDM4B (42), and MIZ-1 (43, 44). In the initiation and progression of cancer, the expression of tumor-suppressing genes is usually repressed. Likewise, the expression of NDRG2 (45), PTEN (46), CDKN2C (46), CDKN1A (46), p21 (47, 48), p15 (48, 49), N-cadherin (48), is repressed by MYC, and therefore suppresses tumor suppressing functions, leading to cancer progression (50). Another key determinant of MYC global transcriptional amplification and systemic activity is its abundance and regulation (2). Patange et al’s investigation reveals that the overexpression of MYC results in prolonged bursts of transcriptional activation by altering the binding affinity of transcription factors involved in the pre-initiation complex to RNA polymerase II (51). Together, the abundance of MYC and a balance of these transactivators or transrepressors, dictate the fate of cancer progression.
Of these hallmarks, cancer metabolism and oncoimmunology have garnered interest from several researchers due to the rising opportunities in therapeutic development. In this direction, MYC is a systemic regulator of diverse functions by employing various interactors. The MYC interactome extends further into oncoimmunology and oncometabolism by transcribing or repressing specific gene targets. Key interacting partners, stability partners, cofactors, and gene targets of MYC involved in tumor progression illustrated in Figure 2, in which their details are summarized in Table 1. Some of these key partners are discussed in the contexts of oncometabolism and oncoimmunology in the next section.
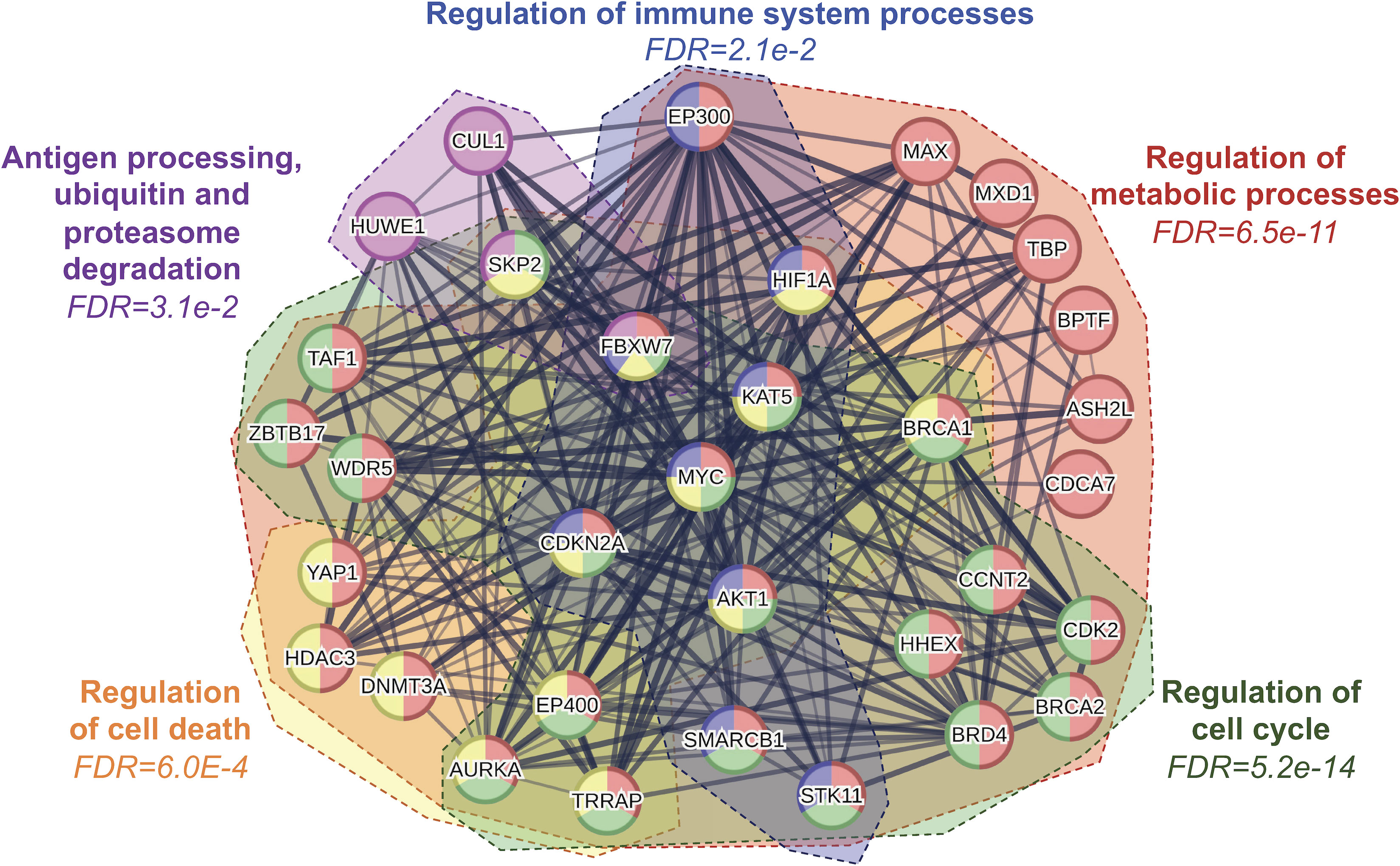
Figure 2 Protein-protein interaction network analysis of MYC and its interacting partners reveal several key regulatory processes including cellular metabolism, immune system, cell cycle and cell death. FDR, false discovery rate.
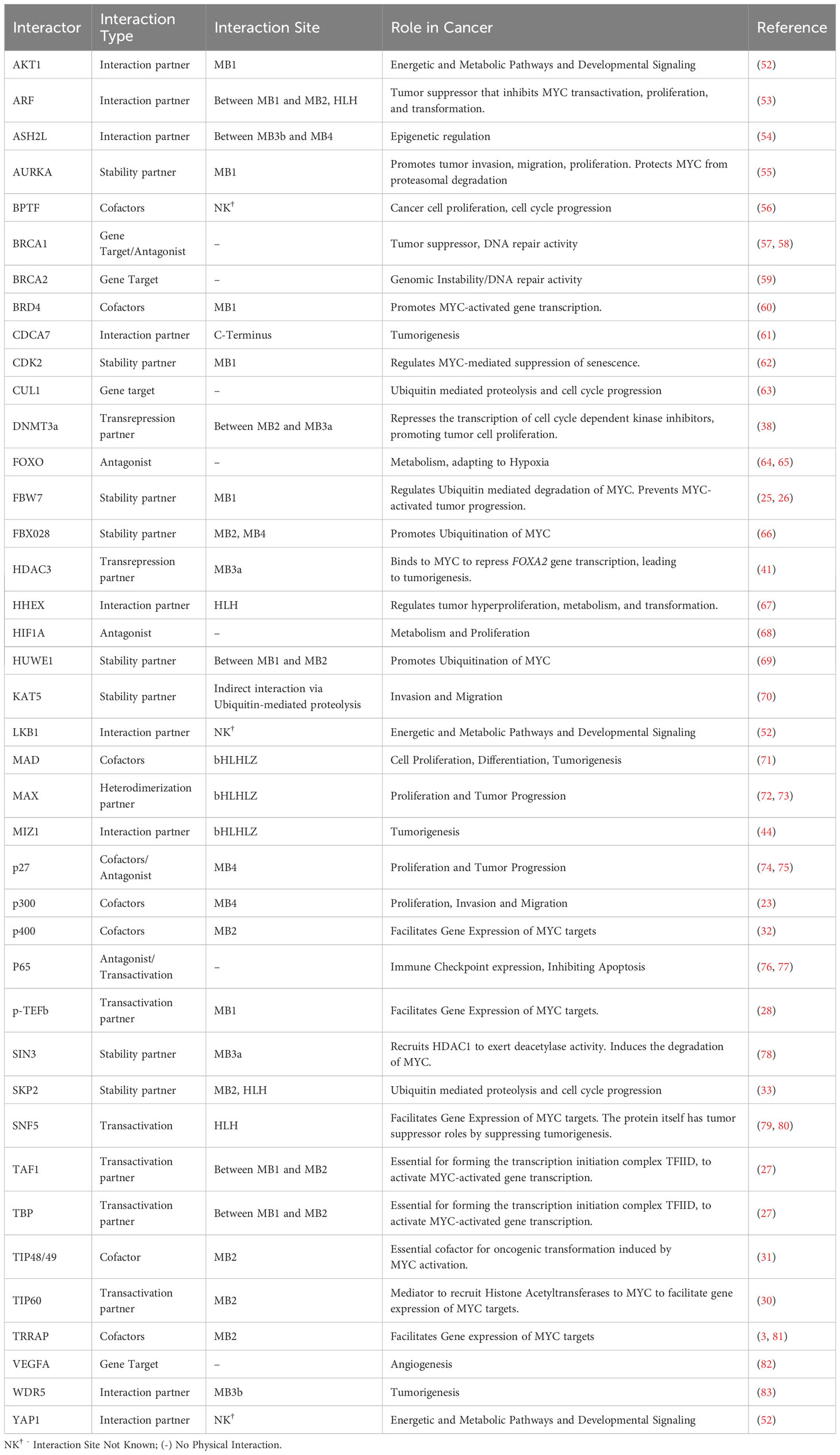
Table 1 Key partners and interactors of MYC involved in cancer.
3 MYC roles in oncometabolism and oncoimmunology
The regulatory network of MYC is extensive, spanning across gene targets and cofactors involved in various aspects of cancer development, including cellular metabolism and immunology. Aberrant cell proliferation not only requires altered energy metabolism but also evasion from immunosurveillance. Recent evidence suggests that metabolism is a key element that controls immune evasion (84–86). The following sections summarize the role of MYC in regulating key elements of oncometabolism and oncoimmunology.
3.1 MYC and cancer metabolic reprogramming
In the case of regulated cell growth and proliferation, nutrient availability is essential. Hence, there needs to be a system in place to “sense” the level of available nutrients, to regulate the metabolism of available resources and maintain the balance of homeostasis. In mammals, systemically, this regulation occurs with the storage of glucose as glycogen in the liver, and the metabolism of fat by lipolysis, in response to starvation. At a cellular level, the availability of nutrients affects the activation of mTOR, and subsequently MYC expression. In the availability of nutrients, mTOR is activated in cells, which phosphorylates PI3K-AKT and therefore inhibits FOXO, a MYC antagonist (65). The activated mTOR also enhances MYC translation and function in transcribing genes favoring cancer progression (87). However, nutrient shortage inhibits mTOR activation, which thereby yields active FOXO, that limits MYC expression and function (65).
In the process of neoplastic transformation, cancer cells require an increase in glucose uptake to energize their rapid proliferation. Interestingly, this glucose is fermented to produce lactate in the presence of oxygen in a process called the Warburg Effect to yield energy in the form of adenosine triphosphate (ATP). Several investigators revealed that this increased consumption of glucose is due to the oncogenic levels of MYC, as evidenced in Burkitt’s lymphoma (88) and MYC-driven liver carcinoma (89). This occurs by MYC upregulating various elements of the glycolytic cycle, such as the expression of glucose transporter, GLUT1 (90), glycolytic enzymes hexokinase 2 (HK2), phosphofructokinase-M1 (PFKM-1) (91), enolase-1 (ENO1) and lactate dehydrogenase A (LDHA) (92). As a result, the increased glucose uptake and metabolic glycolysis driven by MYC, leads to an accumulation of lactate. While often misconstrued as a waste product, tumors take advantage of the lactate produced by the Warburg Effect to promote various pro-oncogenic functions such as immunomodulation and angiogenesis (93). Consequently, a plausible alternative strategy is to inhibit MYC-driven metabolic reprogramming. For instance, Cargill et al. (94) reported the therapeutic potential of a small molecule inhibitor of a glycolytic enzyme, PFKFB3, in inhibiting the downstream effects of MYC in small cell lung cancer. Moreover, Zuo et al. (95), explored the use of vitamin D activated-long noncoding RNA MEG3 to suppress glycolysis by promoting c-MYC degradation in colorectal cancer. As mentioned earlier, MYC exerts control over multiple targets within the glycolytic process, these findings support a potential therapeutic approach by targeting specific components that abate MYC-driven glycolysis. The role of MYC in cancer metabolism is depicted in Figure 3.
