 Xiaohan Li1,2†
Xiaohan Li1,2† Zhang Chen1,2†
Zhang Chen1,2† Weicong Ye1,2†
Weicong Ye1,2† Jizhang Yu1,2
Jizhang Yu1,2 Xi Zhang1,2
Xi Zhang1,2 Yuan Li1,2Yuqing Niu1,2Shuan Ran1,2
Yuan Li1,2Yuqing Niu1,2Shuan Ran1,2 Song Wang1,2Zilong Luo1,2
Song Wang1,2Zilong Luo1,2 Jiulu Zhao1,2Yanglin Hao1,2Junjie Zong1,2Chengkun Xia3*Jiahong Xia1,2,4*
Jiulu Zhao1,2Yanglin Hao1,2Junjie Zong1,2Chengkun Xia3*Jiahong Xia1,2,4* Jie Wu1,2,4*
Jie Wu1,2,4*- 1Department of Cardiovascular Surgery, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 2Center for Translational Medicine, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 3Department of Anesthesiology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 4Key Laboratory of Organ Transplantation, Ministry of Education, National Health Commission (NHC) Key Laboratory of Organ Transplantation, Key Laboratory of Organ Transplantation, Chinese Academy of Medical Sciences, Wuhan, China
Organ transplantation is the gold standard therapy for end-stage organ failure. However, the shortage of available grafts and long-term graft dysfunction remain the primary barriers to organ transplantation. Exploring approaches to solve these issues is urgent, and CRISPR/Cas9-based transcriptome editing provides one potential solution. Furthermore, combining CRISPR/Cas9-based gene editing with an ex vivo organ perfusion system would enable pre-implantation transcriptome editing of grafts. How to determine effective intervention targets becomes a new problem. Fortunately, the advent of high-throughput CRISPR screening has dramatically accelerated the effective targets. This review summarizes the current advancements, utilization, and workflow of CRISPR screening in various immune and non-immune cells. It also discusses the ongoing applications of CRISPR/Cas-based gene editing in transplantation and the prospective applications of CRISPR screening in solid organ transplantation.
1 Introduction
The CRISPR/Cas9 system is a third-generation genome editing technology that has emerged as an advanced replacement for zinc-finger nucleases and transcription activator-like effector nucleases in gene editing due to its improved simplicity and precision (1). CRISPR/Cas systems and their derivatives can be used for single genetic procedures and high-throughput studies, including pooled and arrayed screening (2). Feng et al. (3) and Langer and Sabatini (4) introduced the first high-throughput CRISPR screening in 2014. CRISPR screening has enabled comprehensive transcriptome analyses and improved mechanistic insights into gene regulation networks (5). Over the past decades, numerous genome-scale RNA interference (RNAi) screens have been conducted in biomedical studies. Unlike CRISPR-based technologies, it does not require co-delivery of exogenous proteins (6). However, due to off-target effects as well as differences in knockdown efficiencies, RNA interference (RNAi) screens exhibit limited validation or overlap across studies (6). Fortunately, CRISPR screenings demonstrate more pronounced phenotypic effects, higher validation rates, greater result consistency, reproducible data, and minimal off-target effects (7, 8). CRISPR screening has been used in multiple immunological fields to explore potential targets in drug therapy or cellular immunotherapy, including cancer, infection, and autoimmune disease (9–11). However, CRISPR screening has not been used in the organ transplantation field.
The success rate of organ transplantation has significantly increased in recent years due to advances in surgical techniques, perioperative care, immunosuppressive drugs, and infectious disease management. Organ transplantation has become the primary therapeutic approach for patients with terminal organ failure. However, the lack of available grafts and poor long-term prognosis remain significant challenges to organ transplantation. Therefore, there is an urgent need for alternative approaches to increase both the quantity and longevity of grafts in transplantation. CRISPR/Cas9-mediated transcriptome editing is one potential solution. It provides a promising approach to precisely alter potential intervention targets in multiple cells. Moreover, combining the CRISPR/Cas9 technique with an ex vivo organ perfusion system enables pre-implantation transcriptome editing of grafts (12, 13). How to select effective targets becomes the new challenge. Fortunately, CRISPR screening has facilitated the identification of potential therapeutic targets within immune and non-immune cells in various immune-related diseases (5), offering a novel insight for transplantation.
This review provides a comprehensive overview of the current advancements and utilization of CRISPR screenings in diverse immune and non-immune cells, providing valuable guidance for applying these techniques in organ transplantation research. Then, we summarize the ongoing applications of CRISPR/Cas-based gene editing in transplantation and the prospective applications of CRISPR screening in solid organ transplantation.
2 Current application of high-throughput CRISPR technology in solid organ transplantation
The long-term graft dysfunction mediated by allogeneic rejection remain the primary barriers to organ transplantation. Numerous immunosuppressants have been developed for transplantation, including calcineurin inhibitors and mTOR inhibitors (14, 15). However, most of these have limited therapeutic effects on long-term allograft survival (16). In addition, cellular immunomodulatory therapies have been proven effective in suppressing the immune response in transplantation (17). Regulatory T cells (Tregs) are currently employed to suppress immune responses and promote antigens tolerance in clinical (18). Additionally, multiple suppressor cells, including regulatory macrophages, tolerogenic dendritic cells (DCtols), and mesenchymal stromal cells (MSCs), are under investigation in clinical trials (16). Moreover, chimeric antigen receptor (CAR) T-cell therapy has emerged as a potent therapeutic approach to achieve long-term graft survival. Recent studies have shown that CAR technology can be applied successfully to create Treg cells based on donor HLA molecules, and can overcome the limitations of existing protocols for enriching Tregs based on repetitive stimulation with alloantigen (19). Unfortunately, these therapeutic cells usually cannot maintain the intended function to effectively build the tolerance in solid organ transplantation (16). Enhancing the effector function of therapeutic cells with CRISPR/Cas9 based genome editing, providing one potential solution (20–22). However, current studies have identified limited genetic targets that can effectively regulate the immune cells in transplantation (23). Exploring new intervention targets for immunosuppressant and cellular immunomodulatory therapies is urgent. Furthermore, combining CRISPR/Cas9-based gene editing with ex vivo organ perfusion system would enable pre-implantation transcriptome editing of grafts (12). This is a potential way to solve the shortage of available grafts. However, how to select resistance genes becomes the new challenge.
Fortunately, high-throughput CRISPR screening has dramatically accelerated the effective targets in various immunocytes (2). However, its application in solid organ transplantation is limited. Therefore, we have provided an overview of the current utilization of CRISPR screenings in diverse immune cells, providing insights for screening therapeutic targets of immunocytes in solid organ transplantation. Moreover, CRISPR screens had emerged as a powerful way to explore the potential targets in immune escape mechanism of cancer cells under immune pressure (24). In solid organ transplantation, grafts face a similar immune pressure, specifically mediated by allograft rejection. This provides a novel insight for exploring the resistance genes in solid organ transplantation. A comprehensive discussion of CRISPR screenings in cancer cells is also presented in the subsequent sections, providing guidance for conducting CRISPR screenings in grafts.
3 CRISPR/Cas9 system: a reliable and effective genome editing tool
Precise genetic manipulation techniques in living cells have significantly contributed to advancements in biomedical research and the application of genetic perturbation in clinical medicine (25). Protein-DNA recognition is a critical aspect of genome editing and plays a pivotal role in the precise interaction between genetic editing enzymes and DNA targets (26). However, site-specific nucleic acid targeting is a significant challenge in genome editing strategies based on protein-DNA recognition (27). Numerous studies have elucidated the protein-DNA recognition mechanism (26, 28–30). One of the most significant ongoing discussions in the specific genetic editing field is the CRISPR/Cas system (31–33).
The CRISPR/Cas system is a type of natural immune system in prokaryotes (34). CRISPR stands for “Clustered Regularly Interspaced Short Palindromic Repeats”. Prokaryotes use this mechanism to counteract invading viruses and foreign DNA (35) (Figure 1). Upon viral invasion, certain bacteria can incorporate a viral gene fragment into their DNA within a designated region, known as the CRISPR storage space (36). When reencountering the virus, bacteria initiate CRISPR transcription to produce precursor CRISPR RNA (pre-crRNA), which subsequently undergoes processing to yield crRNAs carrying a sequence complementary to the foreign genes. Upon recognizing sequences homologous to the viral gene, the crRNAs guide Cas nucleases to bind to and cleave the target gene, protecting the bacterial host from viral infiltration (36).
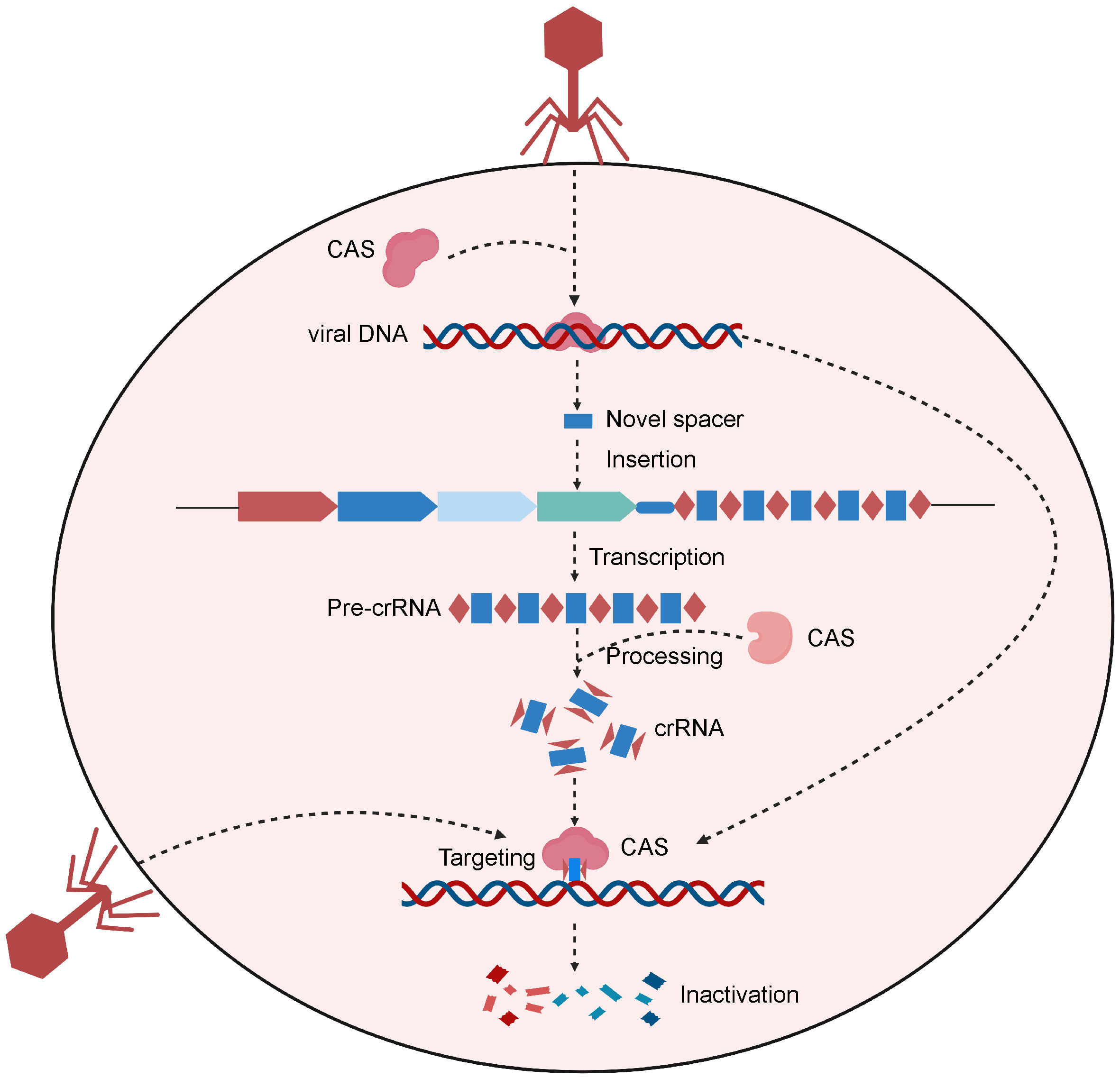
Figure 1 Schematic of the CRISPR-Cas system of the natural prokaryotic immune system. After viral invasion, certain bacteria can incorporate a viral gene fragment into their CRISPR storage space. When reencountering the virus, bacteria initiate CRISPR transcription to produce pre-crRNA, which are then processed to create crRNAs. Upon recognizing sequences homologous to the viral gene, the crRNAs guide Cas protein to bind to and cleave the target gene, protecting the bacterial host from viral infiltration. Created with BioRender.com.
Based on differences in the sequences and structures of Cas proteins, CRISPR/Cas systems can be classified into two classes and six types (37). The class 1 CRISPR/Cas system contains a multisubunit crRNA–effector complex, including the type I, type III and type IV CRISPR/Cas system (38). However, the class 2 CRISPR–Cas system which includes the type I, type III and type IV CRISPR/Cas system just needs a single subunit crRNA–effector module (38). The detailed description of these CRISPR/Cas system is provided in the Table 1. As the simple and flexible genome editing method, the CRISPR/Cas system has revolutionized the biomedical research field (39). The type II CRISPR/Cas system emerged as an simple and efficient genome editing tools for various cells and organisms (40, 41), only requiring a multi-functional Cas9 protein to interfere with the invading genetic elements (42). The type II CRISPR system provided the foundation for the CRISPR/Cas9 gene editing system. However, the efficiency of the CRISPR/Cas9 system in eukaryotes is influenced by the chromosomal status of the target site (6). To efficiently deliver the Cas9 protein into mammalian nuclei, its N- or C-terminus is fused with the eukaryotic nuclear localization signal (43).

Table 1 The summarization of all types of CRISPR/Cas systems.
The CRISPR/Cas9 system comprises two primary components: a single-stranded guide RNA (sgRNA) and a Cas protein. A trans-activating crRNA (tracrRNA) and a crRNA are fused to form a sgRNA. CRISPR/Cas9 technology uses the sgRNA to identify the target genome sequence, guiding the Cas9 endonuclease to precisely cut the DNA double-strand (44). Gene knockout or knock-in events occur during the repair process. Typically, cells use highly efficient non-homologous terminal junctions to repair broken DNA. During the repair process, base insertions or deletions often lead to frameshift mutations, knocking out the target gene (Figure 2A) (45). After a DNA double-strand break, if a DNA repair template enters the cell, the broken part of the gene will undergo homologous recombination repair using the repair template, causing gene knock-in (46).
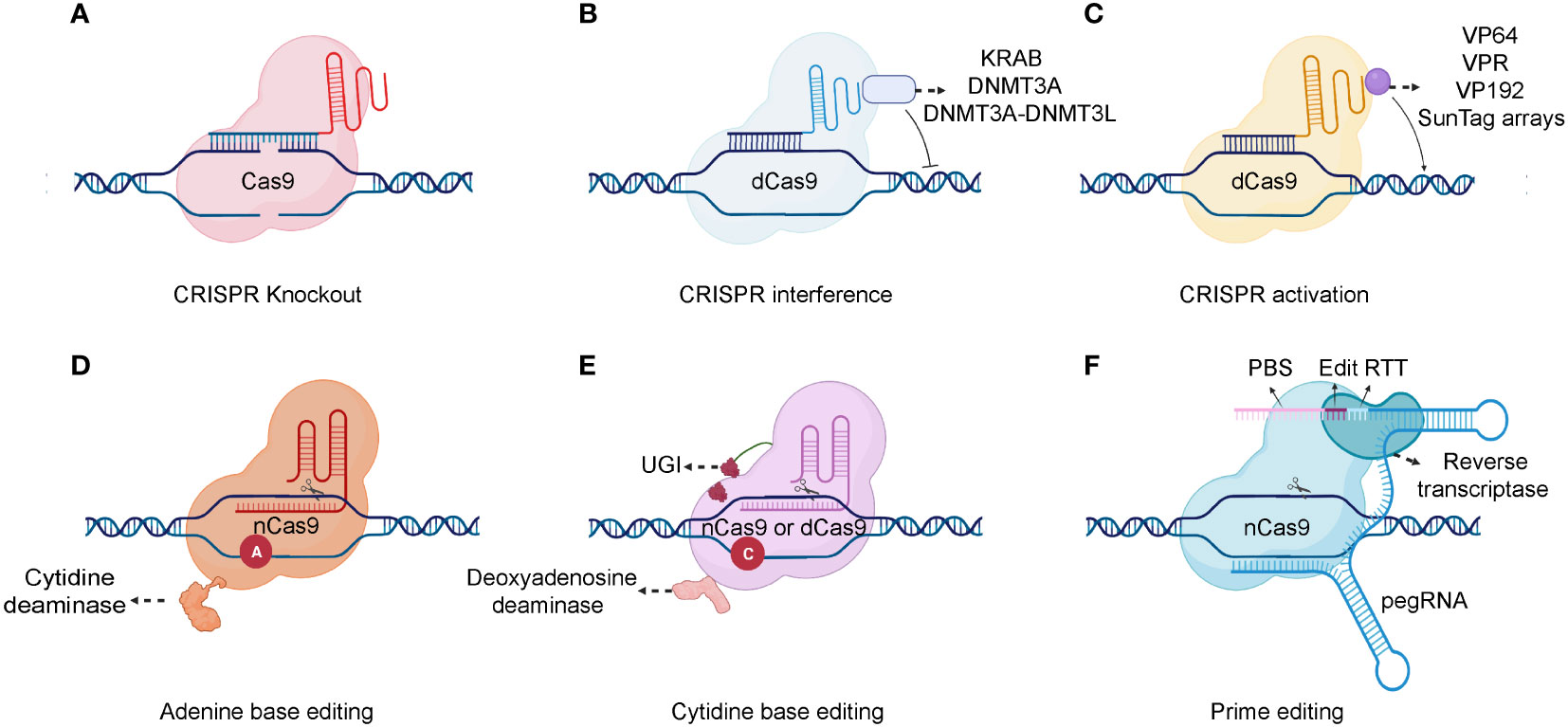
Figure 2 Schematic of different CRISPR-Cas system types. (A) A gRNA is used to identify the target genome sequence, guiding the Cas9 endonuclease to precisely cut the DNA double-strand. (B) For gene repression, dCas9 can be fused to KRAB, DNMT3A, or DNMT3A-DNMT3L. (C) Various transcription activators can be fused to dCas9, including VP64, VP192, SunTag arrays, and the VPR comprising VP64, p65, and Rta. (D, E) Combining dCas9 or nCas9 with cytidine or adenine deaminases makes it possible to directly convert cytidine into uridine or adenine into inosine. (F) As a fusion protein, prime editor is composed of reverse transcriptase and nCas9. Prime editor binds to a specific target DNA sequence under the guidance of prime-editing guide RNA (pegRNA). And a single-strand break is made by the nCas9. There are two main components of pegRNA: the reverse transcription template (RTT) and the protospacer binding sequence (PBS). During reverse transcription, the RTT encodes the desired edits which are primed by PBS and the edits are incorporated into the newly synthesized DNA strand. After DNA repair, the edits are stably incorporated into the genome. Created with BioRender.com.
The Cas9 nickase (nCas9) and dead Cas9 (dCas9) are also applied in the CRISPR based genetic modification. In detail, the nCas9 is a Cas9 variant protein that generated from the mutation in either RuvC nuclease domain or histidine/asparagine (HNH) endonuclease domain (47). While, the dCas9 is another variant Cas9 that generated from the mutation in both endonuclease domains (RuvC nuclease domain and HNH endonuclease domain) (48). Consequently, nCas9 can only induce a single-strand nick in the target DNA rather than a DNA double-strand break (DSB). And, the dCas9 specifically binds to the target gene guided by the sgRNA but lacks DNA cleavage activity. The binding of dCas9 to a gene’s transcriptional start site can obstruct its transcription initiation, impeding its expression (49). The dCas9 protein can also interact with transcriptional suppressors or activators to inhibit or activate downstream target gene transcription (50) (Figures 2B, C). Additionally, combining dCas9 or nCas9 with cytidine or adenine deaminases enables the direct conversion of cytidine into uridine or adenine into inosine (51, 52)(Figures 2D, E). Although base editing can introduce point mutations efficiently, it is difficult to generate precise indels and avoid bystander mutations (53). A recent genome editing technology called prime editing allows point mutations, small insertions, and small deletions to be introduced precisely and effectively with a favorable editing to indel ratios (54) (Figure 2F). Prime editor is composed of reverse transcriptase and nCas9. Prime editor binds to a specific target DNA sequence under the guidance of prime-editing guide RNA (pegRNA) and a single-strand break is made by the nCas9. During a reverse transcription, the desired edits are encoded and are incorporated into the newly synthesized DNA strand. After DNA repair, the edits can be stably incorporated into the genome. In conclusion, the CRISPR/Cas9 system is already significantly contributing to functional genomic experiments and is poised to have an unprecedented impact on experimental biology in the future (55). Moreover, it holds immense potential for diverse gene therapy applications, including blood diseases, tumors, and other genetic disorders.
