 Xiaohan Guo
Xiaohan Guo Yi Wu
Yi Wu Ying Xue1
Ying Xue1 Na Xie
Na Xie Guobo Shen
Guobo Shen- 1West China School of Basic Medical Sciences and Forensic Medicine, Sichuan University, and Collaborative Innovation Center for Biotherapy, Chengdu, China
- 2State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, Sichuan University, and Collaborative Innovation Center for Biotherapy, Chengdu, China
Recent progressions in immunotherapy have transformed cancer treatment, providing a promising strategy that activates the immune system of the patient to find and eliminate cancerous cells. Bispecific antibodies, which engage two separate antigens or one antigen with two distinct epitopes, are of tremendous concern in immunotherapy. The bi-targeting idea enabled by bispecific antibodies (BsAbs) is especially attractive from a medical standpoint since most diseases are complex, involving several receptors, ligands, and signaling pathways. Several research look into the processes in which BsAbs identify different cancer targets such angiogenesis, reproduction, metastasis, and immune regulation. By rerouting cells or altering other pathways, the bispecific proteins perform effector activities in addition to those of natural antibodies. This opens up a wide range of clinical applications and helps patients with resistant tumors respond better to medication. Yet, further study is necessary to identify the best conditions where to use these medications for treating tumor, their appropriate combination partners, and methods to reduce toxicity. In this review, we provide insights into the BsAb format classification based on their composition and symmetry, as well as the delivery mode, focus on the action mechanism of the molecule, and discuss the challenges and future perspectives in BsAb development.
1 Introduction
Currently emerging cancer immunotherapies include cancer vaccines, T cell receptor T cells (TCR-T) or chimeric antigen receptor T cells (CAR-T), cytokine therapies, immune checkpoint blockades (ICBs), and tumor-targeted antibodies (1). Monoclonal antibodies (mAbs), in particular, are powerful tumor-targeted antibodies that have been licensed for use in cancer in the US and Europe for the first time in 2022 (2). However, the complicated pathophysiology of tumors limits the therapeutic efficacy of mAbs (3, 4), while the combination of two or more mAbs may be subject to safety and efficacy issues (5). Bispecific antibodies (BsAbs) have been developed to bind two specific epitopes or target proteins at the same time. These antibodies have improved specificity, increased targeting ability, and reduced off-target toxicity. Moreover, BsAbs have the potential to effectively lower the cost of treatment, revitalizing the field of cancer immunotherapy (6).
The first BsAb was created in the early 1960s and was based on mild reoxidation of binding fragments from two different polyclonal sera (7). Later, based on enzymatic digestion of hybridoma peptides, hybridoma technology allowed the chemical coupling of mAbs or fragment antigen-binding (Fab) fragments (8). With the rapid development of genetic engineering technology, the multifunctional BsAb formats received great attention in clinical application. Mechanically, BsAbs inhibit tumor progression directly or indirectly mainly by redirecting immune effector cells into tumors, delivering radioactive or drug payloads to cancer cells, targeting multiple signaling pathways, and so on. For example, BsAb drug catumaxomab, which contains anti-epithelial cell adhesion molecule (EpCAM) and anti-cluster of differentiation 3 (CD3) molecule, destroys tumors via T cell-driven lysis, cytotoxicity triggered by antibodies, and phagocytosis via helper cells with Fcγ receptors (FcγRs) (9, 10). Four BsAb medications, including tebentafusp, faricimab, mosunetuzumab, and teclistamab, were approved for marketing in 2022 alone, suggesting that BsAbs are promising approaches to develop antitumor therapies (Figure 1).
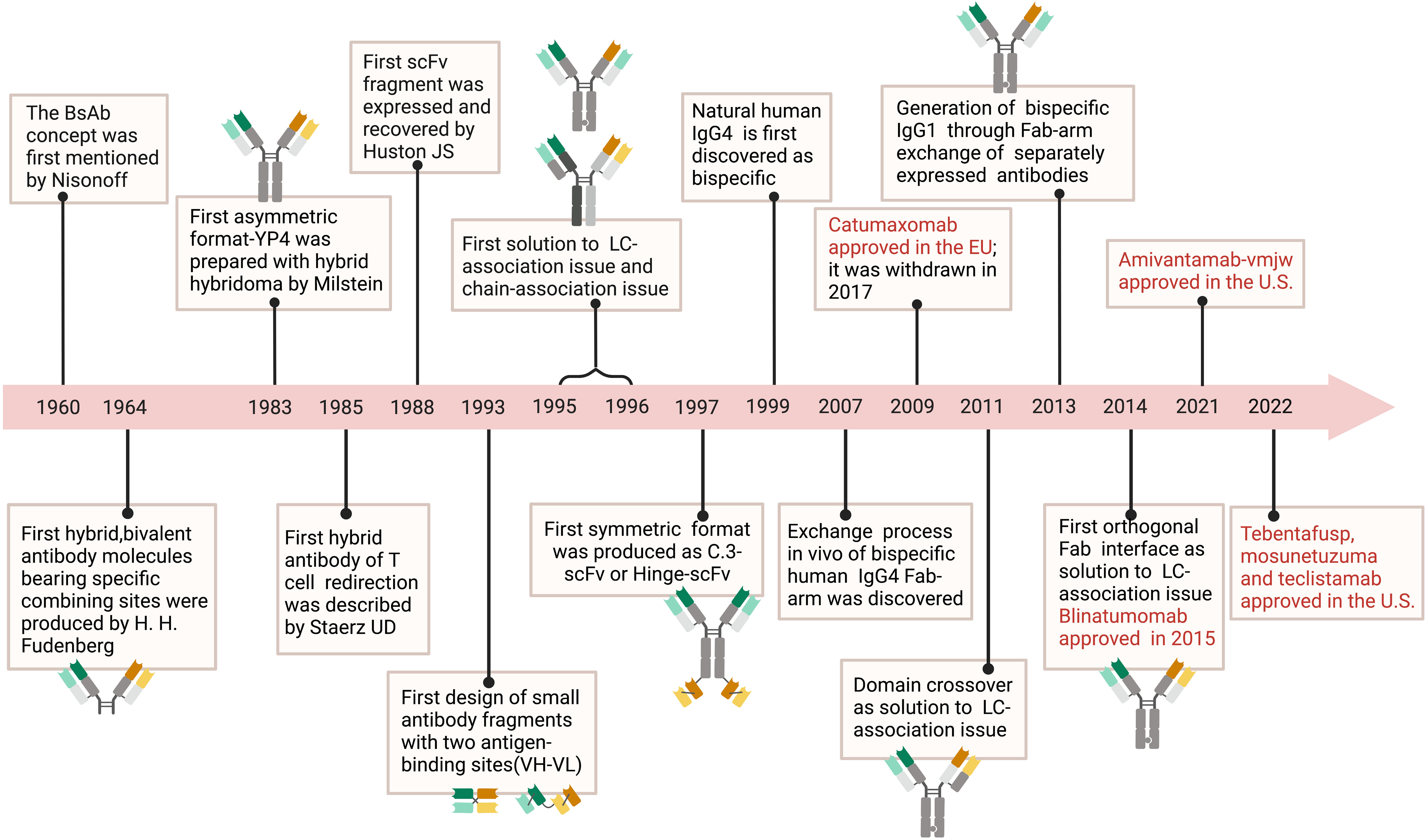
Figure 1 Timeline. The timeline showcases the technical innovations and clinical research in tumor of BsAb. In 1960, the concept of BsAb was proposed (11). In 1964, researchers created molecules with two different binding sites (12). BsAb with asymmetric structure was produced using hybridoma technology in 1983 (13). In 1985, the idea of BsAb that can redirect T cells was proposed (14). Diabody, a small molecule BsAb fragment, was designed in 1993 (15). In 1988, researchers developed scFv fragments (16). From 1995 to 1996, the problem of protein subunit pairing was first solved (17, 18). In 1997, BsAb with symmetric structure was manufactured (19). It was discovered in 1999 that natural human IgG4 molecules were bispecific (20). In 2007, the process of Fab fragment exchange in human IgG4 was explained (21). Catumaxomab was approved by the EU in 2009 but later withdrawn in 2007 (22). In 2011, the problem of light chain pairing was solved through domain swapping strategy (23). Bispecific IgG1 was produced using Fab fragment exchange in 2013 (11). In 2014, the problem of light chain pairing was solved through orthogonal Fab fragments (24). In 2015, Blinatumomab was approved (24). Amivantamab-vmjw was approved in the U.S. in 2021, and in 2022, Tebentafusp, Mosunetuzuma, and Teclistamab were also approved in the U.S (25–27).
In the review, we will systematically cover the antitumor principle and clinical applications of BsAbs in multiple formats. We will also introduce the preparation technology and delivery method of BsAb, and discuss their challenges and prospects in the treatment of solid tumors.
2 Format of BsAbs
The power of BsAbs lies in their capacity to create new activities that demand the union of two binding specificities in a single molecule (28). Their functionality can be greatly impacted by domain composition or “shifting” linker length and unique arrangements of (non-)chemical bonds. The design of BsAbs’ forms can also affect other factors including diffusion and pharmacokinetic activity (29, 30). In addition to expression platform’s stability and output, the presence or absence of undesirable side products is another factor that must be taken into account. The wide range of BsAb formats produced by the numerous designing methods can be categorized by their design elements or functional characteristics (28).
2.1 Classification of BsAbs based on composition
Based on structural components, BsAbs could be roughly classified into BsAbs with Fragment crystallizable (Fc) regions and BsAbs without Fc regions. BsAbs with Fc regions can help activate the immune system via Fc domain’s interaction with cell surface receptors, as well as endow the BsAb molecules with longer half-lives on account of their larger sizes and the neonatal Fc receptors (FcRn)-mediated recycling pathway (31). However, Fc region’s engagement with FcγRs can lead to serious cytotoxicity events, which may be a merit of BsAbs without Fc region (32). The Fragment variable (Fv)-only molecules are also easier to produce. While BsAbs without Fc domains lack interactions with CH (constant heavy chain)1/CL (constant light chain) regions, more techniques must be applied to stabilize the Fab regions.
2.1.1 BsAbs with Fc regions
BsAbs that contain the Fc region include Immunoglobulin G(IgG) constructs such as Duobody (controlled Fab-arm exchange technology) (33, 34), Fabs-In-Tandem Immunoglobulin (FIT-Ig) (35), Cross-Mab (36), scFv-Fc constructs (single chain variable fragment), VHH-Fc constructs, and dual-affinity retargeting (DART)-Fc constructs (37).
Fc region offers BsAbs a number of advantages. The engagement of Fc region with membrane Fc receptors (FcRs) and certain complement system proteins help to activate the immune response. The Fc-directed receptor downregulation and malignant cells apoptosis through monocyte/macrophage trogocytosis is required for the antitumor efficacy of amivantamab (38, 39). These BsAbs have longer half-lives because of their big size and recycling pathways controlled by FcRn (37). An entire IgG antibody has a molecular mass of 150 kDa and is removed by the liver, whereas molecules with a molecular weight less than 60 kDa are filtered by the renal system (40). The combination of homologous variable heavy chain (VH) and variable light chain (VL) domains is further driven by the fusion of a heterodimerizing Fc region, making purification with affinity resins like protein G feasible (41).
However, off-target cytotoxicity and reduced treatment efficiency are associated with Fc-mediated downstream actions. When Fc region of medicinal antibodies interact with FcγRs, serious adverse effects may occur (42). Except for safety concerns, CD3-directing BsAbs with an active core demonstrated less effective in vivo (28, 43). To reduce the aforementioned negative effects, presently available BsAbs targeting CD3 either omit the Fc region or have modified Fc domains to minimize FcγR interaction (28).
2.1.2 BsAbs without Fc regions
BsAbs without Fc region lack the Fc-mediated effector actions mentioned above, but they aid in eliminating the chain-association problem. Moreover, the formats can be produced economically and high-yieldingly by expressing 1–2 peptides strands in simple eukaryotic and prokaryotic protein synthesis platforms (28, 44, 45).
BsAbs without Fc region mainly consist of tandem single-chain variable fragments (scFv2, taFv), bispecific one-domain antibody hybrid proteins, diabodies, and fragment antigen-binding (Fab fusion protein) (30). The taFv, which stands for the minimum BsAb, can be created by joining two scFvs together with a linker and normally ranges 50-60 kDa in size (30). However, these Fv-only moieties are short of the native-like connections with CH1/CL regions which is required for the stability and solubility of Fab regions (46). By creating a disulfide connection between the VH and VL domains, the stability of tandem scFv can be enhanced (30, 47). Bispecific single-domain antibody hybrid molecules can be made by one-domain antibodies, such as VH or VL domains, VHH, variable new antigen receptor (VNAR) and nanobodies (Nbs) (30). Compared to human programmed cell death-ligand 1 (PD-L1)-targeting mAb or vascular endothelial growth factor receptor type 1 domain 2 (VEGFR1D2) fusion protein alone, the HB0025 that combines the VEGFR1D2 and anti-PD-L1 mAb was more effective at preventing the growth of tumor (48). Diabody is a noncovalent heterodimer comprising the VH and VL portions of the scFv fragment linked by a short peptide. Since only some of the potential arrangements and orientations preserved binding potential for both antigens, it is crucial to choose the ideal VH/VL organization and alignment (30, 49, 50). In addition to domain order, the diabody-Ig platform utilize the dimerization domains CH1/CL, heterodimerizing EH Domain Containing 2 (hetEHD2), EH Domain Containing 2 (EHD2), and IgM heavy chain domain 2 (MHD2) to stabilize the diabody (51, 52). Furthermore, the domain connection was modified to promote heterodimerization (30). Fabs can serve as the foundation to which other binding elements are attached (30). A scFv may be attached to the C-terminal of either the light strand or the VH-CH1 (Fd) chains (e.g., bibody Fab-L-scFv, Fab-H-scFv), or to both strands (e.g., tribody, Fab(scFv)2) when Fabs are connected by hinge-regions (30, 45, 53–59).
The antigen-combining abilities of heavy-chain antibodies are entirely preserved in Nbs created from variable heavy-chain segments (VHH) in camelid heavy-chain antibodies (60). The molecule weight of the Nbs is 12–15 kDa, which is considerably less than the molecule weight of typical antibodies (150 kDa) (61, 62). Nbs with hydrophilic interfaces prevent the discrepancies in the heavy and light chain pairing of traditional antibodies, are not bound to light chains, which are vulnerable to polymerization, and are distinguished by tiny molecular mass, excellent solubility, and persistence (62). Nbs exhibit lower immunogenicity and simpler for humanizing and application in the clinic than traditional antibodies (62, 63). BsAbs can be created by modifying two Nbs which hit separate tumour antigens in order to enhance the selectivity of anticancer antibodies and render them optimal antibodies. In the detection and management of infection, cancer, and immunity, BsNbs are a scientific focus due to the improvement of BsNbs binding capacity, lengthening of plasma half-life, decrease in drug resistance, and severe side effects (62, 64). Liu et al. created the anti-CD20/CD3 BsNb by merging the anti-CD20 VHH gene with a thoroughly validated anti-human CD3 VHH built on the acquired anti-CD20 Nbs. After being incubated with human sera at 37°C for 48 hours, the anti-CD20/CD3 BsNb was still able to retain 80% of its binding efficacy. The findings demonstrated the potent anticancer activity of the developed anti-CD20/CD3 BsNb (62). Employing a BsNb which could concurrently target epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 2 (HER2) on cancer cells, Hong et al. created a dual-directed non-IgG form of BsAbs. The absence of Fc effector functional capabilities was restored by site-selective alteration of the EGFR-HER2-targeted BsNb employing the rhamnose (Rha) hapten through sortase A-regulated binding. The adjusted BsNb-Rha combination demonstrated significantly better pharmacokinetics and effective inhibiting actions against in vivo development of xenograft tumors (65). With the application of in silico methods, an improved bispecific design was created that can engage the therapeutically important antigens TNF-α and TNF-α-23 concurrently and, thanks to its increased avidity, efficiently block the death of TNF-α-sensitive L929 cells (66).
2.2 Classification of BsAbs based on symmetry
Through the lens of symmetry, BsAbs can be categorized into the asymmetric ones and the symmetric ones (Figure 2). Asymmetric BsAbs are initially created by combining two antibody-producing cell lines; with the advancement of genetic engineering, the technology is employed to produce BsAb, greatly assisting with the “chain” problems (it will be clarified in the following text) (Figure 3). Another approach to get around the “chain” issues and make the construction simpler is to design symmetric BsAbs, which could be generated by fusing or modifying IgG proteins.
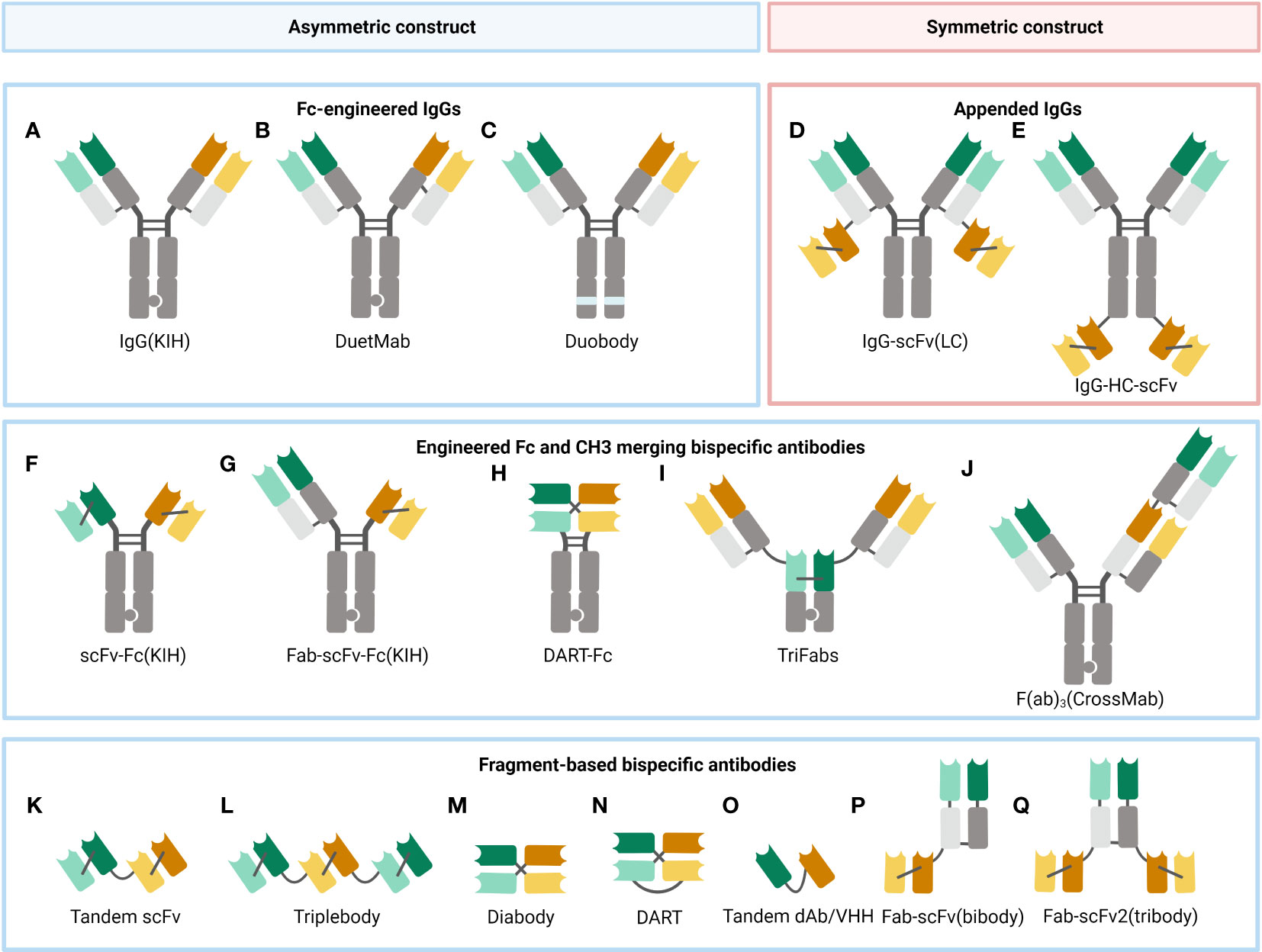
Figure 2 A selection of some common BsAb formats. (A) Fc-modified IgG format, built with the KIH technology to heterodimerize two different heavy chains. (B) DuetMab, improving the efficacy of homologous heavy and light chain coupling by designing a new disulfide bond to substitute the natural one in one of the CH1-CL interfaces (67). (C) Duobody, its Fc region was suppressed by inserting mutations, which circumvents the Fc-mediated cytotoxicity (68). (D) Appended IgG format, IgG- single-chain variable fragment (scFv) (light chain, LC). (E) Appended IgG format, IgG-heavy chain (HC)-scFv. (F) scFv-Fc format, constructed with the KIH approach. (G) Fab-scFv-Fc format, built with the KIH method. (H) DART-Fc construct, a DART protein unites two separate antigen-binding regions in a stable, diabody-like architecture (69). (I) TriFabs, IgG-based BsAbs made up of two normal Fab arms connected by flexible linker peptides to a third Fab-sized interaction unit (70). (J) CrossMab, antibody domain crossover enables the proper connection of generic light chains (71). (K) Tandem scFv (taFv), the minimum BsAb. (L) Triplebody, a construct similar to taFv. (M) Diabody (db), a short protein linker joins the heavy chain variable (VH) and light chain variable (VL) domains of a scFv segment to form a noncovalent heterodimer. (N) DART, made up of two Fv segments that heterodimerize to generate two distinct antigen-binding sites. (O) Tandem single-domain antibody (dAb)/VHH, made up of the antigen-engaging portion of heavy chain-only antibodies (72). (P) Fab-scFv (bibody), a scFv segment is fused to the C-terminus of the Fab framework to produce the bibody. (Q) Fab-scFv (tribody), a format similar to the bibody.
