 Leeana D. Peters1
Leeana D. Peters1 Wen-I Yeh1
Wen-I Yeh1 Juan M. Arnoletti1
Juan M. Arnoletti1 Matthew E. Brown1
Matthew E. Brown1 Amanda L. Posgai1
Amanda L. Posgai1 Clayton E. Mathews1
Clayton E. Mathews1 Todd M. Brusko1,2*†
Todd M. Brusko1,2*†- 1Department of Pathology, Immunology and Laboratory Medicine, College of Medicine, Diabetes Institute, University of Florida, Gainesville, FL, United States
- 2Department of Pediatrics, College of Medicine, Diabetes Institute, University of Florida, Gainesville, FL, United States
The autoimmune pathogenesis of type 1 diabetes (T1D) involves cellular infiltration from innate and adaptive immune subsets into the islets of Langerhans within the pancreas; however, the direct cytotoxic killing of insulin-producing β-cells is thought to be mediated primarily by antigen-specific CD8+ T cells. Despite this direct pathogenic role, key aspects of their receptor specificity and function remain uncharacterized, in part, due to their low precursor frequency in peripheral blood. The concept of engineering human T cell specificity, using T cell receptor (TCR) and chimeric antigen receptor (CAR)-based approaches, has been demonstrated to improve adoptive cell therapies for cancer, but has yet to be extensively employed for modeling and treating autoimmunity. To address this limitation, we sought to combine targeted genome editing of the endogenous TCRα chain gene (TRAC) via CRISPR/Cas9 in combination with lentiviral vector (LV)-mediated TCR gene transfer into primary human CD8+ T cells. We observed that knockout (KO) of endogenous TRAC enhanced de novo TCR pairing, which permitted increased peptide:MHC-dextramer staining. Moreover, TRAC KO and TCR gene transfer increased markers of activation and effector function following activation, including granzyme B and interferon-γ production. Importantly, we observed increased cytotoxicity toward an HLA-A*0201+ human β-cell line by HLA-A*02:01 restricted CD8+ T cells engineered to recognize islet-specific glucose-6-phosphatase catalytic subunit (IGRP). These data support the notion of altering the specificity of primary human T cells for mechanistic analyses of autoreactive antigen-specific CD8+ T cells and are expected to facilitate downstream cellular therapeutics to achieve tolerance induction through the generation of antigen-specific regulatory T cells.
1 Introduction
Autoreactive T cells specific for β-cell antigens have been shown to be integral to type 1 diabetes (T1D) disease pathogenesis in the non-obese diabetic (NOD) mouse and are associated with human disease progression through immune studies and histopathological evidence (1). Murine models of T1D have helped to illuminate numerous antigens targeted during the initiation and propagation of the autoimmune process (2–4). One of these antigens, islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP), has been implicated as a key autoantigen in the non-obese diabetic (NOD) mouse model of T1D (5). Moreover, landmark human tissue studies have visually confirmed the targeting of this and other antigens by CD4+ and CD8+ T cells in situ in T1D with peptide-MHC multimer staining (6).
In order to understand the contribution of autoreactive T cells in driving T1D, we previously generated islet antigen-specific T cell “avatars” via lentiviral-mediated TCR gene transfer to primary CD8+ T cells. Our previous work using the IGRP265-273 reactive (clone 32) (7) T cell “avatars” highlighted the role of innate cytokines IL-12 and IL-18 to synergistically drive a Tc1 phenotype (8), which enhanced the killing of the β-cell line βlox5 (9). Moreover, our group has also utilized this clone to investigate the regulatory mechanism underlying type I interferon-mediated T cell cytotoxicity (10). Importantly, we demonstrated the utility of these avatars in constructing an isogenic system to investigate key disease relevant cellular interactions (11). Despite these advances in assessing antigen-specific T cell function, there remain a number of outstanding questions regarding which receptor specificities are directly pathogenic and thus, important for T1D initiation and progression. Our lab (12) and others (13, 14) have previously demonstrated that despite some repertoire sharing across tissues (up to 24% of clones), the CD8+ T cell repertoire in islet infiltrates and secondary lymphatics remains remarkably diverse, with reactivities against both native and post-translationally modified antigens detected (14). Importantly, the vast majority of CD8+ T cell lines from handpicked islets of a recent onset T1D donor were of unknown reactivities (14), highlighting the need for studies examining a broader range of autoantigen targets and immune receptors in T1D. There is also increasing evidence of divergent patterns of initial targets of autoimmunity, characterized by insulin autoantibody (IAA) or GAD autoantibody (GADA) initial seroconversion, which seems to be linked to age (15) as well as genetic risk at the HLA region (16) (HLA-DR4 and HLA-DR3, respectively). Thus, these potential disease endotypes, coupled with the large impacts of environmental exposures and aging on the immune repertoire (17, 18), as well as heterogeneity in immune phenotypes of CD8+ T cells in T1D (19), all serve to complicate the assessment of antigen-specific phenotype and function.
In vitro modeling of autoimmunity and clinical adoptive cell therapy (ACT) strategies rely on the generation of large numbers of autoreactive T cells, surpassing the frequency obtainable in peripheral blood without extended months of clonal expansion, potentially altering the phenotype from the in vivo state. Despite the success of common methods to generate antigen-specific T cells (20), namely using a viral (often lentiviral) vector to deliver a de novo T cell receptor (TCR), there remain concerns of off-target effects arising from heterologous TCR chain pairing (21, 22). Work in the cancer immunotherapy space have demonstrated the utility of gene editing technologies (i.e., CRISPR/Cas9) in targeting endogenous TCRs to enhance on target-specificity and anti-tumor functionality (21, 22), though to our knowledge this has not been shown previously for T1D autoantigen reactive T cells. We hypothesized that reducing the potential for heterologous chain pairing would translate into enhanced effector phenotype and function of a T1D-associated IGRP265-273 reactive clone (clone 32) (7). We chose to test our workflow in engineered IGRP-reactive CD8+ T cells, as a number of studies have explored the importance of this antigen in T1D as a target for human CD8+ T cells in circulation (5, 23) and in situ (6). Therefore, we used CRISPR/Cas9 to knockout (KO) the endogenous TCRα in engineered IGRP-reactive CD8+ T cells, with the goal of more effectively recapitulating autoreactive CD8+ T cell function in T1D.
2 Materials and methods
2.1 Human subjects
Peripheral blood mononuclear cells (PBMCs) were isolated via density-gradient centrifugation from leukapheresis-processed blood of healthy donors (N=4, median age: 25.5 years, range 21-27 years, 25% male) purchased from LifeSouth Community Blood Centers (Gainesville, FL).
2.2 Cell isolation and expansion
Naïve CD8+ T cells were isolated in a two-step procedure consisting of negative selection followed by positive selection from leukapheresis products using the naïve CD8+ T cell isolation kit (Miltenyi Biotec) and were cultured as reported previously (9). Briefly, cells were seeded at a concentration of 2.5 x 105 cells/mL in complete RPMI formulated as previously described (9) and stimulated with α-CD3/28 coated beads (Dynabeads, Thermo Fisher) at a 1:1 ratio with 100 IU/mL recombinant human IL-2 (Teceleukin, NIH). Media and IL-2 were added assuming consumption every 2-3 days, with 5 ng/mL IL-7 (R&D systems) added every 2-3 days after day 5 of culture.
2.3 Generation of autoreactive T cell avatars
Cells were transduced 48 hours post-activation with a multicistronic (pCCL.TRB.P2A.TRA.T2A.eGFP) lentiviral vector (LV) encoding the HLA-A*02:01 restricted IGRP reactive clone 32 TCR (7) or mock (pCNFW.eGFP) control LV as previously described (24, 25). Briefly, virus was added at 3 transducing units (TU)/cell along with 8 µg/mL protamine sulfate, and cells were spinnoculated at 1000xg for 30 minutes at 32°C. Transduction efficiency was 34.0±4.1%. Cells were expanded, using methods described above, for 5-7 days post-transduction, then assessed by flow cytometry for stable transduction by expression of GFP. Data were acquired on an LSRFortessa flow cytometer (BD Biosciences) and analyzed using FlowJo software v10 (BD).
2.4 CRISPR/Cas9-mediated KO of TRAC
After 5-7 days of expansion post-transduction, α-CD3/28 coated beads were removed, and transduced T cells were electroporated with ribonucleoprotein (RNP) complexes comprised of a single guide RNA (sgRNA, Synthego) and Cas9 (Thermo Fisher Scientific) at a 6:1 molar ratio (26) (assembled at room temperature for 10 minutes) using the Lonza 4D-Nucleofector X Unit and P3 Primary Cell 4D-Nucleofector X Kit (Lonza Bioscience) with the pulse code EH100. For the negative control condition, cells were electroporated without sgRNA or Cas9. The sequence for the TRAC guide used was as follows: GUCAGGGUUCUGGAUAUCUG.