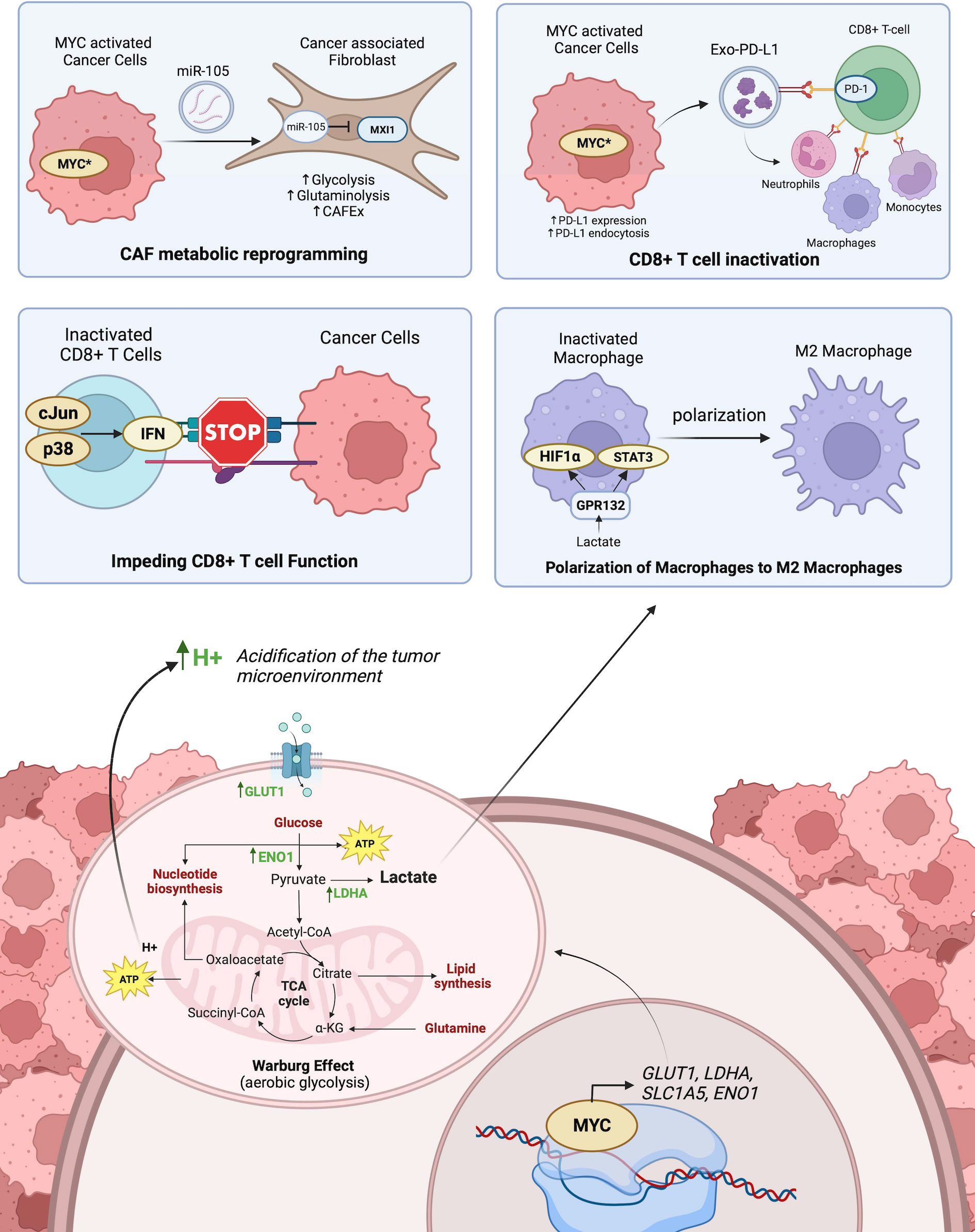
Figure 3 The role of MYC-driven transcriptional activation on cancer and immune cell metabolism and its influence on anti-tumor immunity. Top-left panel shows CAF metabolic reprogramming as a result of activated MYC in tumors exporting miR-105 which is imported into CAFs and inhibits MXI1. Top-right panel shows the inactivation of CD8+ T cells by MYC activated export of PD-L1 from tumor cells bound to PD-1 receptors on CD8+. T cells. Middle left panel shows how the acidification of the microenvironment triggers p38 and c-Jun signaling pathways in CD8+ T cells which promotes interferon-mediated inactivation of CD8+ T cell function. Middle right panel shows lactate released in the tumor microenvironment from tumor cells polarizes the differentiation of M1 macrophages to M2 macrophages. The bottom panel shows how MYC activated transcription of key enzymes promotes Warburg Effect within tumor cells. Created with BioRender.com.
In promoting glucose uptake and metabolism, NAD+ ions are produced as metabolites, which are utilized in amino acid synthesis. Cancer cells exhibit a reliance on amino acids, which promote their survival and proliferation, especially under nutritional constraints. Therefore, malignant cells hijack mechanisms to upregulate amino acid production (96).
Just as MYC is a key driver of the metabolic switch in the presence of oxygen (normoxia), HIF1A is a key driver of the metabolic switch in the absence of oxygen (hypoxia) (97). In hypoxic conditions, MYC activity is usually inhibited by HIF1a by impeding the heterodimerization of MYC/MAX complex. HIF1a and MXI1 bind to MAX, thereby yielding unbound MYC destined for degradation (68). This impediment to MYC activity subsequently affects MYC target genes involved in mitochondrial biogenesis, apoptosis, and metabolic reprogramming (98). HIF1A also impedes MYC activity by upregulating the expression of FOXO3a which binds to MYC gene target promoters (99). Notably, however, when MYC is overexpressed, it overcomes the inhibitory effects of HIF1A. Although MYC and HIF1A antagonize each other functions, they share common gene targets in glycolysis, including HK2, PFK1, ENO1, and LDHA. Additionally, when both MYC and HIF1A are overexpressed, they collaborate in promoting angiogenesis and activating the expression of their gene targets (100). Thus, both HIF1A and MYC are key therapeutic targets for cancer progression.
MYC reprograms amino acid metabolism by activating the serine and glutamine synthesis pathways. Under nutrient-deprived conditions, MYC upregulates the expression of five major enzymes in serine biosynthesis, i.e., phosphoglycerate dehydrogenase (PHGDH), phosphoserine aminotransferase 1 (PSAT1), phosphoserine phosphatase (PSPH), serine hydroxymethyltransferases 1 and 2 (SHMT1 and SHMT2). The transcriptional upregulation of these genes facilitates nucleic acid production and cell cycle progression (101, 102). Another amino acid in high demand during tumor development is glutamine. MYC upregulates glutamine synthetase (GS) to promote glutamine anabolism (103), and paradoxically, it enhances glutamine catabolism by upregulating SLC1A5 and SLC7A5 amino acid transporters (104). To facilitate the conversion of glutamine to glutamate, MYC upregulates the expression of glutaminase (GLS) (105) and represses the expression of miR-23 which interrupts GLS translation (106). The availability of amino acids has emerged as a promising therapeutic target. As a result, there has been a significant focus on developing inhibitors that specifically target enzymes involved in amino acid synthesis. For example, pharmacological inhibition of MYC-driven GLS by CB-839 has recently shown encouraging results in suppressing various cancers in vitro and in vivo (107–109), and currently examined in a phase 1 clinical trial of solid tumors (NCT02071862).
3.2 MYC and cancer immune evasion
The immune system is a highly regulated defense mechanism instated to recognize and eliminate pathogens, or dysregulated cells, to maintain a healthy body. As cancer cells propagate uncontrollably, they acquire traits to evade immune recognition. This happens by downregulating self-antigen presentation, promoting an immunosuppressive TME through the release of cytokines, recruiting pro-tumoral immune cells, and increasing the expression of inhibitory immune checkpoint molecules. MYC is reportedly a grand orchestrator of cancer growth and immune evasion, as it regulates most of these traits by modulating its gene targets (110).
In establishing a tumor-proliferative environment beneath the surveillance of anti-tumor immune cells, tumor cells must recruit and modulate regulatory immune cells. In a lung adenoma model in vivo, Kortlever et al. (72) revealed that MYC cooperated with KRAS to reprogram stromal cells via epithelial-derived CCL9 and IL-23, resulting in CCL9-mediated macrophage recruitment, PD-L1-dependent discrimination of T and B cells, and IL-23 mediated exclusion of adaptive T and B cells and innate immune NK cells. Deactivating MYC was found to reverse this reprogramming and reinstate normal anti-tumor immune function (111). Noted that MYC is upregulated in tumor-associated macrophages (TAMs), which is involved in suppressing immunosurveillance (112). Moreover, in head and neck squamous cell carcinoma (HNSCC), therapeutic inhibition of MYC promoted intrinsic anti-tumor immune responses through the cGAS-STING signaling pathway, and CD8+ T-cell infiltration of HNSCC in vivo (113). Together, this evidence shows how MYC creates the TME through the release of cytokines or modulating gene expression to promote pro-tumoral immune cell infiltration and suppress immunosurveillance.
Importantly, MYC also governs the expression of immune checkpoint molecules and self-antigens to switch off immune cell recognition of tumors. Particularly in osteosarcoma, Jiang et al. (114), observed that pharmacological inhibition of MYC resulted in reprogramming the tumor immune microenvironment through the release of T-cell recruiting chemokines and crosstalk of co-stimulatory immune checkpoint molecules CD40 and CD40L. Moreover, a recent study by Dhanasekaran et al. (115), reported that MYC transcriptionally repressed MHC-1 antigen presentation and therefore repressed T-cell immune response in MYC-driven hepatocellular carcinoma. This phenomenon was pharmacologically reversible by the dual-inhibition of immune checkpoint molecules, PD-L1 and CTLA-4. MYC directly regulates the expression of CD47 and PD-L1 through transcriptional activation (116). Other than upregulating the expression of PD-L1 as cell surface receptors, expressed PD-L1 is also packaged into vesicles for export into exo-PD-L1 (117). This exo-PD-L1 promotes immune escape by PD-1/PD-L1 mediated cytotoxic T-cell inactivation through direct interaction or indirectly by exo-PD-L1 uptake in tumor-promoting immune cells (Figure 3). Recent investigations have reported evidence of exo-PD-L1 in various cancers including prostate, breast, melanoma, and pancreatic cancer (118). Besides the regulation of immune checkpoint molecule expression, MYC is also involved in post-translational modification of immunosuppressive glycans. Smith et al. (119), recently demonstrated that MYC regulates Siglec ligands through the transcriptional regulation of ST6GALNAC4 and the induction of a glycan so-called disialylated Galβ1-3GalNAc (disialyl-T antigen). Disialyl-T functions as an inhibitory glyco-immune checkpoint molecule that “switches off” immune response in T-cells by engaging pro-tumoral macrophages. This shows that MYC systemically extends itself in creating an immunosuppressive environment by recruiting pro-tumoral macrophages, repelling anti-tumor immune cells, releasing cytokines, and modulating immune checkpoint molecules, by transcriptionally activating key gene targets.
