 Alexander T. Fields1
Alexander T. Fields1 Elizabeth A. Andraska2
Elizabeth A. Andraska2 Christof Kaltenmeier2
Christof Kaltenmeier2 Zachary A. Matthay1
Zachary A. Matthay1 Kimberly Herrera1
Kimberly Herrera1 Brenda Nuñez-Garcia1Chayse M. Jones3Katherine D. Wick3,4Silvia Liu5Jian-Hua Luo5Yan-Ping Yu5
Brenda Nuñez-Garcia1Chayse M. Jones3Katherine D. Wick3,4Silvia Liu5Jian-Hua Luo5Yan-Ping Yu5 Michael A. Matthay3,4Carolyn M. Hendrickson3Roland J. Bainton6
Michael A. Matthay3,4Carolyn M. Hendrickson3Roland J. Bainton6 Tessa J. Barrett7
Tessa J. Barrett7 Jeffrey S. Berger7,8,9
Jeffrey S. Berger7,8,9 Matthew D. Neal2
Matthew D. Neal2 Lucy Z. Kornblith1* the COVID-19 Associated Coagulopathy Inflammation Thrombosis (Co-ACIT) Study Group
Lucy Z. Kornblith1* the COVID-19 Associated Coagulopathy Inflammation Thrombosis (Co-ACIT) Study Group- 1Department of Surgery, University of California, San Francisco, Zuckerberg San Francisco General Hospital, San Francisco, CA, United States
- 2Trauma and Transfusion Medicine Research Center, Department of Surgery, University of Pittsburgh, Pittsburgh, PA, United States
- 3Division of Pulmonary, Critical Care, Allergy and Sleep Medicine, Department of Medicine, University of California, San Francisco, San Francisco, CA, United States
- 4Cardiovascular Research Institute, University of California, San Francisco, San Francisco, CA, United States
- 5Department of Pathology, School of Medicine, University of Pittsburgh, Pittsburgh, PA, United States
- 6Department of Anesthesia and Perioperative Care, School of Medicine, University of California, San Francisco, San Francisco, CA, United States
- 7Leon H. Charney Division of Cardiology, Department of Medicine, New York University (NYU) Grossman School of Medicine, New York, NY, United States
- 8New York University (NYU) Center for the Prevention of Cardiovascular Disease, New York University (NYU) Langone Health, New York, NY, United States
- 9Division of Vascular Surgery, Department of Surgery, New York University (NYU) Grossman School of Medicine, New York, NY, United States
Introduction: Thromboinflammatory complications are well described sequalae of Coronavirus Disease 2019 (COVID-19), and there is evidence of both hyperreactive platelet and inflammatory neutrophil biology that contributes to the thromoinflammatory milieu. It has been demonstrated in other thromboinflammatory diseases that the circulating environment may affect cellular behavior, but what role this environment exerts on platelets and neutrophils in COVID-19 remains unknown. We tested the hypotheses that 1) plasma from COVID-19 patients can induce a prothrombotic platelet functional phenotype, and 2) contents released from platelets (platelet releasate) from COVID-19 patients can induce a proinflammatory neutrophil phenotype.
Methods: We treated platelets with COVID-19 patient and disease control plasma, and measured their aggregation response to collagen and adhesion in a microfluidic parallel plate flow chamber coated with collagen and thromboplastin. We exposed healthy neutrophils to platelet releasate from COVID-19 patients and disease controls and measured neutrophil extracellular trap formation and performed RNA sequencing.
Results: We found that COVID-19 patient plasma promoted auto-aggregation, thereby reducing response to further stimulation ex-vivo. Neither disease condition increased the number of platelets adhered to a collagen and thromboplastin coated parallel plate flow chamber, but both markedly reduced platelet size. COVID-19 patient platelet releasate increased myeloperoxidasedeoxyribonucleic acid complexes and induced changes to neutrophil gene expression.
Discussion: Together these results suggest aspects of the soluble environment circulating platelets, and that the contents released from those neutrophil behavior independent of direct cellular contact.
1 Introduction
Thromboinflammatory complications are well described sequalae of Coronavirus Disease 2019 (COVID-19) (1), mediated by cellular and non-cellular thrombotic and inflammatory derangements resulting from SARS-CoV-2 infection (2, 3). Platelets are known as a crucial link between thrombosis and inflammation, and in COVID-19 there is evidence of alterations to platelet behavior including to platelet hemostatic function, cytokine production (4, 5), and effector cell properties demonstrated by induction of neutrophil release of extracellular traps (6, 7) and promotion of an inflammatory hypercoagulable endotheliopathy (3).
Platelets are classically known for their role in thrombus formation and hemostasis; however, evidence of their interaction with and regulation of immune cells and participation in inflammatory processes, both infectious and non-infectious, have been a crucial link in understanding thromboinflammatory diseases (8, 9). In the setting of COVID-19 platelets have been found to have altered functional responses and gene expression profiles (4, 10), as well as induce myeloid and endothelial cell activation (3, 4, 11). As is the case in many inflammatory and infectious diseases, a variety of inflammatory and thrombosis-related signaling molecules are released from platelets and other cell types in COVID-19 patients, including serotonin, platelet factor 4, and monocyte chemoattractant protein 1 (5, 7, 12–14). Increased platelet activity and associated release of cytokines recruit leukocytes, the most abundant being the neutrophil, which can engage in a number of pro-inflammatory immune behaviors contributing to COVID-19 symptoms and outcomes. Biologically, these pro-inflammatory behaviors include formation of neutrophil extracellular traps (NETs), in a process referred to as NETosis (15, 16). NETosis is a link in the thromboinflammatory continuum, as NETs have been observed in microvascular occlusions in COVID-19 patient lung tissue that was also co-enriched in platelet factor 4 (7). Together these observations suggest a high degree of crosstalk between thrombotic and inflammatory systems, and evidence that this crosstalk may not require direct cell-cell interactions, but that the soluble microenvironment may also play a key role. In fact, in other diseases characterized by thromboinflammation, there is also evidence that the circulating soluble environment can alter platelet behavior (17–19). However, what role the circulating soluble environment of COVID-19 has on platelet and neutrophils remains incompletely elucidated. Here we test the hypotheses that 1) plasma from COVID-19 patients can induce a prothrombotic platelet functional phenotype, and 2) contents released from platelets (platelet releasate) from COVID-19 patients can induce a proinflammatory neutrophil phenotype.
2 Methods
2.1 Patient samples
Patient samples were collected with the approval of the University of California, San Francisco’s Institutional Review Board (IRB). Whole blood samples were collected under an initial waiver of consent from adults (>18 years) undergoing evaluation for possible COVID-19 on arrival to the Zuckerberg San Francisco General Hospital (ZSFG) emergency department based on fever, respiratory symptoms (cough, shortness of breath, upper respiratory infection, wheezing), gastrointestinal symptoms (diarrhea, nausea/vomiting), and/or changes in taste or smell as part of the overarching COVID-19 Associated Coagulopathy, Inflammation, and Thrombosis (Co-ACIT) Study. COVID-19 diagnosis in these patients was based on a positive or negative SARS-CoV-2 polymerase chain reaction (PCR) test performed in the course of clinical care. This created tandem cohorts of COVID-19 patients and symptom-matched disease control patients. COVID-19 patients and disease controls were followed until discharge from the emergency department or hospital, and comprehensive patient characteristics, physiological and laboratory parameters, and clinical outcomes were collected.
2.2 Assays of patient samples
On whole blood samples from COVID-19 patients, and disease controls, clotting function was assessed by Thromboelastography (TEG) using a TEG6s analyzer (Haemonetics, Boston, MA) with the global hemostasis and lysis cartridge, which performs a resonance-based quantitative assessment of clot formation, maintenance and breakdown. Platelet aggregation was assessed pre- and post-stimulation with agonists adenosine diphosphate (ADP), collagen, a PAR1 agonist hexapeptide (thrombin-receptor activating peptide-6 (20), arachidonic acid, or ristocetin, using multiple electrode impedance aggregometry via the Multiplate analyzer (Roche, Basel, Switzerland), according to manufacturer instructions. Agonists were purchased from Hart Biologicals (Hartlepool, UK). Biomarkers of thrombosis and inflammation present in plasma were measured by enzyme-linked immunosorbent assays (ELISA).
2.3 Preparation of plasma and platelet releasate
Plasma was isolated by centrifugation at 2960xg for 10 minutes and stored at -80°C. Contents released from platelets (platelet releasate) were generated as previously described (3). Briefly, whole blood was centrifuged at 200xg for 10 minutes, and resulting platelet rich plasma was centrifuged at 1000xg for 10 minutes in the presence of 1:10 acid citrate dextrose solution A anticoagulant. The resulting platelets were resuspended in HEPES-buffered Tyrode’s solution with 1μM Prostaglandin-E1 (Cayman Chemical) and counted. The platelets were pelleted again and resuspended at 5x105/μL in HEPES-buffered Tyrode’s and supplemented with 2mM CaCl2 to allow for content release. The resulting platelet releasate was collected and stored at -80°C. Healthy donor whole blood samples were obtained and processed in the same way for the purpose of having control treatment groups in the ex-vivo experiments described below.