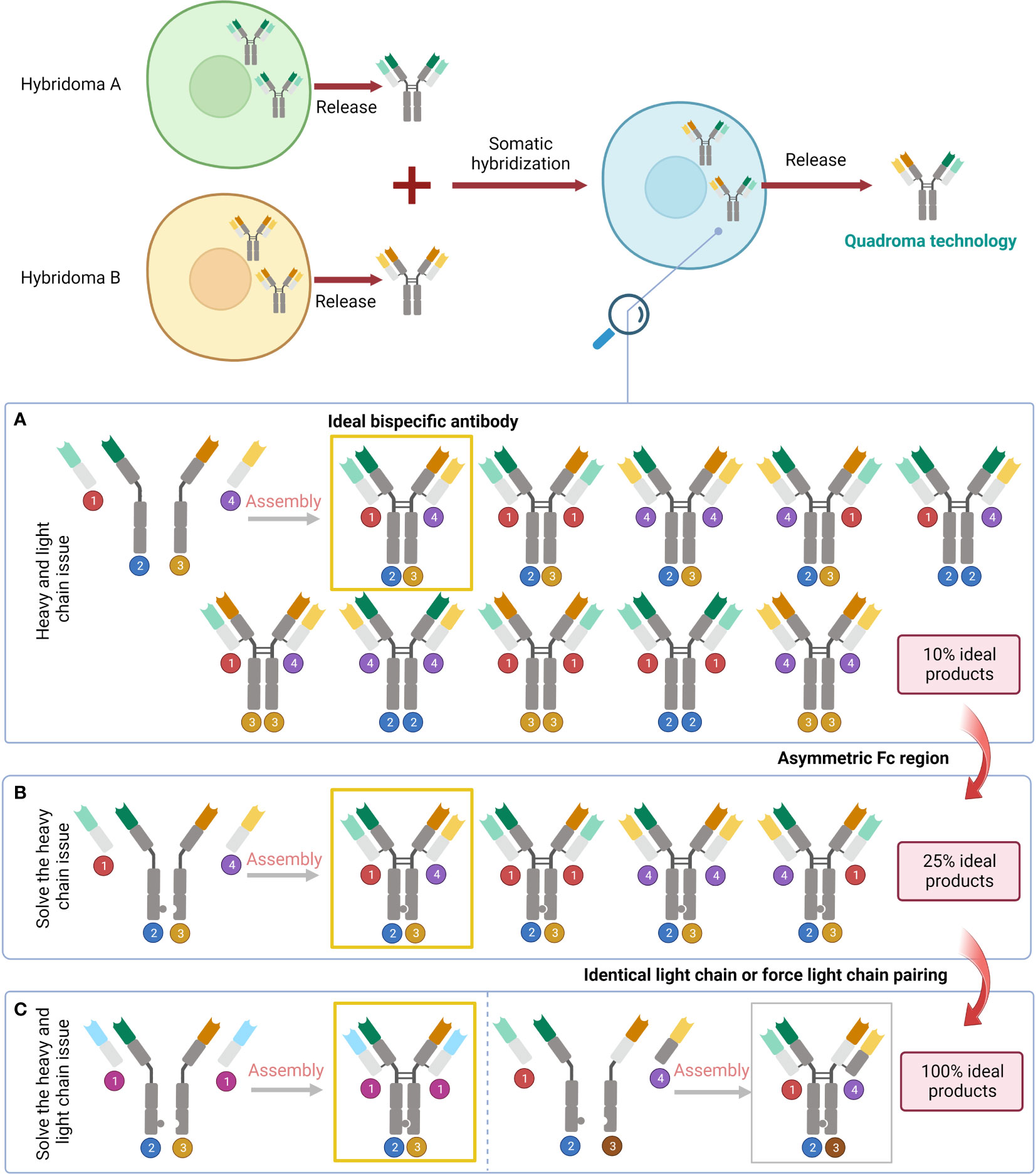
Figure 3 Problems raised by the variety of pairings and the corresponding solutions. (A) The result of simply fusing two different cell lines. (B) Solve the heavy chain problem by designing an asymmetric Fc region. (C) Address the issue with light chains by subsequent design of the identical light chain or force light chain pairing.
2.2.1 BsAbs of an asymmetric architecture
Every bispecific IgG molecule (antibody that is similar to natural immunoglobulins in constitute) is bivalent and has an asymmetric architecture because it contains at least distinct Fv regions (30).
Asymmetric BsAbs can be generated by merging two antibody-generating cell lines, e.g., producing a hybrid-hybridoma by fusing YTH12 and the MGICD19 cell lines (73). However, nine unwanted products will be generated by simply fusing two cell lines together (30). Genetic engineering is an alternative to remedy the issue. Through genetic methods, it is possible to create cell lines that produce two separate heavy and light chains and enable their proper integration (30). The heavy chain issue can be handled by BsAbs featuring an asymmetric Fc domain. Several methods created over the past 20 years leverage specific interchain disulfides, as well as steric or electrostatic steering effects to create a complimentary interface, benefiting heterodimerization against homodimerization (30). The knobs-into-holes (KIHs) strategy is a promising way to create BsAbs by inducing heterodimerization with mutations in the CH3 domain of each half antibody (74, 75). It was found that there was no significant change in the conception kinetics of BsAb produced by the KIHs technique, and the stability was similar to that of the wild-type antibody structure (74). Epcoritamab which recognises CD3 and CD20 and was created via cFAE of a humanized CD3 mAb and the human CD20 mAb7D8 (68). In extremely resistant patients with large B-cell lymphoma, notably those who had previously been exposed to CAR T cells, subcutaneous Epcoritamab produced profound reactions as well as reasonable safety (76). Flexible linker peptides may also be employed to fuse Fabs at their C-termini to a highly hereodimerizing Fab-like molecule (30). For instance, TriFabs are BsAbs with an IgG structure made of two conventional Fab connected by elastic linker protein to a single asymmetrical third Fab-sized interaction unit. The third module is S354C-Y349C disulfides connect CH3 knob-hole heterodimers, which replaced the original Fc region (70).
To maintain the related functions and favorable qualities, the major methods to create the forms in the category aim to preserve the structure of natural antibodies precisely. Nevertheless, in certain formats, the complex architecture to address the chain-association problem may negate these benefits (28). Compared to formats that permit multivalent target binding, asymmetric forms’ lower avidity may influence their strength (28, 77).
2.2.2 BsAbs of a symmetric architecture
Incorporating two particularities into single heavy & light combination or peptide strand will result in symmetrical BsAbs, and can solve the chain-association problem whilst preserving the Fc domain (28). Also, the symmetric form is easier to construct (58).
One strategy is to produce IgG fusion proteins, to be more specific, by fusing scFv, domain antibodies and scaffold proteins, Fab arms, or additional VH and VL domains. For instance, the T cell-stimulating BsAb CLN-049, which binds to CD3 and FLT3, was created as an IgG heavy chain/scFv hybrid (78). Another strategy is to modify IgG molecules. Either the VH and VL domains’ original antigen-binding sites was altered or an extra binding site was transplanted to the Fc fragment’s bottom portion (30). With two unique, regionally separated interaction sites inside the human antibody CDR loops, dual targeting Fab (DutaFab) molecules was developed (79).
While almost mimicking natural antibodies, symmetric forms bear differences in size and organization. These variations may adversely alternate antibodies’ features (eg, consistency and solubility), which could disrupt their physicochemical and/or pharmacokinetic qualities (28, 80, 81). Most clinical test formats feature tetravalent 2+2 configurations due to the symmetric character and thus anticipated to have enhanced affinity against both malignant cells and T cells. However, this was just of secondary significance in terms of therapeutic efficacy. Despite sharing identical neoplasm binding and having improved T cell interaction compared to 2+2, 2+2B (Bispecific T cell engager (BiTE)-Fc) and 2+2HC (IgG-[H]-scFv) both failed to exhibit anticancer efficacy in vivo (28, 58).
3 Preparation technology of BsAbs
Methods of preparation of BsAbs are classified as chemical coupling, hybridoma binding and gene cloning methods (82, 83). Chemical cross-linking is the process of forming disulfide bonds between antibody molecules of different specificities or F(ab’) by using a specific chemical cross-linking agent, thus creating a heterodimer. This can be in the form of coupling between whole antibody molecules, or between F(ab’) and F(ab’)2 (84). BsAbs prepared by this method can directly utilise existing antibodies and has a high yield, but its activity may be affected by damage to the antigen-binding site (85). Hybridoma technology is based on monoclonal antibody technology, in which hybridoma cells secreting two antibodies are fused to produce hybridomas that stably secrete BsAb. Co-expression of the respective immunoglobulin (Ig) genes produces two types of H and L chains, which combine to form a BsAbs with the characteristics of the parental Ig (86). BsAb prepared using the hybridoma method is more random and relatively inefficient, but it has better biological activity and a more stable antibody structure (87). Genetic engineering techniques have opened up new avenues for the preparation of BsAbs, either by cloning the gene encoding the parental antibody and transfecting it into host cells for direct expression of BsAbs, or by gene shearing and constructing a scFv for the preparation of modified BsAbs (30).
A quality technology platform is key to the success of BsAbs development, and several technology platforms are in progress (88). Among the BsAbs technology platforms with Fc are Crossmab/KIH, DVD-Ig (dual variable domain-Ig), IgG-scFv, FIT-Ig, mAb-Trap, duobody ect (48, 89–92). Dual antibody technology platforms without Fc include BiTE, DART, TandAb, ImmTAC, BriKE, etc (93, 94). Developing BsAbs with the aforementioned functions requires careful adjustment of a number of variables in order to attain the ideal practical result. A blend of complementary binders and other elements in formats which allow the required functionality is necessary for the creation of BsAbs (95). Here, we will introduce several promising preparation methods.
3.1 Knob-into-hole technology
BsAbs possess two distinct paratopes on their variable regions that recognize two separate antigens, in contrast to normal mAbs that contain two same antigen-binding or Fab components. Due to this special characteristic, BsAbs can take on more intricate forms, such as homodimers made of two distinct arms or light chain mismatches, among others. The KIH configuration was employed to create BsAbs to foster heavy-chain heterodimer pairing of the two hemi-antibodies (17, 96). Knob and hole mutations shouldn’t affect antigen recognition or Fc activity since they are in the CH3 domain interaction surface. There are multiple ways to prevent light chain mismatching over assembly (97). One method involves expressing two half-antibodies in two separate host cells during an in vitro production process. Following two distinct Protein A specificity grab procedures, the two hemi-antibodies are combined for in vitro synthesis by reduction and oxidation, which is proceeded by subsequent BsAb purifying (98, 99). It was possible to detect chemical alteration sites and evaluate the steadiness and wholeness along with the operation of a BsAb by applying a variety of stressful situations together with dimension isolation chromatography, ion switch chromatography, LC-MS/MS peptides mapping, and practical examination by cell-based tests. Grunert et al. observed that IgG1 KIH CrossMab-engineered BsAbs were significantly more stable than commercially available antibodies (100). Furthermore, Liu et al. discovered that the KIH architecture did not significantly modify the organization or conformation motion, and the structural security is comparable to that of wild-type (WT) IgG4 (apart from a minor change in the CH3 domain) generated in E. coli (74).
The KIH structure and in vitro construction may effectively promote the heterodimerization of the heavy chains; nevertheless, throughout hemi-antibody isolation and BsAb installation, some homodimers (such as knob-knob and hole-hole dimerization) remain detectable (96). In the Fc region of an IgG1 BsAb, Elliott et al. uncovered the molecular specifics for KIH and homodimer engagements (101). The knob-knob and hole-hole Fc component homodimers’ X-ray crystal structures have been resolved, revealing a juxtaposed Fc configuration. Bispecific variations have been identified and quantified via intact mass evaluation (99, 102–104).
3.2 CrossMab technology
Combined with techniques that allow for accurate heavy-chain connection with already-existing pairs of antibodies, CrossMab technology, in conjunction with KIH technology, permits an appropriate antibody light-chain interaction with its corresponding heavy chain in BsAbs (105). The BsAbs are made up of two arms: one altered, and the other is not. Adjustments may be restricted to the VL-VH domain, the whole Fab region, or the CL-CH1 area (23). Due to the modifications, the intended chain interaction is enacted because the unaltered heavy chain could no longer interact with the altered light chain. In terms of structure and purity, the CL-CH1 CrossMAb displayed the best results among the three potential changes (106). Clinical trials are now being conducted to assess a number of BsAbs developed by CrossMAb innovation (71). The BsAbs that have currently been produced using CrossMAb include RO6958688 (CD3, CEA) for carcinoembryonic antigen (CEA)+ solid tumors, RG7221 (Ang2, VEGF), RO7121661 (PD-1, TIM3), and RO5520985 (Ang2, VEGF) (28, 107).
3.3 FORCE technology
Being a high-throughput method, Format Chain Exchange (FORCE) provides effective combined production of BsAbs in various arrangements for screening in ultimate form. The technique is based on the formation of BsAb from monospecific educts carrying various binders in various forms. Input agents for the production of BsAbs are monospecific entities with matching CH3-interface-regulated and imitation chains with affinity tags, analogous to KIH hemi-antibodies. These comprise mutations which result in minor interaction repulsions without affecting the production or biological characteristics of educts. Instability at the CH3-educt interfaces resolves to complete compatibility upon mild reduction of pairings of educts, initiating unprompted chain interchange events. This results in the formation of sizable BsAb matrices including various binders in various forms. Processing automation is made possible by benign biological characteristics, excellent production outputs of educts, and ease of purification. The monospecific input components comprise designed Fc-mimic chains that induce heavy-chain interchange processes that lead to formation. Production automation is made possible by efficiency, sturdiness, and simplicity (including assembly and one-step output purifying), allowing for thorough screens of BsAb binder-format layout spaces (95).
3.4 SEEDBodies technology
By creating strand-exchange engineered domain (SEED) CH3 heterodimers, Davis et al. built a heterodimeric Fc system which facilitates the construction of bispecific and asymmetric hybrid proteins. Human SEED CH3 heterodimers, which are made up of alternate parts of human IgA and IgG CH3 patterns, are produced by the variants of human IgG and IgA CH3 regions. When produced in mammal cells, the resultant pair of SEED CH3 regions selectively interacts to generate heterodimers. SEEDbody (Sb) fused proteins are made up of [IgG1 hinge]-CH2-[SEED CH3], and one or more fused couples could be genetically related to them. Mammal cells producing SEEDbody (Sb) fusing proteins result with large outputs of Sb heterodimers which can be easily separated to get rid of the modest byproducts. To simplify examination of heterodimer production in the current study, fusion companions are usually introduced to the N- or the C-terminal of one Sb chain. The lengthy plasma half-life prolongation characteristic of analogous fusion involving Fc segments and IgG1 standards were visible in the Sb pharmacokinetics after being delivered intravenously to rodents (108).
3.5 Duobody® technology
DuoBody® innovation was created by Engelberts et al. to produce complete IgG1 BsAb employing cFAE. Here, under carefully monitored fabrication circumstances, two original IgG1 mAb with paired single spot mutations in the IgG Fc region rearrange into full-length bispecific IgG1s (33, 109). At both the laboratorial and industrial scales, it was demonstrated that the cFAE technique is a simple and reliable way to produce durable BsAb with a greater output contrasted with other BsAb technologies (110). Additionally, the approach offers the chance to create and screen sizable and different arrays of BsAb, allowing for the identification of BsAb with the best functionality (68). In patients with progressed solid cancers, DuoBody-CD40-4-1BB has the potential to improve anticancer immunity by altering DC and T-cell activities (111). A transformed CD3 mAb and the human CD20 mAb 7D8 were combined to create DuoBody-CD3xCD20 (GEN3013), a BsAb recognizing CD3 and CD20 (76, 112). The subcutaneous injection route might offer a way to lower patients’ peak cytokine levels, as well as a way to ease their medical strain and make more efficient use of the facility’s resources (68).
4 Delivery of BsAbs
Currently, there are two ways to deliver BsAbs to the tumor sites. The first is to administer BsAbs after they have been produced in vitro, a process that is generally costly, time-consuming, and ineffective. The second is to enable the in vivo synthesis of BsAbs, which can counteract the quick kidney elimination of Fc-free forms, rendering a prolonged potent antibody level and can bypass issues with recombinant antibody assembly and long-term preservation (113, 114) (Figure 4).
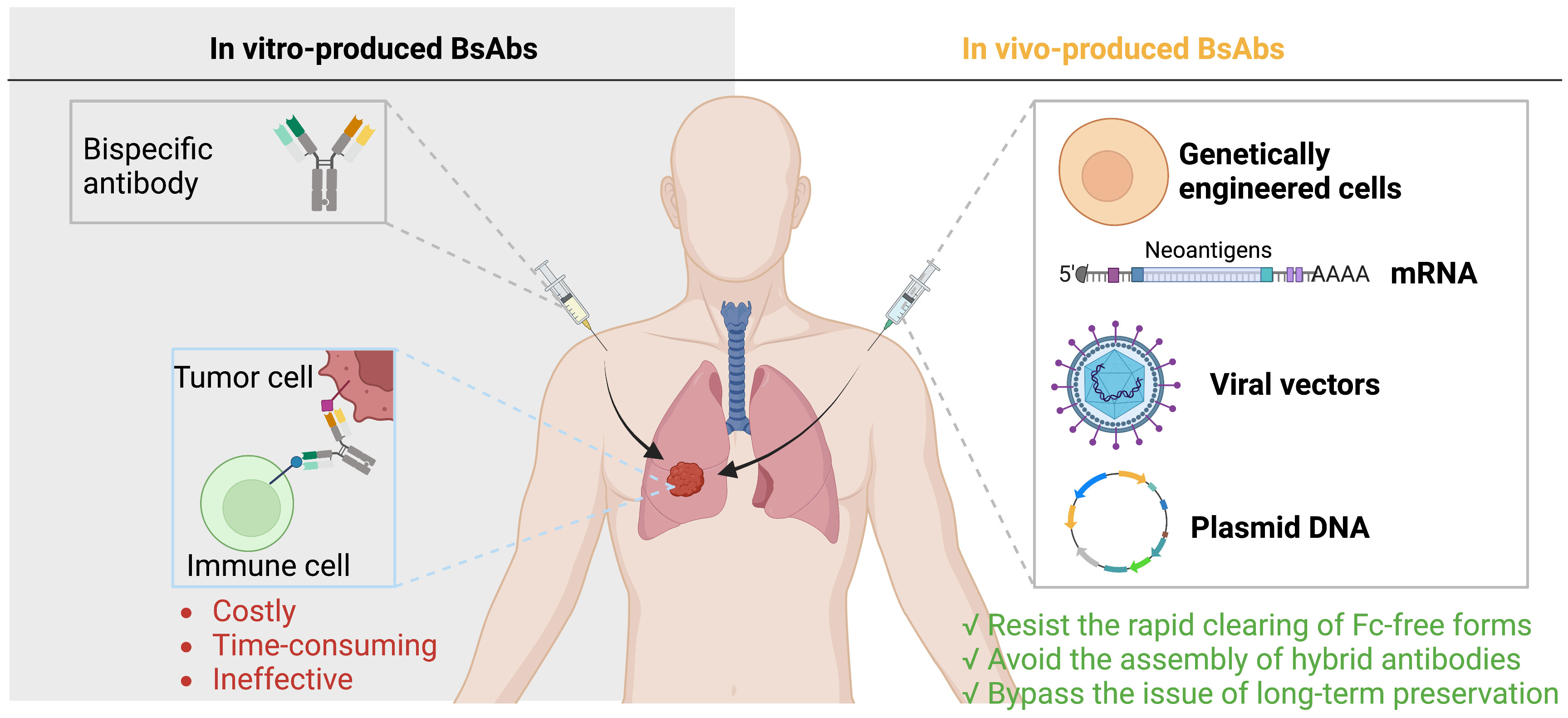
Figure 4 Delivery of BsAbs. There are two ways to deliver BsAbs to tumors: administer pre-made BsAbs, which is expensive and inefficient, or allow for in vivo synthesis of BsAbs, which can avoid elimination assembly, and preservation issues.
4.1 Delivery of in vitro-produced BsAbs
The majority of antibody-based treatments are administered following in vitro synthesis. In terms of the scope of action, these deliveries could be classified into circulatory delivery (e.g., intravenous (IV), intracutaneous (IC), subcutaneous (SC), or intramuscular (IM) delivery) and local delivery. Delivery to cancer is regulated by an intricate interaction of factors: the spot of infusion (e.g., IV, IC, or intratumor), carry via the plasma and lymphatics, permeation across the endothelium and basal lamina into the interstitium, hydrodynamic tension in the blood vs. the carcinoma tissues, and removal of biologics from the scheme (e.g., by renal filtration, hepatic evacuation (115)).
Although the IV method provides 100% bioavailability, physiological obstacles and circulatory dispersion significantly lower the real BsAb level in the target sites (116). Additionally, IV infusion is inconvenient and takes time, as purified BsAbs that require higher concentrations must be supplied by gradual IV in order to prevent injection responses (114). Considering the maximal amount of infusion is limited, accordingly, low BsAb solubility at high densities is the most typical barrier for SC or IM. Clinical-grade BsAbs are very costly and have production difficulties, such as low durability over long-range preservation and a propensity to congregate (117, 118). The BBB is a tough hurdle that prevents drug transport to the brain. The employment of intrinsic brain endothelial delivery channels is a viable strategy to bypassing the biological hurdle via receptor-mediated transcytosis (RMT). BsAbs are the perfect choice for the purpose since treatments engineered demand at least two capabilities, one that aids delivery and the other to give clinical effect (119). The most typical RMT (TfR, InsR, and LRP1 receptor) have been effectively exploited to cross the BBB (120, 121). Since the BBB is disrupted in brain malignancies, it is necessary to particularly look into the production of novel candidate receptors facilitating RMT particularly for the blood–brain tumor barrier (122). Besides getting to the pathways, innovations to effectuate BsAb distribution into the brain should also be taken into account (119).
