 Diana Boraschi
Diana Boraschi Paola Italiani
Paola Italiani Paola Migliorini5
Paola Migliorini5 Paola Bossù
Paola Bossù- 1Shenzhen Institute of Advanced Technology (SIAT), Chinese Academy of Sciences (CAS), Shenzhen, China
- 2Institute of Biochemistry and Cell Biology (IBBC), National Research Council (CNR), Naples, Italy
- 3Stazione Zoologica Anton Dohrn (SZN), Napoli, Italy
- 4China-Italy Joint Laboratory of Pharmacobiotechnology for Medical Immunomodulation, Shenzhen, China
- 5Clinical Immunology and Allergy Unit, Department of Clinical and Experimental Medicine, University of Pisa, Pisa, Italy
- 6Laboratory of Experimental Neuro-psychobiology, Department of Clinical and Behavioral Neurology, Santa Lucia Foundation, Rome, Italy
Cytokines and receptors of the IL-1 family are key mediators in innate immune and inflammatory reactions in physiological defensive conditions, but are also significantly involved in immune-mediated inflammatory diseases. Here, we will address the role of cytokines of the IL-1 superfamily and their receptors in neuroinflammatory and neurodegenerative diseases, in particular Multiple Sclerosis and Alzheimer’s disease. Notably, several members of the IL-1 family are present in the brain as tissue-specific splice variants. Attention will be devoted to understanding whether these molecules are involved in the disease onset or are effectors of the downstream degenerative events. We will focus on the balance between the inflammatory cytokines IL-1β and IL-18 and inhibitory cytokines and receptors, in view of future therapeutic approaches.
1 Introduction
The interleukin-1 (IL-1) superfamily encompasses eleven structurally and evolutionarily related cytokines that mainly act by binding to specific receptors (1). The IL-1 receptor (IL-1R) complexes are formed by a ligand-binding chain and an accessory chain necessary for signal transduction. The IL-1R family comprises ten related transmembrane molecules with extracellular Ig-like domains and intracellular TIR (Toll-Like Receptor/IL-1 Receptor) domains (2). Most IL-1 and IL-1R molecules are involved in inflammation/immunostimulation and its regulation. It is notable that, while IL-1R are derived from invertebrate TIR-containing receptors involved in innate immunity, IL-1 cytokines only appeared in vertebrates (likely linked to the development of adaptive immunity) and the majority of them is only present in mammals (3, 4).
In mammals, eleven cytokines of the IL-1 family have been identified, the inflammatory IL-1α, IL-1β, IL-18, IL-36α, IL-36β, IL-36γ, and the regulatory/anti-inflammatory IL-1Ra, IL-33, IL-36Ra, IL-37 and IL-38 (overview in 1). IL-1 family cytokines mainly act by binding to receptor complexes, composed by a ligand binding chain and an accessory signal transducing chain. Signalling is initiate by the pairing of the intracellular TIR domains of the two receptor chains. Receptors of the IL-1R family are ten structurally related transmembrane proteins, which may also give rise to soluble receptors upon gene splicing or proteolysis of the membrane chains (overview in 2). IL-1R1 binds the agonist ligands IL-1α and IL-1β, and its signalling depends on the engagement of the accessory chain IL-1R3. IL-1R1 also binds IL-1Ra but in this case fails to recruit the IL-1R3, and no activation occurs. Binding of IL-38 has also been reported. IL-1R2 is similar to IL-1R1 in its ligand binding capacity and IL-1R3 engagement, but it lacks the intracellular TIR domain and its therefore an inhibitory receptor. IL-1R4 is the receptor for IL-33, an uses the same IL-1R3 as accessory chain for signalling. IL-1R5 is the receptor for IL-18, and uses its specific accessory chain IL-1R7. It was reported that IL-37 also binds to IL-1R5 but is unable to recruit IL-1R7. IL-1R6 is the ligand binding chain for all the IL-36 isoforms (IL-36α, IL-36β, IL-36γ) and uses the promiscuous IL-1R3 as accessory chain. IL-1R6 also binds the receptor antagonist IL-36Ra and fails to engage the accessory chain. Binding of IL-38 has also been reported. IL-1R8 is an inhibitory receptor, which can interact with several IL-1R complexes and interfere with their signalling and may also act as inhibitory accessory chain for IL-37 bound to IL-1R5 and IL-38 bound to IL-1R6. IL-1R9 is an orphan receptor, although binding to IL-38 has been reported, predominantly expressed in neurons and likely involve in IL-1-independent neuronal functions. Likewise, IL-1R10 is abundantly expressed in the brain and has no IL-1-dependent functions (for complete references, please see the reference lists of 1, 2).
IL-1 and IL-1R molecules expressed in brain have as their main role the modulation of neuronal plasticity and function, in addition to mediating immune protective functions, in both physiologic and pathologic condition (5–8). IL-1, originally described as endogenous pyrogen or leukocytic pyrogen, is mainly produced in the hypothalamus in response to inflammatory agents and can induce fever by upregulating cyclooxygenase-2, the enzyme responsible for the synthesis of the vasoactive and inflammatory prostaglandin E2 (9, 10). A central nervous system (CNS)-restricted expression of splice variants of different IL-1 and IL-1R family members, as truncated forms with regulatory functions, have been identified for both IL-1 (11) and IL-18 (12–14). In the case of IL-37, only one of its five splice variants (IL-37a) is present in the brain (15). Some IL-1R members are only present in the brain, including a particular variant of the accessory receptor chain IL-1R3 (IL-1R3b), expressed in neurons and involved in IL-1-mediated neuroprotection. Notably, the receptor complex for IL-1α and β in the brain can be the canonical one (IL-1R1 plus IL-1R3, present everywhere in the body) that mediates inflammatory effects, or the alternative one (IL-1R1 plus IL-1R3b, specific for the brain) that mediates neuroprotective effects (2, 16–18). Other brain-specific receptors are the two orphan receptors IL-1R9 and IL-1R10, which are mostly expressed in neurons. IL-1R9 is likely involved in memory and learning capacity, as its mutations are linked to X-linked mental retardation and autism (2, 19, 20). The functions of IL-18 in the brain are also peculiar, since the cytokine is active (inflammation and homeostasis) even in the absence of its receptor IL-1R5, while IL-1R5 seems to be involved in the pathogenesis of experimental allergic encephalomyelitis (EAE, the mouse model of multiple sclerosis) in the absence of its IL-18 ligand (21, 22). This suggests that a CNS-specific network of IL-1 family cytokines and receptors is crucial in maintaining brain homeostasis, thus highlighting the limitation of a simplistic inflammatory paradigm to explain the function of these cytokines in neuroinflammatory diseases.
In this perspective, we examine the available experimental and clinical evidence of the involvement of these molecules in two major neurodegenerative diseases, Alzheimer’s disease (AD) and multiple sclerosis (MS), and propose a more global picture of the pathogenic and pathological role of the IL-1/IL-1R system.
2 Alzheimer’s disease
2.1 Disease pathogenesis and inflammation
Alzheimer’s disease (AD) in its sporadic form is the most common dementia diagnosis, characterized by a long progressive process of neuronal damage that affects memory and thinking and ultimately leads to death, in general preceded by a preclinical condition named mild cognitive impairment (MCI) (23).
The main pathological hallmarks of AD are the extracellular accumulation of amyloid β (Aβ) peptides and the intraneuronal neurofibrillary tangles (NFT) formed by aggregation of hyperphosphorylated tau proteins. Neuroinflammation is the third core feature of the disease (24, 25). Many inflammation-related genes have been identified as important AD risk factors (26), and increased inflammatory responses in the brain and periphery have been widely reported in AD (27, 28). Innate immune cells, primarily brain-resident microglia, have a dual role in AD neuroinflammation (29–33). Microglia have a protective function by releasing neurotrophic factors and clearing misfolded proteins, and can degrade Aβ thereby reducing its accumulation. Indeed, a transition of microglia from homeostatic to disease-associated populations endowed with protective potential has been described as a function of the disease progression (34). However, during the course of AD, microglia can be hyperactivated by accumulating danger-associated molecular patterns (DAMPs), mainly Aβ and tau, and secrete neurotoxic and inflammatory cytokines that cause persistent neuroinflammation and initiate/exacerbate neurodegeneration (35–38). The focus on neuroinflammation and inflammasome activation as cause of neuronal damage in AD (39–43) leads to assessing the role of the inflammatory cytokines generated by inflammasome activation, in particular the IL-1 family cytokines IL-1β and IL-18 (44).
2.1.1 IL-1
In the brain, IL-1β is required for learning and memory processes, but, when expressed at aberrant levels, it is involved in infection- and sterile inflammation-induced cognitive dysfunction (45, 46). It has also been suggested that low levels of IL-1 act in the CNS to perform non-immunological functions, while higher concentrations could engage brain non-neuronal cells to produce neuroinflammation (47). Notably, brain IL-1β is produced by and target different cells, ranging from microglia, infiltrating leukocytes, astrocytes, to neurons. By activating different specific cell types, including endothelial cells, neurons and choroid plexus cells, IL-1 may exert different functions of both immunological and non-immunological nature, and neuroinflammation itself may be the result of multiple responses to IL-1 by different brain cell types (8).
In AD, IL-1β is an important mediator of neuroinflammation (48, 49), but also a protective factor (50), able to influence the balance between beneficial and detrimental outcomes (51). In mice lacking the gene encoding the IL-1 inhibitor IL-1Ra the AD-like pathology is exacerbated (52), while the IL-1R1 blockade results in decreased neuroinflammation, attenuated tau pathology and reversal of cognitive deficits (53). Modulation of levels of IL-1Ra or different IL-1R has been observed in brain and blood of AD patients (54–58). Overall, the available experimental and clinical data do not support an exclusively detrimental role of IL-1-driven inflammation in AD.
2.1.2 IL-18
IL-18, its receptors IL-1R5 and IL-1R7 and its inhibitory protein (IL-18BP) are normally expressed in the brain and modulated in both experimental and clinical AD conditions (21). Similar to IL-1, IL-18 could exert both detrimental and protective functions in AD. Thus, while IL-18 dysregulation has been observed in AD patients in association with severity of symptoms and correlated to increased Aβ production in vitro (59–62), a protective function has been also reported in IL-18-deficient mice in both physiological (63) and AD-like conditions (64).
