 Yuanling Zhang
Yuanling Zhang Junhao Chen3†
Junhao Chen3†- 1School of Medicine, Guizhou University, Guiyang, China
- 2Department of Gastrointestinal Surgery, Guizhou Provincial People’s Hospital, Guiyang, China
- 3Graduate School of Zunyi Medical University, Zunyi, China
- 4Department of Medical Cosmetology, Guizhou Provincial People’s Hospital, Guiyang, China
- 5Department of General Surgery, Zhijin County People’s Hospital, Bijie, China
- 6Orthopedics Department, Dongguan Songshan Lake Tungwah Hospital, DongGuan, China
- 7Department of Thyroid and Breast Surgery, Affiliated Hospital of Panzhihua University, Panzhihua, China
Epigenetic modifications may alter the proliferation and differentiation of normal cells, leading to malignant transformation. They can also affect normal stimulation, activation, and abnormal function of immune cells in the tissue microenvironment. Histone methylation, coordinated by histone methylase and histone demethylase to stabilize transcription levels in the promoter area, is one of the most common types of epigenetic alteration, which gained increasing interest. It can modify gene transcription through chromatin structure and affect cell fate, at the transcriptome or protein level. According to recent research, histone methylation modification can regulate tumor and immune cells affecting anti-tumor immune response. Consequently, it is critical to have a thorough grasp of the role of methylation function in cancer treatment. In this review, we discussed recent data on the mechanisms of histone methylation on factors associated with immune resistance of tumor cells and regulation of immune cell function.
Introduction
Over the past decade, immunotherapy, such as immune checkpoint and CAR T cell therapy, has become a promising strategy for treating cancer (1, 2). Cancer treatment is achieved by increasing the number and effectiveness of immune cells, which can recognize tumor cells, collaborating with tumor surface suppressors and soluble factors in the tumor microenvironment to prevent the tumor invasion and metastasis, thus maintaining the immune microenvironment homeostasis of the body, and improving immune response (3–5). However, due to the tumor heterogeneity and primary or acquired treatment resistance, only 10% to 30% of patients can benefit from immunotherapy (6–8). Therefore, identifying the source of low immune reactivity, effectively regulating immune cell and tumor cell therapeutic targets, and improving immunogenicity are of utmost importance.
The oncogenic transformation caused by the accumulation of related oncogene and tumor suppressor gene mutations accompanied by alteration of histone methylation modification has been observed in various human cancers, further emphasizing the importance of histone methylation modification in medical oncology research (9, 10). Many studies have suggested that aberrant methylation of histones can reduce the expression of tumor-associated antigens, hinder antigen presentation, and affect the exercise of anti-tumor immunity by anti-tumor effector T cells, specialized antigen-presenting cells (APCs), and other cells (11, 12). Moreover, it can alter the number and differentiation process of non-specialized APC infiltration, such as myeloid-derived suppressor cells (MDSCs), regulatory T cells (Tregs), and tumor-associated macrophages (TAMs), assisting tumor cell immune escape (13). Given the impact of histone methylation modification on the immune system and tumor cells, it is worth exploring whether targeting these enzymes may alter the tumor immune microenvironment and improve the efficacy of immunotherapy. Our findings showed that enzymes involved in histone methylation regulate tumor immunity, providing innovative strategies for formulating more perfect immunotherapy strategies. In this review, we discussed the effect and mechanism of aberrant histone methylation in the tumor immune microenvironment on immune cells and tumor cells.
Classification and biological functions of histone methyltransferases (HMTs)
The amino terminus of histones can be modified to create a class of “histone codes” that increase the amount of information in the genetic code of genes, resulting in different cell fate and pathological development in the same cases (14). Lysine and arginine residues of certain histones are catalyzed by a family of conserved proteins known as the histone methyltransferases (HMTs), consisting of two species based on their structure and modification sites, i.e., histone lysine methyltransferase (KMT) and protein arginine methyltransferase (PRMT), both of which use N-terminal residues as modification sites, such as H3K4, H3K9, H3K27, H3K36, H3K79, and H4K20 (15). Most KMT contain a conserved catalytic domain, called the SET domain. Accordingly, the KMT family can be divided into SET domain-containing enzymes, including EZH2, G9a, SETD2, SUV39H1, and SET domain-free DOT1-like proteins (16). PRMT is a group of enzymes that use S-adenosine methionine (SAM) as a methyl donor. The PRMT family has nine members (PRMT1-9) that generate a single methyl group, which is added to the target protein to create a monomethylarginine (MMA) tag (17). Based on the catalyzed methylation reaction type, the PRMT family is divided into three isoforms, a class of highly conserved genetic products (18).
HMTs have a major role in the epigenetic regulation of gene expression, especially in the regulation of genes related to tumor invasion and metastasis. HMTs catalyze the lysine and arginine residues of particular histones, which are involved in a variety of biological activities, including packaging of chromosome structures, affecting transcription factor recruitment and binding, initiation and extension factors and target DNA binding, RNA processing, editing, and other processes. They also regulate genome mutations, ultimately leading to cancer (10). These methyltransferases have been demonstrated to have an important role in tumor maturation, carcinogenesis, and maintenance of stem cell components. HMTs act in a closely controlled manner to direct the necessary cellular processes under normal cell physiological settings. However, these enzymes may dysregulate and modify the epigenetic landscape and proteome to drive cell growth and survival in malignant circumstances (18, 19).
Histone lysine methyltransferase (KMT) and tumor immunity
KMT abnormalities in the complex tumor microenvironment cause expression mutations of key immune regulators in tumor cells and effector genes in immune cells, which may lead to antigen presentation suppression, loss of immune tolerance, blocked anti-tumor immunity, and negative effects on immunotherapy. In the following paragraphs, we discuss the regulatory mechanisms of numerous popular histone lysine methylases in tumors and their effect on immune cells, further emphasizing the crucial necessity of inhibiting histone lysine methylases for immunotherapy (Table 1) (Figure 1).