2.5 Confirmation of TCRα KO and MHC-multimer binding
Efficiency of TRAC KO was assessed 5-7 days post CRISPR/Cas9 (culture day 12-14). Briefly, cells were stained with Live/Dead Fixable Near-IR (Thermo Fisher Scientific) for 10 minutes at 4°C, washed with stain buffer (PBS + 2% FBS + 0.05% NaN3), and subsequently stained for 30 minutes at 4°C with anti-human CD3e-PerCP-Cy5.5 (clone UCHT1, BioLegend) and TCRαβ-BV421 (clone IP26, BioLegend) for 30 minutes at 4°C. Endogenous TCRα KO efficiency was assessed by percentage of TCRαβ negative cells within the internal control untransduced (GFP-) cells for each donor culture. To assess changes in alpha chain expression and proper de-novo TCR chain pairing, cells were stained with Live/Dead Fixable Near-IR as above in addition to CD8-BV605 (clone SK1, BioLegend), TCR V alpha 2 (V⍺2, clone F1, Thermo Fisher) conjugated in house using the Zenon Alexa Fluor 647 mouse IgG2a labeling kit (Invitrogen) according to manufacturer instructions, and TCR V beta 20-1-PE (Vβ2, clone MPB2D5, Beckman Coulter) or TCRαβ-BV421 (clone IP26, BioLegend) for 30 minutes at 4°C. To assess changes in functional receptor avidity, IGRP265-273 MHC class I dextramer staining was conducted as per manufacturer’s instructions (Immudex). Briefly, cells were stained with fixable Live Dead NIR as above, followed by incubation with TruStain Fc block (BioLegend) for 5 minutes and incubation with 10 µL dextramer for 10 minutes at room temperature, after which cells were extracellularly stained with anti-human CD8-BV605 (clone SK1, BioLegend) and TCRαβ-BV421 (clone IP26, BioLegend) for 20 minutes at room temperature. Data were acquired on an LSRFortessa flow cytometer (BD Biosciences) and analyzed using FlowJo software v10 (BD).
2.6 Stimulation assays
A derivative of the chronic myelogenous leukemia (CML) cell line K562 engineered to express HLA-A*02:01 (9, 24), kindly provided by Drs. James Riley and Bruce Levine (University of Pennsylvania), was used as a source of artificial antigen presenting cells (aAPCs) for our T cell avatars. K562s were loaded with 10 µg/mL IGRP265-273 peptide (GenScript) at a concentration of 1 x 106cells/mL at 4°C for one hour. IGRP TCR transduced (IGRP) or eGFP mock transduced T cells (mock), with or without TRAC KO, were plated at a 1:1 ratio with peptide-pulsed K562s. Cells were stained to assess expression of activation markers using Live/Dead Fixable Near-IR as above, after which cells were incubated with TruStain Fc block (BioLegend) for 5 minutes at 4°C. Cells were stained for 30 minutes at 4°C with anti-human CD69-BV711 (clone FN50, BioLegend) and CD25-APC (clone BC96, BD Biosciences) at baseline and after 2, 4 and 24 hours of stimulation. To assess the cytokine production profile, cells were stimulated as above for 4 hours at 37°C in the presence of 0.66 µL/mL Golgistop (BD Biosciences), after which cells were stained with anti-human CD8a-APC (clone RPA-T8, BD Biosciences) and anti-GFP-AF488 (clone FM264G, BioLegend) prior to fixation and permeabilization with FOXP3 transcription factor staining buffer set (eBioscience) according to manufacturer’s protocol. Intracellular staining was performed for 30 minutes at room temperature with anti-human GZMB-PE (clone GB11, BD Biosciences), TNFα-BV650 (clone MAb11, BioLegend), Perforin-BV421 (clone dG9, BioLegend), IFNγ-PE-Cy7 (clone 4S.B3, BioLegend), and IL-2-PerCP-Cy5.5 (clone MQ1-17H12, BioLegend).
2.7 Cell-mediated lympholysis
TRAC KO or electroporation only negative control IGRP265-273 reactive CD8+ T cells were assayed for their capacity to kill the HLA-A*02:01-expressing human β-cell line βLox5, as previously described (9). In brief, βLox5 cells were harvested from flasks by incubating with Cell Dissociation Buffer for 5-10 minutes at 37°C and 5% CO2 (Gibco). βLox5 cells were then washed, resuspended at 5-10 x 106 /mL in PBS, and labeled with 5uM CellTrace Violet (Thermo Fisher Scientific) according to manufacturer’s instructions. Labeled βLox5 were plated at a concentration of 30,000 cells per well in a 48-well tissue culture treated plate in complete DMEM (cDMEM) formulated as previously described (27). After 24hr, CD8+ T cell avatars were plated with βLox5 at 0:1, 1:1, and 5:1 effector:target (E:T) ratios in duplicate, and co-cultured for 16 hours at 37°C and 5% CO2. Cells were harvested and stained with a master mix comprised of 88uL Annexin V-Binding Buffer (BioLegend), 2uL Annexin V-AF647 (BioLegend) and 10uL of propidium iodide at 3mg/mL (PI, Invitrogen). Data were acquired on an LSRFortessa flow cytometer (BD Biosciences) and analyzed using FlowJo software v10 (BD).
2.8 Statistical analyses
Statistical testing and visualization were performed in GraphPad Prism Software V9. Data were analyzed by Two-Way ANOVA with Bonferroni correction or Tukey’s multiple comparison test and paired t test as described in the figure legends and text. Data are represented as mean ± standard deviation (SD) unless otherwise specified. P-values <0.05 were considered significant.
3 Results
3.1 KO of the endogenous TCRα enhances pairing of an engineered T1D-associated TCR
We hypothesized that deletion of the endogenous TCRα would reduce heterologous pairing with an exogenously introduced T1D-associated IGRP-reactive TCR, thereby improving on-target specificity. Thus, we generated CD8+ T cell avatars reactive to IGRP265-273 (7) and targeted the TCRα chain constant (TRAC) region via CRISPR/Cas9 (Figure 1A), as has been previously reported to improve on-target efficacy of avatars specific for a melanoma antigen (22). Our IGRP-TCR construct possesses 4 base pair (bp) mismatches as well as a mutated protospacer adjacent motif (PAM) (26) at the location targeted by sgRNA1 (Figure S1), which importantly, prevents cleavage of the de novo TCR by Cas9; hence, we selected this guide for our studies. We first validated the efficiency of endogenous TRAC KO in primary human CD8+ T cells transduced with the IGRP-reactive clone 32 TCR (9, 10). We achieved, on average, an 86% KO efficiency (86.13 ± 5.5) after targeting TRAC via CRISPR/Cas9, as quantified by the percentage of TCRαβ negative cells (Figures 1B, D). Moreover, we noted increased expression of the clone 32 TCR alpha variable region, TCRVα2 in the TRAC KO avatars (IGRP KO (red triangles), 2074.0 ± 122.3) relative to those that received electroporation but not sgRNA or Cas9 (IGRP negative control (NC, blue circles), 541.7 ± 68.1, fold difference=3.8), and mock transduced cells lacking the de novo TCR which received electroporation only (Mock NC (blue), 189.0 ± 32.1, fold difference=10.9) or mock transduced cells deleted for TRAC (Mock KO (red), 163.3 ± 21.7, fold difference=12.7) (Figures 1C, E). We also observed a corresponding increase in cells positive for TCRVα2 in the TRAC KO avatars (52.7 ± 3.8) relative to the negative control cells (15.8 ± 1.3, fold difference=3.3), and mock transduced negative control (3.8 ± 0.8, fold difference=13.9) and KO (0.7 ± 0.3, fold difference=71.2) cells (Figures 1C, F). This increase in TCRα expression occurred independently of differences in total TCRαβ expression between TRAC KO and negative control avatars (Figure 1G), indicative of increased proper TCR pairing following editing. Indeed, we observed increased proper pairing in the TRAC KO avatars (67.0 ± 13.7) relative to negative control avatars (19.6 ± 6.8, fold difference=3.4), while mock controls lacked expression of both chains, when stained with the de novo TCR variable chains TCRVα2 and TCRVβ2 (Figures 1C, H).