2.1.3 IL-33
A third IL-1 family cytokine, IL-33, is being explored in CNS diseases (65). IL-33 is constitutively expressed in astrocytes and to a minor extent in oligodendrocytes, microglia and neurons; its receptor IL-1R4 is expressed on astrocytes, endothelial cells, glial cells and neurons (6). IL-33 can induce microglia cell proliferation and production of inflammatory IL-1β and TNFα, and anti-inflammatory IL-10 (66). In animal models, IL-33 decreases inflammatory responses and improves AD-like pathology (67). In patients, IL-33 expression in the brain is variably modulated relative to healthy subjects (68, 69). Patients with measurable circulating IL-33 levels have preserved cognitive functions, compared with those that do not express the cytokine (70). Eventually, in MCI patients treated with the neuroprotective compound homotaurine, the improvement of cognitive functions correlates with increased IL-33 plasma levels (71), confirming the potential protective role of this cytokine in AD development.
2.2 Mechanisms of IL-1/IL-1R family in AD pathology
IL-1β, IL-18 and IL-33 are dysregulated in AD and likely exerting a dual role: driving the inflammatory pathogenic processes associated with the disease and providing protection to the damaged CNS. The functions of these mediators in brain homeostasis and pathology can be pleiotropic, redundant, synergic and cross-regulating, aiming to switch off or amplify the inflammatory response in a context-dependent manner. In this scenario, the IL-1R signaling could lead to the production of inflammatory cytokines and participate in Aβ-induced inflammasome activation in microglia, while it might even increase the Amyloid Precursor Protein non-amyloidogenic processing in neurons, thereby preventing neurotoxic Aβ generation (72). Overall, whether and when the different molecules belonging to the IL-1/IL-1R family are either beneficial or detrimental to neuronal function in the course of AD, and how their negative feedback mechanisms influence this, is still a matter of intense research (39). However, the general deregulation of IL-1 family members observed in patients consistently reflect the AD-linked activation of the innate immune system, strongly suggesting that an inflammatory condition, although not exclusively detrimental or beneficial, is indisputably present in AD.
It is likely that neuroinflammation is both a response to Aβ and tau NFT that exacerbates their deleterious effects, and a cause of the disease, that increases brain deposit of Aβ and tau phosphorylation starting before onset of symptoms (73, 74). Convincing new data in patients show that microglia activation and consequent neuroinflammation are associated with disease development and progression (75). The infectious hypothesis (76, 77) offers a fascinating explanation of the etiological meaning of inflammation in the disease and the underlying role of IL-1 family cytokines. According to the observation that Aβ has antimicrobial functions (78), several microbial infections (herpesviruses, periodontal bacteria, gut microbiota dysbiosis) have been associated to AD. By inducing inflammasome activation, infectious agents can induce the release of IL-1 family cytokines, which can contribute to blood brain barrier alterations, amplify pre-existing inflammation in the brain, influence Aβ pathology (note that infections upregulate Aβ production) and tau-related neurodegeneration. Indeed, it has been proposed that the AD pathology can be modulated by innate immune mechanisms associated with infectious burden, including long-term innate memory, which affect epigenetic reprogramming of microglia and the release of IL-1 family cytokines (79, 80).
3 Multiple sclerosis
3.1 Pathology and inflammation
Multiple sclerosis (MS) is a chronic inflammatory neurological disorder driven by myelin-targeting CD4+ T cells. The two main disease phenotypes are the relapsing-remitting form (RRMS) and the primary progressive MS, with a steady loss of function (PPMS) (81, 82). RRMS patients may evolve to a progressive disease course, diagnosed as secondary progressive MS (SPMS). The characteristic demyelinating lesions are localized in both white and grey matter, and contain an abundant inflammatory infiltrate with activated macrophages, microglia, CD8+ and CD4+ T cells, B and plasma cells (83). Inflammatory mediators released by innate and adaptive immune cells lead to damage of oligodendrocytes, the cells responsible for myelin deposition, and axonal injury. A wealth of information, in particular in experimental models, suggests a role for the gut microbiota in bidirectionally modulating brain inflammation in MS, with changes in bacterial composition inducing upregulation vs. downregulation of the production of IL-1β and other inflammation-related factors (84–87).
3.1.1 IL-1β
Acting on astrocytes and endothelial cells, IL-1β increases the permeability of the blood-brain barrier and allows leukocyte recruitment (88); acting on T cells, it induces the encephalitogenic phenotype of Th17 (89). IL1B transcript and IL-1β protein are detected in CNS lesions of MS patients (88), and microglia display an intense IL-1β staining (90). Changes in IL-1 levels in sera and cerebro-spinal fluid (CSF) are non-univocal, possibly due to patients’ heterogeneity and the interference of therapies. Elevated serum levels of IL-1β were reported (91), and the difference with normal subjects was higher for untreated RRMS patients with active disease (92). Other authors report normal levels of IL-1α and IL-1β in active patients, who however had received immunosuppressive therapy (93). Conflicting data have been obtained also in the analysis of CSF. Markedly increased levels of IL-1β in CSF were reported by some authors (91, 94) but not confirmed by others (95, 96). The possible relationship between IL-1β levels and disease activity is also challenged by the observation that in a group of active patients neither serum nor CSF levels were affected by high dose steroid treatment (91).
Soluble inhibitors and decoy receptors are also modulated in MS. IL-1Ra, undetectable in normal CNS, is expressed in lesional foamy macrophages, which also express the anti-inflammatory factors IL-10 and Transforming Growth Factor β, and markers typical of anti-inflammatory M2 macrophages (97). IL-1Ra levels are significantly higher in CSF but not in serum, and even if not correlated with disease activity, they are further increased by steroid treatment (91). IL-1Ra levels are strongly related with disability index, suggesting the potential role of IL-1Ra as serum biomarker of disease progression (98). On the contrary, the soluble decoy receptor IL-1R2 is not elevated in serum compared to normal controls, but steroid treatment induces an increase of its levels (91). In CSF, sIL-1R2 is undetectable irrespective of the disease stage. Finally, soluble IL-1R3 levels are significantly elevated in sera and CSF of MS patients, but not related to disease activity and not modulated by therapy.
3.1.2 IL-18
IL-18 levels were elevated in sera (99–101) and also in CSF, especially in patients with active lesions (99). The findings in CSF, however, were not confirmed in other studies (102). An increase in serum IL-18 levels is detected in untreated RRMS (93) but also in SPMS (102). As observed in most inflammatory disorders, the increase in IL-18 serum levels is accompanied by a parallel increase in its soluble inhibitor IL-18BP, aimed at counteracting the IL-18 inflammatory effects. Although free IL-18 was not evaluated, the IL-18BP/IL-18 ratio was higher in RRMS patients than in controls (93).
3.1.3 Other IL-1 family members
IL-33 has been extensively investigated in MS and EAE. The expression levels of both the cytokine and its receptor IL-1R4 are increased in lesions of MS patients and EAE mice (65, 103). Serum levels of IL-33 are elevated in patients and decreased by treatment with IFNβ (103). Longitudinal assessment of peripheral expression of IL-33 in RRMS revealed peak expression after relapses, but a clear correlation with clinical recovery was not demonstrated (104). In EAE mice, administration of IL-33 attenuates the disease, while antibody-mediated or genetic blockade exacerbates it (105–107). Similarly, IL-1R4 deficient mice develop a severe form of EAE. In this model, IL-33 induces M2 macrophage polarization indirectly, by stimulating mast cells to produce IL-13, and M2 cells then limit the expansion of pathogenic Th17 cells (108). The contribution of IL-1R4+ Treg, activated by IL-33, should also be taken into account. Controversial results have been obtained on the direct effect of IL-33 on myelination (109, 110), but on the whole the data are consistent with the notion that IL-33 is involved in neuroprotection and repair.
Interesting data underline the role of IL-37, an anti-inflammatory member of the IL-1 family. Intracellularly, IL-37 acts as an anti-inflammatory transcriptional regulator. Extracellularly, IL-37 signaling is allegedly mediated by a receptor complex formed by IL-1R5 and IL-1R8. IL-37 is barely detectable in PBMC from normal or MS patients; in the brain, its low expression levels are further downregulated in active MS lesions (111). Notably, the level of IL-37 gene expression in PBMC of MS patients is associated with a lower number of relapses (112). IL-1R5 and IL-1R8 are expressed at similar levels in PBMC and brain from normal or MS patients, and the levels of IL-1R5 (which is also the receptor for IL-18) tend to increase in active lesions. In sera, IL-37 is detectable in 8% of RRMS patients in a stable disease phase and 42% in the active stage; its levels are increased by treatment with a disease-modifying agent, fingolimod (112). Data from patients suggest that the anti-inflammatory activity of IL-37 may be defective in MS, as also confirmed by animal experiments. EAE is milder in transgenic mice expressing human IL-37, with reduced inflammatory infiltrate and decreased numbers of Th1 cells and inflammatory macrophages in lesions (111). This beneficial effect of IL-37 on EAE is lost in mice lacking IL-1R8, indicating that the cytokine confers protection acting through the inhibitory receptor IL-1R8. Moreover, administration of recombinant IL-37 reduces neurological deficits and demyelination in EAE mice. Thus, IL-37 is protective in neuroinflammation and may represent a novel treatment in MS.
4 The NLRP3 inflammasome
The NOD-like receptor 3 (NLRP3) inflammasome, a multiprotein complex that regulates production and release of inflammatory cytokines, in the brain is mainly expressed in microglia but also in astrocytes and neurons. Besides being involved in protection from bacterial, fungal and viral agents, NLRP3 plays a role in neurodevelopment and neuroprotection, but its aberrant activation is crucial in neuroinflammation (113).
Different pathways, including impaired autophagy and dysregulated cell death mechanisms, upregulate NLRP3 activity in AD (114). Aβ and tau can directly activate NLRP3 in microglia (115–117), and several compounds that ameliorate AD-associated pathology reduce NLRP3 activity.