4 High-throughput CRISPR technology
4.1 CRISPR screening
Genetic screening is a powerful tool for investigating genes contributing to diverse biological phenotypes and diseases. Numerous genome-wide targeted techniques have emerged with advancements in genetic tools. For example, RNAi libraries were commonly used to screen gene functions due to their efficiency in reducing gene expression at the mRNA level (56). RNAi has been the favored approach for studying gene function in the last decade, particularly in functional genomic screening (6). However, CRISPR has emerged as a more powerful genome editing technique than RNAi, with lower off-target efficiency and higher interference levels (6). Studies have used Cas9 to perform experiments with remarkable results in various cell lines and animal models. However, using one or more sgRNAs for gene knockdown lacks sufficient throughput. Therefore, CRISPR screening was developed based on the RNAi screening technique. Compared to RNAi-based screenings, CRISPR screening provides higher validation rates, greater phenotypic effects, minimal off-target effects, and consistent results (7, 8). Moreover, the comparison between CRISPR based screening and RNAi based screening are detailed described in Table 2.

Table 2 The comparison between CRISPR based screening and RNAi based screening.
4.1.1 CRISPR screening workflow
The general CRISPR screening workflow can be summarized in the following four steps (61) (Figure 3). Firstly, three or more sgRNAs are designed for each gene in a species, which are synthesized with high throughput and cloned into a lentiviral vector. Subsequently, the lentiviral library is packaged with a low multiplicity of infection (MOI; generally <0.3) to infect target cells, ensuring that each cell is infected by only one viral particle. The entire transduced cell pool is divided into two groups. One half serves as the experimental group and is subjected to screening pressure, such as viral infection or drug treatment, while the other serves as the control group. Cells are sorted based on phenotypic traits such as drug resistance, proliferation capacity, and survival capability. Finally, genomes of sorted cells are isolated from both the experimental and control groups. Then, sgRNA fragments are polymerase chain reaction (PCR) amplified, high-throughput sequenced, and bioinformatically analyzed.
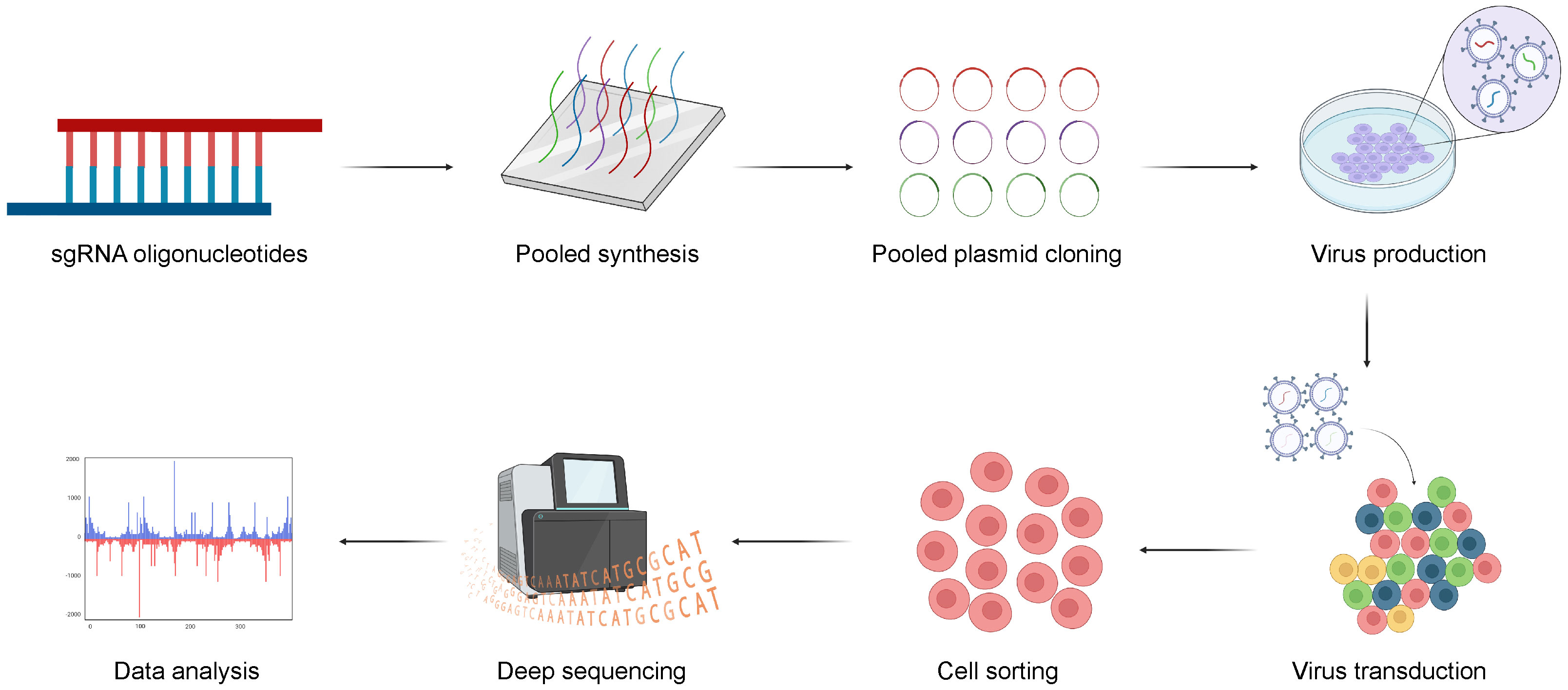
Figure 3 The general workflow of CRISPR screening. Firstly, three or more sgRNAs are designed for each gene, sgRNAs are synthesized with high throughput and cloned into a lentiviral vector. And, the lentiviral library is transduced into target cells and the transduced cells are sorted for sequencing with multiple selective strategies. Finally, the potential genes can be validated with bioinformatic analysis. Created with BioRender.com.
4.1.2 Phenotypic selection in CRISPR screening
CRISPR screening is primarily used to screen functional genes. Depending on the specific screening objectives, it can be categorized into positive and negative selection. Positive selection involves applying selective pressure to a cell library with successfully integrated sgRNA to promote the survival of cells with the desired phenotypes and ultimately enrich key genes (62). Conversely, negative selection involves the survival of cells not exhibiting the desired phenotypes. Determining sgRNA abundances at different time points allows for identifying differences in sgRNA profiles, revealing key genes (57). Negative screening can identify genes responsible for inducing specific functional deficits in cells (63). For example, essential genes required for cell survival can be identified by extending the screening duration. Different cellular phenotypes can also be reflected by fluorescent signals and sorted by fluorescence-activated cell sorting (FACS) (64). The potential targets controlling specific cellular phenotypes can then be identified by deep sequencing. Moreover, the cells for sequencing can also be sorted using magnetic-activated cell sorting (MACS) based on their expression of a specific surface protein (64).
4.1.3 Design tools for sgRNAs
4.1.3.1 CrisprVerse: A new ecosystem of R packages
Various CRISPR technologies, including knockout or knockdown, activation or interference, and base editing, significantly enhance gRNA design and annotation capabilities (65). The core R package, crisprDesign, offers a user-friendly and standardized interface that supports adding off-target annotations, comprehensive gene annotations, and assessments of both on- and off-target activity ratings (65). All nucleases targeting DNA or RNA can access these features, including Cas9, Cas12, and Cas13 (65).
4.1.3.2 CRISPOR
CRISPOR.org is a web-based tool designed explicitly for CRISPR/Cas9 genome editing research. This platform identifies gRNAs within an input sequence and uses various metrics to rank them, evaluating their potential for off-target effects in the target genome and predicting their on-target activity (66). Recent advancements in CRISPOR include the development of next-generation sequencing primers for off-target mutation assessment, customized oligonucleotide creation for guide cloning, and batch design strategies for genome-wide CRISPR screening (66).
4.1.3.3 The UCSC genome browser
The University of California-Santa Cruz (UCSC) genome browser provides diverse information and tools that enhance our understanding of the genomic context across numerous species. It facilitates in-depth data analysis and enables queries of regions of interest within diverse datasets from multiple sources (67). In addition, researchers can include their data in the primary display, facilitating data querying and sharing with others (67). When conducting CRISPR/Cas9 genome editing research, one can select from a pool of reliable gRNA sequences previously generated by focusing on the specific target gene region (68).
4.1.3.4 E-CRISP
E-CRISP is a web application developed for designing genomic RNA sequences that provides experiment-centered design parameters, enabling the construction of multiple libraries and the systematic investigation of the effects in diverse settings (69). E-CRISP identifies target sequences that are complementary to the gRNA and terminate with the N(G) or A(G) protospacer-adjacent motif (PAM). This motif is essential for enabling the Cas9 nuclease to cleave the DNA double-strand upon activation (69). E-CRISP uses a binary interval tree to rapidly annotate potential gRNA target positions and a fast-indexing strategy for locating binding sites (69).
4.1.3.5 CHOPCHOP
CHOPCHOP is a web-based tool to identify sgRNA targets within the CRISPR/Cas system (70). Its guiding principle is to provide an accessible and powerful tool to benefit users of all levels, whether novice or experienced (71). CHOPCHOP provides various advanced options for selecting targets and accommodates various inputs, such as gene IDs, genomic regions, or pasted sequences (72). It applies efficient sequence alignment techniques to reduce search times and duplication by accurately predicting the off-target binding of sgRNAs (72). Each query generates an interactive representation of the gene, displaying potential target sites at their genomic locations, color-coded based on their quality ratings (72).
4.1.3.6 CRISPRscan
The variability in activity among numerous sgRNAs remains a significant problem (73). Moreno-Mateos et al. discovered that increasing guanine and decreasing adenine content improved sgRNA stability and activity (73). They also found that sgRNAs with 5’ mismatches and 1–2 nucleotide truncations could effectively replace conventional sgRNAs (73). Based on their findings, they developed the CRISPRscan predictive sgRNA-scoring algorithm, which effectively captures the sequence attributes that impact CRISPR/Cas9 in vivo activity (73).
4.1.3.7 GuideScan
GuideScan is a serial software that is computationally intensive in building CRISPR guide RNA libraries from big genomes (74). GuideScan generates high-density sets of single- and paired-gRNAs for genome-wide screening (75). GuideScan precisely and efficiently tallies all targetable sequences within a specified genome using a retrieval tree data structure (75). Additional details regarding each gRNA, including its on-target efficiency scores and overlapping genomic features, can be incorporated into its annotation (75).
4.1.4 Analysis tools for CRISPR screening
In this section, we discuss the current analysis tools for CRISPR screening. Moreover, we provide the detailed information of analysis tools in Table 3.
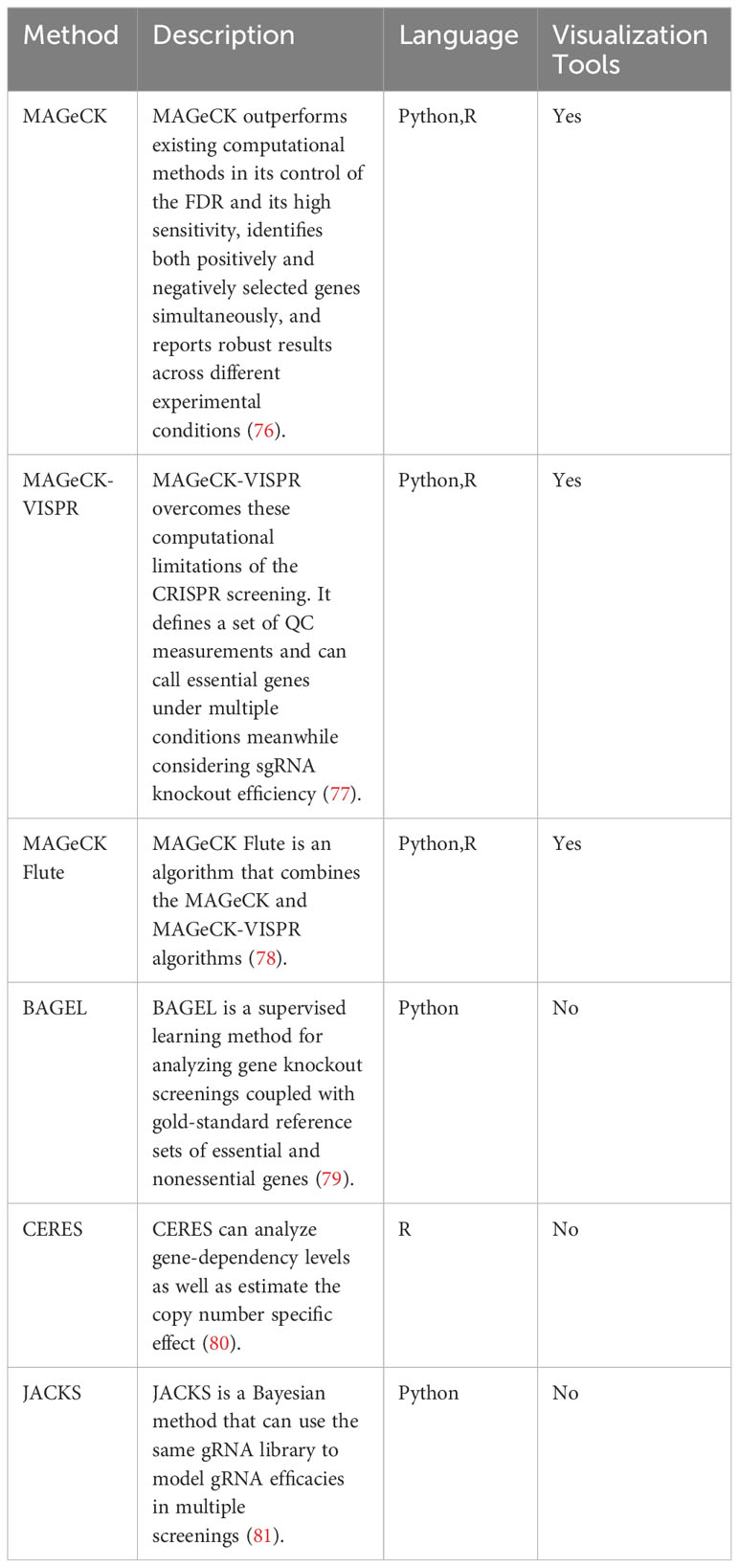
Table 3 Tools for CRISPR screens results analysis.
4.1.4.1 MAGeCK
While not designed explicitly for CRISPR/Cas9 knockout screenings, several established algorithms can still be used to identify significantly enriched sgRNAs or genes in next-generation RNA sequencing, genome-scale small interfering RNA, or shRNA screens (76). The Model-based Analysis of Genome-wide CRISPR/Cas9 Knockout (MAGeCK) method surpasses current computational methods in controlling the false discovery rate and shows high sensitivity. It concurrently identifies genes selected in both positive and negative directions, providing consistent outcomes across various experimental conditions (76). Distinct MAGeCK algorithms typically comprise two key steps (1): Analyzing raw read counts associated with sgRNAs by median normalization and (2) modeling the mean-variance relationship within replicates using a negative binomial distribution to assess the differences between treatments and controls (82).
4.1.4.2 MAGeCK-VISPR
High-throughput sequencing data can only be analyzed by comparing two conditions in most algorithms developed for screening analysis. However, in many cases, screenings are conducted simultaneously across various time points, treatment conditions, or cell lines (77). MAGeCK-VISPR overcomes these computational limitations of CRISPR screening by defining a set of quality control (QC) measurements. In order to iteratively estimate gene essentiality and sgRNA knockout efficiency, the algorithm uses a generalized linear model to deconvolute different effects. This method can identify essential genes under different conditions while considering the sgRNA knockout efficiency (77). In addition, it provides a web-based visualization framework to allow users to interactively explore CRISPR screening QC and analysis results (77).
4.1.4.3 MAGeCKFlute
The MAGeCKFlute algorithm combines the MAGeCK and MAGeCK-VISPR algorithms and offers downstream analysis capabilities (78). MAGeCKFlute includes several functions, including normalization, batch effect removal, and QC (78). A desktop computer with Linux or Mac OS and support for R is sufficient to complete the whole MAGeCKFlute pipeline in approximately two hours. MAGeCKFlute’s comprehensive pipeline includes various modules for analyzing CRISPR screening data, making it unique among competing tools.
4.1.4.4 BAGEL
The Bayesian Analysis of Gene EssentiaLity (BAGEL) method is executed as a Python script, requiring the Python modules numpy and scipy. A gold-standard reference set of essential and non-essential genes is used to analyze gene knockout screens (79). Furthermore, BAGEL2 was developed in 2021, which can detect tumor suppressor genes and uses a multi-target correction to mitigate false positives resulting from off-target CRISPR gRNAs (83). BAGEL2 shows enhanced sensitivity and specificity compared to BAGEL.
4.1.4.5 CERES
The sgRNA-Cas9 complex can target the genomic copy number to affect cell proliferation in genome-scale CRISPR/Cas9 loss-of-function screens (8, 84) mainly through the independent antiproliferative effect of Cas9-mediated DNA cleavage. The CRISPR Engineering for the Rapid Enhancement of Strains (CERES) method enables the analysis of gene-dependency levels and estimation of the copy number-specific effect (80). Moreover, Tsherniak et al. found it could decrease false positives in CRISPR-Cas9 essentiality screens of cancer cell lines (80).
4.1.4.6 JACKS
The signal variability caused by different gRNAs targeting the same gene limits the applications of genome-wide CRISPR/Cas9 knockout screens (81). Joint Analysis of CRISPR/Cas9 Knockout Screens (JACKS) uses a Bayesian approach to model gRNA efficacies across multiple screens using the same gRNA library (81).
4.2 Perturb-seq: combining CRISPR screening and emerging single-cell technologies
Analyzing gene and cellular functions by establishing connections between genetic changes and their phenotypic outcomes is of paramount significance (85). While genetic screenings are powerful tools that can help us identify the inferred gene function in mammalian cells, they cannot effectively investigate complex phenotypes, such as transcription profiles (86). Such screenings focus on individual perturbations, delivering and evaluating them separately (86).
Genetic perturbation has emerged as a tool to elucidate causal connections between genes and phenotypes (87). With the help of perturb-seq, pooled CRISPR screens can be combined with single-cell RNA sequencing to investigate functional CRISPR screens at a single-cell level (87). Additionally, genome-scale Perturb-seq can multi-dimensionally reveal cellular behavior, gene function, and regulatory networks. We can also use the Perturb-seq to obtain rich genotypic and phenotypic maps, realizing systematic genetic and cellular function exploration. For example, Zhang et al. used Perturb-seq in vivo to determine the relationships of autism risk genes with neuronal and glial abnormalities (88). Moreover, Hein and Weissman used Perturb-seq to characterize thousands of CRISPR-modified cells, revealing the roles of host and viral factors (89). Moreover, a more powerful genome-scale Perturb-seq method was developed in 2022, which uses CRISPR interference (CRISPRi) to target all expressed genes across >2.5 million human cells (85).
In conclusion, we can obtain rich biological insights and identify the mechanisms of complex biological responses using Perturb-seq (90). Perturb-seq allows us to extract additional experimental data, contributing to the advancement of biomedical research.
5 CRISPR screening in immune cells
In this section, we discuss current CRISPR screening in immune cells, with a particular focus on human or mouse-derived immune cells. Moreover, we provide a summarization in Tables 4 and 5. We also systematically summarize the different response genes identified in human and mouse-derived immune cells using CRISPR screening in Figure 4 and Table 6.