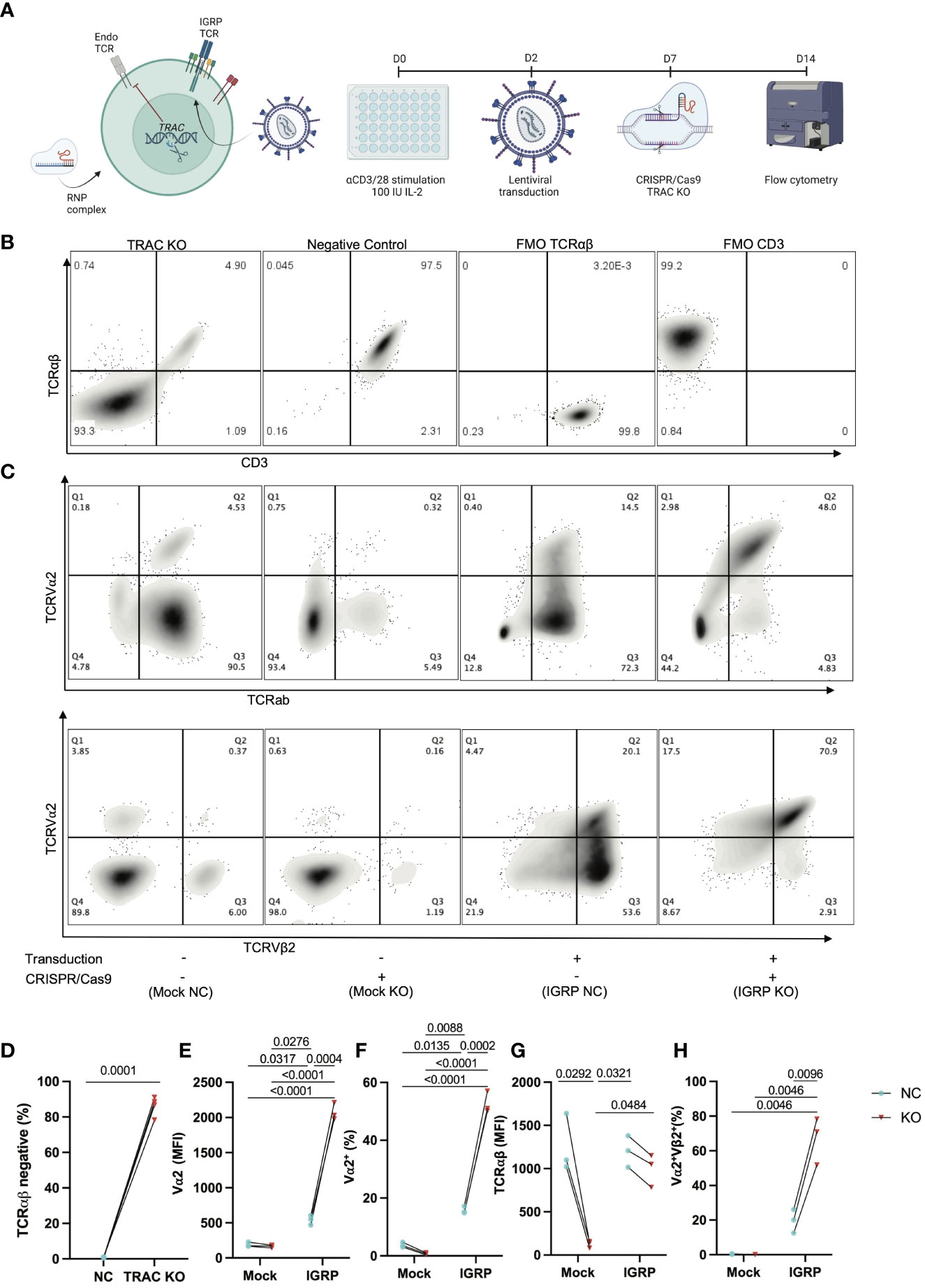
Figure 1 Efficient KO of the endogenous TCR is achieved by targeting TRAC. (A) Schematic of our experimental procedure wherein CD8+ T cells are transduced with a LV encoding the IGRP-reactive clone 32 TCR as reported previously (24), after which CRISPR/Cas9 was performed targeting TRAC using RNP complexes comprised of a 6:1 molar ratio of sgRNA : Cas9. Created with BioRender.com. (B) Representative plots of TCRαβ and CD3 expression in GFP- cells (internal controls within each donor culture which are exposed to editing conditions but are not stably transduced with the de novo TCR) show high efficiency KO as delineated by largely all TCRαβ negative cells (far left) as compared to the electroporation only negative control condition (middle left), as compared to fluorescence minus one (FMO) controls for TCRab (middle right) and CD3 (far right). (C) Representative plot of TCRVα2 and TCRαβ expression (top) and paired TCRVα2 and TCRVβ2 expression (bottom) across all conditions (+ or – transduction with the IGRP reactive clone 32 TCR and + or - CRISPR/Cas9 deletion of endogenous TRAC). (D) The TRAC KO condition possesses significantly more cells that are TCRαβ negative (paired t test, n=4, p value shown on figure). (E) TRAC KO avatars show significantly increased expression of the clone 32 α-chain TCRVα2 (24), as well as (F) increased TCRVα2+ cells (Repeated Measures two-way ANOVA with Tukey’s multiple comparison test, n=3, p values shown on figures), without a significant increase in (G) total TCR expression between clone 32 TCR transduced negative control and KO avatars (Repeated measures two-way ANOVA with Tukey’s multiple comparison test, n=3, p value shown on figure). We also observe an increase in (H) correct de novo TCR pairing with TRAC KO (Repeated measures two-way ANOVA with Tukey’s multiple comparison test, n=3, p value shown on figure).
It has been shown that competition for TCR-CD3 complex assembly between the endogenous and exogenously introduced TCR chains influences pairing and thereby, the functional avidity of each TCR dimer (28). Given that self-reactive T cells are by nature lower affinity to permit escape from thymic negative selection (29, 30), as compared to pathogen-specific T cells, mispairing could limit avidity and thus, functionality of the de novo autoreactive TCR. We hypothesized that deletion of the endogenous TCRα would result in an increase in avidity of the de novo clone 32 TCR to cognate antigen. In order to examine this, we employed peptide-major histocompatibility complex (pMHC)-dextramer reagents, which permit visualization of binding to TCR and can serve as a measure of TCR avidity, as reviewed previously (31). We expected that minimization of TCR chain mispairing would result in enhanced IGRP pMHC dextramer binding capability, as reduced dextramer binding has been shown upon TCR transduction in cells with an intact endogenous TCR (32). Indeed, staining with HLA-A2-IGRP265-273 dextramer revealed an increased percentage of IGRP dextramer+ cells in TRAC KO clone 32 T cell avatars (58.9 ± 13.3%) relative to clone 32 avatars that received electroporation but not sgRNA or Cas9 (negative control, 13.4 ± 10.7%, fold difference = 4.4) and mock transduced T cells lacking the de novo TCR and exposed to electroporation without CRISPR editing (0.21 ± 0.18%, fold difference= 276.5) and with endogenous TCR KO (0.39 ± 0.35%, fold difference = 150.1) (Figures 2A, B). TRAC KO clone 32 T cell avatars also showed increased IGRP dextramer mean fluorescence intensity (MFI) (693.3 ± 437.1) relative to negative control avatars (330.2 ± 342.2, fold difference = 2.1), both indicative of enhanced on target pMHC-TCR binding capacity (Figure 2C).