Strong evidence points to a critical role of inflammasome activation in MS. Caspase-1 is more expressed in peripheral blood mononuclear cells (PBMC) of MS patients than in controls (118). Its serum levels can be considered a biomarker of MS, even if they do not identify more active or more severe patients (101). In untreated patients, upregulation of NLRP3 gene expression and higher caspase 1 activity were detected in circulating monocytes (119). NLRP3 upregulation characterized patients with PPMS and was associated with hyperexpression of several inflammatory chemokines and cytokines. IL-1β was one of the most expressed genes, correlated with faster disease progression in PPMS (119). There is a marked increase in the number of IL-1β, caspase-1, and gasdermin D (GSDMD)-positive cells in white matter of MS versus non-MS patients. GSDMD, activated by inflammatory caspases, forms membrane pores responsible for pyroptosis an inflammasome-driven programmed cell death (120). In MS lesions, inflammasome activation and pyroptosis are detected in microglia and oligodendrocytes, suggesting a role in demyelination. Inhibition of inflammasome activation and caspase-1-dependent IL-1β production have already been exploited in MS therapy: IFNβ treatment reduces NLRP3 activity and induces IL-10, thus decreasing the production of pro-IL-1α, pro-IL-1β and mature cytokines (121).
In experimental autoimmune encephalomyelitis (EAE), the mouse MS model, blockade of caspase-1 expression or activity attenuates neuroinflammation (122). Caspase-1 inhibition also reduces GSDMD activation and pyroptosis, limiting oligodendrocyte death and demyelination (120). Similarly, NLRP3 KO or IL-18 KO mice have a less severe disease course, with a marked reduction of CNS inflammatory infiltrate (123, 124). Microglia-specific deletion of the gene encoding the NFκB regulatory protein A20 leads to microglial cell proliferation, susceptibility to LPS-induced inflammation and increased EAE severity, reversed by NLRP3 or caspase-1 genetic deletion (125), indicating a critical role of microglia in MS, mediated by inflammasome activation. The inflammasome involvement in neuroinflammation is also suggested by data in a cohort of MS patients, in which low penetrance mutations of the NLRP3 gene were detected in 16% of the cases (126). These mutations are associated with mild symptoms (suggestive of autoinflammatory disease) and increased production of IL-1β and IL-18 (127).
5 IL-1 family molecules in other main neurodegenerative diseases
Inflammation is involved in other neurological diseases, both neurodegenerative (see below) and of traumatic or other origin (trauma, stroke, autism, schizophrenia, epilepsy). Focusing on neurodegenerative disorders, these are classified in four main groups: amyloidoses, tauopathies, alpha-synucleinopathies, and TDP (transactivation response DNA binding protein)-43 proteinopathies, all characterised by abnormal protein conformations and the formation of protein aggregates in neuronal cells in different brain areas (128, 129). AD is the most common and prevalent of both amyloidoses and tauopathies, along with frontotemporal dementia with parkinsonism and some forms of prion diseases. Main α-synuclein-related diseases include Parkinson’s disease (PD), Dementia with Lewy Bodies, and Multiple System Atrophy, while Amyotrophic Lateral Sclerosis (ALS) is mainly associated with TDP-43 pathology. The Huntington disease (HD) is associated with mutations in the huntingtin protein, which clumps within neurons provoking cell damage and death (130).
Although it is well known and documented that all neurodegenerative diseases share the chronic aberrant inflammation (131), less is known on the role of IL-1 family cytokines and receptors in specific neurodegenerative diseases (132). In addition to AD and MS, reviewed in the previous sections, here we will briefly examine the involvement of IL-1 family molecules in PD, HD, and ALS.
PD is characterised by the death of dopaminergic neurons in the substantia nigra and by the cytoplasmatic aggregation of fibrillar α-synuclein in Lewy bodies (133). Increase of α-synuclein released outside the cells drives the activation of microglia, and the microglia-dependent production of TNF-α, NO, and IL-1β sustains the neuroinflammatory process in PD (134). Recent results in the 6-OHDA mouse model showed that the increased levels of IL-1β and TNF-α have a different kinetics, with TNF-α appearing in the advanced disease stage, while increased level of IL-1β in serum were already evident with a moderate degree of lesion in substantia nigra, suggesting a prognostic value for this inflammatory cytokine (135). Although the results on the level and role of IL-1β in PD patients are conflicting (136), the activation of the NLRP3 inflammasome seems to be determinant for the development of PD at least in vitro. Indeed, when activated, the inflammasome can cause the aggregation of α-synuclein in a neuronal cell model of PD, as caspase-1 directly cleaves α-synuclein generating a truncated protein highly prone to aggregation and able to provoke cell death (137). The involvement of the NLRP3 inflammasome activation in PD also includes two additional mechanisms, the interaction with the parkin protein, whose mutations are responsible of autosomal recessive familial and sporadic early-onset PD (138, 139); and the association with mitochondrial dysfunction that affects neuron performance and favours neuronal degeneration (140). Although IL-1β is a major product of the NLRP3 inflammasome activation, its role in causing/sustaining inflammation in PD patients is still debated (141).
Increasing evidence suggest that inflammation contributes to the development of HD, but the involvement of IL-1 family is still poorly known. The role of IL-1β has been investigated in mice expressing a mutant and pathogenic huntingtin crossed with animals knocked out for IL-1R1 (142). These mice showed more severe neurological symptoms compared to control mice expressing IL-1R1, suggesting a protective role for IL-1R1 signalling in preventing the HD neuropathology. Whether IL-1β is the molecule that activates IL-1R1 signalling is not known. Although IL-1β has been identified as one of the inflammation-related markers of the disease in the porcine HD model (143), its level does not increase in plasma of HD patients, at variance with IL-6 or other inflammatory factors (144, 145). An involvement of the NLRP3 inflammasome has been also suggested for HD, although evidence is still poor and mostly from pre-clinical studies (146).
ALS is a neuromuscular disorder characterized by the progressive loss of anterior-lateral horn spinal cord motoneurons (147). Sporadic ALS affects the 90-95% of patients, while the familiar form affects the remaining 5-10% of patients. The two most relevant genetic mutations associated with ALS are a defect in the hexanucleotide repeat expansion (HRE) in intron 1 of the C9orf72 gene (which makes a protein that regulates actin dynamics and endosomal recycling of GluR1 at the synapse) (148), and in the free radical scavenging enzyme Cu,Zn-superoxide dismutase (SOD1) gene (149). The involvement of inflammation in the etiopathogenesis of ALS is starting to be investigated, but studies are still limited (150, 151). In a recent study, the authors suggest hyperinflammation and immunodeficiency as primary triggers of motoneuron death in ALS, and underline the complex link between the development ALS and T cell and monocyte profiles of patients, as well as polymorphisms in cytokine and chemokine receptors, suggesting a need for personalised therapies based on immunophenotyping (152). In this regard, recently new polymorphisms in the gene encoding IL-1β and in genes involved in oxidative stress (e.g., SOD2) have been identified as modifiers of ALS progression (153). IL-1β is one of the main biomarkers of inflammation in ALS (151, 152). Even if it is not the best marker of ALS severity (154), its plasma levels negatively correlate with survival in ALS patients with genetic variant C9orf72HRE (155). Among the IL-1 family cytokines and receptors, it has been observed that only IL-18 is associated with sporadic ALS, whereas the serum levels of the cytokines IL-33 and IL-36 and the soluble receptors sIL-1R2 and sIL-1R4 were comparable between ALS patients and healthy controls, and IL-1β, IL-1Ra and IL-37 were below detection (156). The possible pathogenic role of IL-18 in ASL has been also recently underlined by a Whole Genome Sequencing study, which identified a genetic variant in a noncoding region of the gene encoding the IL-18 receptor accessory protein (IL18RAP) able to reduce mRNA stability and motor neuron neurotoxicity, thereby decreasing 5x the risk of developing ALS (157). A protective role has been suggested for another member of IL-1 family, IL-33 (158). In the ALS mouse model transgenic for G93A-superoxide dismutase 1 (SOD1-G93A), long-term IL-33 administration delays disease onset in females but not males, probably through peripheral Th2 response (159).
6 Conclusions and perspectives
The role of IL-1 family cytokines and receptors in the etiology and pathogenesis of neurodegenerative diseases is still poorly understood. However, there is strong evidence of their involvement in the initiation and regulation of inflammation associated to most CNS diseases, and their participation to neuroinflammation has been widely described in AD an MS. This is evident despite the limitations due to the current lack of reliable animal models that realistically recapitulate the pathological features of the human diseases, such as in the case of sporadic AD (160). In humans, longitudinal molecular monitoring of inflammation is also hampered by the reduced accessibility of human brain and the limited availability of clinical studies with extensive follow-ups (161, 162).
From the combined in vivo and in vitro functional and molecular evidence, it is clear that these factors play a homeostatic and neuroprotective role in healthy conditions. It is notable that the IL-1 family system in the CNS includes several molecules and functions that are CNS-restricted, implying tissue-specific needs and consequent functional adaptation. Disease conditions imply an imbalance of the IL-1 system network (Table 1).
