 Ji Wang1,2,3†
Ji Wang1,2,3† Yilin Zhao1,4†Xin Zhang1,5†Wei Tu1,6
Yilin Zhao1,4†Xin Zhang1,5†Wei Tu1,6 Rongjun Wan1,7Yingchun Shen1
Rongjun Wan1,7Yingchun Shen1 Yan Zhang1,7Ruchik Trivedi1
Yan Zhang1,7Ruchik Trivedi1 Peisong Gao1*
Peisong Gao1*- 1Division of Allergy and Clinical Immunology, Johns Hopkins School of Medicine, Baltimore, MD, United States
- 2Department of Respiratory and Critical Care Medicine, West China Hospital, Sichuan University, Chengdu, China
- 3Laboratory of Pulmonary Immunology and Inflammation, Frontiers Science Center for Disease-related Molecular Network, Sichuan University, Chengdu, China
- 4Department of Respiratory Medicine, Xijing Hospital, The Fourth Military Medical University, Xi’an, China
- 5Department of Integrated Traditional Chinese and Western Medicine, West China Hospital, Sichuan University, Chengdu, China
- 6Department of Respirology and Allergy, Third Affiliated Hospital of Shenzhen University, Shenzhen, China
- 7Department of Respiratory Medicine, Xiangya Hospital, Central South University, Changsha, China
Rationale: Aryl hydrocarbon receptor (AhR), a ligand-activated transcription factor, has been considered as an important regulator for immune diseases. We have previously shown that AhR protects against allergic airway inflammation. The underlying mechanism, however, remains undetermined.
Objectives: We sought to determine whether AhR specifically in type II alveolar epithelial cells (AT2) modulates allergic airway inflammation and its underlying mechanisms.
Methods: The role of AhR in AT2 cells in airway inflammation was investigated in a mouse model of asthma with AhR conditional knockout mice in AT2 cells (Sftpc-Cre;AhRf/f). The effect of AhR on allergen-induced autophagy was examined by both in vivo and in vitro analyses. The involvement of autophagy in airway inflammation was analyzed by using autophagy inhibitor chloroquine. The AhR-regulated gene profiling in AT2 cells was also investigated by RNA sequencing (RNA-seq) analysis.
Results: Sftpc-Cre;AhRf/f mice showed exacerbation of allergen-induced airway hyperresponsiveness and airway inflammation with elevated Th2 cytokines in bronchoalveolar lavage fluid (BALF). Notably, an increased allergen-induced autophagy was observed in the lung tissues of Sftpc-Cre;AhRf/f mice when compared with wild-type mice. Further analyses suggested a functional axis of AhR-TGF-β1 that is critical in driving allergic airway inflammation through regulating allergen-induced cellular autophagy. Furthermore, inhibition of autophagy with autophagy inhibitor chloroquine significantly suppressed cockroach allergen–induced airway inflammation, Th2 cytokines in BALFs, and expression of autophagy-related genes LC3 and Atg5 in the lung tissues. In addition, RNA-seq analysis suggests that autophagy is one of the major pathways and that CALCOCO2/NDP52 and S1009 are major autophagy-associated genes in AT2 cells that may contribute to the AhR-mediated cockroach allergen–induced airway inflammation and, subsequently, allergic asthma.
Conclusion: These results suggest that AhR in AT2 cells functions as a protective mechanism against allergic airway inflammation through controlling cell autophagy.
Introduction
Asthma is a leading serious chronic illness of children and adults worldwide, and its prevalence has reached unprecedented levels over the past few decades (1). Environmental allergen exposure has been considered to be one of the major risk factors for asthma (2). Of these, cockroach allergen is one of the major sources of indoor allergens and can give rise to Immunoglobulin E (IgE) sensitization and, subsequently, development of asthma (3–5). Furthermore, a recent study suggested that serum IgE levels specific to cockroach allergen were correlated with Th2 polarization (6). However, it remains unclear about the biological mechanisms underlying the cockroach allergen exposure-induced Th2-associated airway inflammation in asthma. Airway epithelium is the first line of defense against inhaled allergens. Interaction of airway epithelium with allergens leads to the release of inflammatory mediators, resulting in inappropriate recruitment of immune cells and skewing of downstream immune responses. However, the mechanisms underlying the allergen-induced mediator release from epithelial cells are still not completely understood.
The aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor widely expressed in barrier organs, such as airway, gut, and skin (7, 8), and in different cell types [e.g., DCs, Th17 and Treg cells, ILCs, and mesenchymal stem cells (MSCs)] (9–12). AhR is originally characterized for its function in the metabolism of environmental toxicants such as dioxins and many other polycyclic aromatic hydrocarbons (PAHs) (13–16). Recent studies have suggested a central role for AhR as an environmental sensing molecule in cell growth, cell differentiation, cell immune responses, and respiratory function (17–21). Not surprisingly, AhR has emerged as an attractive therapeutic target for different diseases including asthma (14–16, 22). Indeed, AhR has been associated with environmental pollutant–induced allergic airway inflammation (18, 23–25) and reactive oxygen species (ROS)-dependent mast cell degranulation and activation (26, 27). Our previous findings have suggested that benzo(a)pyrene (BaP, a ubiquitous air pollutant) co-exposure with Der f 1 (a common allergen to human) exacerbated AhR signaling pathway that regulates the co-exposure–induced airway epithelial cell oxidative stress and cytokine release (28, 29). Furthermore, our recent study demonstrated that epithelial AhR is essential in protecting against cockroach allergen–induced ROS generation, NLRP3 inflammasome activation, and Muc5ac expression (30). However, the specific role of AhR signaling in regulating allergic airway inflammation and its precise mechanisms has not been fully elucidated.
Autophagy is an the endogenous housekeeping process in maintaining cell homeostatic process by delivering damaged proteins and redundant cellular organelles to lysosomes for degradation during cellular stress (2). Evidence shows that autophagy is critical in shaping cellular immune responses and progression of inflammatory diseases. It was found that autophagy was increased in peripheral blood cells and sputum granulocytes from severe asthmatic patients (31). The increased autophagy has also been associated with asthma pathological processes such as extracellular matrix deposition, airway remodeling, and immune dysregulation (32–35). Previously, we found that inhibition of autophagy led to attenuated cockroach allergen–induced airway epithelial ROS generation, cytokine release, and subsequent allergic airway inflammation, suggesting a role for autophagy in promoting asthma development (36). However, it remains unclear how cellular autophagy is regulated in the pathogenesis of asthma.
In the present study, we focused on type II alveolar epithelial cells (AT2) to determine whether AhR, specifically in AT2 cells, is involved in regulating cellular autophagy and, subsequently, allergic airway inflammation. In particular, we generated AhR conditional knockout mice in AT2 cells (Sftpc-Cre;AhRf/f) and investigated the role of AhR specifically in AT2 cells in cockroach allergen–induced airway inflammation. We then uncovered a unique role for AhR in regulating allergen-induced cellular autophagy by both in vivo and in vitro analyses. We further elucidated an overexpression of autophagy in the lung tissues of asthma mouse model and inhibition of autophagy suppressed allergic airway inflammation. Most importantly, RNA sequencing (RNA-seq) analysis identified autophagy to be one of the most AhR-regulated biological processes in AT2 cells. Further analyses suggest that CALCOCO2 and S100a9 are the major AhR-regulated autophagy-associated genes. Overall, this study provides evidence that AhR regulates allergen-induced autophagy that may contribute to the protective mechanism of AhR signaling against allergic airway inflammation.
Methods
Mice
Sftpc-Cre;AhRf/f mice on the C57BL/6 background were generated by cross-breeding Sftpc-cre with AhRf/f mice in this study. Sftpc-cre mice were provided by Michael A. O’Reilly at the University of Rochester Medical Center (Rochester, NY, USA), and AhRf/f mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). All mice were maintained under specific pathogen–free conditions at the animal facility of the Johns Hopkins University School of Medicine. The animal care and experiments were performed in compliance with the institutional and US National Institutes of Health guidelines and were reviewed and approved by the Johns Hopkins University Animal Care and Use Committee. All mice were used at 6–8 weeks of age, and all experiments used age- and sex-matched controls.
Cockroach allergen–induced asthma mouse model
The cockroach allergen–induced asthma mouse model was established with the protocol as illustrated in Figure 1A. Briefly, mice were sensitized by intra-tracheal (i.t.) inhalation of 20 µg of cockroach extract (CRE, B46, GREER Laboratories) per mouse in 50 μl of Phosphate-Buffered Saline (PBS) under light anesthesia with isoflurane and challenged with the same amount of CRE. Control mice were treated with PBS. Mice were sacrificed, and samples [lung tissues, bronchoalveolar lavage fluid (BALF), and blood] were collected on the next day after the last allergen challenge (30). In particular, BALF was harvested by two consecutive flushes of the lung with 1.0 ml of ice-cold PBS. Blood was taken to screen for serum antibodies against cockroach allergen. In some cases, mice were pre-treated with autophagy inhibitor chloroquine (CLQ; C6628, 60 mg/kg, Sigma-Aldrich, St. Louis, MO, USA) or saline vehicle control by intraperitoneal administration 1 h before every single allergen challenge.
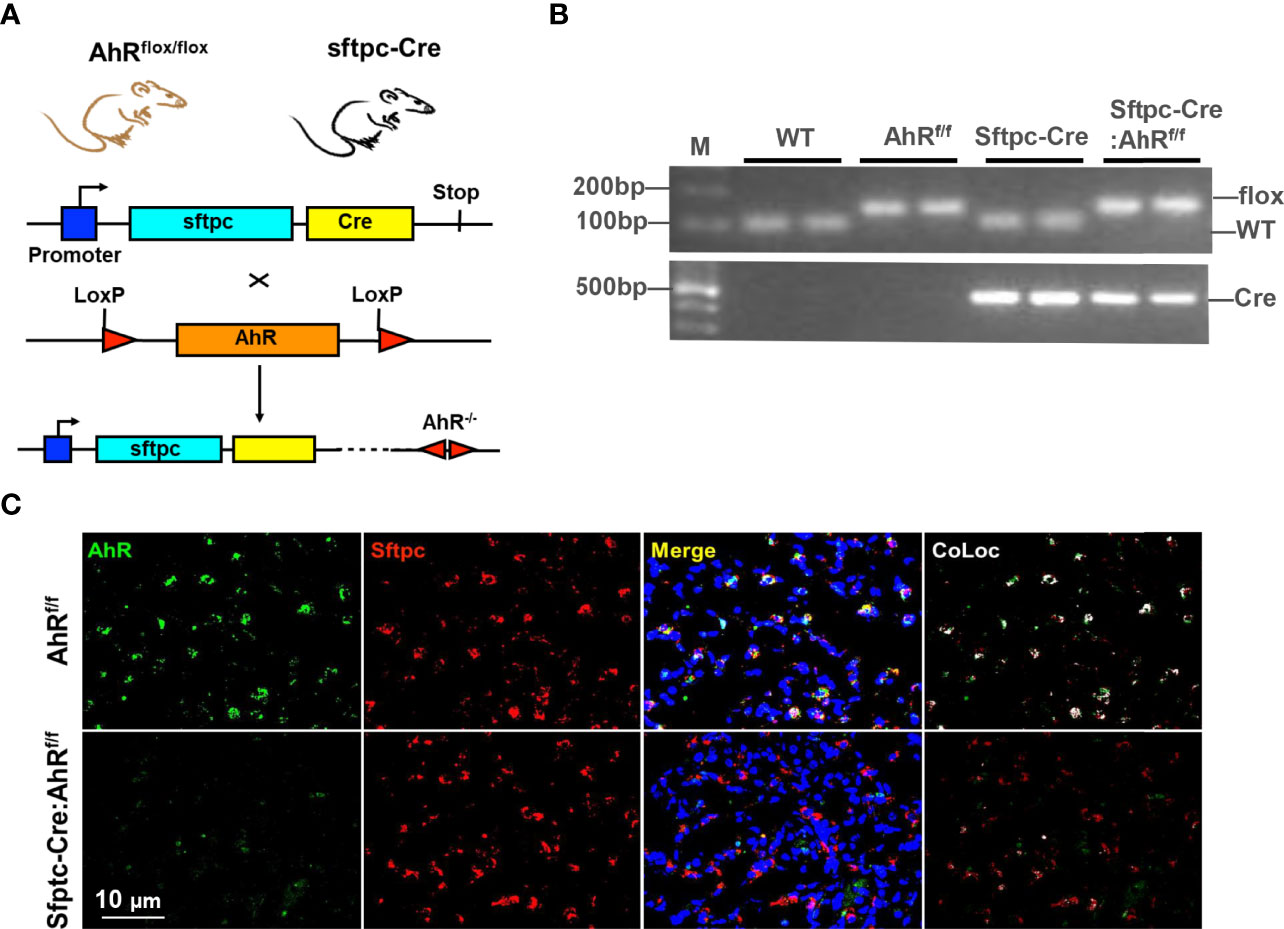
Figure 1 Generation of type II alveolar epithelial cell–specific AhR knockout mice. (A) Schematic representation of the crossbreeding of a floxed AhR mouse (AhRf/f) with a Sftpc-cre mouse. (B) Confirmation of Sftpc-cre: AhRf/f mice by genotyping. (C) AhR deletion in AT2 cells of Sftpc-cre: AhRf/f mice was confirmed by co-immunostaining with Sftpc and AhR in the lung tissues.