2.4 Ex-vivo platelet assays
Ex-vivo platelet assays were performed based on previously described methods (17). Briefly, platelets from healthy donors were isolated by centrifugation at 200xg for 20 minutes to generate platelet rich plasma, which was then centrifuged to generate a platelet pellet, which was washed and resuspended in HEPES-buffered Tyrode’s solution supplemented with 3mg/mL bovine serum albumin. The platelet count was adjusted to 2x105/μL. The platelets were allowed to rest for 30 minutes and then treated with 12% (v/v) patient plasma for 30 minutes (19). Treated platelets were subjected to measurements of adhesion and aggregation by microfluidic adhesion assays and multiple electrode impedance aggregometry, respectively. Microfluidic adhesion assays were performed as previously described (21, 22). Briefly, treated platelets were perfused through a parallel plate flow chamber (Ibidi, Grafelfing, Germany) coated with 100μg/mL of collagen (Rat tail Type I, Corning) and thromboplastin (Extem reagent, Werfen, Barcelona, Spain) at approximate venous shear (100s-1) for 10 minutes. In order to avoid artifactual attachment, unattached platelets were washed out of the chamber with HEPES-buffered Tyrode’s solution at approximate arterial shear (1000s-1) for 20 minutes (22). Attached platelets were counted and imaged under phase contrast at 60x magnification. Area of attached platelets was quantified using FIJI (23). Aggregometry was performed as described above with the agonist collagen (Hart Biologicals) to parallel the collagen coating in the adhesion assays. Data were analyzed using Excel (Microsoft) and R (www.r-project.org). For presentation, all ex-vivo treated platelet data were normalized to autologous healthy control plasma treatment of matched parallel samples of platelets isolated and treated on the same day (i.e., platelets treated with plasma from the same donor).
2.5 Ex vivo neutrophil assays
Neutrophils isolated from healthy donors were exposed to platelet releasate from COVID-19 patients and disease controls, and neutrophil extracellular trap (NET) formation was assessed by ELISA for myeloperoxidase (MPO)-deoxyribonucleic acid (DNA) and gene expression changes by ribonucleic acid (RNA) sequencing.
2.5.1 Neutrophil isolation and stimulation
Whole blood was drawn from a single male donor (to limit donor heterogeneity for sequencing) with sodium heparin, treated with 3% dextran, and then washed at 500g for 5 min. Red blood cells were lysed with H20 for 30 seconds followed by staining with CD66b PE (Thermo Fisher) and CD16 APC (Thermo Fisher). Cells were sorted using a BD FACSAria. Cells were then plated at a density of 150,000 cells per well. Wells rested for one hour and were then treated with platelet releasate (1:2 dilution) for four hours. Positive control wells were treated with PMA (250nM) or Ionomycin (2.5ug/ml). All experiments were performed in duplicates. After four hours of treatment the supernatant was removed for NETosis capture ELISA.
2.5.2 NETosis quantification
NETosis was quantified with an ELISA detecting MPO-DNA complexes (24). After treatment with platelet releasate or positive control, cell supernatant was aspirated and incubated on a 96 well plate from a commercial MPO ELISA kit (Hycult biotech) and then NETs were measured using MPO (Roche) associated with DNA ELISA (Roche). Samples were analyzed on a plate reader at 405nm. Each sample was plated in duplicate, and values were reported as average of the duplicates.
2.5.3 Neutrophil bulk RNA sequencing
Following four hours of treatment with platelet releasate, stimulated neutrophils were harvested and washed twice in PBS (500g x 5 min). RNA was isolated according to the TRIzol Reagent protocol (Invitrogen). RNA integrity (RIN) was determined, and samples were further processed if RIN > 7 and purity of > 1.9 (260nm/280nm). The sequencing core at the University of Pittsburgh was given at least 100ng RNA in duplicates for further sequencing. Samples were sequenced using NovaSeq6000 platform (Illumina). The sequencing reads were then quality controlled. Reads were aligned by STAR aligner. Based on aligned RNA-seq data, gene counts were quantified for each library. Differential expression analysis was performed with R package DESeq2. Differentially expressed genes (DEGs) were defined by false discovery rate (FDR)≤5%, and DEGs with changes larger than 1.5-fold were further applied into Ingenuity Pathway Analysis (Qiagen) to detect pathways enriched by the gene alterations. Significant pathways were defined by FDR≤ 5%.
The above experimental strategies are represented schematically in Figure 1.
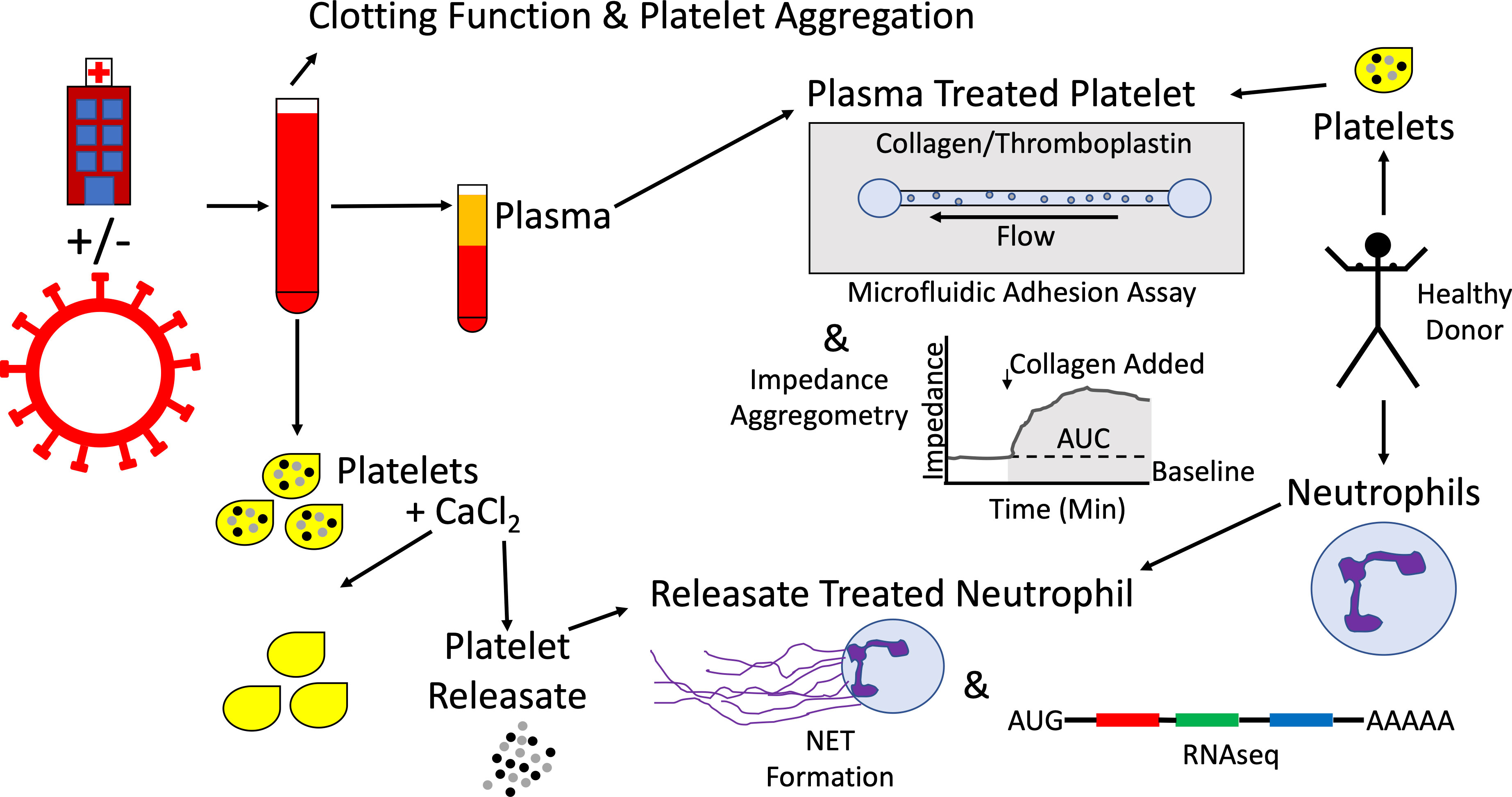
Figure 1 Schematic representation of experimental strategy. Patients being evaluated for COVID-19 were enrolled on arrival to the emergency department, yielding tandem cohorts of COVID-19 patients and symptom-matched disease control patients. Whole blood was collected and clotting function and platelet aggregation were assessed by thromboelastography and multiple electrode impedance aggregometry, respectively. Whole blood was processed into plasma and stored at -80°C. Platelets were isolated, exposed to calcium chloride, and the resulting platelet releasate was collected and stored at -80°C. Separately, platelets were isolated from healthy donors, exposed to plasma samples, and their adhesion and aggregation properties were studied using a microfluidic parallel plate flow chamber and multiple electrode impedance aggregometry, respectively. Neutrophils were isolated from healthy donors and exposed to platelet releasates from COVID-19 patients and disease controls. Neutrophil extracellular trap (NET) formation was assayed and neutrophil RNA was sequenced.