Cumulatively, MYC systemically extends itself in creating an immunosuppressive environment by recruiting pro-tumoral macrophages, repelling anti-tumor immune cells, releasing cytokines, and modulating immune checkpoint molecules, by transcriptionally activating key gene targets.
3.3 MYC at the intersection between oncometabolism and oncoimmunology
Since MYC plays an essential role in regulating key targets involved in both metabolic reprogramming and immune evasion, it is likely that MYC induces one hallmark to influence the activation of another. Studies to date support this notion. Figure 3 summarizes the role of MYC at the interplay between cancer metabolism and oncoimmunology.
MYC promotes the Warburg Effect by upregulating glucose transporters and key glycolytic enzymes, the yield of H+ ions from NADH reduction influences the microenvironment by lowering the pH. This acidic environment facilitates cancer cells to invade the tumor stroma (120). This acidification of the microenvironment also suppresses CD8+ T lymphocyte functions, thereby promoting an immunosuppressive microenvironment. More specifically, this is mediated by activation of the p38, JNK/c-Jun signaling pathways, which promotes interferon production (121). Moreover, the lactate produced from tumors polarizes M2-tumor-associated macrophages (122). This is facilitated by the recognition of extracellular lactate levels with GPR132, and the subsequent upregulation of HIF-1α and activation of STAT3 signaling (123, 124). HIF-1α and MYC reciprocally regulate the expression of each. MYC is often seen to interact with HIF-1α in regulating T-cell metabolism by transcriptionally regulating genes involved in glucose and glutamine transport. Moreover, HIF-1α cooperates with MYC to shape the tumor immune microenvironment (100, 125). Similarly, Marchingo et al. (126), unraveled metabolic proteome changes including SLC7A5 and SLC1A5 during T cell activation governed by MYC. These are some of the ways MYC influences oncoimmunology by promoting glucose or amino acid metabolisms.
Conversely, modulating the tumor immune microenvironment also influences cellular energetics. This is particularly evidenced in the metabolic reprogramming of T lymphocytes after antigen activation. Wang et al. (127), reported the antigen activation of T lymphocytes drove the upregulation of genes encoding enzymes and transporters involved in glycolysis and glutaminolysis as governed by MYC. This antigen-activated MYC-driven metabolic reprogramming is responsible for T cell proliferation. Another investigation by Tsai et al. (128) focused on how immunoediting of the TME in early-stage tumorigenesis reprograms cancer metabolism in a way that supports immune evasion. The results suggested that interferon-gamma (IFNγ) released from T cell immunosurveillance stimulated STAT3-dependent MYC upregulation in melanoma cells, which subsequently activated genes involved in glycolysis and oxidative phosphorylation while suppressing IFNγ-induced cellular senescence (128). Besides T cells, cancer-associated fibroblasts (CAFs) are known to play an important role in regulating antitumoral immunity by recruiting the infiltration of effector T cells and modifying immunosuppressive cells (129, 130). CAFs also influence the metabolism of cancer cells through the secretion of various metabolites that fuel cancer proliferation (131). In breast cancer, MYC promotes this interaction through extracellular vesicles (EVs) containing miR-105 transported from cancer cells to CAFs (132). MiR-105 suppresses the expression of endogenous MYC inhibitor MXI1, thereby sustaining MYC activation in CAFs and subsequently facilitating glucose and glutamine metabolisms (132). Increased metabolism in CAFs yields increased lactate levels in the TME which offers an advantage for cancer cells and impedes effector T-cell function (133). While the influence of cancer metabolism on the immunosuppressive TME is well characterized, there are fewer studies exploring the reciprocal communication between cancer and stromal cells through EV molecular cargos.
Besides the dynamic abundance of MYC effecting global transcriptional changes involved in oncometabolism and oncoimmunology, MYC can modulate gene targets that induce metabolic changes influencing cancer immunity and possess dual roles in cancer development. We have compiled the summarized information in Table 2, highlighting a few notable gene targets of the exhaustive list of MYC-regulated genes involved in both oncometabolism and oncoimmunology. Further studies focusing on the gene targets and MYC-regulated gene network at the immune-metabolic crossroad shall offer novel alternative strategies to attenuate tumor invasiveness and treatment resistance caused by MYC aberration.
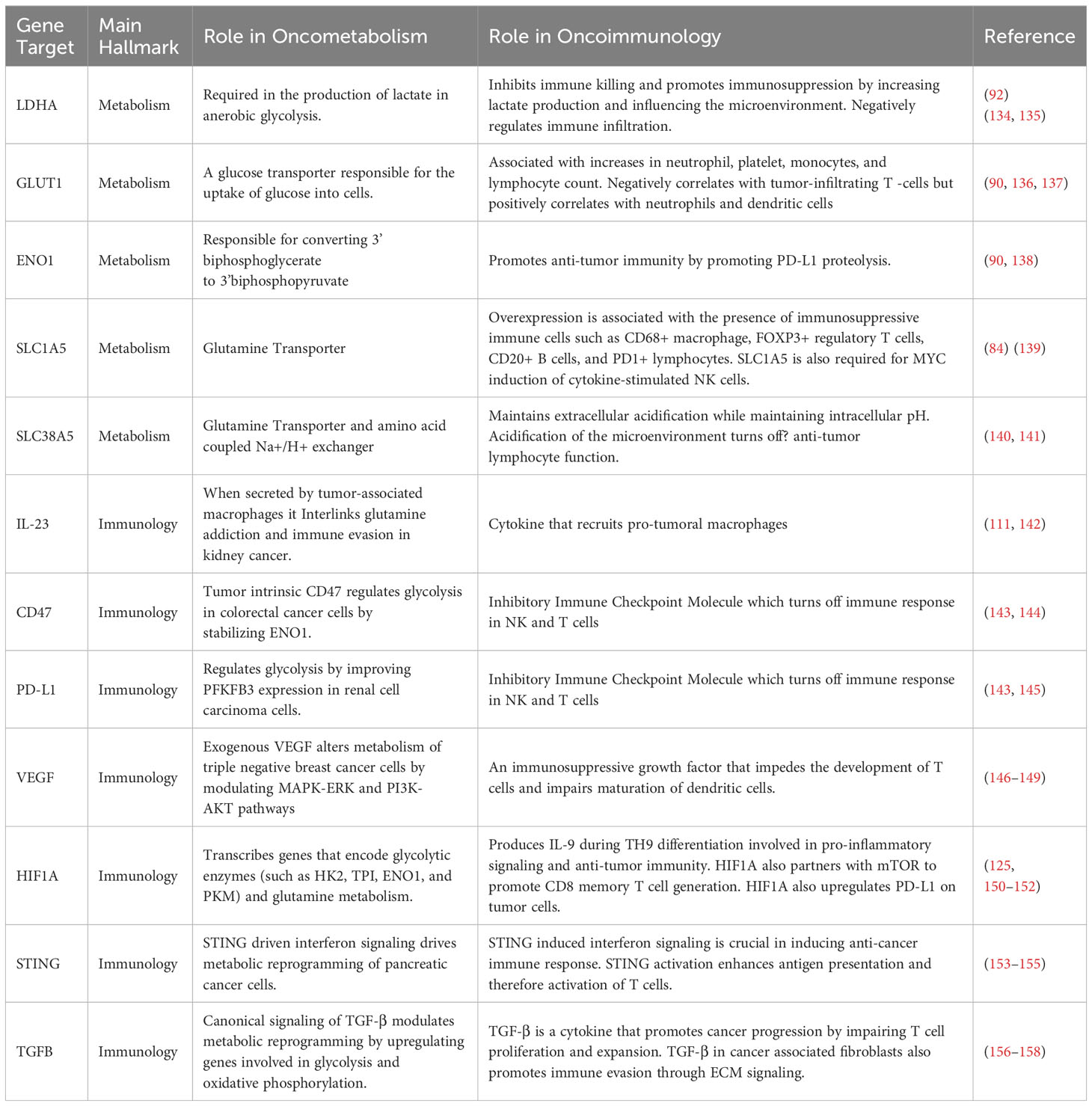
Table 2 Key gene targets of MYC in cancer metabolism and oncoimmunology.
4 Targeting MYC to tackle oncometabolism and oncoimmunology: 2 birds 1 stone?
MYC has previously been labeled “undruggable” due to its lack of an enzymatic active site and inaccessibility to its nuclear localization (20, 159). Various approaches have been employed to address the undruggable MYC through its actionable interacting partners and gene targets as illustrated in Figure 4.
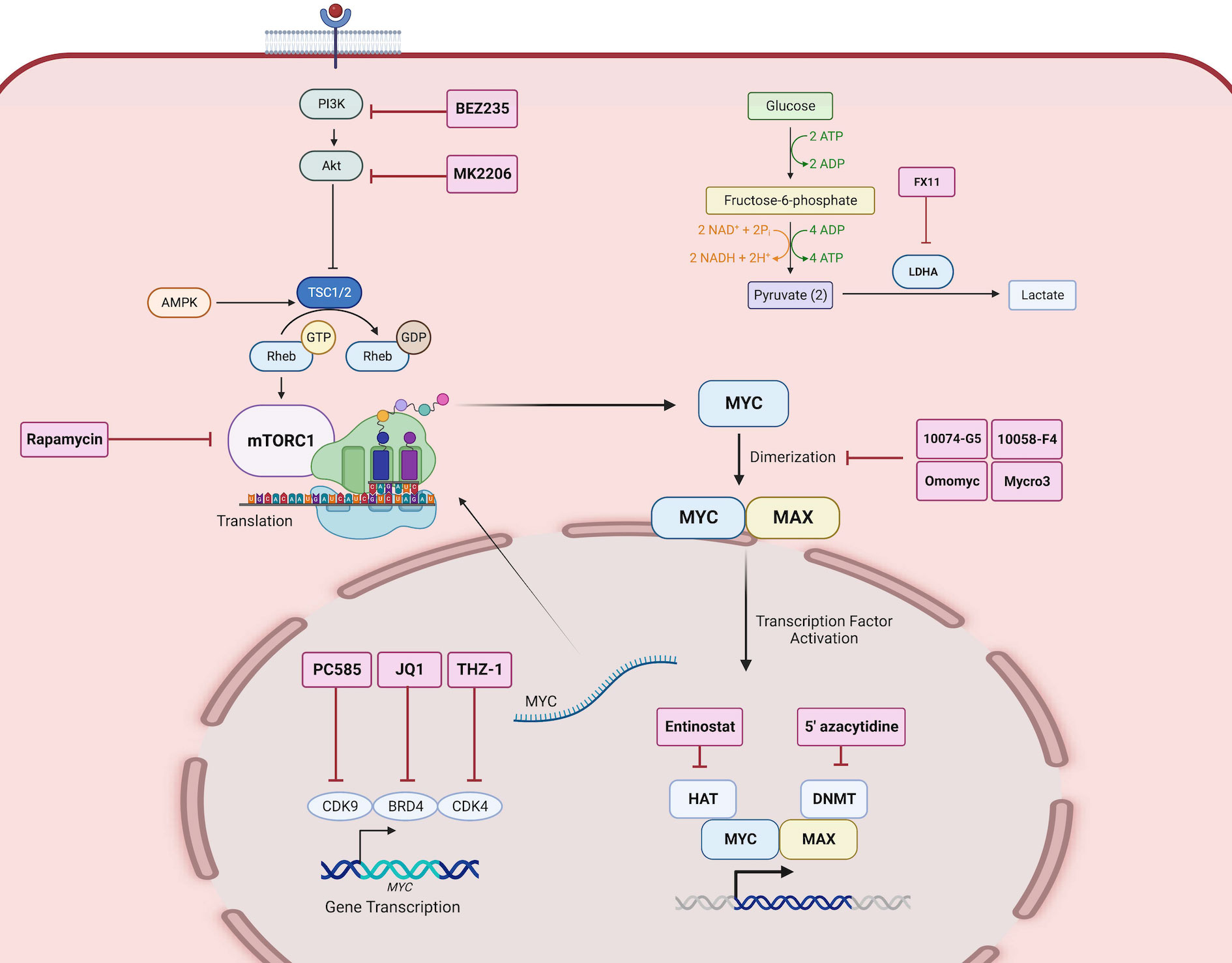
Figure 4 Direct and indirect MYC-targeted therapeutic strategies. Therapeutic inhibitors are depicted as labeled red boxes. PC585 inhibits CDK9, JQ1 inhibits BRD4, and THZ-1 inhibits CDK4, which together are key transcription factors that regulate MYC gene expression. BEZ235 is a PI3K inhibitor, MK2206 is an Akt inhibitor, and Rapamycin is a mTOR inhibitor, which together inhibit the translation of MYC. 10074-G5, 10058-F4, Omomyc, and Mycro3 inhibits the heterodimerization of MYC and MAX. Entinostat inhibits HAT and 5’azacytidine inhibits DNMT which are co-factors that aid in MYC activated transcription of gene targets. FX11 inhibits LDHA, a gene target of MYC, and thus inhibits the downstream function of MYC activation. Created with BioRender.com.