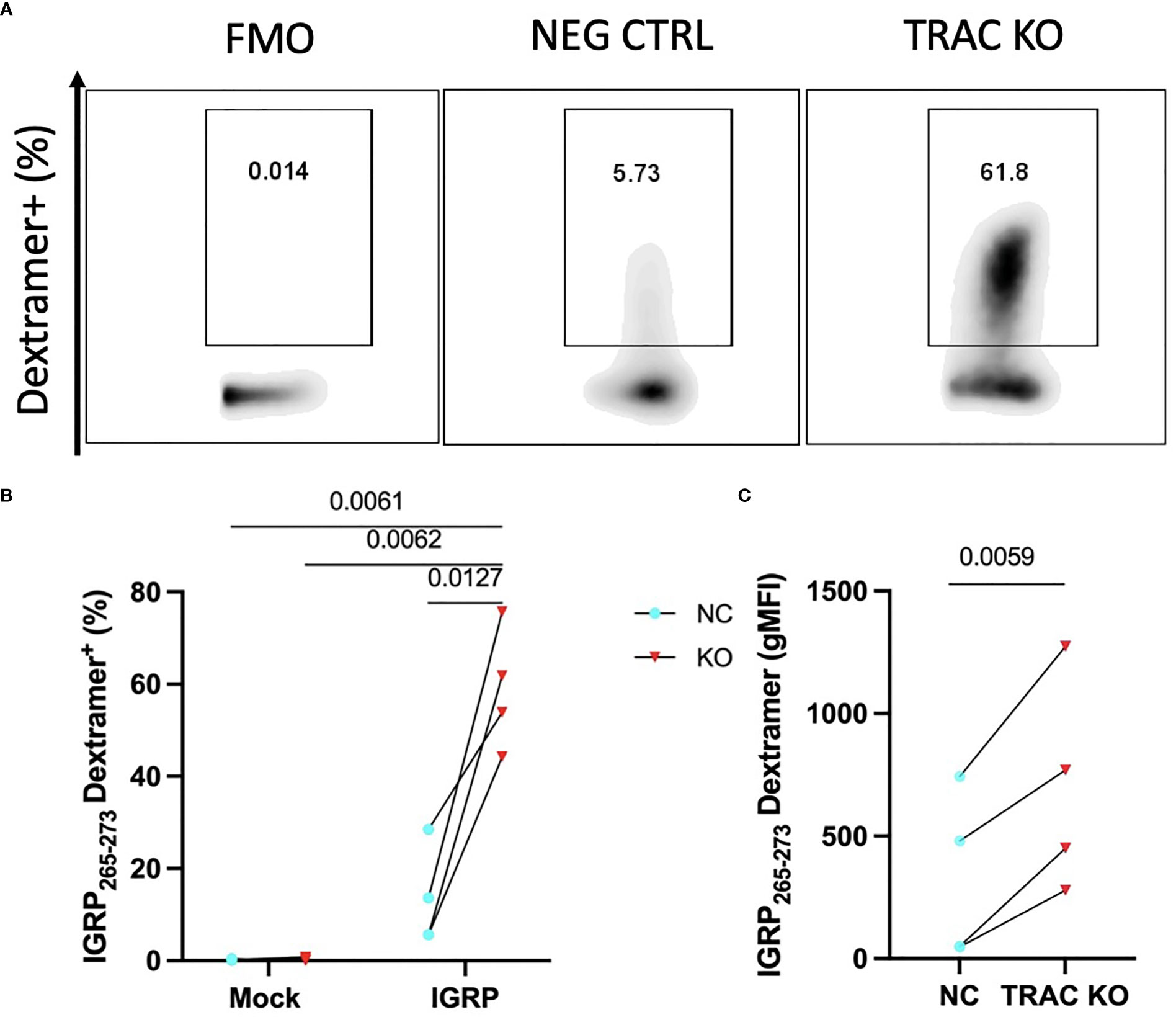
Figure 2 TRAC KO confers increased pMHC avidity. Clone 32 IGRP TCR transduced CD8+ T cells were stained with a pMHC-IGRP265-273 dextramer. (A) Representative plots of dextramer binding cells within the KO as compared to the negative control condition. (B) Significant enhancement of the dextramer binding fraction within the TRAC KO condition as compared to the negative control (NC; transduced with LV TCR, no endogenous TRAC KO) and mock conditions (electroporated with or without CRISPR but not transduced with LV TCR). Repeated measures two-way ANOVA with Tukey’s multiple comparison test, p value shown on figure (n=4). (C) Significant enhancement in dextramer mean fluorescence intensity (MFI) between the TRAC KO and NC conditions. Paired t-test, p value shown on figure (n=4).
3.2 TRAC KO T cell avatars display increased activation in response to autoantigen
TCR affinity and functional avidity have been shown to be linearly correlated with activation by cognate antigen and downstream effector function in the context of TCR:pMHC interactions (33). We investigated whether the increased pMHC avidity observed in our TRAC KO avatars would translate to increased activation in response to cognate antigen. To test this, we loaded HLA-A2+ K562 cells with IGRP265-273 peptide and co-cultured these aAPCs with our TRAC KO or negative control avatars. We observed no significant differences in CD69 expression at 2 hours post-stimulation (Figure S2A), but after 4 hours of co-culture, TRAC KO avatars exhibited a greater percentage of cells expressing the early activation marker CD69 (79.5 ± 7.9%) relative to negative control clone 32 avatars (with intact endogenous TRAC, 44.8 ± 5.8%, fold difference = 1.8) and mock transduced control T cells with (26.1 ± 7.7%, fold difference = 3.0) or without TRAC KO (23.6 ± 11.1%, fold difference = 3.4) (Figures 3A, B), indicative of sustained activation in the IGRP TRAC KO avatars. Moreover, at the 24-hour time point, we observed an increased percentage of IGRP-reactive TRAC KO avatars expressing the late activation marker CD25 (81.2 ± 12.8%) as compared to IGRP-reactive negative control avatars (21.6 ± 6.7%, fold difference = 3.76) and mock transduced cells (mock TRAC KO: 4.1 ± 0.63%, fold difference = 19.8; mock negative control: 5.2 ± 0.83%, fold difference = 15.6) (Figures 3C, D). These data indicate that deletion of the endogenous TCRα permits increased activation by the de novo TCR.
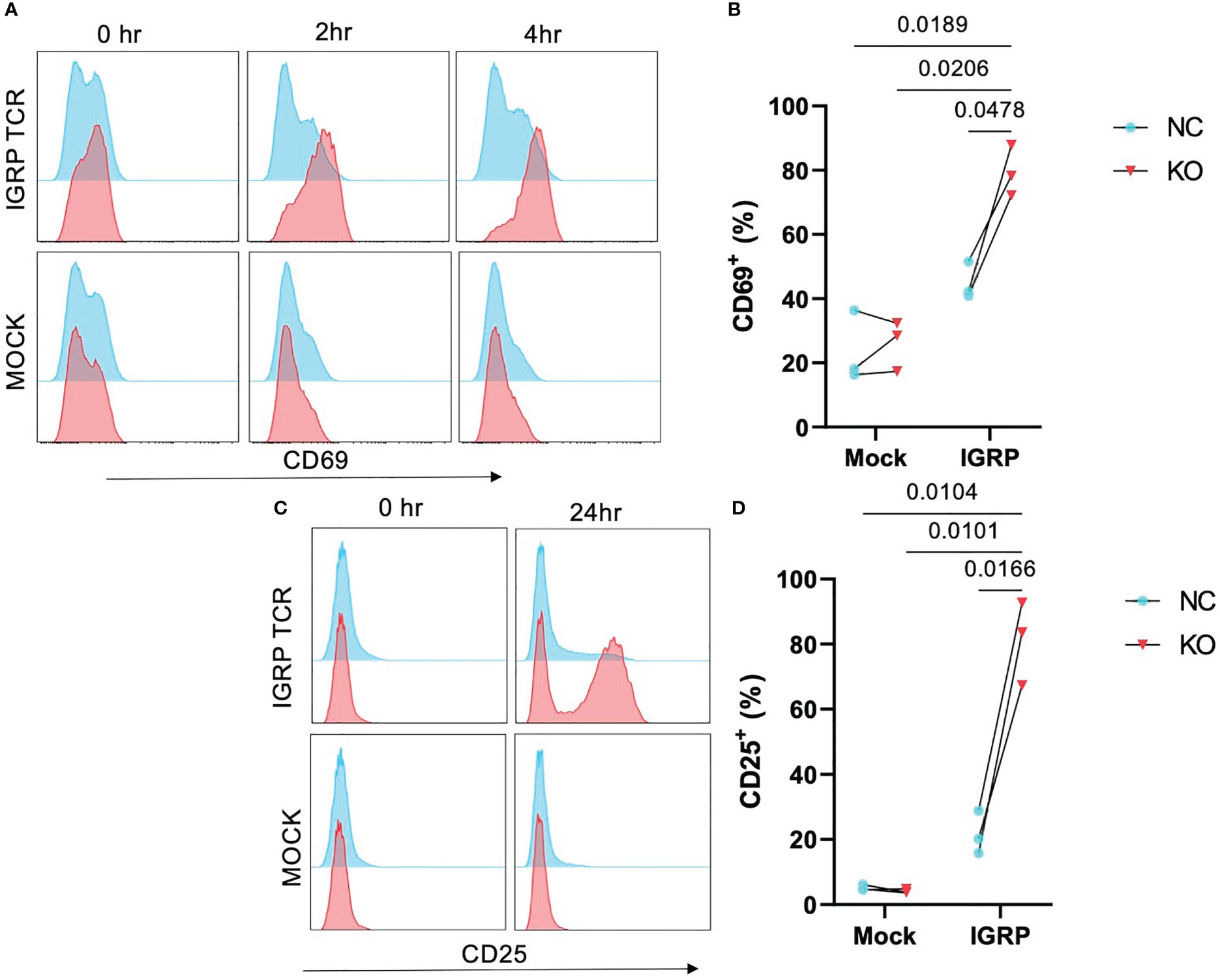
Figure 3 Enhanced activation in TRAC KO avatars. Clone 32 IGRP TCR or mock transduced CD8+ T cells were co-cultured at a 1:1 ratio with IGRP265-273 loaded HLA-A2+ K562 aAPCs for 0, 2, 4, and 24 hours, then assessed for activation markers CD25 and CD69 by flow cytometry. (A) Representative histograms of CD69 expression on TRAC KO IGRP TCR (upper pink) versus TRAC KO mock transduced (lower pink) and negative control (NC; blue) avatars at 0-, 2- and 4-hour time points. (B) Significant enhancement of the CD69+ fraction within the TRAC KO condition as compared to the negative control condition at 4 hours post stimulation. (C) Representative plots of CD25 expression on TRAC KO (upper pink) versus mock transduced (lower pink) and negative control (blue) avatars at 0- and 24-hour timepoints. (D) Significant enhancement of the CD25+ fraction within the TRAC KO condition as compared to the negative control and both mock control conditions. Repeated measures two-way ANOVA with Tukey’s multiple comparison test, p values shown on figure (n=3).