3 Results
3.1 Coagulation and inflammation characteristics of plasma and platelet releasate donors
3.1.1 Plasma donors
The demographics, clinical characteristics, global clotting function, platelet aggregation function, and thrombosis and inflammation biomarkers of the COVID-19 patient and disease control plasma donors are shown in Table 1. In general, COVID-19 patient plasma donors had some evidence of a prothrombotic and proinflammatory biomarker profile relative to disease controls. This is evidenced by trends toward elevated levels of prothrombotic biomarkers such as fibrinogen (median 477.5 vs 355mg/dL), and antiplasmin (median 72 vs 65.7% activity), increased clot strength due to fibrinogen (median TEG Functional Fibrinogen maximum amplitude 35.45 vs 23.4mm), and increases in platelet aggregation in response to stimulation with ADP (median 67.5 vs 40 AUC units) and ristocetin (median 81 vs 56 AUC units) in COVID-19 patients relative to disease controls. Additionally, COVID-19 patients had a trend toward elevated levels of proinflammatory biomarkers such as Interferon γ-inducible protein 10 (IP10, median 450 vs 77.5pg/mL), interleukin 10 (IL-10, 13.6 vs 6.7pg/mL), and receptor for advanced glycation end-products (RAGE, median 3091.6 vs 720.4 pg/mL) relative to disease controls.
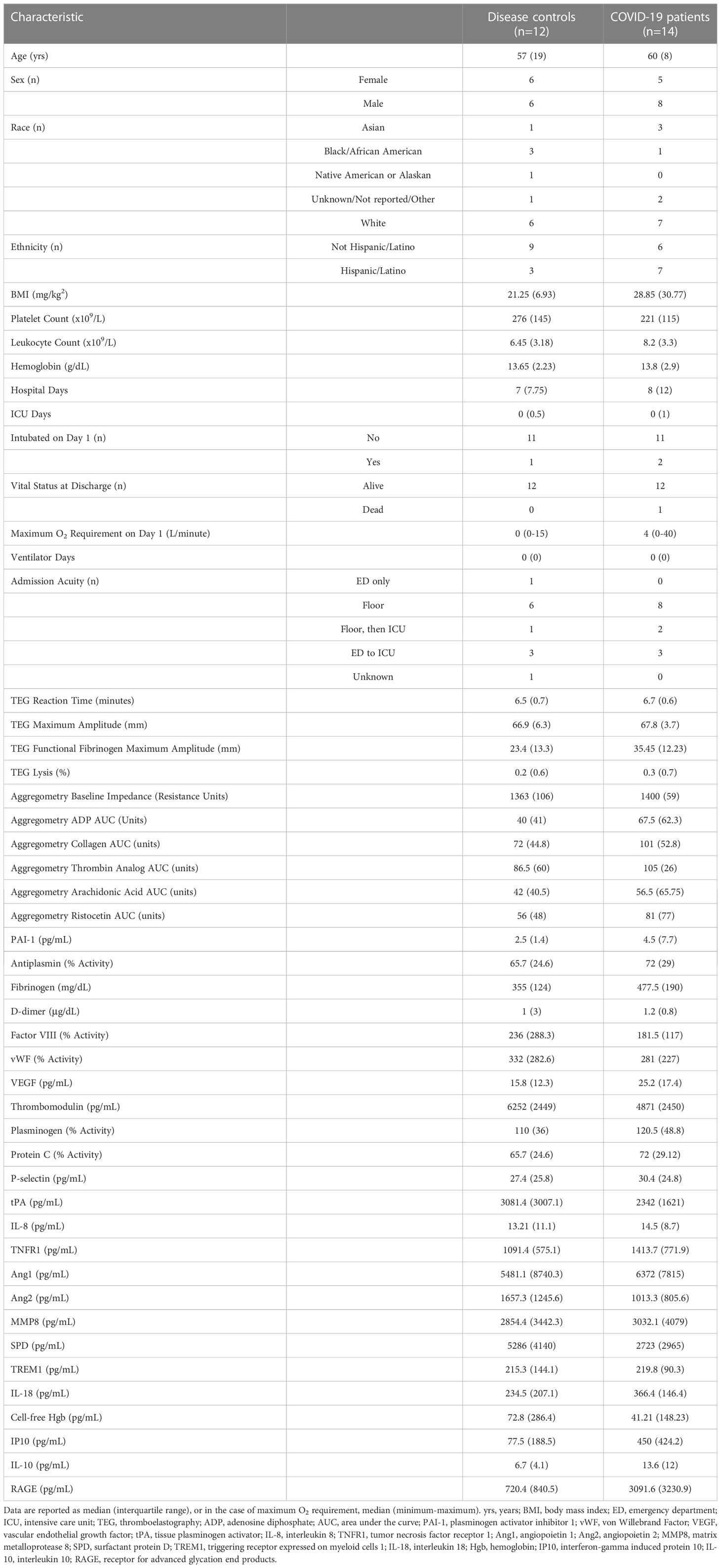
Table 1 Characteristics of plasma donors.
3.1.2 Platelet releasate donors
The demographics, clinical characteristics, global clotting function, platelet aggregation function, and thrombosis and inflammation biomarkers of the COVID-19 patient and disease control platelet releasate donors are shown in Table 2. Overall, similar to the plasma donors, COVID-19 platelet releasate donors had prothrombotic and proinflammatory biomarker profiles compared to disease controls. COVID-19 patients had a trend toward elevated levels of prothrombotic biomarkers such as PAI-1 (median 4.5 vs 2.6pg/mL), plasminogen (median 101.5 vs 78% activity), and fibrinogen (median 393 vs 318mg/dL), evidence of increased clot strength (median TEG overall maximum amplitude 67.3mm vs 63.7mm, and Functional Fibrinogen maximum amplitude 26 vs 20.5mm), decreased clot breakdown (median TEG Lysis 0.1 vs 1.1%), and increased platelet aggregation in response to stimulation with ADP (median 69 vs 58 AUC units) and ristocetin (median 108 vs 59.5 AUC units). Additionally, COVID-19 patients had increases in inflammatory biomarkers such as IP10 (median 429.1 vs 22.4pg/mL), IL-10 (median 9.3 vs. 1.3pg/mL), and RAGE (median 2476 vs. 686pg/mL), when compared to disease controls.

Table 2 Characteristics of platelet releasate donors.
3.2 Plasma treated healthy platelets
To understand the impact of the circulating soluble environment of COVID-19 on platelet function we treated healthy platelets with plasma from COVID-19 patients and disease controls. Following plasma treatment of the platelets, we performed multiple electrode impedance aggregometry (Figure 2), measuring platelet aggregation response prior to stimulation (Figures 2A, B) and in response to collagen stimulation (Figures 2C, D). COVID-19 patient plasma treatment of healthy platelets increased their baseline impedance prior to stimulation with collagen (Figure 2A), and decreased their response to collagen stimulation compared to disease control plasma treatment (Figure 2C). This held true when plasma donor patients were clinically stratified by cut point analysis of degree of illness using the American Physiology and Chronic Health Evaluation (APACHE III) score (Figures 2B, D). Treatment of healthy platelets with plasma from COVID-19 patients with higher APACHE scores compared to lower APACHE scores induced similar increases in baseline impedance prior to stimulation with collagen, but a much larger decrease in response to stimulation with collagen (Figure 2D). Together these data suggest the presence of a platelet activating soluble milieu in COVID-19 that leads to a reduction in the circulating platelet response to exogenous stimulation in ex-vivo assays. This reduction in the response to exogenous stimulation was magnified when platelets were treated with plasma from COVID-19 patients who were sicker as measured by higher APACHE scores.