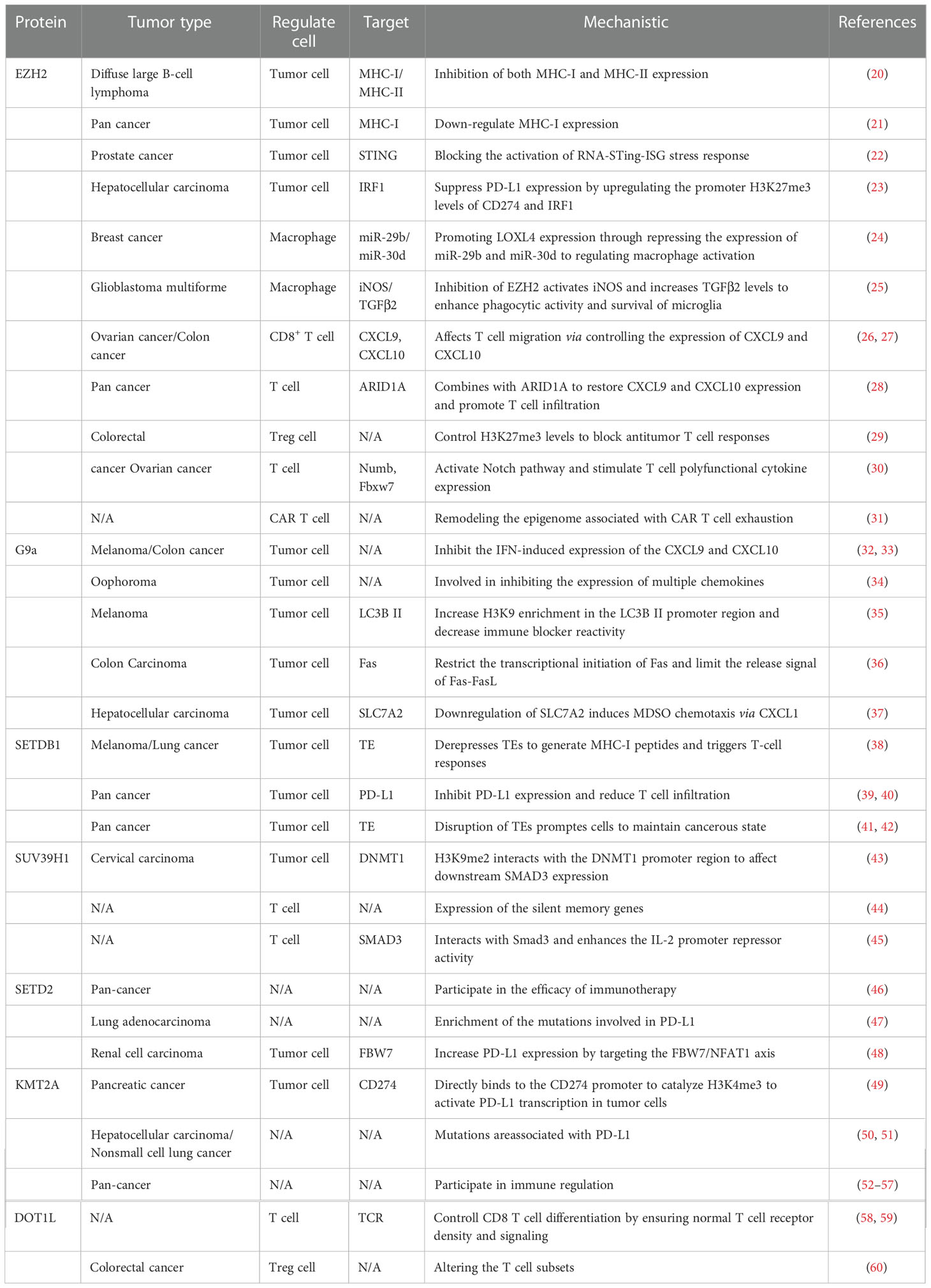
Table 1 Related functions of lysine methylase and tumor immunity.
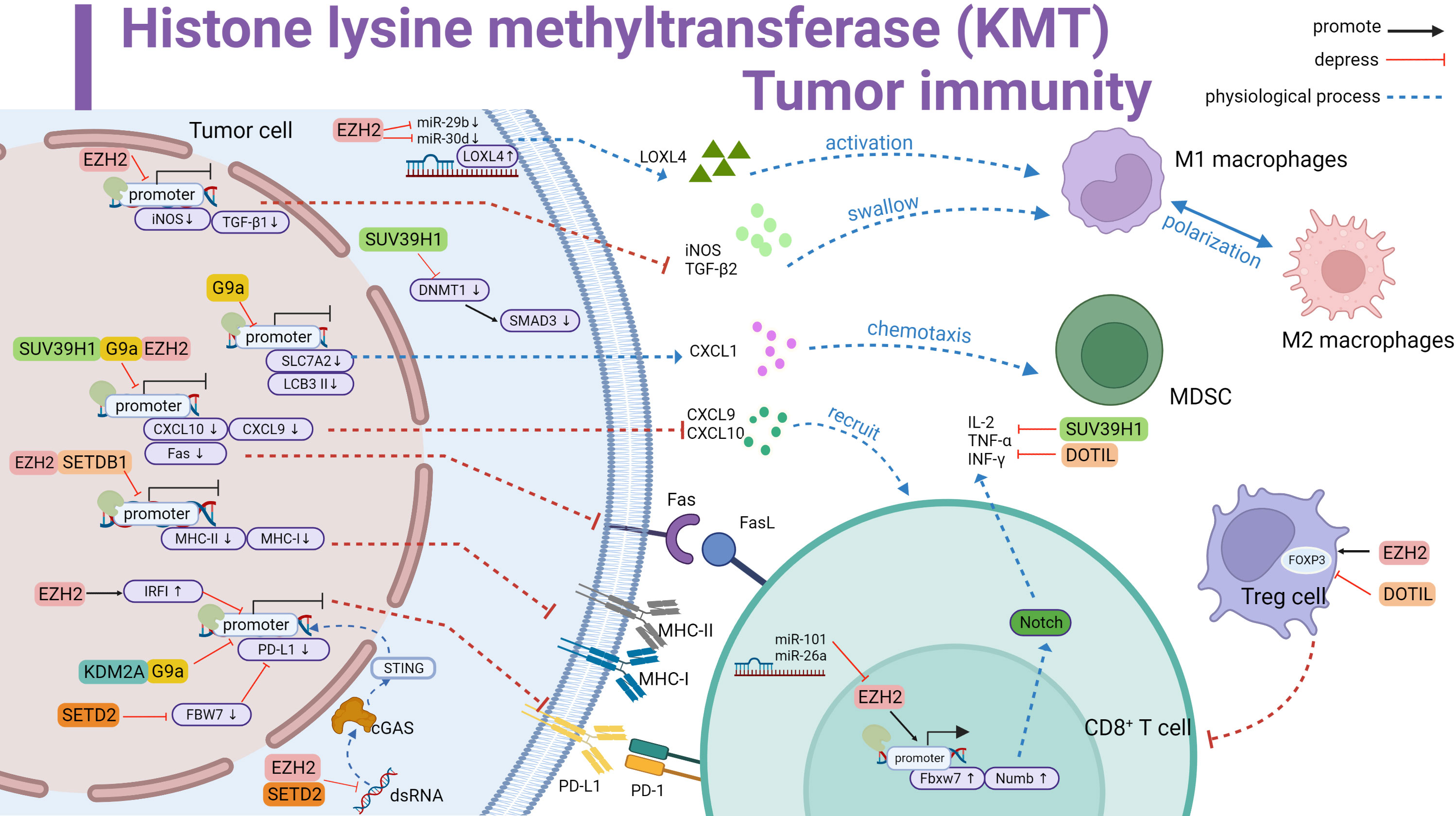
Figure 1 Histone lysine methyltransferase (KMT) involved in tumor immune summary. Promotes LOXL4 upregulation by antagonizing miR-29b and miR-30d to activate macrophage polarization; downregulates iNOS and TGF-β1 and inhibits macrophage phagocytosis. Downregulates iNOS and TGF-β1, inhibits macrophage phagocytosis. G9a inhibits SLC7A2, upregulates CXCL1 and thus recruits MDSC. G9a inhibits LCB 3II transcription and promotes immune escape. SUV39H1, G9a and EZH2 inhibit CXCL10 and CXCL9 transcription and reduce T cell recruitment. Meanwhile inhibit Fas transcription and curb Fas-FasL signaling pathway activation. EZH2 and SETDB1 repress MHC-II and MHC-I to affect antigen recognition. KDM2A and G9a directly repress the initiation of PD-L1 transcription. EZH2 and SETD2 inhibit the dsRNA-cGAS-STING pathway in the cytoplasm affecting PD-L1 transcription. In addition, EZH2 upregulates IRF1 to inhibit PD-L1 transcription. SETD2 downregulates FBW7 to inhibit PD-L1 expression. SUV39H1 inhibits SMAD3 in the cytoplasm and forms immunosuppression. EZH2 promotes FOXP3 transcription and Treg cell suppressor function. In contrast, DOTIL is the opposite. In T cells, EZH2 upregulates IL-2,TNF-αand INF-γ by promoting Fbxw7 and Numb activation of the Notch pathway, a process inhibited by miR-101 and miR-26a antagonism. SUV39H1 and DOTIL suppress the expression of immune factors. Black line represents promotion, red line represents inhibition, and dashed line represents physiological function.
EZH2
The Zeste homology 2 (EZH2) is responsible for modifying the lysine methylation of histone 3 (H3K27me3) to silence the gene (61). Previous studies have shown that EZH2 participates in malignant biological phenotypes such as the cell cycle, proliferation, invasion and metastasis actin, which is an important target for solid tumors and hematological tumors (62, 63). Moreover, several potential molecular mechanisms have revealed that EZH2 enrichment shapes the immunosuppressive tumor microenvironment. In tumor cells, EZH2 mutations down-regulate the expression of tumor antigens, thereby evading specific immune recognition by T cells. Major histocompatibility complex-I (MHC-I) acts as a potent marker for T cells to monitor tumors sensitively, and EZH2 suppresses its normal expression. Treatment with EPZ-6438 or EPZ-011989, EZH2 inhibitor, significantly depleted H3K27me3 and increased the expression of surface MHC-I protein (20, 21). In addition, studies have shown that the overexpression of EZH2 can inhibit programmed cell death protein 1 (PD-L1) in prostate cancer and hepatocellular carcinoma by enhancing the H3K27me3 level of the interferon regulatory factor 1(IRF1) transcription factor (22, 23). The use of EZH2 inhibitors (EPZ) activates the STING stress response to promote INF-γ-induced PD-L1 expression. Furthermore, EZH2 inhibitor combined with PD-1 treatment did not produce resistance or toxicity and had significant therapeutic effects (22).