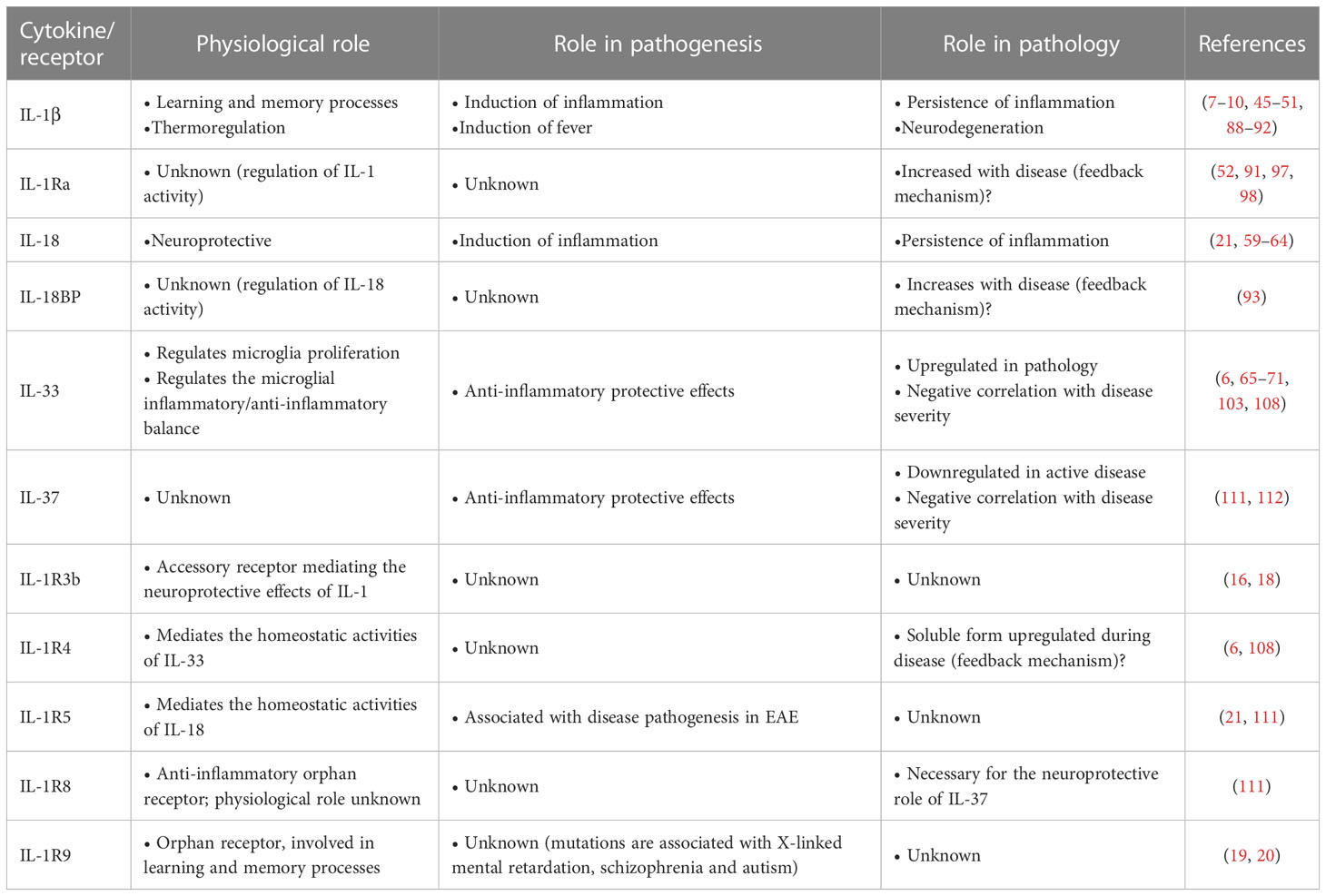
Table 1 IL-1 family cytokines and receptors in brain physiology and pathology.
From the available experimental and clinical evidence, we can draw the following conclusions and suggestions.
• IL-1 family cytokines and receptors are altered in AD and MS and related to the neuroinflammatory conditions.
• Alterations in the amount of produced inflammatory cytokines (IL-1, IL-18) likely switch their role from neuroprotection/homeostasis to pathological inflammation.
• The excess of inflammatory cytokines causes an excessive induction of feedback mechanisms, e.g., the production of downregulating factors (IL-1Ra, IL-18BP, IL-33, IL-37), which however do not succeed in rebalancing the excessive inflammation.
• Unbalanced IL-1 family molecules contribute to alterations in adaptive immune responses, thereby amplifying the autoimmune aspects of neurodegenerative diseases.
• Whether anomalies in the IL-1 family cytokine and receptor network are cause or effect of neuroinflammation and neurodegeneration in AD and MS is not an appropriate question, as it seems that these factors are involved in all steps of disease, from its induction to its establishment and downstream symptoms (Figure 1).
• The CNS-restricted peculiarities of the network of IL-1 family cytokines and receptors suggest a tissue-specific physiological balance and pathological dysregulation. A more thorough understanding of the CNS specificities in the IL-1 family system will open the way to a precision rebalancing approach for therapeutic purposes.
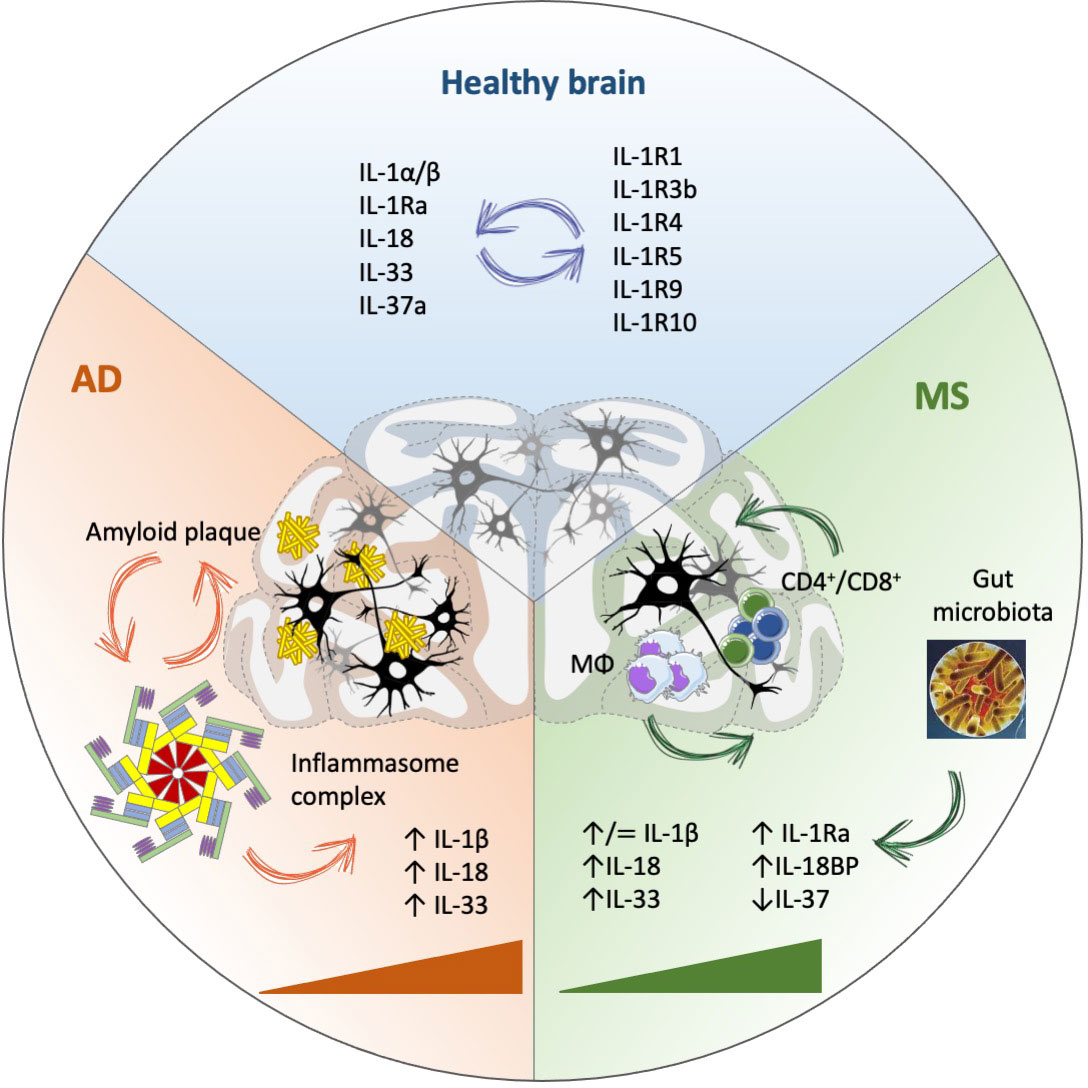
Figure 1 IL-1 family involvement in Alzheimer’s disease (AD) and Multiple Sclerosis (MS). IL-1 family cytokines and receptors are crucial in maintaining brain homeostasis, with an important role in the modulation of neuronal plasticity and function, in addition to mediating immune protective functions. In physiological conditions, many activities of IL-1 family cytokines depend on binding to specific receptors, but independent functions have been observed for both cytokines (e.g., IL-18) and receptors (e.g., IL-1R9). In disease conditions (AD, MS), IL-1 family members are substantially modulated during disease progression (triangles), associated with increased inflammation (partly dependent on inflammasome activation) and likely involved in all disease phases, from initiation and establishment to progression, without a clear association with either disease initiation or progression. MΦ, macrophages.
Author contributions
DB, PM, PI and PB wrote the manuscript and critically revised it. DB and PI contributed to figures and tables. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the EU Commission H2020 projects PANDORA (GA671881) and ENDONANO (GA 812661) (PI, DB), the project MIUR/PRIN-20173ZECCM of the Italian Ministry of Research (PI) and the projects RC 2018-2021, ADIMB, CoEN 2019-Pathfinder IV and RF-2021-12374301 by the Italian Ministry of Health (PB).