Airway hyperresponsiveness
Mice were anesthetized with a ketamine (90 mg/kg)/xylazine (18 mg/kg) mixture, and a tracheotomy tube was inserted. Ventilation was initiated with a volume-cycled ventilator (Flexivent; SCIREQ Scientific) with a positive-end expiratory pressure of 2 cmH2O). Airway responsiveness was monitored by challenging mice with a dose-dependent aerosolized methacholine (0–30 mg/ml). The airway resistance was measured with the Flexivent software and exported to Pulmodyn data-acquisition software (Hugo Sachs Electronic) for data analysis (37).
Histological analysis
Mouse lungs were perfused with 10 ml of ice-cold PBS injected into the right ventricle followed by excision, fixed with 4% formalin, and embedded in paraffin. Sections (4 µm) were then prepared from these paraffin-embedded lungs and subjected to hematoxylin and eosin (H&E) and periodic acid–Schiff (PAS) staining to evaluate general morphology and mucus production as previously described (30).
Flow cytometry analysis
Single-cell suspensions were prepared from BALFs, and total cells in BALFs were counted by Countless II (ThermoFisher). Cellular differential percentages in BALFs were measured by flow cytometry on an Accuri C6 Plus Flow Cytometer (BD Biosystems), and the data were analyzed with FlowJo software (Tree Star Inc.) as previously described (38).
Immunofluorescence staining
Immunofluorescence staining was performed as previously reported (37). Briefly, sectioned lung tissues were first blocked using 5% w/v BSA at room temperature for 1 h, followed by incubation with the primary antibodies as listed in the online repository (Supplementary Table 1) overnight at 4°C. The sample sections were then incubated with secondary antibodies conjugated with fluorescence at room temperature for 1 h. Isotype-matched negative control antibodies were used under the same conditions. The sections were mounted with the Fluoromount Mounting medium (Sigma) with DAPI (4′,6-diamidino-2-phenylindole) (ThermoFisher) and then observed by a NIKON ECLIPSE Ti-U microscope equipped with DS-Fi2 camera (NIKON, USA).
Enzyme-linked immunosorbent assay
Interleukin-4 (IL-4), IL-5, Interferon-gamma (IFN-γ), IL-17A, IL-25, and Transforming growth factor-beta 1 (TGF-β1) in cell-free BALF or supernatant were quantified by using the Ready-Set-Go! ELISA (ThermoFisher) according to the manufacturer’s instructions. Serum levels of cockroach allergen–specific IgE and IgG1 were analyzed by Enzyme-linked immunosorbent assay (ELISA) as previously described (36).
Cell culture and treatment
Human bronchial epithelial cells (HBEC3-KT, ATCC CRL-4051) were cultured in Ham’s F-12K (Kaighn’s) medium supplemented with 10% v/v FBS and 1% penicillin‐streptomycin. The cells were maintained at 37°C in a humidified atmosphere at 5% CO2. HBECs were treated with cockroach extract (CRE, Greer Laboratory) alone or pre-treated with AhR agonist TCDD (Sigma, 45899; 1.0 µM) or antagonist CH-223191 (Sigma, C8124; 10 µM) or vehicle controls 1 h before CRE treatment.
Isolation of mouse alveolar type 2 cells
Mouse AT2 cells were isolated from AhRfl/fl mice and Sftpc-Cre;AhRflox/flox mice as previously reported (38). Briefly, mice were euthanized with ketamine and xylazine intraperitoneally. Lung tissues from these mice were digested with Dispase (50 units/ml, Gibco) at room temperature for 45 min and then incubated with AT2 isolation medium consisting of a 1:1 mixture of Dulbecco’s modified Eagle’s medium (DMEM) and Ham’s F-12 (DMEM/F-12; Sigma) supplemented with 0.01% DNase I (Sigma), sodium penicillin G (100 U/ml), and streptomycin (100 µg/ml). The mixed cells were filtered through cell strainers; stained with biotinylated anti-CD16/32, anti-CD45, anti-Ter119, and anti-Sca-1 (BioLegend); and incubated at 37°C shaking incubator for 30 min. These cells were further negatively selected with streptavidin-conjugated magnetic beads for 30 min at room temperature. Cells were centrifuged for 10 min at 300g at 4°C and incubated on dishes pre-coated with mouse IgG. After incubation for 2 h, non-adherent cells were collected, centrifuged, and re-suspended with AT2 isolation medium.
RNA-seq analysis
RNA-seq libraries were prepared for sequencing using an Illumina TruSeq stranded mRNA sample preparation kit following the manufacturer’s recommended procedure. Briefly, total RNA was extracted using the RNeasy Mini Kit (QIAGEN). mRNA was enriched using oligo dT beads and then fragmented chemically by incubating at 94°C for 8 min. cDNA was synthesized with SuperScript II. After purification using AMPure XP beads, the double-stranded cDNA was ligated to TruSeq RNA adapters followed by 15 cycles of amplification and library purification. Sequencing was performed on an Illumina NextSeq6000. RNA-seq reads were aligned to the mouse reference genome GRCm38 using STAR aligner version 2.7.2b. BAM file outputs from STAR were annotated using Partek Genomic Suite (v6.6) and the RefSeq database. The gene expression levels were defined as counts by Salmon (39) and then normalized by log2-transformed counts per million (CPM, edgeR version 3.34.1) (40). Different expression analyses were performed with multiple linear regression models and empirical Bayes moderation embedded in the limma-trend approach (limma version 3.48.3) (40). Genes with absolute log2 fold change [abs(log2FC)] > 0.5 and p < 0.05 were defined as differentially expressed genes (DEGs). Gene set enrichment analyses was performed by the clusterProfiler 4.0 version 4.0.5 (41). Up- or downregulated DEGs were visualized by ComplexHeatmap (version 2.8.0) and ggplot2 (version 3.3.5) (42). All RNA-seq–based downstream analyses and visualizations were performed in R version 4.1.2. (see RNA-seq data used for analysis in Supplementary Materials).
Transfection of siRNA
AhR knockdown in HBECs was accomplished by using a pre-designed MISSION siRNA small interfering RNA pair (Sigma-Aldrich, SASI_Hs01_00140198). siRNA transfection was performed with Lipofectamine™ RNAiMAX (ThermoFisher) following the manufacturer’s instruction, whereas control cells received negative control siRNA. Cells were cultured for 48 h, and transfection efficiency was evaluated by quantitative real-time PCR and Western blot.
RNA isolation and quantitative real-time PCR analysis
Total RNA from lung tissues or HBECs was isolated using the Monarch Total RNA Miniprep Kit (New England Biolabs), and cDNAs were synthesized with the High-Capacity cDNA Reverse Transcription Kit (ThermoFisher). Quantitative real-time PCR analysis was performed using the Power SYBR Green PCR Master Mix (ThermoFisher) on an ABI Prism 7300 detection system. Data were analyzed using the 2−ΔΔCT method relative to the housekeeping gene Actin (43). Primer sequences are provided in the Online Repository (see Table E1).
Western blotting
Tissues or cells were lysed with an radioimmunoprecipitation assay (RIPA) buffer containing protease and phosphatase inhibitor cocktails (Sigma-Aldrich). Protein concentrations were measured by using a BCA kit (Thermo Fisher). An equal amount of total protein (20–50 µg) was loaded onto a 4%–12% Tris-Glycine Gel in NuPAGE MOPS SDS Running Buffer (Thermo Fisher Scientific) and then transferred using the iBlot2 NC Stack System (ThermoFisher). The membranes were blocked in 5% non-fat milk in Tris buffered saline with Tween® 20 (TBST) for 1 h at room temperature and then probed with primary antibodies overnight at 4°C. Species-appropriate secondary antibodies conjugated to IRDye 680RD or IRdye 800CW (LI-COR Biosciences) were used according to the manufacturer’s instructions. Detection was performed using iBright 1500 Imaging Systems, and fluorescent intensity was quantified using the system built in IBright Analysis Software (Thermo Fisher Scientific).
Statistical analysis
All statistical analyses were carried out using GraphPad Prism version 8.2.1 (GraphPad Software, La Jolla, CA, USA). All data are presented as means ± SEM. Comparison of two groups was performed by student’s two-tailed unpaired t-test. Comparison of more than two groups was performed by ordinary one-way ANOVA followed by Tukey’s post hoc test. P < 0.05 was considered statistically significant.
Results
Generation of type II alveolar epithelial cell–specific AhR knockout mice
Airway epithelial cells are the first line of defense against inhaled particulate antigens, and epithelial activation is one of the characteristics of asthma (44). We have found that expression of AhR was significantly increased in the airways of asthmatic patients (45) and the mouse model of asthma (12). To determine the significance of epithelial AhR in cockroach allergen–induced asthma, we deleted AhR selectively from type II alveolar airway epithelium (AT2) by crossing floxed AhR mice (AhRf/f) with Sftpc-cre mice that express the Cre recombinase in AT2 cells from the endogenous promoter/enhancer elements of the surfactant protein C (Sftpc) locus (Sftpc-Cre; AhRf/f, Figure 1A). The mice were confirmed by genotyping (Figure 1B). The deletion of AhR in AT2 cells was further confirmed by co-immunostaining with AhR and Sftpc in the lung tissues (Figure 1C). Thus, these mice represent an ideal mouse model to examine the functional role of AhR in AT2 cells in allergic airway inflammation.
Type II alveolar epithelial cell AhR protects against allergic airway inflammation
Next, we used these newly generated Sftpc-Cre;AhRf/f mice to create an asthma mouse model by using our previous protocol as illustrated in Figure 2A. Histological analysis demonstrated that CRE-treated Sftpc-Cre;AhRf/f mice had an increased recruitment of inflammatory cells to the lung with dense peribronchial infiltrates (Figure 2B, upper panel) and goblet cell hyperplasia (Figure 2B, lower panel) as compared with AhRf/f mice. Consistently, these Sftpc-Cre;AhRf/f mice showed a significant increase in airway resistance (Figure 2C) and decrease in airway compliance (Figure 2D). Compared with AhRf/f mice, the total inflammatory cells, especially eosinophils, were remarkably increased in the BALFs of Sftpc-Cre;AhRf/f mice (Figure 2E). Higher levels of cockroach allergen–specific IgE and IgG1 were observed in serum of CRE-treated Sftpc-Cre;AhRf/f (Figure 2F). Furthermore, Sftpc-Cre;AhRf/f mice had higher levels of IL-4 and IL-5 in BALFs (Figure 2G). In contrast, no clear change was observed for IFN-γ, IL-17, and IL-25. Collectively, these findings indicate that AhR in AT2 cells protects against cockroach allergen–induced airway hyperresponsiveness and Th2-associated allergic airway inflammation.