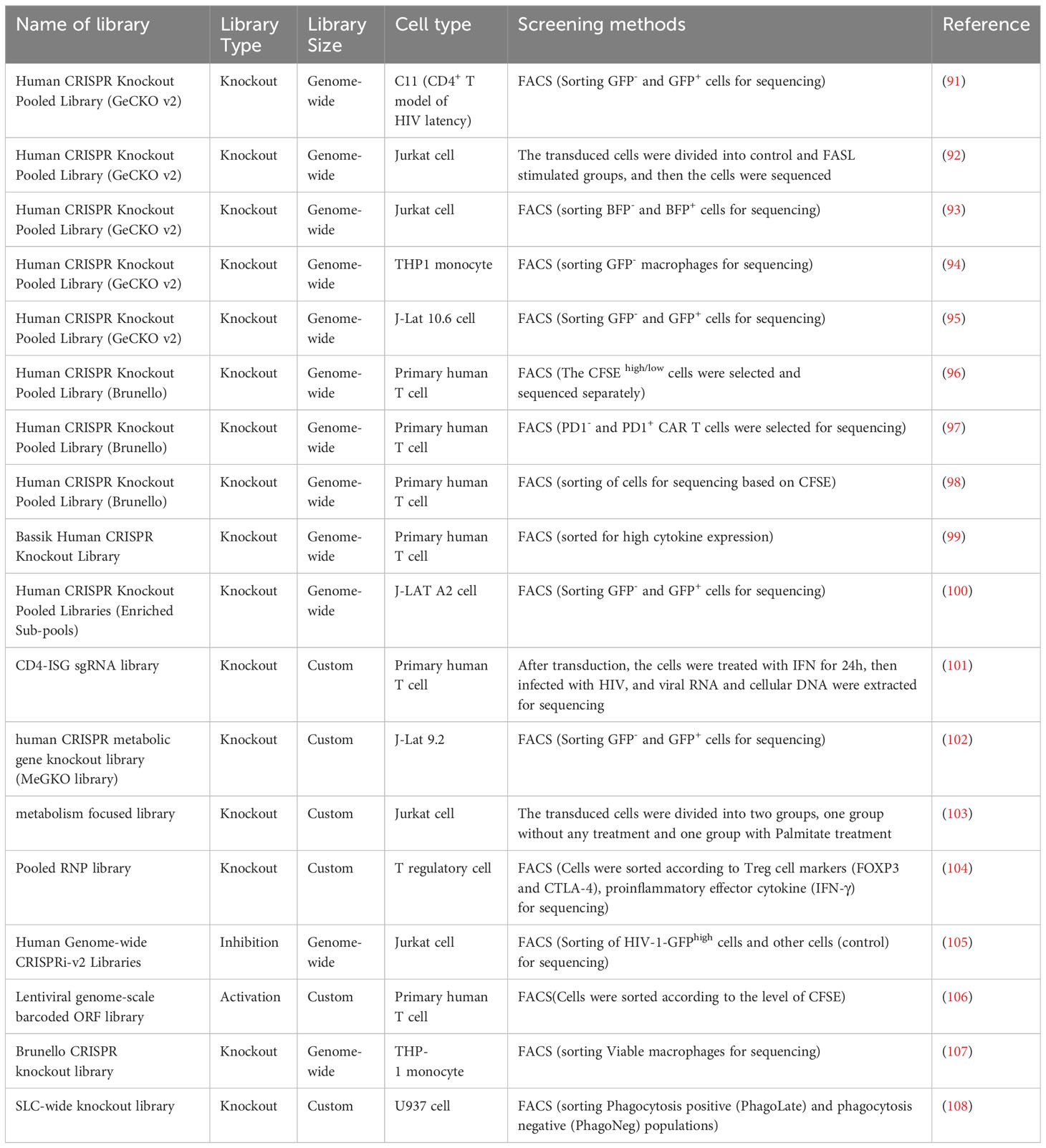
Table 4 CRISPR screening studies in human immune cells.
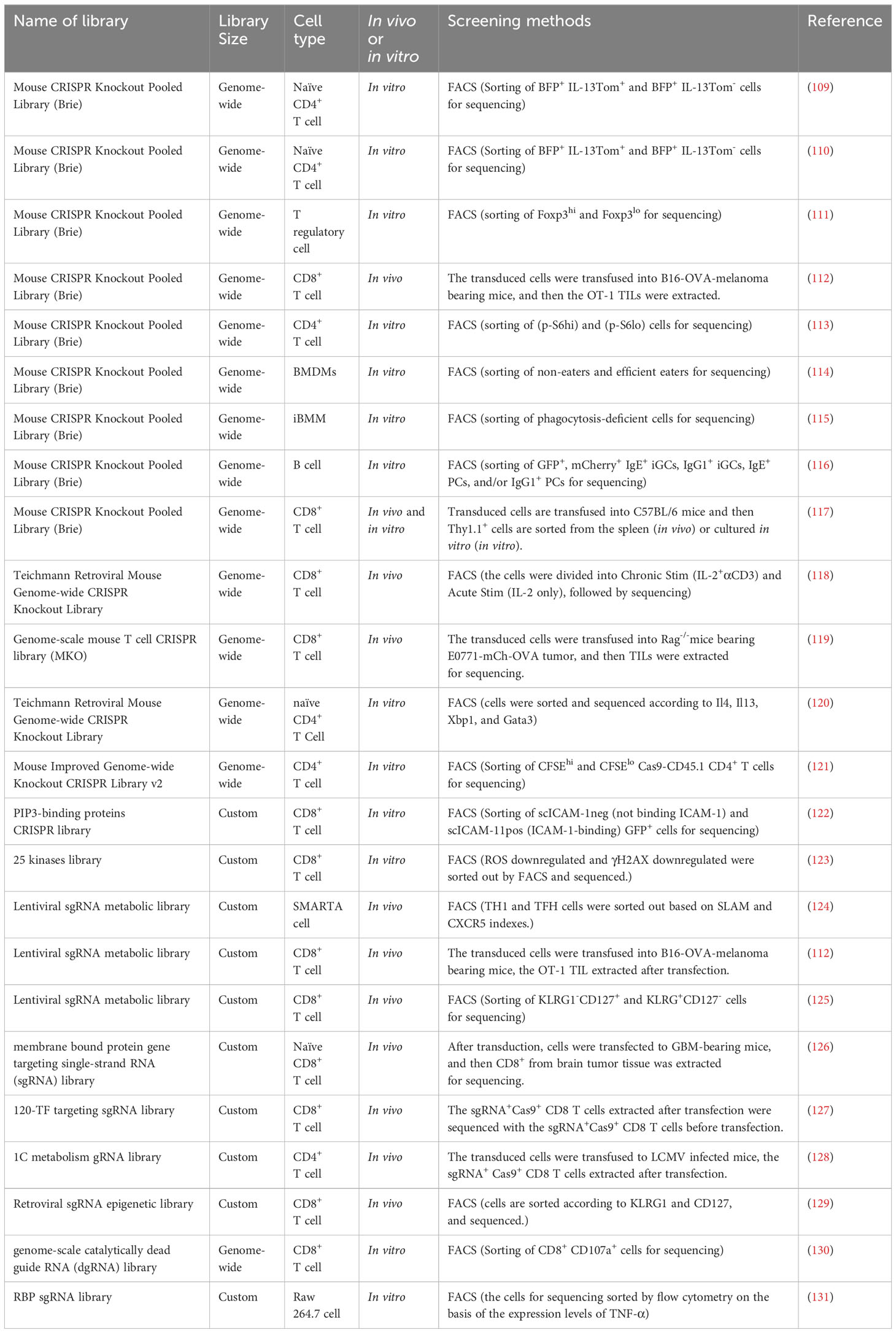
Table 5 CRISPR screening studies in murine immune cells.
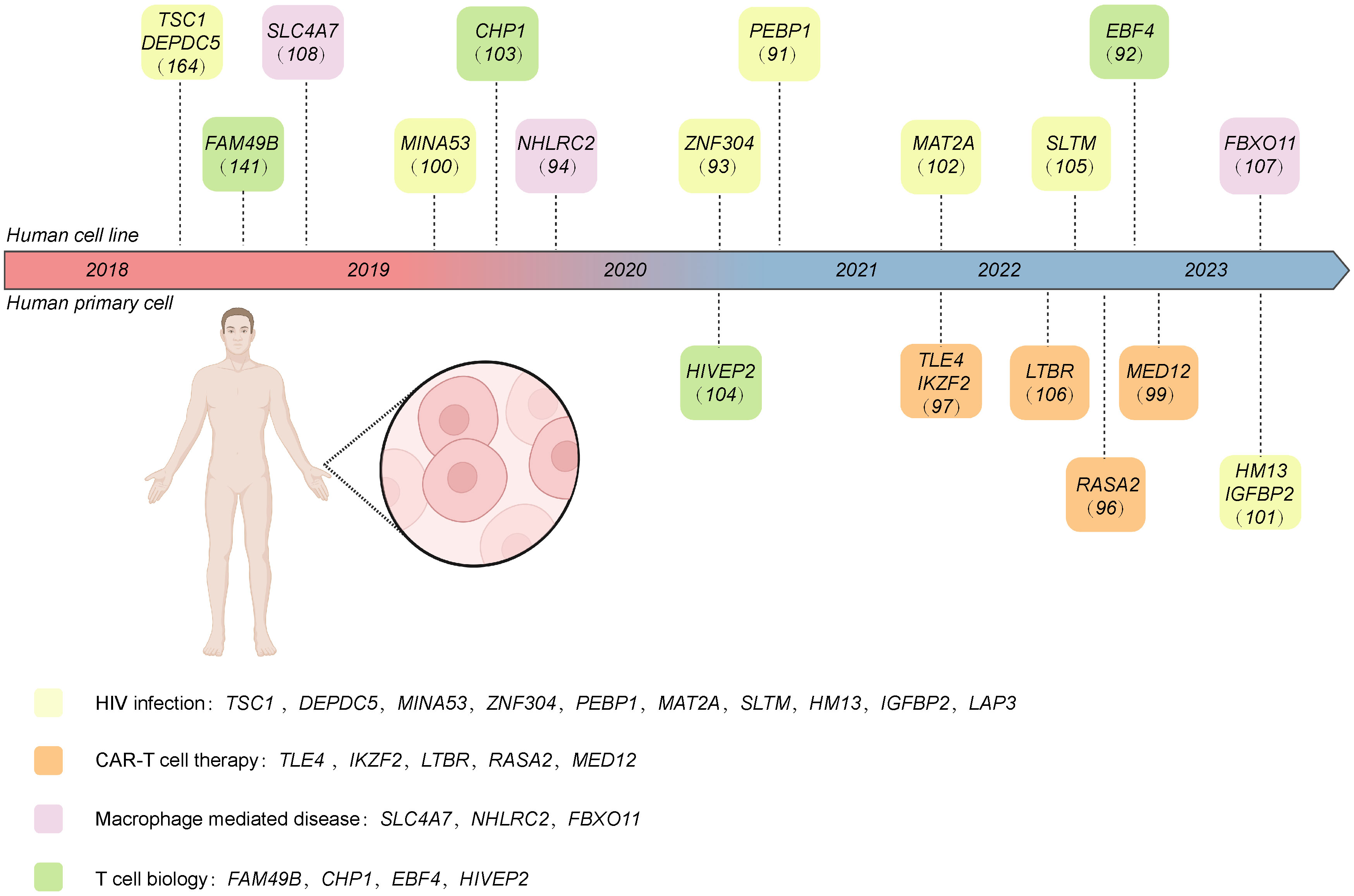
Figure 4 The target genes of CRISPR screening performed in human-derived immune cells discussed in this review. These genes are classified into different research areas, including CAR T therapy, HIV infection, macrophage-mediated disease, and T cell biology. Created with BioRender.com.
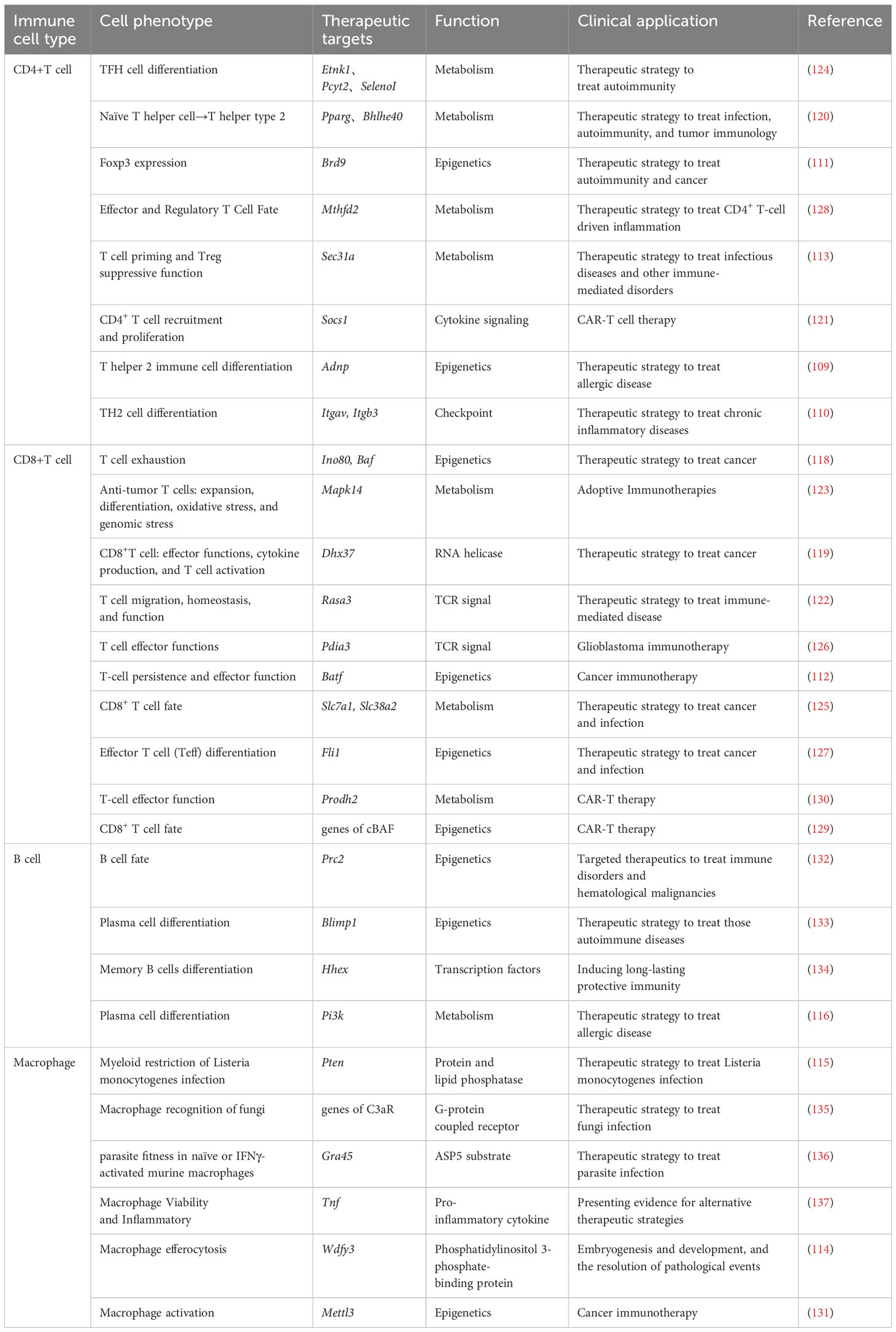
Table 6 The therapeutic targets in murine immune cells identified by CRISPR screening.
5.1 CRISPR screening in various areas based on human immune cells
5.1.1 Chimeric antigen receptor - T cell therapy
Chimeric antigen receptor (CAR)-T cell treatments have become a pivotal therapeutic approach for various hematological cancers (138). However, CAR-T cell therapy has various limitations, including antigen escape, off-target effects, CAR-T cell trafficking, tumor infiltration, and CAR-T cell-associated toxicity (139). Genome-scale CRISPR screenings in primary human T cells can comprehensively explore potential targets in CAR-T therapy (98). However, conducting CRISPR screens in primary human cells has been challenging. In 2018, Marson et al. introduced a novel method called sgRNA lentiviral infection with Cas9 protein electroporation (SLICE) that can precisely and efficiently disrupt target genes in primary human cells (98). They introduced the Brunello human CRISPR knockout pooled library into primary human T cells using the SLICE method (96). The transduced cells were cultured under a series of immunosuppressive conditions to identify genes causing T cell dysfunction in the tumor microenvironment (TME) (96). Their findings suggest that RASA2 is a prospective target for improving T cell therapy effectiveness and persistence in treating cancer.
In addition, inadequate T cell potency is a significant obstacle to T cell immunotherapy progress (99). Recently, Mackall et al. used the Bassik human CRISPR knockout library to target primary T cells, which were then transformed into CAR-T cells and co-cultured with tumor cells (99). MED12 deletion enhanced CAR T cells’ antitumor ability and sustained effector phenotype. Moreover, glioblastoma multiform (GBM), a primary brain tumor, is characterized by its uniform lethality and resistance to conventional treatments (140). However, CAR T cells show a limited general therapeutic effect in GBM. After transduction with a whole-genome knockout CRISPR/Cas9 library, CAR T cells were co-cultured with glioblastoma stem cells. Then, PD1+ and PD1− CAR T cells were individually sorted and sequenced (97). Notably, TLE4 and IKZF2 were identified as potential targets, and their knockout enhanced CAR antitumor efficacy. Nevertheless, a major limitation of CRISPR-based loss-of-function screens is that they only focus on negative regulators of T-cell function and permanent genomic modifications. Sanjana et al. conducted a gain-of-function screening in primary human T cells using a gRNA library targeting human open reading frames (106). Overexpressing LTBR induced significant epigenomic and transcriptional remodeling, enhancing T cell effector capabilities and conferring resistance to exhaustion under chronic stimulation conditions.
5.1.2 T cell biology
Despite decades of research on T-cell activation mechanisms, the exact mechanism remains elusive, posing a significant impediment to immunotherapy of T-cell tumors. Wang et al. conducted a genome-wide CRISPR screening in Jurkat cells. The transduced cells were sorted for sequencing based on their CD69 expression after T cell receptor (TCR) stimulation (141). FAM49B was found to prevent T cell activation by suppressing Rac activity and controlling cytoskeleton rearrangement.
Apoptosis, a programmed cell death mechanism in various cell types, is pivotal for immune system homeostasis, including removing activated immune cells (142). Interestingly, it was recently discovered that apoptosis is associated with the cytotoxic immune function of immune cells (143). In 2022, Lenardo et al. conducted a genome-wide CRISPR knockout screen for regulators of FAS/APO-1/CD95-mediated T cell death in Jurkat cells. The transduced cells were divided into control and Fas ligand (FASL)-stimulated groups (92). Their findings showed that EBF4 was indispensable for the cytotoxic functions of natural killer and CD8+ T cells by regulating TBX21, EOMES, granzymes, and perforin.
Moreover, increasing evidence shows that triglyceride and fatty acid metabolism are indispensable in regulating T cells (144). For example, exposure to circulating fatty acids can alter T cell function through metabolic reprogramming. In 2019, Birsoy et al. conducted a CRISPR knockout screen in Jurkat cells with two gRNA libraries: a metabolism-focused library and a genome-wide CRISPR library. The Jurkat cells were divided into control and palmitate-treated groups (103). Interestingly, they found that CHP1 regulated endoplasmic reticulum glycerolipid synthesis by binding and activating GPAT4 to catalyze the initial rate-limiting step in glycerolipid synthesis.
A key component of immune homeostasis and humoral immunity is the Treg (145). While the transcription factor (TF) FOXP3 is a known lineage-specific factor for Tregs, the key TFs controlling Treg gene expression are poorly understood (104). Marson et al. conducted a pooled and arrayed Cas9 ribonucleoprotein screen in primary human Tregs to identify the TFs involved in regulating critical proteins in human Tregs (104). Transduced cells were sorted for sequencing based on their protein expression under diverse cytokine conditions (104). Sequencing of single-cell transcriptomes revealed the distinct control of gene networks by FOXP3 and PRDM1. HIVEP2 was found to co-regulate a separate gene network with SATB1 in Treg cell-mediated immunosuppression.
5.1.3 HIV infection
Antiretroviral therapy (ART) advancements have substantially reduced morbidity and mortality in individuals infected with the human immunodeficiency virus (HIV) (146). Individuals with HIV can effectively manage their infections over an extended period through daily pills or monthly injections (147). However, ART is not a curative method and must be taken for life. One of the major barriers to ART is the latent reservoir of HIV (148). HIV-1 preferentially infects the activated CD4+ T cells, and most infected cells die quickly (149). A few infected cells exist in a resting state, and the viral genome will integrate into their genome indefinitely (149).
Several inflammation-related genes are regulated by the canonical NF-κB and MAPK signaling pathways (150). In 2020, Zhu et al. conducted a genome-wide knockout screen in the C11 cell line using a human CRISPR knockout pooled library (GeCKOv2) (91). They discovered that PEBP1 could inactivate the MAPK/NF-κB signaling, inhibiting HIV transcription and inducing HIV latency. Moreover, ubiquitination is crucial in regulation of NF-κB signaling, such as ubiquitination mediated by E3 ubiquitin ligases (151). Ho et al. conducted a genome-wide knockout screen in the J-Lat 10.6 cell line using a human CRISPR knockout pooled library (95). The J-Lat 10.6 cell line contains an integrated replication-incompetent HIV-1 gene with a green fluorescent protein (GFP) reporter, with GFP expression correlating with HIV-1 reactivation (152). Combining the GFP expression and CRISPR screening results, they identified several deubiquitinases, including UCH37, USP14, OTULIN, and USP5, that may be HIV-1 latency regulators (95). This study was the first comprehensive investigation of HIV latency mechanisms centered on deubiquitinases.