Investigators exploited the heterodimerization between MYC and MAX to inactivate MYC-activated transcription. One study showed that pharmacological inhibition of MYC by 10058-F4 resulted in changes in lipid and amino acid metabolism in neuroblastoma cell lines (160). Additionally, another MYC-MAX perturbagen, Mycro3 resulted in enhanced CD8+T cell function in surveilling cancer cells and inducing anti-tumor immune response (161). Another approach is the inhibition of MYC transcription by bromodomain-containing 4 (BRD4), using inhibitors such as JQ1 and OTX-015 (162). In medulloblastoma, the transcriptional inhibition of MYC by OTX-015 alters cancer glycolysis and amino acid metabolism (163). Moreover, the use of JQ1 in neuroblastoma, melanoma cells promoted tumor immunogenicity and potentiated immune checkpoint blockade therapy (164). However, over the decades, MYC-targeted strategies against cancers have yet to see success in clinical trials due to the half-life of MYC and the rapid metabolism of the small-molecule inhibitors (20, 165). One significant challenge has been translating in vitro findings in vivo (166), until recently.
In the advent of overcoming the limitations of current MYC inhibitor designs, Omomyc, a 90 amino acid mutant MYC peptide that disrupts the MYC-MAX dimerization, rose to clinical development (167). Omomyc has exuded various pro-apoptotic effects in various cancers, and the potential of immune reprogramming of tumors (168). However, the effect of Omomyc treatment on the metabolic reprogramming of cancers is yet to be determined. Because of its potent reduction of tumor burden, Omomyc stands as the first direct MYC inhibitor to ascend in dose-escalated phase 1 and phase 2 clinical trials of patients with non-small cell lung, colorectal, and breast cancer (NCT04808362). More recently, another phase 1 clinical trial (NCT06059001) has been initiated in metastatic pancreatic cancer. This success should encourage further improvements in this design to effectively target MYC and systemically shut down MYC-driven oncogenic pathways.
The growing body of evidence of the vastness of the “onco-MYC network” and its grave implications on cancer progression point to MYC being an ideal therapeutic target. Considering the overlap in function of the gene targets of MYC between oncometabolism and oncoimmunology, we believe that targeting MYC directly or indirectly may systemically impact both hallmarks. Several investigators have untangled the MYC network to identify indirect putative targets to combat MYC-driven effects. For example, the inhibition of LDHA, a direct gene target of MYC, by FX11, not only suppresses MYC but also inhibits MYC-induced metabolic changes (169). Moreover, inhibition of MYC-regulated glutaminase (GLS) by CB-839 also has a similar effect in reversing MYC-driven metabolic changes such as nucleotide metabolism in ovarian and glioblastoma (109, 170). Moreover, this has shown promise for clinical development in various cancers including colorectal and leukemic cancers (NCT02861300; NCT02071927).
The approach of tackling MYC gene targets has also been successful in modulating the immune evasive nature of tumors. For instance, dual inhibition of MYC targets PD-L1 and CTLA-4 reverses MYC-driven immunosuppression through pro-inflammatory macrophages in hepatocellular carcinoma (115). Moreover, MYC partners with epigenetic modulators such as histone acetylases (HAT) and DNA methylases (DNMT), in the transcriptional activation of immunosuppressive gene targets of MYC (171). Targeting, MYC-epigenetic modulators may reverse this phenomenon and exude anticancer effects. In this direction, Topper et al. (172), tested this hypothesis by combining epigenetic modulators including 5’-azacytidine and entinostat to assess its effect on tumor burden. As a result, this combination increased the number CD8+T and natural killer cells in the TME, promoted immunosurveillance of tumors, and reduced MYC-driven interferon signaling. These indirect pharmacological inhibitions are effective in modulating the downstream effects of MYC-driven tumors (172). These indirect pharmacological inhibitions are effective in modulating the downstream effects of MYC aberration as aforementioned, the therapeutic potentials of targeting gene targets at the crossroad between oncometabolism and oncoimmunology (Table 2) warrants further investigations.
5 Challenges and perspectives
Despite the success of Omomyc in preclinical models, the development of MYC-targeted therapy has miles to go until we reach the growing demand of patients who require effective treatment. The main challenge posed against all small molecule inhibitors against MYC is the rapid metabolism of the drug, and the quick half-life of MYC regeneration. One reported limitation of Omomyc is the fast distribution and catabolism, thereby limiting its use in preclinical and in vivo models (173). Other challenges include the multiple disordered conformations of the putative binding regions of MYC (174). Thus, this warrants further development in the design of MYC inhibition. Recent investigations approach this issue by using in silico tools to facilitate drug design. Using in silico tools offers a wealth of information to guide the development of a MYC-targeted therapeutic strategy. This ranges from identifying potential binding sites on MYC and predicting different drug binding conformations using molecular docking to identifying close targets or partners upstream or downstream of MYC. For instance, Yu et al. employed conformational simulation of intrinsically disordered MYC to identify binding sites and “multi-conformational” molecular docking. This guided the identification of seven compounds that bind to MYC in vitro and inhibited cell proliferation in c-MYC overexpressing cell lines (175). Moreover, in 2018, a novel inhibitor, 7594-0035 was reported to specifically target MYC indicated for the treatment of refractory multiple myeloma. The novel inhibitor was identified using the drug database ChemDiv and molecularly docked to the crystallized structure of the MYC-MAX heterodimer complexed with DNA (PDB ID: 1NKP) (176). This evidence shows promise in unmasking the elusive binding pockets of MYC by simulating the interaction between the MYC-MAX heterodimer and small molecule structures, to develop better direct inhibitors of the MYC oncoprotein.
The advent of machine learning and artificial intelligence opens opportunities for investigators to design novel peptides, predict novel binding sites on MYC, and explore indirect key partners or regulators of MYC that may be therapeutically targeted alternatively. One successful example of this approach is the discovery of novel inhibitors by Xing et al. (177). that target BRD4 which regulates the transcription of the MYC gene (Figure 4). In their investigation, a structure-based virtual screening approach with machine-learning algorithms was performed to learn the structure of the BRD4 protein and predict the likelihood of the compound inhibiting BRD4 based on its binding pattern. This led to the discovery of 15 new BRD4 inhibitors which were experimentally validated (177). This approach could be extended by integrating machine learning and molecular docking to identify binding pockets within MYC at which predicted inhibitor structures may bind. Another approach utilizes novel in silico tools to predict miRNAs capable of regulating MYC and its partners; nonetheless, only a few miRNA regulators of MYC expression, such as miR-19, have been validated (178). This presents an avenue of research yet to be claimed to expand the available therapeutic options for inhibiting MYC.
A promising approach in employing in silico tools to discover pharmacological inhibition of MYC is using pharmacogenomic connectivity analysis of cancer transcriptomes and drug sensitivity data. To this effect, the iLINCS consortium facilitates “pharmaco-multi-Omics” analysis by integrating data from transcriptomic, proteomic, phospho-proteomic, and genomic sources to drug sensitivity data from chemical perturbation or gene knockdown signatures (179). This approach may not only supplement our understanding of the potential interactors of MYC, but also of the potential mechanism of action of these small molecules against MYC. An example of this approach being successful is seen in an excellent investigation led by Howard et al. (180). In the interest of repositioning pharmacological inhibitors toward the inhibition of eIF4A1 against triple-negative breast cancer, Howard et al. (180) surveyed and screened the Prestwick Chemical Library for potential therapeutics against eIF4A1, where iLINCS pharmacogenomics was implemented to elucidate the mechanism of action of these candidate molecules. They identified that in the inhibition of eIF4A1, c-MYC is also suppressed, thus warranting further exploration of the interaction between eIF4A1 and c-MYC (180). While this investigation showed how c-MYC itself is an indirect target of some small molecules, future investigations may build on this information and identify other small molecules that impede c-MYC activity.
6 Conclusion
MYC activation is characteristic of various aggressive tumor types. This aggression is typically mediated by the crosstalk of cancer metabolism and cancer immunity. MYC is central to both hallmarks by partnering with various cofactors or transcription factors and by its gene targets. This thus presents MYC as a promising therapeutic target for cancer therapy. This review explores how MYC bridges these hallmarks by inducing metabolic reprogramming that influences an immunosuppressive microenvironment, and conversely, promoting immune evasive markers to influence immune cell and cancer cell metabolism. Moreover, the gene targets of MYC are often seen to be involved in both hallmarks and would therefore present as ideal alternative targets to combat MYC-driven effects. Direct inhibition of MYC has been challenging due to the short half-life of MYC oncoprotein and the high metabolism of the small molecule inhibitors, which has impeded the development of MYC inhibitors in clinical trials. However, Omomyc overcame these limitations, exuded potent anti-cancer effects, and has ascended toward clinical development for multiple cancers. This review surmises that MYC inhibition would be beneficial in systemically combating metabolic reprogramming and immune evasion in various cancers. Thus, we encourage more pharmacological strategies should be centered around MYC inhibition. Moreover, future investigation attention should be drawn toward elucidating the molecular mechanism behind MYC inhibition in both oncometabolism and oncoimmunology.