Conflict of interest
The reviewer JB declared a shared affiliation, with no collaboration, with one of the authors, DB, to the handling editor at the time of the review.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Dinarello CA. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev (2018) 281:8–27. doi: 10.1111/imr.12621
2. Boraschi D, Italiani P, Weil S, Martin MU. The family of the interleukin-1 receptors. Immunol Rev (2018) 281:197–232. doi: 10.1111/imr.12606
3. Rivers-Auty J, Daniels MJD, Colliver I, Robertson DL, Brough D. Redefining the ancestral origins of the interleukin-1 superfamily. Nat Commun (2018) 9:1156. doi: 10.1038/s1467-018-03362-1
4. Boraschi D. What is IL-1 for? the functions of IL-1 across evolution. Front Immunol (2021) 13:872155. doi: 10.3389/fimmu.2022.872155
5. Alboni S, Cervia D, Sugama S, Conti B. Interleukin 18 in the CNS. J Neuroinflamm (2010) 7:9. doi: 10.1186/1742-2094-7-9
6. Fairlie-Clarke K, Barbour M, Wilson C, Hridi SU, Allan D, Jiang H-R. Expression and function of IL-33/ST2 axis in the central nervous system under normal and diseased conditions. Front Immunol (2018) 9:2596. doi: 10.3389/fimmu.2018.02596
7. Liu X, Quan N. Microglia and CNS interleukin-1: beyond immunological concepts. Front Neurol (2018) 9:8. doi: 10.3389/fneur.2018.00008
8. Liu X, Nemeth DP, McKim DB, Zhu L, DiSabato DJ, Berdysz O, et al. Cell-type-specific interleukin 1 receptor 1 signaling in the brain regulates distinct neuroimmune activities. Immunity (2019) 50:317. doi: 10.1016/j.immuni.2018.12.012
9. Dinarello CA. Cytokines as endogenous pyrogens. J Infect Dis (1999) 179:S294–304. doi: 10.1086/513856
10. El-Rahi AS. Pathogenesis of fever. In: Clinical manual of fever in children, nature public health emergency collection (2019). Springer, Cham. p. 53–68. doi: 10.1007/978-3-319-92336-9_3
11. Smith DE, Lipsky BP, Russell C, Ketchem RR, Kirchner J, Hensley K, et al. A novel CNS-restricted isoform of the IL-1R accessory protein modulates neuronal responses to IL-1. Immunity (2009) 30:817. doi: 10.1016/j.immuni.2009.03.020
12. Andre R, Wheeler RD, Collins PD, Luheshi GN, Pickering-Brown S, Kimber I, et al. Identification of a truncated IL-18Rβ mRNA: a putative regulator of IL-18 expressed in rat brain. J Neuroimmunol (2003) 145:40–5. doi: 10.1016/j.jneuroim.2003.09.005
13. Alboni S, Cervia D, Ross B, Montanari C, Gonzalez AS, Sanchez-Alavez M, et al. Mapping of the full length and the truncated interleukin-18 receptor alpha in the mouse brain. J Neuroimmunol (2009) 214:43–54. doi: 10.1016/j.jneuroim.2009.06.016
14. Alboni S, Montanari C, Benatti C, Blom JMC, Simone ML, Brunello N, et al. Constitutive and LPS-regulated expression of interleukin-18 receptor beta variants in the mouse brain. Brain Behav Immun (2011) 25:483–93. doi: 10.1016/j.bbi.2010.11.011
15. Boraschi D, Lucchesi D, Hainzl S, Leitner M, Maier E, Mangelberger D, et al. IL-37: an anti-inflammatory cytokine of the IL-1 family. Eur Cytokine Netw (2011) 22:127–47. doi: 10.1684/ecn.2011.0288
16. Smith DE, Lipsky BP, Russell C, Ketchem RR, Kirchner J, Hensley K, et al. A central nervous system-restricted isoform of the interleukin-1 receptor accessory protein modulates neuronal responses to interleukin-1. Immunity (2009) 30:817–31. doi: 10.1016/j.immuni.2009.03.020
17. Nguyen L, Rothwell NJ, Pinteaux E, Boutin H. Contribution of interleukin-1 receptor accessory protein b to interleukin-1 actions in neuronal cells. Neurosignals (2011) 19:222–30. doi: 10.1159/000330803
18. Huang Y, Smith DE, Ibáñez-Sandoval O, Sims JE, Fridman WJ. Neuron-specific effects of interleukin-1β are mediated by a novel isoform of the IL-1 receptor accessory protein. J Neurosci (2011) 31:18048–59. doi: 10.1523/JNEUROSCI.4067-11.201
19. Carrié A, Jun L, Bienvenu T, Vinet MC, McDonell N, Couvert P, et al. A new member of the IL-1 receptor family highly expressed in hippocampus and involved in X-linked mental retardation. Nat Genet (1999) 23:25–31. doi: 10.1038/12623
20. Piton A, Michaud JL, Peng H, Aradhya S, Gauthier J, Mottron L, et al. Mutations in the calcium-related gene IL1RAPL1 are associated with autism. Hum Mol Genet (2008) 17:3965–74. doi: 10.1093/hmg/ddn300
21. Bossù P, Ciaramella A, Salani F, Vanni D, Palladino I, Caltagirone C, et al. Interleukin-18, from neuroinflammation to alzheimer’s disease. Curr Pharm Des (2010) 16:4213–24. doi: 10.2174/138161210794519147
22. Gutcher I, Urich E, Wolter K, Prinz M, Becher B. Interleukin 18-independent engagement of interleukin 18 receptor-α is required for autoimmune inflammation. Nat Immunol (2006) 7:946–53. doi: 10.1038/ni1377
23. 2022 alzheimer’s disease facts and figures. Alzheimer’s Dementia (2022) 18:700–89. doi: 10.1002/alz.12638
24. McGeer PL, McGeer EG. The amyloid cascade-inflammatory hypothesis of Alzheimer disease: implications for therapy. Acta Neuropathol (2013) 126:479–97. doi: 10.1007/s00401-013-1177-7
25. Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a central mechanism in alzheimer’s disease. Alzheimers Dement (2018) 4:575–90. doi: 10.1016/j.trci.2018.06.014
26. Bellenguez C, Küçükali F, Jansen IE, Kleineidam L, Moreno-Grau S, Amin N, et al. New insights into the genetic etiology of alzheimer’s disease and related dementias. Nat Genet (2022) 54:412–36. doi: 10.1038/s41588-022-01024-z
27. Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in alzheimer’s disease. Lancet Neurol (2015) 14:388–405. doi: 10.1016/S1474-4422(15)70016-5
28. Ennerfelt HE, Lukens JR. The role of innate immunity in alzheimer’s disease. Immunol Rev (2020) 297:225–46. doi: 10.1111/imr.12896
29. Rezai-Zadeh K, Gate D, Town T. CNS infiltration of peripheral immune cells: d-day for neurodegenerative disease? J Neuroimmune Pharmacol (2009) 4:462–75. doi: 10.1007/s11481-009-9166-2
30. Mildner A, Schlevogt B, Kierdorf K, Bottcher C, Erny D, Kummer MP, et al. Distinct and non-redundant roles of microglia and myeloid subsets in mouse models of alzheimer’s disease. J Neurosci (2011) 31:11159–71. doi: 10.1523/JNEUROSCI.6209-10.2011
31. Le Page A, Dupuis G, Frost EH, Larbi A, Pawelec G, Witkowski JM, et al. Role of the peripheral innate immune system in the development of alzheimer’s disease. Exp Gerontol (2018) 107:59–66. doi: 10.1016/j.exger.2017.12.019
32. Fani Maleki A, Rivest S. Innate immune cells: monocytes, monocyte-derived macrophages and microglia as therapeutic targets for alzheimer’s disease and multiple sclerosis. Front Cell Neurosci (2019) 13:355. doi: 10.3389/fncel.2019.00355
33. Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol (2020) 17:157–72. doi: 10.1038/s41582-020-00435-y
34. Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A unique microglia type associated with restricting development of alzheimer's disease. Cell (2017) 169:1276–90.e17. doi: 10.1016/j.cell.2017.05.018
35. Cuello AC. Early and late CNS inflammation in alzheimer’s disease: two extremes of a continuum? Trends Pharmacol Sci (2017) 38:956–66. doi: 10.1016/j.tips.2017.07.005
36. Hickman S, Izzy S, Sen P, Morsett L, El Khoury J. Microglia in neurodegeneration. Nat Neurosci (2018) 21:1359–69. doi: 10.1038/s41593-018-0242-x
37. Hansen DV, Hanson JE, Sheng M. Microglia in alzheimer’s disease. J Cell Biol (2018) 217:459–72. doi: 10.1083/jcb.201709069
38. Cai Y, Liu J, Wang B, Sun M, Yang H. Microglia in the neuroinflammatory pathogenesis of alzheimer’s disease and related therapeutic targets. Front Immunol (2022) 13:856376. doi: 10.3389/fimmu.2022.856376
39. Lau S-F, Fu AKY, Ip NY. Cytokine signaling convergence regulates the microglial state transition in alzheimer’s disease. Cell Mol Life Sci (2021) 78:4703–12. doi: 10.1007/s00018-021-03810-0
40. Passaro AP, Lebos AL, Yao Y, Stice SL. Immune response in neurological pathology: emerging role of central and peripheral immune crosstalk. Front Immunol (2021) 12:676621. doi: 10.3389/fimmu.2021.676621
41. Bai H, Zhang Q. Activation of NLRP3 inflammasome and onset of alzheimer’s disease. Front Immunol (2021) 12:701282. doi: 10.3389/fimmu.2021.701282
42. Milner MT, Maddugoda M, Götz J, Burgener SS, Schroder K. The NLRP3 inflammasome triggers sterile neuroinflammation and alzheimer’s disease. Curr Opin Immunol (2021) 68:116–24. doi: 10.1016/j.coi.2020.10.011
43. Liang T, Zhang Y, Wu S, Chen Q, Wang L. The role of NLRP3 inflammasome in alzheimer’s disease and potential therapeutic targets. Front Pharmacol (2022) 13:845185. doi: 10.3389/fphar.2022.845185
44. Chan AH, Schroder K. Inflammasome signaling and regulation of interleukin-1 family cytokines. J Exp Med (2019) 217:e20190314. doi: 10.1084/jem.20190314
45. Yirmiya R, Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun (2011) 25:181–213. doi: 10.1016/j.bbi.2010.10.015
46. Skelly DT, Griffin ÉW, Murray CL, Harney S, O’Boyle C, Hennessy E, et al. Acute transient cognitive dysfunction and acute brain injury induced by systemic inflammation occur by dissociable IL-1-dependent mechanisms. Mol Psychiatry (2019) 24:1533–48. doi: 10.1038/s41380-018-0075-8