Type I interferons (IFN-Is) are crucial in immune regulation during viral persistence, modulating various environmental and cellular functions (153). IFN-stimulated genes (ISGs) can prevent lab-adapted HIV infections in cells (153). However, their restrictive mechanism in primary CD4+T Cells, the HIV target cells, is poorly understood. Overbaugh et al. conducted a CRISPR-knockout screen in primary T cells using a CD4-ISG sgRNA library (101). They identified several previously unknown HIV-restrictive ISGs, including HM13, IGFBP2, and LAP3. This study represents one of the few articles investigating the HIV infection mechanism in primary T cells.
Several epigenetic regulators have also been found to contribute to HIV latency regulation through retrovirus and retrotransposon silencing (154–159). In 2019, Zhu et al. used a lentiviral sgRNA knockout sub-library to target nuclear proteins in J-LAT A2 cells hosting the latently infected “LTR-Tat-IRES-GFP” HIV-1 minigenome (100). The transduced cells were selected for sequencing based on their GFP expression level. They found that knockdown of the MINA53 gene can reactivate the latent HIV consistently. Furthermore, Ho et al. conducted a CRISPRi screen in Jurkat cells using human genome-wide CRISPRi-v2 libraries. They found that SLTM knockdown promoted the reactivation of latent HIV in Jurkat cells by increasing the chromatin accessibility of HIV-1 (105). Moreover, the mechanisms of transcriptional repression acting on the integrated viral promoter are crucial for HIV latency regulation. Taube et al. conducted a CRISPR genome-wide knockout screen in Jurkat cells. They found that ZNF304 and TRIM28 could combine and be recruited to the viral promoter heterochromatin-inducing methyltransferases to silence HIV gene transcription (93).
Additionally, sufficient evidence suggests that metabolism changes in CD4+ T cells affect HIV infection (160–163), although the mechanism is poorly understood. In 2018, Xu et al. explored the host factors required for HIV latency using a genome-wide CRISPR knockout screen (164). They used an HIV-1 latent infection cell line and sorted cells for sequencing based on their GFP expression level (164). They found that knocking out TSC1 or DEPDC5 promoted HIV-1 reactivation in the T-cell line by influencing the mechanistic target of rapamycin complex 1 (mTORC1) pathway, a key driver of cell metabolism. Subsequently, in 2021, Deng et al. conducted a CRISPR knockout screen in an HIV-1 latently infected cell line (J-Lat 9.2) with a human CRISPR metabolic gene knockout library. They found that MAT2A-mediated one-carbon metabolism contributes to regulating HIV latency (102).
5.1.4 Macrophages-mediated diseases
THP-1 is a human peripheral blood mononuclear cell line originally derived from a patient with acute monocytic leukemia. It is a suspension cell line and suitable for transfection or infection experiments. In 2023, Naderer et al. conducted a FACS-based genome-wide CRISPR/Cas9 screen in the THP-1 cell line to explore the mechanism between Panton–Valentine leukocidin (PVL) and complement C5a receptor 1 (C5aR1) (107). FBXO11 was identified as the potential target in macrophages for inflammation. Moreover, in 2019, a study performed a genome-scale CRISPR knockout library screen in THP-1 cells to identify potential genes involved in resistance to Salmonella uptake (94). NHLRC2 was identified as the target gene involved in both Salmonella invasion and macrophage differentiation.
Additionally, the U-937 is another cell line that be used to study monocyte behavior and differentiation. It was originally isolated from a 37-year-old male patient with histiocytic lymphoma. In 2018, Superti-Furga et al. conducted a FACS-based CRISPR screen with a solute carrier (SLC)-knockout library in U-937 cells to specifically explore SLCs (108). SLCs participated in metabolic changes associated with phagocytosis of macrophages. They determined that SLC4A7 inhibits human intracellular microbicidal activity and limits the acidification of phagocytosed beads.
5.2 CRISPR screening in murine immune cells
5.2.1 CD4+ T cells
5.2.1.1 Regulatory T cells
Tregs are a small subset of immune cells that play a pivotal role in maintaining immune homeostasis. Dysregulation or dysfunction of Tregs can give rise to inflammatory disorders, including graft-versus-host disease (GvHD), transplant rejection, and autoimmune diseases (165). In 2022, Rathmell et al. designed a 1C metabolism gRNA library based on the Brie CRISPR knockout pooled library and conducted a CRISPR knockout screen in an ovalbumin (OVA)-induced lung inflammation model (128). They showed that MTHFD2 participates in proliferation and inflammatory cytokine production by regulating de novo purine synthesis and activation signals.
Moreover, one major barrier to immunotherapy in cancer is the immunosuppressive effect of the Tregs (128). Gaining insight into the mechanism governing Treg function would significantly contribute to identifying novel therapeutic targets for cancer treatment. Moreover, transcriptional regulator FOXP3 has been identified as a master regulator of Tregs and is activated throughout their development of the thymus (166). In 2020, Zheng et al. conducted a genome-wide knockout screen to explore the FOXP3 regulator in mouse primary Tregs using the Brie mouse CRISPR knockout pooled library. The cells were sorted for sequencing based on their Foxp3 expression level (111). The Brd9-containing non-canonical BAF complex was identified as a therapeutic target for manipulating Treg function by promoting Foxp3 expression.
Additionally, studies have increasingly found that nutrients interact with immunological signals to activate mTORC1, a key regulator of cell metabolism (167–170). Understanding the mechanism of nutrient signaling processes may be a novel insight into therapeutic target discover for immune-mediated disorders. In 2021, a study conducted a genome-wide CRISPR knockout screen in the Tregs using the Brie genome-scale sgRNA library. The cells were sorted for sequencing based on their phosphorylation of S6 (p-S6) level (113). Sec31a was found to interact with the GATOR2 component Sec13 to promote mTORC1 activation.
5.2.1.2 Non-regulatory T cells
Recently, some studies used CRISPR screening to explore cell fate in non-regulatory T cells at the transcriptional and epigenetic levels, refining our understanding of CD4+ T cell biology. In 2021, Teichmann et al. conducted a genome-wide CRISPR screen in mouse naïve CD4+ T cells with the Teichmann retroviral mouse genome-wide CRISPR knockout library. The cells were sorted for sequencing based on different factors, including IL4 and IL13, XBP1, and GATA3 (120). They found that PPARG and BHLHE40 were the central networks regulating the differentiation of T helper 2 (Th2) cells. Additionally, the networks of lineage-specifying TFs controlling the specific differentiation of Th-cell subsets were affected by epigenetic processes (171). These epigenetic processes could change the genetic expression programs in Th cells (172). In 2023, a genome-wide CRISPR knockout screen was conducted in naive CD4+ T cells using a sgRNA library targeting 1,131 TFs to explore the differentiation mechanism of Th cells (109). The retrovirally transduced naïve CD4+ T cells of IL13-reporter mice (Rosa26Cas9EGFP Il13tdTomato) were induced through the TCR and IL4 signaling pathway to shift their differentiation toward Th2 cells. The cells were sorted for sequencing based on their IL13Tom expression (109). By acting as a crucial link between pioneer TFs and chromatin remodeling, the ADNP was found to be essential for immunological responses to allergens. Moreover, sufficient evidence suggests that metabolism is the guiding force in the differentiation of CD4+ T cells, which will undergo different metabolism changes throughout their life (173). One study used an in vivo screen to investigate the mechanisms underlying post-transcriptional and metabolic programs in T follicular helper (TFH) cell differentiation (124). Using a LCMV-Arm infection model, this study identified ETNK1, PCYT2, and SELENOI as specific post-transcriptional regulators selectively influencing TFH cell differentiation by enhancing CXCR5 surface expression and functional effects.
Additionally, CRISPR screening has also dramatically accelerated the therapeutic targets discovery in Th cells-mediated diseases in the last few years. Adoptive T-cell therapy (ATCT) contributes significantly to the therapeutic developments for cancer. Some studies have already found that coinjecting CD4+ and CD8+ T cells can improve and prolong antitumor activity (174, 175). While CD8+ T cells can clonally proliferate after antigen stimulation in vivo, intrinsic factors limit the proliferation of CD4+ T cells. In 2021, Menger et al. conducted a genome-wide CRISPR knockout screen in mouse CD4+ T cells. The transduced cells were sorted for sequencing based on their CFSE expression (121). They found that the SOCS1 acts as a nonredundant checkpoint inhibiting in vivo genome-wide CRISPR screens identify SOCS1 as intrinsic checkpoint of Th 1 cell proliferation. Moreover, systemic lupus erythematosus (SLE) is an autoimmune rheumatic disorder characterized by CD4+ T cell-mediated inflammatory responses that can result in tissue infiltration and organ damage (176). Recent studies have demonstrated a potential association between SLE pathogenesis and iron metabolism (177–179). In 2023, Rathmell et al. conducted a CRISPR knockout screen in naïve CD4+ T cells using a library targeting genes involved in iron metabolism (180). The transferrin receptor (TFRC/CD71) was identified as an activator of Th effector functions, including IL10 production, and an inhibitor of Tregs. Furthermore, the pathogenesis of helminth infections and allergic diseases is closely associated with Th2-mediated protective type 2 immune responses (181, 182) and Th2-dependent inflammatory responses (183–185). One study conducted a genome-wide CRISPR knockout screen in CD4+ T cells from IL13-reporter mice (Rosa26Cas9 Il13tdTomato) using the Brie mouse CRISPR knockout pooled library. The transduced cells were sorted for sequencing into brain finger protein BFP+ IL13Tom+ and BFP+ IL13Tom− (110). They found that αvβ3-expressing Th2 cells could promote and enhance the Th2 response by forming multicellular factories such as T-T cell clustering and IL2/CD25/STAT5 signaling.
5.2.2 CD8+ T cells
The studies based on CRISPR screens in mouse CD8+ T cells have mainly focused on the adoptive cell therapy (ACT). ACT can provide long-term benefits to cancer immunotherapy by isolating and engineering living T cells (186). The engineering of CD8+ T cells has sparked significant interest in ACT research due to its improved prognosis and specific cytotoxic capabilities (187–189).
The tumor model is mainly used in in vivo CRISPR screens to explore potential targets for ACT. Chen et al. conducted a genome-wide CRISPR knockout screen in OT1 cells using Rag−/− mice bearing E0771-mCh-OVA tumors, finding that DHX37 is a functional regulator of CD8+ T cells (119). In 2019, they also conducted an in vivo CRISPR screen to identify membrane targets for improving immunotherapy in GBM by transducing an adeno-associated virus (AAV)-surf library into naïve CD8+ T cells and transfusing them into GBM-bearing mice (126). In addition, another study transduced a lentiviral sgRNA metabolic sublibrary into activated OT1 cells and transfused them into B16-Ova melanoma-bearing mice to explore potential targets in ACT (112). They found that Reg1-deficiency promoted the persistence and effector function of the CD8+ T cells in tumors. Recently, Satpathy et al. also conducted a CRISPR knockout screen in the OT1 cell using a mini pool library, and the transduced cells were transfused into the Rag−/− mice six days after tumor inoculation (118). By sequencing the T cells from the tumors and spleen of the Rag−/− mice, they found that chromatin remodeling factors limit T cell persistence in vivo.
Moreover, the in vivo infectious model is another effective model in CRISPR screening research. In 2021, a study transduced a retroviral library into Cas9 CD8+ T cells, which have the P14-transgenic TCRs specific for LCMV gp3333–41, and then transfused the transduced cells into LCMV-infected mice (127). They found that genetic deletion of Fli1 enhanced effector T cell (TEFF) differentiation in infections and cancer. Additionally, Chi et al. conducted a CRISPR knockout screen based on the in vivo LCMV infectious model to systematically investigate metabolic factors in TEFF and memory T cell (TMEM) fate determination. The cells were sorted for sequencing based on their KLRG1 and CD127 expression (125). They demonstrated that amino acid transporters SLC7A1 and SLC38A2 inhibit TMEM differentiation by modulating mTORC1 signaling. Furthermore, in 2022, Green et al. transduced naïve OT-I cells with a knockout library targeting epigenetic regulators and transfused them into the Listeria monocytogenes-OVA infected mice to explore TMEM mechanisms (129). They identified the cBAF complex as a negative determinant of TMEM cells.
While the above in vivo CRISPR screens allow for exploring cell mechanisms at a general level, they are highly susceptible to interference by various complex factors. Indeed, in vitro CRISPR screens are also an essential component in high-throughput cell biological research. In 2020, a study conducted a CRISPR knockout screen in CD8+ T cells using a gRNA library targeting 25 kinases known to maintain their activity in T cells for up to 16 hours after TCR stimulation (123). Cells were categorized into distinct phenotypes based on their expression of diverse molecular markers. The p38 Kinase was identified as a potential therapeutic target for adoptive immunotherapies. Moreover, Chen et al. conducted a comprehensive genome-wide CRISPR activation screen within a cytotoxicity assay (130). SIINFEKL peptide-incubated E0771 cells were co-cultured with transduced CD8+ T cells, with CD8+ CD107a+ T cells subsequently sorted for sequencing. They identified PRODH2 as a potential target for promoting CAR-T cell efficacy. LFA1 can interact with ICAM1 and ICAM2 to participate in T cell migration, adhesion, and activation (190). Schwartzberg et al. conducted a CRISPR knockout screen in CD8+ T cells using a PIP3-binding protein CRISPR library to identify genes influencing the binding ability of primary mouse T cells to ICAM1 (122). RASA3 was identified as the potential target gene by comparing sgRNA frequencies between scICAM1neg (not binding ICAM1) and scICAM1pos (ICAM1-binding) cells.
5.2.3 B cells
Plasma cells are the primary source of antibodies, and the mechanism underlying their divergence from B cells has been thoroughly investigated (191–193). Disruptions in the typical antibody-secreting cell (ASC) formation pathway can cause immune-related diseases (132). The MAC-seq, which combines genetic analysis with quantitative methodologies, has been used to screen for drugs impacting the epigenetic machinery of B cells while assessing alterations in humoral immunity (132). Polycomb repressive complex 2 (PRC2) inhibitors were discovered that can support ASC differentiation in murine and human B cells in vitro. In addition, MYBL1, MYOF, GAS7, and ATOH8 were identified by functional dissection of downstream PRC2 effectors using an arrayed CRISPR screen (132).
Additionally, using a CRISPR/Cas9 screen, Wu et al. identified TCF3 and TCF4 and IRF4 as downstream effectors of p38 that govern plasma cell differentiation via Blimp1 transcription (133). Moreover, in 2020, Laidlaw et al. identified HHEX as a TF controlling memory B cell differentiation using an inducible CRISPR/Cas9 screening strategy (134). Furthermore, aiming to better understand the IgE-BCR-mediated modulation of IgE responses, Newman and Tolar conducted a whole-genome CRISPR screen to compare the needs of IgE+ and IgG1+ B cells for plasma cell proliferation, survival, and differentiation (116). Interestingly, they found that the absence of calcium-calcineurin-NFAT pathway components improved IgE+ plasma cell development, whereas IgE+ plasma cells depended on the phosphoinositide 3-kinase PI3K-mTOR axis (116).
5.2.4 Macrophages
Macrophages are a major component of the innate immune system and play a critical role in host-pathogen interactions (194). Microbial pathogens have evolved extensive defense systems to avoid being killed by macrophages (195). L. monocytogenes is a Gram-positive intracellular pathogen that can cause serious invasive infections (196). In 2023, Reniere et al. conducted an unbiased CRISPR/Cas9 screen using the Brie sgRNA library in immortalized bone marrow-derived macrophages (iBMDMs) to identify host factors important for L. monocytogenes infection (115). Tumor suppressor PTEN was found to promote macrophage phagocytosis of L. monocytogenes. Additionally, Mycobacterium abscessus was found to cause increasing community- and hospital-acquired infections in humans (197). In 2023, a study transduced murine macrophages with a genome-wide knockout library to generally define and characterize important regulators in macrophage-M. abscessus interactions, such as sulfated glycosaminoglycans(sGAG) (198). They found that macrophage-M. abscessus interactions were mainly regulated by sGAG. Moreover, Histoplasma capsulatum is a common pathogenic fungus that can live inside macrophage phagosomes (199) and causes the most common fungal respiratory disease in the United States (200). In 2022, Sil et al. performed a host-directed CRISPR/Cas9 screen in the mouse mononuclear macrophage cell J774A.1 cell line to explore the interaction mechanism between H. capsulatum and macrophages (135). They found that the C3a receptor (C3aR) was important in macrophages capturing pathogenic fungi.
We can also use CRISPR screening to explore the molecular mechanisms of different pathways involved in macrophage activation and function. IFN-γ promotes the proinflammatory responses of macrophages during infection (201). In 2020, Saeij et al. conducted a genome-wide CRISPR screen in bone-marrow-derived macrophages (BMDMs), discovering 353 toxoplasma genes determining parasite fitness in naїve or IFNγ-activated murine macrophages (136). They also found that parasites missing the dense granule protein GRA45 were less severe and more vulnerable to IFN-mediated growth suppression in mice. Moreover, in 2020, a study conducted a CRISPR screen in iBMDMs to explore genes essential for macrophage viability (137). The tumor necrosis factor (TNF) signaling cascade was found to have a suppressive effect on macrophages in an autocrine manner. Additionally, efferocytosis, which involves the clearance of apoptotic cells, is crucial for various processes, such as immune cell proliferation, tissue turnover, and organ regeneration (202). In 2022, Zhang et al. conducted a FACS-based genome-wide CRISPR knockout screen in primary mouse macrophages. They identified WDFY3 as a potential target for efferocytosis in macrophages (114). Futhermore, the N6-methyladenosine RNA modification can occur post-transcriptionally in almost all biological processes (203). However, its function in the innate immune cells is unknown. Li et al. identified major m6 A “writers” as the top candidate genes controlling macrophage activation in an RNA binding protein-focused CRISPR screen (131). They also discovered that Mettl3 deficiency could inhibit TNF-α production in macrophages after lipopolysaccharide stimulation in vitro.
6 CRISPR screening in cancer immunology
Cancer is a genetic disease caused by cumulative genetic/epigenetic aberrations (204). An essential characteristic of cancer is immune evasion, primarily governed by checkpoints (205), which poses a substantial hurdle to cancer therapy. Fortunately, we can precisely identify the candidate genes whose mutation causes cancer with the help of the CRISPR screening, enabling us to explore appropriate drug targets and helping us understand cancer genomics (52). We summarized the current applications of CRISPR screens in the cancer cells and provided a summarization in Table 7.
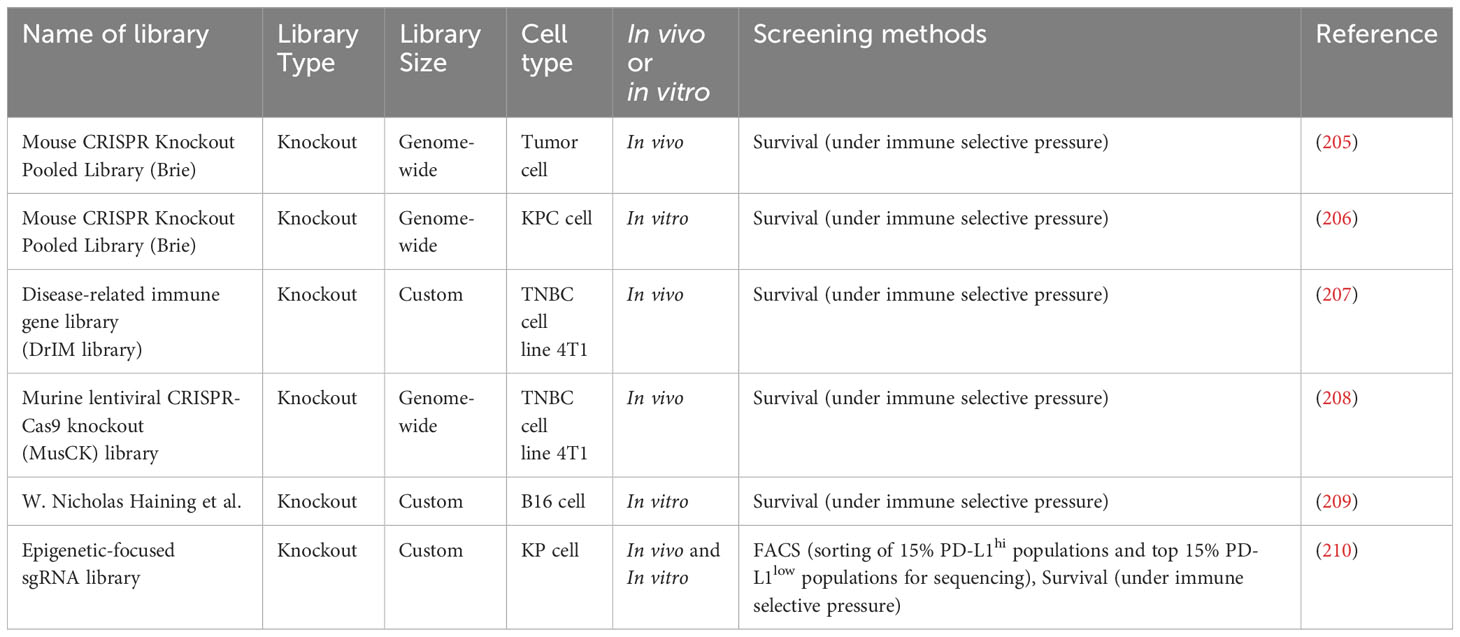
Table 7 CRISPR screening studies in cancer immunology.