EZH2 can also drive tumor cells to release certain mediators to affect the transport and activity of immune cells. LOXL4 is an important chemical inducer of macrophages. It was reported that EZH2 regulates macrophage activation through the miR-29b/miR-30d-LOXL4 axis and enhances tumor-associated macrophage (TAM) infiltration in breast cancer (24). In glioblastoma multiforme (GBM), iNOS and TGF-β2 can impaire engulfing and viability of macrophages (25). The number of infiltrating cells and the lethality of T cells represent the improved anticancer immunity of the body. Genome-wide studies showed that EZH2 levels are negatively correlated with CD8+ T cells, mainly inhibiting the production of tumor TH1-type chemokines CXCL9 and CXCL10 and thus reducing the recruitment of T cells (22, 26, 27), while the binding of carboxyl structure of ARID1A to EZH2 can reverse this step (28). Animal experiments have shown that the synergistic treatment of ovarian cancer with GSK126 (EZH2 inhibitor) and DNMT inhibitor improves the therapeutic efficacy of anti-PD-L1 therapy and overt T-cell therapy (27). In additional, the use of CPI-1205 (EZH2 inhibitor) in a mouse colorectal cancer tumor (MC38) model had a synergistic effect on the immunotherapeutic modality (29). Meanwhile, the activity of EZH2 in Treg cells maintains the stability of FOXP3 protein, increases the number of tumor-infiltrating FOXP3+ Tregs, alters the homeostatic balance with tumor effector T cells in the microenvironment and impairs the anti-tumor immune response (29). In contrast, EZH2 in CD8+ T cell can activate the Notch pathway, promote the release of cytokines in T cells, and maintain its good antineoplastic activity (30). Moreover, EZH2 is also involved in genome remodeling related to T-cell failure and promotes functional recovery (31). However, the tumor microenvironment can limit the conversion of oxidative phosphorylation to aerobic glycolysis by maintaining high expression of microRNA101 and microRNA26a, and limit the expression of EZH2 in T cells by controlling glucose metabolism. This hinders the normal expression of multifunctional cytokines (30). In overview, EZH2 has an important regulatory role on immune microenvironment components. Several clinical trials are currently recruiting to test the CPI-1205 or tazemetostat (an EZH2-targeted agent) in combination with Pembrolizumab in solid tumors (NCT03854474 and NCT03337698).