3.3 TRAC KO T cell avatars demonstrate enhanced GZMB-dependent β-cell cytotoxicity
TCR signal strength has been shown to largely influence the efficiency of cytotoxic T lymphocyte (CTL) killing (34). After demonstrating that endogenous TRAC KO enhanced the avidity of the clone 32 TCR, evidenced by increased pMHC-dextramer binding and activation, we reasoned that this would translate to augmented killing of IGRP-expressing target cells. We utilized the HLA-A2+ β-cell line βLox5 (11) as such targets in order to test the in vitro killing capacity of our TRAC KO avatars. After 16 hours of co-culture, we observed significant increases in killing between the 5:1 vs the 1:1 E:T ratios in both TRAC KO (red, p=0.0074) and negative control (blue, p=0.0217) conditions, as expected (Figures 4A, B). Importantly, we also observed a significant increase in killing capacity in the 5:1 E:T TRAC KO condition as compared to the 5:1 E:T negative control condition (p=0.0207) (Figure 4B).
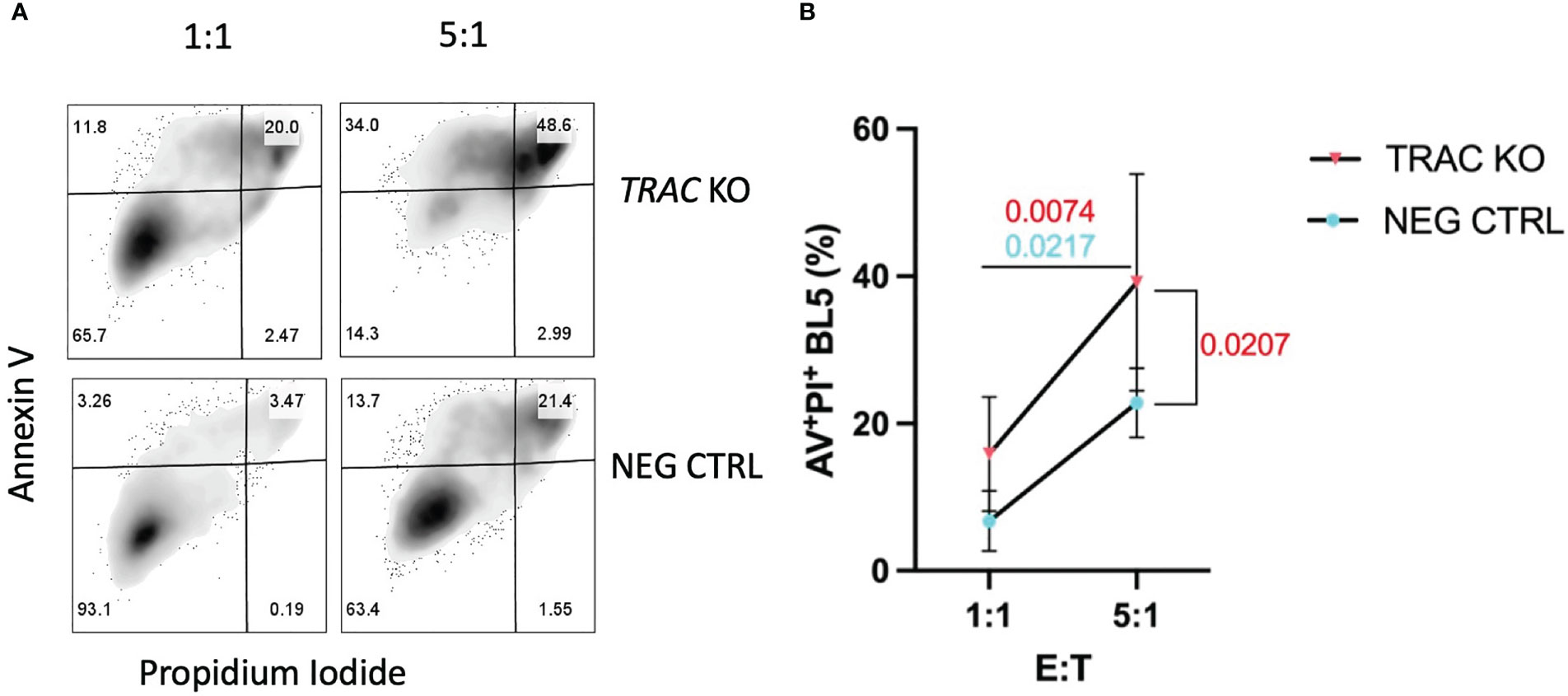
Figure 4 TRAC KO avatars display increased cytotoxicity against a human β-cell line. Clone 32 IGRP TCR transduced CD8+ T cells were co-cultured at a 1:1 and 5:1 effector:target (E:T) ratio with the HLA-A2+ βLox5 cell line for 16 hours and assessed for apoptosis and cell death with Annexin V and propidium iodide (PI) staining. (A) Representative contour plot of Annexin V and PI staining of βLox5 cultured with TRAC KO (upper) or negative control T cell avatars (lower). (B) Increased cell mediated lysis of βLox5 in the 5:1 vs the 1:1 effector: target (E:T) ratio for both TRAC KO (red, p=0.0074) and negative control (blue, p=0.0217) avatars, as well as a significant increase in killing capacity in the 5:1 E:T TRAC KO condition as compared to the 5:1 E:T negative control condition (red, p=0.0207). Repeated measures two-way ANOVA with Bonferroni correction, p values shown on figure (n=4).
The mechanisms underlying CD8+ T cell dependent β-cell cytotoxicity have been shown to include cytolysis by perforin and granzyme B (10), as well as the induction of stress through the production of soluble effector molecules such as interferon gamma (IFN-γ) and tumor necrosis factor alpha (TNF-α) (29, 35). Thus, we assayed for the production of cytolytic molecules, namely TNF-α, IFN-γ, perforin, and GZMB, as well as IL-2 as a readout of mitogenic capacity (36), after stimulation with aAPCs presenting cognate antigen for four hours in the presence of a protein transport inhibitor. While we observed no differences in the production of TNF-α, IL-2 or perforin (Figures S2B–D), we observed notably enhanced production of GZMB (47.7 ± 25.5) and IFN-γ (10.2 ± 1.7) by the TRAC KO versus negative control (23.2 ± 15.8, fold difference = 2.1 and 1.2 ± 0.5, fold difference = 8.2, respectively) clone 32 avatars (Figures 5A, B). This enhancement was found to be specific to cells possessing the clone 32 TCR as there was no difference in production of these molecules between the TRAC KO and negative control conditions in internal control GFP- T cells (i.e., derived from the same cultures as the GFP+ cells but lacking the de novo TCR; Figures S2E, F).
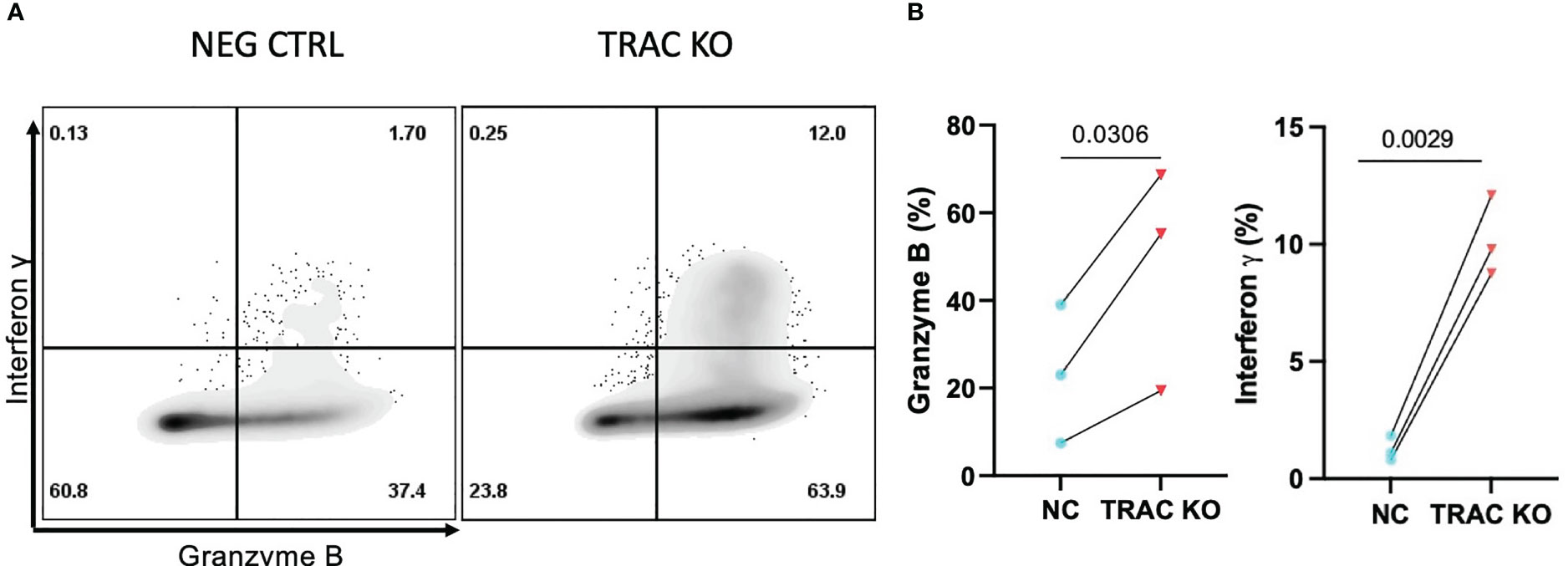
Figure 5 Enhanced inflammatory cytokine and cytolytic molecule production by TRAC KO avatars. Clone 32 IGRP TCR transduced CD8+ T cells were co-cultured at a 1:1 ratio with the HLA-A2+ K562 cell line for four hours in the presence of GolgiStop and assessed for production of effector molecules granzyme B (GZMB) and interferon-γ (IFN- γ). (A) Representative contour plot of IFN-γ and GZMB expression by TRAC KO or negative control T cell avatars. (B) Significant increase in the percent of cells that express GZMB (left) and IFN- γ (right) in the TRAC KO condition as compared to the negative control condition. Paired t test, p value shown on figures (n=3).