Pancreatic ductal adenocarcinoma (PDA) is characterized by inherent immune cell deficiency and a distinctive TME composed of desmoplastic stroma (211) and suppressive immune cells (212). Frey et al. conducted in vitro and in vivo CRISPR screening to systematically explore PDA’s intrinsic mechanisms of immune evasion (206). They discovered that VPS4B and RNF31 are important in escaping CD8+ T cell killing. Additionally, immunotherapy is limited in patients with PDA due to inadequate T-cell infiltration and activation within the TME (213). Moreover, the CRISPR screens conducted by Fan et al. identified KDM3A as a potent epigenetic regulator of the immunotherapy response in patients with PDA (214). Furthermore, triple-negative breast cancer (TNBC) has the highest mortality and recurrence rates of all breast cancer types (215). Ji et al. conducted two-step customized in vivo CRISPR screens targeting disease-related immune genes (207). They identified LGALS2 as a potential immune escape regulator in different mouse models with multifaceted immune-deficiency characteristics.
Immune checkpoint blockade (ICB) is the primary immunotherapy in cancer care, and it has a considerable curative effect in many solid tumor types (216). Dubrot et al. conducted genome-scale CRISPR screens across cancer models treated with ICB, discovering that immune escape could be facilitated by inhibitory checkpoints involving IFN-mediated upregulation of classical and non-classical major histocompatibility complex (MHC) class I (205). However, the ICB is not appropriate for everyone with TNBC, and it can also cause additional side effects (217). Wang et al. conducted in vivo CRISPR screens, discovering that knockout of the Cop1 in cancer cells decreased macrophage-associated chemokine secretion and tumor macrophage infiltration but enhanced antitumor immunity and ICB response in TNBC (208). An immune therapy blocking the PD1 checkpoint also had durable antitumor effects in different cancer types (218). However, it is only effective in a minority of patients (209). Manguso et al. conducted a CRISPR/Cas9 genome editing in transplantable mouse tumors treated with immunotherapy, confirming that defective IFN-γ signaling leads to immunotherapy resistance (209). Moreover, TSC1 and TSC2 are frequently mutated in non-small cell lung cancer (219). However, their functions in antitumor immunity remain unexplored. Huang et al. conducted in vitro and in vivo CRISPR screening in a murine KrasG12D/Trp53−/− lung cancer model to systemically explore cell-intrinsic regulators of antitumor immunity (210). TSC1 and TSC2 were found to regulate PD-L1 expression in vitro. Additionally, they influenced the sensitivity to anti-PD1 treatment.
In conclusion, we can systematically explore genetic targets contributing to the immune escape mechanism using high-throughput CRISPR technology, which may provide new insights into the immune tolerance building of transplantation.
7 CRISPR technology in solid organ transplantation
7.1 Genomic modification of immune cells in organ recipients
Advancements in surgical techniques, high-quality perioperative care, efficient immunosuppressive agents, and effective post-transplant management of infectious diseases have substantially increased the success rate of organ transplantation. Therefore, organ transplantation has become the primary therapeutic approach for end-stage organ failure. However, the limited lifetime of the allograft is the primary barrier in organ transplantation. Twenty percent of recipients will lose their graft within five years, and 50% within 10–12 years (220). Allografts rejection, primarily regulated by the immune system, represents the central problem to the long-term survival of grafts. The interplay between the innate and adaptive immune systems, mediated by diverse immune cells, constitutes a key component of allografts rejection (221, 222). Furthermore, these immune cells can induce more vigorous immunological rejection in xenotransplantation. In recent years, numbers of studies have focused on the CRISPR genome editing of immune cells in recipients.
Firstly, Tregs have shown significant potential in ACT due to their feasibility, tolerability, and potential efficacy (165). Moreover, Tregs are pivotal target cells for establishing transplant tolerance (223). Until now, Treg therapy has consistently been shown to be a safe therapeutic method (224). It is important to minimize morbidity and mortality among solid organ transplant recipients and reduce the need for pan-immunosuppressive drugs (19). The first application of Treg therapy was in GvHD, and its therapeutic effect was demonstrated with data from preclinical models (165). In recent years, more and more studies used the CRISPR technology to modify the immune-modulating function of Tregs (225). In 2016, Levings et al. created an A2-CAR (HLA-A2–specific CAR) that markedly enhanced the therapeutic potential of Tregs in transplantation using CAR technology (225). Additionally, in 2017, Yoshimura et al. edited the Foxp3 gene in mouse primary T cells using CRISPR/dCas9 (21). Moreover, in 2019, Horvat et al. used an activator-domain fusion-based dCas9 transcription activator to achieve prolonged FOXP3 expression in Jurkat cells (22).
Besides Tregs, dendritic cells (DCs) are another potential gene-editing target for immune tolerance in organ transplantation. In 2019, Wang et al. used CRISPR/Cas9-based nanomedicine technology to directly cause CD40 deficiency in DCs in vitro and in vivo (226). They successfully induced a protective effect on graft survival by regulating T-cell activation.
In conclusion, the CRISPR genome editing technology might be a potential way to precisely and effectively intervene the immune cell function of organ recipients.
7.2 Genomic modification in organ donors
Another tricky issue for developing organ transplantation is the insufficient supply of donor organs. In 2020, the Organ Procurement and Transplantation Network reported that 116,577 patients were on waiting lists for organ transplantation in the USA (227). On average, 20 patients on the waiting list die every day because of the shortage of donor organs (228). Pre-implantation transcriptome modulation in allografts or xenografts have been proven a potential way to expand the donor pool in solid organ transplantation (229, 230).
In 2023, Keshavjee et al. used CRISPR/Cas technologies to upregulate IL10 expression in the donor lung and transplanted it into recipient rats (231). In addition, human and rat cell lines were tested for potency, titratability, and multiplexibility (231). This study has proved the feasibility of pre-implantation transcriptome modulation using CRISPR technology to expand the donor pool and improve transplantation outcomes. Moreover, using CRISPR gene editing, Schrepfer et al. removed the MHC I and II molecules and inserted CD47 into mouse and human induced pluripotent stem cells (iPSCs) to create hypoimmunogenic iPSCs that retain their pluripotent stem cell potential and differentiation capacity (232). In totally MHC mismatched allogeneic recipients, endothelial cells, smooth muscle cells, and cardiomyocytes created from hypoimmunogenic mouse or human iPSCs reliably evaded immune rejection and survived over the long term without needing treatment (232). These findings suggest the possibility of creating hypoimmunogenic cell grafts for future universal transplantation.
In the xenotransplantation field, increasing evidence suggests that pigs may be the best xenograft source (230) because of their size, availability, breeding characteristics, and physiological similarities to humans (228). However, the major problem with xenotransplantation is the immune barriers between pigs and humans (233, 234). In recent years, advances in the genetic modification of pigs with CRISPR-Cas9 have contributed significantly to the advancement of xenotransplantation (233). In 2014, Zhou et al. created the first CRISPR/Cas9-engineered pig and developed an efficient one-step method to generate genome-modified pigs by zygote injection of the CRISPR/Cas system (235). This method suggests the potential of CRISPR technology to swiftly establish substantial animal models for human research. Moreover, subsequent studies have used CRISPR technology to knockout glycosyltransferase and other genes (PERV, GHR, ULBP1, and SLA-1) or knock-in one gene (an anti-CD2 monoclonal antibody transgene into GGTA1) in pigs (236). Overall, there is ample evidence to demonstrate that CRISPR technology has emerged as an effective tool in xenotransplantation research.
8 Prospective applications of CRISPR technology in solid organ transplantation
As mentioned above, rejection caused by immune cells and the shortage of donor organs are the two significant barriers to developing organ transplantation. Impeding the function of immune cells in rejection and genetically modifying the graft to escape the immune system are the major tasks in organ transplantation. Additionally, with the rapid development of CRISPR technology, we can make precise and stable genome edits in immune cells and grafts. However, current studies have identified few genetic targets that can effectively build immune tolerance in transplantation. Advancements in high-throughput CRISPR technology have accelerated the discovery of potential therapeutic targets in immune cells and cancer cells. This provides a novel insight into identifying therapeutic targets in transplantation. Genome-wide screening of the immune or non-immune cells and intervening these targets using the CRISPR/Cas9 system may be a novel horizon in transplantation research.
8.1 Conducting CRISPR screening in solid organ transplantation
8.1.1 CRISPR screening in immunocytes of organ recipients
Immunocyte-mediated rejection is the primary barrier to building long-term graft survival. There is an urgent need to explore effective approaches to modulate immunocytes in transplantation. However, few treatments are available for immunocyte intervention despite significant advances in transplantation in recent decades. Most immunosuppressive agents have successfully improved short-term graft survival, but long-term graft survival has stagnated. Fortunately, advancements in CRISPR/Cas9 genome editing technology make it possible to conduct precision medicine based on immunocytes in transplantation. In the last decade, CRISPR screening has been widely used in different immunological diseases to explore many potential targets in immunocytes, providing novel insights into transplantation.
8.1.1.1 CRISPR screening in alloreactive T cells
As mentioned above, CRISPR screening has been widely applied to T cells with a particular focus on improving immune function in cancer (237), suppressing the inflammatory effect in autoimmune disease (11), and enhancing antiviral function in infectious diseases (238). Sufficient evidence demonstrates that CRISPR screening is an effective method for detecting potential targets in T cells. Based on the clinical experience, the therapeutic agents that can modulate the function of alloreactive T cells have the considerable effects on long-term graft survival. By understanding how alloreactive T lymphocytes recognize donor antigens, we can intervene them more effectively in solid organ transplantation (239). Conducting CRISPR screens in alloreactive T cells may be a novel approach to exploring potential targets for long-term graft survival.
The current applications of CRISPR screening in T cells have mainly focused on Tregs, Th cells, and cytotoxic T cells. These cells also play an important role in transplant rejection and can be the suitable cell models for CRISPR screening in transplantation. With advances in CRISPR screening, we can use multiple approaches to explore the genetic mechanism underlying different effector cell phenotypes in transplantation, including cell fate, effector function, and cell exhaustion.
For in vitro CRISPR screening, we can use a suitable gRNA library to target alloreactive T cells and apply different intervention factors to the transduced cells. Then, the different alloreactive T cells phenotypes can be selected by FACS or MACS based on the multiple cell markers. After sequencing, the genetic mechanism underlying different phenotypes can be elaborated. For in vivo CRISPR screening, the alloreactive T cells can be transduced with the gRNA library in vitro. Then, the transduced cells can be transferred into various animal transplantation models. The different intervention approaches can also be conducted in animal models. Finally, the cells for sequencing can be sorted based on different cell makers, and the target genes can be validated using the sequencing results.
8.1.1.2 CRISPR screening in alloantigen APCs
By directly interacting with T cells, APCs orchestrate distinct functional outcomes of the immune response (240). Therefore, exploring effective approaches to modulate APCs is one major task for transplantation. In transplantation, dying graft cells release inflammatory molecules that trigger APC maturation and T-cell response (241). As mentioned above, one study successfully used the CRISPR/Cas9-based nanomedicine technology to directly cause CD40 deficiency in DCs to create a protective effect on graft survival. This study showed that CRISPR-based genomic editing may be an effective method to make precise genomic edits in APCs. Moreover, using CRISPR screening, we can explore the potential targets at the genome-wide scale. The CRISPR screens conducted in APCs have mainly focused on the host-pathogen interaction of macrophages. Macrophages have been shown to be a reliable and stable cell model for CRISPR screening in APCs. In transplantation, after antigen stimulation (in vitro or in vivo), the transduced cells can be sorted for sequencing using different cell markers, such as the MHC. Then, the candidate genes are validated using the sequencing results. Finally, multiple antigen presentation assays can be used to validate the candidate genes in APCs.
8.1.2 CRISPR screening in organ donors
CRISPR screens in non-immune cells have mainly focused on the immune escape mechanism of cancer cells under immune pressure. In organ transplantation, allograft rejection-mediated immune pressure is also an important factor affecting graft survival, significantly shrinking the donor pool for organ transplantation. Performing CRISPR screens in the grafts will accelerate the discovery of resistance target genes to immune pressure. Additionally, combining donor organ ex vivo perfusion systems and CRISPR gene editing technology increases the time window and enhances the capability for pre-implantation transcriptome editing of grafts. Identifying resistance target genes using CRISPR screens and then using CRISPR technology to modify grafts may be a novel approach for transplantation.
For in vitro CRISPR screens in grafts, the transduced cell can be applied with a killing assay mediated by the cytotoxic cells in allograft rejection. Then, the surviving cells can be sorted for sequencing to explore the resistance genes for allograft rejection. Additionally, Pu et al. successfully conducted an in vivo CRISPR screening in Cas9 mutagenesis mice using an AAV gRNA library to explore potential targets for cardiomyocyte maturation (242). This approach may be a reliable tool for conducting in vivo CRISPR screens in grafts. After transducing the grafts with the AAV gRNA library, they could be transplanted into an allogeneic animal model to create immune pressure in vivo. Then, the graft cells could be sorted for sequencing to identify the potential therapeutic targets. Additionally, various biomedical methods can be used to validate the therapeutic effects of the candidate genes.
8.2 The overview of CRISPR technology in solid organ transplantation
By combining CRISPR screening with traditional CRISPR technology in transplantation research, we conceive an overview specific to solid organ transplantation studies. The main protocol steps are provided in Figure 5. First, we should have a scientific question. Second, an appropriate model should be chosen based on this question. Third, an optimized gRNA library (custom or genome-wide) should be designed, followed by pooled sgRNA synthesis, plasmid cloning, and viral particle production. Fourth, after transducing the gRNA library into the target cells, they should be sorted for sequencing based on positive or negative selection or different phenotypes. Fifth, the genomic DNA (gDNA), which contains the global genetic information of sorted cells, need be isolated and amplified for further analysis. Sixth, various methods should be used to validate candidate genes identified from the sequencing results. A mini-pool gRNA library can be designed based on these candidate genes to enable their more precise targeting. Finally, after identifying candidate genes, we can genetically modify grafts or immune cells to build immune tolerance in clinical applications or animal models.
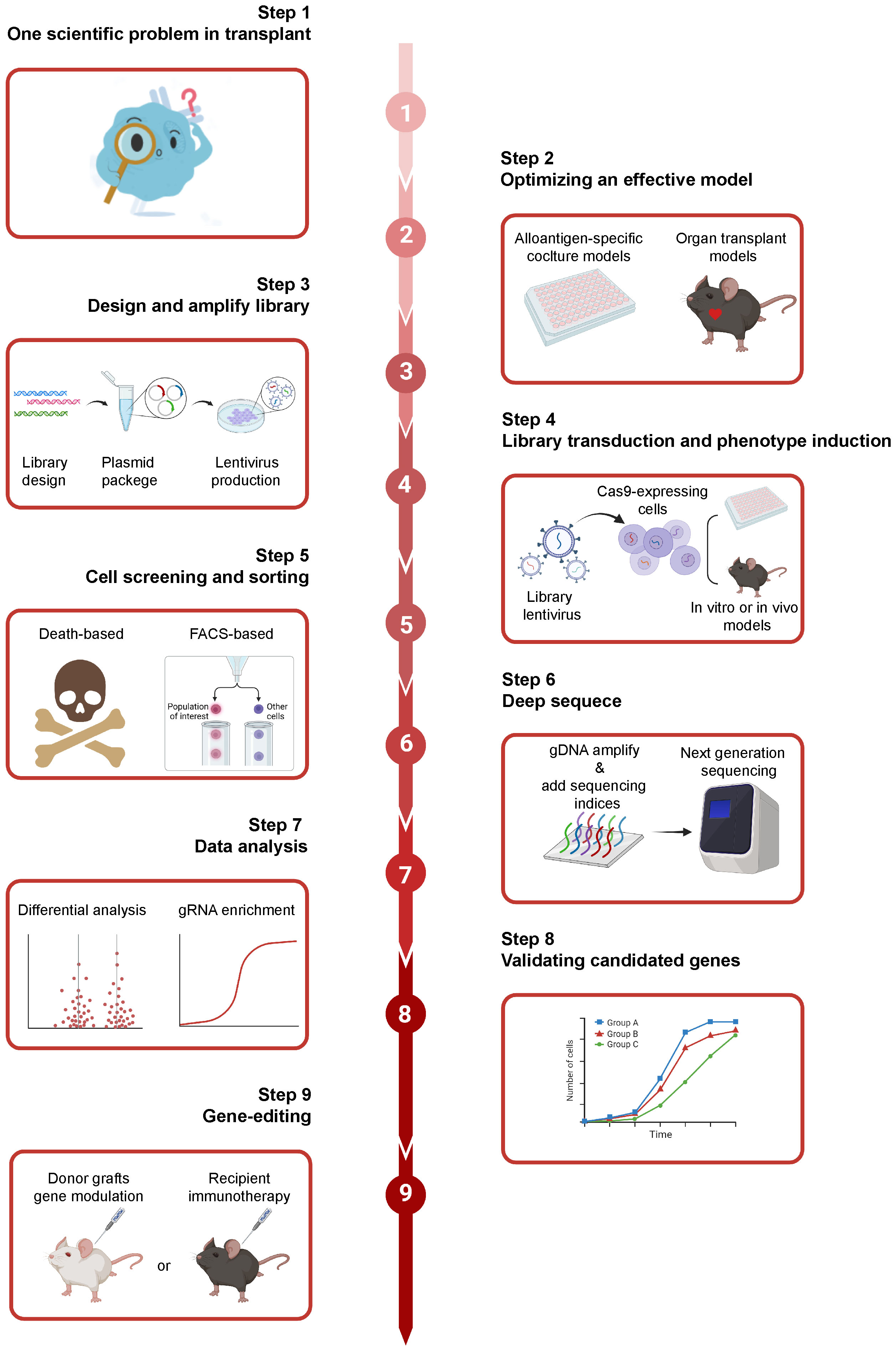
Figure 5 The overview of CRISPR screening in solid organ transplantation. Firstly, we should have a scientific question. Subsequently, an appropriate model can be chosen based on the scientific question. And, an optimized gRNA library (custom or genome-wide) should be designed, followed by pooled sgRNA synthesis, plasmid cloning, and viral particle production. Additionally, after transducing the gRNA library into the target cells, they can be sorted for sequencing based on positive or negative selection or different phenotypes. The genomic DNA (gDNA), the global genetic information of sorted cells, need to be isolated and amplified for further analysis. Finally, the potential targets in the donor grafts and immunotherapy can be intervened with the CRISPR/Cas 9 based genome editing, giving the novel insights for clinical problems. Created with BioRender.com.
8.3 The challenges of CRISPR technology in solid organ transplantation
Firstly, the delivery of Cas9 to certain cell types is often constrained by low transfection and transduction efficiency, particularly in primary immune cells and in vivo (243). Up to now, electroporation may be a potential way to solve this. Additionally, in CRISPR knockout screenings, certain perturbed cells might retain target protein function due to in-frame editing (244). And, verifying individual cell edits is usually not possible which increases the screen’s noise signals. Off-target effects are also a major potential problem for CRISPR genome editing (245). Incorrect gRNA binding will lead to undesired off-target mutations, potentially leading to genome instability and disrupting the normal functions of genes. CRISPR/Cas9 genome editing can also lead to massive base loss, which is potentially a more severe outcome than off-target effects (246). Furthermore, on-target mutagenesis in double-strand breaks which are caused by single-guided RNA/Cas9 can produce the long-range transcriptional consequences and pathogenic consequences (247). Finding the efficient tools that can make more precise genome edits is urgent in the field of CRISPR genome editing.