G9a
G9a (Euchromatic histone-lysine N-methyltransferase 2, EHMT2) is frequently upregulated in different types of cancer (64). G9a overexpression enhances H3K9me2 deposition, silencing and inhibiting tumor suppressor genes, and promoting tumor proliferation and migration through the Wnt pathway and epithelial-to-mesenchymal transformation (EMT), which can be a useful target for anticancer therapy (65). Notably, the special effects of G9a and the tumor microenvironment (TME) may explain the poor immunogenicity in specific cancers. For example, G9a is inversely associated with CD8+ T cell infiltration in melanoma and colon cancer. Moreover, it can inhibit the activated of Th1 cytokines/chemokines (32, 33). Further investigation revealed that Ga9 induces chromatin variability in chemokine-related genes, involved in homing of intratumoral effector lymphocytes and natural killer cells (34). In clinical cases, immunohistochemistry showed high intensity of G9a staining in 12 melanoma patients who did not respond to anti-PD-1 or anti-CTLA-4 treatment. Mouse melanoma resistance models treated with UNC0642 (a G9a inhibitor) in combination with anti-PD-1 therapy significantly reduced H3K9 levels in the LC3B II promoter region activating cellular autophagic responses and increasing PD-L1 levels, enhancing the blockade response to PD-1 immune checkpoint inhibitors (35).
G9a can also influence the methylation levels of multiple activated molecules of immune-related pathways. A previous study showed that G9a enhances H3K9me3 enrichment in the Fas promoter, restricts Fas-fasL release signals, and inhibits the tumor immune surveillance of host T cells (36). Moreover, in hepatocellular carcinoma (HCC), G9a silences SLC7A2 expression to induce CXCL1, promoting the recruitment of bone marrow-derived suppressor cells (MDSC) to the microenvironment (37). Given the above regulatory mechanisms, inhibition of G9a can remodel active tumor antigens and substantially modulate the tumor immune microenvironment. The combination of G9a inhibitors and immunotherapy strategies may be able to convert some “cold” immune tumors into “hot” tumors to achieve good immunotherapeutic results.
SETDB1
The Forked histone lysine methyltransferase 1 (SETDB1) containing the SET domain is responsible for the di-and trimethylation of the H3K9 residues. It is abnormally amplified and overexpressed in tumors (66). Yet, the underlying mechanisms of SETD2 gene mutations or loss of function leading to the corresponding dysfunction of tumor tissue proteins remain largely unexplored. Animal experiments showed that accumulation of SETDB1 mutations downregulates MHC-I-associated antigen presentation, thus preventing CD8+ T from correctly recognizing tumor cells and affecting sensitivity to PD-1/CTLA-4 treatment (38). On the other hand, SETDB1 in tumor cells forms a complex with TRIM28 or acts together with KDM5B that interferes with PD-L1 expression by blocking double-stranded RNA (dsRNA) production through the endogenous retroviral (ERV) pathway (39, 40). The loss of the SETDB1 gene also triggers type I interferon-induced PD-L1 expression through the cyclic GMP–AMP synthase (cGAS)–stimulator of interferon genes (STING) pathway and enhances anti-PD-L1 immune checkpoint blockade for antitumor effects (39–42). cGAS-STING pathway, an important pathway regulating host innate immunity, has been successively validated in various tumor models where SETD2 is an important epigenetic regulator. Thus, SETD2 is an attractive target for promoting immunotherapeutic responses.
SUV39H1
The variant suppressor 39 homolog 1 (SUV39H1), also known as KMT1A, is responsible for the introduction of the dimethylation and trimethylation of histone 3 lysine 9 (H3K9me3) (67). It mainly disrupts some important gene regulatory elements in tumor cells and reduces the sensitivity to immune response. In cervical cancer, SMAD3 is a key mediator of activation of multiple immune signaling pathways. SUV39H1 negatively regulates DNMT1 and reduces the direct binding of DNMT1 to the promoter region of the SMAD3 gene, thus inhibiting the activation of signaling by multiple downstream immune signaling pathways (43). In colon cancer, SUV39H1 negatively regulates Fas transcription and impairs the sensitivity of tumor cells to CTL Fas L-mediated cytotoxicity (35). More importantly, SUV39H1 has a non-negligible role in the dysfunction of tumor-infiltrating cells (CTL). It deprives effector T cells of their long-term memory reprogramming capacity (44) and induces SMAD2/3 inhibition of T cells to produce IL-2-mediated immune modulation (45). In conclusion, the inhibition of tumor cell gene expression by SUV39H1 under pathological conditions and its central role in suppressing the killing and memory functions of effector T cells provide new evidence in support of its effectiveness.