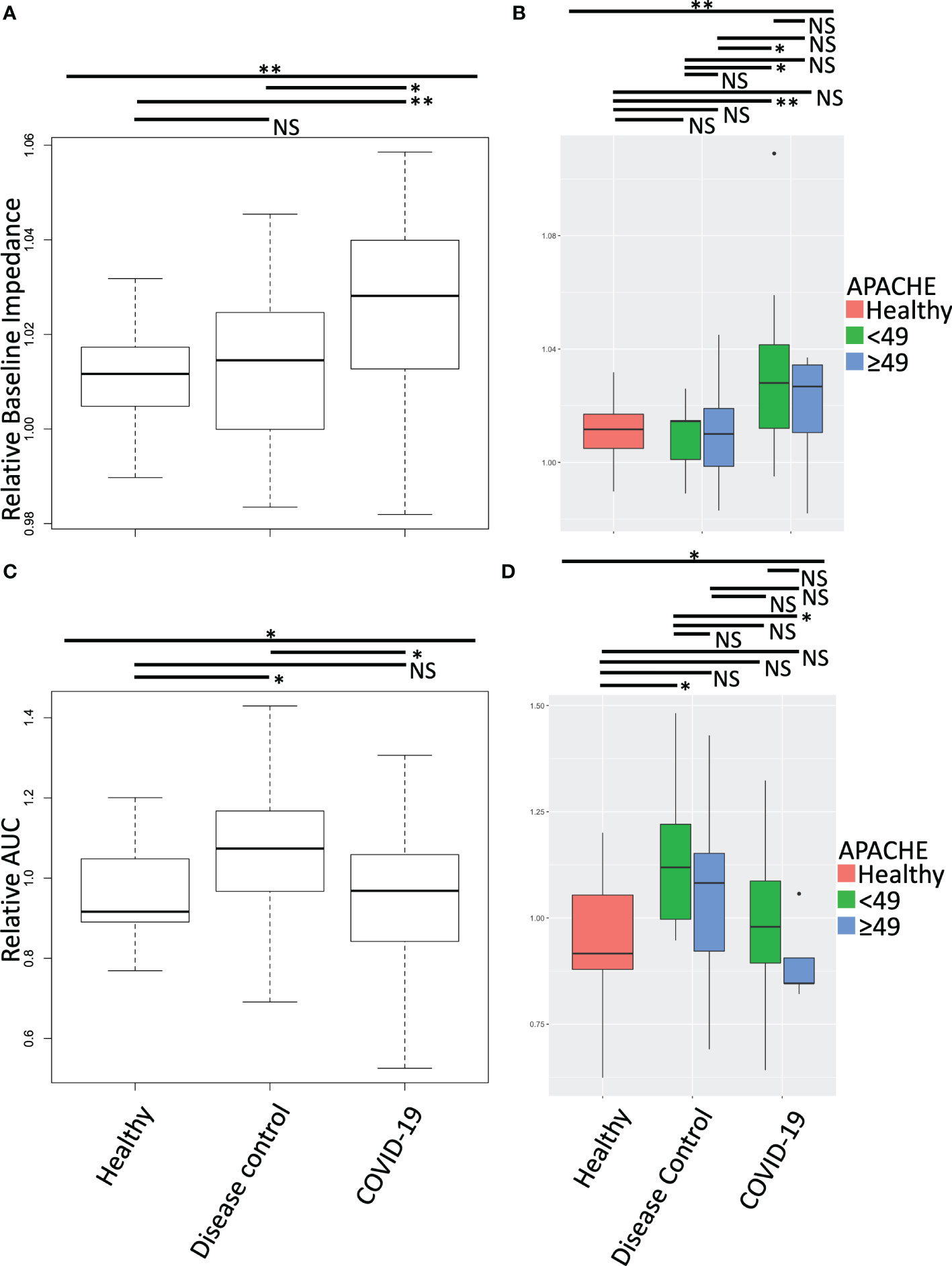
Figure 2 Aggregation effect of healthy platelets treated with plasma from COVID-19 patients and disease controls was measured by multiple electrode impedance aggregometry. Following treatment of isolated healthy donor platelets with plasma from COVID-19 patients and disease controls, multiple electrode impedance aggregometry was performed and (A, B) platelet aggregation prior to stimulation (baseline impedance), and (C, D) following collagen stimulation were measured. All data were normalized to autologous healthy donor plasma treatment performed in tandem, and compared to treatment with heterologous healthy donor plasma. (A, C) Treatment of healthy platelets with plasma from COVID-19 patients induced a statistically significant increase in platelet aggregation prior to stimulation (baseline impedance), but a decrease in response to collagen stimulation. Together these data suggest a platelet activating circulating milieu in COVID-19 that leads to a reduction in the responsiveness of platelets to further exogenous stimulation in ex-vivo assays. (B, D) To stratify patients, we performed a cut point analysis based on APACHE score of the plasma donors. The reduction in the response to exogenous stimulation was magnified following treatment with plasma from COVID-19 patients with higher compared to lower APACHE scores. Platelets from one of two male or two female healthy donors were treated with plasma from seven healthy donors, 12 disease control, and 14 COVID-19 patients. All plasma treatments were normalized to autologous healthy donor plasma treatment collected in tandem. Data were tested for normal distribution and statistical significance was assessed by one-way ANOVA followed by Tukey’s HSD post hoc. NS, not statistically significant; *, p ≤ 0.05; **, p ≤ 0.01.
Plasma treated healthy platelets were also subjected to adhesion assays in a collagen and thromboplastin coated parallel plate microfluidic flow chamber (Figure 3). There was no significant difference in adhesion of healthy platelets treated with plasma from COVID-19 patients, disease controls, and healthy controls (Figure 3B). While the number of adhered platelets did not differ, the morphology of the adhered platelets differed (Figure 3A). Compared to healthy plasma treated platelets, treatment with plasma from both COVID-19 patients and disease controls induced platelets to adopt small and rounded phenotypes. As a means of quantifying the observed morphologic changes, we measured the area of adhered platelets (Figure 3C). Adhered platelets were significantly smaller following treatment with plasma from both COVID-19 patients and disease controls compared to healthy controls. Although there was not a significant quantitative difference in the measured area of adhered platelets between those treated with plasma from COVID-19 patients compared to disease controls, treatment with plasma from COVID-19 patients qualitatively induced increased process development compared to disease controls.
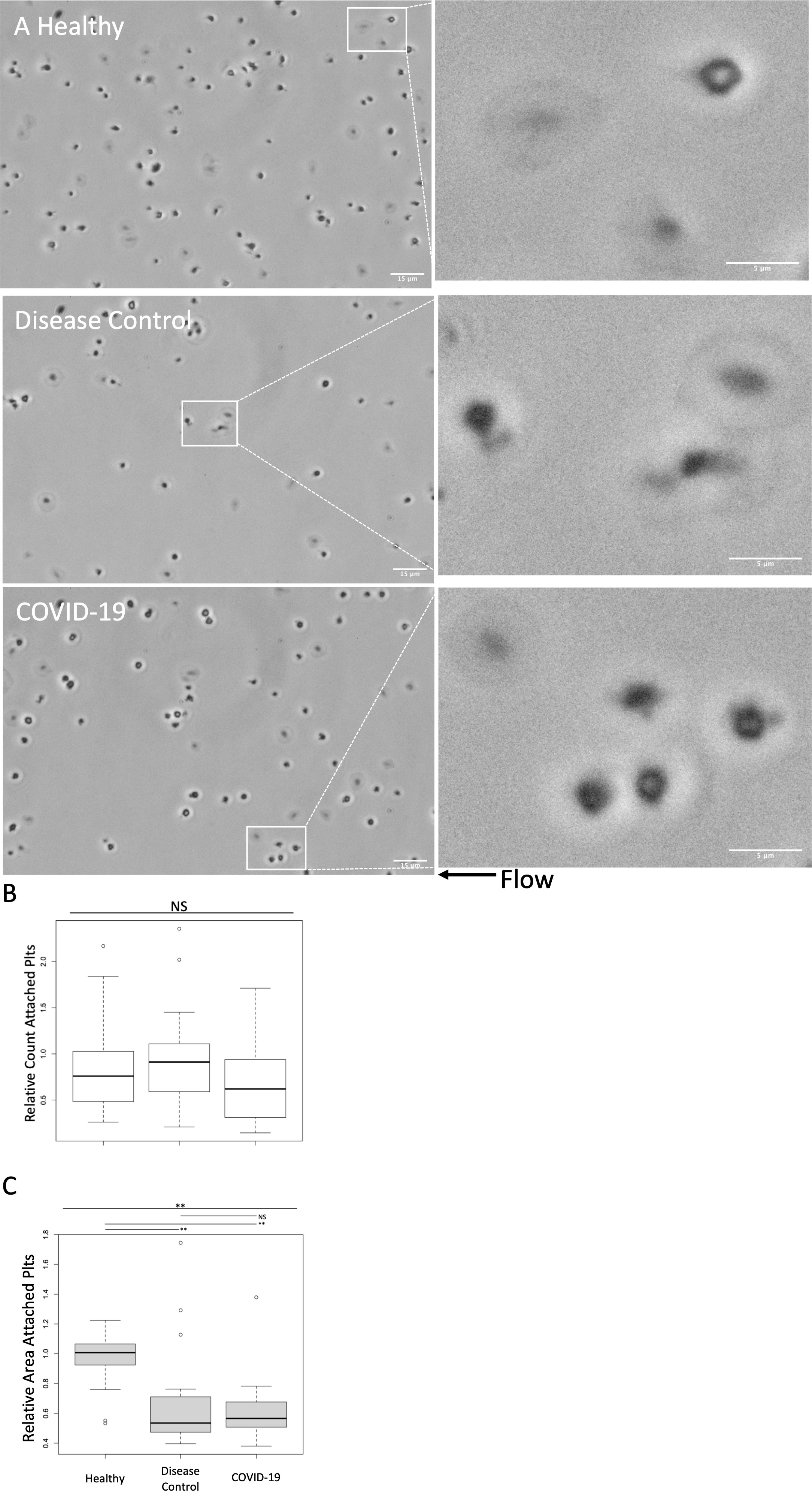
Figure 3 Adhesion and morphologic effects of healthy platelets treated with plasma from COVID-19 patients and disease controls was measured in a microfluidic adhesion assay. Healthy donor platelets were treated with plasma from COVID-19 patients, and healthy and disease controls, and then were passed through a collagen and thromboplastin coated chamber at 100s-1 (approximately venous shear). Unattached platelets were washed out at 1000s-1 (approximately arterial shear). Attached platelets were examined by phase contrast microscopy at 60X. (A) Representative images. In general, compared to healthy plasma treated platelets, treatment with plasma from both COVID-19 patients and disease controls induced platelets to adopt small and rounded phenotypes. Scale Bars are 15μm and 5μm for zoomed out and zoomed in panels, respectively (B) Attached platelets in five 60X fields were counted and counts were normalized to an autologous plasma treated group performed in tandem. The number of adhered platelets did not differ significantly between groups (C) Area of attached platelets was quantified to further examine morphological changes. Adhered platelets were significantly smaller following treatment with plasma from both COVID-19 patients and disease controls compared to healthy. There was not, however, a significant quantitative difference between treatment with plasma from COVID-19 patients compared to disease controls. Overall, the morphologic changes suggest that the circulating milieu in both COVID-19 patients and disease controls induced similar adhesive effects on healthy platelets. Treatment with plasma from COVID-19 patients did induce a qualitative increase in morphologic process development. Platelets from one of two male or two female healthy donors were treated with plasma from seven healthy donors, 12 disease control, and 14 COVID-19 patients. All plasma treatments were normalized to autologous healthy donor plasma treatment collected in tandem. Statistical significance was assessed using the Kruskal-Wallis test followed by pairwise Wilcoxon Signed-Rank tests. NS, not statistically significant; *, p ≤ 0.05; **, p ≤ 0.01.