Regional administration can improve potency and lessen overall contact for various ailments. In certain malignant situations, intratumoral or intraperitoneal injection of BiTEs may confine effects to malignancies (123–125). An efficient and feasible approach is to use solid implants, granules, or injected storage made of biodegradable and biocompatible polymers to entangle BsAbs and unleash them as the polymer breaks down. PEG-PLA copolymers, a depot-injectable polymeric platform created in situ, were utilized to transport BiJ591 (a BsAb targeting CD3+ T cell and prostate-specific membrane antigen (PSMA) on prostate cancer cell) against prostate tumor. The transport method could regulate BiJ591’s discharge while preserving its durability and activity, and the treatment efficiency was higher than IV delivery (126, 127).
Transport techniques utilizing nanoparticles also showed impressive performance. Contrasted to XA-1 protein alone, lipid nanoparticle-encased mRNA-expressed XA-1 displayed greater potential anti-cancer effectiveness (128). Administration platforms like liposomes and cell-infiltrating peptides have also been demonstrated to be more efficient than the use of a solitary drug (127).
4.2 Delivery of in vivo-produced BsAbs
In vivo gene treatment was created, in an effort to strike a balance between potency and security. The two major schemes are the in vitro inoculation of genetically engineered cells and straight gene transport via vectors, mRNA, and plasmid DNA (127).
Both viral and nonviral vectors could be employed to convey the genetic material encoded for BsAb in vivo, while utilizing mRNA or plasmid DNA, direct in vivo administration of synthesized nucleic acid-coded BsAbs suggests new methods (114, 117, 129, 130). A CaPO-nanoneedle/minicircle DNA platform generated BsAb (EpCAM/CD3-targeted) resulting in a considerable slowdown of tumor development and a prolongation of animal life-span with minimal toxicity in an intraperitoneal xenograft model with human ovarian carcinoma cell line SKOV3 (131). A synthesized HER2 plasmid DNA-coded BiTE effectively recruited T cells to identify and ruin HER2+ melanoma cells, and it exhibited potent anti-cancer effects in vivo (127, 132). Additionally, numerous oncolytic viruses were equipped with expression cassettes generating BiTEs, proving that when straight oncolysis and T cell-regulated destruction are combined, anticancer potency is increased in contrast with the original equivalent in syngenic and xenograft malignancy models (114, 127, 129, 132). The strategy may speed up the clinical progression of new BsAbs as it is quick to produce pharmaceutical-grade mRNA and DNA. Moreover, the temperature tolerance of DNA could make it simpler to carry and administer to larger populations due to its long-term preservation and temperature durability. Also, the in vivo synthesis could maintain an efficacious protein level, allaying worries about a quick kidney clearance (129, 130, 133, 134).
It is possible to in vitro transmit genetic information into cells obtained from patients, after which the BsAb-releasing cells are infused back to the patient. Compared to straight gene transfer methods, tumor infiltration and general on-target/off-cancer cytotoxicity problems may be addressed by the tumor anchoring of injected cells and ensuing intratumoral release (114, 135). New methods centered on modified cells secreting BiTEs (STAb cells) natively are now being developed (135). Research detailed the creation of anti-CEA and anti-CD3 dAb-releasing human T cells and revealed that the intratumoral delivery of lentivirally transfected STAb-T cells dramatically decreased in vivo cancer progression in human HCT-116 colon malignancy xenografts (135).
5 Anti-tumor mechanism of BsAbs
BsAbs could executive its antitumor effect in three major ways, including redirecting immune effector cells to tumors, delivering radioactive or drug payloads to carcinoma cells, targeting multiple signaling pathways to suppress tumor progression directly or indirectly. The classification of BsAb clinical applications based on target antigens is presented in Table 1.
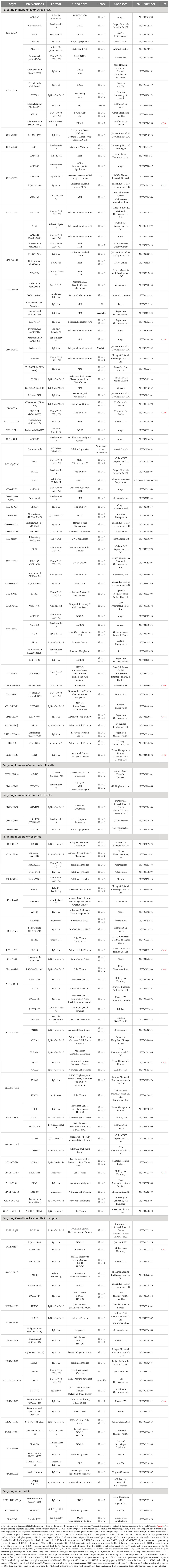
Table 1 Clinical-stage BsAbs for cancer indications.
5.1 Reorientating immune effector cells
In the case of cancer treatment, one arm of the BsAb targets a tumor-associated antigen (TAA), while the other arm targets a molecule present on immune cells. Via targeting both the TAA on tumor cells and immune cell molecules, the BsAb brings the immune cell in close proximity to the tumor cell, resulting in the immune cell’s stimulation and then destroying the tumor cell (Figure 5).
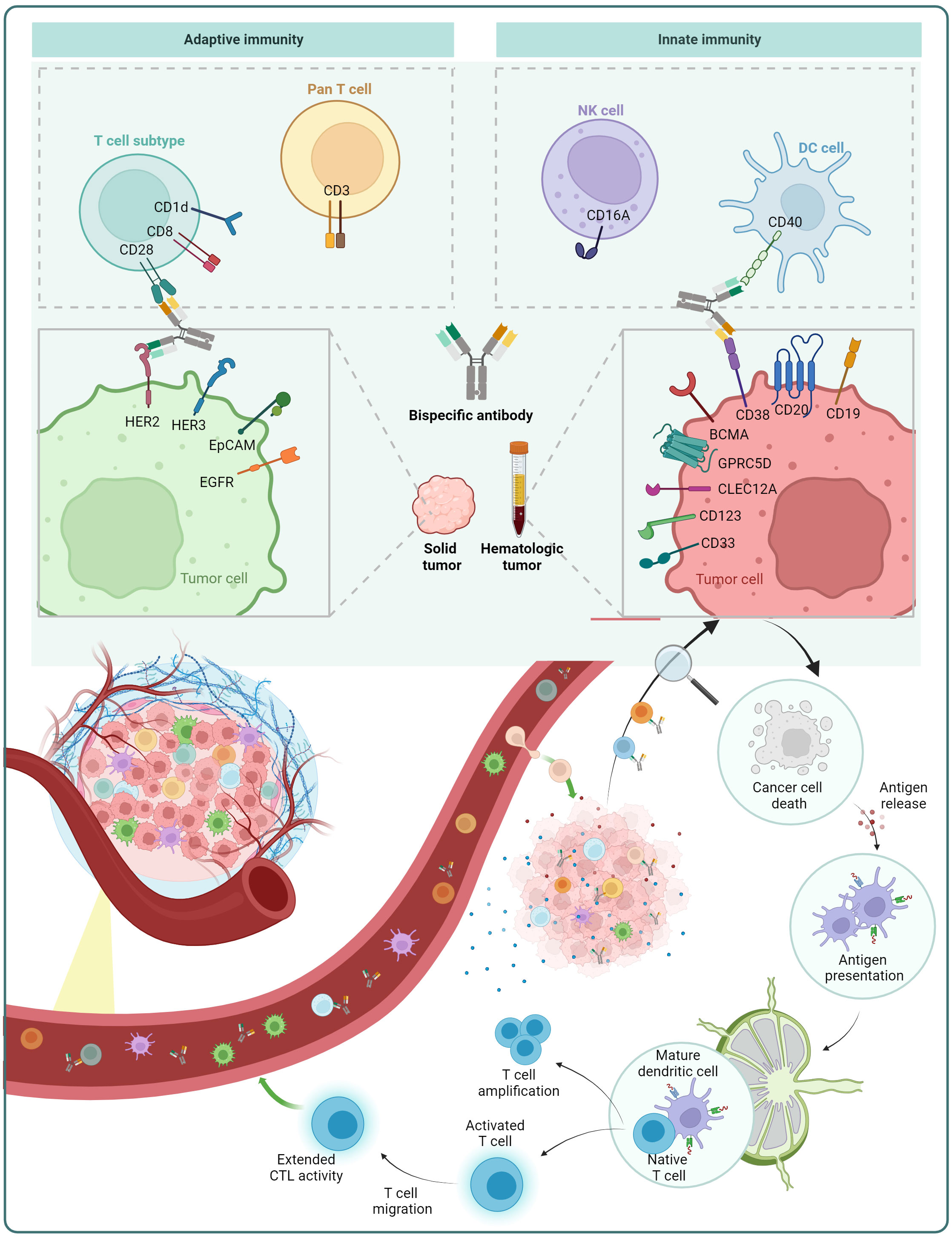
Figure 5 Mechanism of action of BsAb: redirection of immune cells. BsAb can redirect pan T cells and T cell subsets from the adaptive immune system, as well as natural killer (NK) cells and dendritic cells (DCs) in the innate immune system. The figure also displays some of the immune cell surface antigens and tumor cell surface antigens that are already under investigation.
Under microhomeostatic circumstances, anticancer immunity is one of the essential cancer therapy techniques. However, in order to survive and propagate, tumor cells are able to escape the “cancer-immunity cycle” which describes how the innate and adaptive immune systems collaborate to prevent malignancy genesis through sequential events (149). This is accomplished by mechanisms that suppress anti-tumor immunity, such as increased expression of molecules like PD-L1 that block T-cell action or decreased production of human leucocyte antigen (HLA) class I molecules that hinder antigen presentation (150–153).
Substantial therapy outcomes can be attained by reviving and strengthening the latent immune cells, which has been demonstrated by the development of several mAb immune checkpoint inhibitors (ICIs) that bind PD1, PD-L1, CTLA-4, etc. during the past ten years (154–157). Major improvements in total and advancement-free survival have been obtained in melanoma, lung carcinoma, and urothelial cancer (58, 130).
5.1.1 Reorientating the cells of the adaptative immune system
As of October 2023, the majority of BsAbs bridge cells as their primary action mode and have T cell reorientation as their shared thread. Rerouting effector T cells with cytotoxic activity to destroy malignant cells is a classic paradigm of these BsAbs (28).
BiTEs primarily stimulate T cells via interaction with CD3ϵ in the T-cell receptor (TCR) complex, thus are defined as pan-T-cell engagers (158, 159). The high affinity between BiTEs and TAA/CD3a renders a huge proportion of activation receptors (TCR/CD3 complexes) gather between cells, resulting in effectual T-cell excitation with just one receptor-ligand interplay and the killing of cancer cells through releasing perforin and granzymes (130, 160, 161). Most CD3-targeted pan-T-cell stimulators at the clinical phase are designed to treat blood tumors, such as targeting CD19 and CD20 for non-Hodgkin lymphoma (NHL), targeting B cell maturation antigen (BCMA), GPRC5D and CD38 for multiple myeloma (MM), targeting CD33, CD123, and CLEC12A for acute myeloid leukemia (AML) (162). Although these targets are widely expressed on healthy blood cells as well, their depletion can be handled without eliciting serious negative impacts (114). A relatively small amount of TCRs target MHC-presented TAAs in solid tumors (163). Phase I clinical studies are being conducted with AMV564, a TandAb with two CD3 binding sites and two CD33 binding sites respectively (162).
The TAA selection is crucial for BiTEs to perform properly. The performance of BiTEs is associated with expression ratios of targets, as was the case with BiTEs targeting EpCAM, CD33, and HER2 (164). Three distinct cell lines exhibiting high (EOL-1), medium (MOLM-13), and low (MV4-11) rates of FMS-like tyrosine kinase 3 (FLT3) expression were employed to analyze the influence of receptor density on the potency of FLT3 BsAb in vivo. Compared with the MOLM-13 model, the EOL-1 model demonstrated total potency at a lower dose of 7370, which is aligned with EOL-1’s higher membrane FLT3 expression (165). BiTEs’ activity is also influenced by TAA’s characteristics, such as size and mobility on cell membrane (166). Chinese hamster ovary (CHO) cells presenting small surface target antigens were typically more effectively lysed than those with bigger antigens (167). The antigen’s affinity to candidates is also an important determinant for the strength of BiTE. High affinity for HER2 was essential for the HER2/CD3-targeted BiTEs’ ability to destroy cancer cells. Nevertheless, a worse safety profile, such as cytokine release and impairment to HER2-expressing tissues, was also linked to increased HER2 affinity. Adopting a dose-fractionation method could enhance the HER2/CD3-targeted tolerance (168).
Architecture of CD3-binding part impacts the biodistribution of BiTEs. Despite BiTEs with high CD3 affinity demonstrated better efficacy in co-culture tests in vitro, a reduced affinity of the CD3-binding domain is preferred to enable effective tumor diffusion in vivo without triggering rapid CD3-regulated plasma elimination or antibody entrapment in organs which store T cells (169–173). Many BiTEs only have one CD3-binding domain, whereas some clinical-stage BsAbs possess two CD3-targeting sites. However, whether such formats could functionally connect CD3 bivalently is unclear, which is essential for antigenic regulation and tolerance evoked by CD3-mAbs (28, 174). A monovalent CD3 interplay is favored because bivalent CD3 binding may crosslink the TCR/CD3 complex even if it is not simultaneously bound to TAA-expressing cells, resulting in systemic T-cell stimulation and cytokine release syndrome (CRS) (114, 175).
There are several FDA-authorized BiTEs for tumor therapy. In 2009, catumaxomab was granted clinical approval as the first BiTE. This antibody has two distinct antigen-targeting sites—one for the CD3 antigen on T-cells and another for the EpCAM on tumor cells—and also binds to accessory cell FcγR via its preserved Fc region (9). However, the IV injection of catumaxomab was linked to serious harmful effects that were ascribed to the active Fc site’s off-target adhesion to other immune cells expressing FcγRs, causing CRS and T cell-mediated hepatic damage (114, 176, 177). In 2014, Blinatumomab, a BiTE created by Amgen Inc. for the treating blood malignancies derived from B-cell lines (178), was authorized by the FDA for the therapy of acute lymphoblastic leukemia (ALL). Blinatumomab is a tiny BsAb with a molecular weight of around 55 kDa and a brief plasma half-life of 1.25±0.63 hours in vivo (28, 179–181). In this regard, the switch from sporadic IV infusion to constant IV infusion was a crucial choice in the clinical development of blinatumomab, which not only elevated security but also permitted more sustained T cell activity by preserving effective drug serum rates for the duration of a treatment cycle (182). Motivated by the promising clinical data of binatumomab, a variety of BiTEs with multivalent TAA affinity and monovalent CD3 binding, as well as the DART format have been developed to improve tumor selectivity and reduce off-target side effects (28, 114). In 2021, zenocutuzumab, an innovative IgG1 class HER2/HER3 BsAb utilizing the “dock-and-block” strategy, was granted the Fast Track Designation for NRG1+ metastatic neoplasms. Owing to its selectivity for HER2’s domain 1, zenocutuzumab can suppress HER2/HER3 signals regardless of the presence of HER2. Furthermore, it has no synergistic toxicity on cardiac myocytes conducted by HER2/HER4, thanks to its selectivity in blocking HER2/HER3 dimerization (183, 184).
Despite their potential, certain investigations revealed that T cells activated by BiTEs become less potent over time since they deplete more quickly (130, 185). As is the case with blinatumomab and catumaxomab, the administration of BiTEs is linked to CRS, which is indicated by abrupt elevations in the serum amounts of inflammatory cytokines such interleukins-6 (IL-6), tumour necrosis factor (TNF), and interferons (IFNs) (186–188). In an intriguing study applying an anti-PSMA T-BsAb, the scFv-Fc-scFv T-BsAb design allowed for the generation of powerful T-cell-dependent cellular cytotoxicity (TDCC) in vitro while limiting cytokine release, indicating that carcinoma cytotoxicity and cytokine storm might be distinct events or that an ideal balance between efficacy and toxicity can be realized by altering BiTE layout (189). Another significant drawback of CD3-targeted BiTEs is a significant fraction of the T-cell population is awakened. Therefore, compared to existing CD3-targeted pan-T-cell activators, BiTEs specifically activating distinct T-cell subtypes would be advantageous (114).
Targeting particular T cell subgroups render BsAb more effective in the destruction of tumor cells (28). Blinatumomab could stimulate Tregs in vitro, which inhibited effector T cells’ cytotoxicity (190). Additionally, in 42 patients with B cell ALL, the number of Tregs in the peripheral blood before blinatumomab administration negatively predicted response (190). Therefore, one of the objectives to construct a CD8+ T cell and prostate stem cell antigen binding tandem scFv was to avoid the induction of Tregs (40, 191). Vγ9Vδ2-T cells are a small and conserved T-cell fraction with a powerful inherent immunotherapeutic prospective. Vγ9Vδ2-T cell concentration and powerful CD1d-reliant tumor lysis are made possible by a bispecific Vγ9Vδ2-T cell engager (192).
Obstructing inhibitory checkpoints can revive weary neoplasm-permeating T cells (114). Inhibitory receptors such as PD1, mucindomain containing3 (TIM3), and lymphocyteactivation gene 3 (LAG3) are abundantly expressed when T cells are in a worn-out condition, which results in defective effector outcomes (193–195). Immune checkpoint-targeting BsAbs are arising following the therapeutic efficacy of anti-CTLA4 (cytotoxic T lymphocyte antigen 4), anti-programmed cell death protein 1 (PD-1), and anti-PD-L1 antibodies, while the enhanced therapeutic effect seen in coupled research using mAbs that engage the checkpoints serves as justification for concurrently engaging two immune checkpoints (28, 196). The majority of the BsAbs inhibit the PD-1/PD-L1 pathway with one arm while blocking CTLA-4, LAG3, or TIM3 with the other (28, 114). Fc-silenced BsAbs were developed to block the PD-1 cascade via high-affinity PD-1 interaction whilst obstructing CTLA4 with a low-affinity binding domain in order to enhance the safety aspect of simultaneous engagement of PD-1 and CTLA4 (28).
5.1.2 Reorientate cells of the innate immune system
BsAbs are also evolving as a substitute therapy strategy geared at the induction of intrinsic immune effector cell toxicity versus cancers with prospects for therapeutic potency and reduced therapeutic toxicity (197, 198). The bulk of BsAbs regarding the innate immune system focuses on dendritic cell (DCs), natural killer (NK) cells, and phagocytes (114, 199).
DCs are professional antigen-presenting cells (APCs). BsAbs with intact Fc domain can be employed to boost the chances of DCs and T cells coming into contact (130). In this regard, a BsAb which concurrently and agonistically activated CD28 on naïve T cells and CD40 on AML-DC was developed. In addition to improving CD28-mediated messaging, it was proposed that the ensuing cellular cross-linking would strengthen and prolong T cell/AML-DC contacts, thus boosting T cells’ sensitivity to AML antigens (200).
NKs may identify and destroy stressed cells, triggering an immune response much more quickly without antibodies or MHC. Tandem scFv, also known as “bispecific killer cell engager” (BiKE) or “trispecific killer cell engager” (TriKE), is a technique for directing NK cells toward cancer cells (201, 202). The innate cell engager (ICE®) AFM13 is a tetravalent BsAb that targets CD16A, the main FcR on NK cells, and CD30, which is prevalent in blood malignancies. In individuals with recurrent or resistant Hodgkin lymphoma, it has exhibited early clinical efficacy without significant therapeutical toxicity (197, 203–207). NK cells can also be recruited to cancer cells based on a scFv-Fc-scFv format. RO7297089, a bispecific BCMA/CD16A-targeted ICE® intended to cause BCMA+ MM cell lysis via strong affinity interaction of CD16A and redirection of NK cell toxicity and macrophage phagocytosis, promotes antibody-dependent cell-mediated cytotoxicity (ADCC) and ADCP against myeloma cells effectively, as well as pharmacodynamic efficacy in cynomolgus monkeys (197).
The modification of in vitro activated or expanded immune cells with BsAbs represents a new therapy for cancer treatment. The first clinical report of this approach emerged in 1990. Nitta et al. applied the method to malignant gliomas by using anti-CD3/glioma BsAb-modified lymphokine-activated killer (LAK) cells, which exhibited a favorable anti-tumour effect (208). Following the emergence of cytokine-induced killer (CIK) technology, modified CIK cells with BsAbs have been introduced into clinical studies (209). In nude mice, the use of BsAb-CIK cells resulted in a significant (p<0.05) reduction in CD133 (high) tumour growth (210). Golay et al. utilized CIK cells from cryopreserved cord blood units along with blinatumomab for the treatment of CD19 malignancies, which showed meaningful therapeutic effects with no evidence of toxicity or graft-versus-host disease (210). BsAbs ex vivo armed T cells (EAT) could potentially overcome certain limitations of chimeric antigen receptor-modified T cells (CAR-T), including cytokine release syndrome and neurotoxicity (211). Park et al. developed EAT utilizing the IgG-[L]-scFv platform BsAb. This resulted in a more efficient infiltration of tumors and a lower concentration of TNF-α released compared to the use of BsAb or T-cell injections alone (212).
5.2 Delivery of medicines
The distribution of payloads such as medicines, radiolabels, and nanoparticles is an intriguing deployment of BsAbs (97). A payload comprising an isotope or a medication is directly attached to a BsAb in this method (40).
5.2.1 Radioactive payload
Inferior therapeutic indices (TI) cause the majority of radioligand treatments to have unforeseen dose-limiting toxicities to crucial organs, which leads to many patients receiving subtherapeutic dosage of treatment. BsAbs can be applied to enhance radioactive payloads in neoplasm areas, which can greatly increase the tumor/blood percentage and serum retention duration (97, 213–215) (Figure 6A).