SETD2
SETD2 is the only human gene responsible for the trimethylation of histone H3 lysine 36 (H3K36me3) that interacts with RNA polymerase II (68, 69). Although there is clear evidence that SETD2 is abnormally expressed in various tumors, its causal relationship with tumorigenesis is still unclear. In the analysis of clinical sample, mutations in SETD2 led to the enrichment of tumor cell surface mutation-specific neoantigens, such as mutational load (TMB) microsatellite instability-high (dMMR/MSI-H). In addition, these patients with SETD2 mutated cancer were accompanied by transcriptional upregulation of genes associated with immune activity (46). Another clinical analysis of lung adenocarcinoma found many SETD2 gene mutations and significantly higher IFN-γ expression in the PD-L1 high-expression group (47). Furthermore, an experimental study in renal cell carcinoma found that SETD2 acts as a transcription factor regulating E3 ubiquitin ligase FBW7 target gene expression, causing altered PD-L1 expression levels and promoting CD4+ and CD8+ T cell infiltration and enhancing the anti-tumor effects of PD-1 antibodies (48). Based on the above studies, mutations in SETD2 are significantly correlated with tumor immune-specific genes and can drive tumor immunophenotypic alterations. However, extensive experimental studies are still needed to identify specific regulatory mechanisms of SETD2 on immune-related factors, which could provide new insights into the heterogeneous immune treatment of individual tumor patients.
KMT2 family
The histone-lysine N-methyltransferase 2 (KMT2) family of proteins is one of the most common mutations in human genome and confers the key functions of chromatin modifiability and DNA accessibility by modifying lysine 4 (H3K4) in the H3 tail of histone H3 (70). The current anti-tumor effects involving the KMT2 family are mainly focused on investigating immune checkpoints. In pancreatic cancer, inhibition of MLL1(KMT2A)activity or silencing expression reduces H3K4me3 levels in the CD274 promoter region and downregulates PD-L1 expression. Moreover, a KMT2A inhibitor combined with anti-PD-L1 or anti-PD-1 antibodies can effectively restrain the growth of a mouse model of pancreatic tumor in a Fas L- and CTL-dependent manner (49). Also, KMT2D is the main mutated gene in PD-L1-positive patients with hepatocellular carcinoma, whose large accumulation may lead to the ineffective response of PD-1 reagents (50). Frequent mutations in KMT2D have also been observed in non-small-cell carcinomas, along with mutations in TP53 (51). The response to immune checkpoint inhibitor (ICI) therapy is mainly influenced by intracellular tumor factors (e.g., tumor mutational load and microsatellite instability) and the tumor microenvironment. In an analysis of the immune assessment of ICI-treated patients through the Biocredit database, KMT2D was identified to have a critical role in a variety of tumor such as bladder cancer (52), esophageal cancer (53), gastric adenocarcinoma (54), lymphoma (56), and head and neck cancer (57, 71). These findings confirm that the KMT2 family is one of the drivers of immune escape. Alterations in its family-related genes may serve as predictive biomarkers for immunotherapy and help us to understand the prognostic effect of immune checkpoint therapy.
DOT1L
DOT1L (telomere silencing interference; also known as KMT4), which mainly catalyzes the methylation of H3K79, leads to gene mutations and impairs the interaction between Sir2 and Sir3 in the telomeric region (71). Inhibition of its catalytic activity has been widely used in cancer therapy. Recent studies have suggested that DOT1L is a central player in CD8+ T cell physiology, ensuring the activation of normal T cell receptor signaling and related signaling pathways that control CD8+ T cell differentiation. In the CD4-CRE transgenic mouse model, deletion of the DOT1L gene inhibited CD8+ Tcells apoptosis, as well as TNF and INF-γ expression. Furthermore, inhibition of DOT1L increased the threshold for TCR activation in T cells (58). Another study suggested that the loss of DOT1L directly impairs TCR/CD3 expression, resulting in an impaired immune response (59). Furthermore, DOT1L controls the subset differentiation of Foxp3+ regulatory T cells during carcinogenesis, reducing local inflammatory production in the microenvironment (60). The above results suggest that DOT1L is an important epigenetic target for regulating allogeneic T-cell responses, affecting the amount of immune cell infiltration, the direction of cell differentiation, and the secretion of immunomodulatory factors.
Protein arginine methyltransferase (PRMT) and tumor immunity
As a common post-translational modification, PRMT can catalyze the transfer of methyl groups from S-adenosine methionine (AdoMet) to the guanidine nitrogen atom of arginine. It can also affect the methylation status of the cancer genome, leading to activation or inhibitory recruitment of transcriptional mechanisms that are dysregulated in most tumors (72). In recent years, the development of PRMT-targeted drugs has been widely used in cancer therapy. Considering that PRMT1, PRMT4, and PRMT5 have the highest expression in cancer, their immunosuppressive effect have been well investigated (Table 2) (Figure 2).