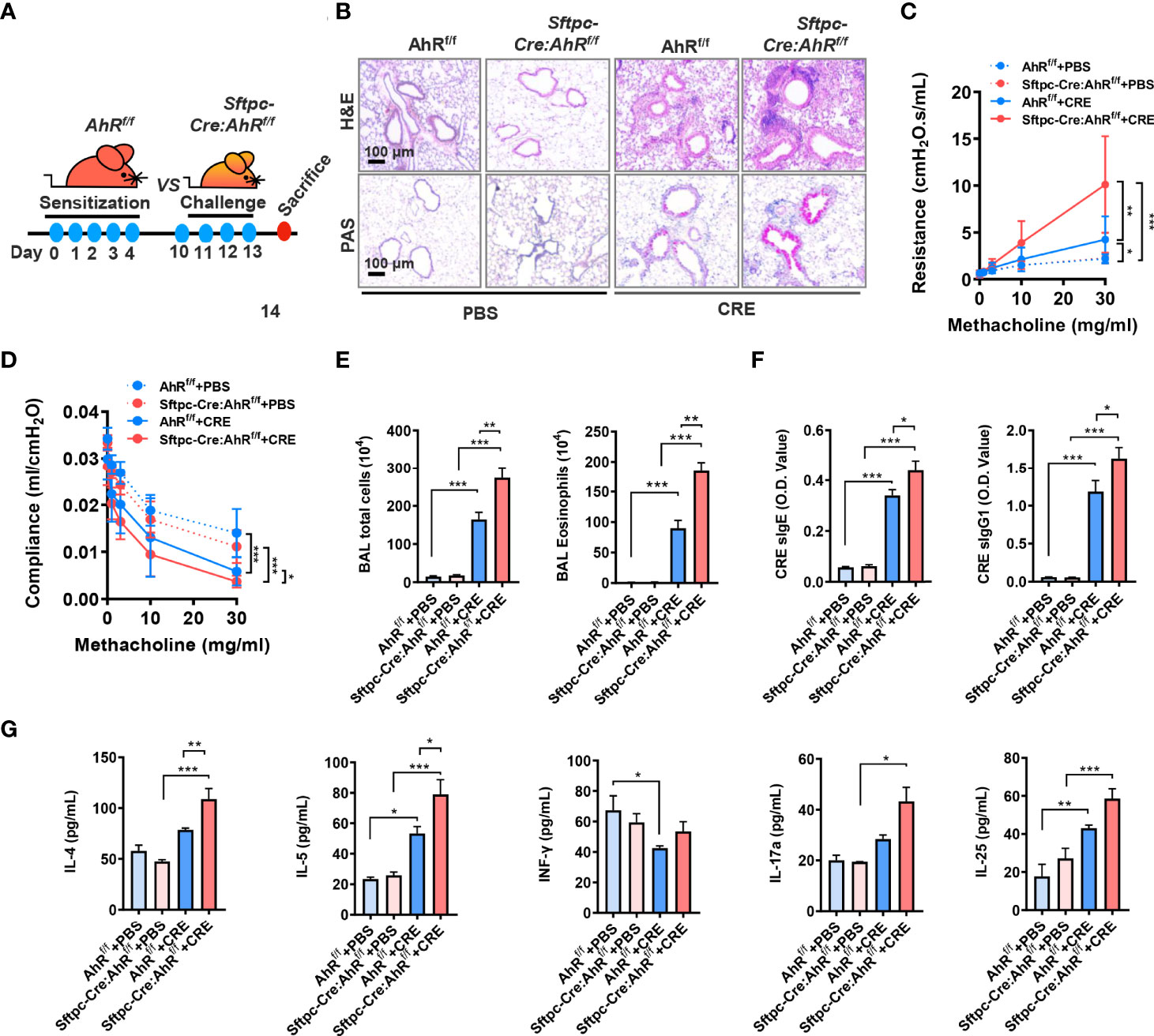
Figure 2 Type II alveolar epithelial cell AhR protects against airway hyperresponsiveness and allergic airway inflammation. (A) Experimental scheme for cockroach allergen–induced mouse model of asthma. (B) Histological examination of mouse paraffin lung sections stained with hematoxylin and eosin (H&E, upper panel) and Alcian Blue periodic acid–Schiff (AB-PAS, lower panel). (C, D) Lung resistance (C) and compliance (D) in response to increasing concentrations of methacholine using the forced oscillation technique (FlexiVent, SCIREQ). (E) Bronchoalveolar lavage (BAL) fluid total and eosinophil cell counts as assessed by flow cytometry. (F) Serum levels of cockroach allergen–specific IgE and IgG1. (G) Levels of cytokines in BALFs. n = 8–10. Data represent means ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001.
Type II alveolar epithelial cell AhR protects against allergen-induced autophagy in a mouse model of asthma
Previously, we have demonstrated that excessive autophagy is critical in cockroach allergen–induced ROS generation, cytokine release, and subsequent allergic airway inflammation (36). To determine whether epithelial AhR regulates cockroach allergen–induced autophagy, we detected expression of autophagy-related genes in the lung tissues of CRE-induced mouse model with AhRf/f or Sftpc-Cre;AhRf/f mice. Compared with PBS-treated mice, increased expression of LC3A, Atg5, and p62 was seen in the lung tissues of CRE-sensitized and challenged mice as analyzed by RT-PCR (Figure 3A). Interestingly, compared with AhRf/f mice, the increased expression of ILC3A, LC3B, and Beclin-1 was further enhanced in CRE-treated Sftpc-Cre;AhRf/f mice. The increased autophagy was specifically confirmed in AT2 cells by co-immunofluorescent staining. The CRE-treated Sftpc-Cre;AhRf/f mice showed a significantly increased expression of LC3B in AT2 cells compared with AhRf/f mice (Figure 3B). Similar patterns were noted for Beclin-1 and ATG5, indicating that AhR in AT2 cells protects against cockroach allergen–induced autophagy.
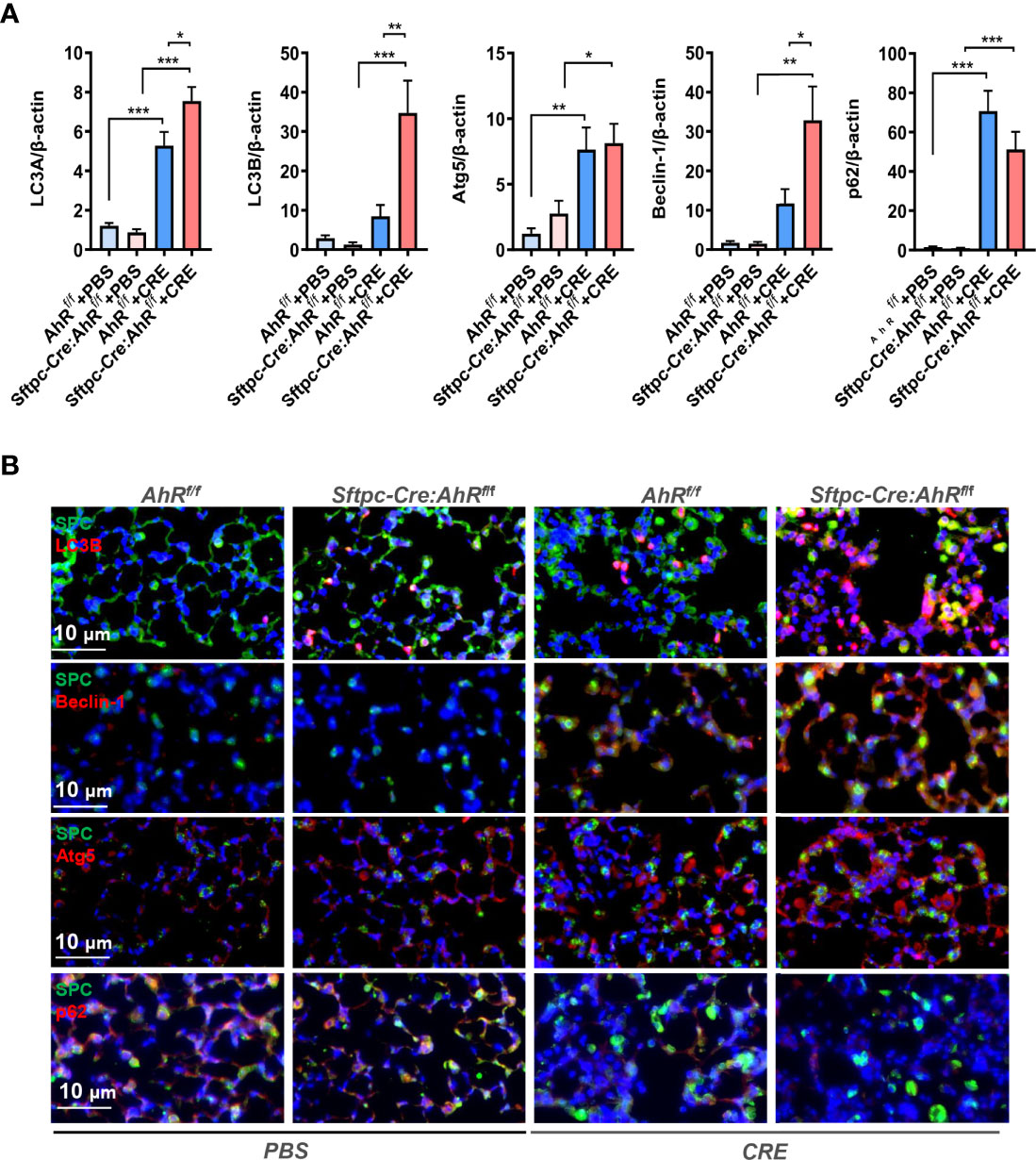
Figure 3 Type II alveolar epithelial cell AhR protects against allergen-induced autophagy in a mouse model of asthma. (A) RT-PCR analyses of autophagy-related genes in the lung tissues of asthma mouse model. n = 8–12. (B) Representative images of co-immunofluorescence staining of autophagy-related genes with AT2 marker SPC. n = 6. Data in (A) represent means ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001.
Aryl hydrocarbon receptor modulates allergen-induced autophagy in human bronchial epithelial cells
To further confirm the functional significance of AhR in modulating autophagy, we investigated whether AhR knockdown can affect cockroach allergen–induced autophagy by in vitro analysis. AhR was knocked down in HBECs by using different doses of siRNA, and siRNA at 100 pmol showed the best deletion of AhR as assessed by RT-PCR (Figure 4A). AhR knockdown was further confirmed by Western blot (Figure 4B). As expected, immunostaining analysis illustrated that CRE exposure induced autophagy in HBECs as assessed by the increased expression of LC3 (Figures 4C, D), ATG5 (Figures 4C, E), and Beclin-1 (Figures 4C, F) but decreased expression of p62 (Figures 4C, G). Of interest, the increased expression of LC3 (Figures 4C, D), ATG5 (Figures 4C, E), and Beclin-1 (Figures 4C, F) was further enhanced in HBECs with AhR knockdown. To visualize the regulation of autophagy by AhR, we transfected either a GFP-LC3 or p62-mCherry construct in HBECs and then treated them with AhR antagonist CH223191. As shown in Figure 4H, the autophagic flux induced by CRE was increased according to the number of LC3 puncta (Figures 4H, I). The increased autophagy was potentiated in HBECs pre-treated with AhR antagonist CH223191. In contrast, expression of p62 was significantly reduced in HBECs treated with CRE in relative to PBS. No statistical difference was observed for HBECs with or without CH223191 pre-treatment (Figures 4H, J). Taken together, our in vitro analysis supports that epithelial AhR regulates cockroach allergen–induced autophagy.
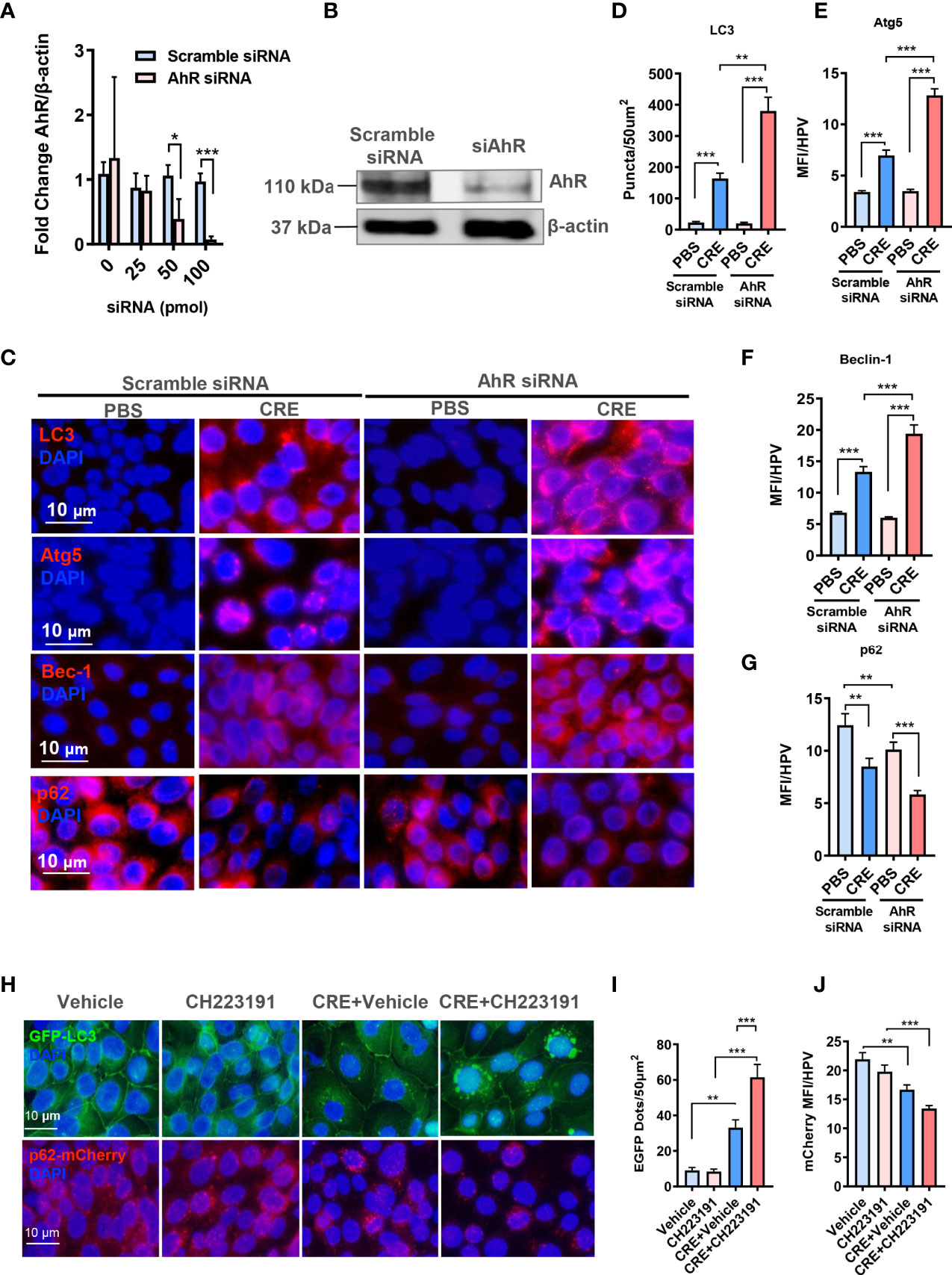
Figure 4 Aryl hydrocarbon receptor modulates allergen-induced autophagy in human bronchial epithelial cells (HBECs). (A) RT-PCR confirmation of AhR knockdown in HBECs by using different doses of AhR siRNA. (B) AhR knockdown in HBECs with 100-pmol AhR siRNA was further confirmed by Western blot. (C) Representative images of immunofluorescence staining of autophagy-related genes in HBECs with or without AhR knockdown. n = 6. (D–G) Quantification of immunofluorescence staining for autophagy-related genes LC3 (D), Atg5 (E), Beclin-1 (F), and p62 (G) in HBECs with or without AhR knockdown. n = 12. (H) Representative immunofluorescence images of CRE-treated HBECs expressing GFP-LC3 or p62-mCherry in the presence or absence of AhR antagonist CH223191. (I) Quantitative analysis of autophagic flux according to the number of LC3 puncta (GFP dots/50 µm2). (J) Quantitative analysis of p62-mCherry florescent signal in (H). n = 12 [four different high-power fields (hpfs) of three independent experiments]. Data represent means ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001.