4 Discussion
In our study, we utilized CRISPR/Cas9 to delete the endogenous TCRα of engineered autoreactive CD8+ T cell avatars, as has been previously performed with LV transduced cancer-reactive T cells (22). To our knowledge, ours is the first study to illustrate these improvements in T cell avidity and cytotoxic function as it pertains to β-cell antigen specificity, though others have utilized a similar approach when designing chimeric antigen receptor (CAR) regulatory T cells (Tregs) targeting the HLA-A2 molecule (37). We first demonstrated the utility of this approach in enhancing the ability of the de novo IGRP-reactive TCR to bind pMHC-dextramer reagents. While dextramer reagents demonstrate increased capacity for binding lower affinity TCRs than tetramer reagents (38), previous studies have shown that the pMHC-TCR affinity threshold for multimer binding to be lower than that required for activation (39), resulting in the possibility for low affinity and recently activated T cells which have downregulated the CD3/TCRαβ complex (40) to be underestimated using this technique. However, enhancing the capacity of the de novo TCR to bind these reagents through endogenous TRAC KO could be helpful in a clinical setting to monitor the persistence of an ACT product. We also demonstrated that deletion of the endogenous TCRα served to augment the activation of our T cell avatars and consequently, the killing capacity of a human β-cell line. We note that at the earlier activation timepoint (2 hours) there was no difference in activation, while at the later timepoints (4 and 24 hours) we observed increased expression of activation markers CD69 and CD25, respectively. Future experiments will be required to determine the impact on TCR activation kinetics and to elucidate if the resulting upregulation of activation markers is the consequence of increased magnitude or duration of activation, or both. Furthermore, while the TCR utilized in this manuscript has been shown to be reactive to IGRP265-273, it has also been indicated that this receptor might possess promiscuity towards other epitopes presented in the context of HLA-A2 (7). While a certain degree of cross-reactivity likely is advantageous in that the limited TCR repertoire within an individual can provide protection against a vast array of foreign exposures, it is possible that this promiscuity may also contribute to autoreactivity. Indeed, studies examining autoreactive receptors have shown promiscuity to a wide variety of endogenous and foreign epitopes (41, 42). One particular clone of note is the 1E6 TCR clone, which is reactive to PPI15-24 and has been shown to mediate β-cell killing (35). In a comprehensive set of experiments testing TCR degeneracy, this clone was shown to recognize more than one million peptides in the context of HLA-A2 (41). Thus, it is apparent that cross-reactivity could be a key feature of autoreactive receptors and an important mechanism of autoimmunity that warrants further investigation. With isogenic systems becoming an attractive means to model diseases (11), we propose that the use of gene edited T cell avatars, such as those presented herein, will aid in deciphering the mechanisms underlying dysregulation in the form of cell-cell interactions, chemotaxis, and phenotype and function of antigen specific T cells. As such, experiments are ongoing to track the movement of these T cells within an engineered islet niche (43). Further genetic engineering of T cell avatars and their targets will provide options to modulate key candidate genes and single nucleotide polymorphisms (SNPs) (10, 39) predicted to be important for phenotypes, immune cell or otherwise.
Though islet antigen specific CD8+ T cells have been implicated as key players in T1D disease pathogenesis (6, 44), we still know little about their phenotype and function in vivo. Data resulting from clinical trials support the critical need to understand CD8+ T cell signatures in order to unravel mechanisms central to autoimmune β-cell destruction. In particular, response signatures of CD8+ T cell exhaustion following teplizumab therapy (45) would indicate that inducing this phenotype could preserve β-cell function. However, other studies have observed human T1D antigen specific CD8+ T cells as having a “self-renewing” stem cell memory phenotype (46, 47), which would indicate that these cells are not as susceptible to exhaustion or tolerization as one would expect from cells that are chronically exposed to antigen (48). Bridging these gaps in understanding necessitates optimal in vitro modeling of autoreactive CD8+ T cell function to identify targetable molecules and pathways to reduce immune-mediated pathology through pharmacological or biological interventions.
ACT are at the forefront of clinical innovation, with an increased focus on genetic engineering to enhance the efficacy and safety of these cell products. In T1D, there is great interest in developing antigen-specific Treg ACT products (49, 50). Islet antigen-specific Tregs have the potential to suppress diabetogenic T cells in vitro (24) and in mouse models of T1D (51), but these cells have yet to effectively be used to combat disease in humans. Optimizing the activation of Tregs specific for β-cell antigens is of particular importance, as we have previously shown that engineered GAD555-567 specific Tregs possessing the higher affinity clone R164 TCR outperformed the lower affinity clone 4.13 TCR Tregs, which recognize the same peptide, in antigen-specific suppressive capacity (24). While both low- and high-avidity TCR interactions in Tregs appear to be important for suppression of autoimmunity (52), Tregs possessing higher affinity receptors have been shown to preferentially home to the pancreatic lymph node and islets, and express inhibitory receptors to a greater degree than lower affinity Tregs (52). Therefore, enhancing the avidity of an antigen-specific Treg cell product via endogenous TRAC KO may confer several functional advantages.
4.1 Limitations of the study
Our study focuses on a single HLA class I restricted receptor, thus the impact of endogenous TCR deletion on receptors of different specificities, HLA restrictions, and affinities in T1D is unknown, and though others have seen success with higher affinity tumor-reactive receptors (21, 22), recapitulating the phenotype and function of a lower affinity receptor may be more challenging. Additionally, we utilized an immortalized β-cell line, which is not functionally identical to primary human islets, thus future efforts testing this workflow with primary human islet material will be necessary, particularly for specificities such as derivatives of preproinsulin, or post-translationally modified epitopes (14). Moreover, in our study we have targeted the TRAC locus, as the paired TCRαβ dimer is necessary for full function of the TCR (53), though we acknowledge the possibility of residual mispairing between the endogenous β- and de novo α-chain, as well as potential alloreactivity due to incomplete HLA matching. Additionally, the field of genome editing is rapidly evolving, and newer methodologies allowing for targeted TCR delivery and regulation via the endogenous promoter (54), as well as base editing strategies enabling induction of a stop codon with minimal DNA damage (55), though lower efficiency at the time of writing, may soon provide more context appropriate results or products for ACT. We also recognize that utilizing humanized animal models or cell and tissue engineering strategies will likely be necessary to confirm in vivo functionality (56) and obtain a complete picture of islet-immune interactions (43). These studies may include investigating autoantigen specific T cell function, homing, and long-term engraftment. Along a similar vein, T1D is a highly polygenic disease (57), and this study was not designed to investigate the influence of T1D risk loci on T cell avatar function, which is known to alter TCR signals (25). We are currently working to establish isogenic systems in order to appropriately test the contributions of key SNPs which may modulate TCR signaling and cytotoxic T cell function (11).
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
Ethical review and approval was not required for studies using purchased human blood products in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required as these studies were conducted using blood products acquired from LifeSouth Blood Centers under Institutional Review Board exempt protocols in accordance with the national legislation and institutional requirements.
Author contributions
LDP designed the studies, conducted experiments, acquired data, analyzed data, acquired funding, and wrote the manuscript. W-IY designed the studies, conducted experiments, acquired data, analyzed data, and reviewed/edited the manuscript. JMA conducted experiments, acquired data, and reviewed/edited the manuscript. MEB conducted experiments, acquired data, and reviewed/edited the manuscript. ALP contributed to discussion and reviewed/edited the manuscript. CEM contributed to discussion and reviewed/edited the manuscript. TMB conceptualized the project, designed studies, provided reagents, acquired funding, and reviewed/edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
These studies were supported by grants from the National Institutes of Health (UH3 DK122638, P01 AI042288, and R01 DK106191 to TMB; T32 DK108736 and F31 DK129004 to LDP), as well as programmatic support from The Leona M. and Harry B. Helmsley Charitable Trust.