Overall, the circulating milieu in both COVID-19 patients and disease controls induced similar aggregatory and adhesive effects on healthy platelets, but treatment with plasma from COVID-19 patients induced a reduction in the platelet aggregation response to exogenous stimulation compared to disease controls, and a qualitative increase in morphologic process development.
3.3 Platelet releasate treated healthy neutrophils
To determine if the contents released from platelets in COVID-19 patients induce a proinflammatory neutrophil phenotype, we isolated platelets from patients with COVID-19 and disease controls and collected releasate from those platelets following stimulation with CaCl2. We then treated healthy neutrophils with these platelet releasates. Treatment with platelet releasate from COVID-19 patients caused healthy neutrophils to produce more MPO-DNA complexes (Figure 4A). Additionally, treatment with platelet releasate from COVID-19 patients caused healthy neutrophils to have distinct differentially expressed genes when compared to those treated with platelet releasate from disease controls (Figure 4B). Genes in the complement pathway were significantly downregulated in healthy neutrophils treated with platelet releasate from COVID-19 patients compared to disease controls (p<0.05) (Figures 4C, D). Overall, this suggests that immunomodulatory paracrine factors are released from platelets in COVID-19 patients distinct from disease controls.
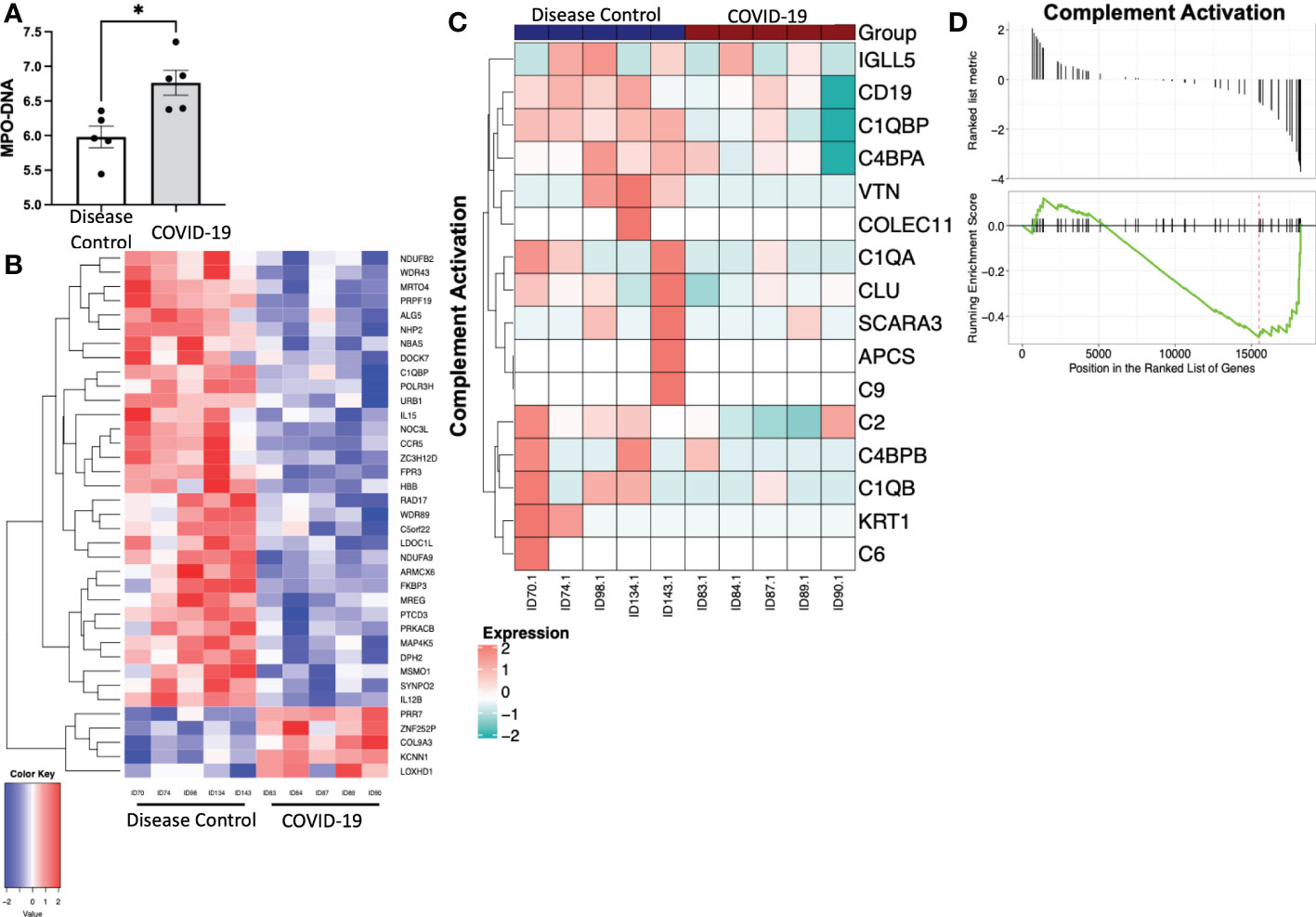
Figure 4 Treatment of healthy neutrophils with platelet releasate from COVID-19 patients and disease controls. (A) Myeloperoxidase-deoxy ribonucleic acid (MPO-DNA) complexes were quantified from COVID-19 patient and disease control platelet releasate treated neutrophils, measured with a capture ELISA. Data presented as optical density standardized to negative control. *, p<0.05.(B) Heatmap with hierarchical clustering displaying transcriptomic changes between treatment with platelet releasate from COVID-19 patients and disease controls. Bulk RNA sequencing results of gene changes requiring a fold change >2 and p-value <.05. There are 5 upregulated genes and 32 downregulated genes. N = 5. (C) Heatmap and (D) plot displaying the gene set enrichment pathway analysis of complement activation (Enrichment score=-0.493, p=0.038). N = 5.
4 Discussion
COVID-19 is associated with prothrombotic proinflammatory biologic profiles thought to mediate the thromboinflammatory clinical complications. Although evidence has accumulated that platelets and neutrophils are both crucial cellular contributors to the thromboinflammation of COVID-19 (25), therapeutic targeting of these cellular contributors to improve morbidity and mortality of COVID-19 has proven difficult (26). Further, the effect of the circulating soluble environment on this platelet-neutrophil axis has not been fully elucidated. As such, we tested the hypotheses that 1) plasma from patients with COVID-19 can induce a prothrombotic platelet functional phenotype, and that 2) contents released from platelets (platelet releasate) from COVID-19 patients can induce a proinflammatory neutrophil phenotype. To explore this, we prospectively collected early plasma samples from COVID-19 patients (before receipt of any therapeutics) and in parallel collected plasma samples from symptom-matched COVID-19 negative disease controls to assess the effects of COVID-19 plasma on healthy platelet aggregation, adhesion, and morphology. In addition, we created platelet releasates from platelets isolated from COVID-19 patients and disease controls to assess the indirect non-contact mediated effects of COVID-19 platelets on healthy neutrophil NET-formation and transcriptomic profiles.
First, we found that when healthy platelets were exposed to these plasma environments, the COVID-19 patient plasma induced platelets to have an auto-aggregatory profile and altered adhesion characteristics similar to treatment with disease control plasma, with one notable difference. COVID-19 patient plasma induced a functional phenotype consistent with what has been described as a ‘functional exhaustion’ in other disease states (27–29). This was evidenced by the induction of significantly increased platelet aggregation in response to the plasma treatment alone prior to exogenous stimulation with agonists and increased morphologic process development, yet a weaker aggregation response following collagen stimulation. These ex-vivo experimental observations suggest that the circulating soluble thromboinflammatory milieu of COVID-19 elicits a stronger functional effect on platelets than that of other disease, capable of reducing or exhausting the ability of platelets to aggregate further in response to additional surface stimulation. This biology could be hypothesized to contribute to an overall lack of success in decreasing microthrombosis associated complications in COVID-19 clinical trials with anti-platelet therapies (30–32). If the circulating platelets are in an environment that causes a ‘functional exhaustion’, it may be that further platelet inhibition may not be beneficial.