Inhibition of autophagy attenuates allergic airway inflammation
Given the significance of autophagy in AT2 cell AhR-modulated allergic airway inflammation, we asked whether inhibition of autophagy can suppress cockroach allergen–induced airway inflammation by using autophagy inhibitor CLQ in our mouse model following the protocol illustrated in (Figure 5A). CLQ is the most widely used classic inhibitor of autophagy that inhibits the last stage of autophagy (46). Consistent with our previous or current findings, CRE exposure induced airway inflammation when compare with PBS treatment. When these CRE-treated mice were pre-exposed to CLQ, the CRE-induced airway inflammation was significantly attenuated. In particular, CLQ pre-treatment inhibited CRE-induced peribronchial inflammation (H&E) (Figure 5B, upper panel) and goblet cell hyperplasia (PAS) (Figure 5B, lower panel). These CLQ-treated mice also showed reduced numbers of total inflammatory cells, particularly eosinophils in BALFs (Figure 5C). Moreover, cockroach allergen specific IgE and IgG1 in serum (sIgE and sIgG1) were reduced in mice treated with CLQ (Figure 5D). Furthermore, CLQ treatment led to the significant reduction in the levels of IL-4, IL-5, IL13, IFNγ, and TGF-β1 in BALFs (Figure 5E). Furthermore, as expected, treatment with CLQ attenuated CRE-induced expression of LC3A, LC3B, and Atg5 but led to the accumulation of p62 (Figure 5F). These results further support that inhibition of autophagy can prevent cockroach allergen–induced Th2-associated airway inflammation.
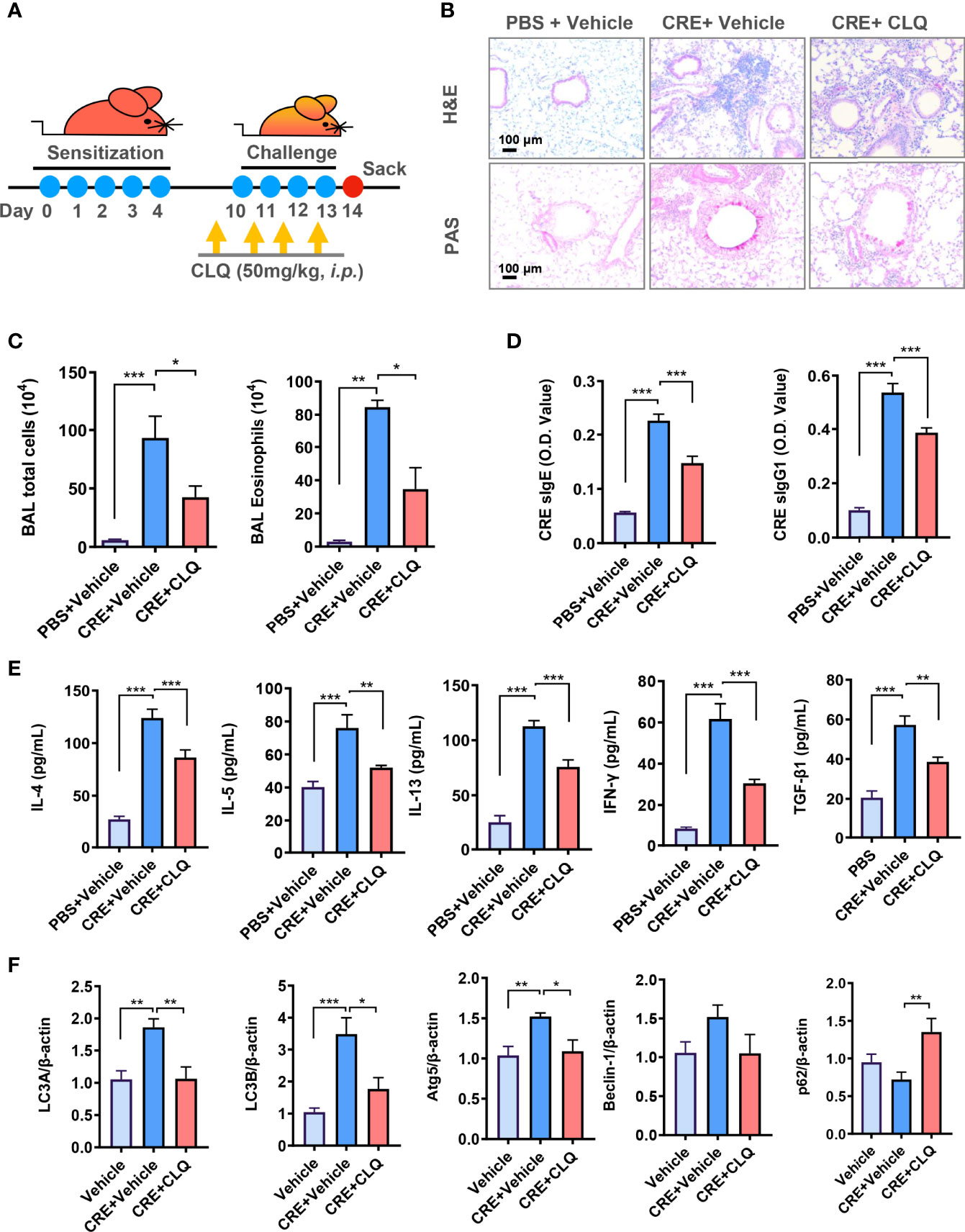
Figure 5 Inhibition of autophagy attenuates allergic airway inflammation. (A) Experimental scheme for cockroach allergen–induced mouse model of asthma in the presence or absence of autophagy inhibitor chloroquine (CLQ). (B) Histological examination of mouse paraffin lung sections stained with hematoxylin and eosin (H&E, upper panel) and periodic acid–Schiff (PAS, lower panel). (C) Bronchoalveolar lavage (BAL) fluid total and eosinophil cell counts as assessed by flow cytometry. (D) Serum levels of cockroach allergen–specific IgE and IgG1. (E) Levels of cytokines in BALFs. (F) RT-PCR analyses of autophagy-related genes in the lung tissues of asthma mouse model. n = 10. Data represent means ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001.
TGF-β1 is required in AhR-mediated cockroach allergen–induced autophagy
We have previously reported that AhR modulates allergen-induced epithelial TGF-β1 secretion and signaling activation (29). We hypothesized that TGF-β1 may be a key player in AhR-mediated cockroach allergen–induced autophagy. AhR in HEBCs was knocked down by siRNA s illustrated in (Figures 4A, B). These HBECs were exposed to CRE for 24 h, and levels of TGF-β1 in supernatants were measured by ELISA. As noted, cockroach allergen exposure induced significant levels of TGF-β1 release from HBECs (Figure 6A). Of interest, the TGF-β1 release was enhanced in HEBCs with AhR knockdown. These findings provide further evidence that AhR modulates cockroach allergen–induced epithelial TGF-β1 secretion. To determine the role of TGF-β1 in autophagy, HBECs were treated with different doses of recombinant TGF-β1 for 24 h, the expression of autophagy-associated genes was detected by RT-PCR. Significantly increased LC3A and Atg5, but reduced p62 expression was observed in TGF-β1–treated HBECs (Figure 6B). No clear change was noted for Beclin-1. To further investigate whether TGF-β1 is required in cockroach allergen–induced autophagy, TGF-β1 neutralizing antibody (α-TGF-β1) was used to remove TGF-β1 in supernatants of CRE-treated HBECs. Intriguingly, the CRE-induced increased expression of LC3A, LC3B, Atg5, and Beclin-1 was almost completely attenuated in α-TGF-β1 pre-treated HBECs (Figure 6C). In contrast, α-TGF-β1 treatment induced increased expression of p62 that was reduced in CRE-treated HBECs. These findings support the hypothesis that TGF-β1 is essential in AhR-mediated cockroach allergen–induced autophagy.
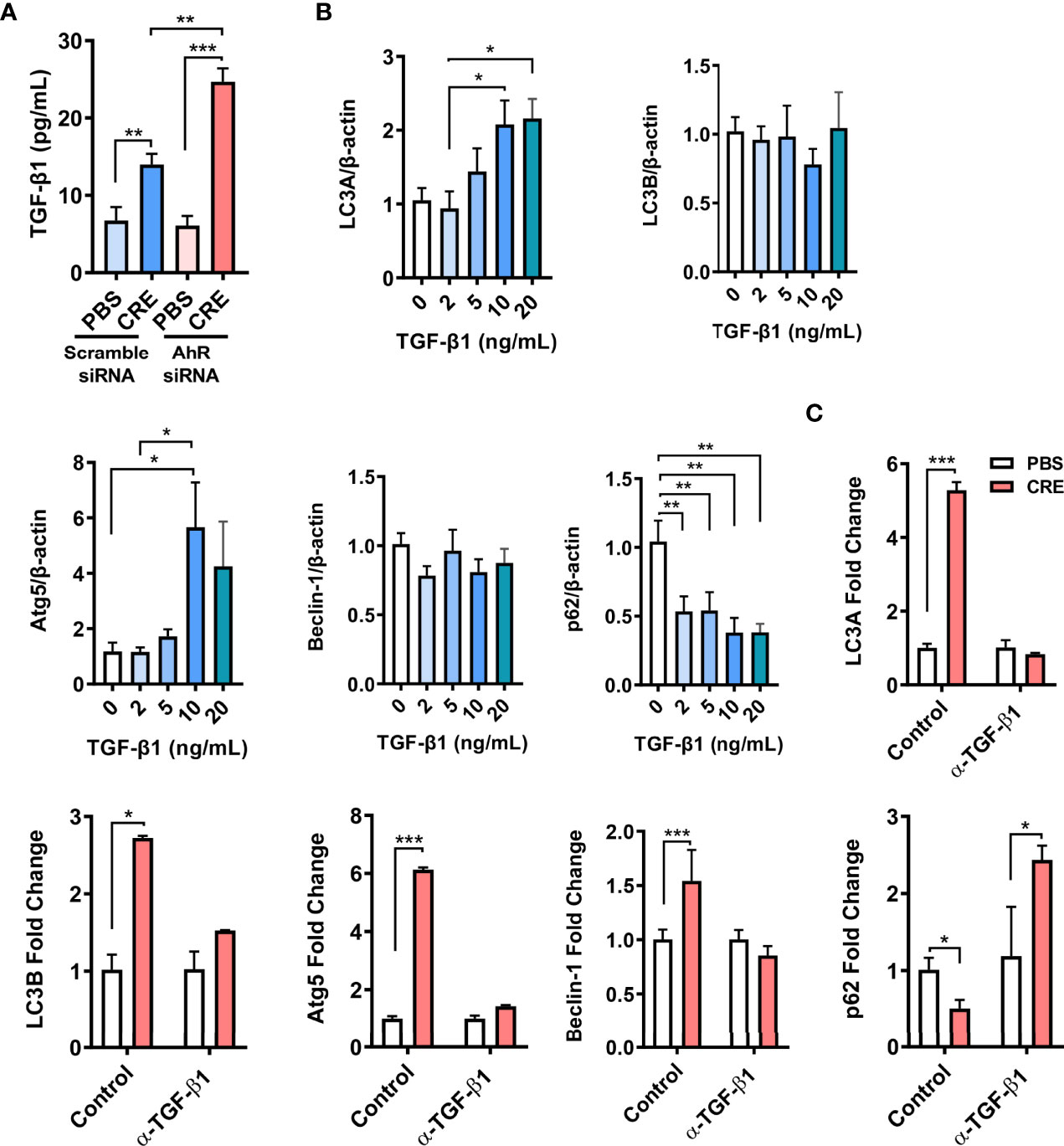
Figure 6 TGF-β1 is required in AhR-mediated cockroach allergen–induced autophagy. (A) ELISA analysis of TGF-β1 levels in supernatants of cockroach allergen (CRE)–treated HEBCs with or without AhR siRNA knockdown. (B) Expression of LC3A, LC3B, Atg5, Beclin-1, and p62 in the recombinant TGF-β1–treated HBECs at different concentrations. (C) Expression of LC3A, LC3B, Atg5, Beclin-1, and p62 in cockroach allergen (CRE-treated) HBECs in the presence or absence of TGF-β1 neutralizing antibody (α-TGF-β1). Data represent means ± SEM of at least two independent experiments. *p < 0.05, **p < 0.01, and ***p < 0.001.