Author contributions
SV: Data curation, Methodology, Visualization, Writing – original draft. BB: Data curation, Methodology, Writing – review & editing. CT: Resources, Supervision, Writing – review & editing. JM: Resources, Supervision, Writing – review & editing. RT: Funding acquisition, Resources, Supervision, Writing – review & editing. SC: Conceptualization, Methodology, Supervision, Visualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research has received funding support from the NSRF via the Program Management Unit for Human Resources and Institutional Development, Research, and Innovation grant number B36G660002 (to RT) and the Mahidol Postdoctoral Fellowship grant (to SV).
Acknowledgments
All figures were created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV. MYC, metabolism, and cancer. Cancer Discovery (2015) 5:1024–39. doi: 10.1158/2159-8290.CD-15-0507
2. Das SK, Lewis BA, Levens D. MYC: a complex problem. Trends Cell Biol (2022) 33:235–46. doi: 10.1016/j.tcb.2022.07.006
3. Kalkat M, Resetca D, Lourenco C, Chan PK, Wei Y, Shiah YJ, et al. MYC protein interactome profiling reveals functionally distinct regions that cooperate to drive tumorigenesis. Mol Cell (2018) 72:836–848 e7. doi: 10.1016/j.molcel.2018.09.031
4. Marinkovic T, Marinkovic D. Obscure involvement of MYC in neurodegenerative diseases and neuronal repair. Mol Neurobiol (2021) 58:4169–77. doi: 10.1007/s12035-021-02406-w
5. Trop-Steinberg S, Azar Y. Is myc an important biomarker? Myc expression in immune disorders and cancer. Am J Med Sci (2018) 355:67–75. doi: 10.1016/j.amjms.2017.06.007
6. Gabay M, Li Y, Felsher DW. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb Perspect Med (2014) 4:a014241. doi: 10.1101/cshperspect.a014241
7. Wilson A, Murphy MJ, Oskarsson T, Kaloulis K, Bettess MD, Oser GM, et al. c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev (2004) 18:2747–63. doi: 10.1101/gad.313104
8. Liu H, Radisky DC, Yang D, Xu R, Radisky ES, Bissell MJ, et al. MYC suppresses cancer metastasis by direct transcriptional silencing of alphav and beta3 integrin subunits. Nat Cell Biol (2012) 14:567–74. doi: 10.1038/ncb2491
9. Eberlin LS, Gabay M, Fan AC, Gouw AM, Tibshirani RJ, Felsher DW, et al. Alteration of the lipid profile in lymphomas induced by MYC overexpression. Proc Natl Acad Sci USA (2014) 111:10450–5. doi: 10.1073/pnas.1409778111
10. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell (2000) 100:57–70. doi: 10.1016/S0092-8674(00)81683-9
11. Casey SC, Baylot V, Felsher DW. MYC: master regulator of immune privilege. Trends Immunol (2017) 38:298–305. doi: 10.1016/j.it.2017.01.002
12. Mossafa H, Damotte D, Jenabian A, Delarue R, Vincenneau A, Amouroux I, et al. Non-Hodgkin’s lymphomas with Burkitt-like cells are associated with c-Myc amplification and poor prognosis. Leuk Lymp (2006) 47:1885–93. doi: 10.1080/10428190600687547
13. Vita M, Henriksson M. The myc oncoprotein as A therapeutic target for human cancer. Semin Cancer Biol (2006) 16:318–30. doi: 10.1016/j.semcancer.2006.07.015
14. Qu J, Zhao X, Wang J, Liu X, Yan Y, Liu L, et al. MYC overexpression with its prognostic and clinicopathological significance in breast cancer. Oncotarget (2017) 8:93998–4008. doi: 10.18632/oncotarget.21501
15. De Jonge AV, Mutis T, Roemer MGM, Scheijen B, Chamuleau MED. Impact of MYC on anti-tumor immune responses in aggressive B cell non-hodgkin lymphomas: consequences for cancer immunotherapy. Cancers (Basel) (2020) 12:3052. doi: 10.3390/cancers12103052
16. Dong Y, Tu R, Liu H, Qing G. Regulation of cancer cell metabolism: oncogenic MYC in the driver’s seat. Signal Transduct Target Ther (2020) 5:124. doi: 10.1038/s41392-020-00235-2
17. Podsypanina K, Politi K, Beverly LJ, Varmus HE. Oncogene cooperation in tumor maintenance and tumor recurrence in mouse mammary tumors induced by Myc and mutant Kras. Proc Natl Acad Sci USA (2008) 105:5242–7. doi: 10.1073/pnas.0801197105
18. Tang HY, Goldman AR, Zhang X, Speicher DW, Dang CV. Measuring MYC-mediated metabolism in tumorigenesis. Methods Mol Biol (2021) 2318:231–9. doi: 10.1007/978-1-0716-1476-1_11
19. Carabet LA, Rennie PS, Cherkasov A. Therapeutic inhibition of myc in cancer. Structural bases and computer-aided drug discovery approaches. Int J Mol Sci (2018) 20:120. doi: 10.3390/ijms20010120
20. Llombart V, Mansour MR. Therapeutic targeting of “undruggable”. MYC. EBioMed (2022) 75:103756. doi: 10.1016/j.ebiom.2021.103756
21. Soucek L, Whitfield JR, Sodir NM, Masso-Valles D, Serrano E, Karnezis AN, et al. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev (2013) 27:504–13. doi: 10.1101/gad.205542.112
22. Conacci-Sorrell M, Mcferrin L, Eisenman RN. An overview of MYC and its interactome. Cold Spring Harb Perspect Med (2014) 4:a014357. doi: 10.1101/cshperspect.a014357
23. Cheng C, He T, Chen K, Cai Y, Gu Y, Pan L, et al. P300 interacted with N-myc and regulated its protein stability via altering its post-translational modifications in neuroblastoma. Mol Cell Proteomics (2023) 22:100504. doi: 10.1016/j.mcpro.2023.100504
24. Hann SR. MYC cofactors: molecular switches controlling diverse biological outcomes. Cold Spring Harb Perspect Med (2014) 4:a014399. doi: 10.1101/cshperspect.a014399
25. Welcker M, Orian A, Jin J, Grim JE, Harper JW, Eisenman RN, et al. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci USA (2004) 101:9085–90. doi: 10.1073/pnas.0402770101
26. Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, et al. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J (2004) 23:2116–25. doi: 10.1038/sj.emboj.7600217
27. Wei Y, Resetca D, Li Z, Johansson-Akhe I, Ahlner A, Helander S, et al. Multiple direct interactions of TBP with the MYC oncoprotein. Nat Struct Mol Biol (2019) 26:1035–43. doi: 10.1038/s41594-019-0321-z
28. Gargano B, Amente S, Majello B, Lania L. P-TEFb is a crucial co-factor for Myc transactivation. Cell Cycle (2007) 6:2031–7. doi: 10.4161/cc.6.16.4554
29. Kenneth NS, Ramsbottom BA, Gomez-Roman N, Marshall L, Cole PA, White RJ. TRRAP and GCN5 are used by c-Myc to activate RNA polymerase III transcription. Proc Natl Acad Sci USA (2007) 104:14917–22. doi: 10.1073/pnas.0702909104
30. Frank SR, Parisi T, Taubert S, Fernandez P, Fuchs M, Chan HM, et al. MYC recruits the TIP60 histone acetyltransferase complex to chromatin. EMBO Rep (2003) 4:575–80. doi: 10.1038/sj.embor.embor861
31. Wood MA, Mcmahon SB, Cole MD. An ATPase/helicase complex is an essential cofactor for oncogenic transformation by c-Myc. Mol Cell (2000) 5:321–30. doi: 10.1016/S1097-2765(00)80427-X
32. Tworkowski KA, Chakraborty AA, Samuelson AV, Seger YR, Narita M, Hannon GJ, et al. Adenovirus E1A targets p400 to induce the cellular oncoprotein Myc. Proc Natl Acad Sci USA (2008) 105:6103–8. doi: 10.1073/pnas.0802095105
33. Kim SY, Herbst A, Tworkowski KA, Salghetti SE, Tansey WP. Skp2 regulates Myc protein stability and activity. Mol Cell (2003) 11:1177–88. doi: 10.1016/S1097-2765(03)00173-4
34. Hermeking H, Rago C, Schuhmacher M, Li Q, Barrett JF, Obaya AJ, et al. Identification of CDK4 as a target of c-MYC. Proc Natl Acad Sci USA (2000) 97:2229–34. doi: 10.1073/pnas.050586197
35. Galaktionov K, Chen X, Beach D. Cdc25 cell-cycle phosphatase as a target of c-myc. Nature (1996) 382:511–7. doi: 10.1038/382511a0
36. Shi Y, Xu X, Zhang Q, Fu G, Mo Z, Wang GS, et al. tRNA synthetase counteracts c-Myc to develop functional vasculature. Elife (2014) 3:e02349. doi: 10.7554/eLife.02349
37. Yan S, Zhou C, Lou X, Xiao Z, Zhu H, Wang Q, et al. PTTG overexpression promotes lymph node metastasis in human esophageal squamous cell carcinoma. Cancer Res (2009) 69:3283–90. doi: 10.1158/0008-5472.CAN-08-0367
38. Brenner C, Deplus R, Didelot C, Loriot A, Vire E, De Smet C, et al. Myc represses transcription through recruitment of DNA methyltransferase corepressor. EMBO J (2005) 24:336–46. doi: 10.1038/sj.emboj.7600509
39. Tian X, Pelton A, Shahsafaei A, Dorfman DM. Differential expression of enhancer of zeste homolog 2 (EZH2) protein in small cell and aggressive B-cell non-Hodgkin lymphomas and differential regulation of EZH2 expression by p-ERK1/2 and MYC in aggressive B-cell lymphomas. Mod Pathol (2016) 29:1050–7. doi: 10.1038/modpathol.2016.114
40. Satou A, Taira T, Iguchi-Ariga SM, Ariga H. A novel transrepression pathway of c-Myc. Recruitment of a transcriptional corepressor complex to c-Myc by MM-1, a c-Myc-binding protein. J Biol Chem (2001) 276:46562–7. doi: 10.1074/jbc.M104937200
41. Kurland JF, Tansey WP. Myc-mediated transcriptional repression by recruitment of histone deacetylase. Cancer Res (2008) 68:3624–9. doi: 10.1158/0008-5472.CAN-07-6552
42. Yang J, Altahan AM, Hu D, Wang Y, Cheng PH, Morton CL, et al. The role of histone demethylase KDM4B in Myc signaling in neuroblastoma. J Natl Cancer Inst (2015) 107:djv080. doi: 10.1093/jnci/djv080
43. Staller P, Peukert K, Kiermaier A, Seoane J, Lukas J, Karsunky H, et al. Repression of p15INK4b expression by Myc through association with Miz-1. Nat Cell Biol (2001) 3:392–9. doi: 10.1038/35070076
44. Van Riggelen J, Muller J, Otto T, Beuger V, Yetil A, Choi PS, et al. The interaction between Myc and Miz1 is required to antagonize TGFbeta-dependent autocrine signaling during lymphoma formation and maintenance. Genes Dev (2010) 24:1281–94. doi: 10.1101/gad.585710
45. Yao L, Zhang J, Liu X. NDRG2: a Myc-repressed gene involved in cancer and cell stress. Acta Biochim Biophys Sin (Shanghai) (2008) 40:625–35. doi: 10.1111/j.1745-7270.2008.00434.x