47. An Y, Chen Q, Quan N. Interleukin-1 exerts distinct actions on different cell types of the brain in vitro. J Inflammation Res (2011) 2011:11. doi: 10.2147/JIR.S15357
48. Mrak RE, Griffin WST. Interleukin-1 and the immunogenetics of Alzheimer disease. J Neuropathol Exp Neurol (2000) 59:471. doi: 10.1093/jnen/59.6.471
49. Allan SM, Tyrrell PJ, Rothwell NJ. Interleukin-1 and neuronal injury. Nat Rev Immunol (2005) 5:629–40. doi: 10.1038/nri1664
50. Shaftel SS, Kyrkanides S, Olschowka JA, Miller JH, Johnson RE, O’Banion MK. Sustained hippocampal IL-1β overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J Clin Invest (2007) 117:1595. doi: 10.1172/JCI31450
51. Shaftel SS, Griffin WST, O’Banion MK. The role of interleukin-1 in neuroinflammation and Alzheimer disease: an evolving perspective. J Neuroinflamm (2008) 5:1–12. doi: 10.1186/1742-2094-5-7
52. Craft JM, Watterson DM, Hirsch E, Van Eldik LJ. Interleukin 1 receptor antagonist knockout mice show enhanced microglial activation and neuronal damage induced by intracerebroventricular infusion of human beta-amyloid. J Neuroinflamm (2005) 2:15. doi: 10.1186/1742-2094-2-15
53. Kitazawa M, Cheng D, Tsukamoto MR, Koike MA, Wes PD, Vasilevko V, et al. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β-catenin pathway function in an alzheimer's disease model. J Immunol (2011) 187:6539–49. doi: 10.4049/jimmunol.1100620
54. Yasuhara O, Matsuo A, Terai K, Walker DG, Berger AE, Akiguchi I, et al. Expression of interleukin-1 receptor antagonist protein in post-mortem human brain tissues of alzheimer’s disease and control cases. Acta Neuropathol (1997) 93:414–20. doi: 10.1007/s004010050633
55. Taipa R, das Neves SP, Sousa AL, Fernandes J, Pinto C, Correia AP, et al. Proinflammatory and anti-inflammatory cytokines in the CSF of patients with alzheimer’s disease and their correlation with cognitive decline. Neurobiol Aging (2019) 76:125–32. doi: 10.1016/j.neurobiolaging.2018.12.019
56. Garlind A, Brauner A, Höjeberg B, Basun H, Schultzberg M. Soluble interleukin-1 receptor type II levels are elevated in cerebrospinal fluid in alzheimer’s disease patients. Brain Res (1999) 826:112–6. doi: 10.1016/S0006-8993(99)01092-6
57. Italiani P, Puxeddu I, Napoletano S, Scala E, Melillo D, Manocchio S, et al. Circulating levels of IL-1 family cytokines and receptors in alzheimer’s disease: new markers of disease progression? J Neuroinflamm (2018) 15:342. doi: 10.1186/s12974-018-1376-1
58. Zettergren A, Höglund K, Kern S, Thorvaldsson V, Johan Skoog M, Hansson O, et al. Association of IL1RAP-related genetic variation with cerebrospinal fluid concentration of Alzheimer-associated tau protein. Sci Rep (2019) 9:2460. doi: 10.1038/s41598-018-36650-3
59. Motta M, Imbesi R, Di Rosa M, Stivala F, Malaguarnera L. Altered plasma cytokine levels in alzheimer’s disease: correlation with the disease progression. Immunol Lett (2007) 114:46–51. doi: 10.1016/j.imlet.2007.09.002
60. Bossù P, Ciaramella A, Salani F, Bizzoni F, Varsi E, Di Iulio F, et al. Interleukin-18 produced by peripheral blood cells is increased in alzheimer’s disease and correlates with cognitive impairment. Brain Behav Immun (2008) 22:487–92. doi: 10.1016/j.bbi.2007.10.001
61. Ojala J, Alafuzoff I, Herukka S-K, van Groen T, Tanila H, Pirttilä T. Expression of interleukin-18 is increased in the brains of alzheimer’s disease patients. Neurobiol Aging (2009) 30:198–209. doi: 10.1016/j.neurobiolaging.2007.06.006
62. Sutinen EM, Pirttilä T, Anderson G, Salminen A, Ojala JO. Pro-inflammatory interleukin-18 increases alzheimer’s disease-associated amyloid-β production in human neuron-like cells. J Neuroinflamm (2012) 9:199. doi: 10.1186/1742-2094-9-199
63. Yamanishi K, Doe N, Mukai K, Ikubo K, Hashimoto T, Uwa N, et al. Interleukin-18-deficient mice develop hippocampal abnormalities related to possible depressive-like behaviors. Neuroscience (2019) 408:147–60. doi: 10.1016/j.neuroscience.2019.04.003
64. Tzeng T-C, Hasegawa Y, Iguchi R, Cheung A, Caffrey DR, Thatcher EJ, et al. Inflammasome-derived cytokine IL18 suppresses amyloid-induced seizures in Alzheimer-prone mice. Proc Natl Acad Sci USA (2018) 115:9002–7. doi: 10.1073/pnas.1801802115
65. Sun Y, Wen Y, Wang L, Wen L, You W, Wei S, et al. Therapeutic opportunities of interleukin-33 in the central nervous system. Front Immunol (2021) 12:654626. doi: 10.3389/fimmu.2021.654626
66. Yasuoka S, Kawanokuchi J, Parajuli B, Jin S, Doi Y, Noda M, et al. Production and functions of IL-33 in the central nervous system. Brain Res (2011) 1385:8–17. doi: 10.1016/j.brainres.2011.02.045
67. Fu AKY, Hung K-W, Yuen MYF, Zhou X, Mak DSY, Chan ICW, et al. IL-33 ameliorates alzheimer’s disease-like pathology and cognitive decline. Proc Natl Acad Sci USA (2016) 113:E2705–13. doi: 10.1073/pnas.1604032113
68. Chapuis J, Hot D, Hansmannel F, Kerdraon O, Ferreira S, Hubans C, et al. Transcriptomic and genetic studies identify IL-33 as a candidate gene for alzheimer’s disease. Mol Psychiatry (2009) 14:1004–16. doi: 10.1038/mp.2009.10
69. Xiong Z, Thangavel R, Kempuraj D, Yang E, Zaheer S, Zaheer A. Alzheimer’s disease: evidence for the expression of interleukin-33 and its receptor ST2 in the brain. J Alzheimers Dis (2014) 40:297–308. doi: 10.3233/JAD-132081
70. Liang C-S, Su K-P, Tsai C-L, Lee J-T, Chu C-S, Yeh T-C, et al. The role of interleukin-33 in patients with mild cognitive impairment and alzheimer’s disease. Alzheimers Res Ther (2020) 12:86. doi: 10.1186/s13195-020-00652-z
71. Toppi E, Sireno L, Lembo M, Banaj N, Messina B, Golesorkhtafti S, et al. IL-33 and IL-10 serum levels increase in MCI patients following homotaurine treatment. Front Immunol (2022) 13:813951. doi: 10.3389/fimmu.2022.813951
72. Wang H, Wang X. Interleukin 1 receptor and alzheimer’s disease-related neuroinflammation. in mechanisms of neuroinflammation. InTech (2017) 2017. doi: 10.5772/intechopen.69067
73. Zhang B, Gaiteri C, Bodea L-G, Wang Z, McElwee J, Podtelezhnikov AA, et al. Integrated systems approach identifies genetic nodes and networks in late-onset alzheimer’s disease. Cell (2013) 153:707. doi: 10.1016/j.cell.2013.03.030
74. Zhao J, Zhao D, Wang J, Luo X, Guo R. Inflammation–cause or consequence of late onset alzheimer’s disease or both? a review of the evidence. Eur J Inflammation (2022) 20. doi: 10.1177/1721727X221095383
75. Pascoal TA, Benedet AL, Ashton NJ, Kang MS, Therriault J, Chamoun M, et al. Microglial activation and tau propagate jointly across braak stages. Nat Med (2021) 27:1592–9. doi: 10.1038/s41591-021-01456-w
76. Fulop T, Witkowski JM, Bourgade K, Khalil A, Zerif E, Larbi A, et al. Can an infection hypothesis explain the beta amyloid hypothesis of alzheimer’s disease? Front Aging Neurosci (2018) 10:224. doi: 10.3389/fnagi.2018.00224
77. Itzhaki RF, Golde TE, Heneka MT, Readhead B. Do infections have a role in the pathogenesis of Alzheimer disease? Nat Rev Neurol (2020) 16:193–7. doi: 10.1038/s41582-020-0323-9
78. Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, et al. The alzheimer’s disease-associated amyloid β-protein is an antimicrobial peptide. PLoS One (2010) 5:e9505. doi: 10.1371/journal.pone.0009505
79. Wendeln A-C, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G, et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature (2018) 556:332–8. doi: 10.1038/s41586-018-0023-4
80. Salani F, Sterbini V, Sacchinelli E, Garramone M, Bossù P. Is innate memory a double-edge sword in alzheimer’s disease? a reappraisal of new concepts and old data. Front Immunol (2019) 10:1768. doi: 10.3389/fimmu.2019.01768
81. Dobson R, Giovannoni G. Multiple sclerosis: a review. Eur J Neurol (2019) 26:27–40. doi: 10.1111/ene.13819
82. Cree BAC, Arnold DL, Chataway J, Chitnis T, Fox RJ, Pozo Ramajo A, et al. Secondary progressive multiple sclerosis. New Insights Neurol (2021) 97:378–88. doi: 10.1212/WNL.0000000000012323
83. Attfield KE, Torp Jensen L, Kaufmann M, Friese MA, Fugger L. The immunology of multiple sclerosis. Nat Rev Immunol (2022) 22:734–50. doi: 10.1038/s41577-022-00718-z
84. Chen J, Chia N, Kalari KR, Yao JZ, Novitna M, Soldan MMP, et al. Multiple sclerosis patients have a distinct gut microbiota compare to healthy controls. Sci Rep (2016) 6:1–10. doi: 10.1038/srep28484
85. Cekanaviciute E, Yoo BB, Runia TF, Debelius JW, Singh S, Nelson CA, et al. Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc Natl Acad Sci USA (2017) 114:10713–8. doi: 10.1073/pnas.1711235114
86. Rutsch A, Kantsjö JB, Ronchi F. The gut-brain axis: how microbiota and hist inflammasome influence brain physiology and pathology. Front Immunol (2020) 11:604179. doi: 10.3389/fimmu.2020.604179
87. Mestre L, Carrillo-Salinas FJ, Feliú A, Mecha M, Alonso G, Espejo C, et al. How oral probiotics affect the severity of an experimental model of progressive multiple sclerosis? bringing commensal bacteria into the neurodegenerative process. Gut Microbes (2020) 12:1813532. doi: 10.1080/19490976.2020.1813532