RNA-seq analysis identifies autophagy as one of the major AhR-regulated signaling pathways in AT2 cells
To further examine the underlying mechanisms by which epithelial AhR modulates allergic airway inflammation, we analyzed the transcriptional profiles of AT2 cells isolated from the lungs of AhRf/f and Sftpc-cre;AhRf/f mice that were treated with CRE (50 µg/ml) for 24 h. The pattern of the up- and downregulated genes is illustrated by heatmap. First comparisons were made in AT2 cells treated with either PBS or CRE. Results revealed approximately 863 mRNAs that were differentially expressed in AT2 cells from AhRf/f mice, including 357 downregulated (e.g., Siglec-F) and 506 upregulated genes [Ccl20, Cxcl2, NOXO1, Duox2, and Slc26a4 (38)] (Figure 7A). Approximately 685 mRNAs were differentially expressed in AT2 cells from Sftpc-Cre;AhRf/f mice, including 318 downregulated and 377 upregulated genes (Figure 7B). Next comparisons were made in AT2 cells between AhRf/f and Sftpc-Cre;AhRf/f mice. Results showed a total of 845 mRNAs differentially expressed in PBS-treated AT2 cells, including 364 downregulated (e.g., Cyp1a1 and Cyp1b1) and 481 upregulated genes (Figure 7C). Approximately 717 mRNAs were differentially expressed in CRE-treated AT2 cells, including 356 downregulated [e.g., Ahrr, Cyp1b1, and Ptgs2 (also called COX-2) (47)] and 361 upregulated genes [e.g., Slc26a4 (38)] (Figure 7D). The DEGs in CRE-treated AT2 cells were further grouped into functional groups using the Gene Ontology (GO) functional enrichment analysis. Of interest, autophagy was identified to be one of the most enriched GO terms (Figure 7E). Several other significant GO terms were also identified, including ubiquitin-protein transferase activity, cell activation, and cilium-related terms (e.g., movement, assembly, organization, and cilium) (Figure 7E). Among all these autophagy-associated genes, sh3bp4, Calcoco2, Prkd1, Fbxl2, Usp10, and Trim34b were significantly upregulated, but Trim34a, Trim12a, Trim30d, Trim5, S100a9, and BCL21ll were downregulated (Figure 7F). These results support our previous findings that autophagy is one of the major AhR-regulated biological processes in AT2 cells.
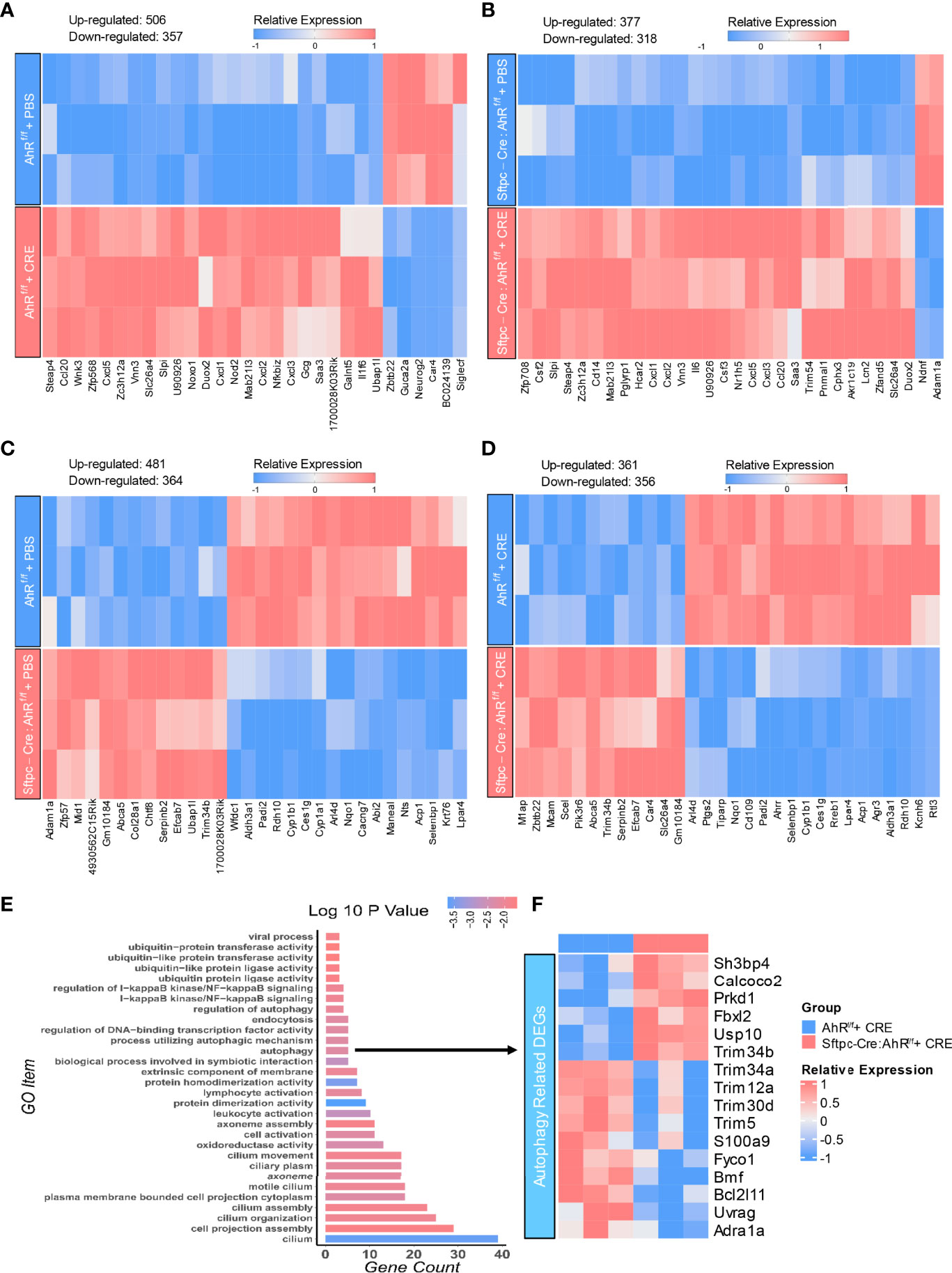
Figure 7 AhR-regulated transcriptional profiles in AT2 cells. (A, B) Heatmap of differentially expressed mRNAs in AT2 cells of AhRf/f (A) or Sftpc-cre;AhRf/f (B) mice treated with PBS verses cockroach allergen (CRE, 50 µg/ml) for 24 h (C, D) Heatmap of differentially expressed mRNAs in AT2 cells of AhRf/f verses Sftpc-cre;AhRf/f mice treated with PBS (C) or cockroach allergen CRE, 50 µg/ml) (D) for 24 h n = 3 per group (A–D). (E) Gene Ontology (GO) functional enrichment analysis of the differentially expressed genes in (D). (F) Heatmap of differentially expressed genes (DEG) that are involved in cellular autophagy.
CALCOCO2 and S100a9 are the major AhR-regulated autophagy-associated genes in AT2 cells
RT-PCR validation further narrowed down the gene list by focusing on several highly selected genes, including Calcoco2, sh3bp4, Prkd1, Usp10, and S100a9. AT2 cells were isolated from AhRfl/fl or Sftpc-Cre;AhRf/f mice and then confirmed by immunostaining (Figure 8A). AhR deficiency in AT2 cells derived from Sftpc-Cre;AhRf/f mice was further confirmed by RT-PCR (Figure 8B). Of these selected genes, Calcoco2 showed a significant increase in CRE-treated AT2 cells from Sftpc-cre;RhoAf/f mice as compared with those from AhRf/f mice (Figure 8C). Calcoco2 has been shown to initiate selective autophagy through recruiting Unc-51-like kinase (ULK) and TANK-binding kinase 1 (TBK1) kinase complexes (48, 49). Further analysis demonstrated an increased expression of Calcoco2 in the lung tissues of our asthma mouse model as assessed by immunostaining (Figure 8D). The increased Calcoco2 was further enhanced in Sftpc-cre;RhoAf/f mice as compared with control mice. Notably, the same pattern was observed for Calcoco2 expression specifically in AT2 cells. In contrast to Calcoco2, RT-PCR analysis showed that S100a9 expression was significantly reduced in AT2 cells from Sftpc-cre;RhoAf/f mice compared with those from AhRf/f mice (Figure 8C). S100a9 (S100 calcium binding protein A9) is a pro-inflammatory alarmin associated with inflammation-related diseases. Suppression of S100a9 has been implicated to protect against LPS-induced lung injury (50). Together, these data suggest a possible mechanism that AhR regulates cellular autophagy through modulating Calcoco2/S100a9 expression.
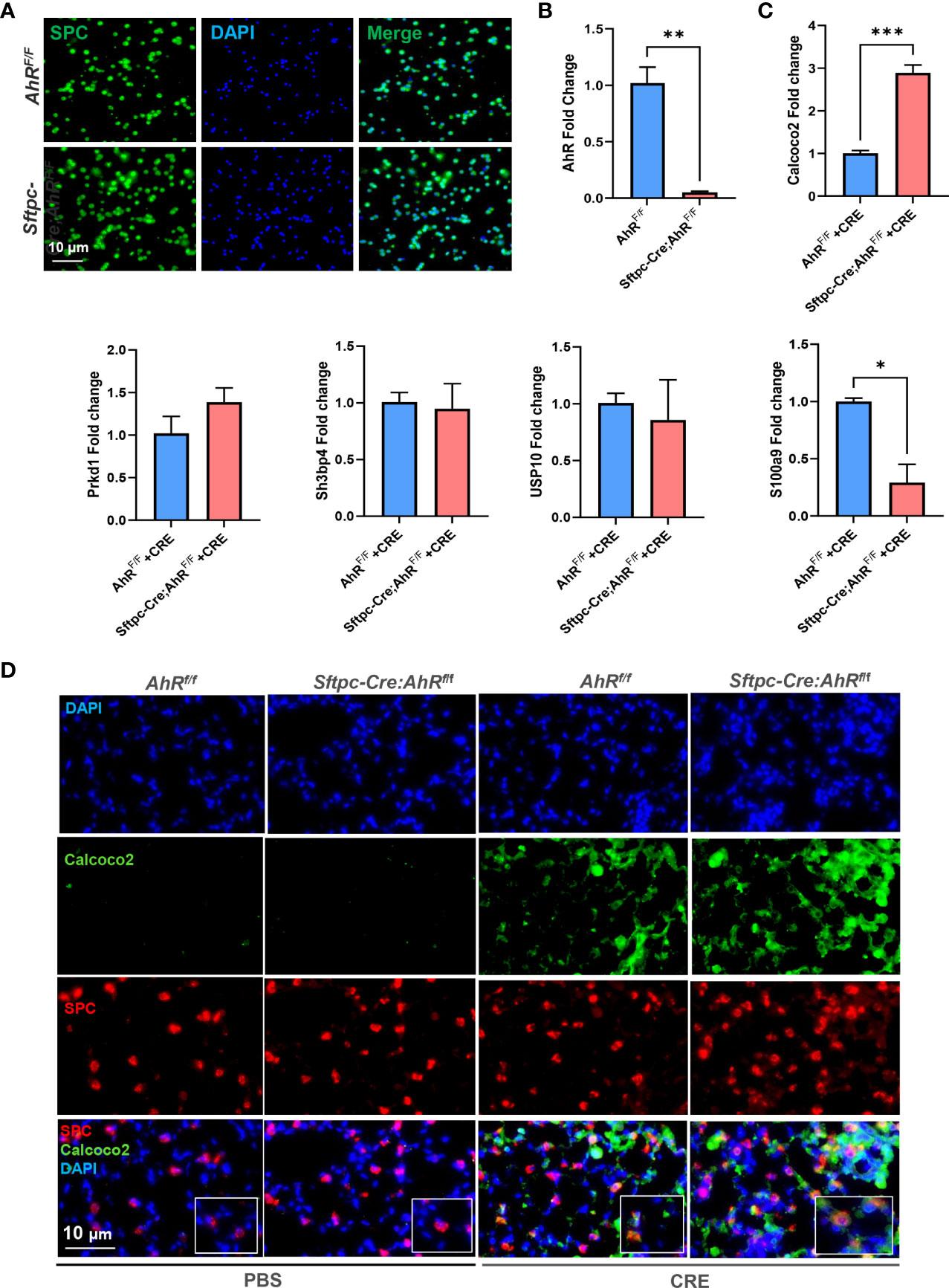
Figure 8 Calcoco2 and S100a9 are the major AhR-regulated autophagy-associated genes in AT2 cells. (A) Confirmation of AT2 cells isolated from AhRf/f or Sftpc-Cre;AhRf/f mice by immunostaining. (B) Confirmation of AhR deficiency in AT2 cells derived from Sftpc-Cre;AhRf/f mice by RT-PCR. (C) RT-PCR analysis of highly selected differentially expressed autophagy-associated genes of in AT2 cells. (D) Representative images of co-immunofluorescence staining of Calcoco2 with AT2 marker SPC. Data represent means ± SEM of at least two independent experiments. *p < 0.05, **p < 0.01, and ***p < 0.001.