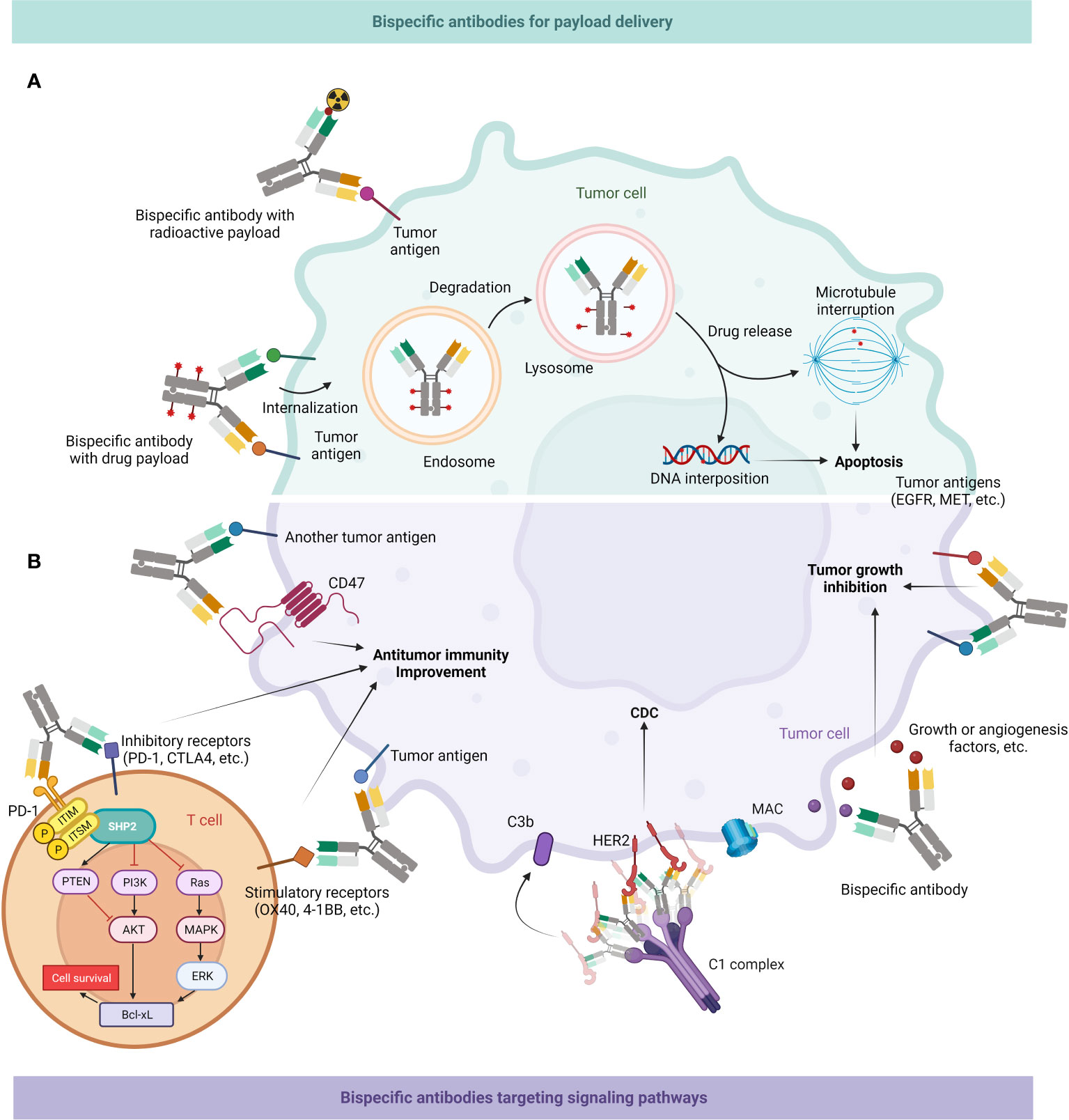
Figure 6 Mechanism of action of BsAb: delivery of payloads & targeting tumor-associated signaling pathways. (A) BsAb can enrich radioactive substances around tumor cells; they might also load drugs and then enter cancer cells through endocytosis, first into endosomes and then into lysosomes, and then release drugs to interfere with microtubule formation or DNA replication, ultimately leading to apoptosis of tumor cells. (B) BsAb can directly inhibit tumor growth. It can interfere with multiple signaling pathways related to neoplastic growth, as well as inhibit growth factors or angiogenic factors in the tumor microenvironment (TME). BsAb may also facilitate antitumor immune activity. It can bind CD47 on the surface of tumor cells to promote macrophages-mediated phagocytosis of them, or target inhibitory or stimulatory receptors on T cell surfaces to enhance the effector function of T cells. BsAb can also form hexamers by binding to receptors like HER2, recruiting the C1 complex, and subsequently mediating tumor cell death through the CDC effect (216).
A BsAb with specificity for both the TAA and the load can be cultured with the load before infusion (40). Optionally, pre-targeting strategies can be applied to avoid sustained exposure of normal tissue to the payload, reducing toxicity and side effects, which involve first injecting the BsAb, followed by administering the payload to deliver radioactive loads to a malignancy (40, 217). Such BsAbs with a radioactive payload could be utilized for radioimmunotherapy as well as cancer imaging. Pilot research revealed that pretargeted immunological positron emission tomography (immuno-PET) employing a CEA/IMP288-targeted BsAb and a [68Ga] Ga-labelled hapten was secure and practical with encouraging diagnostic accuracy (218). A creative cancer-targeted DOTA-hapten pre-targeted radioimmunotherapy platform, Pr, is composed of a vacant DOTA-chelate for 225Ac, which is connected to a lutetium-composited DOTA for picomolar DOTA-bound chelate scFv adhering through a short polyethylene glycol linker. In three solid patient tumor xenograft models for neuroblastoma (GD2), colorectal cancer (GPA33), and breast cancer (HER2), extended general survival, complete responses, and histologic healing were noted. Also, [225Ac]Pr has a significantly higher security profile in contrast with RIT with tumor-targeted IgG antibodies (219).
5.2.2 Drug payload
By combining targeted treatment with a strong cytotoxic payload to antibodies, antibody-drug conjugate (ADC) medicines destroy malignant cells through a pharmaceutical Trojan horse approach (37) (Figure 6A). The monoclonal antibody-based ADCs, including the anti-CD33 drug gemtuzumab ozogamicin, the first ADC authorized by the FDA, and the anti-HER2 drug adotrastuzumab emtansine to treat advanced breast carcinoma, have achieved a significant progress (220).
However, the potency of therapy may be restricted gradually since tumors may evolve defences against drug effects. Downregulation of the antigen on the cell membrane, which makes the ADC comparatively less likely to perform the cytotoxic activity, may be one cause of resistance (221, 222). Bispecific or biparatopic mAbs, which bind to non-overlapping epitopes on a single target antigen or two distinct antigens simultaneously, are novel strategies to circumvent antigen-specific rejection mechanisms (223). M1231 is an experimental ADC combining a BsAb which concurrently targets MUC1 and EGFR with a payload linked to hemiasterlin. Patients with progressive solid tumors, such as esophageal cancer, and non-small-cell carcinoma (NSCLC) are currently being studied using the BsAb as a monotherapy in phase I trials (224).
It is vital to internalize proteins effectively and direct them to lysosomes where proteolysis can occur. Nevertheless, the amplitude of these activities for many cell membrane proteins and carbohydrate compounds on tumor cells is inadequate to support an efficient ADC strategy. One approach is to incorporate BsAbs into ADCs, where one binding region would offer malignant affinity while the other binding site would enable localization to the lysosome (224). A bispecific ADC that targets both HER2 and the lysosome membrane protein CD63 appears to increase lysosomal aggregation as well as cargo delivery (225, 226).
5.2.3 Targeted delivery of nanoparticles
Delivery of nanoparticles with BsAbs can overcome the inefficiency of the enhanced permeability and retention (EPR) effect of nanomaterials and improve the active targeting and delivery efficiency of the drug itself. At the same time, BsAbs can enhance the anti-tumour activity of nanoparticles and reduce the amount of drug used, thus reducing off-target cytotoxicity.
Active targeting of nanomaterials can be achieved through coupling BsAbs to the surface of the nanomaterials. BsAbs and nanoparticles are linked via antibody-antigen interactions, which are more specific than random coupling methods (227). The vast majority of connections between BsAbs and nanoparticles are non-covalent, and upon cellular endocytosis of drugs, these bonds are readily broken, thus increasing drug release and improving delivery efficiency (228). Furthermore, the selectivity of BsAbs allows for the specific targeting of tumour tissues through nanomedicines, holding immense significance in the treatment of solid tumors. EGFR-targeted EDV nanocells loaded with paclitaxel are currently part of clinical studies and can be safely administered to patients with advanced solid tumors at a maximum tolerated dose (MTD) of 1×1010 microcells per dosage (229). Pre-targeted therapies are another method of delivering nanomedicines based on BsAbs (230). A CD20 Ab-mPEG scFv was able to target CD20-expressing Raji cells while carrying simultaneously grasping polyethylene glycolated liposomal DiD with a 24% increase in internalization capacity (60 h). It showed a nine-fold increase in tumour cytotoxicity (LC50: 3.45 nM) and improved anti-liquid tumour efficacy (p=0.005) compared to CD20 Ab-mPEG scFv and PLD alone (231). Another study showed that BsAbs pre-targeted delivery of D-Dox-PGA reduced off-target toxicity in human breast cancer xenografts and had antitumor efficacy comparable to Dox therapy (232).
5.3 Signaling pathway targeting
Some cancer cells can develop resistance to single-target therapies by activating alternative signaling pathways. BsAbs can co-target several tumor-related receptors on the cell membrane (Figure 6B), providing powerful anticancer strategy and reversing resistance, eliciting a more potent and effective response than single-target antibodies.
5.3.1 Bridge receptors
Resistance is a main downside of RTKs, such as EGFR and HER2. Resistance typically entails the activation of parallel signal transduction by upregulating other RTKs that avoid targeted receptor blockage. For example, the MET oncogene is amplified to induce HER3 and the PI3K-AKT cell survival pathway, enabling NSCLC to escape the suppression of EGFR-TKIs (226, 233). Several BsAbs co-targeting several RTKs were developed and are currently undergoing clinical trials (233).
The members of the ErbB family, EGFR, HER2, and HER3, are typical targets for BsAbs that interrupt two signals because of their interaction (40, 234–236). Zenocutuzumab targets the HER2 and HER3 cytoplasmic regions. By blocking Neuregulin 1 (NRG1) binding to HER3 and preventing HER3 from going through the conformational alteration necessary for heterodimerization with HER2 and possibly with EGFR, zenocutuzumab inhibit downstream oncogenic signals and the phosphorylation of HER3’s cytoplasmic region (148, 183). Clinical success was verified in patients with pancreatic and lung malignancies induced by NRG1 rearrangements (148). Other oncogenes such as MET can also become targets. Amivantamab (JNJ-372) is a first-in-class, completely humanized, IgG1 BsAb that targets both EGFR and MET synchronically. It hinders the stimulation of ligand-binding receptors, encourages their internalization and destruction to impede downstream signaling cascades. It can involve Fc-mediated attachment to macrophages and then trigger a potent antibody-reliant ADCC which is vital for the blockage of both EGFR and MET pathways (38, 39, 237). Additional targets under research include lysosomal internalization-related receptors like CD63 and death receptors like death receptor 5 (DR5) (40). Shivange et al. develop antibodies that limit DR5-mediated apoptotic activity toward FOLR1-expressing ovarian tumor cells yet minimizing the need for ADCC to trigger cell destruction. It turned out that these antibodies operated better than DR5 agonist antibodies reported from clinical testing (238).
Besides, the activation of the complement-dependent cytotoxicity (CDC) system is regarded as a method for enhancing the clinical efficiency of anticancer Abs (239). It comprises one of the identified modes of action (MOA) for B-cell specific monoclonal Abs such as rituximab and atumumab (240–242). The capacity of the antigen-Ab combination to adopt a shape which permits effective C1q interaction is influenced by an array of variables, such as antigen diameter and density, which regulate activation of the classical complement system (243). Hexameric arrangement of Ab Fc regions in the Ab-antigen complexes is necessary for optimum CDC action (244). In preclinical investigations, techniques that improve CDC, such as Ab hexamerization and Fc alterations, have showed potential for improving anticancer efficacy. For instance, hexamerization was employed to create a biparatopic anti-CD37 B-cell specific Ab with increased in vitro CDC efficacy (216, 245). Utilizing BsAbs which antagonize the C-mediators CD55 and CD59 to promote C-regulated activities, Macor et al. devised an innovative method to improve CDC (246). Although trastuzumab, pertuzumab, and tras+pert exert anticancer action via a variety of MOAs, they are unable to induce CDC in HER2-exhibiting cells when human serum is present (247–249). According to Weisser et al.’s hypothesis, a biparatopic Ab designed to increase receptor crosslinking and gathering might offer the receptor-Ab complex needed to connect C1q and activate the effective anti-cancer mechanism, as well as preserve and/or improve all other identifiable anti-cancer MOA assigned to authorized anti-HER2 Ab therapies (216).
5.3.2 Biparatopic BsAbs
BsAbs might be constructed to concurrently attach to two non-overlapping epitopes on the same target rather than two distinct antigens. Biparatopic targeting mimics the actions of antibody cocktails and polyclonal antibodies by enhancing binding efficiency via antigen crosslinking and accumulation (28). Earlier research has demonstrated that it is crucial for biparatopic BsAb to structurally identify two epitopes or to have a strong specificity for the antigen (250). Therefore, the membrane protein, which has extensive extracellular domains and has been the subject of a few mAbs, is a viable target for BsAbs (251). ZW25 is a BsAb that adheres to two HER2 epitopes concurrently, one is the pertuzumab (Perjeta; Genentech) binding site ECD2, and one is the trastuzumab binding site ECD4. According to preclinical studies, ZW25 are able to mute HER2 signaling more efficiently than trastuzumab or pertuzumab and exhibit high anticancer efficacy at a variety of HER2 expression rates. Currently, the drug is being evaluated in a phase I basket test for HER2-positive tumors (11).
5.3.3 BsAb targeting redundant ligand
Another area of concern for BsAbs is tackling redundancy for numerous growth or angiogenesis factors in the tumor microenvironment (TME). By reducing angiogenin-2 (Ang-2) and vascular endothelial growth factor-A (VEGF-A) in the TME, the CrossMab construction vanucizumab prevents angiogenesis, while VEGF and delta-like ligand 4 are the objectives of the BsAb OMP-305B83. In these architectures, both BsAbs carry Fc as a lengthy half-life is essential for efficient factor elimination (40). Moreover, ZVEGFR2 Bp2, which has anti-angiogenic impacts equivalent to the clinically authorized Ramucirumab, was regarded as the best candidate for further assessment because of the slow dissociation level and the benefits of a shorter linker in a study to test the potential of four biparatopic constructs (252).
6 Challenges and prospects in solid tumor
The application of BsAbs in cancer has also encountered numerous bottlenecks, particularly in solid tumors, which are mainly attributed to the more complex microenvironment of solid tumors. In this section, we will discuss the challenges of identifying suitable targets within solid tumors, penetrating the tumor, as well as the difficulties related to tolerance, side effects, and large-scale production of BsAb therapy.
6.1 Identifying the promising targets in solid tumor
For solid malignancies, the first step to effective immunotherapy is identifying the right targets, which are probably TSAs uniformly expressed on cancer cells and are crucial in the growth of tumors (253). Although both immune cell-redirecting and antigen-bridging BsAbs are being investigated, over three-quarters of these investigations focus on the second one. Bridged antigen pairings can be single targets like HER2/HER2 (biparatopic BsAb) or associations of two separate antigens like PD-1/CTLA4, PD-1/PD-L1, VEGF/Ang-2, IGF-1/IGF-2, etc. These BsAbs primarily target double signaling transductions to produce reinforced inhibited or activated impacts in lymphocytes or tumor cells. Immune cell-redirecting, CD3-targeted/TAA-targeted BsAbs have used TAAs such as EGFR, HER2, and c-Met, whereas TSAs like EGFRvIII (254), p95HER2 (255), and RAS G12V are more desirable targets to prevent on-target, off-tumor impacts caused by target expression on healthy cells. In fact, hemopoietic differentiation antigens like BCMA, CD123, and CD20 satisfy these requirements, while there are few TSAs accessible for solid tumors and, if exists, antigen heterogeneity would be an issue (256).
6.2 Penetration of the immune cells
Only a modest percentage (less than 1%) of migrated T cells were ultimately able to penetrate solid neoplasm tissues, according to preclinical research (257). Therefore, in order to maximize the efficacy of immunotherapies in cancers, T cell penetration must be improved (258). Macromolecules like BiTEs force out via transvascular holes with diameters ranging from 200 nm to 1.2 μm inside the solid tumor vasculature and permeate into the neoplasm (259). TILs are present in a variety of solid malignancies, and their number is closely correlated with cancer reaction to ICIs and prognosis (260–262). In malignancies lacking TILs (263), it is depended on the BiTEs’ capacity to draw T cells from the blood. The TME presents a variety of difficulties for BiTE medication, as it does for other macromolecule treatments for solid malignancies. A solid tumor’s physical hurdle prevents trafficking and penetration, greatly lowering the quantity of both BiTEs and T cells involved (264). An alluring remedy is the gene editing of an oncolytic virus to generate a BsAb following its invasion of a tumor cell. After selectively infecting tumor cells, the virus releases its offspring into the surroundings. In the vicinity of the tumour, the virus releases BiTEs that trigger a T cell activation during the viral cycle. A study reveals the viability and efficiency of using an erythropoietinproducing human hepatocellular carcinoma A2 receptor (EpHA2)-targeted BiTE in combination with oncolytic adenoviruses and adenovirus-delivered gene treatment; this design can provide a robust tumor-specific cytotoxic response that brings long-term management of pediatric high-grade gliomas (265). Adenoviruses and other oncolytic viruses can also destroy cells immediately via oncolysis, which might cause the liberation of neoantigens from the ruptured cells. The APCs’ further exposure of them may serve as an in situ vaccine, boosting the precise immune reaction (266). Comparable methods are reported by Gardell et al., who administered a retrovirally edited macrophage that could release a BiTE selective for EGFRvIII to mouse models of glioma. The macrophages would stay in the solid malignancy and continue to secrete IL-12 and BiTE, which would strengthen the T cell activity (267, 268). Given that the effectiveness of BsAbs in the therapy of solid tumors appears to be constrained by low tumor infiltration and on-target, off-tumor activity ending in dose limiting toxicity (DLTs), BiTE-armed oncolytic viruses represent an attractive new strategy for enhanced performance (187, 269–272).
6.3 Resistance to BsAb therapy
Another concern is the emergence of resistance to BsAb treatment. One typical tactic of resistance is the downregulation or removal of BsAb-specific TAAs on carcinoma cells, which results in cancer immune evasion. ICI resistance is linked to cancer-intrinsic deficiencies in antigen presentation, such as lack of β2 microglobulin needed for MHCI protein production on the cell membrane. Immune checkpoint overexpression, a key component of the immunosuppressive TME, could help with BsAb resistance. Although ICIs have transformed lung carcinoma therapy and are now the standard of care for patients with advanced solid cancers, but the general reaction is still poor, which reveals significant unsatisfied therapeutic needs (37, 273–275). A monovalent IgG1-based BsAb named MEDI5752 was developed to specifically inhibit the PD-1 cascade and CTLA-4 on PD-1+ stimulated T cells. Notably, in contrast to the individual PD-1-targeted or CTLA-4-targeted mAbs, MEDI5752 was discovered to promote swift internalization and breakdown of PD-1 and to concentrate selectively in malignancies, generating stronger anti-cancer efficacy (276). OX40/CTLA-4-specific BsAbs are in the preliminary stages of research. Agonist mAbs have been developed to stimulate the immunological costimulatory protein OX40. A number of BsAbs that can selectively attach to OX40 and CTLA-4 to target Tregs in the TME have been invented. These BsAbs are reported to have anticancer impact on bladder, pancreatic, and colon cancers and to work in conjunction with anti-PD-1 antibodies to control colorectal tumor (277, 278).
6.4 Side toxicity of BsAbs
The two major issues associated with the cytotoxicity of BsAbs are CRS and neurotoxicity. Exhaustion, headache, and fever are common signs of CRS, as well as more serious symptoms like hypotension, tachycardia, acute respiratory distress syndrome, and circulatory failure. Sequential dosage, premedication with dexamethasone, and SC dosing might reduce the frequency and intensity of CRS (37, 68, 163). The symptoms of neurotoxicity comprise headache, intention tremor, speech difficulties, cognitive problems, and seizures (187). Step dosage and prophylactic corticosteroid usage are among the methods for managing neurotoxicity (37, 279), but there are also ideas to utilize anti-adhesive medicines attempting to avoid neurotoxicities. According to Klinger et al., T-cell adherence to endothelium is a required but inadequate precursor to the emergence of blinatumomab-related neurological negative effects, and inhibiting adherence is a potential mitigating strategy (279).
6.5 Large-scale manufacturing of BsAbs
For a number of factors, producing BsAbs is difficult. For instance, challenges with protein production, stability, or the adoption of atypical production techniques may lead to low yields or unsuitable facility design (110). Since the essential methods for regulated Fab-arm interchange are consistent with typical procedures for commercial synthesis of conventional human IgG1, this technology seems to be well suited for large-scale manufacture. The adaptability was validated by the large-scale application of the bench-scale technique, which did not require extensive tuning or result in a decline of output quality (33). CHO cells were used to produce two original antibodies, each of which had a single matching point alteration in the CH3 domain. These antibodies were then produced at a volume of 1000 L utilizing a system fed-batch and purifying procedure created for conventional antibody manufacturing. By combining the two mother molecules in carefully monitored reduction circumstances, the BsAb was produced, featuring an effective Fab-arm interchange of >95% at kilogram scale. By using diafiltration to eliminate the reductant, the disulfide between chains spontaneously reoxidized. In addition to the molecule’s bispecificity, in-depth evaluation showed that the IgG1 structural coherence was preserved, involving functionality and durability (110).