Second, platelets themselves contribute to the soluble circulating environment as they release a rich repertoire of contents that are capable of noncellular and cellular control of coagulation and inflammation (5, 7). As such, we explored the contribution of platelets to the soluble environment by creating platelet releasates from platelets isolated from COVID-19 patients and disease controls and treating neutrophils with these releasates to assess the paracrine platelet-neutrophil axis. Treating healthy neutrophils with platelet releasate from COVID-19 patients induced evidence of NETosis and differential regulation of complement pathway genes. These findings were pronounced in comparison to the treatment of healthy neutrophils with platelet releasate from disease controls.
NETs are known pro-thrombotic scaffolds (33). Previous work has suggested that NETosis in COVID-19 is due to binding of SARS-CoV-2 to neutrophil angiotensin converting enzyme 2 (15, 20), and also due to interactions with platelets (7). NETs have been found in occlusions of micro-vessels in COVID-19 (34). Further, platelet factor-4 has been found to be elevated in COVID-19 patient plasma, and evidence of NETosis was found in COVID-19 patient lung tissue that was also rich in platelet factor-4 (7). Platelets, themselves, have been shown to be hyperreactive in COVID-19 patients (4, 35). There has been some suggestion that this could potentially result from direct contact between SARS-CoV-2 and platelets (36). However, evidence for SARS-CoV-2 internalization in platelets is scant (4, 37). The actual mechanism more likely involves multiple inputs. Classically, neutrophils (and other leukocytes) are viewed as being recruited to sites of inflammation or endothelial damage by platelets through a combination of chemoattractant and direct cell-cell interaction, to allow them to extravasate and carry out immune-inflammatory functions (38). In-vivo, this is likely to be highly complex and include a process of paracrine signaling from platelets to leukocytes (39, 40). Overall, our observations expand on these existing findings by demonstrating that the soluble circulating environment of COVID-19 may also have important effects on the described adhesion and aggregation behavior of platelets, and further that those platelets are critical effector cells of neutrophils via a cell-cell contact independent paracrine line of communication in COVID-19 patients. Together our data further highlight a link between thrombotic and inflammatory mediators in COVID-19 that may be more pronounced and/or unique compared to other diseases. With this insight, it is possible that platelets’ contribution to thrombosis in COVID-19 could leverage the inflammatory side of the thromboinflammatory continuum. As previously mentioned, platelet inhibition has not been a panacea in the context of COVID-19. However, platelets’ role as an inflammatory paracrine effector cell may represent a therapeutic avenue.
There are limitations to these studies. Regarding the ex-vivo assays, the sample size of platelet releasate treated neutrophils is low. A single neutrophil donor was chosen to limit donor heterogeneity in sequencing experiments, as the primary goal was to compare the effects of platelet releasate from COVID-19 patients and disease controls on neutrophils, thus the differences observed should only be due to treatment. However, the disease control releasates were skewed towards male. Regarding the plasma treated platelets, measuring baseline impedance is a proxy for auto-aggregation, but we do not have confirmatory surface marker expression or other measures of their activation or aggregation status. Finally, regarding our plasma and platelet releasate donors, it should be noted that although the data demonstrate evidence of a generally prothrombotic proinflammatory phenotype of the COVID-19 patients relative to the disease controls, the study was not powered to detect differences in these biomarkers, and the differences were small overall. This may be important given that much of the COVID-19 literature lacks parallel prospectively collected disease control samples for comparison, and either rely on previously collected specimens for disease controls or use healthy control comparisons alone. As such, these methods may risk overestimation of the procoagulant and proinflammatory phenotypes of COVID-19. Importantly, all samples included were collected on emergency department presentation for both COVID-19 patients and disease controls, which is novel compared to much of the COVID-19 literature in which samples were collected in admitted patients later in their disease courses and often after initiation of anti-viral and anti-inflammatory therapies, another aspect that could contribute to the small differences overall from the disease controls.
Finally, it is not clear what is in the plasma and released from the platelets that is responsible for the cell-cell contact independent effector cell biology that we identified. Unfortunately, we were unable to perform characterization studies of the platelet releasate samples described here. Platelet releasates were prepared in very limited volume because they were made from whole blood obtained under an initial waiver of consent, which was central to obtaining these samples prior to any therapeutics in sick patients. As such, we were able to prepare only very limited quantities that were used in the experiments. Of note, among the biomarkers we measured in the plasma, PAI-1 was different between COVID-19 patients and disease control donors. PAI-1 has anti-fibrinolytic function, is differentially regulated by diverse cytokines including those involved in inflammation (41). PAI-1 is a driver of hypofibrinolysis in COVID-19 (42). Interestingly, PAI-1 has been suggested to regulate and be regulated by inflammatory biomarkers, namely IL-6, thus linking thrombotic and inflammatory processes (43, 44). A large reservoir of PAI-1 resides in platelets themselves, and active PAI-1 is displayed on the platelet membrane on activation (45–47). Exposure to PAI-1 has been shown to have pleiotropic effects on neutrophils, including promotion of tissue infiltration and ischemia reperfusion injury (48–50). This may be relevant to our findings that platelet releasate from patients with COVID-19 caused healthy neutrophils to increase NET formation and alter their transcriptional program related to complement. Testing the involvement of PAI-1 in our observations is warranted. In addition, several biomarkers of inflammation were upregulated in COVID-19 patients compared to the disease controls donors, namely IP-10, IL-10, and RAGE. IP-10 is best known for its role as a leukocyte chemoattractant and inhibitor of endothelial cell proliferation (51). Although the canonical role of IL-10 is as an anti-inflammatory cytokine, there has been some evidence that in COVID-19 it may exert a pro-inflammatory effect (52). RAGE is expressed in alveolar epithelium and is associated with lung injury, acute respiratory distress syndrome, and, more recently, COVID-19 severity (53–55). Importantly, the biomarker profiles of the plasma used to treat platelets were similar to those in the plasma of patients whose platelets were used to generate platelet releasate samples.
In summary, our work suggests that in COVID-19, platelets are exposed to a procoagulant proinflammatory environment that leads to auto-aggregation, morphologic change, and may induce a ‘functional exhaustion’. Further, our data supports that the platelets themselves may contribute to the soluble circulating milieu and via cell-cell contact independent paracrine effects can alter neutrophils at both the transcriptional and functional levels. Other investigators have noted PAI-1 as well as vWF decorating NETs in the setting of ischemic stroke (56), and NETs have a well know role in thrombus formation (57). Given our observations of increased plasma levels of PAI-1 in COVID-19 patients, and increased NET formation following treatment of healthy neutrophils with platelet releasate from COVID-19 patients, there is potential that platelets may also play a cell-cell contact independent paracrine role in COVID-19 hypercoagulability via signaling to neutrophils.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: Gene Expression Omnibus (GEO) with accession ID: GSE225217.
Ethics statement
The studies involving human participants were reviewed and approved by Institutional Review Board/Human Research Protection Program of the University of California, San Francisco. The patients/participants provided their written informed consent to participate in this study.
Author contributions
All authors participated in the writing and editing of the manuscript. AF assisted in study design, designed experiments, performed experiments, analyzed data, and prepared the manuscript. EA designed experiments, performed experiments, and analyzed data. CK designed experiments, performed experiments, and analyzed data. ZM assisted in study design, designed experiments, performed experiments analyzed data. KH performed experiments. BN-G managed clinical sample and data collection. CH assisted in study design. RB assisted in study design, designed experiments, performed experiments, analyzed data. CJ performed experiments. KW performed experiments and analyzed data. SL performed experiments, and analyzed data. J-HL performed experiments, and analyzed data. Y-PY performed experiments, and analyzed data. MM assisted in study design, and designed experiments. TB assisted in study design. JB assisted in study design. MN assisted in study design, and designed experiments. LK assisted in study design, designed experiments, analyzed data, and prepared the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was, in part, funded by the National Institutes of Health (NIH) Agreement 1OT2HL156812 through the NHLBI Collaborating Network of Networks for Evaluating COVID-19 and Therapeutic Strategies (CONNECTS) Program. The views and conclusions contained in this document are those of the authors and should not be interpreted as representing the official policies, either expressed or implied, of the NIH.
Acknowledgments
We are grateful for the support of the patients and their families who participated in this trial and want to acknowledge the NHLBI Collaborating Network of Networks for Evaluating COVID-19 and Therapeutic Strategies (CONNECTS) Program.
Additional Co-ACIT Study Group members
Biniam Ambachew1, Sarah Cary4, Lauren Chalwell9, Christopher Colwell9, Clayton Josephy3, Deanna Lee1, Matthieu LeGrand4, Juan Carlos Montoy9, Aaron E. Kornblith9, Philip Kurien4, Brenda Nunez-Garcia1, Viet Nguyen1, John J. Park1, Arun Prakash4, Brittany Robinson1, India Shelley1.