Discussion
AhR as a receptor for environmental contaminants has been shown to protect against allergic airway information and remodeling in allergic asthma by using global AhR knockout mice (18, 23–25, 51, 52). In the present study, we specially focused on AT2 cells by generating AhR knockout mice in AT2 cells (Sftpc-Cre;AhRf/f) and provided supporting evidence that AhR in AT2 cells prevents allergen-induced airway hyperresponsiveness and Th2-associated airway inflammation.
AT2 cells are recognized as progenitor cells in response to lung injury and have a unique function in providing pulmonary surfactant and a variety of inflammatory mediators needed for the special microenvironment in the alveolus (53, 54). AT2 cell damage has been implicated in lung fibrosis (55) and inflammation (56). However, the genetic regulation and functional significance of AT2 cells in allergic immune responses to environmental allergens in asthma remain unknown. Here, we generated the Sftpc-Cre;AhRf/f mice to determine whether AhR specifically in AT2 cells play an important role in allergic airway inflammation. Sftpc-Cre;AhRf/f mice with AhR deletion selectively from AT2 cells were generated by crossing AhRf/f with Sftpc-cre mice. We selected Sftpc-cre mice for AhR deletion in AT2 cells because Sftpc is highly expressed by AT2 cells and has been widely used as a genetic marker for AT2 cells (57–59). However, we recognize that Sftpc may not be uniquely expressed in AT2 cells (60–62), and future studies are thus needed to identify unique markers to further define the role of AT2 cells in allergic airway inflammation.
Using the newly generated Sftpc-Cre;AhRf/f mice, we found that AT2 cell–specific deletion of AhR led to an exacerbation of allergen-induced airway hyperresponsiveness and airway inflammation with elevated eosinophils and Th2 cytokines in BALFs. These results were consistent with our previously report with global AhR knockout mice. Especially, cockroach allergen–treated AhR−/− mice showed exacerbation of airway inflammation when compared with WT mice, which was further supported by using an AhR agonist 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) (12). Furthermore, Chang et al. have also reported that AhR−/− mice had exacerbated ovalbumin (OVA)–induced asthma symptoms, including airway inflammation, mucus production, and airway remodeling, and suggested that AhR is required in maintaining normal lung function and mediating disease severity (51). In contrast, Traboulsi et al. found that OVA-induced mouse model with AhR−/− mice showed significantly enhanced eosinophilia and lymphocyte infiltrates into the airways with increased IL-4 and IL-5 in the airways, but no changes were observed for airway hyperresponsiveness (52). These discoveries highlight the complexity of AhR activation and the need for the investigation into how AhR specifically in AT2 cells regulates airway inflammation and airway hyperresponsiveness.
Our studies have suggested several underlying mechanisms as to how AhR regulates airway inflammation. We have provided evidence that AhR protects lungs from allergen-induced inflammation by modulating MSC recruitment and their immune-suppressive activity (12). Further studies suggest that active AhR signaling in MSCs regulates MSC suppressive activity by polarizing macrophages into anti-inflammatory M2 in asthma (63). Our very recent studies uncover a key functional axis of AhR-ROS-NLRP3 inflammasome that plays an important role in the development of allergic airway inflammation (30). In particular, we demonstrated that epithelial AhR plays a role in cockroach allergen–induced ROS generation and NLRP3 inflammasome activation that are essential in the development of allergic airway inflammation and mucus production. Intriguingly, MCC950, a NLRP3-specifc inhibitor, showed significant inhibition of cockroach allergen–induced airway inflammation and Muc5ac expression. In the current study, we focused on autophagy and demonstrated that AhR regulates allergen-induced cellular autophagy, and, subsequently, allergic airway inflammation. Autophagy is a homeostatic process in which eukaryotic cell encapsulates damaged proteins or organelles for lysosomal degradation and recycling (64). It has also been shown to shape cellular immune responses initiated by environmental pollutants, allergens, and respiratory tract infections (3, 34, 65–71). Abnormality in autophagy has been associated with several major asthma phenotypes, including airway hyperresponsiveness (66), inflammation (72), mucus hyperproduction (73), and remodeling (32). Accumulating evidence suggests that autophagy may be detrimental or beneficial in asthma that depends on the cell types involved (74). We have previously demonstrated that cockroach allergen can induce autophagy in human airway epithelial cells and excessive autophagy may contribute to cockroach allergen–induced airway epithelial ROS generation, cytokine release, and allergic airway inflammation (36). Importantly, autophagy has been shown to regulate NLRP3 inflammasome and secretion of proinflammatory cytokines (75), raising the possibility that autophagy may be a part of the AhR-regulated axis of ROS–inflammasome, which would be of interest to explore in the future.
In this study, we provide a critical but previously unrecognized mechanism that AhR may protect lung from allergic inflammation via regulating allergen-induced autophagy in AT2 cells. In particular, we found that Sftpc-Cre;AhRf/f mice showed an increased cockroach allergen–induced autophagy compared with AhRf/f mice. The increased autophagy was confirmed in AT2 cells of those mice. Although no human AT2 cells were available, the regulation of AhR in allergen-induced autophagy was further confirmed in HBECs with or without AhR knockdown, suggesting that the AhR regulation on AT2 cell autophagy might also be true for the bronchial airway epithelial cells. Importantly, this finding was further supported by direct monitoring of autophagic flux according to the number of GFP-LC3 puncta under fluorescence microscopy. CRE-treated HBECs showed an increased autophagic flux, which was further enhanced by the treatment with AhR antagonist CH223191. Most importantly, pre-treatment of mice with autophagy inhibitor CLQ abrogated cockroach allergen–induced airway inflammation, Th2-associated cytokines, and autophagy-related gene expression. CLQ is a classic inhibitor of autophagy that blocks the binding of autophagosomes to lysosomes (46). Collectively, these studies highlight a critical role for AhR in AT2 cells in preventing allergen-induced autophagy.
Next, we explored the possible mechanisms underlying the AhR-regulated autophagy. We have previously reported that AhR modulates allergen-induced epithelial TGF-β1 secretion and activation (29, 45). This finding led us to hypothesize that TGF-β1 may be a major player in AhR-mediated autophagy. Indeed, we found increased levels of TGF-β1 in supernatants of cockroach allergen–treated HBECs, which were further enhanced in HBECs with AhR knockdown. Furthermore, we investigated whether the AhR-regulated TGF-β1 can directly induce autophagy. As expected, HBECs showed an increased autophagy after exposed to different doses of recombinant TGF-β1. In contrast, we found that cockroach allergen–induced autophagy was almost completely blocked when TGF-β1 was deleted by using TGF-β1 neutralizing antibody (α-TGF-β1). These studies suggest that TGF-β1 is required in AhR-regulated cockroach allergen–induced autophagy.
In addition to TGF-β1, we expanded our studies to identify novel genes and pathways that are involved in AhR-mediated autophagy and airway inflammation. We analyzed the transcriptional profiles of AT2 cells isolated from the lungs of AhRf/f and Sftpc-cre;AhRf/f mice. Although there were different comparisons between groups, the major comparison was between CRE-treated AT2 cells derived from Sftpc-cre;AhRf/f mice and AhRf/f mice with a total of 717 differentially expressed mRNAs identified. Of these, both AhR downstream genes Ahrr and Cyp1b1 showed significant reduction in AT2 cells of Sftpc-cre;AhRf/f mice, supporting the AhR deficiency in AT2 cells and proper experimental approaches we used in this study. In addition, we found our previously identified genes COX-2 (47) and Slc26a4 (38) that are associated with allergic airway inflammation. Furthermore, GO functional enrichment analysis based on these DEGs identified autophagy as one of the most enriched GO terms. Among these autophagy-related genes, Calcoco2, also named as NDP52 (nuclear dot protein 52), is an essential selective autophagy adaptor that initiates selective autophagy through recruiting ULK and TBK1 kinase complexes (48). Recruitment of Calcoco2 has also been suggested to amplify PTEN Induced Kinase 1 (PINK1)/Parkin mitophagy signaling (49). Our study demonstrated that Calcoco2 was highly expressed in the lung tissues and AT2 cells of our asthma mouse model as determined by immunofluorescence staining. In contrast to Calcoco2, S100a9 is downregulated in AT2 cells of Sftpc-cre;AhRf/f mice compared with AhRf/f mice. S100a9 is known as damage-associated molecular pattern, which can be induced in many cells under inflammatory condition (76). Interestingly, studies have highlighted a role for S100a9 in inducing pro-inflammatory cytokine release and in the ROS-dependent activation of the NLRP3 inflammasome (76). However, it remains unclear whether S100a9 is also involved in the allergen-induced and ROS-dependent activation of autophagy. In addition, we have also identified several other significant GO terms, including ubiquitin-protein transferase activity, cell activation, and cilium-related terms (e.g., movement, assembly, organization, and cilium). Those identified biological processes are clearly worth to be investigated in the future for their contributions to the AhR-mediated airway inflammation.
Taken together, we used our newly generated Sftpc-Cre;AhRf/f mice and provided a strong evidence that AhR in AT2 cells protects against cockroach allergen–induced airway hyperresponsiveness and Th2-associated airway inflammation. Further in vivo and in vitro studies suggest that cockroach allergen can induce autophagy in AT2 cells, and AhR in AT2 cells can prevent cockroach allergen–induced autophagy. Most intriguingly, autophagy inhibition attenuated cockroach allergen–induced airway inflammation and Th2-associated cytokines. Mechanistically, we identified a functional axis of AhR-TGF-β1 that is critical in driving allergen-induced cellular autophagy. Furthermore, our GO functional enrichment analysis on these DEGs from the RNA-seq analysis suggested that autophagy is one of the most enriched GO terms, and the autophagy adaptor CALCOCO2 and S100a9 are the major AhR-regulated autophagy genes in cockroach allergen–treated AT2 cells (Figure 9, graphic summary). However, the regulation of AhR on these identified autophagy genes and the exact role of these genes in the pathogenesis of asthma remain unclear, thus warranting further, in-depth investigations. One of the major limitations is the lack of primary human AT2 cells. Instead, we used HBECs, an immortalized cell line under submerged cultures, for in vitro studies, which may not truly reflect the real changes seen in AT2 cells. However, our findings in AT2 cells may also apply to the bronchial epithelial cells. Collectively, these findings highlight a critical role for AhR in AT2 cells in preventing allergic airway inflammation through regulating cellular autophagy.