46. Liu M, Yao B, Gui T, Guo C, Wu X, Li J, et al. PRMT5-dependent transcriptional repression of c-Myc target genes promotes gastric cancer progression. Theranostics (2020) 10:4437–52. doi: 10.7150/thno.42047
47. Wu S, Cetinkaya C, Munoz-Alonso MJ, Von Der Lehr N, Bahram F, Beuger V, et al. Myc represses differentiation-induced p21CIP1 expression via Miz-1-dependent interaction with the p21 core promoter. Oncogene (2003) 22:351–60. doi: 10.1038/sj.onc.1206145
48. Zeller KI, Jegga AG, Aronow BJ, O’donnell KA, Dang CV. An integrated database of genes responsive to the Myc oncogenic transcription factor: identification of direct genomic targets. Genome Biol (2003) 4:R69. doi: 10.1186/gb-2003-4-10-r69
49. Seoane J, Le HV, Massague J. Myc suppression of the p21(Cip1) Cdk inhibitor influences the outcome of the p53 response to DNA damage. Nature (2002) 419:729–34. doi: 10.1038/nature01119
50. Adhikary S, Eilers M. Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol (2005) 6:635–45. doi: 10.1038/nrm1703
51. Patange S, Ball DA, Wan Y, Karpova TS, Girvan M, Levens D, et al. Myc amplifies gene expression through global changes in transcription factor dynamics. Cell Rep (2022) 38:110292. doi: 10.1016/j.celrep.2021.110292
52. Mo X, Qi Q, Ivanov AA, Niu Q, Luo Y, Havel J, et al. AKT1, LKB1, and YAP1 revealed as MYC interactors with nanoLuc-based protein-fragment complementation assay. Mol Pharmacol (2017) 91:339–47. doi: 10.1124/mol.116.107623
53. Zhang Q, Spears E, Boone DN, Li Z, Gregory MA, Hann SR. Domain-specific c-Myc ubiquitylation controls c-Myc transcriptional and apoptotic activity. Proc Natl Acad Sci USA (2013) 110:978–83. doi: 10.1073/pnas.1208334110
54. Ullius A, Luscher-Firzlaff J, Costa IG, Walsemann G, Forst AH, Gusmao EG, et al. The interaction of MYC with the trithorax protein ASH2L promotes gene transcription by regulating H3K27 modification. Nucleic Acids Res (2014) 42:6901–20. doi: 10.1093/nar/gku312
55. Richards MW, Burgess SG, Poon E, Carstensen A, Eilers M, Chesler L, et al. Structural basis of N-Myc binding by Aurora-A and its destabilization by kinase inhibitors. Proc Natl Acad Sci USA (2016) 113:13726–31. doi: 10.1073/pnas.1610626113
56. Richart L, Carrillo-De Santa Pau E, Rio-Machin A, De Andres MP, Cigudosa JC, Lobo VJS, et al. BPTF is required for c-MYC transcriptional activity and in vivo tumorigenesis. Nat Commun (2016) 7:10153. doi: 10.1038/ncomms10153
57. Wang Q, Zhang H, Kajino K, Greene MI. BRCA1 binds c-Myc and inhibits its transcriptional and transforming activity in cells. Oncogene (1998) 17:1939–48. doi: 10.1038/sj.onc.1202403
58. Chen Y, Xu J, Borowicz S, Collins C, Huo D, Olopade OI. c-Myc activates BRCA1 gene expression through distal promoter elements in breast cancer cells. BMC Cancer (2011) 11:246. doi: 10.1186/1471-2407-11-246
59. Luoto KR, Meng AX, Wasylishen AR, Zhao H, Coackley CL, Penn LZ, et al. Tumor cell kill by c-MYC depletion: role of MYC-regulated genes that control DNA double-strand break repair. Cancer Res (2010) 70:8748–59. doi: 10.1158/0008-5472.CAN-10-0944
60. Devaiah BN, Mu J, Akman B, Uppal S, Weissman JD, Cheng D, et al. MYC protein stability is negatively regulated by BRD4. Proc Natl Acad Sci USA (2020) 117:13457–67. doi: 10.1073/pnas.1919507117
61. Gill RM, Gabor TV, Couzens AL, Scheid MP. The MYC-associated protein CDCA7 is phosphorylated by AKT to regulate MYC-dependent apoptosis and transformation. Mol Cell Biol (2013) 33:498–513. doi: 10.1128/MCB.00276-12
62. Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev (2000) 14:2501–14. doi: 10.1101/gad.836800
63. O’hagan RC, Ohh M, David G, De Alboran IM, Alt FW, Kaelin WG, et al. Myc-enhanced expression of Cul1 promotes ubiquitin-dependent proteolysis and cell cycle progression. Genes Dev (2000) 14:2185–91. doi: 10.1101/gad.827200
64. Bouchard C, Lee S, Paulus-Hock V, Loddenkemper C, Eilers M, Schmitt CA. FoxO transcription factors suppress Myc-driven lymphomagenesis via direct activation of Arf. Genes Dev (2007) 21:2775–87. doi: 10.1101/gad.453107
65. Peck B, Ferber EC, Schulze A. Antagonism between FOXO and MYC regulates cellular powerhouse. Front Oncol (2013) 3:96. doi: 10.3389/fonc.2013.00096
66. Cepeda D, Ng HF, Sharifi HR, Mahmoudi S, Cerrato VS, Fredlund E, et al. CDK-mediated activation of the SCF(FBXO) (28) ubiquitin ligase promotes MYC-driven transcription and tumourigenesis and predicts poor survival in breast cancer. EMBO Mol Med (2013) 5:1067–86. doi: 10.1002/emmm.201202341
67. Marfil V, Blazquez M, Serrano F, Castell JV, Bort R. Growth-promoting and tumourigenic activity of c-Myc is suppressed by Hhex. Oncogene (2015) 34:3011–22. doi: 10.1038/onc.2014.240
68. Koshiji M, Kageyama Y, Pete EA, Horikawa I, Barrett JC, Huang LE. HIF-1alpha induces cell cycle arrest by functionally counteracting Myc. EMBO J (2004) 23:1949–56. doi: 10.1038/sj.emboj.7600196
69. Adhikary S, Marinoni F, Hock A, Hulleman E, Popov N, Beier R, et al. The ubiquitin ligase HectH9 regulates transcriptional activation by Myc and is essential for tumor cell proliferation. Cell (2005) 123:409–21. doi: 10.1016/j.cell.2005.08.016
70. Wei X, Cai S, Boohaker RJ, Fried J, Li Y, Hu L, et al. KAT5 promotes invasion and metastasis through C-MYC stabilization in ATC. Endocr Relat Cancer (2019) 26:141–51. doi: 10.1530/ERC-18-0193
71. Amati B, Land H. Myc-Max-Mad: a transcription factor network controlling cell cycle progression, differentiation and death. Curr Opin Genet Dev (1994) 4:102–8. doi: 10.1016/0959-437X(94)90098-1
72. Blackwood EM, Eisenman RN. Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science (1991) 251:1211–7. doi: 10.1126/science.2006410
73. Castell A, Yan Q, Fawkner K, Hydbring P, Zhang F, Verschut V, et al. A selective high affinity MYC-binding compound inhibits MYC : MAX interaction and MYC-dependent tumor cell proliferation. Sci Rep (2018) 8:10064. doi: 10.1038/s41598-018-28107-4
74. Bouchard C, Thieke K, Maier A, Saffrich R, Hanley-Hyde J, Ansorge W, et al. Direct induction of cyclin D2 by Myc contributes to cell cycle progression and sequestration of p27. EMBO J (1999) 18:5321–33. doi: 10.1093/emboj/18.19.5321
75. Garcia-Gutierrez L, Bretones G, Molina E, Arechaga I, Symonds C, Acosta JC, et al. Myc stimulates cell cycle progression through the activation of Cdk1 and phosphorylation of p27. Sci Rep (2019) 9:18693. doi: 10.1038/s41598-019-54917-1
76. La Rosa FA, Pierce JW, Sonenshein GE. Differential regulation of the c-myc oncogene promoter by the NF-kappa B rel family of transcription factors. Mol Cell Biol (1994) 14:1039–44. doi: 10.1128/MCB.14.2.1039
77. Huang H, Ma L, Li J, Yu Y, Zhang D, Wei J, et al. NF-kappaB1 inhibits c-Myc protein degradation through suppression of FBW7 expression. Oncotarget (2014) 5:493–505. doi: 10.18632/oncotarget.1643
78. Garcia-Sanz P, Quintanilla A, Lafita MC, Moreno-Bueno G, Garcia-Gutierrez L, Tabor V, et al. Sin3b interacts with Myc and decreases Myc levels. J Biol Chem (2014) 289:22221–36. doi: 10.1074/jbc.M113.538744
79. Cheng SW, Davies KP, Yung E, Beltran RJ, Yu J, Kalpana GV. c-MYC interacts with INI1/hSNF5 and requires the SWI/SNF complex for transactivation function. Nat Genet (1999) 22:102–5. doi: 10.1038/8811
80. Weissmiller AM, Wang J, Lorey SL, Howard GC, Martinez E, Liu Q, et al. Inhibition of MYC by the SMARCB1 tumor suppressor. Nat Commun (2019) 10:2014. doi: 10.1038/s41467-019-10022-5
81. Mcmahon SB, Van Buskirk HA, Dugan KA, Copeland TD, Cole MD. The novel ATM-related protein TRRAP is an essential cofactor for the c-Myc and E2F oncoproteins. Cell (1998) 94:363–74. doi: 10.1016/S0092-8674(00)81479-8
82. Baudino TA, Mckay C, Pendeville-Samain H, Nilsson JA, Maclean KH, White EL, et al. c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev (2002) 16:2530–43. doi: 10.1101/gad.1024602
83. Thomas LR, Wang Q, Grieb BC, Phan J, Foshage AM, Sun Q, et al. Interaction with WDR5 promotes target gene recognition and tumorigenesis by MYC. Mol Cell (2015) 58:440–52. doi: 10.1016/j.molcel.2015.02.028
84. Loftus RM, Assmann N, Kedia-Mehta N, O’brien KL, Garcia A, Gillespie C, et al. Amino acid-dependent cMyc expression is essential for NK cell metabolic and functional responses in mice. Nat Commun (2018) 9:2341. doi: 10.1038/s41467-018-04719-2
85. Kim S, Jang JY, Koh J, Kwon D, Kim YA, Paeng JC, et al. Programmed cell death ligand-1-mediated enhancement of hexokinase 2 expression is inversely related to T-cell effector gene expression in non-small-cell lung cancer. J Exp Clin Cancer Res (2019) 38:462. doi: 10.1186/s13046-019-1407-5
86. Cruz-Bermudez A, Laza-Briviesca R, Casarrubios M, Sierra-Rodero B, Provencio M. The role of metabolism in tumor immune evasion: novel approaches to improve immunotherapy. Biomedicines (2021) 9:361. doi: 10.3390/biomedicines9040361
87. Csibi A, Lee G, Yoon SO, Tong H, Ilter D, Elia I, et al. The mTORC1/S6K1 pathway regulates glutamine metabolism through the eIF4B-dependent control of c-Myc translation. Curr Biol (2014) 24:2274–80. doi: 10.1016/j.cub.2014.08.007
88. Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab (2012) 15:110–21. doi: 10.1016/j.cmet.2011.12.009
89. Yuneva MO, Fan TW, Allen TD, Higashi RM, Ferraris DV, Tsukamoto T, et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab (2012) 15:157–70. doi: 10.1016/j.cmet.2011.12.015
90. Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, et al. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem (2000) 275:21797–800. doi: 10.1074/jbc.C000023200