7 Conclusion and future perspectives
Recent advances in cancer immunotherapy have highlighted the enormous potential of BsAbs as innovative targeted therapy tools. Over the past two decades, the development of BsAbs has accelerated significantly, benefiting from groundbreaking innovations in antibody formatting. Notably, strategies such as modifying the Fc region have proven effective in mitigating adverse effects (68, 280), while antibody tandem techniques and dimerization enhanced the stability of BsAbs without involving the Fc region (173, 281, 282). However, the paradox between stability and adverse effects associated with the presence or absence of the Fc fragment remains a complex issue requiring further exploration through deliberate design strategies (173). Emerging gene therapies such as genetically engineered cells or nucleic acid drugs hold promise for improving the efficacy and safety paradox.
The landscape of BsAb therapies continues to expand with some preclinical and clinical studies suggesting macrophages and neutrophils as potential therapeutic targets (283). These innate immune cells possess unique cytotoxic properties that can be leveraged to eliminate cancer cells (284). Notably, tumor-associated neutrophils (TANs), especially tumor-associated macrophages (TAMs), are already abundantly present in the TME, precluding the need for external recruitment (285–288). By stimulating these immune effectors in situ, BsAb therapies offer the potential for highly efficient tumor cell clearance (289). The strategy of combining BsAbs with other antitumor approaches such as radiotherapy, chemotherapy, mAb, Small-molecule inhibitors or other BsAbs, can overcome the limitations of monotherapy. ATIM-05 (anti-CD3 and anti-EGFR) used prior to radiation/temozolomide, can overcome the highly immunosuppressive glioblastoma (GBM) microenvironment (290). A clinical study (NCT02892123) has shown that ZW25 is well tolerated in combination with chemotherapeutic agents (paclitaxel, capecitabine, or vinorelbine). This approach has excellent and long-lasting antitumor activity in patients with HER2+ BC (291). The clinical study (NCT04626635) has demonstrated that RENG7075 used in tandem with cemiplimab boosts the anti-tumour effectiveness of cemiplimab immunotherapy by targeting CD28 with BsAbs (292). Combining BsAb treatment with immune checkpoint blockade provides another avenue for enhanced anti-tumor responses (293). Such combinations take advantage of BsAbs preferential targeting of specific cell types like CD47 and PD-L1 expressing cells to ensure selective cancer cell elimination while protecting normal cells (294, 295). V-aCD3Mu in combination with ICIs can eliminate solid tumors in different cancer models such as B16-F10, 4T1 and CT26 Models (296). The combination of two BsAbs, such as AK117 and AK112 was used in the humanized PD-1 mice models, showing good anti-tumour effects (297). These combined approaches present a promising therapeutic option that provides a wider outlook for BsAbs.
Despite significant progress, BsAb development in oncology remains unfinished. While several BsAbs have gained marketing approval, the rapid emergence of trispecific and multispecific antibodies highlights their dynamic potential (298–301). Robust validation through larger phase II-III clinical trials across cancer types and individualized treatment regimens will be key to fully harnessing the advantages of BsAbs. However, in the realm of solid tumor treatment, BsAbs still face many challenges including but not limited to off-target effects, immune cell penetration, toxicity, resistance mechanisms, manufacturing complexity, and cost (188, 302, 303).
In summary, BsAbs represent a promising direction for designing and developing novel anti-cancer drugs, and their continued R&D will be an integral part of biotherapeutic oncology. Meanwhile, optimizing BsAb pharmacokinetics, manufacturability, deeply understanding factors impacting their penetration and activity in the TME, developing rational combination therapies to improve response rates, and evaluating their engagement with other immune cells like NK cells and CAR-T cells, while elucidating resistance mechanisms, will be critical to advancing BsAbs clinical translation and realizing their anti-tumor potential.
Author contributions
GS: Conceptualization, Writing – original draft, Writing – review & editing. XG: Writing – original draft, Writing – review & editing. YW: Writing – original draft, Writing – review & editing. YX: Writing – original draft. NX: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from theNational Key R&D Program of China (2020YFA0509400), Guangdong Basic and Applied Basic Research Foundation (2019B030302012), National Natural Science Foundation of China (81821002, 82130082, 82341004), and 1·3·5 project for disciplines of excellence, West China Hospital, Sichuan University (ZYGD22007), Sichuan Science and Technology Program (2021YFH0002, 2023NSFSC1878).
Acknowledgments
BioRender was used to create the figures.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
References
1. Liu Z, Shi M, Ren Y, Xu H, Weng S, Ning W, et al. Recent advances and applications of CRISPR-Cas9 in cancer immunotherapy. Mol Cancer (2023) 22:35. doi: 10.1186/s12943-023-01738-6
2. Kaplon H, Crescioli S, Chenoweth A, Visweswaraiah J, Reichert JM. Antibodies to watch in 2023. MAbs (2023) 15:2153410. doi: 10.1080/19420862.2022.2153410
3. Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol (2005) 23:1011–27. doi: 10.1200/JCO.2005.06.081
4. Barron CC, Stefanova I, Cha Y, Elsolh K, Zereshkian A, Gaafour N, et al. Chronic immune-related adverse events in patients with cancer receiving immune checkpoint inhibitors: a systematic review. J Immunother Cancer (2023) 11:e006500. doi: 10.1136/jitc-2022-006500
5. Henricks LM, Schellens JH, Huitema AD, Beijnen JH. The use of combinations of monoclonal antibodies in clinical oncology. Cancer Treat Rev (2015) 41:859–67. doi: 10.1016/j.ctrv.2015.10.008
6. Zhu Y, Choi SH, Shah K. Multifunctional receptor-targeting antibodies for cancer therapy. Lancet Oncol (2015) 16:e543–54. doi: 10.1016/S1470-2045(15)00039-X
7. Nisonoff A, Wissler FC, Lipman LN. Properties of the major component of a peptic digest of rabbit antibody. Science (1960) 132:1770–1. doi: 10.1126/science.132.3441.1770
8. Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature (1975) 256:495–7. doi: 10.1038/256495a0
9. Seimetz D, Lindhofer H, Bokemeyer C. Development and approval of the trifunctional antibody catumaxomab (anti-EpCAM x anti-CD3) as a targeted cancer immunotherapy. Cancer Treat Rev (2010) 36:458–67. doi: 10.1016/j.ctrv.2010.03.001
10. Frampton JE. Catumaxomab: in Malignant ascites. Drugs (2012) 72:1399–410. doi: 10.2165/11209040-000000000-00000
11. Meric-Bernstam F, Beeram M, Blum MA, Hausman DF, Infante JR, Patnaik A, et al. Phase 1 dose escalation of ZW25, a HER2-targeted bispecific antibody, in patients (pts) with HER2-expressing cancers. J Clin Oncol (2017) 35:1035. doi: 10.1200/JCO.2017.35.15_suppl.1035
12. Fudenberg HH, Drews G, Nisonoff A. Serologic demonstration of dual specificity of rabbit bivalent hybrid antibody. J Exp Med (1964) 119:151–66. doi: 10.1084/jem.119.1.151
13. Milstein C, Cuello AC. Hybrid hybridomas and their use in immunohistochemistry. Nature (1983) 305:537–40. doi: 10.1038/305537a0
14. Staerz UD, Kanagawa O, Bevan MJ. Hybrid antibodies can target sites for attack by T cells. Nature (1985) 314:628–31. doi: 10.1038/314628a0
15. Holliger P, Prospero T, Winter G. “Diabodies”: small bivalent and bispecific antibody fragments. Proc Natl Acad Sci U S A (1993) 90:6444–8. doi: 10.1073/pnas.90.14.6444
16. Durruthy-Durruthy R, Sperry ED, Bowen ME, Attardi LD, Heller S, Martin DM. Single cell transcriptomics reveal abnormalities in neurosensory patterning of the chd7 mutant mouse ear. Front Genet (2018) 9:473. doi: 10.3389/fgene.2018.00473
17. Ridgway JB, Presta LG, Carter P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. (1996) 9:617–21. doi: 10.1093/protein/9.7.617
18. Lindhofer H, Mocikat R, Steipe B, Thierfelder S. Preferential species-restricted heavy/light chain pairing in rat/mouse quadromas. Implications for a single-step purification of bispecific antibodies. J Immunol (1995) 155:219–25. doi: 10.4049/jimmunol.155.1.219
19. Coloma MJ, Morrison SL. Design and production of novel tetravalent bispecific antibodies. Nat Biotechnol (1997) 15:159–63. doi: 10.1038/nbt0297-159
20. Schuurman J, Van Ree R, Perdok GJ, Van Doorn HR, Tan KY, Aalberse RC. Normal human immunoglobulin G4 is bispecific: it has two different antigen-combining sites. Immunology (1999) 97:693–8. doi: 10.1046/j.1365-2567.1999.00845.x
21. van der Neut Kolfschoten M, Schuurman J, Losen M, Bleeker WK, Martinez-Martinez P, Vermeulen E, et al. Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science (2007) 317:1554–7. doi: 10.1126/science.1144603
22. Wu C, Ying H, Bose S, Miller R, Medina L, Santora L, et al. Molecular construction and optimization of anti-human IL-1alpha/beta dual variable domain immunoglobulin (DVD-Ig) molecules. MAbs (2009) 1:339–47. doi: 10.4161/mabs.1.4.8755
23. Schaefer W, Regula JT, Bahner M, Schanzer J, Croasdale R, Durr H, et al. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc Natl Acad Sci U S A. (2011) 108:11187–92. doi: 10.1073/pnas.1019002108
24. Lewis SM, Wu X, Pustilnik A, Sereno A, Huang F, Rick HL, et al. Generation of bispecific IgG antibodies by structure-based design of an orthogonal Fab interface. Nat Biotechnol (2014) 32:191–8. doi: 10.1038/nbt.2797
28. Labrijn AF, Janmaat ML, Reichert JM, Parren P. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discovery (2019) 18:585–608. doi: 10.1038/s41573-019-0028-1
29. Seckinger A, Delgado JA, Moser S, Moreno L, Neuber B, Grab A, et al. Target expression, generation, preclinical activity, and pharmacokinetics of the BCMA-T cell bispecific antibody EM801 for multiple myeloma treatment. Cancer Cell (2017) 31:396–410. doi: 10.1016/j.ccell.2017.02.002
30. Brinkmann U, Kontermann RE. The making of bispecific antibodies. MAbs (2017) 9:182–212. doi: 10.1080/19420862.2016.1268307
31. Chen W, Yuan Y, Jiang X. Antibody and antibody fragments for cancer immunotherapy. J Control Release. (2020) 328:395–406. doi: 10.1016/j.jconrel.2020.08.021
32. Wang L, Hoseini SS, Xu H, Ponomarev V, Cheung NK. Silencing fc domains in T cell-engaging bispecific antibodies improves T-cell trafficking and antitumor potency. Cancer Immunol Res (2019) 7:2013–24. doi: 10.1158/2326-6066.CIR-19-0121
33. Labrijn AF, Meesters JI, de Goeij BE, van den Bremer ET, Neijssen J, van Kampen MD, et al. Efficient generation of stable bispecific IgG1 by controlled Fab-arm exchange. Proc Natl Acad Sci U S A. (2013) 110:5145–50. doi: 10.1073/pnas.1220145110
34. van der Horst HJ, de Jonge AV, Hiemstra IH, Gelderloos AT, Berry D, Hijmering NJ, et al. Epcoritamab induces potent anti-tumor activity against Malignant B-cells from patients with DLBCL, FL and MCL, irrespective of prior CD20 monoclonal antibody treatment. Blood Cancer J (2021) 11:38. doi: 10.1038/s41408-021-00430-6
35. Gong S, Ren F, Wu D, Wu X, Wu C. Fabs-in-tandem immunoglobulin is a novel and versatile bispecific design for engaging multiple therapeutic targets. MAbs (2017) 9:1118–28. doi: 10.1080/19420862.2017.1345401
36. Brunker P, Wartha K, Friess T, Grau-Richards S, Waldhauer I, Koller CF, et al. RG7386, a novel tetravalent FAP-DR5 antibody, effectively triggers FAP-dependent, avidity-driven DR5 hyperclustering and tumor cell apoptosis. Mol Cancer Ther (2016) 15:946–57. doi: 10.1158/1535-7163.MCT-15-0647
37. Lim SM, Pyo KH, Soo RA, Cho BC. The promise of bispecific antibodies: Clinical applications and challenges. Cancer Treat Rev (2021) 99:102240. doi: 10.1016/j.ctrv.2021.102240
38. Yun J, Lee SH, Kim SY, Jeong SY, Kim JH, Pyo KH, et al. Antitumor activity of amivantamab (JNJ-61186372), an EGFR-MET bispecific antibody, in diverse models of EGFR exon 20 insertion-driven NSCLC. Cancer Discovery (2020) 10:1194–209. doi: 10.1158/2159-8290.CD-20-0116
39. Park K, Haura EB, Leighl NB, Mitchell P, Shu CA, Girard N, et al. Amivantamab in EGFR exon 20 insertion-mutated non-small-cell lung cancer progressing on platinum chemotherapy: initial results from the CHRYSALIS phase I study. J Clin Oncol (2021) 39:3391–402. doi: 10.1200/JCO.21.00662
40. Suurs FV, Lub-de Hooge MN, de Vries EGE, de Groot DJA. A review of bispecific antibodies and antibody constructs in oncology and clinical challenges. Pharmacol Ther (2019) 201:103–19. doi: 10.1016/j.pharmthera.2019.04.006
41. Rau A, Kocher K, Rommel M, Kuhl L, Albrecht M, Gotthard H, et al. A bivalent, bispecific Dab-Fc antibody molecule for dual targeting of HER2 and HER3. MAbs (2021) 13:1902034. doi: 10.1080/19420862.2021.1902034
42. Elter A, Yanakieva D, Fiebig D, Hallstein K, Becker S, Betz U, et al. Protease-activation of fc-masked therapeutic antibodies to alleviate off-tumor cytotoxicity. Front Immunol (2021) 12:715719. doi: 10.3389/fimmu.2021.715719
43. Labrijn AF, Meesters JI, Bunce M, Armstrong AA, Somani S, Nesspor TC, et al. Efficient generation of bispecific murine antibodies for pre-clinical investigations in syngeneic rodent models. Sci Rep (2017) 7:2476. doi: 10.1038/s41598-017-02823-9
44. Birch JR, Racher AJ. Antibody production. Adv Drug Delivery Rev (2006) 58:671–85. doi: 10.1016/j.addr.2005.12.006
45. Lou H, Cao X. Antibody variable region engineering for improving cancer immunotherapy. Cancer Commun (Lond). (2022) 42:804–27. doi: 10.1002/cac2.12330
46. Wu X, Sereno AJ, Huang F, Lewis SM, Lieu RL, Weldon C, et al. Fab-based bispecific antibody formats with robust biophysical properties and biological activity. MAbs (2015) 7:470–82. doi: 10.1080/19420862.2015.1022694
47. Yu L, Huang N, Ge L, Sun H, Fu Y, Liu C, et al. Structural design of tetravalent T-cell engaging bispecific antibodies: improve developability by engineering disulfide bonds. J Biol Eng. (2021) 15:18. doi: 10.1186/s13036-021-00272-7
48. Cui X, Jia H, Xin H, Zhang L, Chen S, Xia S, et al. A novel bispecific antibody targeting PD-L1 and VEGF with combined anti-tumor activities. Front Immunol (2021) 12:778978. doi: 10.3389/fimmu.2021.778978
49. Lu D, Jimenez X, Witte L, Zhu Z. The effect of variable domain orientation and arrangement on the antigen-binding activity of a recombinant human bispecific diabody. Biochem Biophys Res Commun (2004) 318:507–13. doi: 10.1016/j.bbrc.2004.04.060
50. Jang S, Song J, Kim N, Bak J, Jung K, Park YW, et al. Development of an antibody-like T-cell engager based on VH-VL heterodimer formation and its application in cancer therapy. Biomaterials (2021) 271:120760. doi: 10.1016/j.biomaterials.2021.120760
51. Seifert O, Rau A, Beha N, Richter F, Kontermann RE. Diabody-Ig: a novel platform for the generation of multivalent and multispecific antibody molecules. MAbs (2019) 11:919–29. doi: 10.1080/19420862.2019.1603024
52. Hsiue EH, Wright KM, Douglass J, Hwang MS, Mog BJ, Pearlman AH, et al. Targeting a neoantigen derived from a common TP53 mutation. Science (2021) 371:eabc8697. doi: 10.1126/science.abc8697
53. Schoonjans R, Willems A, Schoonooghe S, Fiers W, Grooten J, Mertens N. Fab chains as an efficient heterodimerization scaffold for the production of recombinant bispecific and trispecific antibody derivatives. J Immunol (2000) 165:7050–7. doi: 10.4049/jimmunol.165.12.7050
54. Lu D, Jimenez X, Zhang H, Bohlen P, Witte L, Zhu Z. Fab-scFv fusion protein: an efficient approach to production of bispecific antibody fragments. J Immunol Methods (2002) 267:213–26. doi: 10.1016/s0022-1759(02)00148-5
55. Qu Z, Goldenberg DM, Cardillo TM, Shi V, Hansen HJ, Chang CH. Bispecific anti-CD20/22 antibodies inhibit B-cell lymphoma proliferation by a unique mechanism of action. Blood (2008) 111:2211–9. doi: 10.1182/blood-2007-08-110072
56. Kellner C, Bruenke J, Horner H, Schubert J, Schwenkert M, Mentz K, et al. Heterodimeric bispecific antibody-derivatives against CD19 and CD16 induce effective antibody-dependent cellular cytotoxicity against B-lymphoid tumor cells. Cancer Lett (2011) 303:128–39. doi: 10.1016/j.canlet.2011.01.020
57. Wang S, Peng L, Xu W, Zhou Y, Zhu Z, Kong Y, et al. Preclinical characterization and comparison between CD3/CD19 bispecific and novel CD3/CD19/CD20 trispecific antibodies against B-cell acute lymphoblastic leukemia: targeted immunotherapy for acute lymphoblastic leukemia. Front Med (2022) 16:139–49. doi: 10.1007/s11684-021-0835-8
58. Santich BH, Park JA, Tran H, Guo HF, Huse M, Cheung NV. Interdomain spacing and spatial configuration drive the potency of IgG-[L]-scFv T cell bispecific antibodies. Sci Transl Med (2020) 12:eaax1315. doi: 10.1126/scitranslmed.aax1315
59. Passariello M, Yoshioka A, Takahashi K, Hashimoto SI, Inoue T, Nakamura K, et al. Novel tri-specific tribodies induce strong T cell activation and anti-tumor effects in vitro and in vivo. J Exp Clin Cancer Res (2022) 41:269. doi: 10.1186/s13046-022-02474-3
60. Biesemann N, Margerie D, Asbrand C, Rehberg M, Savova V, Agueusop I, et al. Additive efficacy of a bispecific anti-TNF/IL-6 nanobody compound in translational models of rheumatoid arthritis. Sci Transl Med (2023) 15:eabq4419. doi: 10.1126/scitranslmed.abq4419
61. Arbabi-Ghahroudi M. Camelid single-domain antibodies: historical perspective and future outlook. Front Immunol (2017) 8:1589. doi: 10.3389/fimmu.2017.01589