Moreover, the immune response is another severe issue in CRISPR genome editing. The gRNAs and DNA plasmids can activate the pattern recognition receptors that sense the foreign nucleic acids. For example, a triphosphate group at the 5’ end of gRNAs in the ribonucleoprotein (RNP) complex can be recognized as a foreign viral RNA by the RIG-I receptor, inducing the type I IFN-mediated immune response that mediates a cytotoxic effect (248, 249). Moreover, adenoviral vectors can also induce an inflammatory response, impeding the development of in vivo genome intervention (250–252). Thus, safe and efficient delivery systems is urgent in therapeutic genome editing in transplantation.
Conclusion
In recent years, CRISPR/Cas9 has proven a promising strategy for precisely altering multiple genetic targets in previously untreatable disorders. It has already shown potential therapeutic value in solid organ transplantation, such as CAR-T therapy for Allograft rejection and genetic modification in xenotransplantation. Especially, combining CRISPR/Cas9-based gene editing with an ex vivo organ perfusion system would enable pre-implantation transcriptome editing of grafts. Additionally, a high-throughput CRISPR screening technology has shown a powerful ability to determine the genetic mechanisms behind specific phenotypes in the genomic level. Genome-wide screening of the immune or non-immune cells and intervening these targets using the CRISPR/Cas9 system may be a novel horizon in transplantation research.
Author contributions
XL: Writing – original draft. ZC: Writing – original draft. WY: Writing – original draft. JY: Writing – review & editing. XZ: Writing – review & editing. YL: Writing – review & editing. YN: Writing – review & editing. SR: Writing – review & editing. SW:Writing – review & editing. ZL: Writing – review & editing. JLZ: Writing – review & editing. YH: Writing – review & editing. JJZ: Writing – review & editing. CX: Writing – review & editing. JX: Writing – review & editing. JW: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Fundamental Research Funds for the Central Universities [2021GCRC037 to JX, and YCJJ20230232 to SR], the National Natural Science Foundation of China [82241217 and 82071803 to JW, 82271811, 82170504, and 81730015 to JX], the project funded by China Postdoctoral Science Foundation [2021M691155 to JY, and 2023M741285 to XZ], and the Science foundation of union hospital [2021xhyn096 to JY].
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
References
1. Janik E, Niemcewicz M, Ceremuga M, Krzowski L, Saluk-Bijak J, Bijak M. Various aspects of a gene editing system-CRISPR-cas9. Int J Mol Sci (2020) 21:9604. doi: 10.3390/ijms21249604
2. Zhou X, Renauer PA, Zhou L, Fang SY, Chen S. Applications of CRISPR technology in cellular immunotherapy. Immunol Rev (2023) 320:199–216. doi: 10.1111/imr.13241
3. Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science (2014) 343:84–7. doi: 10.1126/science.1247005
4. Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science (2014) 343:80–4. doi: 10.1126/science.1246981
5. Shi H, Doench JG, Chi H. CRISPR screens for functional interrogation of immunity. Nat Rev Immunol (2023) 23:363–80. doi: 10.1038/s41577-022-00802-4
6. Schuster A, Erasimus H, Fritah S, Nazarov PV, van Dyck E, Niclou SP, et al. RNAi/CRISPR screens: from a pool to a valid hit. Trends Biotechnol (2019) 37:38–55. doi: 10.1016/j.tibtech.2018.08.002
7. Evers B, Jastrzebski K, Heijmans JP, Grernrum W, Beijersbergen RL, Bernards R. CRISPR knockout screening outperforms shRNA and CRISPRi in identifying essential genes. Nat Biotechnol (2016) 34:631–33. doi: 10.1038/nbt.3536
8. Munoz DM, Cassiani PJ, Li L, Billy E, Korn JM, Jones MD, et al. CRISPR screens provide a comprehensive assessment of cancer vulnerabilities but generate false-positive hits for highly amplified genomic regions. Cancer Discovery (2016) 6:900–13. doi: 10.1158/2159-8290.CD-16-0178
9. Huang A, Garraway LA, Ashworth A, Weber B. Synthetic lethality as an engine for cancer drug target discovery. Nat Rev Drug Discovery (2020) 19:23–38. doi: 10.1038/s41573-019-0046-z
10. Rousset F, Bikard D. CRISPR screens in the era of microbiomes. Curr Opin Microbiol (2020) 57:70–7. doi: 10.1016/j.mib.2020.07.009
11. Ding J, Frantzeskos A, Orozco G. Functional interrogation of autoimmune disease genetics using CRISPR/Cas9 technologies and massively parallel reporter assays. Semin Immunopathol (2022) 44:137–47. doi: 10.1007/s00281-021-00887-4
12. Kauke-Navarro M, Noel OF, Knoedler L, Knoedler S, Panayi AC, Stoegner VA, et al. Novel strategies in transplantation: genetic engineering and vascularized composite allotransplantation. J Surg Res (2023) 291:176–86. doi: 10.1016/j.jss.2023.04.028
13. Lian J, Meng X, Zhang X, Hu H. Establishment and genetic manipulation of murine hepatocyte organoids. J Vis Exp (2022) e62438. doi: 10.3791/62438
14. Carter NM, Pomerantz JL. Calcineurin inhibitors target Lck activation in graft-versus-host disease. J Clin Invest (2021) 131:e149934. doi: 10.1172/JCI149934
15. Lim S, Kirkiles-Smith NC, Pober JS, Bothwell A, Choi JM. Regulation of human T cell responses by dNP2-ctCTLA-4 inhibits human skin and microvessel graft rejection. Biomaterials (2018) 183:128–38. doi: 10.1016/j.biomaterials.2018.08.049
16. Husain I, Luo X. Novel approaches to immunomodulation for solid organ transplantation. Annu Rev Med (2023) 75:4.1–4.12. doi: 10.1146/annurev-med-050522-034012
17. Nash A, Lokhorst N, Veiseh O. Localized immunomodulation technologies to enable cellular and organoid transplantation. Trends Mol Med (2023) 29:635–45. doi: 10.1016/j.molmed.2023.05.008
18. Mikami N, Sakaguchi S. Regulatory T cells in autoimmune kidney diseases and transplantation. Nat Rev Nephrol (2023) 19:544–57. doi: 10.1038/s41581-023-00733-w
19. Cremoni M, Massa F, Sicard A. Overcoming barriers to widespread use of CAR-Treg therapy in organ transplant recipients. Hla (2022) 99:565–72. doi: 10.1111/tan.14591
20. MacDonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, Orban PC, et al. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Invest (2016) 126:1413–24. doi: 10.1172/JCI82771
21. Okada M, Kanamori M, Someya K, Nakatsukasa H, Yoshimura A. Stabilization of Foxp3 expression by CRISPR-dCas9-based epigenome editing in mouse primary T cells. Epigenet Chromatin (2017) 10:24. doi: 10.1186/s13072-017-0129-1
22. Forstneric V, Oven I, Ogorevc J, Lainscek D, Praznik A, Lebar T, et al. CRISPRa-mediated FOXP3 gene upregulation in mammalian cells. Cell Biosci (2019) 9:93. doi: 10.1186/s13578-019-0357-0
23. Zubair H, Azim S, Maluf DG, Mas VR, Martins PN. Contribution of proteomics in transplantation: identification of injury and rejection markers. Transplantation (2023) 107:2143–54. doi: 10.1097/TP.0000000000004542
24. LaFleur MW, Sharpe AH. CRISPR screens to identify regulators of tumor immunity. Annu Rev Cancer Biol (2022) 6:103–22. doi: 10.1146/annurev-cancerbio-070120-094725
25. Chen PJ, Liu DR. Prime editing for precise and highly versatile genome manipulation. Nat Rev Genet (2023) 24:161–77. doi: 10.1038/s41576-022-00541-1
26. Bogdanove AJ, Bohm A, Miller JC, Morgan RD, Stoddard BL. Engineering altered protein-DNA recognition specificity. Nucleic Acids Res (2018) 46:4845–71. doi: 10.1093/nar/gky289
27. Knott GJ, Doudna JA. CRISPR-Cas guides the future of genetic engineering. Science (2018) 361:866–69. doi: 10.1126/science.aat5011
28. Wetzel JL, Zhang K, Singh M. Learning probabilistic protein-DNA recognition codes from DNA-binding specificities using structural mappings. Genome Res (2022) 32:1776–86. doi: 10.1101/gr.276606.122
29. Afek A, Shi H, Rangadurai A, Sahay H, Senitzki A, Xhani S, et al. DNA mismatches reveal conformational penalties in protein-DNA recognition. Nature (2020) 587:291–96. doi: 10.1038/s41586-020-2843-2
30. Etheve L, Martin J, Lavery R. Decomposing protein-DNA binding and recognition using simplified protein models. Nucleic Acids Res (2017) 45:10270–83. doi: 10.1093/nar/gkx627
31. Schuler G, Hu C, Ke A. Structural basis for RNA-guided DNA cleavage by IscB-omegaRNA and mechanistic comparison with Cas9. Science (2022) 376:1476–81. doi: 10.1126/science.abq7220
32. Xiao R, Wang S, Han R, Li Z, Gabel C, Mukherjee IA, et al. Structural basis of target DNA recognition by CRISPR-Cas12k for RNA-guided DNA transposition. Mol Cell (2021) 81:4457–66. doi: 10.1016/j.molcel.2021.07.043
33. Zhang B, Lin J, Perculija V, Li Y, Lu Q, Chen J, et al. Structural insights into target DNA recognition and cleavage by the CRISPR-Cas12c1 system. Nucleic Acids Res (2022) 50:11820–33. doi: 10.1093/nar/gkac987
34. Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science (2007) 315:1709–12. doi: 10.1126/science.1138140
35. Horvath P, Barrangou R. CRISPR/cas, the immune system of bacteria and archaea. Sci (American Assoc Advancement Science) (2010) 327:167–70. doi: 10.1126/science.1179555
36. Amitai G, Sorek R. CRISPR-Cas adaptation: insights into the mechanism of action. Nat Rev Microbiol (2016) 14:67–76. doi: 10.1038/nrmicro.2015.14
37. Makarova KS, Wolf YI, Iranzo J, Shmakov SA, Alkhnbashi OS, Brouns S, et al. Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nat Rev Microbiol (2020) 18:67–83. doi: 10.1038/s41579-019-0299-x
38. Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, et al. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol (2015) 13:722–36. doi: 10.1038/nrmicro3569
39. Yan J, Kang DD, Dong Y. Harnessing lipid nanoparticles for efficient CRISPR delivery. Biomater Sci (2021) 9:6001–11. doi: 10.1039/d1bm00537e
40. Jiang W, Marraffini LA. CRISPR-cas: new tools for genetic manipulations from bacterial immunity systems. Annu Rev Microbiol (2015) 69:209–28. doi: 10.1146/annurev-micro-091014-104441
41. Takasugi PR, Wang S, Truong KT, Drage EP, Kanishka SN, Higbee MA, et al. Orthogonal CRISPR-Cas tools for genome editing, inhibition, and CRISPR recording in zebrafish embryos. Genetics (2022) 220:iyab196. doi: 10.1093/genetics/iyab196
42. Khoshandam M, Soltaninejad H, Mousazadeh M, Hamidieh AA, Hosseinkhani S. Clinical applications of the CRISPR/Cas9 genome-editing system: Delivery options and challenges in precision medicine. Genes Dis (2024) 11:268–82. doi: 10.1016/j.gendis.2023.02.027
43. Hu P, Zhao X, Zhang Q, Li W, Zu Y. Comparison of various nuclear localization signal-fused cas9 proteins and cas9 mRNA for genome editing in zebrafish. G3 (Bethesda) (2018) 8:823–31. doi: 10.1534/g3.117.300359
44. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science (2012) 337:816–21. doi: 10.1126/science.1225829
45. Azhagiri M, Babu P, Venkatesan V, Thangavel S. Homology-directed gene-editing approaches for hematopoietic stem and progenitor cell gene therapy. Stem Cell Res Ther (2021) 12:500. doi: 10.1186/s13287-021-02565-6
46. Shy BR, Vykunta VS, Ha A, Talbot A, Roth TL, Nguyen DN, et al. High-yield genome engineering in primary cells using a hybrid ssDNA repair template and small-molecule cocktails. Nat Biotechnol (2022) 41:521–31. doi: 10.1038/s41587-022-01418-8
47. Zhao Z, Shang P, Mohanraju P, Geijsen N. Prime editing: advances and therapeutic applications. Trends Biotechnol (2023) 41:1000–12. doi: 10.1016/j.tibtech.2023.03.004
48. Anzalone AV, Koblan LW, Liu DR. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol (2020) 38:824–44. doi: 10.1038/s41587-020-0561-9
49. Koopal B, Kruis AJ, Claassens NJ, Nobrega FL, van der Oost J. Incorporation of a Synthetic Amino Acid into dCas9 Improves Control of Gene Silencing. ACS Synth Biol (2019) 8:216–22. doi: 10.1021/acssynbio.8b00347
50. Jensen TI, Mikkelsen NS, Gao Z, Fosselteder J, Pabst G, Axelgaard E, et al. Targeted regulation of transcription in primary cells using CRISPRa and CRISPRi. Genome Res (2021) 31:2120–30. doi: 10.1101/gr.275607.121
51. Wang B, Chen JZ, Luo XQ, Wan GH, Tang YL, Wang QP. The application of genome-wide CRISPR-Cas9 screens to dissect the molecular mechanisms of toxins. Comput Struct Biotechnol J (2022) 20:5076–84. doi: 10.1016/j.csbj.2022.09.012
52. Wang SW, Gao C, Zheng YM, Yi L, Lu JC, Huang XY, et al. Current applications and future perspective of CRISPR/Cas9 gene editing in cancer. Mol Cancer (2022) 21:57. doi: 10.1186/s12943-022-01518-8
53. Molla KA, Sretenovic S, Bansal KC, Qi Y. Precise plant genome editing using base editors and prime editors. Nat Plants (2021) 7:1166–87. doi: 10.1038/s41477-021-00991-1
54. Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature (2019) 576:149–57. doi: 10.1038/s41586-019-1711-4
55. Doudna JA, Charpentier E. The new frontier of genome engineering with CRISPR-Cas9. Science (2014) 346:1077. doi: 10.1126/science.1258096
56. Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell (2006) 124:1283–98. doi: 10.1016/j.cell.2006.01.040
57. Shalem O, Sanjana NE, Zhang F. High-throughput functional genomics using CRISPR-Cas9. Nat Rev Genet (2015) 16:299–311. doi: 10.1038/nrg3899
58. Housden BE, Perrimon N. Comparing CRISPR and RNAi-based screening technologies. Nat Biotechnol (2016) 34:621–23. doi: 10.1038/nbt.3599
59. Weber J, Braun CJ, Saur D, Rad R. In vivo functional screening for systems-level integrative cancer genomics. Nat Rev Cancer (2020) 20:573–93. doi: 10.1038/s41568-020-0275-9
60. Shi H, Doench JG, Chi H. CRISPR screens for functional interrogation of immunity. Nat Rev Immunol (2022) 23:363–80. doi: 10.1038/s41577-022-00802-4
61. Chulanov V, Kostyusheva A, Brezgin S, Ponomareva N, Gegechkori V, Volchkova E, et al. CRISPR screening: molecular tools for studying virus–host interactions. Viruses (2021) 13:2258. doi: 10.3390/v13112258
62. Reczek CR, Birsoy K, Kong H, Martinez-Reyes I, Wang T, Gao P, et al. A CRISPR screen identifies a pathway required for paraquat-induced cell death. Nat Chem Biol (2017) 13:1274–79. doi: 10.1038/nchembio.2499
63. Hanna RE, Hegde M, Fagre CR, DeWeirdt PC, Sangree AK, Szegletes Z, et al. Massively parallel assessment of human variants with base editor screens. Cell (2021) 184:1064–80. doi: 10.1016/j.cell.2021.01.012
64. Li K, Ouyang M, Zhan J, Tian R. CRISPR-based functional genomics screening in human-pluripotent-stem-cell-derived cell types. Cell Genom (2023) 3:100300. doi: 10.1016/j.xgen.2023.100300
65. Asmamaw M, Zawdie B. Mechanism and applications of CRISPR/cas-9-mediated genome editing. Biologics (2021) 15:353–61. doi: 10.2147/BTT.S326422
66. Concordet JP, Haeussler M. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res (2018) 46:W242–45. doi: 10.1093/nar/gky354
67. Mangan ME, Williams JM, Kuhn RM, Lathe WR. The UCSC genome browser: what every molecular biologist should know. Curr Protoc Mol Biol (2014) 107:19. doi: 10.1002/0471142727.mb1909s107
68. Pallares MM, Knodlseder N, Guell M. CRISPR-gRNA design. Methods Mol Biol (2019) 1961:3–11. doi: 10.1007/978-1-4939-9170-9_1
69. Heigwer F, Kerr G, Boutros M. E-CRISP: fast CRISPR target site identification. Nat Methods (2014) 11:122–23. doi: 10.1038/nmeth.2812
70. Labun K, Montague TG, Krause M, Torres CY, Tjeldnes H, Valen E. CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res (2019) 47:W171–74. doi: 10.1093/nar/gkz365
71. Labun K, Montague TG, Gagnon JA, Thyme SB, Valen E. CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res (2016) 44:W272–76. doi: 10.1093/nar/gkw398
72. Montague TG, Cruz JM, Gagnon JA, Church GM, Valen E. CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res (2014) 42:W401–07. doi: 10.1093/nar/gku410
73. Moreno-Mateos MA, Vejnar CE, Beaudoin JD, Fernandez JP, Mis EK, Khokha MK, et al. CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat Methods (2015) 12:982–88. doi: 10.1038/nmeth.3543
74. Li T, Wang S, Luo F, Wu FX, Wang J. MultiGuideScan: a multi-processing tool for designing CRISPR guide RNA libraries. Bioinformatics (2020) 36:920–21. doi: 10.1093/bioinformatics/btz616
75. Perez AR, Pritykin Y, Vidigal JA, Chhangawala S, Zamparo L, Leslie CS, et al. GuideScan software for improved single and paired CRISPR guide RNA design. Nat Biotechnol (2017) 35:347–49. doi: 10.1038/nbt.3804
76. Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol (2014) 15:554. doi: 10.1186/s13059-014-0554-4
77. Li W, Koster J, Xu H, Chen CH, Xiao T, Liu JS, et al. Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome Biol (2015) 16:281. doi: 10.1186/s13059-015-0843-6
78. Wang B, Wang M, Zhang W, Xiao T, Chen CH, Wu A, et al. Integrative analysis of pooled CRISPR genetic screens using MAGeCKFlute. Nat Protoc (2019) 14:756–80. doi: 10.1038/s41596-018-0113-7
79. Hart T, Moffat J. BAGEL: a computational framework for identifying essential genes from pooled library screens. BMC Bioinf (2016) 17:164. doi: 10.1186/s12859-016-1015-8
80. Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet (2017) 49:1779–84. doi: 10.1038/ng.3984
81. Allen F, Behan F, Khodak A, Iorio F, Yusa K, Garnett M, et al. JACKS: joint analysis of CRISPR/Cas9 knockout screens. Genome Res (2019) 29:464–71. doi: 10.1101/gr.238923.118
82. Colic M, Hart T. Common computational tools for analyzing CRISPR screens. Emerg Top Life Sci (2021) 5:779–88. doi: 10.1042/ETLS20210222
83. Kim E, Hart T. Improved analysis of CRISPR fitness screens and reduced off-target effects with the BAGEL2 gene essentiality classifier. Genome Med (2021) 13:2. doi: 10.1186/s13073-020-00809-3