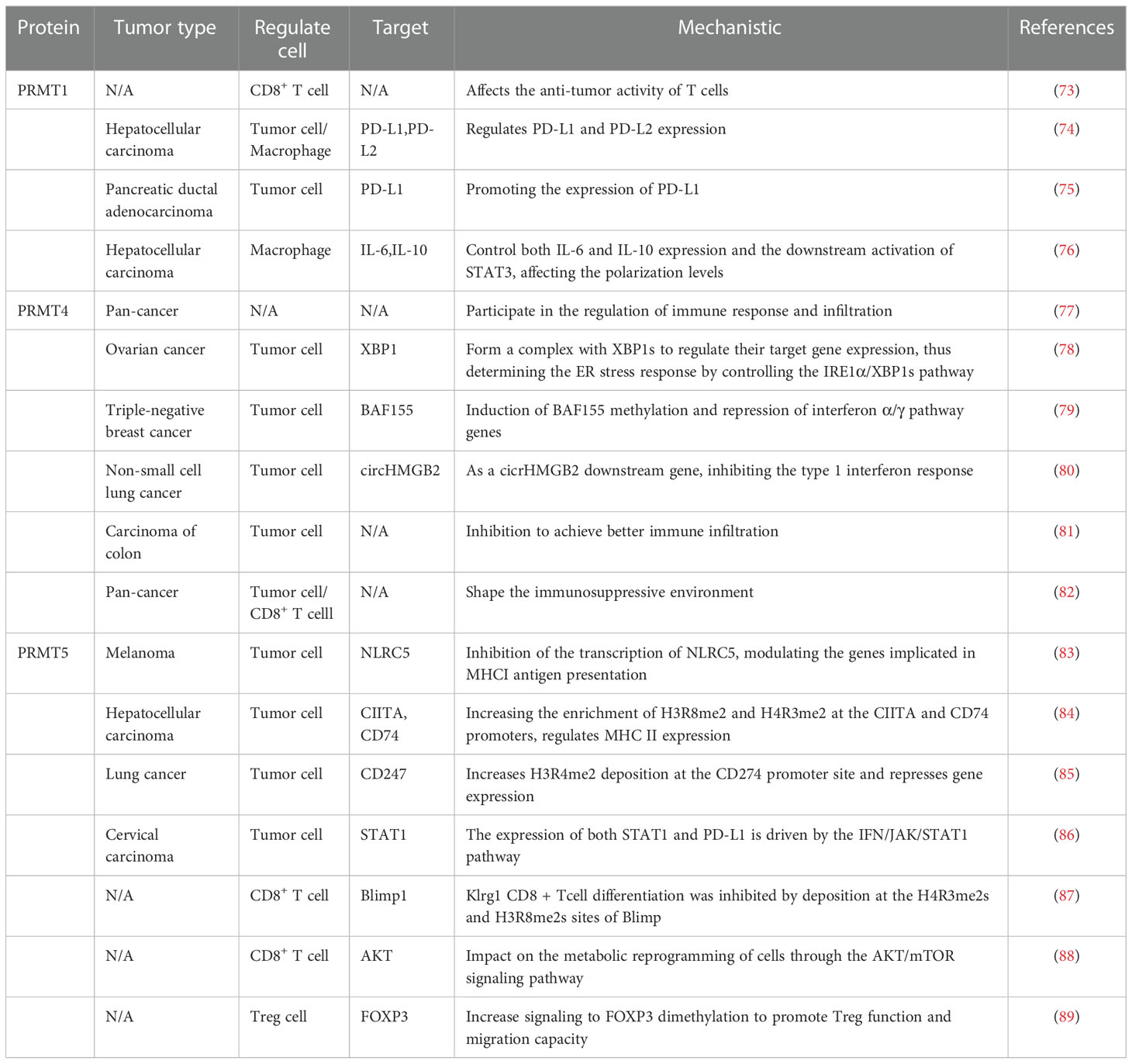
Table 2 Related functions of arginine methylase and tumor immunity.
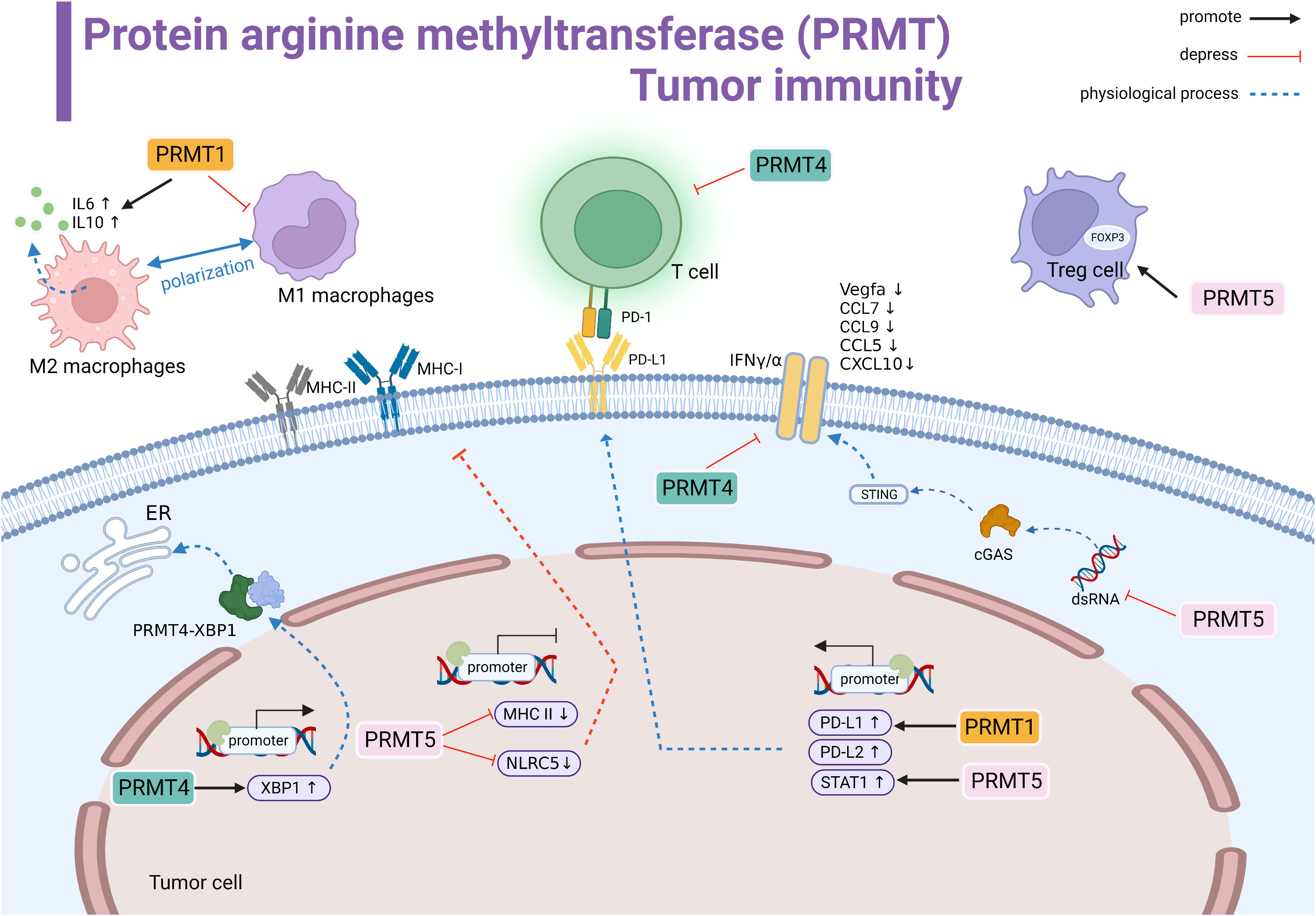
Figure 2 Protein arginine methyltransferase (PRMT) involved in tumor immune summary. PRMT1 regulates M2 macrophage polarization and promotes the transcription of IL-6 and IL-10. PRMT1 promotes the transcriptional level of PD-L1. PRMT4 negatively regulates T cells. PRMT4 promotes the transcription of XBP1 and forms the PRMT4-XBP1 complex to activate the endoplasmic reticulum stress pathway. PRMT5 inhibits the transcription of MHCI and MHCII and suppresses antigen recognition. PRMT5 promotes STAT1 expression to promote PD-L1 expression levels. In the cytoplasm, PRMT5 inhibits the dsRNA-cGAS-STING pathway, downregulates the interferon pathway, and downstream genes Vegfa, CCL7, CCL9, CCL5, and CCL10 expression are suppressed.PRMT4 inhibits IFNγ/α.PRMT5 promotes the immunosuppressive function of Foxp3 regulatory T cells. Black line indicates promotion, red line indicates suppression, and dashed line indicates physiological function.