91. Kim JW, Gao P, Liu YC, Semenza GL, Dang CV. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol Cell Biol (2007) 27:7381–93. doi: 10.1128/MCB.00440-07
92. Shim H, Dolde C, Lewis BC, Wu CS, Dang G, Jungmann RA, et al. c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proc Natl Acad Sci USA (1997) 94:6658–63. doi: 10.1073/pnas.94.13.6658
93. Perez-Tomas R, Perez-Guillen I. Lactate in the tumor microenvironment: an essential molecule in cancer progression and treatment. Cancers (Basel) (2020) 12:3244. doi: 10.3390/cancers12113244
94. Cargill KR, Stewart CA, Park EM, Ramkumar K, Gay CM, Cardnell RJ, et al. Targeting MYC-enhanced glycolysis for the treatment of small cell lung cancer. Cancer Metab (2021) 9:33. doi: 10.1186/s40170-021-00270-9
95. Zuo S, Wu L, Wang Y, Yuan X. Long Non-coding RNA MEG3 Activated by Vitamin D Suppresses Glycolysis in Colorectal Cancer via Promoting c-Myc Degradation. Front Oncol (2020) 10:274. doi: 10.3389/fonc.2020.00274
96. Wei Z, Liu X, Cheng C, Yu W, Yi P. Metabolism of amino acids in cancer. Front Cell Dev Biol (2020) 8:603837. doi: 10.3389/fcell.2020.603837
97. Lee P, Chandel NS, Simon MC. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat Rev Mol Cell Biol (2020) 21:268–83. doi: 10.1038/s41580-020-0227-y
98. Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, et al. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell (2007) 11:407–20. doi: 10.1016/j.ccr.2007.04.001
99. Jensen KS, Binderup T, Jensen KT, Therkelsen I, Borup R, Nilsson E, et al. FoxO3A promotes metabolic adaptation to hypoxia by antagonizing Myc function. EMBO J (2011) 30:4554–70. doi: 10.1038/emboj.2011.323
100. Li Y, Sun XX, Qian DZ, Dai MS. Molecular crosstalk between MYC and HIF in cancer. Front Cell Dev Biol (2020) 8:590576. doi: 10.3389/fcell.2020.590576
101. Haggerty TJ, Zeller KI, Osthus RC, Wonsey DR, Dang CV. A strategy for identifying transcription factor binding sites reveals two classes of genomic c-Myc target sites. Proc Natl Acad Sci USA (2003) 100:5313–8. doi: 10.1073/pnas.0931346100
102. Sun L, Song L, Wan Q, Wu G, Li X, Wang Y, et al. cMyc-mediated activation of serine biosynthesis pathway is critical for cancer progression under nutrient deprivation conditions. Cell Res (2015) 25:429–44. doi: 10.1038/cr.2015.33
103. Bott AJ, Peng IC, Fan Y, Faubert B, Zhao L, Li J, et al. Oncogenic myc induces expression of glutamine synthetase through promoter demethylation. Cell Metab (2015) 22:1068–77. doi: 10.1016/j.cmet.2015.09.025
104. Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell (2009) 136:521–34. doi: 10.1016/j.cell.2008.11.044
105. Wise DR, Deberardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA (2008) 105:18782–7. doi: 10.1073/pnas.0810199105
106. Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature (2009) 458:762–5. doi: 10.1038/nature07823
107. Jacque N, Ronchetti AM, Larrue C, Meunier G, Birsen R, Willems L, et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood (2015) 126:1346–56. doi: 10.1182/blood-2015-01-621870
108. Luengo A, Gui DY, Vander Heiden MG. Targeting metabolism for cancer therapy. Cell Chem Biol (2017) 24:1161–80. doi: 10.1016/j.chembiol.2017.08.028
109. Shen YA, Hong J, Asaka R, Asaka S, Hsu FC, Suryo Rahmanto Y, et al. Inhibition of the MYC-regulated glutaminase metabolic axis is an effective synthetic lethal approach for treating chemoresistant ovarian cancers. Cancer Res (2020) 80:4514–26. doi: 10.1158/0008-5472.CAN-19-3971
110. Dhanasekaran R, Deutzmann A, Mahauad-Fernandez WD, Hansen AS, Gouw AM, Felsher DW. The MYC oncogene - the grand orchestrator of cancer growth and immune evasion. Nat Rev Clin Oncol (2022) 19:23–36. doi: 10.1038/s41571-021-00549-2
111. Kortlever RM, Sodir NM, Wilson CH, Burkhart DL, Pellegrinet L, Brown Swigart L, et al. Myc cooperates with ras by programming inflammation and immune suppression. Cell (2017) 171:1301–1315 e14. doi: 10.1016/j.cell.2017.11.013
112. Gao FY, Li XT, Xu K, Wang RT, Guan XX. c-MYC mediates the crosstalk between breast cancer cells and tumor microenvironment. Cell Commun Signal (2023) 21:28. doi: 10.1186/s12964-023-01043-1
113. Liu S, Qin Z, Mao Y, Zhang W, Wang Y, Jia L, et al. Therapeutic targeting of MYC in head and neck squamous cell carcinoma. Oncoimmunology (2022) 11:2130583. doi: 10.1080/2162402X.2022.2130583
114. Jiang K, Zhang Q, Fan Y, Li J, Zhang J, Wang W, et al. MYC inhibition reprograms tumor immune microenvironment by recruiting T lymphocytes and activating the CD40/CD40L system in osteosarcoma. Cell Death Discovery (2022) 8:117. doi: 10.1038/s41420-022-00923-8
115. Dhanasekaran R, Hansen AS, Park J, Lemaitre L, Lai I, Adeniji N, et al. MYC overexpression drives immune evasion in hepatocellular carcinoma that is reversible through restoration of proinflammatory macrophages. Cancer Res (2023) 83:626–40. doi: 10.1158/0008-5472.CAN-22-0232
116. Marinkovic D, Marinkovic T. The new role for an old guy: MYC as an immunoplayer. J Cell Physiol (2021) 236:3234–43. doi: 10.1002/jcp.30123
117. Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W, et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature (2018) 560:382–6. doi: 10.1038/s41586-018-0392-8
118. Liu J, Peng X, Yang S, Li X, Huang M, Wei S, et al. Extracellular vesicle PD-L1 in reshaping tumor immune microenvironment: biological function and potential therapy strategies. Cell Commun Signal (2022) 20:14. doi: 10.1186/s12964-021-00816-w
119. Smith BAH, Deutzmann A, Correa KM, Delaveris CS, Dhanasekaran R, Dove CG, et al. MYC-driven synthesis of Siglec ligands is a glycoimmune checkpoint. Proc Natl Acad Sci USA (2023) 120:e2215376120. doi: 10.1073/pnas.2215376120
120. Estrella V, Chen T, Lloyd M, Wojtkowiak J, Cornnell HH, Ibrahim-Hashim A, et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res (2013) 73:1524–35. doi: 10.1158/0008-5472.CAN-12-2796
121. Ippolito L, Morandi A, Giannoni E, Chiarugi P. Lactate: A metabolic driver in the tumour landscape. Trends Biochem Sci (2019) 44:153–66. doi: 10.1016/j.tibs.2018.10.011
122. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature (2014) 513:559–63. doi: 10.1038/nature13490
123. Chen P, Zuo H, Xiong H, Kolar MJ, Chu Q, Saghatelian A, et al. Gpr132 sensing of lactate mediates tumor-macrophage interplay to promote breast cancer metastasis. Proc Natl Acad Sci USA (2017) 114:580–5. doi: 10.1073/pnas.1614035114
124. Wang L, Sun L, Sun H, Xing Y, Zhou S, An G, et al. GPR65 as a potential immune checkpoint regulates the immune microenvironment according to pan-cancer analysis. Heliyon (2023) 9:e13617. doi: 10.1016/j.heliyon.2023.e13617
125. Gnanaprakasam JNR, Sherman JW, Wang R. MYC and HIF in shaping immune response and immune metabolism. Cytokine Growth Factor Rev (2017) 35:63–70. doi: 10.1016/j.cytogfr.2017.03.004
126. Marchingo JM, Sinclair LV, Howden AJ, Cantrell DA. Quantitative analysis of how Myc controls T cell proteomes and metabolic pathways during T cell activation. Elife (2020) 9:e53725. doi: 10.7554/eLife.53725
127. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity (2011) 35:871–82. doi: 10.1016/j.immuni.2011.09.021
128. Tsai CH, Chuang YM, Li X, Yu YR, Tzeng SF, Teoh ST, et al. Immunoediting instructs tumor metabolic reprogramming to support immune evasion. Cell Metab (2023) 35:118–133 e7. doi: 10.1016/j.cmet.2022.12.003
129. Liu T, Han C, Wang S, Fang P, Ma Z, Xu L, et al. Cancer-associated fibroblasts: an emerging target of anti-cancer immunotherapy. J Hematol Oncol (2019) 12:86. doi: 10.1186/s13045-019-0770-1
130. Mao X, Xu J, Wang W, Liang C, Hua J, Liu J, et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol Cancer (2021) 20:131. doi: 10.1186/s12943-021-01428-1
131. Martinez-Outschoorn UE, Lisanti MP, Sotgia F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin Cancer Biol (2014) 25:47–60. doi: 10.1016/j.semcancer.2014.01.005
132. Yan W, Wu X, Zhou W, Fong MY, Cao M, Liu J, et al. Cancer-cell-secreted exosomal miR-105 promotes tumour growth through the MYC-dependent metabolic reprogramming of stromal cells. Nat Cell Biol (2018) 20:597–609. doi: 10.1038/s41556-018-0083-6
133. Comito G, Iscaro A, Bacci M, Morandi A, Ippolito L, Parri M, et al. Lactate modulates CD4(+) T-cell polarization and induces an immunosuppressive environment, which sustains prostate carcinoma progression via TLR8/miR21 axis. Oncogene (2019) 38:3681–95. doi: 10.1038/s41388-019-0688-7
134. Serganova I, Cohen IJ, Vemuri K, Shindo M, Maeda M, Mane M, et al. LDH-A regulates the tumor microenvironment via HIF-signaling and modulates the immune response. PloS One (2018) 13:e0203965. doi: 10.1371/journal.pone.0203965
135. Van Wilpe S, Koornstra R, Den Brok M, De Groot JW, Blank C, De Vries J, et al. Lactate dehydrogenase: A marker of diminished antitumor immunity. Oncoimmunology (2020) 9:1731942. doi: 10.1080/2162402X.2020.1731942
136. Li F, He C, Yao H, Liang W, Ye X, Ruan J, et al. GLUT1 Regulates the Tumor Immune Microenvironment and Promotes Tumor Metastasis in Pancreatic Adenocarcinoma via ncRNA-mediated Network. J Cancer (2022) 13:2540–58. doi: 10.7150/jca.72161
137. Park SY, Cho DG, Shim BY, Cho U. Relationship between systemic inflammatory markers, GLUT1 expression, and maximum 18F-fluorodeoxyglucose uptake in non-small cell lung carcinoma and their prognostic significance. Diagn (Basel) (2023) 13:1013. doi: 10.3390/diagnostics13061013