84. Aguirre AJ, Meyers RM, Weir BA, Vazquez F, Zhang CZ, Ben-David U, et al. Genomic copy number dictates a gene-independent cell response to CRISPR/cas9 targeting. Cancer Discovery (2016) 6:914–29. doi: 10.1158/2159-8290.CD-16-0154
85. Replogle JM, Saunders RA, Pogson AN, Hussmann JA, Lenail A, Guna A, et al. Mapping information-rich genotype-phenotype landscapes with genome-scale Perturb-seq. Cell (2022) 185:2559–75. doi: 10.1016/j.cell.2022.05.013
86. Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, et al. Perturb-seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell (2016) 167:1853–66. doi: 10.1016/j.cell.2016.11.038
87. Schraivogel D, Gschwind AR, Milbank JH, Leonce DR, Jakob P, Mathur L, et al. Targeted Perturb-seq enables genome-scale genetic screens in single cells. Nat Methods (2020) 17:629–35. doi: 10.1038/s41592-020-0837-5
88. Jin X, Simmons SK, Guo A, Shetty AS, Ko M, Nguyen L, et al. In vivo Perturb-Seq reveals neuronal and glial abnormalities associated with autism risk genes. Science (2020) 370:eaaz6063. doi: 10.1126/science.aaz6063
89. Hein MY, Weissman JS. Functional single-cell genomics of human cytomegalovirus infection. Nat Biotechnol (2022) 40:391–401. doi: 10.1038/s41587-021-01059-3
90. Adamson B, Norman TM, Jost M, Cho MY, Nunez JK, Chen Y, et al. A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell (2016) 167:1867–82. doi: 10.1016/j.cell.2016.11.048
91. Yang X, Wang Y, Lu P, Shen Y, Zhao X, Zhu Y, et al. PEBP1 suppresses HIV transcription and induces latency by inactivating MAPK/NF-kappaB signaling. EMBO Rep (2020) 21:e49305. doi: 10.15252/embr.201949305
92. Kubo S, Kataria R, Yao Y, Gabrielski JQ, Zheng L, Markowitz TE, et al. Early B cell factor 4 modulates FAS-mediated apoptosis and promotes cytotoxic function in human immune cells. Proc Natl Acad Sci U S (2022) 119:e2086445177. doi: 10.1073/pnas.2208522119
93. Krasnopolsky S, Kuzmina A, Taube R. Genome-wide CRISPR knockout screen identifies ZNF304 as a silencer of HIV transcription that promotes viral latency. PloS Pathog (2020) 16:e1008834. doi: 10.1371/journal.ppat.1008834
94. Yeung A, Choi YH, Lee A, Hale C, Ponstingl H, Pickard D, et al. A genome-wide knockout screen in human macrophages identified host factors modulating salmonella infection. Mbio (2019) 10:e02169-19. doi: 10.1128/mBio.02169-19
95. Rathore A, Iketani S, Wang P, Jia M, Sahi V, Ho DD. CRISPR-based gene knockout screens reveal deubiquitinases involved in HIV-1 latency in two Jurkat cell models. Sci Rep (2020) 10:5350. doi: 10.1038/s41598-020-62375-3
96. Carnevale J, Shifrut E, Kale N, Nyberg WA, Blaeschke F, Chen YY, et al. RASA2 ablation in T cells boosts antigen sensitivity and long-term function. Nature (2022) 609:174–82. doi: 10.1038/s41586-022-05126-w
97. Wang D, Prager BC, Gimple RC, Aguilar B, Alizadeh D, Tang H, et al. CRISPR screening of CAR T cells and cancer stem cells reveals critical dependencies for cell-based therapies. Cancer Discovery (2021) 11:1192–211. doi: 10.1158/2159-8290.CD-20-1243
98. Shifrut E, Carnevale J, Tobin V, Roth TL, Woo JM, Bui CT, et al. Genome-wide CRISPR screens in primary human T cells reveal key regulators of immune function. Cell (2018) 175:1958–71. doi: 10.1016/j.cell.2018.10.024
99. Freitas KA, Belk JA, Sotillo E, Quinn PJ, Ramello MC, Malipatlolla M, et al. Enhanced T cell effector activity by targeting the Mediator kinase module. Science (2022) 378:n5647. doi: 10.1126/science.abn5647
100. Huang H, Kong W, Jean M, Fiches G, Zhou D, Hayashi T, et al. A CRISPR/Cas9 screen identifies the histone demethylase MINA53 as a novel HIV-1 latency-promoting gene (LPG). Nucleic Acids Res (2019) 47:7333–47. doi: 10.1093/nar/gkz493
101. Itell HL, Humes D, Overbaugh J. Several cell-intrinsic effectors drive type I interferon-mediated restriction of HIV-1 in primary CD4(+) T cells. Cell Rep (2023) 42:112556. doi: 10.1016/j.celrep.2023.112556
102. Yang X, Huang T, Wang T, Gao H, Zhang H, Peng W, et al. MAT2A-mediated S-adenosylmethionine level in CD4(+) T cells regulates HIV-1 latent infection. Front Immunol (2021) 12:745784. doi: 10.3389/fimmu.2021.745784
103. Zhu XG, Nicholson PS, Shen Y, La K, Ozlu C, Wang T, et al. CHP1 regulates compartmentalized glycerolipid synthesis by activating GPAT4. Mol Cell (2019) 74:45–58. doi: 10.1016/j.molcel.2019.01.037
104. Schumann K, Raju SS, Lauber M, Kolb S, Shifrut E, Cortez JT, et al. Functional CRISPR dissection of gene networks controlling human regulatory T cell identity. Nat Immunol (2020) 21:1456–66. doi: 10.1038/s41590-020-0784-4
105. Pedersen SF, Collora JA, Kim RN, Yang K, Razmi A, Catalano AA, et al. Inhibition of a chromatin and transcription modulator, SLTM, increases HIV-1 reactivation identified by a CRISPR inhibition screen. J Virol (2022) 96:e57722. doi: 10.1128/jvi.00577-22
106. Legut M, Gajic Z, Guarino M, Daniloski Z, Rahman JA, Xue X, et al. A genome-scale screen for synthetic drivers of T cell proliferation. Nature (2022) 603:728–35. doi: 10.1038/s41586-022-04494-7
107. Jeon Y, Chow SH, Stuart I, Weir A, Yeung AT, Hale C, et al. FBXO11 governs macrophage cell death and inflammation in response to bacterial toxins. Life Sci Alliance (2023) 6:e202201735. doi: 10.26508/lsa.202201735
108. Sedlyarov V, Eichner R, Girardi E, Essletzbichler P, Goldmann U, Nunes-Hasler P, et al. The bicarbonate transporter SLC4A7 plays a key role in macrophage phagosome acidification. Cell Host Microbe (2018) 23:766–74. doi: 10.1016/j.chom.2018.04.013
109. Ferreira A, Szeto A, Clark PA, Crisp A, Kozik P, Jolin HE, et al. Neuroprotective protein ADNP-dependent histone remodeling complex promotes T helper 2 immune cell differentiation. Immunity (2023) 56:1468–84. doi: 10.1016/j.immuni.2023.05.010
110. Szeto A, Ferreira A, Mannion J, Clark PA, Sivasubramaniam M, Heycock M, et al. An alphavbeta3 integrin checkpoint is critical for efficient T(H)2 cell cytokine polarization and potentiation of antigen-specific immunity. Nat Immunol (2023) 24:123–35. doi: 10.1038/s41590-022-01378-w
111. Loo CS, Gatchalian J, Liang Y, Leblanc M, Xie M, Ho J, et al. A genome-wide CRISPR screen reveals a role for the non-canonical nucleosome-remodeling BAF complex in foxp3 expression and regulatory T cell function. Immunity (2020) 53:143–57. doi: 10.1016/j.immuni.2020.06.011
112. Wei J, Long L, Zheng W, Dhungana Y, Lim SA, Guy C, et al. Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature (2019) 576:471–76. doi: 10.1038/s41586-019-1821-z
113. Long L, Wei J, Lim SA, Raynor JL, Shi H, Connelly JP, et al. CRISPR screens unveil signal hubs for nutrient licensing of T cell immunity. Nature (2021) 600:308–13. doi: 10.1038/s41586-021-04109-7
114. Shi J, Wu X, Wang Z, Li F, Meng Y, Moore RM, et al. A genome-wide CRISPR screen identifies WDFY3 as a regulator of macrophage efferocytosis. Nat Commun (2022) 13:7929. doi: 10.1038/s41467-022-35604-8
115. Glover RC, Schwardt NH, Leano SE, Sanchez ME, Thomason MK, Olive AJ, et al. A genome-wide screen in macrophages identifies PTEN as required for myeloid restriction of Listeria monocytogenes infection. PloS Pathog (2023) 19:e1011058. doi: 10.1371/journal.ppat.1011058
116. Newman R, Tolar P. Chronic calcium signaling in IgE(+) B cells limits plasma cell differentiation and survival. Immunity (2021) 54:2756–71. doi: 10.1016/j.immuni.2021.11.006
117. Zhao H, Liu Y, Wang L, Jin G, Zhao X, Xu J, et al. Genome-wide fitness gene identification reveals Roquin as a potent suppressor of CD8 T cell expansion and anti-tumor immunity. Cell Rep (2021) 37:110083. doi: 10.1016/j.celrep.2021.110083
118. Belk JA, Yao W, Ly N, Freitas KA, Chen YT, Shi Q, et al. Genome-wide CRISPR screens of T cell exhaustion identify chromatin remodeling factors that limit T cell persistence. Cancer Cell (2022) 40:768–86. doi: 10.1016/j.ccell.2022.06.001
119. Dong MB, Wang G, Chow RD, Ye L, Zhu L, Dai X, et al. Systematic immunotherapy target discovery using genome-scale in vivo CRISPR screens in CD8 T cells. Cell (2019) 178:1189–204. doi: 10.1016/j.cell.2019.07.044
120. Henriksson J, Chen X, Gomes T, Ullah U, Meyer KB, Miragaia R, et al. Genome-wide CRISPR screens in T helper cells reveal pervasive crosstalk between activation and differentiation. Cell (2019) 176:882–96. doi: 10.1016/j.cell.2018.11.044
121. Sutra DGA, Menegatti S, Fuentealba J, Lucibello F, Perrin L, Helft J, et al. In vivo genome-wide CRISPR screens identify SOCS1 as intrinsic checkpoint of CD4(+) T(H)1 cell response. Sci Immunol (2021) 6:e8219. doi: 10.1126/sciimmunol.abe8219
122. Johansen KH, Golec DP, Huang B, Park C, Thomsen JH, Preite S, et al. A CRISPR screen targeting PI3K effectors identifies RASA3 as a negative regulator of LFA-1-mediated adhesion in T cells. Sci Signal (2022) 15:l9169. doi: 10.1126/scisignal.abl9169
123. Gurusamy D, Henning AN, Yamamoto TN, Yu Z, Zacharakis N, Krishna S, et al. Multi-phenotype CRISPR-cas9 screen identifies p38 kinase as a target for adoptive immunotherapies. Cancer Cell (2020) 37:818–33. doi: 10.1016/j.ccell.2020.05.004
124. Fu G, Guy CS, Chapman NM, Palacios G, Wei J, Zhou P, et al. Metabolic control of T(FH) cells and humoral immunity by phosphatidylethanolamine. Nature (2021) 595:724–29. doi: 10.1038/s41586-021-03692-z
125. Huang H, Zhou P, Wei J, Long L, Shi H, Dhungana Y, et al. In vivo CRISPR screening reveals nutrient signaling processes underpinning CD8(+) T cell fate decisions. Cell (2021) 184:1245–61. doi: 10.1016/j.cell.2021.02.021
126. Ye L, Park JJ, Dong MB, Yang Q, Chow RD, Peng L, et al. In vivo CRISPR screening in CD8 T cells with AAV-Sleeping Beauty hybrid vectors identifies membrane targets for improving immunotherapy for glioblastoma. Nat Biotechnol (2019) 37:1302–13. doi: 10.1038/s41587-019-0246-4
127. Chen Z, Arai E, Khan O, Zhang Z, Ngiow SF, He Y, et al. In vivo CD8(+) T cell CRISPR screening reveals control by Fli1 in infection and cancer. Cell (2021) 184:1262–80. doi: 10.1016/j.cell.2021.02.019
128. Sugiura A, Andrejeva G, Voss K, Heintzman DR, Xu X, Madden MZ, et al. MTHFD2 is a metabolic checkpoint controlling effector and regulatory T cell fate and function. Immunity (2022) 55:65–81. doi: 10.1016/j.immuni.2021.10.011
129. Guo A, Huang H, Zhu Z, Chen MJ, Shi H, Yuan S, et al. cBAF complex components and MYC cooperate early in CD8(+) T cell fate. Nature (2022) 607:135–41. doi: 10.1038/s41586-022-04849-0
130. Ye L, Park JJ, Peng L, Yang Q, Chow RD, Dong MB, et al. A genome-scale gain-of-function CRISPR screen in CD8 T cells identifies proline metabolism as a means to enhance CAR-T therapy. Cell Metab (2022) 34:595–614. doi: 10.1016/j.cmet.2022.02.009
131. Tong J, Wang X, Liu Y, Ren X, Wang A, Chen Z, et al. Pooled CRISPR screening identifies m(6)A as a positive regulator of macrophage activation. Sci Adv (2021) 7:eabd4742. doi: 10.1126/sciadv.abd4742
132. Kong IY, Trezise S, Light A, Todorovski I, Arnau GM, Gadipally S, et al. Epigenetic modulators of B cell fate identified through coupled phenotype-transcriptome analysis. Cell Death Differ (2022) 29:2519–30. doi: 10.1038/s41418-022-01037-5
133. Wu J, Yang K, Cai S, Zhang X, Hu L, Lin F, et al. A p38alpha-BLIMP1 signalling pathway is essential for plasma cell differentiation. Nat Commun (2022) 13:7321. doi: 10.1038/s41467-022-34969-0
134. Laidlaw BJ, Duan L, Xu Y, Vazquez SE, Cyster JG. The transcription factor Hhex cooperates with the corepressor Tle3 to promote memory B cell development. Nat Immunol (2020) 21:1082–93. doi: 10.1038/s41590-020-0713-6
135. Cohen A, Jeng EE, Voorhies M, Symington J, Ali N, Rodriguez RA, et al. Genome-scale CRISPR screening reveals that C3aR signaling is critical for rapid capture of fungi by macrophages. PloS Pathog (2022) 18:e1010237. doi: 10.1371/journal.ppat.1010237
136. Wang Y, Sangare LO, Paredes-Santos TC, Hassan MA, Krishnamurthy S, Furuta AM, et al. Genome-wide screens identify Toxoplasma gondii determinants of parasite fitness in IFNgamma-activated murine macrophages. Nat Commun (2020) 11:5258. doi: 10.1038/s41467-020-18991-8
137. Covarrubias S, Vollmers AC, Capili A, Boettcher M, Shulkin A, Correa MR, et al. High-throughput CRISPR screening identifies genes involved in macrophage viability and inflammatory pathways. Cell Rep (2020) 33:108541. doi: 10.1016/j.celrep.2020.108541
138. Cappell KM, Kochenderfer JN. Long-term outcomes following CAR T cell therapy: what we know so far. Nat Rev Clin Oncol (2023) 20:359–71. doi: 10.1038/s41571-023-00754-1
139. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J (2021) 11:69. doi: 10.1038/s41408-021-00459-7
140. Miska J, Chandel NS. Targeting fatty acid metabolism in glioblastoma. J Clin Invest (2023) 133:e163448. doi: 10.1172/JCI163448
141. Shang W, Jiang Y, Boettcher M, Ding K, Mollenauer M, Liu Z, et al. Genome-wide CRISPR screen identifies FAM49B as a key regulator of actin dynamics and T cell activation. Proc Natl Acad Sci U S (2018) 115:E4051–60. doi: 10.1073/pnas.1801340115
142. Kaufmann T, Simon HU. Pharmacological induction of granulocyte cell death as therapeutic strategy. Annu Rev Pharmacol Toxicol (2023) 63:231–47. doi: 10.1146/annurev-pharmtox-051921-115130
143. Afonina IS, Cullen SP, Martin SJ. Cytotoxic and non-cytotoxic roles of the CTL/NK protease granzyme B. Immunol Rev (2010) 235:105–16. doi: 10.1111/j.0105-2896.2010.00908.x
144. Reilly NA, Lutgens E, Kuiper J, Heijmans BT, Wouter JJ. Effects of fatty acids on T cell function: role in atherosclerosis. Nat Rev Cardiol (2021) 18:824–37. doi: 10.1038/s41569-021-00582-9
145. Wing JB, Lim EL, Sakaguchi S. Control of foreign Ag-specific Ab responses by Treg and Tfr. Immunol Rev (2020) 296:104–19. doi: 10.1111/imr.12888
146. O'Brien KE, Riddell NE, Gomez-Olive FX, Rae DE, Scheuermaier K, von Schantz M. Sleep disturbances in HIV infection and their biological basis. Sleep Med Rev (2022) 65:101571. doi: 10.1016/j.smrv.2021.101571
147. Landovitz RJ, Scott H, Deeks SG. Prevention, treatment and cure of HIV infection. Nat Rev Microbiol (2023) 21:657–70. doi: 10.1038/s41579-023-00914-1
148. Chun TW, Finzi D, Margolick J, Chadwick K, Schwartz D, Siliciano RF. In vivo fate of HIV-1-infected T cells: quantitative analysis of the transition to stable latency. Nat Med (1995) 1:1284–90. doi: 10.1038/nm1295-1284
149. Cohn LB, Chomont N, Deeks SG. The biology of the HIV-1 latent reservoir and implications for cure strategies. Cell Host Microbe (2020) 27:519–30. doi: 10.1016/j.chom.2020.03.014
150. Ruland J, Hartjes L. CARD-BCL-10-MALT1 signalling in protective and pathological immunity. Nat Rev Immunol (2019) 19:118–34. doi: 10.1038/s41577-018-0087-2
151. Harhaj EW, Dixit VM. Deubiquitinases in the regulation of NF-kappaB signaling. Cell Res (2011) 21:22–39. doi: 10.1038/cr.2010.166
152. Jordan A, Bisgrove D, Verdin E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J (2003) 22:1868–77. doi: 10.1093/emboj/cdg188
153. Snell LM, McGaha TL, Brooks DG. Type I interferon in chronic virus infection and cancer. Trends Immunol (2017) 38:542–57. doi: 10.1016/j.it.2017.05.005
154. Wolf D, Goff SP. TRIM28 mediates primer binding site-targeted silencing of murine leukemia virus in embryonic cells. Cell (2007) 131:46–57. doi: 10.1016/j.cell.2007.07.026
155. Tchasovnikarova IA, Timms RT, Matheson NJ, Wals K, Antrobus R, Gottgens B, et al. GENE SILENCING. Epigenetic silencing by the HUSH complex mediates position-effect variegation in human cells. Science (2015) 348:1481–85. doi: 10.1126/science.aaa7227
156. Zhu Y, Wang GZ, Cingoz O, Goff SP. NP220 mediates silencing of unintegrated retroviral DNA. Nature (2018) 564:278–82. doi: 10.1038/s41586-018-0750-6
157. Yurkovetskiy L, Guney MH, Kim K, Goh SL, McCauley S, Dauphin A, et al. Primate immunodeficiency virus proteins Vpx and Vpr counteract transcriptional repression of proviruses by the HUSH complex. Nat Microbiol (2018) 3:1354–61. doi: 10.1038/s41564-018-0256-x
158. Chougui G, Munir-Matloob S, Matkovic R, Martin MM, Morel M, Lahouassa H, et al. HIV-2/SIV viral protein X counteracts HUSH repressor complex. Nat Microbiol (2018) 3:891–97. doi: 10.1038/s41564-018-0179-6
159. Liu N, Lee CH, Swigut T, Grow E, Gu B, Bassik MC, et al. Selective silencing of euchromatic L1s revealed by genome-wide screens for L1 regulators. Nature (2018) 553:228–32. doi: 10.1038/nature25179
160. Valle-Casuso JC, Angin M, Volant S, Passaes C, Monceaux V, Mikhailova A, et al. Cellular metabolism is a major determinant of HIV-1 reservoir seeding in CD4(+) T cells and offers an opportunity to tackle infection. Cell Metab (2019) 29:611–26. doi: 10.1016/j.cmet.2018.11.015
161. Loisel-Meyer S, Swainson L, Craveiro M, Oburoglu L, Mongellaz C, Costa C, et al. Glut1-mediated glucose transport regulates HIV infection. Proc Natl Acad Sci U S (2012) 109:2549–54. doi: 10.1073/pnas.1121427109
162. Zhou YH, Sun L, Chen J, Sun WW, Ma L, Han Y, et al. Tryptophan metabolism activates aryl hydrocarbon receptor-mediated pathway to promote HIV-1 infection and reactivation. Mbio (2019) 10:e02591-19. doi: 10.1128/mBio.02591-19
163. Baldauf HM, Pan X, Erikson E, Schmidt S, Daddacha W, Burggraf M, et al. SAMHD1 restricts HIV-1 infection in resting CD4(+) T cells. Nat Med (2012) 18:1682–87. doi: 10.1038/nm.2964
164. Jin S, Liao Q, Chen J, Zhang L, He Q, Zhu H, et al. TSC1 and DEPDC5 regulate HIV-1 latency through the mTOR signaling pathway. Emerg Microbes Infect (2018) 7:138. doi: 10.1038/s41426-018-0139-5