PRMT1
Protein arginine methyltransferase 1 (PRMT1) is the main type I PRMT. Many experimental studies have shown that PRMT1 is overexpression or has an shear state in many cancer types (90). Using a genome-wide CRISPR immune screening system to screen for tumor-intrinsic factors that modulate tumor cell sensitivity to T cell-mediated killing, Hou J et al. identified PRMT1 as an intrinsic factor affecting T cell transport and lethality. The possible mechanism is the altered RNA levels of the cytokines/chemokines (73). In some tumor types, PRMT1 is an important regulator of the immune checkpoint pathway. In human hepatocellular carcinoma (HCC), PRMT1 expression is positively correlated with both PD-L1 and PD-L2 immune checkpoint expression (74). Similarly, PT1001B (PRMT1 inhibitor) enhances antitumor immunity by inhibiting PD-L1 expression on tumor cells, upregulating tumor-infiltrating CD8+ T lymphocytes. When the anti-PD-L1 monoclonal antibody was combined with PT1001B, the proportion of tumor-infiltrating effector cells was significantly increased in mice, and resistance to anti-PD-L1 treatment was well reversed (75). In addition, PRMT1 can protect the tumor cells, which can induce macrophages to assist in immune escape. Inhibition of PRMT1 in mice led to the inhibition of IL6 signaling and downstream STAT3 activation and decreased the number of tumor cells and M2 type macrophages (76). Taken together, these studies suggested that effective inhibition of PRMT1 can control T cell-mediated tumor killing and can effectively remodel the tumor immune microenvironment.
PRMT4
Protein arginine methyltransferase 4 (PRMT4), also known as coactivator-associated arginine methyltransferase 1 (CARM1), has a carcinogenic role in human cancer and is closely involved in the process of tumor growth and immune tolerance (91). CARM1 is overexpressed in different tumors and negatively associated with CD8+ T cells. It can also be used as a potent biomarker for pan-cancer prediction (77). In ovarian cancer, CARM1 acts as a transcriptional activator to promote XBP1 target gene expression. CARM1 and interacts with XBP1 to modulatie the ER stress response in the IRE1α/XBP1 pathway, triggering an immunosuppressive environment (78). Furthermore, CARM1 mainly targets BAF155 in triple-negative breast cancer by inhibiting the interferon pathway to inhibit the host immune response (79). Similarly, CARM1 is positively regulated by circHMGB2, which inhibits type I interferon responses and downstream genes. EZM2302 (a CARM1 inhibitor) and anti-PD-1 antibody significantly inhibited the immunosuppressive environment in vivo shaped by tumor growth in mice and reduced the efficacy of anti-PD-1 monotherapy in non-small cell lung cancer (80). In a mouse colon cancer model, inhibitors targeting CARM1 were effective in arresting solid tumor progression and enhancing immune infiltration (81). In addition, the inactivation of the CARM1 gene in T cells can increase the number of specific memory-like T cell populations in the microenvironment, allowing the body to maintain a continuous and effective immune attack against tumors. EZM2302 (CARM1) enhances the checkpoint blockade sensitivity of CTLA-4 mAb in a synergistic manner (82). Overall, the inhibition of the activity against CARM1 suppresses tumor progression, promotes T-cell infiltration and sustained immune memory, and may be an effective for immunotherapy of drug-resistant tumors.
PRMT5
PRMT5 is the major type II arginine methyltransferase, active in a variety of cellular activities, that achieve tumor-promoting effects through methylation-mediated transcription repression, including inhibition of normal expression of the tumor surface antigen proteins in different tumor types (92). For example, in melanoma, PRMT5 activity inhibits NLRC5 transcription and changes the regulation of the expression of genes involved in the presentation of the major histocompatibility complex class I (MHCI) antigen. Meanwhile, PRMT5 interfere with the dsRNA-cGAS-STING pathway to affect type I interferon responses, promoting immune escape (83). In addition, inhibition of PRMT5 promotes the expression of MHC II (84). Treatment with GSK3326595 (PRMT5 inhibitor) plus anti-PD-1 antibody enhanced the anti-tumor response in the mouse organism (83, 84). Thus, targeting PRMT5 may synergize with immune checkpoint therapy to improve therapeutic efficacy. PD-L1 is a key molecule highly expressed in tumor cells that interacts with immune cells to constitute an immunosuppressive environment. In lung cancer, GSK591 drug inhibits PRMT5-induced PD-L1 expression, which then trigger immune resistance (85). Thus, the combination with PD-1 treatment and inhibition and elimination of PRMT5 may promote synergistic inhibition. In contrast, in cervical cancer, PRMT5 promotes cancer progression by increasing the expression of histone H3R2 symmetric dimethylation (H3R2me2s), which is enriched in the promoter region of STAT1 to enhance transcription and drive up-regulation of PD-L1 expression (86).