88. Lin CC, Edelson BT. New insights into the role of IL-1β in experimental autoimmune encephalomyelitis and multiple sclerosis. J Immunol (2017) 198:4553–60. doi: 10.4049/jimmunol.1700263
89. Lukens JR, Barr MJ, Chaplin DD, Chi H, Kanneganti TD. Inflammasome-derived IL-1β regulates the production of GM-CSF by CD4(+) T cells and γδ T cells. J Immunol (2012) 188:3107–15. doi: 10.4049/jimmunol.1103308
90. Burm SM, Peferoen LA, Zuiderwijk-Sick EA, Haanstra KG, 't Hart BA, van der Valk P, et al. Expression of IL-1β in rhesus EAE and MS lesions is mainly induced in the CNS itself. J Neuroinflamm (2016) 13:138. doi: 10.1186/s12974-016-0605-8
91. Dujmovic I, Mangano K, Pekmezovic T, Quattrocchi C, Mesaros S, Stojsavljevic N, et al. The analysis of IL-1β and its naturally occurring inhibitors in multiple sclerosis: the elevation of IL-1 receptor antagonist and IL-1 receptor type II after steroid therapy. J Neuroimmunol (2009) 207:101–6. doi: 10.1016/j.neuroim.2008.11.004
92. D'Angelo C, Reale M, Costantini E, Di Nicola M, Porfilio I, de Andrés C, et al. Profiling of canonical and non-traditional cytokine levels in interferon-β-treated relapsing-remitting-multiple sclerosis patients. Front Immunol (2018) 9:1240. doi: 10.3389/fimmu.2018.01240
93. Thöne J, Kleiter I, Stahl A, Ellrichmann G, Gold R, Hellwig K. Relevance of endoglin, IL-1α, IL-1β and anti-ovarian antibodies in females with multiple sclerosis. J Neurol Sci (2016) 362:240–3. doi: 10.1016/j.jns.2016.01.057
94. Hauser SL, Doolittle TH, Lincoln R, Brown RH, Dinarello CA. Cytokine accumulations in CSF of multiple sclerosis patients: frequent detection of interleukin-1 and tumor necrosis factor but not interleukin-6. Neurology (1990) 40:1735–9. doi: 10.1212/wnl.40.11.1735
95. Maimone D, Gregory S, Arnason BG, Reder AT. Cytokine levels in the cerebrospinal fluid and serum of patients with multiple sclerosis. J Neuroimmunol (1991) 32:67–74. doi: 10.1016/0165-5728(91)90073-g
96. Tsukada N, Miyagi K, Matsuda M, Yanagisawa N, Yone K. Tumor necrosis factor and interleukin-1 in the CSF and sera of patients with multiple sclerosis. J Neurol Sci (1991) 104:230–4. doi: 10.1016/0022-510x(91)90315-x
97. Boven LA, Van Meurs M, Van Zwam M, Wierenga-Wolf A, Hintzen RQ, Boot RG, et al. Myelin-laden macrophages are anti-inflammatory, consistent with foam cells in multiple sclerosis. Brain (2006) 129:517–26. doi: 10.1093/brain/awh707
98. Blandford SN, Galloway DA, Williams JB, Arsenault S, Brown J, MacLean G, et al. Interleukin-1 receptor antagonist: an exploratory plasma biomarker that correlates with disability and provides pathophysiological insights in relapsing-remitting multiple sclerosis. Mult Scler Relat Disord (2021) 52:103006. doi: 10.1016/j.msard.2021.103006
99. Chen YC, Chen SD, Miao L, Liu ZG, Li W, Zhao ZX, et al. Serum levels of interleukin (IL)-18, IL-23 and IL-17 in Chinese patients with multiple sclerosis. J Neuroimmunol (2012) 243:56–60. doi: 10.1016/j.jneuroim.2011.12.008
100. Losy J, Niezgoda A. IL-18 in patients with multiple sclerosis. Acta Neurol Scand (2001) 104:171–3. doi: 10.1034/j.1600-0404.2001.00356.x
101. Keane RW, Dietrich WD, de Rivero Vaccari JP. Inflammasome proteins as biomarkers of multiple sclerosis. Front Neurol (2018) 9:135. doi: 10.3389/fneur.2018.00135
102. Nicoletti F, Di Marco R, Mangano K, Patti F, Reggio E, Nicoletti A, et al. Increased serum levels of interleukin-18 in patients with multiple sclerosis. Neurology (2001) 57:342–4. doi: 10.1212/wnl.57.2.342
103. Christophi GP, Gruber RC, Panos M, Christophi RL, Jubelt B, Massa PT. Interleukin-33 upregulation in peripheral leukocytes and CNS of multiple sclerosis patients. Clin Immunol (2012) 142:308–19. doi: 10.1016/j.clim.2011.11.007
104. Sriram S, Shaginurova G, Tossberg JT, Natarajan C, Spurlock CF 3rd, Aune TM. Longitudinal changes in the expression of IL-33 and IL-33 regulated genes in relapsing remitting MS. PloS One (2018) 13:e0208755. doi: 10.1371/journal.pone.0208755
105. Chen H, Sun Y, Lai L, Wu H, Xiao Y, Ming B, et al. Interleukin-33 is released in spinal cord and suppresses experimental autoimmune encephalomyelitis in mice. Neuroscience (2015) 308:157–68. doi: 10.1016/j.neuroscience.2015.09.019
106. Jiang HR, Milovanović M, Allan D, Niedbala W, Besnard AG, Fukada SY, et al. IL-33 attenuates EAE by suppressing IL-17 and IFN-γ production and inducing alternatively activated macrophages. Eur J Immunol (2012) 42:1804–14. doi: 10.1002/eji.201141947
107. Xiao Y, Lai L, Chen H, Shi J, Zeng F, Li J, et al. Interleukin-33 deficiency exacerbated experimental autoimmune encephalomyelitis with an influence on immune cells and glia cells. Mol Immunol (2018) 101:550–63. doi: 10.1016/j.molimm.2018.08.026
108. Finlay CM, Cunningham KT, Doyle B, Mills KHG. IL-33-stimulated murine mast cells polarize alternatively activated macrophages, which suppress T cells that mediate experimental autoimmune encephalomyelitis. J Immunol (2020) 205:1909–19. doi: 10.4049/jimmunol.1901321
109. Allan D, Fairlie-Clarke KJ, Elliott C, Schuh C, Barnett SC, Lassmann H, et al. Role of IL-33 and ST2 signalling pathway in multiple sclerosis: expression by oligodendrocytes and inhibition of myelination in central nervous system. Acta Neuropathol Commun (2016) 4:75. doi: 10.1186/s40478-016-0344-1
110. Natarajan C, Yao SY, Sriram S. TLR3 agonist poly-IC induces IL-33 and promotes myelin repair. PloS One (2016) 11:e0152163. doi: 10.1371/journal.pone.0152163
111. Sánchez-Fernández A, Zandee S, Amo-Aparicio J, Charabati M, Prat A, Garlanda C, et al. IL-37 exerts therapeutic effects in experimental autoimmune encephalomyelitis through the receptor complex IL-1R5/IL-1R8. Theranostics (2021) 11:1–13. doi: 10.7150/thno.47435
112. Cavalli E, Mazzon E, Basile MS, Mammana S, Pennisi M, Fagone P, et al. In silico and in vivo analysis of IL-37 in multiple sclerosis reveals its probable homeostatic role on the clinical activity, disability, and treatment with fingolimod. Molecules (2019) 25:20. doi: 10.3390/molecules25010020
113. Jha D, Bakker ENTP, Kumar R. Mechanistic and therapeutic role of NLRP3 inflammasome in the pathogenesis of alzheimer's disease. J Neurochem (2023) 00:1–25. doi: 10.1111/jnc.15788
114. Kumar S, Budhathoki S, Oliveira CB, Kahle AD, Calhan OY, Lukens JR, et al. Role of the caspase-8/RIPK3 axis in alzheimer’s disease pathogenesis and aβ-induced NLRP3 inflammasome activation. JCI Insight (2023) 8:e157433. doi: 10.1172/jci.insight.157433
115. Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. NLRP3 is activated in alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature (2013) 493:674–8. doi: 10.1038/nature11729
116. Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A, et al. NLRP3 inflammasome activation drives tau pathology. Nature (2019) 575:669–73. doi: 10.1038/s41586-019-1769-z
117. Tejera D, Mercan D, Sanchez-Caro JM, Hanan M, Greenberg D, Soreq H, et al. Systemic inflammation impairs microglial aβ clearance through NLRP3 inflammasome. EMBO J (2019) 38:e101064. doi: 10.15252/embj.2018101064
118. Huang WX, Huang P, Hillert J. Increased expression of caspase-1 and interleukin-18 in peripheral blood mononuclear cells in patients with multiple sclerosis. Mult Scler (2004) 10:482–7. doi: 10.1191/1352458504ms1071oa
119. Malhotra S, Cost C, Eixarch H, Keller CW, Amman L, Martinez-Banaclocha H, et al. NLRP3 inflammasome as prognostic factor and therapeutic target in primary progressive multiple sclerosis patients. Brain (2020) 143:1414–30. doi: 10.1093/brain/awaa084
120. McKenzie BA, Mamik MK, Saito LB, Boghozian R, Monaco MC, Major EO, et al. Caspase-1 inhibition prevents glial inflammasome activation and pyroptosis in models of multiple sclerosis. Proc Natl Acad Sci USA (2018) 115:E6065–74. doi: 10.1073/pnas.1722041115
121. Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Förster I, et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity (2011) 34:213–23. doi: 10.1016/j.immuni.2011.02.006
122. Furlan R, Martino G, Galbiati F, Poliani PL, Smiroldo S, Bergami A, et al. Caspase-1 regulates the inflammatory process leading to autoimmune demyelination. J Immunol (1999) 163:2403–9. doi: 10.4049/jimmunol.163.5.2403
123. Gris D, Ye Z, Iocca HA, Wen H, Craven RR, Gris P, et al. NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J Immunol (2010) 185:974–81. doi: 10.4049/jimmunol.0904145
124. Inoue M, Williams KL, Gunn MD, Shinohara ML. NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA (2012) 109:10480–5. doi: 10.1073/pnas.1201836109
125. Voet S, Mc Guire C, Hagemeyer N, Martens A, Schroeder A, Wieghofer P, et al. A20 critically controls microglia activation and inhibits inflammasome-dependent neuroinflammation. Nat Commun (2018) 9:2036. doi: 10.1038/s41467-018-04376-5