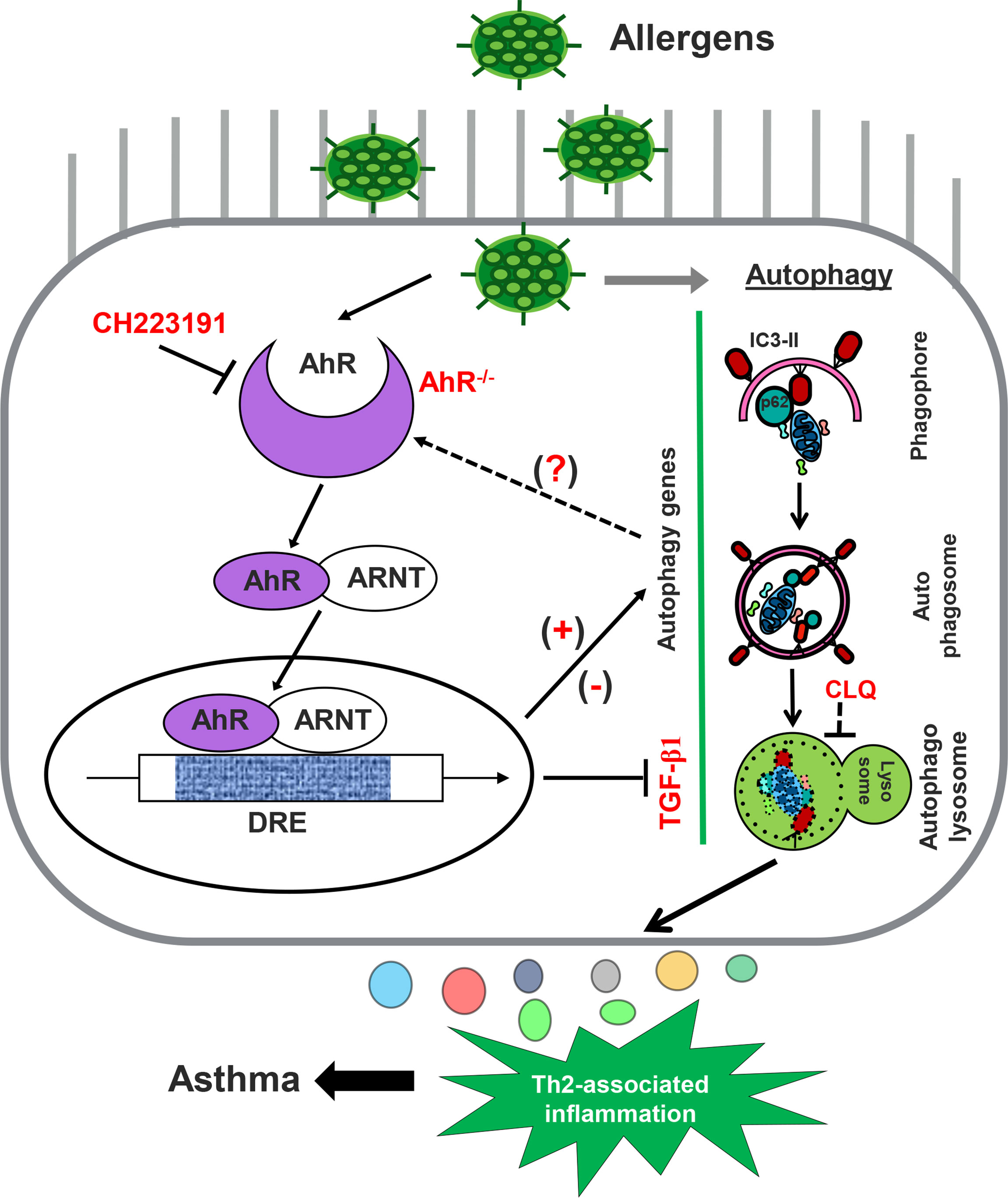
Figure 9 Graphic summary. AhR in AT2 cells regulates cockroach allergen–induced autophagy. Cockroach allergen can induce autophagy that is critical in controlling proinflammatory mediator release, Th2-associated airway inflammation, and, subsequently, allergic asthma. AhR in AT2 cells inhibits cockroach allergen–induced autophagy through either controlling TGF-β1 release or AhR-regulated autophagy-associated genes [either upregulated (e.g., CALCOCO2) or downregulated (e.g., S100a9)].
Data availability statement
The datasets presented in this study can be found in online repositories. The name of the repository and accession number can be found below: National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO), https://www.ncbi.nlm.nih.gov/geo/, GSE205818.
Ethics statement
The animal study was reviewed and approved by The Johns Hopkins University Animal Care and Use Committee.
Author contributions
JW, YLZ, XZ, WT, RW, YS, and YZ performed experiments, analyzed data, and review the manuscript. PG designed and supervised the study, and wrote the manuscript. All authors read and approved the final version of the manuscript.
Funding
This work was supported by grants from the US National Institutes of Health (NIH) (1R01AI153331 and R01AI141642 to PG). This study was also supported by grants from the National Natural Science Foundation of China (31900667 to WT) and Shenzhen Scientific Technology Basic Research Project (JCYJ20190812171617278 to WT).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.964575/full#supplementary-material
Abbreviations
CRE, cockroach extract; AhR, aryl hydrocarbon receptor; WT, wild-type; BALF, bronchoalveolar lavage fluid; PAS, Alcian Blue periodic acid–Schiff; RT-PCR, quantitative real-time PCR; AHR, airway hyperresponsiveness; BSA, bovine serum albumin; FBS, fetal bovine serum; H&E, hematoxylin and eosin; AT2, type II alveolar epithelial cells; HBEC, human bronchial epithelial cell; CLQ, chloroquine; GO, Gene Ontology; NLRP3, nucleotide-binding oligomerization domain (NOD)–like receptor family pyrin domain-containing 3.
References
1. Akinbami LJ, Moorman JE, Bailey C, Zahran HS, King M, Johnson CA, et al. Trends in asthma prevalence, health care use, and mortality in the united states, 2001-2010. NCHS Data Brief (2012) 94):1–8.
2. Sachdeva K, Do DC, Zhang Y, Hu X, Chen J, Gao P. Environmental exposures and asthma development: Autophagy, mitophagy, and cellular senescence. Front Immunol (2019) 10:2787. doi: 10.3389/fimmu.2019.02787
3. Do DC, Zhao Y, Gao P. Cockroach allergen exposure and risk of asthma. Allergy (2016) 71(4):463–74. doi: 10.1111/all.12827
4. Glesner J, Filep S, Vailes LD, Wunschmann S, Chapman MD, Birrueta G, et al. Allergen content in German cockroach extracts and sensitization profiles to a new expanded set of cockroach allergens determine in vitro extract potency for IgE reactivity. J Allergy Clin Immunol (2019) 143(4):1474–81 e8. doi: 10.1016/j.jaci.2018.07.036
5. Pomes A, Glesner J, Calatroni A, Visness CM, Wood RA, O’Connor GT, et al. Cockroach allergen component analysis of children with or without asthma and rhinitis in an inner-city birth cohort. J Allergy Clin Immunol (2019) 144(4):935–44. doi: 10.1016/j.jaci.2019.05.036
6. da Silva Antunes R, Sutherland A, Frazier A, Schulten V, Pomes A, Glesner J, et al. Heterogeneity of magnitude, allergen immunodominance, and cytokine polarization of cockroach allergen-specific T cell responses in allergic sensitized children. Clin Transl Allergy (2021) 11(8):e12073. doi: 10.1002/clt2.12073
7. Di Meglio P, Duarte JH, Ahlfors H, Owens ND, Li Y, Villanova F, et al. Activation of the aryl hydrocarbon receptor dampens the severity of inflammatory skin conditions. Immunity (2014) 40(6):989–1001. doi: 10.1016/j.immuni.2014.04.019
8. Stockinger B, Di Meglio P, Gialitakis M, Duarte JH. The aryl hydrocarbon receptor: multitasking in the immune system. Annu Rev Immunol (2014) 32:403–32. doi: 10.1146/annurev-immunol-032713-120245
9. Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, et al. Control of t(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature (2008) 453(7191):65–71. doi: 10.1038/nature06880
10. Nguyen NT, Kimura A, Nakahama T, Chinen I, Masuda K, Nohara K, et al. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc Natl Acad Sci United States America (2010) 107(46):19961–6. doi: 10.1073/pnas.1014465107
11. Nguyen NT, Nakahama T, Le DH, Van Son L, Chu HH, Kishimoto T. Aryl hydrocarbon receptor and kynurenine: recent advances in autoimmune disease research. Front Immunol (2014) 5:551. doi: 10.3389/fimmu.2014.00551
12. Xu T, Zhou Y, Qiu L, Do DC, Zhao Y, Cui Z, et al. Aryl hydrocarbon receptor protects lungs from cockroach allergen-induced inflammation by modulating mesenchymal stem cells. J Immunol (2015) 195(12):5539–50. doi: 10.4049/jimmunol.1501198
13. Nguyen LP, Bradfield CA. The search for endogenous activators of the aryl hydrocarbon receptor. Chem Res Toxicol (2008) 21(1):102–16. doi: 10.1021/tx7001965
14. Totlandsdal AI, Cassee FR, Schwarze P, Refsnes M, Lag M. Diesel exhaust particles induce CYP1A1 and pro-inflammatory responses via differential pathways in human bronchial epithelial cells. Particle Fibre Toxicol (2010) 7:41. doi: 10.1186/1743-8977-7-41
15. Manners S, Alam R, Schwartz DA, Gorska MM. A mouse model links asthma susceptibility to prenatal exposure to diesel exhaust. J Allergy Clin Immunol (2014) 134(1):63–72. doi: 10.1016/j.jaci.2013.10.047
16. Stevens EA, Mezrich JD, Bradfield CA. The aryl hydrocarbon receptor: a perspective on potential roles in the immune system. Immunology (2009) 127(3):299–311. doi: 10.1111/j.1365-2567.2009.03054.x
17. Suen JL, Hsu SH, Hung CH, Chao YS, Lee CL, Lin CY, et al. A common environmental pollutant, 4-nonylphenol, promotes allergic lung inflammation in a murine model of asthma. Allergy (2013) 68(6):780–7. doi: 10.1111/all.12156
18. Xia M, Viera-Hutchins L, Garcia-Lloret M, Noval Rivas M, Wise P, McGhee SA, et al. Vehicular exhaust particles promote allergic airway inflammation through an aryl hydrocarbon receptor-notch signaling cascade. J Allergy Clin Immunol (2015) 136(2):441–53. doi: 10.1016/j.jaci.2015.02.014
19. Huang SK, Zhang Q, Qiu Z, Chung KF. Mechanistic impact of outdoor air pollution on asthma and allergic diseases. J Thorac Dis (2015) 7(1):23–33. doi: 10.3978/j.issn.2072-1439.2014.12.13
20. Li X-m, Peng J, Gu W, Guo X-j. TCDD-induced activation of aryl hydrocarbon receptor inhibits Th17 polarization and regulates non-eosinophilic airway inflammation in asthma. PloS One (2016) 11(3). 1-12doi: 10.1371/journal.pone.0150551
21. Morales-Rubio R, Amador-Munoz O, Rosas-Perez I, Sanchez-Perez Y, Garcia-Cuellar C, Segura-Medina P, et al. PM2.5 induces airway hyperresponsiveness and inflammation via the AhR pathway in a sensitized Guinea pig asthma-like model. Toxicology (2022) 465:153026. doi: 10.1016/j.tox.2021.153026
22. Yang Y, Chan WK. Selective autophagy maintains the aryl hydrocarbon receptor levels in HeLa cells: A mechanism that is dependent on the p23 Co-chaperone. Int J Mol Sci (2020) 21(10):3449. doi: 10.3390/ijms21103449
23. Wong TH, Lee CL, Su HH, Lee CL, Wu CC, Wang CC, et al. A prominent air pollutant, Indeno[1,2,3-cd]pyrene, enhances allergic lung inflammation via aryl hydrocarbon receptor. Sci Rep (2018) 8(1):5198. doi: 10.1038/s41598-018-23542-9
24. Xia M, Harb H, Saffari A, Sioutas C, Chatila TA. A jagged 1-notch 4 molecular switch mediates airway inflammation induced by ultrafine particles. J Allergy Clin Immunol (2018) 142(4):1243–56 e17. doi: 10.1016/j.jaci.2018.03.009
25. Vogel CFA, Van Winkle LS, Esser C, Haarmann-Stemmann T. The aryl hydrocarbon receptor as a target of environmental stressors - implications for pollution mediated stress and inflammatory responses. Redox Biol (2020) 34:101530. doi: 10.1016/j.redox.2020.101530
26. Zhou Y, Tung HY, Tsai YM, Hsu SC, Chang HW, Kawasaki H, et al. Aryl hydrocarbon receptor controls murine mast cell homeostasis. Blood (2013) 121(16):3195–204. doi: 10.1182/blood-2012-08-453597
27. Wang H, Do DC, Liu J, Wang B, Qu J, Ke X, et al. Functional role of kynurenine and aryl hydrocarbon receptor axis in chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol (2018) 141(2):586–600 e6. doi: 10.1016/j.jaci.2017.06.013
28. Wang E, Liu X, Tu W, Do DC, Yu H, Yang L, et al. Benzo(a)pyrene facilitates dermatophagoides group 1 (Der f 1)-induced epithelial cytokine release through aryl hydrocarbon receptor in asthma. Allergy (2019) 74(9):1675–90. doi: 10.1111/all.13784
29. Wang E, Tu W, Do DC, Xiao X, Bhatti SB, Yang L, et al. Benzo(a)pyrene enhanced dermatophagoides group 1 (Der f 1)-induced TGFbeta1 signaling activation through the aryl hydrocarbon receptor-RhoA axis in asthma. Front Immunol (2021) 12:643260. doi: 10.3389/fimmu.2021.643260
30. Hu X, Shen Y, Zhao Y, Wang J, Zhang X, Tu W, et al. Epithelial aryl hydrocarbon receptor protects from mucus production by inhibiting ROS-triggered NLRP3 inflammasome in asthma. Front Immunol (2021) 12:767508:. doi: 10.3389/fimmu.2021.767508
31. Ban GY, Pham DL, Trinh TH, Lee SI, Suh DH, Yang EM, et al. Autophagy mechanisms in sputum and peripheral blood cells of patients with severe asthma: a new therapeutic target. Clin Exp Allergy (2016) 46(1):48–59. doi: 10.1111/cea.12585
32. McAlinden KD, Deshpande DA, Ghavami S, Xenaki D, Sohal SS, Oliver BG, et al. Autophagy activation in asthma airways remodeling. Am J Respir Cell Mol Biol (2019) 60(5):541–53. doi: 10.1165/rcmb.2018-0169OC