62. Liu Y, Ao K, Bao F, Cheng Y, Hao Y, Zhang H, et al. Development of a bispecific nanobody targeting CD20 on B-cell lymphoma cells and CD3 on T cells. Vaccines (Basel) (2022) 10:1335. doi: 10.3390/vaccines10081335
63. Ackaert C, Smiejkowska N, Xavier C, Sterckx YGJ, Denies S, Stijlemans B, et al. Immunogenicity risk profile of nanobodies. Front Immunol (2021) 12:632687. doi: 10.3389/fimmu.2021.632687
64. Yu YJ, Atwal JK, Zhang Y, Tong RK, Wildsmith KR, Tan C, et al. Therapeutic bispecific antibodies cross the blood-brain barrier in nonhuman primates. Sci Transl Med (2014) 6:261ra154. doi: 10.1126/scitranslmed.3009835
65. Hong H, Lin H, Li D, Gong L, Zhou K, Li Y, et al. Chemoenzymatic synthesis of a rhamnose-functionalized bispecific nanobody as a bispecific antibody mimic for cancer immunotherapy. Angew Chem Int Ed Engl (2022) 61:e202208773. doi: 10.1002/anie.202208773
66. Bai Z, Wang J, Li J, Yuan H, Wang P, Zhang M, et al. Design of nanobody-based bispecific constructs by in silico affinity maturation and umbrella sampling simulations. Comput Struct Biotechnol J (2023) 21:601–13. doi: 10.1016/j.csbj.2022.12.021
67. Mazor Y, Oganesyan V, Yang C, Hansen A, Wang J, Liu H, et al. Improving target cell specificity using a novel monovalent bispecific IgG design. MAbs (2015) 7:377–89. doi: 10.1080/19420862.2015.1007816
68. Engelberts PJ, Hiemstra IH, de Jong B, Schuurhuis DH, Meesters J, Beltran Hernandez I, et al. DuoBody-CD3xCD20 induces potent T-cell-mediated killing of Malignant B cells in preclinical models and provides opportunities for subcutaneous dosing. EBioMedicine (2020) 52:102625. doi: 10.1016/j.ebiom.2019.102625
69. Huang L, Shah K, Barat B, Lam CK, Gorlatov S, Ciccarone V, et al. Multispecific, multivalent antibody-based molecules engineered on the DART(R) and TRIDENT(TM) platforms. Curr Protoc Immunol (2020) 129:e95. doi: 10.1002/cpim.95
70. Mayer K, Baumann AL, Grote M, Seeber S, Kettenberger H, Breuer S, et al. TriFabs–trivalent igG-shaped bispecific antibody derivatives: design, generation, characterization and application for targeted payload delivery. Int J Mol Sci (2015) 16:27497–507. doi: 10.3390/ijms161126037
71. Klein C, Schaefer W, Regula JT. The use of CrossMAb technology for the generation of bi- and multispecific antibodies. MAbs (2016) 8:1010–20. doi: 10.1080/19420862.2016.1197457
72. Bever CS, Dong JX, Vasylieva N, Barnych B, Cui Y, Xu ZL, et al. VHH antibodies: emerging reagents for the analysis of environmental chemicals. Anal Bioanal Chem (2016) 408:5985–6002. doi: 10.1007/s00216-016-9585-x
73. Haagen IA, van de Griend R, Clark M, Geerars A, Bast B, de Gast B. Killing of human leukaemia/lymphoma B cells by activated cytotoxic T lymphocytes in the presence of a bispecific monoclonal antibody (alpha CD3/alpha CD19). Clin Exp Immunol (1992) 90:368–75. doi: 10.1111/j.1365-2249.1992.tb05853.x
74. Liu P, Gao X, Lundin V, Shi C, Adem Y, Lin K, et al. Probing the impact of the knob-into-hole mutations on the structure and function of a therapeutic antibody. Anal Chem (2020) 92:1582–8. doi: 10.1021/acs.analchem.9b04855
75. Bogen JP, Carrara SC, Fiebig D, Grzeschik J, Hock B, Kolmar H. Expeditious generation of biparatopic common light chain antibodies via chicken immunization and yeast display screening. Front Immunol (2020) 11:606878. doi: 10.3389/fimmu.2020.606878
76. Teeling JL, French RR, Cragg MS, van den Brakel J, Pluyter M, Huang H, et al. Characterization of new human CD20 monoclonal antibodies with potent cytolytic activity against non-Hodgkin lymphomas. Blood (2004) 104:1793–800. doi: 10.1182/blood-2004-01-0039
77. Wec AZ, Nyakatura EK, Herbert AS, Howell KA, Holtsberg FW, Bakken RR, et al. A “Trojan horse” bispecific-antibody strategy for broad protection against ebolaviruses. Science (2016) 354:350–4. doi: 10.1126/science.aag3267
78. Mehta NK, Pfluegler M, Meetze K, Li B, Sindel I, Vogt F, et al. A novel IgG-based FLT3xCD3 bispecific antibody for the treatment of AML and B-ALL. J Immunother Cancer (2022) 10:e003882. doi: 10.1136/jitc-2021-003882
79. Beckmann R, Jensen K, Fenn S, Speck J, Krause K, Meier A, et al. DutaFabs are engineered therapeutic Fab fragments that can bind two targets simultaneously. Nat Commun (2021) 12:708. doi: 10.1038/s41467-021-20949-3
80. Chames P, Baty D. Bispecific antibodies for cancer therapy. Curr Opin Drug Discovery Devel. (2009) 12:276–83. doi: 10.4161/mabs.1.6.10015
81. Chan AC, Carter PJ. Therapeutic antibodies for autoimmunity and inflammation. Nat Rev Immunol (2010) 10:301–16. doi: 10.1038/nri2761
82. Szijj P, Chudasama V. The renaissance of chemically generated bispecific antibodies. Nat Rev Chem (2021) 5:78–92. doi: 10.1038/s41570-020-00241-6
83. Alejandra WP, Miriam Irene JP, Fabio Antonio GS, Patricia RR, Elizabeth TA, Aleman-Aguilar JP, et al. Production of monoclonal antibodies for therapeutic purposes: A review. Int Immunopharmacol. (2023) 120:110376. doi: 10.1016/j.intimp.2023.110376
84. Dimasi N, Kumar A, Gao C. Generation of bispecific antibodies using chemical conjugation methods. Drug Discovery Today Technol (2021) 40:13–24. doi: 10.1016/j.ddtec.2021.08.006
85. Kinch MS, Kraft Z, Schwartz T. Immunoconjugates and bispecific antibodies: Trends in therapeutic success and commercial focus. Drug Discovery Today (2023) 28:103462. doi: 10.1016/j.drudis.2022.103462
86. Patke S, Li J, Wang P, Slaga D, Johnston J, Bhakta S, et al. bisFabs: Tools for rapidly screening hybridoma IgGs for their activities as bispecific antibodies. MAbs (2017) 9:430–7. doi: 10.1080/19420862.2017.1281504
87. Chen M, Liu X, Peng N, Zhang T, Mou J, He H, et al. Construction of CD19 targeted dual- and enhanced dual-antibodies and their efficiency in the treatment of B cell Malignancy. Exp Hematol Oncol (2023) 12:64. doi: 10.1186/s40164-023-00423-0
88. Ma J, Mo Y, Tang M, Shen J, Qi Y, Zhao W, et al. Bispecific antibodies: from research to clinical application. Front Immunol (2021) 12:626616. doi: 10.3389/fimmu.2021.626616
89. Klein C, Schaefer W, Regula JT, Dumontet C, Brinkmann U, Bacac M, et al. Engineering therapeutic bispecific antibodies using CrossMab technology. Methods (2019) 154:21–31. doi: 10.1016/j.ymeth.2018.11.008
90. Mohammadi M, Jeddi-Tehrani M, Golsaz-Shirazi F, Arjmand M, Bahadori T, Judaki MA, et al. A novel anti-HER2 bispecific antibody with potent tumor inhibitory effects in vitro and in vivo. Front Immunol (2020) 11:600883. doi: 10.3389/fimmu.2020.600883
91. Ku Z, Xie X, Lin J, Gao P, Wu B, El Sahili A, et al. Engineering SARS-CoV-2 specific cocktail antibodies into a bispecific format improves neutralizing potency and breadth. Nat Commun (2022) 13:5552. doi: 10.1038/s41467-022-33284-y
92. Gong S, Wu C. Efficient production of bispecific antibodies-optimization of transfection strategy leads to high-level stable cell line generation of a Fabs-in-tandem immunoglobin. Antib Ther (2023) 6:170–9. doi: 10.1093/abt/tbad013
93. Konishi T, Ochi T, Maruta M, Tanimoto K, Miyazaki Y, Iwamoto C, et al. Reinforced anti-myeloma therapy via dual-lymphoid activation mediated by a panel of antibodies armed with Bridging-BiTE. Blood (2023), blood.2022019082. doi: 10.1182/blood.2022019082
94. Luke JJ, Patel MR, Blumenschein GR, Hamilton E, Chmielowski B, Ulahannan SV, et al. The PD-1- and LAG-3-targeting bispecific molecule tebotelimab in solid tumors and hematologic cancers: a phase 1 trial. Nat Med (2023), 1–11. doi: 10.1038/s41591-023-02593-0
95. Dengl S, Mayer K, Bormann F, Duerr H, Hoffmann E, Nussbaum B, et al. Format chain exchange (FORCE) for high-throughput generation of bispecific antibodies in combinatorial binder-format matrices. Nat Commun (2020) 11:4974. doi: 10.1038/s41467-020-18477-7
96. Atwell S, Ridgway JB, Wells JA, Carter P. Stable heterodimers from remodeling the domain interface of a homodimer using a phage display library. J Mol Biol (1997) 270:26–35. doi: 10.1006/jmbi.1997.1116
97. Fan G, Wang Z, Hao M, Li J. Bispecific antibodies and their applications. J Hematol Oncol (2015) 8:130. doi: 10.1186/s13045-015-0227-0
98. Spiess C, Merchant M, Huang A, Zheng Z, Yang NY, Peng J, et al. Bispecific antibodies with natural architecture produced by co-culture of bacteria expressing two distinct half-antibodies. Nat Biotechnol (2013) 31:753–8. doi: 10.1038/nbt.2621
99. Zhang HM, Li C, Lei M, Lundin V, Lee HY, Ninonuevo M, et al. Structural and functional characterization of a hole-hole homodimer variant in a “Knob-into-hole” Bispecific antibody. Anal Chem (2017) 89:13494–501. doi: 10.1021/acs.analchem.7b03830
100. Grunert I, Heinrich K, Ernst J, Hingar M, Briguet A, Leiss M, et al. Detailed analytical characterization of a bispecific igG1 crossMab antibody of the knob-into-hole format applying various stress conditions revealed pronounced stability. ACS Omega. (2022) 7:3671–9. doi: 10.1021/acsomega.1c06305
101. Elliott JM, Ultsch M, Lee J, Tong R, Takeda K, Spiess C, et al. Antiparallel conformation of knob and hole aglycosylated half-antibody homodimers is mediated by a CH2-CH3 hydrophobic interaction. J Mol Biol (2014) 426:1947–57. doi: 10.1016/j.jmb.2014.02.015
102. Macchi FD, Yang F, Li C, Wang C, Dang AN, Marhoul JC, et al. Absolute quantitation of intact recombinant antibody product variants using mass spectrometry. Anal Chem (2015) 87:10475–82. doi: 10.1021/acs.analchem.5b02627
103. Woods RJ, Xie MH, Von Kreudenstein TS, Ng GY, Dixit SB. LC-MS characterization and purity assessment of a prototype bispecific antibody. MAbs (2013) 5:711–22. doi: 10.4161/mabs.25488
104. Yin Y, Han G, Zhou J, Dillon M, McCarty L, Gavino L, et al. Precise quantification of mixtures of bispecific IgG produced in single host cells by liquid chromatography-Orbitrap high-resolution mass spectrometry. MAbs (2016) 8:1467–76. doi: 10.1080/19420862.2016.1232217
105. Surowka M, Schaefer W, Klein C. Ten years in the making: application of CrossMab technology for the development of therapeutic bispecific antibodies and antibody fusion proteins. MAbs (2021) 13:1967714. doi: 10.1080/19420862.2021.1967714
106. Fenn S, Schiller CB, Griese JJ, Duerr H, Imhof-Jung S, Gassner C, et al. Crystal structure of an anti-Ang2 CrossFab demonstrates complete structural and functional integrity of the variable domain. PloS One (2013) 8:e61953. doi: 10.1371/journal.pone.0061953
107. Hosseini SS, Khalili S, Baradaran B, Bidar N, Shahbazi MA, Mosafer J, et al. Bispecific monoclonal antibodies for targeted immunotherapy of solid tumors: Recent advances and clinical trials. Int J Biol Macromol. (2021) 167:1030–47. doi: 10.1016/j.ijbiomac.2020.11.058
108. Davis JH, Aperlo C, Li Y, Kurosawa E, Lan Y, Lo KM, et al. SEEDbodies: fusion proteins based on strand-exchange engineered domain (SEED) CH3 heterodimers in an Fc analogue platform for asymmetric binders or immunofusions and bispecific antibodies. Protein Eng Des Sel. (2010) 23:195–202. doi: 10.1093/protein/gzp094
109. Labrijn AF, Meesters JI, Priem P, de Jong RN, van den Bremer ET, van Kampen MD, et al. Controlled Fab-arm exchange for the generation of stable bispecific IgG1. Nat Protoc (2014) 9:2450–63. doi: 10.1038/nprot.2014.169
110. Gramer MJ, van den Bremer ET, van Kampen MD, Kundu A, Kopfmann P, Etter E, et al. Production of stable bispecific IgG1 by controlled Fab-arm exchange: scalability from bench to large-scale manufacturing by application of standard approaches. MAbs (2013) 5:962–73. doi: 10.4161/mabs.26233
111. Muik A, Adams HC 3rd, Gieseke F, Altintas I, Schoedel KB, Blum JM, et al. DuoBody-CD40x4-1BB induces dendritic-cell maturation and enhances T-cell activation through conditional CD40 and 4-1BB agonist activity. J Immunother Cancer (2022) 10:e004322. doi: 10.1136/jitc-2021-004322
112. Teeling JL, Mackus WJ, Wiegman LJ, van den Brakel JH, Beers SA, French RR, et al. The biological activity of human CD20 monoclonal antibodies is linked to unique epitopes on CD20. J Immunol (2006) 177:362–71. doi: 10.4049/jimmunol.177.1.362
113. Sanz L, Blanco B, Alvarez-Vallina L. Antibodies and gene therapy: teaching old ‘magic bullets’ new tricks. Trends Immunol (2004) 25:85–91. doi: 10.1016/j.it.2003.12.001
114. Blanco B, Dominguez-Alonso C, Alvarez-Vallina L. Bispecific immunomodulatory antibodies for cancer immunotherapy. Clin Cancer Res (2021) 27:5457–64. doi: 10.1158/1078-0432.CCR-20-3770
115. Bannas P, Hambach J, Koch-Nolte F. Nanobodies and nanobody-based human heavy chain antibodies as antitumor therapeutics. Front Immunol (2017) 8:1603. doi: 10.3389/fimmu.2017.01603
116. Schweizer D, Serno T, Goepferich A. Controlled release of therapeutic antibody formats. Eur J Pharm Biopharm. (2014) 88:291–309. doi: 10.1016/j.ejpb.2014.08.001
117. Sanchez-Martin D, Sanz L, Alvarez-Vallina L. Engineering human cells for in vivo secretion of antibody and non-antibody therapeutic proteins. Curr Opin Biotechnol (2011) 22:924–30. doi: 10.1016/j.copbio.2011.03.001
118. Velasquez MP, Bonifant CL, Gottschalk S. Redirecting T cells to hematological Malignancies with bispecific antibodies. Blood (2018) 131:30–8. doi: 10.1182/blood-2017-06-741058
119. Watts RJ, Dennis MS. Bispecific antibodies for delivery into the brain. Curr Opin Chem Biol (2013) 17:393–9. doi: 10.1016/j.cbpa.2013.03.023
120. Freskgard PO, Urich E. Antibody therapies in CNS diseases. Neuropharmacology (2017) 120:38–55. doi: 10.1016/j.neuropharm.2016.03.014
121. Han L, Jiang C. Evolution of blood-brain barrier in brain diseases and related systemic nanoscale brain-targeting drug delivery strategies. Acta Pharm Sin B (2021) 11:2306–25. doi: 10.1016/j.apsb.2020.11.023
122. Pathan N, Shende P. Tailoring of P-glycoprotein for effective transportation of actives across blood-brain-barrier. J Control Release. (2021) 335:398–407. doi: 10.1016/j.jconrel.2021.05.046
123. Arvedson T, Bailis JM, Urbig T, Stevens JL. Considerations for design, manufacture, and delivery for effective and safe T-cell engager therapies. Curr Opin Biotechnol (2022) 78:102799. doi: 10.1016/j.copbio.2022.102799
124. Wei PS, Chen YJ, Lin SY, Chuang KH, Sheu MT, Ho HO. In situ subcutaneously injectable thermosensitive PEG-PLGA diblock and PLGA-PEG-PLGA triblock copolymer composite as sustained delivery of bispecific anti-CD3 scFv T-cell/anti-EGFR Fab Engager (BiTEE). Biomaterials (2021) 278:121166. doi: 10.1016/j.biomaterials.2021.121166
125. Nolan-Stevaux O. Abstract DDT02-03: AMG 509: A novel, humanized, half-Life extended, bispecific STEAP1 × CD3 T cell recruiting XmAb® 2 + 1 antibody. Cancer Res (2020) 80:DDT02–03. doi: 10.1158/1538-7445.AM2020-DDT02-03
126. Leconet W, Liu H, Guo M, Le Lamer-Dechamps S, Molinier C, Kim S, et al. Anti-PSMA/CD3 bispecific antibody delivery and antitumor activity using a polymeric depot formulation. Mol Cancer Ther (2018) 17:1927–40. doi: 10.1158/1535-7163.MCT-17-1138
127. Xu G, Luo Y, Wang H, Wang Y, Liu B, Wei J. Therapeutic bispecific antibodies against intracellular tumor antigens. Cancer Lett (2022) 538:215699. doi: 10.1016/j.canlet.2022.215699
128. Wu L, Wang W, Tian J, Qi C, Cai Z, Yan W, et al. Engineered mRNA-expressed bispecific antibody prevent intestinal cancer via lipid nanoparticle delivery. Bioengineered (2021) 12:12383–93. doi: 10.1080/21655979.2021.2003666
129. de Sostoa J, Fajardo CA, Moreno R, Ramos MD, Farrera-Sal M, Alemany R. Targeting the tumor stroma with an oncolytic adenovirus secreting a fibroblast activation protein-targeted bispecific T-cell engager. J Immunother Cancer. (2019) 7:19. doi: 10.1186/s40425-019-0505-4
130. Ordonez-Reyes C, Garcia-Robledo JE, Chamorro DF, Mosquera A, Sussmann L, Ruiz-Patino A, et al. Bispecific antibodies in cancer immunotherapy: A novel response to an old question. Pharmaceutics (2022) 14:1243. doi: 10.3390/pharmaceutics14061243
131. Zhao J, Chen G, Pang X, Zhang P, Hou X, Chen P, et al. Calcium phosphate nanoneedle based gene delivery system for cancer genetic immunotherapy. Biomaterials (2020) 250:120072. doi: 10.1016/j.biomaterials.2020.120072
132. Speck T, Heidbuechel JPW, Veinalde R, Jaeger D, von Kalle C, Ball CR, et al. Targeted biTE expression by an oncolytic vector augments therapeutic efficacy against solid tumors. Clin Cancer Res (2018) 24:2128–37. doi: 10.1158/1078-0432.CCR-17-2651
133. de Miguel M, Umana P, Gomes de Morais AL, Moreno V, Calvo E. T-cell-engaging therapy for solid tumors. Clin Cancer Res (2021) 27:1595–603. doi: 10.1158/1078-0432.CCR-20-2448
134. Huang C, Duan X, Wang J, Tian Q, Ren Y, Chen K, et al. Lipid nanoparticle delivery system for mRNA encoding B7H3-redirected bispecific antibody displays potent antitumor effects on Malignant tumors. Adv Sci (Weinh). (2023) 10:e2205532. doi: 10.1002/advs.202205532
135. Blanco B, Compte M, Lykkemark S, Sanz L, Alvarez-Vallina L. T cell-redirecting strategies to ‘STAb’ Tumors: beyond CARs and bispecific antibodies. Trends Immunol (2019) 40:243–57. doi: 10.1016/j.it.2019.01.008
136. Qin Y, Song Y, Shen Z, Du X, Ji W, Hsu W, et al. Safety and efficacy of obinutuzumab in Chinese patients with B-cell lymphomas: a secondary analysis of the GERSHWIN trial. Cancer Commun (2018) 38:1–9. doi: 10.1186/s40880-018-0300-5
137. Gaudet F, Attar R, Elsayed Y, Packman K, Wu S-J, Jiao Q, et al. A novel C2 domain binding CD33xCD3 bispecific antibody with potent T-cell redirection activity against acute myeloid leukemia. Blood Advances. (2020) 4:906–19. doi: 10.1182/bloodadvances.2019001188
138. Topp MS, Duell J, Zugmaier G, Attal M, Moreau P, Langer C, et al. Anti–B-cell maturation antigen biTE molecule AMG 420 induces responses in multiple myeloma. J Clin Oncol (2020) 38:775–83. doi: 10.1200/jco.19.02657