138. Zhang C, Zhang K, Gu J, Ge D. ENO1 promotes antitumor immunity by destabilizing PD-L1 in NSCLC. Cell Mol Immunol (2021) 18:2045–7. doi: 10.1038/s41423-021-00710-y
139. Ansari RE, Craze ML, Althobiti M, Alfarsi L, Ellis IO, Rakha EA, et al. Enhanced glutamine uptake influences composition of immune cell infiltrates in breast cancer. Br J Cancer (2020) 122:94–101. doi: 10.1038/s41416-019-0626-z
140. Flinck M, Kramer SH, Pedersen SF. Roles of pH in control of cell proliferation. Acta Physiol (Oxf) (2018) 223:e13068. doi: 10.1111/apha.13068
141. Wang ZH, Peng WB, Zhang P, Yang XP, Zhou Q. Lactate in the tumour microenvironment: From immune modulation to therapy. EBioMedicine (2021) 73:103627. doi: 10.1016/j.ebiom.2021.103627
142. Fu Q, Xu L, Wang Y, Jiang Q, Liu Z, Zhang J, et al. Tumor-associated macrophage-derived interleukin-23 interlinks kidney cancer glutamine addiction with immune evasion. Eur Urol (2019) 75:752–63. doi: 10.1016/j.eururo.2018.09.030
143. Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science (2016) 352:227–31. doi: 10.1126/science.aac9935
144. Hu T, Liu H, Liang Z, Wang F, Zhou C, Zheng X, et al. Tumor-intrinsic CD47 signal regulates glycolysis and promotes colorectal cancer cell growth and metastasis. Theranostics (2020) 10:4056–72. doi: 10.7150/thno.40860
145. Yu Y, Liang Y, Li D, Wang L, Liang Z, Chen Y, et al. Glucose metabolism involved in PD-L1-mediated immune escape in the Malignant kidney tumour microenvironment. Cell Death Discovery (2021) 7:15. doi: 10.1038/s41420-021-00401-7
146. Ohm JE, Gabrilovich DI, Sempowski GD, Kisseleva E, Parman KS, Nadaf S, et al. VEGF inhibits T-cell development and may contribute to tumor-induced immune suppression. Blood (2003) 101:4878–86. doi: 10.1182/blood-2002-07-1956
147. Yang J, Yan J, Liu B. Targeting VEGF/VEGFR to modulate antitumor immunity. Front Immunol (2018) 9:978. doi: 10.3389/fimmu.2018.00978
148. Bose D, Banerjee S, Singh RK, Wise LM, Robertson ES. Vascular endothelial growth factor encoded by Parapoxviruses can regulate metabolism and survival of triple negative breast cancer cells. Cell Death Dis (2020) 11:996. doi: 10.1038/s41419-020-03203-4
149. Zalpoor H, Aziziyan F, Liaghat M, Bakhtiyari M, Akbari A, Nabi-Afjadi M, et al. The roles of metabolic profiles and intracellular signaling pathways of tumor microenvironment cells in angiogenesis of solid tumors. Cell Commun Signal (2022) 20:186. doi: 10.1186/s12964-022-00951-y
150. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing cd8 T-cell memory by modulating fatty acid metabolism. Nature (2009) 460:103–7. doi: 10.1038/nature08097
151. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med (2011) 208:1367–76. doi: 10.1084/jem.20110278
152. Wang Y, Bi Y, Chen X, Li C, Li Y, Zhang Z, et al. Histone deacetylase SIRT1 negatively regulates the differentiation of interleukin-9-producing CD4(+) T cells. Immunity (2016) 44:1337–49. doi: 10.1016/j.immuni.2016.05.009
153. Zhu Y, An X, Zhang X, Qiao Y, Zheng T, Li X. STING: a master regulator in the cancer-immunity cycle. Mol Cancer (2019) 18:152. doi: 10.1186/s12943-019-1087-y
154. Liang K, Abt ER, Le TM, Cho A, Dann AM, Cui J, et al. STING-driven interferon signaling triggers metabolic alterations in pancreas cancer cells visualized by [(18)F]FLT PET imaging. Proc Natl Acad Sci USA (2021) 118:e2105390118. doi: 10.1073/pnas.2105390118
155. Wu SY, Xiao Y, Wei JL, Xu XE, Jin X, Hu X, et al. MYC suppresses STING-dependent innate immunity by transcriptionally upregulating DNMT1 in triple-negative breast cancer. J Immunother Cancer (2021) 9:e002528. doi: 10.1136/jitc-2021-002528
156. Chakravarthy A, Khan L, Bensler NP, Bose P, De Carvalho DD. TGF-beta-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat Commun (2018) 9:4692. doi: 10.1038/s41467-018-06654-8
157. Xue VW, Chung JY, Cordoba CAG, Cheung AH, Kang W, Lam EW, et al. Transforming growth factor-beta: A multifunctional regulator of cancer immunity. Cancers (Basel) (2020) 12:3099. doi: 10.3390/cancers12113099
158. Angioni R, Sanchez-Rodriguez R, Viola A, Molon B. TGF-beta in cancer: metabolic driver of the tolerogenic crosstalk in the tumor microenvironment. Cancers (Basel) (2021) 13:401. doi: 10.3390/cancers13030401
159. Wang C, Zhang J, Yin J, Gan Y, Xu S, Gu Y, et al. Alternative approaches to target Myc for cancer treatment. Signal Transduct Target Ther (2021) 6:117. doi: 10.1038/s41392-021-00500-y
160. Zirath H, Frenzel A, Oliynyk G, Segerstrom L, Westermark UK, Larsson K, et al. MYC inhibition induces metabolic changes leading to accumulation of lipid droplets in tumor cells. Proc Natl Acad Sci USA (2013) 110:10258–63. doi: 10.1073/pnas.1222404110
161. Yang C, Liu Y, Hu Y, Fang L, Huang Z, Cui H, et al. Myc inhibition tips the immune balance to promote antitumor immunity. Cell Mol Immunol (2022) 19:1030–41. doi: 10.1038/s41423-022-00898-7
162. Chen H, Liu H, Qing G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct Target Ther (2018) 3:5. doi: 10.1038/s41392-018-0008-7
163. Graziani V, Garcia AR, Alcolado LS, Le Guennec A, Henriksson MA, Conte MR. Metabolic rewiring in MYC-driven medulloblastoma by BET-bromodomain inhibition. Sci Rep (2023) 13:1273. doi: 10.1038/s41598-023-27375-z
164. Wu X, Nelson M, Basu M, Srinivasan P, Lazarski C, Zhang P, et al. MYC oncogene is associated with suppression of tumor immunity and targeting Myc induces tumor cell immunogenicity for therapeutic whole cell vaccination. J Immunother Cancer (2021) 9:e001388. doi: 10.1136/jitc-2020-001388
165. Clausen DM, Guo J, Parise RA, Beumer JH, Egorin MJ, Lazo JS, et al. In vitro cytotoxicity and in vivo efficacy, pharmacokinetics, and metabolism of 10074-G5, a novel small-molecule inhibitor of c-Myc/Max dimerization. J Pharmacol Exp Ther (2010) 335:715–27. doi: 10.1124/jpet.110.170555
166. Camarda R, Williams J, Goga A. In vivo reprogramming of cancer metabolism by MYC. Front Cell Dev Biol (2017) 5:35. doi: 10.3389/fcell.2017.00035
167. Masso-Valles D, Soucek L. Blocking myc to treat cancer: reflecting on two decades of omomyc. Cells (2020) 9:883. doi: 10.3390/cells9040883
168. Beaulieu ME, Jauset T, Masso-Valles D, Martinez-Martin S, Rahl P, Maltais L, et al. Intrinsic cell-penetrating activity propels Omomyc from proof of concept to viable anti-MYC therapy. Sci Transl Med (2019) 11:eaar5012. doi: 10.1126/scitranslmed.aar5012
169. Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, et al. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci USA (2010) 107:2037–42. doi: 10.1073/pnas.0914433107
170. De Los Santos-Jimenez J, Rosales T, Ko B, Campos-Sandoval JA, Alonso FJ, Marquez J, et al. Metabolic adjustments following glutaminase inhibition by CB-839 in glioblastoma cell lines. Cancers (Basel) (2023) 15:531. doi: 10.3390/cancers15020531
171. Poole CJ, Van Riggelen J. MYC-master regulator of the cancer epigenome and transcriptome. Genes (Basel) (2017) 8:142. doi: 10.3390/genes8050142
172. Topper MJ, Vaz M, Chiappinelli KB, Destefano Shields CE, Niknafs N, Yen RC, et al. Epigenetic therapy ties MYC depletion to reversing immune evasion and treating lung cancer. Cell (2017) 171:1284–1300 e21. doi: 10.1016/j.cell.2017.10.022
173. Demma MJ, Mapelli C, Sun A, Bodea S, Ruprecht B, Javaid S, et al. Omomyc reveals new mechanisms to inhibit the MYC oncogene. Mol Cell Biol (2019) 39:e00248-19. doi: 10.1128/MCB.00248-19
174. Michel J, Cuchillo R. The impact of small molecule binding on the energy landscape of the intrinsically disordered protein C-myc. PloS One (2012) 7:e41070. doi: 10.1371/journal.pone.0041070
175. Yu C, Niu X, Jin F, Liu Z, Jin C, Lai L. Structure-based inhibitor design for the intrinsically disordered protein c-myc. Sci Rep (2016) 6:22298. doi: 10.1038/srep22298
176. Yao R, Sun X, Xie Y, Sun X, Yao Y, Li H, et al. Identification of a novel c-Myc inhibitor with antitumor effects on multiple myeloma cells. Biosci Rep (2018) 38:BSR20181027. doi: 10.1042/BSR20181027
177. Xing J, Lu W, Liu R, Wang Y, Xie Y, Zhang H, et al. Machine-learning-assisted approach for discovering novel inhibitors targeting bromodomain-containing protein 4. J Chem Inf Model (2017) 57:1677–90. doi: 10.1021/acs.jcim.7b00098
178. Mu P, Han YC, Betel D, Yao E, Squatrito M, Ogrodowski P, et al. Genetic dissection of the miR-17~92 cluster of microRNAs in Myc-induced B-cell lymphomas. Genes Dev (2009) 23:2806–11. doi: 10.1101/gad.1872909
179. Pilarczyk M, Fazel-Najafabadi M, Kouril M, Shamsaei B, Vasiliauskas J, Niu W, et al. Connecting omics signatures and revealing biological mechanisms with iLINCS. Nat Commun (2022) 13:4678. doi: 10.1038/s41467-022-32205-3
Keywords: MYC, metabolism, oncoimmunology, cancer, immune evasion
Citation: Venkatraman S, Balasubramanian B, Thuwajit C, Meller J, Tohtong R and Chutipongtanate S (2024) Targeting MYC at the intersection between cancer metabolism and oncoimmunology. Front. Immunol. 15:1324045. doi: 10.3389/fimmu.2024.1324045
Received: 18 October 2023; Accepted: 26 January 2024;
Published: 08 February 2024.
Edited by:
Fabrizio Mattei, National Institute of Health (ISS), ItalyReviewed by:
Markus A. N. Hartl, University of Innsbruck, AustriaIvana Samarzija, Rudjer Boskovic Institute, Croatia
Copyright © 2024 Venkatraman, Balasubramanian, Thuwajit, Meller, Tohtong and Chutipongtanate. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rutaiwan Tohtong, rutaiwan.toh@mahidol.ac.th; Somchai Chutipongtanate, chutipsi@ucmail.uc.edu