Acknowledgments
We would like to thank the individuals who donated blood to make these studies possible, as well as all members of the Brusko laboratory, especially Thinzar Myint, for help with coordination and planning, and Dr. Melanie Shapiro for insightful commentary. We would also like to thank colleagues in partnership with the HIRN Consortium for Human Islet Biomimetics for helpful discussions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1142648/full#supplementary-material
References
1. Peters L, Posgai A, Brusko TM. Islet–immune interactions in type 1 diabetes: the nexus of beta cell destruction. Clin Exp Immunol (2019) 198(3):326–40. doi: 10.1111/cei.13349
2. Baker RL, Delong T, Barbour G, Bradley B, Nakayama M, Haskins K. Cutting edge: CD4 T cells reactive to an islet amyloid polypeptide peptide accumulate in the pancreas and contribute to disease pathogenesis in nonobese diabetic mice. J Immunol (2013) 191(8):3990–4. doi: 10.4049/jimmunol.1301480
3. Crawford F, Stadinski B, Jin N, Michels A, Nakayama M, Pratt P, et al. Specificity and detection of insulin-reactive CD4 +T cells in type 1 diabetes in the nonobese diabetic (NOD) mouse. Proc Natl Acad Sci U S A (2011) 108(40):16729–34. doi: 10.1073/pnas.1113954108
4. Jarchum I, Baker JC, Yamada T, Takaki T, Marron MP, Serreze DV, et al. In vivo Cytotoxicity of insulin-specific CD8+ T-cells in HLA-A*0201 transgenic NOD mice. Diabetes (2007) 56(10):2551–60. doi: 10.2337/db07-0332
5. Jarchum I, Nichol L, Trucco M, Santamaria P, DiLorenzo TP. Identification of novel IGRP epitopes targeted in type 1 diabetes patients. Clin Immunol (2008) 127(3):359–65. doi: 10.1016/j.clim.2008.01.015
6. Coppieters KT, Dotta F, Amirian N, et al. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J Exp Med (2012) 209(1). doi: 10.1084/jem.20111187
7. Babad J, Mukherjee G, Follenzi A, Campbell PD, Kay TW, Atkinson MA, et al. Generation of β cell-specific human cytotoxic T cells by lentiviral transduction and their survival in immunodeficient human leucocyte antigen-transgenic mice. Clin Exp Immunol (2015) 179(3):51–60. doi: 10.1111/cei.12465
8. Kemp RA, Bäckström BT, Ronchese F. The phenotype of type 1 and type 2 CD8+ T cells activated in vitro is affected by culture conditions and correlates with effector activity. Immunology (2005) 115(3):398–413. doi: 10.1111/j.1365-2567.2005.02168.x
9. Dean JW, Peters LD, Fuhrman CA, Seay HR, Posgai AL, Stimpson SE, et al. Innate inflammation drives NK cell activation to impair treg activity. J Autoimmun (2020) 108:102417. doi: 10.1016/j.jaut.2020.102417
10. Newby BN, Brusko TM, Zou B, Atkinson MA, Clare-Salzler M, Mathews CE. Type 1 interferons potentiate human cd8+ t-cell cytotoxicity through a stat4-and granzyme b-dependent pathway. Diabetes (2017) 66(12):3061–71. doi: 10.2337/db17-0106
11. Armitage LH, Stimpson SE, Santostefano KE, Sui L, Ogundare S, Newby BN, et al. Use of induced pluripotent stem cells to build isogenic systems and investigate type 1 diabetes. Front Endocrinol (Lausanne) (2021) 12:737276. doi: 10.3389/fendo.2021.737276
12. Seay HR, Yusko E, Rothweiler SJ, Zhang L, Posgai AL, Campbell-Thompson M, et al. Tissue distribution and clonal diversity of the T and b cell repertoire in type 1 diabetes. JCI Insight (2016) 1(20):e88242. doi: 10.1172/jci.insight.88242
13. Anderson AM, Landry LG, Alkanani AA, Pyle L, Powers AC, Atkinson MA, et al. Human islet T cells are highly reactive to preproinsulin in type 1 diabetes. Proc Natl Acad Sci U S A (2021) 118(41):e2107208118. doi: 10.1073/pnas.2107208118
14. Babon JAB, Denicola ME, Blodgett DM, Crèvecoeur I, Buttrick TS, Maehr R, et al. Analysis of self-antigen specificity of islet-infiltrating T cells from human donors with type 1 diabetes. Nat Med (2016) 22(12):264. doi: 10.1038/nm.4203
15. Bauer W, Veijola R, Lempainen J, Kiviniemi M, Härkönen T, Toppari J, et al. Age at seroconversion, HLA genotype, and specificity of autoantibodies in progression of islet autoimmunity in childhood. J Clin Endocrinol Metab (2019) 104(10):4521–30. doi: 10.1210/jc.2019-00421
16. Krischer JP, Lynch KF, Lernmark A, Hagopian WA, Rewers MJ, She JX, et al. Genetic and environmental interactions modify the risk of diabetes-related autoimmunity by 6 years of age: the teddy study. Diabetes Care (2017) 40(9):1194–202. doi: 10.2337/dc17-0238
17. Krishna C, Chowell D, Gönen M, Elhanati Y, Chan TA. Genetic and environmental determinants of human TCR repertoire diversity. Immun Ageing (2020) 17(1). doi: 10.1186/s12979-020-00195-9
18. Lanfermeijer J, de Greef PC, Hendriks M, Vos M, van Beek J, Borghans JAM, et al. Age and CMV-infection jointly affect the EBV-specific CD8+ T-cell repertoire. Front Aging (2021) 2:665637. doi: 10.3389/fragi.2021.665637
19. Laban S, Suwandi JS, van Unen V, Pool J, Wesselius J, Höllt T, et al. Heterogeneity of circulating CD8 T-cells specific to islet, neo-antigen and virus in patients with type 1 diabetes mellitus. PloS One (2018) 13(8):e0200818. doi: 10.1371/journal.pone.0200818
20. Cohen CJ, Li YF, El-Gamil M, Robbins PF, Rosenberg SA, Morgan RA. Enhanced antitumor activity of T cells engineered to express T-cell receptors with a second disulfide bond. Cancer Res (2007) 67(8). doi: 10.1158/0008-5472.CAN-06-3986
21. Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, et al. CRISPR-engineered T cells in patients with refractory cancer. Science (1979) (2020) 367(6481):3898–903. doi: 10.1126/science.aba7365
22. Roth TL, Puig-Saus C, Yu R, Shifrut E, Carnevale J, Li PJ, et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature (2018) 559(7714):405–9. doi: 10.1038/s41586-018-0326-5
23. Standifer NE, Ouyang Q, Panagiotopoulos C, Verchere CB, Tan R, Greenbaum CJ, et al. Identification of novel HLA-A*0201-restricted epitopes in recent-onset type 1 diabetic subjects and antibody-positive relatives. Diabetes (2006) 55(11):3061–7. doi: 10.2337/db06-0066
24. Yeh WI, Seay HR, Newby B, Posgai AL, Moniz FB, Michels A, et al. Avidity and bystander suppressive capacity of human regulatory T cells expressing de novo autoreactive T-cell receptors in type 1 diabetes. Front Immunol (2017) 26(8):1313. doi: 10.3389/fimmu.2017.01313
25. Perry DJ, Peters LD, Lakshmi PS, Zhang L, Han Z, Wasserfall CH, et al. Overexpression of the PTPN22 autoimmune risk variant LYP-620W fails to restrain human CD4 + T cell activation. J Immunol (2021) 207(3):849–59. doi: 10.4049/jimmunol.2000708
26. Seki A, Rutz S. Optimized RNP transfection for highly efficient CRI SPR/Cas9-mediated gene knockout in primary T cells. J Exp Med (2018) 215(3):985–97. doi: 10.1084/jem.20171626
27. Lightfoot YL, Chen J, Mathews CE. Role of the mitochondria in immune-mediated apoptotic death of the human pancreatic β cell line βLox5. PloS One (2011) 6(6). doi: 10.1371/journal.pone.0020617
28. Schaft N, Willemsen RA, de Vries J, Lankiewicz B, Essers BW, Gratama JW, et al. Peptide fine specificity of anti-glycoprotein 100 CTL is preserved following transfer of engineered TCRαβ genes into primary human T lymphocytes. J Immunol (2003) 170(4):2186-94. doi: 10.4049/jimmunol.170.4.2186
29. Madley R, Nauman G, Danzl N, Borsotti C, Khosravi Maharlooei M, et al. Negative selection of human T cells recognizing a naturally-expressed tissue-restricted antigen in the human thymus. J Transl Autoimmun (2020) 3:100061. doi: 10.1016/j.jtauto.2020.100061