139. Bacac M, Fauti T, Sam J, Colombetti S, Weinzierl T, Ouaret D, et al. A novel carcinoembryonic antigen T-cell bispecific antibody (CEA TCB) for the treatment of solid tumors. Clin Cancer Res (2016) 22:3286–97. doi: 10.1158/1078-0432.Ccr-15-1696
140. Harding JJ, Garrido-Laguna I, Chen X, Basu C, Dowlati A, Forgie A, et al. A phase 1 dose-escalation study of PF-06671008, a bispecific T-cell-engaging therapy targeting P-cadherin in patients with advanced solid tumors. Front Immunol (2022) 13:845417. doi: 10.3389/fimmu.2022.845417
141. Johnson M, Lakhani N, Girda E, Olszanski A, Fong L, Han H, et al. 735 A phase 1/2 study of REGN7075 (EGFRxCD28 costimulatory bispecific antibody) in combination with cemiplimab (anti–PD-1) in patients with advanced solid tumors: initial dose-escalation results. Journal for ImmunoTherapy of Cancer (2022) 10:A766–7. doi: 10.1136/jitc-2022-SITC2022.0735
142. Gaspar M, Pravin J, Rodrigues L, Uhlenbroich S, Everett KL, Wollerton F, et al. CD137/OX40 bispecific antibody induces potent antitumor activity that is dependent on target coengagement. Cancer Immunol Res (2020) 8:781–93. doi: 10.1158/2326-6066.Cir-19-0798
143. Lin W, Zhang Y, Yang Y, Lin B, Zhu M, Xu J, et al. Anti-PD-1/her2 bispecific antibody IBI315 enhances the treatment effect of her2-positive gastric cancer through gasdermin B-cleavage induced pyroptosis. Advanced Sci (2023) 10:2303908. doi: 10.1002/advs.202303908
144. Peper-Gabriel JK, Pavlidou M, Pattarini L, Morales-Kastresana A, Jaquin TJ, Gallou C, et al. The PD-L1/4-1BB bispecific antibody–anticalin fusion protein PRS-344/S095012 elicits strong T-cell stimulation in a tumor-localized manner. Clin Cancer Res (2022) 28:3387–99. doi: 10.1158/1078-0432.Ccr-21-2762
145. Lakins MA, Koers A, Giambalvo R, Munoz-Olaya J, Hughes R, Goodman E, et al. FS222, a CD137/PD-L1 tetravalent bispecific antibody, exhibits low toxicity and antitumor activity in colorectal cancer models. Clin Cancer Res (2020) 26:4154–67. doi: 10.1158/1078-0432.Ccr-19-2958
146. Hellmann MD, Bivi N, Calderon B, Shimizu T, Delafontaine B, Liu ZT, et al. Safety and immunogenicity of LY3415244, a bispecific antibody against TIM-3 and PD-L1, in patients with advanced solid tumors. Clin Cancer Res (2021) 27:2773–81. doi: 10.1158/1078-0432.Ccr-20-3716
147. Liu L, Zeng W, Chedid M, Zeng Y, Tschang S-H, Tian Y, et al. Abstract 873: A novel MET-EGFR bispecific antibody LY3164530 shows advantage over combining MET and EGFR antibodies in tumor inhibition and overcome resistance. Cancer Res (2016) 76:873–3. doi: 10.1158/1538-7445.Am2016-873
148. Schram AM, Odintsov I, Espinosa-Cotton M, Khodos I, Sisso WJ, Mattar MS, et al. Zenocutuzumab, a HER2xHER3 bispecific antibody, is effective therapy for tumors driven by NRG1 gene rearrangements. Cancer Discovery (2022) 12:1233–47. doi: 10.1158/2159-8290.CD-21-1119
149. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity (2013) 39:1–10. doi: 10.1016/j.immuni.2013.07.012
150. Sanchez-Paulete AR, Teijeira A, Cueto FJ, Garasa S, Perez-Gracia JL, Sanchez-Arraez A, et al. Antigen cross-presentation and T-cell cross-priming in cancer immunology and immunotherapy. Ann Oncol (2017) 28:xii44–55. doi: 10.1093/annonc/mdx237
151. Strickland M, Quek L, Psaila B. The immune landscape in BCR-ABL negative myeloproliferative neoplasms: inflammation, infections and opportunities for immunotherapy. Br J Haematol (2022) 196:1149–58. doi: 10.1111/bjh.17850
152. Akinboro O, Vallejo JJ, Nakajima EC, Ren Y, Mishra-Kalyani PS, Larkins EA, et al. Outcomes of anti–PD-(L)1 therapy with or without chemotherapy (chemo) for first-line (1L) treatment of advanced non–small cell lung cancer (NSCLC) with PD-L1 score ≥ 50%: FDA pooled analysis. Journal of Clinical Oncology (2022) 40:9000–9000. doi: 10.1200/JCO.2022.40.16_suppl.9000
153. Messaoudene M, Mourikis TP, Michels J, Fu Y, Bonvalet M, Lacroix-Trikki M, et al. T-cell bispecific antibodies in node-positive breast cancer: novel therapeutic avenue for MHC class I loss variants. Ann Oncol (2019) 30:934–44. doi: 10.1093/annonc/mdz112
154. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med (2018) 24:541–50. doi: 10.1038/s41591-018-0014-x
156. Glitza Oliva IC, Ferguson SD, Bassett R, Foster AP, John I, Hennegan TD, et al. Concurrent intrathecal and intravenous nivolumab in leptomeningeal disease: phase 1 trial interim results. Nat Med (2023) 29:898–905. doi: 10.1038/s41591-022-02170-x
157. Zhu A, Garcia JA, Faltas B, Grivas P, Barata P, Shoag JE. Immune checkpoint inhibitors and long-term survival of patients with metastatic urothelial cancer. JAMA Netw Open (2023) 6:e237444. doi: 10.1001/jamanetworkopen.2023.7444
158. Pai JA, Satpathy AT. High-throughput and single-cell T cell receptor sequencing technologies. Nat Methods (2021) 18:881–92. doi: 10.1038/s41592-021-01201-8
159. Kamakura D, Asano R, Yasunaga M. T cell bispecific antibodies: an antibody-based delivery system for inducing antitumor immunity. Pharm (Basel) (2021) 14:1172. doi: 10.3390/ph14111172
160. Holland CJ, Crean RM, Pentier JM, de Wet B, Lloyd A, Srikannathasan V, et al. Specificity of bispecific T cell receptors and antibodies targeting peptide-HLA. J Clin Invest. (2020) 130:2673–88. doi: 10.1172/JCI130562
161. Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol (2015) 93:290–6. doi: 10.1038/icb.2014.93
162. Tian Z, Liu M, Zhang Y, Wang X. Bispecific T cell engagers: an emerging therapy for management of hematologic Malignancies. J Hematol Oncol (2021) 14:75. doi: 10.1186/s13045-021-01084-4
163. Clynes RA, Desjarlais JR. Redirected T cell cytotoxicity in cancer therapy. Annu Rev Med (2019) 70:437–50. doi: 10.1146/annurev-med-062617-035821
164. Lopez-Albaitero A, Xu H, Guo H, Wang L, Wu Z, Tran H, et al. Overcoming resistance to HER2-targeted therapy with a novel HER2/CD3 bispecific antibody. Oncoimmunology (2017) 6:e1267891. doi: 10.1080/2162402X.2016.1267891
165. Yeung YA, Krishnamoorthy V, Dettling D, Sommer C, Poulsen K, Ni I, et al. An optimized full-length FLT3/CD3 bispecific antibody demonstrates potent anti-leukemia activity and reversible hematological toxicity. Mol Ther (2020) 28:889–900. doi: 10.1016/j.ymthe.2019.12.014
166. Tapia-Galisteo A, Alvarez-Vallina L, Sanz L. Bi- and trispecific immune cell engagers for immunotherapy of hematological Malignancies. J Hematol Oncol (2023) 16:83. doi: 10.1186/s13045-023-01482-w
167. Bluemel C, Hausmann S, Fluhr P, Sriskandarajah M, Stallcup WB, Baeuerle PA, et al. Epitope distance to the target cell membrane and antigen size determine the potency of T cell-mediated lysis by BiTE antibodies specific for a large melanoma surface antigen. Cancer Immunol Immunother. (2010) 59:1197–209. doi: 10.1007/s00262-010-0844-y
168. Staflin K, Zuch de Zafra CL, Schutt LK, Clark V, Zhong F, Hristopoulos M, et al. Target arm affinities determine preclinical efficacy and safety of anti-HER2/CD3 bispecific antibody. JCI Insight (2020) 5:e133757. doi: 10.1172/jci.insight.133757
169. Leong SR, Sukumaran S, Hristopoulos M, Totpal K, Stainton S, Lu E, et al. An anti-CD3/anti-CLL-1 bispecific antibody for the treatment of acute myeloid leukemia. Blood (2017) 129:609–18. doi: 10.1182/blood-2016-08-735365
170. Bortoletto N, Scotet E, Myamoto Y, D’Oro U, Lanzavecchia A. Optimizing anti-CD3 affinity for effective T cell targeting against tumor cells. Eur J Immunol (2002) 32:3102–7. doi: 10.1002/1521-4141(200211)32:11<3102::AID-IMMU3102>3.0.CO;2-C
171. List T, Neri D. Biodistribution studies with tumor-targeting bispecific antibodies reveal selective accumulation at the tumor site. MAbs (2012) 4:775–83. doi: 10.4161/mabs.22271
172. Mandikian D, Takahashi N, Lo AA, Li J, Eastham-Anderson J, Slaga D, et al. Relative target affinities of T-cell-dependent bispecific antibodies determine biodistribution in a solid tumor mouse model. Mol Cancer Ther (2018) 17:776–85. doi: 10.1158/1535-7163.MCT-17-0657
173. Ravi G, Costa LJ. Bispecific T-cell engagers for treatment of multiple myeloma. Am J Hematol (2023) 98 Suppl 2:S13–21. doi: 10.1002/ajh.26628
174. Chatenoud L. CD3-specific antibody-induced active tolerance: from bench to bedside. Nat Rev Immunol (2003) 3:123–32. doi: 10.1038/nri1000
175. Chatenoud L, Ferran C, Reuter A, Legendre C, Gevaert Y, Kreis H, et al. Systemic reaction to the anti-T-cell monoclonal antibody OKT3 in relation to serum levels of tumor necrosis factor and interferon-gamma [corrected]. N Engl J Med (1989) 320:1420–1. doi: 10.1056/NEJM198905253202117
176. Linke R, Klein A, Seimetz D. Catumaxomab: clinical development and future directions. MAbs (2010) 2:129–36. doi: 10.4161/mabs.2.2.11221
177. Borlak J, Langer F, Spanel R, Schondorfer G, Dittrich C. Immune-mediated liver injury of the cancer therapeutic antibody catumaxomab targeting EpCAM, CD3 and Fcgamma receptors. Oncotarget (2016) 7:28059–74. doi: 10.18632/oncotarget.8574
178. Sanford M. Blinatumomab: first global approval. Drugs (2015) 75:321–7. doi: 10.1007/s40265-015-0356-3
179. Loffler A, Kufer P, Lutterbuse R, Zettl F, Daniel PT, Schwenkenbecher JM, et al. A recombinant bispecific single-chain antibody, CD19 x CD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes. Blood (2000) 95:2098–103. doi: 10.1182/blood.V95.6.2098
180. Klinger M, Brandl C, Zugmaier G, Hijazi Y, Bargou RC, Topp MS, et al. Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell-engaging CD19/CD3-bispecific BiTE antibody blinatumomab. Blood (2012) 119:6226–33. doi: 10.1182/blood-2012-01-400515
181. Zhao J, Song Y, Liu D. Recent advances on blinatumomab for acute lymphoblastic leukemia. Exp Hematol Oncol (2019) 8:28. doi: 10.1186/s40164-019-0152-y
182. Nagorsen D, Kufer P, Baeuerle PA, Bargou R. Blinatumomab: a historical perspective. Pharmacol Ther (2012) 136:334–42. doi: 10.1016/j.pharmthera.2012.07.013
183. Geuijen CAW, De Nardis C, Maussang D, Rovers E, Gallenne T, Hendriks LJA, et al. Unbiased combinatorial screening identifies a bispecific igG1 that potently inhibits HER3 signaling via HER2-guided ligand blockade. Cancer Cell (2018) 33:922–936 e10. doi: 10.1016/j.ccell.2018.04.003
184. Nagasaka M, Ou SI. NRG1 and NRG2 fusion positive solid tumor Malignancies: a paradigm of ligand-fusion oncogenesis. Trends Cancer. (2022) 8:242–58. doi: 10.1016/j.trecan.2021.11.003
185. Meermeier EW, Welsh SJ, Sharik ME, Du MT, Garbitt VM, Riggs DL, et al. Tumor burden limits bispecific antibody efficacy through T cell exhaustion averted by concurrent cytotoxic therapy. Blood Cancer Discovery (2021) 2:354–69. doi: 10.1158/2643-3230.BCD-21-0038
186. Teachey DT, Rheingold SR, Maude SL, Zugmaier G, Barrett DM, Seif AE, et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood (2013) 121:5154–7. doi: 10.1182/blood-2013-02-485623
187. Goebeler ME, Bargou RC. T cell-engaging therapies - BiTEs and beyond. Nat Rev Clin Oncol (2020) 17:418–34. doi: 10.1038/s41571-020-0347-5
188. Espinosa-Cotton M, Cheung NV. Bispecific antibodies for the treatment of neuroblastoma. Pharmacol Ther (2022) 237:108241. doi: 10.1016/j.pharmthera.2022.108241
189. Hernandez-Hoyos G, Sewell T, Bader R, Bannink J, Chenault RA, Daugherty M, et al. MOR209/ES414, a novel bispecific antibody targeting PSMA for the treatment of metastatic castration-resistant prostate cancer. Mol Cancer Ther (2016) 15:2155–65. doi: 10.1158/1535-7163.MCT-15-0242
190. Duell J, Dittrich M, Bedke T, Mueller T, Eisele F, Rosenwald A, et al. Frequency of regulatory T cells determines the outcome of the T-cell-engaging antibody blinatumomab in patients with B-precursor ALL. Leukemia (2017) 31:2181–90. doi: 10.1038/leu.2017.41
191. Michalk I, Feldmann A, Koristka S, Arndt C, Cartellieri M, Ehninger A, et al. Characterization of a novel single-chain bispecific antibody for retargeting of T cells to tumor cells via the TCR co-receptor CD8. PloS One (2014) 9:e95517. doi: 10.1371/journal.pone.0095517
192. de Weerdt I, Lameris R, Ruben JM, de Boer R, Kloosterman J, King LA, et al. A bispecific single-domain antibody boosts autologous vgamma9Vdelta2-T cell responses toward CD1d in chronic lymphocytic leukemia. Clin Cancer Res (2021) 27:1744–55. doi: 10.1158/1078-0432.CCR-20-4576
193. Baitsch L, Baumgaertner P, Devevre E, Raghav SK, Legat A, Barba L, et al. Exhaustion of tumor-specific CD8(+) T cells in metastases from melanoma patients. J Clin Invest (2011) 121:2350–60. doi: 10.1172/JCI46102
194. Arnone CM, Polito VA, Mastronuzzi A, Carai A, Diomedi FC, Antonucci L, et al. Oncolytic adenovirus and gene therapy with EphA2-BiTE for the treatment of pediatric high-grade gliomas. J Immunother Cancer (2021) 9:e001930. doi: 10.1136/jitc-2020-001930
195. Lin X, Li F, Guan J, Wang X, Yao C, Zeng Y, et al. Janus silica nanoparticle-based tumor microenvironment modulator for restoring tumor sensitivity to programmed cell death ligand 1 immune checkpoint blockade therapy. ACS Nano. (2023) 17:14494–507. doi: 10.1021/acsnano.3c01019
196. Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med (2017) 377:1345–56. doi: 10.1056/NEJMoa1709684
197. Kakiuchi-Kiyota S, Ross T, Wallweber HA, Kiefer JR, Schutten MM, Adedeji AO, et al. A BCMA/CD16A bispecific innate cell engager for the treatment of multiple myeloma. Leukemia (2022) 36:1006–14. doi: 10.1038/s41375-021-01478-w
198. Del Bano J, Chames P, Baty D, Kerfelec B. Taking up cancer immunotherapy challenges: bispecific antibodies, the path forward? Antibodies (Basel) (2015) 5:1. doi: 10.3390/antib5010001
199. Shimasaki N, Jain A, Campana D. NK cells for cancer immunotherapy. Nat Rev Drug Discovery (2020) 19:200–18. doi: 10.1038/s41573-019-0052-1
200. Houtenbos I, Santegoets S, Westers TM, Waisfisz Q, Kipriyanov S, Denkers F, et al. The novel bispecific diabody alphaCD40/alphaCD28 strengthens leukaemic dendritic cell-induced T-cell reactivity. Br J Haematol (2008) 142:273–83. doi: 10.1111/j.1365-2141.2008.06990.x
201. Singer H, Kellner C, Lanig H, Aigner M, Stockmeyer B, Oduncu F, et al. Effective elimination of acute myeloid leukemic cells by recombinant bispecific antibody derivatives directed against CD33 and CD16. J Immunother. (2010) 33:599–608. doi: 10.1097/CJI.0b013e3181dda225
202. Felices M, Lenvik TR, Davis ZB, Miller JS, Vallera DA. Generation of biKEs and triKEs to improve NK cell-mediated targeting of tumor cells. Methods Mol Biol (2016) 1441:333–46. doi: 10.1007/978-1-4939-3684-7_28
203. Bartlett NL, Herrera AF, Domingo-Domenech E, Mehta A, Forero-Torres A, Garcia-Sanz R, et al. A phase 1b study of AFM13 in combination with pembrolizumab in patients with relapsed or refractory Hodgkin lymphoma. Blood (2020) 136:2401–9. doi: 10.1182/blood.2019004701
204. Reiners KS, Kessler J, Sauer M, Rothe A, Hansen HP, Reusch U, et al. Rescue of impaired NK cell activity in hodgkin lymphoma with bispecific antibodies in vitro and in patients. Mol Ther (2013) 21:895–903. doi: 10.1038/mt.2013.14
205. Rothe A, Sasse S, Topp MS, Eichenauer DA, Hummel H, Reiners KS, et al. A phase 1 study of the bispecific anti-CD30/CD16A antibody construct AFM13 in patients with relapsed or refractory Hodgkin lymphoma. Blood (2015) 125:4024–31. doi: 10.1182/blood-2014-12-614636
206. Wu J, Fu J, Zhang M, Liu D. AFM13: a first-in-class tetravalent bispecific anti-CD30/CD16A antibody for NK cell-mediated immunotherapy. J Hematol Oncol (2015) 8:96. doi: 10.1186/s13045-015-0188-3
207. Wu J, Fu J, Zhang M, Liu D. Blinatumomab: a bispecific T cell engager (BiTE) antibody against CD19/CD3 for refractory acute lymphoid leukemia. J Hematol Oncol (2015) 8:104. doi: 10.1186/s13045-015-0195-4
208. Nitta T, Sato K, Yagita H, Okumura K, Ishii S. Preliminary trial of specific targeting therapy against Malignant glioma. Lancet (1990) 335:368–71. doi: 10.1016/0140-6736(90)90205-j
209. Introna M. CIK as therapeutic agents against tumors. J Autoimmun (2017) 85:32–44. doi: 10.1016/j.jaut.2017.06.008
210. Huang J, Li C, Wang Y, Lv H, Guo Y, Dai H, et al. Cytokine-induced killer (CIK) cells bound with anti-CD3/anti-CD133 bispecific antibodies target CD133(high) cancer stem cells in vitro and in vivo. Clin Immunol (2013) 149:156–68. doi: 10.1016/j.clim.2013.07.006
211. Park JA, Santich BH, Xu H, Lum LG, Cheung NV. Potent ex vivo armed T cells using recombinant bispecific antibodies for adoptive immunotherapy with reduced cytokine release. J Immunother Cancer (2021) 9:e002222. doi: 10.1136/jitc-2020-002222
212. Park JA, Espinosa-Cotton M, Guo HF, Monette S, Cheung NV. Targeting tumor vasculature to improve antitumor activity of T cells armed ex vivo with T cell engaging bispecific antibody. J Immunother Cancer (2023) 11:e006680. doi: 10.1136/jitc-2023-006680
213. Goldenberg DM, Rossi EA, Sharkey RM, McBride WJ, Chang CH. Multifunctional antibodies by the Dock-and-Lock method for improved cancer imaging and therapy by pretargeting. J Nucl Med (2008) 49:158–63. doi: 10.2967/jnumed.107.046185
214. McBride WJ, Sharkey RM, Karacay H, D’Souza CA, Rossi EA, Laverman P, et al. A novel method of 18F radiolabeling for PET. J Nucl Med (2009) 50:991–8. doi: 10.2967/jnumed.108.060418
215. Metz S, Haas AK, Daub K, Croasdale R, Stracke J, Lau W, et al. Bispecific digoxigenin-binding antibodies for targeted payload delivery. Proc Natl Acad Sci U S A. (2011) 108:8194–9. doi: 10.1073/pnas.1018565108
216. Weisser NE, Sanches M, Escobar-Cabrera E, O’Toole J, Whalen E, Chan PWY, et al. An anti-HER2 biparatopic antibody that induces unique HER2 clustering and complement-dependent cytotoxicity. Nat Commun (2023) 14:1394. doi: 10.1038/s41467-023-37029-3
217. Boerman OC, van Schaijk FG, Oyen WJ, Corstens FH. Pretargeted radioimmunotherapy of cancer: progress step by step. J Nucl Med (2003) 44:400–11.