33. Xia F, Deng C, Jiang Y, Qu Y, Deng J, Cai Z, et al. IL4 (interleukin 4) induces autophagy in b cells leading to exacerbated asthma. Autophagy (2018) 14(3):450–64. doi: 10.1080/15548627.2017.1421884
34. Poon AH, Chouiali F, Tse SM, Litonjua AA, Hussain SN, Baglole CJ, et al. Genetic and histologic evidence for autophagy in asthma pathogenesis. J Allergy Clin Immunol (2012) 129(2):569–71. doi: 10.1016/j.jaci.2011.09.035
35. Martin LJ, Gupta J, Jyothula SS, Butsch Kovacic M, Biagini Myers JM, Patterson TL, et al. Functional variant in the autophagy-related 5 gene promotor is associated with childhood asthma. PloS One (2012) 7(4):e33454. doi: 10.1371/journal.pone.0033454
36. Zhang Y, Do DC, Hu X, Wang J, Zhao Y, Mishra S, et al. CaMKII oxidation regulates cockroach allergen-induced mitophagy in asthma. J Allergy Clin Immunol (2021) 147(4):1464–77 e11. doi: 10.1016/j.jaci.2020.08.033
37. Do DC, Mu J, Ke X, Sachdeva K, Qin Z, Wan M, et al. miR-511-3p protects against cockroach allergen-induced lung inflammation by antagonizing CCL2. JCI Insight (2019) 4(20):e126832. doi: 10.1172/jci.insight.126832
38. Do DC, Zhang Y, Tu W, Hu X, Xiao X, Chen J, et al. Type II alveolar epithelial cell-specific loss of RhoA exacerbates allergic airway inflammation through SLC26A4. JCI Insight (2021) 6(14):e148147. doi: 10.1172/jci.insight.148147
39. Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods (2017) 14(4):417–9. doi: 10.1038/nmeth.4197
40. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res (2015) 43(7):e47. doi: 10.1093/nar/gkv007
41. Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (N Y) (2021) 2(3):100141. 10.1016/j.xinn.2021.100141
42. Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics (2016) 32(18):2847–9. doi: 10.1093/bioinformatics/btw313
43. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods (2001) 25(4):402–8. doi: 10.1006/meth.2001.1262
44. Wang Y, Bai C, Li K, Adler KB, Wang X. Role of airway epithelial cells in development of asthma and allergic rhinitis. Respir Med (2008) 102(7):949–55. doi: 10.1016/j.rmed.2008.01.017
45. Zhou Y, Mirza S, Xu T, Tripathi P, Plunkett B, Myers A, et al. Aryl hydrocarbon receptor (AhR) modulates cockroach allergen-induced immune responses through active TGFbeta1 release. Mediators Inflammation (2014) 2014:591479. doi: 10.1155/2014/591479
46. Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr M, Hijlkema KJ, et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy (2018) 14(8):1435–55. doi: 10.1080/15548627.2018.1474314
47. Qiu L, Zhang Y, Do DC, Ke X, Zhang S, Lambert K, et al. miR-155 modulates cockroach allergen- and oxidative stress-induced cyclooxygenase-2 in asthma. J Immunol (2018) 201(3):916–29. doi: 10.4049/jimmunol.1701167
48. Boyle KB, Ravenhill BJ, Randow F. CALCOCO2/NDP52 initiates selective autophagy through recruitment of ULK and TBK1 kinase complexes. Autophagy (2019) 15(9):1655–6. doi: 10.1080/15548627.2019.1628548
49. Padman BS, Nguyen TN, Uoselis L, Skulsuppaisarn M, Nguyen LK, Lazarou M. LC3/GABARAPs drive ubiquitin-independent recruitment of optineurin and NDP52 to amplify mitophagy. Nat Commun (2019) 10(1):408. doi: 10.1038/s41467-019-08335-6
50. Zhao B, Lu R, Chen J, Xie M, Zhao X, Kong L. S100A9 blockade prevents lipopolysaccharide-induced lung injury via suppressing the NLRP3 pathway. Respir Res (2021) 22(1):45. doi: 10.1186/s12931-021-01641-y
51. Chang YD, Li CH, Tsai CH, Cheng YW, Kang JJ, Lee CC. Aryl hydrocarbon receptor deficiency enhanced airway inflammation and remodeling in a murine chronic asthma model. FASEB J (2020) 34(11):15300–13. doi: 10.1096/fj.202001529R
52. Traboulsi H, de Souza AR, Allard B, Haidar Z, Sorin M, Moarbes V, et al. Differential regulation of the asthmatic phenotype by the aryl hydrocarbon receptor. Front Physiol (2021) 12:720196. doi: 10.3389/fphys.2021.720196
53. Sucre JMS, Jetter CS, Loomans H, Williams J, Plosa EJ, Benjamin JT, et al. Successful establishment of primary type II alveolar epithelium with 3D organotypic coculture. Am J Respir Cell Mol Biol (2018) 59(2):158–66. doi: 10.1165/rcmb.2017-0442MA
54. Olajuyin AM, Zhang X, Ji HL. Alveolar type 2 progenitor cells for lung injury repair. Cell Death Discovery (2019) 5:63. doi: 10.1038/s41420-019-0147-9
55. Parimon T, Yao C, Stripp BR, Noble PW, Chen P. Alveolar epithelial type II cells as drivers of lung fibrosis in idiopathic pulmonary fibrosis. Int J Mol Sci (2020) 21(7):2269. doi: 10.3390/ijms21072269
56. Post S, Heijink IH, Hesse L, Koo HK, Shaheen F, Fouadi M, et al. Characterization of a lung epithelium specific e-cadherin knock-out model: Implications for obstructive lung pathology. Sci Rep (2018) 8(1):13275. doi: 10.1038/s41598-018-31500-8
57. Finn J, Sottoriva K, Pajcini KV, Kitajewski JK, Chen C, Zhang W, et al. Dlk1-mediated temporal regulation of notch signaling is required for differentiation of alveolar type II to type I cells during repair. Cell Rep (2019) 26(11):2942–54 e5. doi: 10.1016/j.celrep.2019.02.046
58. Wu H, Yu Y, Huang H, Hu Y, Fu S, Wang Z, et al. Progressive pulmonary fibrosis is caused by elevated mechanical tension on alveolar stem cells. Cell (2020) 180(1):107–21 e17. doi: 10.1016/j.cell.2019.11.027
59. Choi J, Park JE, Tsagkogeorga G, Yanagita M, Koo BK, Han N, et al. Inflammatory signals induce AT2 cell-derived damage-associated transient progenitors that mediate alveolar regeneration. Cell Stem Cell (2020) 27(3):366–82 e7. doi: 10.1016/j.stem.2020.06.020
60. Eblaghie MC, Reedy M, Oliver T, Mishina Y, Hogan BL. Evidence that autocrine signaling through Bmpr1a regulates the proliferation, survival and morphogenetic behavior of distal lung epithelial cells. Dev Biol (2006) 291(1):67–82. doi: 10.1016/j.ydbio.2005.12.006
61. Thomas SY, Whitehead GS, Takaku M, Ward JM, Xu X, Nakano K, et al. MyD88-dependent dendritic and epithelial cell crosstalk orchestrates immune responses to allergens. Mucosal Immunol (2018) 11(3):796–810. doi: 10.1038/mi.2017.84
62. Jiang M, Roth MG, Chun-On P, Sullivan DI, Alder JK. Phenotypic diversity caused by differential expression of SFTPC-Cre-Transgenic alleles. Am J Respir Cell Mol Biol (2020) 62(6):692–8. doi: 10.1165/rcmb.2019-0416MA
63. Cui Z, Feng Y, Li D, Li T, Gao P, Xu T. Activation of aryl hydrocarbon receptor (AhR) in mesenchymal stem cells modulates macrophage polarization in asthma. J Immunotoxicol (2020) 17(1):21–30. doi: 10.1080/1547691X.2019.1706671
64. Deretic V, Klionsky DJ. Autophagy and inflammation: A special review issue. Autophagy (2018) 14(2):179–80. doi: 10.1080/15548627.2017.1412229
65. Miraglia del Giudice M, Allegorico A, Parisi G, Galdo F, Alterio E, Coronella A, et al. Risk factors for asthma. Ital J Pediatrics (2014) 40(Suppl 1):A77–A. doi: 10.1186/1824-7288-40-S1-A77
66. Liu JN, Suh DH, Trinh HK, Chwae YJ, Park HS, Shin YS. The role of autophagy in allergic inflammation: a new target for severe asthma. Exp Mol Med (2016) 48(7):e243. doi: 10.1038/emm.2016.38
67. Rowlands DJ. Mitochondria dysfunction: A novel therapeutic target in pathological lung remodeling or bystander? Pharmacol Ther (2016) 166:96–105. doi: 10.1016/j.pharmthera.2016.06.019
68. Prakash YS, Pabelick CM, Sieck GC. Mitochondrial dysfunction in airway disease. Chest (2017) 152(3):618–26. doi: 10.1016/j.chest.2017.03.020
69. Wu J, Dong F, Wang RA, Wang J, Zhao J, Yang M, et al. Central role of cellular senescence in TSLP-induced airway remodeling in asthma. PloS One (2013) 8(10):e77795. doi: 10.1371/journal.pone.0077795
70. Puddicombe SM, Torres-Lozano C, Richter A, Bucchieri F, Lordan JL, Howarth PH, et al. Increased expression of p21(waf) cyclin-dependent kinase inhibitor in asthmatic bronchial epithelium. Am J Respir Cell Mol Biol (2003) 28(1):61–8. doi: 10.1165/rcmb.4715
71. Barnes PJ, Baker J, Donnelly LE. Autophagy in asthma and chronic obstructive pulmonary disease. Clin Sci (Lond) (2022) 136(10):733–46. doi: 10.1042/CS20210900
72. Lee J, Kim HS. The role of autophagy in eosinophilic airway inflammation. Immune Netw (2019) 19(1):e5. doi: 10.4110/in.2019.19.e5
73. Chen ZH, Wu YF, Wang PL, Wu YP, Li ZY, Zhao Y, et al. Autophagy is essential for ultrafine particle-induced inflammation and mucus hyperproduction in airway epithelium. Autophagy (2016) 12(2):297–311:7314. doi: 10.1080/15548627.2015.1124224
74. Theofani E, Xanthou G. Autophagy: A friend or foe in allergic asthma? Int J Mol Sci (2021) 22(12). doi: 10.3390/ijms22126314
75. Maneechotesuwan K, Kasetsinsombat K, Wongkajornsilp A, Barnes PJ. Role of autophagy in regulating interleukin-10 and the responses to corticosteroids and statins in asthma. Clin Exp Allergy: J Br Soc Allergy Clin Immunol (2021) 51(12):1553–65. doi: 10.1111/cea.13825
Keywords: asthma, cockroach allergen, aryl hydrocarbon receptor, autophagy, AT2 cells
Citation: Wang J, Zhao Y, Zhang X, Tu W, Wan R, Shen Y, Zhang Y, Trivedi R and Gao P (2022) Type II alveolar epithelial cell aryl hydrocarbon receptor protects against allergic airway inflammation through controlling cell autophagy. Front. Immunol. 13:964575. doi: 10.3389/fimmu.2022.964575
Received: 08 June 2022; Accepted: 28 June 2022;
Published: 22 July 2022.
Edited by:
Bao-Hui Cheng, Institute of ENT and Shenzhen Key Laboratory of ENT, ChinaReviewed by:
Devendra K. Agrawal, Western University of Health Sciences, United StatesErwin William Gelfand, University of Colorado Anschutz Medical Campus, United States
Copyright © 2022 Wang, Zhao, Zhang, Tu, Wan, Shen, Zhang, Trivedi and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peisong Gao, pgao1@jhmi.edu
†These authors have contributed equally to this work