30. Schuster C, Gerold KD, Schober K, Probst L, Boerner K, Kim MJ, et al. The autoimmunity-associated gene CLEC16A modulates thymic epithelial cell autophagy and alters T cell selection. Immunity (2015) 42(5):942–52. doi: 10.1016/j.immuni.2015.04.011
31. Campillo-Davo D, Flumens D, Lion E. The quest for the best: how TCR affinity, avidity, and functional avidity affect TCR-engineered T-cell antitumor responses. Cells (2020) 9(7). doi: 10.3390/cells9071720
32. van Loenen MM, de Boer R, Amir AL, Hagedoorn RS, Volbeda GL, Willemze R, et al. Mixed T cell receptor dimers harbor potentially harmful neoreactivity. Proc Natl Acad Sci U S A (2010) 107(24):10972–7. doi: 10.1073/pnas.1005802107
33. Zhong S, Malecek K, Johnson LA, Yu Z, Vega-Saenz de Miera E, Darvishian F, et al. T-Cell receptor affinity and avidity defines antitumor response and autoimmunity in T-cell immunotherapy. Proc Natl Acad Sci U S A (2013) 110(17):6973–8. doi: 10.1073/pnas.1221609110
34. Frazer GL, Gawden-Bone CM, Dieckmann NMG, Asano Y, Griffiths GM. Signal strength controls the rate of polarization within ctls during killing. J Cell Biol (2021) 220(10). doi: 10.1083/jcb.202104093
35. Skowera A, Ellis RJ, Varela-Calviño R, Arif S, Huang GC, Van-Krinks C, et al. CTLs are targeted to kill β cells in patients with type 1 diabetes through recognition of a glucose-regulated preproinsulin epitope. J Clin Invest (2008) 118(10):3390–402. doi: 10.1172/JCI35449
36. Redeker A, Welten SPM, Baert MRM, Vloemans SA, Tiemessen MM, Staal FJ, et al. The quantity of autocrine IL-2 governs the expansion potential of CD8 + T cells. J Immunol (2015) 195(10):4792–801. doi: 10.4049/jimmunol.1501083
37. Muller YD, Ferreira LMR, Ronin E, Ho P, Nguyen V, Faleo G, et al. Precision engineering of an anti-HLA-A2 chimeric antigen receptor in regulatory T cells for transplant immune tolerance. Front Immunol (2021) 12:686439. doi: 10.3389/fimmu.2021.686439
38. Dolton G, Lissina A, Skowera A, Ladell K, Tungatt K, Jones E, et al. Comparison of peptide-major histocompatibility complex tetramers and dextramers for the identification of antigen-specific T cells. Clin Exp Immunol (2014) 177(1):47–63. doi: 10.1111/cei.12339
39. Laugel B, van den Berg HA, Gostick E, Cole DK, Wooldridge L, Boulter J, et al. Different T cell receptor affinity thresholds and CD8 coreceptor dependence govern cytotoxic T lymphocyte activation and tetramer binding properties. J Biol Chem (2007) 282(33):23799–810. doi: 10.1074/jbc.M700976200
40. Kao C, Daniels MA, Jameson SC. Loss of CD8 and TCR binding to class I MHC ligands following T cell activation. Int Immunol (2005) 17(12):1607–17. doi: 10.1093/intimm/dxh340
41. Wooldridge L, Ekeruche-Makinde J, Van Den Berg HA, Skowera A, Miles JJ, Tan MP, et al. A single autoimmune T cell receptor recognizes more than a million different peptides. J Biol Chem (2012) 287(2):1168–77. doi: 10.1074/jbc.M111.289488
42. Hansen BE, Rasmussen AH, Jakobsen BK, Ryder LP, Svejgaard A. Extraordinary cross-reactivity of an autoimmune T-cell receptor recognizing specific peptides both on autologous and on allogeneic HLA class II molecules. Tissue Antigens (2007) 70(1):42–52. doi: 10.1111/j.1399-0039.2007.00849.x
43. Patel S, Mathews C, Chandler R, Stabler C. The foundation for engineering a pancreatic islet niche. Front Endocrinol (Lausanne) (2022) 13:881525. doi: 10.3389/fendo.2022.881525
44. Culina S, Lalanne AI, Afonso G, Cerosaletti K, Pinto S, Sebastiani G, et al. Islet-reactive CD8+ T cell frequencies in the pancreas, but not in blood, distinguish type 1 diabetic patients from healthy donors. Sci Immunol (2018) 3(20):eaao4013. doi: 10.1126/sciimmunol.aao4013
45. Long SA, Thorpe J, DeBerg HA, Gersuk V, Eddy J, Harris KM, et al. Partial exhaustion of CD8 T cells and clinical response to teplizumab in new-onset type 1 diabetes. Sci Immunol (2016) 1(5):eaai7793. doi: 10.1126/sciimmunol.aai7793
46. Vignali D, Cantarelli E, Bordignon C, Canu A, Citro A, Annoni A, et al. Detection and characterization of CD8+ autoreactive memory stem T cells in patients with type 1 diabetes. Diabetes (2018) 67(5):936–45. doi: 10.2337/db17-1390
47. Abdelsamed HA, Zebley CC, Nguyen H, Rutishauser RL, Fan Y, Ghoneim HE, et al. Beta cell-specific CD8+ T cells maintain stem cell memory-associated epigenetic programs during type 1 diabetes. Nat Immunol (2020) 21(5):578–87. doi: 10.1038/s41590-020-0633-5
48. Mueller SN, Ahmed R. High antigen levels are the cause of T cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A (2009) 106(21):8623–8. doi: 10.1073/pnas.0809818106
49. Marshall GP II, Cserny J, Perry DJ, Yeh WI, Seay HR, Elsayed AG, et al. Clinical applications of regulatory T cells in adoptive cell therapies. Cell Gene Ther Insights (2018) 4(1):405–29. doi: 10.18609/cgti.2018.042
50. Dong S, Hiam-Galvez KJ, Mowery CT, Herold KC, Gitelman SE, Esensten JH, et al. The effect of low-dose IL-2 and treg adoptive cell therapy in patients with type 1 diabetes. JCI Insight (2021) 6(18):e147474. doi: 10.1172/jci.insight.147474
51. Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, et al. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med (2004) 199(11):1455–65. doi: 10.1084/jem.20040139
52. Sprouse ML, Shevchenko I, Scavuzzo MA, Joseph F, Lee T, Blum S, et al. Cutting edge: low-affinity TCRs support regulatory T cell function in autoimmunity. J Immunol (2018) 200(3):909–14. doi: 10.4049/jimmunol.1700156
53. Kamali E, Rahbarizadeh F, Hojati Z, Frödin M. CRISPR/Cas9-mediated knockout of clinically relevant alloantigenes in human primary T cells. BMC Biotechnol (2021) 21(1). doi: 10.1186/s12896-020-00665-4
54. Müller TR, Jarosch S, Hammel M, Leube J, Grassmann S, Bernard B, et al. Targeted T cell receptor gene editing provides predictable T cell product function for immunotherapy. Cell Rep Med (2021) 2(8):100374. doi: 10.1016/j.xcrm.2021.100374
55. Newby GA, Yen JS, Woodard KJ, Mayuranathan T, Lazzarotto CR, Li Y, et al. Base editing of haematopoietic stem cells rescues sickle cell disease in mice. Nature (2021) 595(7866):295–302. doi: 10.1038/s41586-021-03609-w
56. Tan S, Li Y, Xia J, Jin CH, Hu Z, Duinkerken G, et al. Type 1 diabetes induction in humanized mice. Proc Natl Acad Sci U S A (2017) 114(41):10954–9. doi: 10.1073/pnas.1710415114
57. Johnson MB, Patel KA, de Franco E, Houghton JAL, McDonald TJ, Ellard S, et al. A type 1 diabetes genetic risk score can discriminate monogenic autoimmunity with diabetes from early-onset clustering of polygenic autoimmunity with diabetes. Diabetologia (2018) 61(4):862–9. doi: 10.1007/s00125-018-4551-0
Keywords: CD8 T cell, T cell receptor knockout, type 1 diabetes, gene editing, autoimmunity
Citation: Peters LD, Yeh W-I, Arnoletti JM, Brown ME, Posgai AL, Mathews CE and Brusko TM (2023) Modeling cell-mediated immunity in human type 1 diabetes by engineering autoreactive CD8+ T cells. Front. Immunol. 14:1142648. doi: 10.3389/fimmu.2023.1142648
Received: 11 January 2023; Accepted: 13 April 2023;
Published: 30 May 2023.
Edited by:
Leonardo M. R. Ferreira, Medical University of South Carolina, United StatesReviewed by:
Remi J. Creusot, Columbia University, United StatesNikolaos Skartsis, Mayo Clinic, United States
Copyright © 2023 Peters, Yeh, Arnoletti, Brown, Posgai, Mathews and Brusko. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Todd M. Brusko, tbrusko@ufl.edu
†ORCID: Todd M. Brusko, orcid.org/0000-0003-2878-9296