218. Touchefeu Y, Bailly C, Frampas E, Eugene T, Rousseau C, Bourgeois M, et al. Promising clinical performance of pretargeted immuno-PET with anti-CEA bispecific antibody and gallium-68-labelled IMP-288 peptide for imaging colorectal cancer metastases: a pilot study. Eur J Nucl Med Mol Imaging. (2021) 48:874–82. doi: 10.1007/s00259-020-04989-3
219. Cheal SM, McDevitt MR, Santich BH, Patel M, Yang G, Fung EK, et al. Alpha radioimmunotherapy using (225)Ac-proteus-DOTA for solid tumors - safety at curative doses. Theranostics (2020) 10:11359–75. doi: 10.7150/thno.48810
220. Tarantino P, Carmagnani Pestana R, Corti C, Modi S, Bardia A, Tolaney SM, et al. Antibody-drug conjugates: Smart chemotherapy delivery across tumor histologies. CA Cancer J Clin (2022) 72:165–82. doi: 10.3322/caac.21705
221. Fu Z, Li S, Han S, Shi C, Zhang Y. Antibody drug conjugate: the “biological missile” for targeted cancer therapy. Signal Transduct Target Ther (2022) 7:93. doi: 10.1038/s41392-022-00947-7
222. Garcia-Alonso S, Ocana A, Pandiella A. Resistance to antibody-drug conjugates. Cancer Res (2018) 78:2159–65. doi: 10.1158/0008-5472.CAN-17-3671
223. Dean AQ, Luo S, Twomey JD, Zhang B. Targeting cancer with antibody-drug conjugates: Promises and challenges. MAbs (2021) 13:1951427. doi: 10.1080/19420862.2021.1951427
224. Yu J, Fang T, Yun C, Liu X, Cai X. Antibody-drug conjugates targeting the human epidermal growth factor receptor family in cancers. Front Mol Biosci (2022) 9:847835. doi: 10.3389/fmolb.2022.847835
225. de Goeij BE, Vink T, Ten Napel H, Breij EC, Satijn D, Wubbolts R, et al. Efficient payload delivery by a bispecific antibody-drug conjugate targeting HER2 and CD63. Mol Cancer Ther (2016) 15:2688–97. doi: 10.1158/1535-7163.MCT-16-0364
226. Drago JZ, Modi S, Chandarlapaty S. Unlocking the potential of antibody-drug conjugates for cancer therapy. Nat Rev Clin Oncol (2021) 18:327–44. doi: 10.1038/s41571-021-00470-8
227. Howard CB, Fletcher N, Houston ZH, Fuchs AV, Boase NR, Simpson JD, et al. Overcoming instability of antibody-nanomaterial conjugates: next generation targeted nanomedicines using bispecific antibodies. Adv Healthc Mater (2016) 5:2055–68. doi: 10.1002/adhm.201600263
228. Fatima SW, Khare SK. Benefits and challenges of antibody drug conjugates as novel form of chemotherapy. J Control Release. (2022) 341:555–65. doi: 10.1016/j.jconrel.2021.12.013
229. Solomon BJ, Desai J, Rosenthal M, McArthur GA, Pattison ST, Pattison SL, et al. A first-time-in-human phase I clinical trial of bispecific antibody-targeted, paclitaxel-packaged bacterial minicells. PloS One (2015) 10:e0144559. doi: 10.1371/journal.pone.0144559
230. Huckaby JT, Parker CL, Jacobs TM, Schaefer A, Wadsworth D, Nguyen A, et al. Engineering polymer-binding bispecific antibodies for enhanced pretargeted delivery of nanoparticles to mucus-covered epithelium. Angew Chem Int Ed Engl (2019) 58:5604–8. doi: 10.1002/anie.201814665
231. Ho KW, Chen IU, Cheng YA, Liao TY, Liu ES, Chen HJ, et al. Double attack strategy for leukemia using a pre-targeting bispecific antibody (CD20 Ab-mPEG scFv) and actively attracting PEGylated liposomal doxorubicin to enhance anti-tumor activity. J Nanobiotechnology. (2021) 19:16. doi: 10.1186/s12951-020-00752-w
232. Khaw BA, Gada KS, Patil V, Panwar R, Mandapati S, Hatefi A, et al. Bispecific antibody complex pre-targeting and targeted delivery of polymer drug conjugates for imaging and therapy in dual human mammary cancer xenografts: targeted polymer drug conjugates for cancer diagnosis and therapy. Eur J Nucl Med Mol Imaging. (2014) 41:1603–16. doi: 10.1007/s00259-014-2738-2
233. Cappuzzo F, Janne PA, Skokan M, Finocchiaro G, Rossi E, Ligorio C, et al. MET increased gene copy number and primary resistance to gefitinib therapy in non-small-cell lung cancer patients. Ann Oncol (2009) 20:298–304. doi: 10.1093/annonc/mdn635
234. Fitzgerald JB, Johnson BW, Baum J, Adams S, Iadevaia S, Tang J, et al. MM-141, an IGF-IR- and ErbB3-directed bispecific antibody, overcomes network adaptations that limit activity of IGF-IR inhibitors. Mol Cancer Ther (2014) 13:410–25. doi: 10.1158/1535-7163.MCT-13-0255
235. Huang S, Li C, Armstrong EA, Peet CR, Saker J, Amler LC, et al. Dual targeting of EGFR and HER3 with MEHD7945A overcomes acquired resistance to EGFR inhibitors and radiation. Cancer Res (2013) 73:824–33. doi: 10.1158/0008-5472.CAN-12-1611
236. McDonagh CF, Huhalov A, Harms BD, Adams S, Paragas V, Oyama S, et al. Antitumor activity of a novel bispecific antibody that targets the ErbB2/ErbB3 oncogenic unit and inhibits heregulin-induced activation of ErbB3. Mol Cancer Ther (2012) 11:582–93. doi: 10.1158/1535-7163.MCT-11-0820
237. Passiglia F, Malapelle U, Normanno N, Pinto C. Optimizing diagnosis and treatment of EGFR exon 20 insertions mutant NSCLC. Cancer Treat Rev (2022) 109:102438. doi: 10.1016/j.ctrv.2022.102438
238. Shivange G, Urbanek K, Przanowski P, Perry JSA, Jones J, Haggart R, et al. A single-agent dual-specificity targeting of FOLR1 and DR5 as an effective strategy for ovarian cancer. Cancer Cell (2018) 34:331–345 e11. doi: 10.1016/j.ccell.2018.07.005
239. Gancz D, Fishelson Z. Cancer resistance to complement-dependent cytotoxicity (CDC): Problem-oriented research and development. Mol Immunol (2009) 46:2794–800. doi: 10.1016/j.molimm.2009.05.009
240. Di Gaetano N, Cittera E, Nota R, Vecchi A, Grieco V, Scanziani E, et al. Complement activation determines the therapeutic activity of rituximab in vivo. J Immunol (2003) 171:1581–7. doi: 10.4049/jimmunol.171.3.1581
241. Manches O, Lui G, Chaperot L, Gressin R, Molens JP, Jacob MC, et al. In vitro mechanisms of action of rituximab on primary non-Hodgkin lymphomas. Blood (2003) 101:949–54. doi: 10.1182/blood-2002-02-0469
242. Beurskens FJ, Lindorfer MA, Farooqui M, Beum PV, Engelberts P, Mackus WJ, et al. Exhaustion of cytotoxic effector systems may limit monoclonal antibody-based immunotherapy in cancer patients. J Immunol (2012) 188:3532–41. doi: 10.4049/jimmunol.1103693
243. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I - molecular mechanisms of activation and regulation. Front Immunol (2015) 6:262. doi: 10.3389/fimmu.2015.00262
244. Diebolder CA, Beurskens FJ, de Jong RN, Koning RI, Strumane K, Lindorfer MA, et al. Complement is activated by IgG hexamers assembled at the cell surface. Science (2014) 343:1260–3. doi: 10.1126/science.1248943
245. Oostindie SC, van der Horst HJ, Kil LP, Strumane K, Overdijk MB, van den Brink EN, et al. DuoHexaBody-CD37(®), a novel biparatopic CD37 antibody with enhanced Fc-mediated hexamerization as a potential therapy for B-cell Malignancies. Blood Cancer J (2020) 10:30. doi: 10.1038/s41408-020-0292-7
246. Macor P, Secco E, Mezzaroba N, Zorzet S, Durigutto P, Gaiotto T, et al. Bispecific antibodies targeting tumor-associated antigens and neutralizing complement regulators increase the efficacy of antibody-based immunotherapy in mice. Leukemia (2015) 29:406–14. doi: 10.1038/leu.2014.185
247. Prang N, Preithner S, Brischwein K, Göster P, Wöppel A, Müller J, et al. Cellular and complement-dependent cytotoxicity of Ep-CAM-specific monoclonal antibody MT201 against breast cancer cell lines. Br J Cancer. (2005) 92:342–9. doi: 10.1038/sj.bjc.6602310
248. Mamidi S, Cinci M, Hasmann M, Fehring V, Kirschfink M. Lipoplex mediated silencing of membrane regulators (CD46, CD55 and CD59) enhances complement-dependent anti-tumor activity of trastuzumab and pertuzumab. Mol Oncol (2013) 7:580–94. doi: 10.1016/j.molonc.2013.02.011
249. Wang Y, Yang YJ, Wang Z, Liao J, Liu M, Zhong XR, et al. CD55 and CD59 expression protects HER2-overexpressing breast cancer cells from trastuzumab-induced complement-dependent cytotoxicity. Oncol Lett (2017) 14:2961–9. doi: 10.3892/ol.2017.6555
250. Steinhardt JJ, Guenaga J, Turner HL, McKee K, Louder MK, O’Dell S, et al. Rational design of a trispecific antibody targeting the HIV-1 Env with elevated anti-viral activity. Nat Commun (2018) 9:877. doi: 10.1038/s41467-018-03335-4
251. Watanabe Y, Tanabe A, Hamakubo T, Nagatoishi S, Tsumoto K. Development of biparatopic bispecific antibody possessing tetravalent scFv-Fc capable of binding to ROBO1 expressed in hepatocellular carcinoma cells. J Biochem (2021) 170:307–15. doi: 10.1093/jb/mvab049
252. Fleetwood F, Guler R, Gordon E, Stahl S, Claesson-Welsh L, Lofblom J. Novel affinity binders for neutralization of vascular endothelial growth factor (VEGF) signaling. Cell Mol Life Sci (2016) 73:1671–83. doi: 10.1007/s00018-015-2088-7
253. Yu S, Li A, Liu Q, Yuan X, Xu H, Jiao D, et al. Recent advances of bispecific antibodies in solid tumors. J Hematol Oncol (2017) 10:155. doi: 10.1186/s13045-017-0522-z
254. Gedeon PC, Choi BD, Hodges TR, Mitchell DA, Bigner DD, Sampson JH. An EGFRvIII-targeted bispecific T-cell engager overcomes limitations of the standard of care for glioblastoma. Expert Rev Clin Pharmacol (2013) 6:375–86. doi: 10.1586/17512433.2013.811806
255. Rius Ruiz I, Vicario R, Morancho B, Morales CB, Arenas EJ, Herter S, et al. p95HER2-T cell bispecific antibody for breast cancer treatment. Sci Transl Med (2018) 10:eaat1445. doi: 10.1126/scitranslmed.aat1445
256. Furnari FB, Cloughesy TF, Cavenee WK, Mischel PS. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat Rev Cancer. (2015) 15:302–10. doi: 10.1038/nrc3918
257. Moon EK, Carpenito C, Sun J, Wang LC, Kapoor V, Predina J, et al. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin Cancer Res (2011) 17:4719–30. doi: 10.1158/1078-0432.CCR-11-0351
258. Zhou S, Meng F, Du S, Qian H, Ding N, Sha H, et al. Bifunctional iRGD-anti-CD3 enhances antitumor potency of T cells by facilitating tumor infiltration and T-cell activation. J Immunother Cancer (2021) 9:e001925. doi: 10.1136/jitc-2020-001925
259. Hobbs SK, Monsky WL, Yuan F, Roberts WG, Griffith L, Torchilin VP, et al. Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc Natl Acad Sci U S A. (1998) 95:4607–12. doi: 10.1073/pnas.95.8.4607
260. Gajewski TF, Woo SR, Zha Y, Spaapen R, Zheng Y, Corrales L, et al. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr Opin Immunol (2013) 25:268–76. doi: 10.1016/j.coi.2013.02.009
261. Gooden MJ, de Bock GH, Leffers N, Daemen T, Nijman HW. The prognostic influence of tumour-infiltrating lymphocytes in cancer: a systematic review with meta-analysis. Br J Cancer. (2011) 105:93–103. doi: 10.1038/bjc.2011.189
262. Schalck A, Sakellariou-Thompson D, Forget MA, Sei E, Hughes TG, Reuben A, et al. Single-cell sequencing reveals trajectory of tumor-infiltrating lymphocyte states in pancreatic cancer. Cancer Discovery (2022) 12:2330–49. doi: 10.1158/2159-8290.CD-21-1248
263. Spranger S. Mechanisms of tumor escape in the context of the T-cell-inflamed and the non-T-cell-inflamed tumor microenvironment. Int Immunol (2016) 28:383–91. doi: 10.1093/intimm/dxw014
264. Wang S, Chen K, Lei Q, Ma P, Yuan AQ, Zhao Y, et al. The state of the art of bispecific antibodies for treating human Malignancies. EMBO Mol Med (2021) 13:e14291. doi: 10.15252/emmm.202114291
265. Arnone CM, Polito VA, Mastronuzzi A, Carai A, Diomedi FC, Antonucci L, et al. Oncolytic adenovirus and gene therapy with EphA2-BiTE for the treatment of pediatric high-grade gliomas. J Immunother Cancer (2021) 9:e001930. doi: 10.1136/jitc-2020-001930
266. Mazor R, Onda M, Pastan I. Immunogenicity of therapeutic recombinant immunotoxins. Immunol Rev (2016) 270:152–64. doi: 10.1111/imr.12390
267. Singh K, Hotchkiss KM, Mohan AA, Reedy JL, Sampson JH, Khasraw M. For whom the T cells troll? Bispecific T-cell engagers in glioblastoma. J Immunother Cancer (2021) 9:e003679. doi: 10.1136/jitc-2021-003679
268. Gardell JL, Matsumoto LR, Chinn H, DeGolier KR, Kreuser SA, Prieskorn B, et al. Human macrophages engineered to secrete a bispecific T cell engager support antigen-dependent T cell responses to glioblastoma. J Immunother Cancer (2020) 8:e001202. doi: 10.1136/jitc-2020-001202
269. Scott EM, Duffy MR, Freedman JD, Fisher KD, Seymour LW. Solid tumor immunotherapy with T cell engager-armed oncolytic viruses. Macromol Biosci (2018) 18:1700187. doi: 10.1002/mabi.201700187
270. Rosewell Shaw A, Suzuki M. Oncolytic viruses partner with T-cell therapy for solid tumor treatment. Front Immunol (2018) 9:2103. doi: 10.3389/fimmu.2018.02103
271. Fajardo CA, Guedan S, Rojas LA, Moreno R, Arias-Badia M, de Sostoa J, et al. Oncolytic adenoviral delivery of an EGFR-targeting T-cell engager improves antitumor efficacy. Cancer Res (2017) 77:2052–63. doi: 10.1158/0008-5472.CAN-16-1708
272. Freedman JD, Hagel J, Scott EM, Psallidas I, Gupta A, Spiers L, et al. Oncolytic adenovirus expressing bispecific antibody targets T-cell cytotoxicity in cancer biopsies. EMBO Mol Med (2017) 9:1067–87. doi: 10.15252/emmm.201707567
273. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell (2017) 168:707–23. doi: 10.1016/j.cell.2017.01.017
274. Zhou F, Qiao M, Zhou C. The cutting-edge progress of immune-checkpoint blockade in lung cancer. Cell Mol Immunol (2021) 18:279–93. doi: 10.1038/s41423-020-00577-5
275. Saxena A. Combining radiation therapy with immune checkpoint blockade for the treatment of small cell lung cancer. Semin Cancer Biol (2023) 90:45–56. doi: 10.1016/j.semcancer.2023.02.004
276. Dovedi SJ, Elder MJ, Yang C, Sitnikova SI, Irving L, Hansen A, et al. Design and efficacy of a monovalent bispecific PD-1/CTLA4 antibody that enhances CTLA4 blockade on PD-1(+) activated T cells. Cancer Discovery (2021) 11:1100–17. doi: 10.1158/2159-8290.CD-20-1445
277. Perez-Santos M, Anaya-Ruiz M, Herrera-Camacho I, Millan-Perez Pena L, Rosas-Murrieta NH. Bispecific anti-OX40/CTLA-4 antibodies for advanced solid tumors: a patent evaluation of WO2018202649. Expert Opin Ther Pat. (2019) 29:921–4. doi: 10.1080/13543776.2019.1681400
278. Lisi L, Lacal PM, Martire M, Navarra P, Graziani G. Clinical experience with CTLA-4 blockade for cancer immunotherapy: From the monospecific monoclonal antibody ipilimumab to probodies and bispecific molecules targeting the tumor microenvironment. Pharmacol Res (2022) 175:105997. doi: 10.1016/j.phrs.2021.105997
279. Klinger M, Zugmaier G, Nagele V, Goebeler ME, Brandl C, Stelljes M, et al. Adhesion of T cells to endothelial cells facilitates blinatumomab-associated neurologic adverse events. Cancer Res (2020) 80:91–101. doi: 10.1158/0008-5472.CAN-19-1131
280. Yuan D, Zhang Y, Lim KH, Leung SKP, Yang X, Liang Y, et al. Site-selective lysine acetylation of human immunoglobulin G for immunoliposomes and bispecific antibody complexes. J Am Chem Soc (2022) 144:18494–503. doi: 10.1021/jacs.2c07594
281. Nikkhoi SK, Li G, Eleya S, Yang G, Vandavasi VG, Hatefi A. Bispecific killer cell engager with high affinity and specificity toward CD16a on NK cells for cancer immunotherapy. Front Immunol (2022) 13:1039969. doi: 10.3389/fimmu.2022.1039969
282. Santich BH, Cheal SM, Ahmed M, McDevitt MR, Ouerfelli O, Yang G, et al. A self-assembling and disassembling (SADA) bispecific antibody (BsAb) platform for curative two-step pretargeted radioimmunotherapy. Clin Cancer Res (2021) 27:532–41. doi: 10.1158/1078-0432.CCR-20-2150
283. Tichet M, Wullschleger S, Chryplewicz A, Fournier N, Marcone R, Kauzlaric A, et al. Bispecific PD1-IL2v and anti-PD-L1 break tumor immunity resistance by enhancing stem-like tumor-reactive CD8(+) T cells and reprogramming macrophages. Immunity (2023) 56:162–179 e6. doi: 10.1016/j.immuni.2022.12.006
284. Yi M, Niu M, Wu Y, Ge H, Jiao D, Zhu S, et al. Combination of oral STING agonist MSA-2 and anti-TGF-beta/PD-L1 bispecific antibody YM101: a novel immune cocktail therapy for non-inflamed tumors. J Hematol Oncol (2022) 15:142. doi: 10.1186/s13045-022-01363-8
285. Wang Y, Tiruthani K, Li S, Hu M, Zhong G, Tang Y, et al. mRNA delivery of a bispecific single-domain antibody to polarize tumor-associated macrophages and synergize immunotherapy against liver Malignancies. Adv Mater (2021) 33:e2007603. doi: 10.1002/adma.202007603
286. Li W, Wang F, Guo R, Bian Z, Song Y. Targeting macrophages in hematological Malignancies: recent advances and future directions. J Hematol Oncol (2022) 15:110. doi: 10.1186/s13045-022-01328-x
287. Xiao N, Zhu X, Li K, Chen Y, Liu X, Xu B, et al. Blocking siglec-10(hi) tumor-associated macrophages improves anti-tumor immunity and enhances immunotherapy for hepatocellular carcinoma. Exp Hematol Oncol (2021) 10:36. doi: 10.1186/s40164-021-00230-5
288. Pittet MJ, Michielin O, Migliorini D. Clinical relevance of tumour-associated macrophages. Nat Rev Clin Oncol (2022) 19:402–21. doi: 10.1038/s41571-022-00620-6
289. Heemskerk N, Gruijs M, Temming AR, Heineke MH, Gout DY, Hellingman T, et al. Augmented antibody-based anticancer therapeutics boost neutrophil cytotoxicity. J Clin Invest (2021) 131:e134680. doi: 10.1172/JCI134680
290. Camilo Fadul JK, Thakur A, Purow B, Lopes M-B, Schiff D, Kassay-McAllister J, et al. ATIM-05. A PHASE I STUDY TARGETING NEWLY DIAGNOSED GLIOBLASTOMA WITH ANTI-CD3 × ANTI-EGFR BISPECIFIC ANTIBODY ARMED T CELLS (EGFR BATS) IN COMBINATION WITH RADIATION AND TEMOZOLOMIDE. Neuro-Oncology (2019) 21:vi2. doi: 10.1093/neuonc/noz175.005
291. Beeram, Hamilton E, Chaves J, Cobleigh M, Mwatha T, Woolery J, et al. Abstract P2-13-07: Zanidatamab (ZW25), a HER2-targeted bispecific antibody, in combination with chemotherapy (chemo) for HER2-positive breast cancer (BC): Results from a phase 1 study. Cancer Research (2022) 82:10. doi: 10.1158/1538-7445.SABCS21-P2-13-07
292. Lakhani N, Johnson M, Groisberg R, Han H, Casey K, Li S, et al. 535 A phase I/II study of REGN7075 (EGFRxCD28 costimulatory bispecific antibody) in combination with cemiplimab (anti–PD-1) in patients with advanced solid tumors. J ImmunoTherapy Cancer (2021) 9:A565–5. doi: 10.1136/jitc-2021-SITC2021.535
293. Zhao Y, Ma Y, Zang A, Cheng Y, Zhang Y, Wang X, et al. First-in-human phase I/Ib study of QL1706 (PSB205), a bifunctional PD1/CTLA4 dual blocker, in patients with advanced solid tumors. J Hematol Oncol (2023) 16:50. doi: 10.1186/s13045-023-01445-1
294. Ma Y, Xue J, Zhao Y, Zhang Y, Huang Y, Yang Y, et al. Phase I trial of KN046, a novel bispecific antibody targeting PD-L1 and CTLA-4 in patients with advanced solid tumors. J Immunother Cancer (2023) 11:e006654. doi: 10.1136/jitc-2022-006654
295. Wang R, Zhang C, Cao Y, Wang J, Jiao S, Zhang J, et al. Blockade of dual immune checkpoint inhibitory signals with a CD47/PD-L1 bispecific antibody for cancer treatment. Theranostics (2023) 13:148–60. doi: 10.7150/thno.79367
296. Skeltved N, Nordmaj MA, Berendtsen NT, Dagil R, Stormer EMR, Al-Nakouzi N, et al. Bispecific T cell-engager targeting oncofetal chondroitin sulfate induces complete tumor regression and protective immune memory in mice. J Exp Clin Cancer Res (2023) 42:106. doi: 10.1186/s13046-023-02655-8
297. Qu T, Zhong T, Pang X, Huang Z, Jin C, Wang ZM, et al. Ligufalimab, a novel anti-CD47 antibody with no hemagglutination demonstrates both monotherapy and combo antitumor activity. J Immunother Cancer (2022) 10:e005517. doi: 10.1136/jitc-2022-005517
298. Zhao L, Li S, Wei X, Qi X, Liu D, Liu L, et al. A novel CD19/CD22/CD3 trispecific antibody enhances therapeutic efficacy and overcomes immune escape against B-ALL. Blood (2022) 140:1790–802. doi: 10.1182/blood.2022016243
299. Tapia-Galisteo A, Compte M, Alvarez-Vallina L, Sanz L. When three is not a crowd: trispecific antibodies for enhanced cancer immunotherapy. Theranostics (2023) 13:1028–41. doi: 10.7150/thno.81494
300. Xue F, Yao H, Cui L, Huang Y, Shao C, Shen N, et al. An fc binding peptide-based facile and versatile build platform for multispecific antibodies. Nano Lett (2023) 23:4191–200. doi: 10.1021/acs.nanolett.3c00071
301. Gauthier L, Morel A, Anceriz N, Rossi B, Blanchard-Alvarez A, Grondin G, et al. Multifunctional natural killer cell engagers targeting NKp46 trigger protective tumor immunity. Cell (2019) 177:1701–1713 e16. doi: 10.1016/j.cell.2019.04.041
302. van de Donk N, Zweegman S. T-cell-engaging bispecific antibodies in cancer. Lancet (2023) 402:142–58. doi: 10.1016/S0140-6736(23)00521-4
Keywords: bispecific antibodies, immunotherapy, targeted therapy, tumor microenvironment, cancer
Citation: Guo X, Wu Y, Xue Y, Xie N and Shen G (2023) Revolutionizing cancer immunotherapy: unleashing the potential of bispecific antibodies for targeted treatment. Front. Immunol. 14:1291836. doi: 10.3389/fimmu.2023.1291836
Received: 10 September 2023; Accepted: 08 November 2023;
Published: 01 December 2023.
Edited by:
Prakash Radhakrishnan, University of Nebraska Medical Center, United StatesReviewed by:
Jeong A. Park, Inha University Hospital, Republic of KoreaLiang Gong, Chinese Academy of Sciences (CAS), China
Copyright © 2023 Guo, Wu, Xue, Xie and Shen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guobo Shen, shenguobo@126.com; Na Xie, naxie@scu.edu.cn
†These authors have contributed equally to this work