165. Ferreira L, Muller YD, Bluestone JA, Tang Q. Next-generation regulatory T cell therapy. Nat Rev Drug Discovery (2019) 18:749–69. doi: 10.1038/s41573-019-0041-4
166. Kanamori M, Nakatsukasa H, Okada M, Lu Q, Yoshimura A. Induced regulatory T cells: their development, stability, and applications. Trends Immunol (2016) 37:803–11. doi: 10.1016/j.it.2016.08.012
167. Kim J, Guan KL. mTOR as a central hub of nutrient signalling and cell growth. Nat Cell Biol (2019) 21:63–71. doi: 10.1038/s41556-018-0205-1
168. Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol (2020) 21:183–203. doi: 10.1038/s41580-019-0199-y
169. Huang H, Long L, Zhou P, Chapman NM, Chi H. mTOR signaling at the crossroads of environmental signals and T-cell fate decisions. Immunol Rev (2020) 295:15–38. doi: 10.1111/imr.12845
170. Shi H, Chapman NM, Wen J, Guy C, Long L, Dhungana Y, et al. Amino Acids License Kinase mTORC1 Activity and Treg Cell Function via Small G Proteins Rag and Rheb. Immunity (2019) 51:1012–27. doi: 10.1016/j.immuni.2019.10.001
171. Wilson CB, Rowell E, Sekimata M. Epigenetic control of T-helper-cell differentiation. Nat Rev Immunol (2009) 9:91–105. doi: 10.1038/nri2487
172. Ellmeier W, Seiser C. Author Correction: Histone deacetylase function in CD4(+) T cells. Nat Rev Immunol (2019) 19:266. doi: 10.1038/s41577-019-0153-4
173. Jung J, Zeng H, Horng T. Metabolism as a guiding force for immunity. Nat Cell Biol (2019) 21:85–93. doi: 10.1038/s41556-018-0217-x
174. Linnemann C, Schumacher TN, Bendle GM. T-cell receptor gene therapy: critical parameters for clinical success. J Invest Dermatol (2011) 131:1806–16. doi: 10.1038/jid.2011.160
175. Sadelain M. CAR therapy: the CD19 paradigm. J Clin Invest (2015) 125:3392–400. doi: 10.1172/JCI80010
176. Lorenzo-Vizcaya A, Isenberg DA. Clinical trials in systemic lupus erythematosus: the dilemma-Why have phase III trials failed to confirm the promising results of phase II trials? Ann Rheum Dis (2023) 82:169–74. doi: 10.1136/ard-2022-222839
177. Chen Q, Wang J, Xiang M, Wang Y, Zhang Z, Liang J, et al. The potential role of ferroptosis in systemic lupus erythematosus. Front Immunol (2022) 13:855622. doi: 10.3389/fimmu.2022.855622
178. Wincup C, Sawford N, Rahman A. Pathological mechanisms of abnormal iron metabolism and mitochondrial dysfunction in systemic lupus erythematosus. Expert Rev Clin Immunol (2021) 17:957–67. doi: 10.1080/1744666X.2021.1953981
179. Rubinstein T, Pitashny M, Putterman C. The novel role of neutrophil gelatinase-B associated lipocalin (NGAL)/Lipocalin-2 as a biomarker for lupus nephritis. Autoimmun Rev (2008) 7:229–34. doi: 10.1016/j.autrev.2007.11.013
180. Voss K, Sewell AE, Krystofiak ES, Gibson-Corley KN, Young AC, Basham JH, et al. Elevated transferrin receptor impairs T cell metabolism and function in systemic lupus erythematosus. Sci Immunol (2023) 8:q178. doi: 10.1126/sciimmunol.abq0178
181. Harris NL, Loke P. Recent advances in type-2-cell-mediated immunity: insights from helminth infection. Immunity (2017) 47:1024–36. doi: 10.1016/j.immuni.2017.11.015
182. Walker JA, McKenzie A. T(H)2 cell development and function. Nat Rev Immunol (2018) 18:121–33. doi: 10.1038/nri.2017.118
183. Gay A, Nawijn MC. See no allergen, hear no allergen, speak no allergen! Sci Immunol (2023) 8:h597. doi: 10.1126/sciimmunol.adh0597
184. Chen K, Hao Y, Guzman M, Li G, Cerutti A. Antibody-mediated regulation of basophils: emerging views and clinical implications. Trends Immunol (2023) 44:408–23. doi: 10.1016/j.it.2023.04.003
185. Harker JA, Lloyd CM. T helper 2 cells in asthma. J Exp Med (2023) 220:e20221094. doi: 10.1084/jem.20221094
186. Olson DJ, Odunsi K. Adoptive cell therapy for nonhematologic solid tumors. J Clin Oncol (2023) 41:3397–407. doi: 10.1200/JCO.22.01618
187. Arnouk S, De Groof T, Van Ginderachter JA. Imaging and therapeutic targeting of the tumor immune microenvironment with biologics. Adv Drug Delivery Rev (2022) 184:114239. doi: 10.1016/j.addr.2022.114239
188. Jiang X, Xu J, Liu M, Xing H, Wang Z, Huang L, et al. Adoptive CD8(+) T cell therapy against cancer:Challenges and opportunities. Cancer Lett (2019) 462:23–32. doi: 10.1016/j.canlet.2019.07.017
189. Nelson MA, Ngamcherdtrakul W, Luoh SW, Yantasee W. Prognostic and therapeutic role of tumor-infiltrating lymphocyte subtypes in breast cancer. Cancer Metastasis Rev (2021) 40:519–36. doi: 10.1007/s10555-021-09968-0
190. Bleijs DA, Geijtenbeek TB, Figdor CG, van Kooyk Y. DC-SIGN and LFA-1: a battle for ligand. Trends Immunol (2001) 22:457–63. doi: 10.1016/s1471-4906(01)01974-3
191. Rosser EC, Mauri C. Regulatory B cells: origin, phenotype, and function. Immunity (2015) 42:607–12. doi: 10.1016/j.immuni.2015.04.005
192. Laidlaw BJ, Cyster JG. Transcriptional regulation of memory B cell differentiation. Nat Rev Immunol (2021) 21:209–20. doi: 10.1038/s41577-020-00446-2
193. Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM. The generation of antibody-secreting plasma cells. Nat Rev Immunol (2015) 15:160–71. doi: 10.1038/nri3795
194. Marelli G, Morina N, Portale F, Pandini M, Iovino M, Di Conza G, et al. Lipid-loaded macrophages as new therapeutic target in cancer. J Immunother Cancer (2022) 10:e004584. doi: 10.1136/jitc-2022-004584
195. Leseigneur C, Le-Bury P, Pizarro-Cerda J, Dussurget O. Emerging evasion mechanisms of macrophage defenses by pathogenic bacteria. Front Cell Infect Microbiol (2020) 10:577559. doi: 10.3389/fcimb.2020.577559
196. Koopmans MM, Brouwer MC, Vazquez-Boland JA, van de Beek D. Human listeriosis. Clin Microbiol Rev (2023) 36:e6019. doi: 10.1128/cmr.00060-19
197. Medjahed H, Gaillard JL, Reyrat JM. Mycobacterium abscessus: a new player in the mycobacterial field. Trends Microbiol (2010) 18:117–23. doi: 10.1016/j.tim.2009.12.007
198. Gilliland HN, Beckman OK, Olive AJ. A genome-wide screen in macrophages defines host genes regulating the uptake of mycobacterium abscessus. Msphere (2023) 8:e66322. doi: 10.1128/msphere.00663-22
199. Klein BS. Microbiology. Turning up the heat on Histoplasma capsulatum. Science (2000) 290:1311–12. doi: 10.1126/science.290.5495.1311
200. Sebghati TS, Engle JT, Goldman WE. Intracellular parasitism by Histoplasma capsulatum: fungal virulence and calcium dependence. Science (2000) 290:1368–72. doi: 10.1126/science.290.5495.1368
201. Kann O, Almouhanna F, Chausse B. Interferon gamma: a master cytokine in microglia-mediated neural network dysfunction and neurodegeneration. Trends Neurosci (2022) 45:913–27. doi: 10.1016/j.tins.2022.10.007
202. Trzeciak A, Wang YT, Perry J. First we eat, then we do everything else: The dynamic metabolic regulation of efferocytosis. Cell Metab (2021) 33:2126–41. doi: 10.1016/j.cmet.2021.08.001
203. Liu Y, Yang D, Liu T, Chen J, Yu J, Yi P. N6-methyladenosine-mediated gene regulation and therapeutic implications. Trends Mol Med (2023) 29:454–67. doi: 10.1016/j.molmed.2023.03.005
204. Chen M, Mao A, Xu M, Weng Q, Mao J, Ji J. CRISPR-Cas9 for cancer therapy: Opportunities and challenges. Cancer Lett (2019) 447:48–55. doi: 10.1016/j.canlet.2019.01.017
205. Dubrot J, Du PP, Lane-Reticker SK, Kessler EA, Muscato AJ, Mehta A, et al. In vivo CRISPR screens reveal the landscape of immune evasion pathways across cancer. Nat Immunol (2022) 23:1495–506. doi: 10.1038/s41590-022-01315-x
206. Frey N, Tortola L, Egli D, Janjuha S, Rothgangl T, Marquart KF, et al. Loss of Rnf31 and Vps4b sensitizes pancreatic cancer to T cell-mediated killing. Nat Commun (2022) 13:1804. doi: 10.1038/s41467-022-29412-3
207. Ji P, Gong Y, Jin ML, Wu HL, Guo LW, Pei YC, et al. In vivo multidimensional CRISPR screens identify Lgals2 as an immunotherapy target in triple-negative breast cancer. Sci Adv (2022) 8:l8247. doi: 10.1126/sciadv.abl8247
208. Wang X, Tokheim C, Gu SS, Wang B, Tang Q, Li Y, et al. In vivo CRISPR screens identify the E3 ligase Cop1 as a modulator of macrophage infiltration and cancer immunotherapy target. Cell (2021) 184:5357–74. doi: 10.1016/j.cell.2021.09.006
209. Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature (2017) 547:413–18. doi: 10.1038/nature23270
210. Huang Q, Li F, Hu H, Fang Z, Gao Z, Xia G, et al. Loss of TSC1/TSC2 sensitizes immune checkpoint blockade in non-small cell lung cancer. Sci Adv (2022) 8:i9533. doi: 10.1126/sciadv.abi9533
211. Pfeifer E, Burchell JM, Dazzi F, Sarker D, Beatson R. Apoptosis in the pancreatic cancer tumor microenvironment-the double-edged sword of cancer-associated fibroblasts. Cells (2021) 10:1653. doi: 10.3390/cells10071653
212. Malik S, Westcott JM, Brekken RA, Burrows FJ. CXCL12 in pancreatic cancer: its function and potential as a therapeutic drug target. Cancers (Basel) (2021) 14:86. doi: 10.3390/cancers14010086
213. Fan JQ, Wang MF, Chen HL, Shang D, Das JK, Song J. Current advances and outlooks in immunotherapy for pancreatic ductal adenocarcinoma. Mol Cancer (2020) 19:32. doi: 10.1186/s12943-020-01151-3
214. Li J, Yuan S, Norgard RJ, Yan F, Sun YH, Kim IK, et al. Epigenetic and transcriptional control of the epidermal growth factor receptor regulates the tumor immune microenvironment in pancreatic cancer. Cancer Discovery (2021) 11:736–53. doi: 10.1158/2159-8290.CD-20-0519
215. Leon-Ferre RA, Goetz MP. Advances in systemic therapies for triple negative breast cancer. Bmj (2023) 381:e71674. doi: 10.1136/bmj-2022-071674
216. Malmberg R, Zietse M, Dumoulin DW, Hendrikx J, Aerts J, van der Veldt A, et al. Alternative dosing strategies for immune checkpoint inhibitors to improve cost-effectiveness: a special focus on nivolumab and pembrolizumab. Lancet Oncol (2022) 23:e552–61. doi: 10.1016/S1470-2045(22)00554-X
217. Isaacs J, Anders C, McArthur H, Force J. Biomarkers of immune checkpoint blockade response in triple-negative breast cancer. Curr Treat Options Oncol (2021) 22:38. doi: 10.1007/s11864-021-00833-4
218. Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science (2018) 362:eaar3593. doi: 10.1126/science.aar3593
219. Ryu JS, Lim JH, Kim HJ, Kim MJ, Park MH, Kim JS. Survival prediction of tuberous sclerosis complex gene variant in patients with advanced non-small-cell lung cancer treated with platinum doublet. Biosci Rep (2019) 39:BSR20181426. doi: 10.1042/BSR20181426
220. Valenzuela NM, Reed EF. Antibody-mediated rejection across solid organ transplants: manifestations, mechanisms, and therapies. J Clin Invest (2017) 127:2492–504. doi: 10.1172/JCI90597
221. Mori DN, Kreisel D, Fullerton JN, Gilroy DW, Goldstein DR. Inflammatory triggers of acute rejection of organ allografts. Immunol Rev (2014) 258:132–44. doi: 10.1111/imr.12146
222. DeWolf S, Sykes M. Alloimmune T cells in transplantation. J Clin Invest (2017) 127:2473–81. doi: 10.1172/JCI90595
223. Shaban E, Bayliss G, Malhotra DK, Shemin D, Wang LJ, Gohh R, et al. Targeting regulatory T cells for transplant tolerance: new insights and future perspectives. Kidney Dis (Basel) (2018) 4:205–13. doi: 10.1159/000490703
224. Esensten JH, Muller YD, Bluestone JA, Tang Q. Regulatory T-cell therapy for autoimmune and autoinflammatory diseases: The next frontier. J Allergy Clin Immunol (2018) 142:1710–18. doi: 10.1016/j.jaci.2018.10.015
225. MacDonald KN, Salim K, Levings MK. Manufacturing next-generation regulatory T-cell therapies. Curr Opin Biotechnol (2022) 78:102822. doi: 10.1016/j.copbio.2022.102822
226. Zhang Y, Shen S, Zhao G, Xu CF, Zhang HB, Luo YL, et al. In situ repurposing of dendritic cells with CRISPR/Cas9-based nanomedicine to induce transplant tolerance. Biomaterials (2019) 217:119302. doi: 10.1016/j.biomaterials.2019.119302
227. (2023). Available at: http://optn.transplant.hrsa.gov/ (Accessed 2023/9/3).
228. Sykes M, Sachs DH. Progress in xenotransplantation: overcoming immune barriers. Nat Rev Nephrol (2022) 18:745–61. doi: 10.1038/s41581-022-00624-6
229. Dangi A, Yu S, Luo X. Emerging approaches and technologies in transplantation: the potential game changers. Cell Mol Immunol (2019) 16:334–42. doi: 10.1038/s41423-019-0207-3
230. Sykes M, Sachs DH. Transplanting organs from pigs to humans. Sci Immunol (2019) 4:eaau6298. doi: 10.1126/sciimmunol.aau6298
231. Mesaki K, Juvet S, Yeung J, Guan Z, Wilson GW, Hu J, et al. Immunomodulation of the donor lung with CRISPR-mediated activation of IL-10 expression. J Heart Lung Transplant (2023) 42:1363–77. doi: 10.1016/j.healun.2023.06.001
232. Deuse T, Hu X, Gravina A, Wang D, Tediashvili G, De C, et al. Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat Biotechnol (2019) 37:252–58. doi: 10.1038/s41587-019-0016-3
233. Eisenson DL, Hisadome Y, Yamada K. Progress in xenotransplantation: immunologic barriers, advances in gene editing, and successful tolerance induction strategies in pig-to-primate transplantation. Front Immunol (2022) 13:899657. doi: 10.3389/fimmu.2022.899657
234. Lu T, Yang B, Wang R, Qin C. Xenotransplantation: current status in preclinical research. Front Immunol (2019) 10:3060. doi: 10.3389/fimmu.2019.03060
235. Hai T, Teng F, Guo R, Li W, Zhou Q. One-step generation of knockout pigs by zygote injection of CRISPR/Cas system. Cell Res (2014) 24:372–75. doi: 10.1038/cr.2014.11
236. Cowan PJ, Hawthorne WJ, Nottle MB. Xenogeneic transplantation and tolerance in the era of CRISPR-Cas9. Curr Opin Organ Transplant (2019) 24:5–11. doi: 10.1097/MOT.0000000000000589
237. Li M, Sun J, Shi G. Application of CRISPR screen in mechanistic studies of tumor development, tumor drug resistance, and tumor immunotherapy. Front Cell Dev Biol (2023) 11:1220376. doi: 10.3389/fcell.2023.1220376
238. Chulanov V, Kostyusheva A, Brezgin S, Ponomareva N, Gegechkori V, Volchkova E, et al. CRISPR screening: molecular tools for studying virus-host interactions. Viruses (2021) 13:2258. doi: 10.3390/v13112258
239. Duneton C, Winterberg PD, Ford ML. Activation and regulation of alloreactive T cell immunity in solid organ transplantation. Nat Rev Nephrol (2022) 18:663–76. doi: 10.1038/s41581-022-00600-0
240. Abramson J, Dobes J, Lyu M, Sonnenberg GF. The emerging family of RORgammat(+) antigen-presenting cells. Nat Rev Immunol (2023). doi: 10.1038/s41577-023-00906-5
241. Oberbarnscheidt MH, Lakkis FG. Innate allorecognition. Immunol Rev (2014) 258:145–49. doi: 10.1111/imr.12153
242. VanDusen NJ, Lee JY, Gu W, Butler CE, Sethi I, Zheng Y, et al. Massively parallel in vivo CRISPR screening identifies RNF20/40 as epigenetic regulators of cardiomyocyte maturation. Nat Commun (2021) 12:4442. doi: 10.1038/s41467-021-24743-z
243. Mandal PK, Ferreira LM, Collins R, Meissner TB, Boutwell CL, Friesen M, et al. Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell (2014) 15:643–52. doi: 10.1016/j.stem.2014.10.004
244. Bock C, Datlinger P, Chardon F, Coelho MA, Dong MB, Lawson KA, et al. High-content CRISPR screening. Nat Rev Methods Primers (2022) 2:9. doi: 10.1038/s43586-022-00098-7
245. Tian R, Cao C, He D, Dong D, Sun L, Liu J, et al. Massively parallel CRISPR off-target detection enables rapid off-target prediction model building. Med (N Y) (2023) 4:478–92. doi: 10.1016/j.medj.2023.05.005
246. Cullot G, Boutin J, Toutain J, Prat F, Pennamen P, Rooryck C, et al. CRISPR-Cas9 genome editing induces megabase-scale chromosomal truncations. Nat Commun (2019) 10:1136. doi: 10.1038/s41467-019-09006-2
247. Kosicki M, Tomberg K, Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat Biotechnol (2018) 36:765–71. doi: 10.1038/nbt.4192
248. Kim S, Koo T, Jee HG, Cho HY, Lee G, Lim DG, et al. CRISPR RNAs trigger innate immune responses in human cells. Genome Res (2018) 28:367–73. doi: 10.1101/gr.231936.117
249. Wienert B, Shin J, Zelin E, Pestal K, Corn JE. In vitro-transcribed guide RNAs trigger an innate immune response via the RIG-I pathway. PloS Biol (2018) 16:e2005840. doi: 10.1371/journal.pbio.2005840
250. de Perrot M, Fischer S, Liu M, Imai Y, Martins S, Sakiyama S, et al. Impact of human interleukin-10 on vector-induced inflammation and early graft function in rat lung transplantation. Am J Respir Cell Mol Biol (2003) 28:616–25. doi: 10.1165/rcmb.2002-0109OC
251. Yeung JC, Wagnetz D, Cypel M, Rubacha M, Koike T, Chun YM, et al. Ex vivo adenoviral vector gene delivery results in decreased vector-associated inflammation pre- and post-lung transplantation in the pig. Mol Ther (2012) 20:1204–11. doi: 10.1038/mt.2012.57
Keywords: solid organ transplantation, CRISPR/Cas9, transcriptome editing, CRISPR screening, high-throughput
Citation: Li X, Chen Z, Ye W, Yu J, Zhang X, Li Y, Niu Y, Ran S, Wang S, Luo Z, Zhao J, Hao Y, Zong J, Xia C, Xia J and Wu J (2024) High-throughput CRISPR technology: a novel horizon for solid organ transplantation. Front. Immunol. 14:1295523. doi: 10.3389/fimmu.2023.1295523
Received: 16 September 2023; Accepted: 12 December 2023;
Published: 04 January 2024.
Edited by:
Stanislaw Stepkowski, University of Toledo, United StatesReviewed by:
Ranjeet Kumar, Rutgers, The State University of New Jersey, United StatesJie Xu, University of Michigan, United States
Copyright © 2024 Li, Chen, Ye, Yu, Zhang, Li, Niu, Ran, Wang, Luo, Zhao, Hao, Zong, Xia, Xia and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jie Wu, wujie426@hust.edu.cn; Jiahong Xia, jiahong.xia@mail.hust.edu.cn; Chengkun Xia, 31695450@qq.com
†These authors have contributed equally to this work