1 Department of Surgery, University of California, San Francisco; Zuckerberg San Francisco General Hospital; San Francisco, CA, USA
2 Department of Surgery, University of Pittsburgh; Pittsburgh, PA, USA
3 Division of Pulmonary, Critical Care, Allergy and Sleep Medicine, Department of Medicine, University of California, San Francisco; San Francisco, CA, USA
4 Department of Anesthesia and Perioperative Care, School of Medicine, University of California, San Francisco; San Francisco, CA, USA
5 Cardiovascular Research Institute, University of California, San Francisco; San Francisco, CA, USA
6 Leon H. Charney Division of Cardiology, Department of Medicine, NYU Grossman School of Medicine; New York, NY, USA
7 NYU Center for the Prevention of Cardiovascular Disease, NYU Langone Health; New York, NY, USA
8 Division of Vascular Surgery, Department of Surgery, NYU Grossman School of Medicine; New York, NY, USA
9 Department of Emergency Medicine, University of California, San Francisco; San Francisco, CA, USA
Conflict of interest
LK is on the scientific advisory board for Cerus and Gamma-Prime Fibrinogen, and is a family member of a founder of Capture Diagnostics. MN serves on the scientific advisory board of Haima Therapeutics, has received personal fees from Haemonetics, Takeda, and Janssen Pharmaceuticals, and has received travel support from Meredian Biosciences. He has received grants from Haemonetics and Instrumentation Laboratories.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bikdeli B, Madhavan MV, Jimenez D, Chuich T, Dreyfus I, Driggin E, et al. COVID-19 and thrombotic or thromboembolic disease: Implications for prevention, antithrombotic therapy, and follow-up: JACC state-of-the-Art review. J Am Coll Cardiol (2020) 75(23):2950–73. doi: 10.1016/j.jacc.2020.04.031
2. Portier I, Campbell RA, Denorme F. Mechanisms of immunothrombosis in COVID-19. Curr Opin Hematol (2021) 28(6):445–53. doi: 10.1097/MOH.0000000000000666
3. Barrett TJ, Cornwell M, Myndzar K, Rolling CC, Xia Y, Drenkova K, et al. Platelets amplify endotheliopathy in COVID-19. Sci Adv (2021) 7(37):eabh2434. doi: 10.1126/sciadv.abh2434
4. Manne BK, Denorme F, Middleton EA, Portier I, Rowley JW, Stubben C, et al. Platelet gene expression and function in patients with COVID-19. Blood (2020) 136(11):1317–29. doi: 10.1182/blood.2020007214
5. Zaid Y, Puhm F, Allaeys I, Naya A, Oudghiri M, Khalki L, et al. Platelets can associate with SARS-Cov-2 RNA and are hyperactivated in COVID-19. Circ Res (2020) 127(11):1404–18. doi: 10.1161/CIRCRESAHA.120.317703
6. Caudrillier A, Kessenbrock K, Gilliss BM, Nguyen JX, Marques MB, Monestier M, et al. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest (2012) 122(7):2661–71. doi: 10.1172/JCI61303
7. Middleton EA, He XY, Denorme F, Campbell RA, Ng D, Salvatore SP, et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood (2020) 136(10):1169–79. doi: 10.1182/blood.2020007008
8. Martinod K, Deppermann C. Immunothrombosis and thromboinflammation in host defense and disease. Platelets (2021) 32(3):314–24. doi: 10.1080/09537104.2020.1817360
9. Guo L, Rondina MT. The era of thromboinflammation: Platelets are dynamic sensors and effector cells during infectious diseases. Front Immunol (2019) 10:2204. doi: 10.3389/fimmu.2019.02204
10. Barrett TJ, Bilaloglu S, Cornwell M, Burgess HM, Virginio VW, Drenkova K, et al. Platelets contribute to disease severity in COVID-19. J Thromb Haemost (2021) 19(12):3139–53. doi: 10.1111/jth.15534
11. Hottz ED, Azevedo-Quintanilha IG, Palhinha L, Teixeira L, Barreto EA, Pão CRR, et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood (2020) 136(11):1330–41. doi: 10.1182/blood.2020007252
12. Panigada M, Bottino N, Tagliabue P, Grasselli G, Novembrino C, Chantarangkul V, et al. Hypercoagulability of COVID-19 patients in intensive care unit: A report of thromboelastography findings and other parameters of hemostasis. J Thromb Haemost (2020) 18(7):1738–42. doi: 10.1111/jth.14850
13. Patterson BK, Seethamraju H, Dhody K, Corley MJ, Kazempour K, Lalezari J, et al. Disruption of the CCL5/RANTES-CCR5 pathway restores immune homeostasis and reduces plasma viral load in critical COVID-19. medRxiv (2020). 2020.05.02.20084673. doi: 10.1101/2020.05.02.20084673
14. Caillon A, Trimaille A, Favre J, Jesel L, Morel O, Kauffenstein G. Role of neutrophils, platelets, and extracellular vesicles and their interactions in COVID-19-associated thrombopathy. J Thromb Haemost (2022) 20(1):17–31. doi: 10.1111/jth.15566
15. Veras FP, Pontelli MC, Silva CM, Toller-Kawahisa JE, de Lima M, Nascimento DC, et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J Exp Med (2020) 217(12). doi: 10.1084/jem.20201129
16. Maxwell AJ, Ding J, You Y, Dong Z, Chehade H, Alvero A, et al. Identification of key signaling pathways induced by SARS-CoV2 that underlie thrombosis and vascular injury in COVID-19 patients. J Leukoc Biol (2021) 109(1):35–47. doi: 10.1002/JLB.4COVR0920-552RR
17. Fields AT, Matthay ZA, Nunez-Garcia B, Matthay EC, Bainton RJ, Callcut RA, et al. Good platelets gone bad: The effects of trauma patient plasma on healthy platelet aggregation. Shock (2021) 55(2):189–97. doi: 10.1097/SHK.0000000000001622
18. Mitchell TA, Herzig MC, Fedyk CG, Salhanick MA, Henderson AT, Parida BK, et al. Traumatic hemothorax blood contains elevated levels of microparticles that are prothrombotic but inhibit platelet aggregation. Shock (2017) 47(6):680–7. doi: 10.1097/SHK.0000000000000819
19. Verni CC, Davila A Jr., Balian S, Sims CA, Diamond SL. Platelet dysfunction during trauma involves diverse signaling pathways and an inhibitory activity in patient-derived plasma. J Trauma Acute Care Surg (2019) 86(2):250–9. doi: 10.1097/TA.0000000000002140
20. Arcanjo A, Logullo J, Menezes CCB, de Souza Carvalho Giangiarulo TC, Dos Reis MC, de Castro GMM, et al. The emerging role of neutrophil extracellular traps in severe acute respiratory syndrome coronavirus 2 (COVID-19). Sci Rep (2020) 10(1):19630. doi: 10.1038/s41598-020-76781-0
21. Matthay ZA, Fields AT, Nunez-Garcia B, Park JJ, Jones C, Leligdowicz A, et al. Importance of catecholamine signaling in the development of platelet exhaustion after traumatic injury. J Thromb Haemost (2022) 20(9):2109–18. doi: 10.1111/jth.15763
22. DeCortin ME, Brass LF, Diamond SL. Core and shell platelets of a thrombus: A new microfluidic assay to study mechanics and biochemistry. Res Pract Thromb Haemost (2020) 4(7):1158–66. doi: 10.1002/rth2.12405
23. Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods (2012) 9(7):676–82. doi: 10.1038/nmeth.2019
24. Kessenbrock K, Krumbholz M, Schönermarck U, Back W, Gross WL, Werb Z, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med (2009) 15(6):623–5. doi: 10.1038/nm.1959
25. Falcinelli E, Petito E, Gresele P. The role of platelets, neutrophils and endothelium in COVID-19 infection. Expert Rev Hematol (2022) 15(8):727–45. doi: 10.1080/17474086.2022.2110061
26. Zong X, Wang X, Liu Y, Li Z, Wang W, Wei D, et al. Antiplatelet therapy for patients with COVID-19: Systematic review and meta-analysis of observational studies and randomized controlled trials. Front Med (Lausanne) (2022) 9:965790. doi: 10.3389/fmed.2022.965790
27. Pareti FI, Capitanio A, Mannucci L, Ponticelli C, Mannucci PM. Acquired dysfunction due to the circulation of “exhausted” platelets. Am J Med (1980) 69(2):235–40. doi: 10.1016/0002-9343(80)90383-6
28. Starr NE, Matthay ZA, Fields AT, Nunez-Garcia B, Callcut RA, Cohen MJ, et al. Identification of injury and shock driven effects on ex vivo platelet aggregometry: A cautionary tale of phenotyping. J Trauma Acute Care Surg (2020) 89(1):20–8. doi: 10.1097/TA.0000000000002707