126. Schuh E, Lohse P, Ertl-Wagner B, Witt M, Krumbholz M, Frankenberger M, et al. Expanding spectrum of neurologic manifestations in patients with NLRP3 low-penetrance mutations. Neurol Neuroimmunol Neuroinflamm (2015) 2:e109. doi: 10.1212/NXI.0000000000000109
127. Verma D, Särndahl E, Andersson H, Eriksson P, Fredrikson M, Jönsson JI, et al. The Q705K polymorphism in NLRP3 is a gain-of-function alteration leading to excessive interleukin-1β and IL-18 production. PLoS One (2012) 7:e34977. doi: 10.1371/journal.pone.0034977
128. Kovacs GG. Molecular pathological classification of neurodegenerative diseases: turning towards precision medicine. Int J Mol Sci (2016) 17:189. doi: 10.3390/ijms17020189
129. Dugger BN, Dickson DW. Pathology of neurodegenerative diseases. Cold Spring Harb Perspect Biol (2017) 9:a028035. doi: 10.1101/cshperspect.a028035
130. Nopoulos PC. Huntington disease: a single-gene degenerative disorder of the striatum. Dialogues Clin Neurosci (2016) 18:91–8. doi: 10.31887/DCNS.2016.18.1/pnopoulos
131. Guzman-Martinez L, Maccioni RB, Andrade V, Navarrete LP, Pastor MG, Ramos-Escobar N. Neuroinflammation as a common feature of neurodegenerative disorders. Front Pharmacol (2019) 10:1008. doi: 10.3389/fphar.2019.01008
132. Luís JP, Simões CJV, Brito RMM. The therapeutic prospects of targeting IL-1R1 for the modulation of neuroinflammation in central nervous system disorders. Int J Mol Sci (2022) 23:1731. doi: 10.3390/ijms23031731
133. Petrucci S, Consoli F, Valente EM. Parkinson Disease genetics: a “continuum” from mendelian to multifactorial inheritance. Curr Mol Med (2014) 14:1079–88. doi: 10.2174/1566524014666141010155509
134. Hirsch EC, Hunot S. Neuroinflammation in parkinson's disease: a target for neuroprotection? Lancet Neurol (2009) 8:382–97. doi: 10.1016/S1474-4422(09)70062-6
135. Sophiabadi M, Rastgoo N, Haghdoost-Yazdi H. Dopaminergic neuronal death in substantia nigra associates with serum levels of total bilirubin, selenium, and zinc: evidences from 6-hydroxydopamine animal model of parkinson's disease. Biol Trace Elem Res (2022) 200:4058–67. doi: 10.1007/s12011-021-03012-6
136. Saghazadeh A, Ferrari CC, Rezaei N. Deciphering variability in the role of interleukin-1β in parkinson's disease. Rev Neurosci (2016) 27:635–50. doi: 10.1515/revneuro-2015-0059
137. Wang W, Nguyen LT, Burlak C, Chegini F, Guo F, Chataway T, et al. Caspase-1 causes truncation and aggregation of the parkinson's disease-associated protein α-synuclein. Proc Natl Acad Sci U S A (2016) 113:9587–92. doi: 10.1073/pnas.1610099113
138. Yan YQ, Fang Y, Zheng R, Pu JL, Zhang BR. NLRP3 inflammasomes in parkinson's disease and their regulation by parkin. Neuroscience (2020) 446:323–34. doi: 10.1016/j.neuroscience.2020.08.004
139. Ahmed S, Kwatra M, Ranjan Panda S, Murty USN, Naidu VGM. Andrographolide suppresses NLRP3 inflammasome activation in microglia through induction of parkin-mediated mitophagy in in vitro and in vivo models of Parkinson disease. Brain Behav Immun (2021) 91:142–58. doi: 10.1016/j.bbi.2020.09.017
140. Khot M, Sood A, Tryphena KP, Khan S, Srivastava S, Singh SB, et al. NLRP3 inflammasomes: a potential target to improve mitochondrial biogenesis in parkinson's disease. Eur J Pharmacol (2022) 934:175300. doi: 10.1016/j.ejphar.2022.175300
141. Piancone F, La Rosa F, Marventano I, Saresella M, Clerici M. The role of the inflammasome in neurodegenerative ddiseases. Molecules (2021) 26:953. doi: 10.3390/molecules26040953
142. Wang CE, Li S, Li XJ. Lack of interleukin-1 type 1 receptor enhances the accumulation of mutant huntingtin in the striatum and exacerbates the neurological phenotypes of huntington's disease mice. Mol Brain (2010) 3:33. doi: 10.1186/1756-6606-3-33
143. Valekova I, Jarkovska K, Kotrcova E, Bucci J, Ellederova Z, Juhas S, et al. Revelation of the IFNα, IL-10, IL-8 and IL-1β as promising biomarkers reflecting immuno-pathological mechanisms in porcine huntington's disease model. J Neuroimmunol (2016) 293:71–81. doi: 10.1016/j.jneuroim.2016.02.012
144. Corey-Bloom J, Fischer RS, Kim A, Snell C, Parkin GM, Granger DA, et al. Levels of interleukin-6 in saliva, but not plasma, correlate with clinical metrics in huntington's disease patients and healthy control subjects. Int J Mol Sci (2020) 21:6363. doi: 10.3390/ijms21176363
145. Bjorkqvist M, Wild EJ, Thiele J, Silvestroni A, Andre R, Lahiri N, et al. A novel pathogenic pathway of immune activation detectable before clinical onset in huntington’s disease. J Exp Med (2008) 205:1869–77. doi: 10.1084/jem.20080178
146. de Oliveira Furlam T, Roque IG, Machado da Silva EW, Vianna PP, Costa Valadão PA, Guatimosim C, et al. Inflammasome activation and assembly in huntington's disease. Mol Immunol (2022) 151:134–42. doi: 10.1016/j.molimm.2022.09.002
147. Hardiman O, Al-Chalabi A, Chio A, Corr EM, Logroscino G, Robberecht W, et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers (2017) 5:17071. doi: 10.1038/nrdp.2017.71
148. Smeyers J, Banchi E-G and Latouche M. C9ORF72: what it is, what it does, and why it matters. Front Cell Neurosci (2021) 15:661447. doi: 10.3389/fncel.2021.661447
149. Zou ZY, Zhou ZR, Che CH, Liu CY, He RL, Huang HP. Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry (2017) 88:540–9. doi: 10.1136/jnnp-2016-315018
150. Lyon MS, Wosiski-Kuhn M, Gillespie R, Caress J, Milligan C. Inflammation, immunity, and amyotrophic lateral sclerosis: i. etiology and pathology. Muscle Nerve (2019) 59:10–22. doi: 10.1002/mus.26289
151. Staats KA, Borchelt DR, Tansey MG, Wymer J. Blood-based biomarkers of inflammation in amyotrophic lateral sclerosis. Mol Neurodegeneration (2022) 17:11. doi: 10.1186/s13024-022-00515-1
152. Béland LC, Markovinovic A, Jakovac H, De Marchi F, Bilic E, Mazzini L, et al. Immunity in amyotrophic lateral sclerosis: blurred lines between excessive inflammation and inefficient immune responses. Brain Commun (2020) 2:fcaa124. doi: 10.1093/braincomms/fcaa124
153. Ravnik-Glavač M, Goričar K, Vogrinc D, Koritnik B, Lavrenčič JG, Glavač D, et al. Genetic variability of inflammation and oxidative stress genes affects onset, progression of the disease and survival of patients with amyotrophic lateral sclerosis. Genes (2022) 13:757. doi: 10.3390/genes13050757
154. Brodovitch A, Boucraut J, Delmont E, Parlanti A, Grapperon AM, Attarian S, et al. Combination of serum and CSF neurofilament-light and neuroinflammatory biomarkers to evaluate ALS. Sci Rep (2021) 11:703. doi: 10.1038/s41598-020-80370-6
155. Olesen MN, Wuolikainen A, Nilsson AC, Wirenfeldt M, Forsberg K, Madsen JS, et al. Inflammatory profiles relate to survival in subtypes of amyotrophic lateral sclerosis. Neurol Neuroimmunol Neuroinflamm (2020) 7:e697. doi: 10.1212/NXI.0000000000000697
156. Italiani P, Carlesi C, Giungato P, Puxeddu I, Borroni B, Bossù P, et al. Evaluating the levels of interleukin-1 family cytokines in sporadic amyotrophic lateral sclerosis. J Neuroinflamm (2014) 11:94. doi: 10.1186/1742-2094-11-94
157. Eitan C, Siany A, Barkan E, Olender T, van Eijk KR, Moisse M, et al. Whole-genome sequencing reveals that variants in the interleukin 18 receptor accessory protein 3’UTR protect against ALS. Nat Neurosci (2022) 25:433–45. doi: 10.1038/s41593-022-01040-6
158. Lin CY, Pfluger CM, Henderson RD, McCombe PA. Reduced levels of interleukin 33 and increased levels of soluble ST2 in subjects with amyotrophic lateral sclerosis. J Neuroimmunol (2012) 249:93–5. doi: 10.1016/j.jneuroim.2012.05.001
159. Korhonen P, Pollari E, Kanninen KM, Savchenko E, Lehtonen S, Wojciechowski S, et al. Long-term interleukin-33 treatment delays disease onset and alleviates astrocytic activation in a transgenic mouse model of amyotrophic lateral sclerosis. IBRO Rep (2019) 6:74–86. doi: 10.1016/j.ibror.2019.01.005
160. Dawson TM, Golde TE, Tourenne CL. Animal models of neurodegenerative diseases. Nat Neurosci (2018) 21:1370. doi: 10.1038/s41593-018-0236-8
161. Zhou R, Ji B, Kong Y, Qin L, Ren W, Guan Y, et al. PET imaging of neuroinflammation in alzheimer’s disease. Front Immunol (2021) 12:739130. doi: 10.3389/fimmu.2021.739130
Keywords: IL-1 family cytokines, IL-1 family receptors, neuroinflammation, neurodegeneration, Alzheimer’s disease, multiple sclerosis
Citation: Boraschi D, Italiani P, Migliorini P and Bossù P (2023) Cause or consequence? The role of IL-1 family cytokines and receptors in neuroinflammatory and neurodegenerative diseases. Front. Immunol. 14:1128190. doi: 10.3389/fimmu.2023.1128190
Received: 20 December 2022; Accepted: 11 April 2023;
Published: 08 May 2023.
Edited by:
Bodduluri Haribabu, University of Louisville, United StatesReviewed by:
Robert Friedland, University of Louisville, United StatesJin Bao, The Brain Cognition and Brain Disease Institute, Chinese Academy of Sciences (CAS), China
Copyright © 2023 Boraschi, Italiani, Migliorini and Bossù. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Diana Boraschi, diana.boraschi@itb.cnr.it; Paola Bossù, p.bossu@hsantalucia.it