29. Moore EE, Moore HB, Kornblith LZ, Neal MD, Hoffman M, Mutch NJ, et al. Trauma-induced coagulopathy. Nat Rev Dis Primers (2021) 7(1):30. doi: 10.1007/978-3-030-53606-0
30. Bradbury CA, Lawler PR, Stanworth SJ, McVerry BJ, McQuilten Z, Higgins AM, et al. Effect of antiplatelet therapy on survival and organ support-free days in critically ill patients with COVID-19: A randomized clinical trial. Jama (2022) 327(13):1247–59. doi: 10.1001/jama.2022.2910
31. Berger JS, Kornblith LZ, Gong MN, Reynolds HR, Cushman M, Cheng Y, et al. Effect of P2Y12 inhibitors on survival free of organ support among non-critically ill hospitalized patients with COVID-19: A randomized clinical trial. Jama (2022) 327(3):227–36. doi: 10.1001/jama.2021.23605
32. Group RC. Aspirin in patients admitted to hospital with COVID-19 (RECOVERY): A randomised, controlled, open-label, platform trial. Lancet (2022) 399(10320):143–51. doi: 10.1016/S0140-6736(21)01825-0
33. Thålin C, Hisada Y, Lundström S, Mackman N, Wallén H. Neutrophil extracellular traps: Villains and targets in arterial, venous, and cancer-associated thrombosis. Arterioscler Thromb Vasc Biol (2019) 39(9):1724–38. doi: 10.1161/ATVBAHA.119.312463
34. Leppkes M, Knopf J, Naschberger E, Lindemann A, Singh J, Herrmann I, et al. Vascular occlusion by neutrophil extracellular traps in COVID-19. EBioMedicine (2020) 58:102925. doi: 10.1016/j.ebiom.2020.102925
35. Comer SP, Cullivan S, Szklanna PB, Weiss L, Cullen S, Kelliher S, et al. COVID-19 induces a hyperactive phenotype in circulating platelets. PloS Biol (2021) 19(2):e3001109. doi: 10.1371/journal.pbio.3001109
36. Zhang S, Liu Y, Wang X, Yang L, Li H, Wang Y, et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J Hematol Oncol (2020) 13(1):120. doi: 10.1186/s13045-020-00954-7
37. Bury L, Camilloni B, Castronari R, Piselli E, Malvestiti M, Borghi M, et al. Search for SARS-CoV-2 RNA in platelets from COVID-19 patients. Platelets (2021) 32(2):284–7. doi: 10.1080/09537104.2020.1859104
38. Rayes J, Bourne JH, Brill A, Watson SP. The dual role of platelet-innate immune cell interactions in thrombo-inflammation. Res Pract Thromb Haemost (2020) 4(1):23–35. doi: 10.1002/rth2.12266
39. Eligini S, Barbieri SS, Arenaz I, Tremoli E, Colli S. Paracrine up-regulation of monocyte cyclooxygenase-2 by platelets: Role of transforming growth factor-β1. Cardiovasc Res (2007) 74(2):270–8. doi: 10.1016/j.cardiores.2006.12.013
40. Hug S, Bernhard S, Stratmann AEP, Erber M, Wohlgemuth L, Knapp CL, et al. Activation of neutrophil granulocytes by platelet-activating factor is impaired during experimental sepsis. Front Immunol (2021) 12:642867. doi: 10.3389/fimmu.2021.642867
41. Sillen M, Declerck PJ. Targeting PAI-1 in cardiovascular disease: Structural insights into PAI-1 functionality and inhibition. Front Cardiovasc Med (2020) 7. doi: 10.3389/fcvm.2020.622473
42. Whyte CS, Simpson M, Morrow GB, Wallace CA, Mentzer AJ, Knight JC, et al. The suboptimal fibrinolytic response in COVID-19 is dictated by high PAI-1. J Thromb Haemostasis (2022) 20(10):2394–406. doi: 10.1111/jth.15806
43. Huang P, Zuo Q, Li Y, Oduro PK, Tan F, Wang Y, et al. A vicious cycle: In severe and critically ill COVID-19 patients. Front Immunol (2022) 13. doi: 10.3389/fimmu.2022.930673
44. Kang S, Tanaka T, Inoue H, Ono C, Hashimoto S, Kioi Y, et al. IL-6 trans-signaling induces plasminogen activator inhibitor-1 from vascular endothelial cells in cytokine release syndrome. Proc Natl Acad Sci U S A (2020) 117(36):22351–6. doi: 10.1073/pnas.2010229117
45. Booth NA, Simpson AJ, Croll A, Bennett B, MacGregor IR. Plasminogen activator inhibitor (PAI-1) in plasma and platelets. Br J Haematol (1988) 70(3):327–33. doi: 10.1111/j.1365-2141.1988.tb02490.x
46. Brogren H, Wallmark K, Deinum J, Karlsson L, Jern S. Platelets retain high levels of active plasminogen activator inhibitor 1. PloS One (2011) 6(11):e26762. doi: 10.1371/journal.pone.0026762
47. Gael BM, Claire SW, Nicola JM. Functional plasminogen activator inhibitor 1 is retained on the activated platelet membrane following platelet activation. Haematologica (2019) 105(12):2824–33. doi: 10.3324/haematol.2019.230367
48. Park YJ, Liu G, Lorne EF, Zhao X, Wang J, Tsuruta Y, et al. PAI-1 inhibits neutrophil efferocytosis. Proc Natl Acad Sci U S A (2008) 105(33):11784–9. doi: 10.1073/pnas.0801394105
49. Zmijewski JW, Bae HB, Deshane JS, Peterson CB, Chaplin DD, Abraham E. Inhibition of neutrophil apoptosis by PAI-1. Am J Physiol Lung Cell Mol Physiol (2011) 301(2):L247–54. doi: 10.1152/ajplung.00075.2011
50. Praetner M, Zuchtriegel G, Holzer M, Uhl B, Schaubächer J, Mittmann L, et al. Plasminogen activator inhibitor-1 promotes neutrophil infiltration and tissue injury on ischemia–reperfusion. Arteriosclerosis Thrombosis Vasc Biol (2018) 38(4):829–42. doi: 10.1161/ATVBAHA.117.309760
51. van den Borne P, Quax PHA, Hoefer IE, Pasterkamp G. The multifaceted functions of CXCL10 in cardiovascular disease. BioMed Res Int (2014) 2014:893106. doi: 10.1155/2014/893106
52. Islam H, Chamberlain TC, Mui AL, Little JP. Elevated interleukin-10 levels in COVID-19: Potentiation of pro-inflammatory responses or impaired anti-inflammatory action? Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.677008
53. Uchida T, Shirasawa M, Ware LB, Kojima K, Hata Y, Makita K, et al. Receptor for advanced glycation end-products is a marker of type I cell injury in acute lung injury. Am J Respir Crit Care Med (2006) 173(9):1008–15. doi: 10.1164/rccm.200509-1477OC
54. Jabaudon M, Blondonnet R, Pereira B, Cartin-Ceba R, Lichtenstern C, Mauri T, et al. Plasma sRAGE is independently associated with increased mortality in ARDS: A meta-analysis of individual patient data. Intensive Care Med (2018) 44(9):1388–99. doi: 10.1007/s00134-018-5327-1
55. Wick KD, Siegel L, Neaton JD, Oldmixon C, Lundgren J, Dewar RL, et al. RAGE has potential pathogenetic and prognostic value in nonintubated hospitalized patients with COVID-19. JCI Insight (2022) 7(9). doi: 10.1172/jci.insight.157499
56. Zhang S, Cao Y, Du J, Liu H, Chen X, Li M, et al. Neutrophil extracellular traps contribute to tissue plasminogen activator resistance in acute ischemic stroke. FASEB J (2021) 35(9):e21835. doi: 10.1096/fj.202100471RR
Keywords: blood platelets, neutrophil extracellular traps, COVID-19, thromboinflammation, plasma
Citation: Fields AT, Andraska EA, Kaltenmeier C, Matthay ZA, Herrera K, Nuñez-Garcia B, Jones CM, Wick KD, Liu S, Luo J-H, Yu Y-P, Matthay MA, Hendrickson CM, Bainton RJ, Barrett TJ, Berger JS, Neal MD, Kornblith LZ and the COVID-19 Associated Coagulopathy Inflammation and Thrombosis (Co-ACIT) Study Group (2023) Effects of the circulating environment of COVID-19 on platelet and neutrophil behavior. Front. Immunol. 14:1130288. doi: 10.3389/fimmu.2023.1130288
Received: 23 December 2022; Accepted: 23 February 2023;
Published: 14 March 2023.
Edited by:
Robert Lindsay Medcalf, Monash University, AustraliaReviewed by:
Isao Nagaoka, Juntendo University, JapanErzsebet Komorowicz, Semmelweis University, Hungary
Copyright © 2023 Fields, Andraska, Kaltenmeier, Matthay, Herrera, Nuñez-Garcia, Jones, Wick, Liu, Luo, Yu, Matthay, Hendrickson, Bainton, Barrett, Berger, Neal, Kornblith and the COVID-19 Associated Coagulopathy Inflammation and Thrombosis (Co-ACIT) Study Group. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lucy Z. Kornblith, lucy.kornblith@ucsf.edu