Furthermore, PRMT5 also acts directly on the host immune cells to maintain cellular physiology and homeostasis, especially on the effector CD8+ T cells. PRMT5 can affect the deposition of H4R3me2s and H3R8me2s at the Blimp1 locus and force the differentiation of transient effector CD8+ T cells, resulting in a substantial loss of CD8+ T cell numbers and function (87). Inhibition of PRMT5 is a “double-edged sword”, its inhibition causes reduced AKT/mTOR signaling, which impairs glycolysis and increases fatty acid utilization after human CD8+ Tcells’stimulation leading to metabolic reprogramming (88). In addition, PRMT5 can interact with the FOXP3 transcription factor in Tregs to maintain the functional stabilization of Treg cells (89). In conclusion, given the selective role of PRMT5 in the tumor microenvironment, more attention should be paid to the mechanism of side effects in immune cells, and combined immunotherapy may maximize the efficacy.
Classification and biological functions of histone demethylases(HDMs)
With the progress of science and technology, almost all histone lysine methylation sites have been found to be reversible. To date, two classes of histone demethylases have been identified, mainly the lysine-specific demethylase-1 (LSD1) family and the jumonji (JmjC) domain-containing family (93). LSD1, which was identified first acts only on monomethylated and dimethylated lysines (94). The JmjC family is another class of JmjC domain-containing Fe (II). Ketoglutarate-dependent enzymes are divided into different species according to the sequence homology of the JmjC domain and the overall structure of the related motifs. Thus far, those active against H3K4, H3K9, H3K27, H3K36, and H4K20 have been identified (95). Their special structure allows them to function together with many other biological macromolecules (96).
Histone demethylases do not change the DNA sequence, and dynamically regulate in specific chromatin regions. They are important regulators of the physiological functions of embryonic development, gene regulation, cell reprogramming and other physiological functions, and they maintain genome integrity and epigenetic stability (97). Their role in cancer is particularly important, and it is closely related to the pathogenesis of the disease, including the demethylation of the oncogenes/tumor suppressor genes for mastering the cell fate, the enrichment of transcription factors, gene copy number alterations, and increased mutations. Targeting partial demethylases opens up an emerging field for anticancer therapy. In this process, some enzymes also have a prominent role in regulating the immune microenvironment (Table 3) (Figure 3).
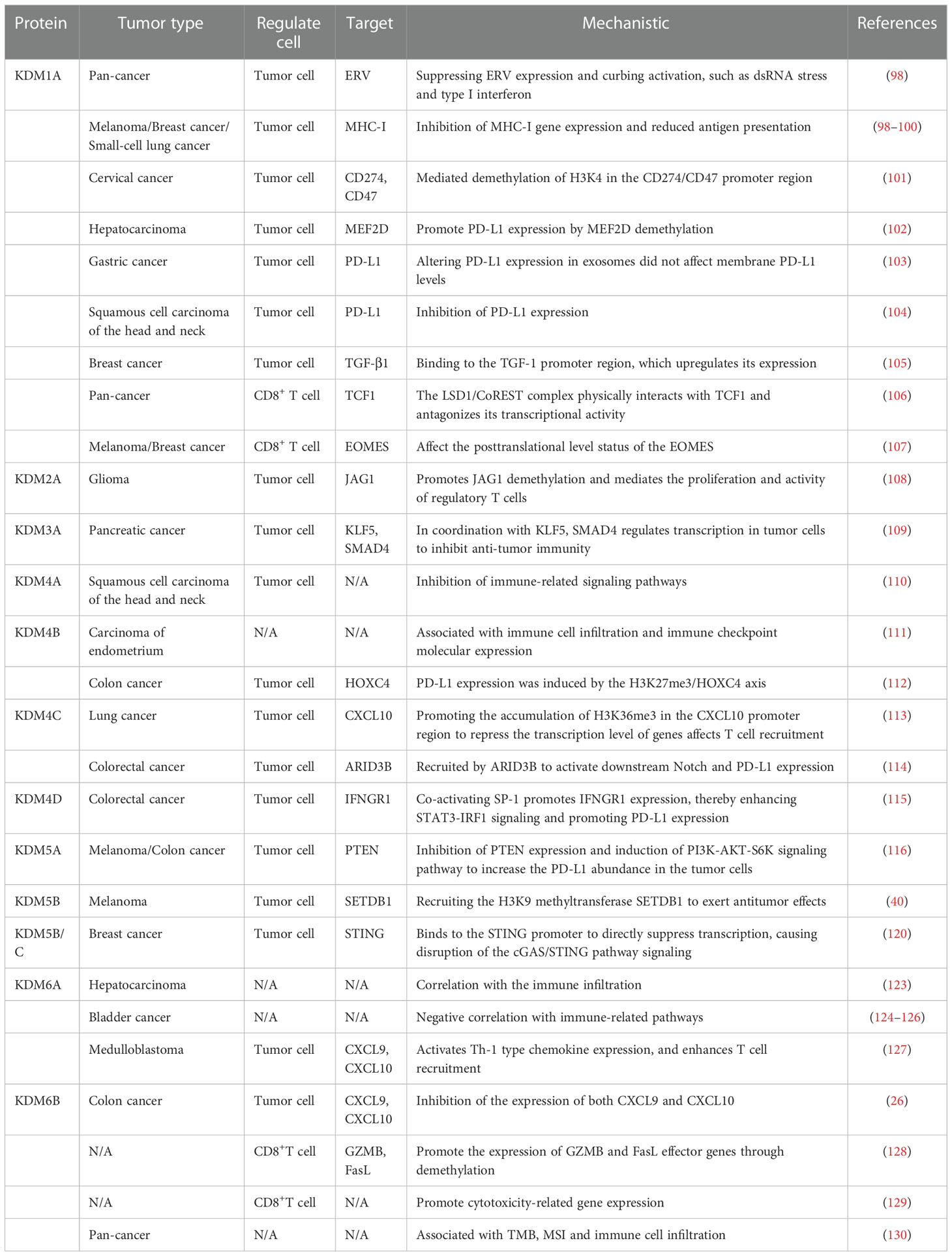
Table 3 Related functions of lysine demethylase and tumor immunity.
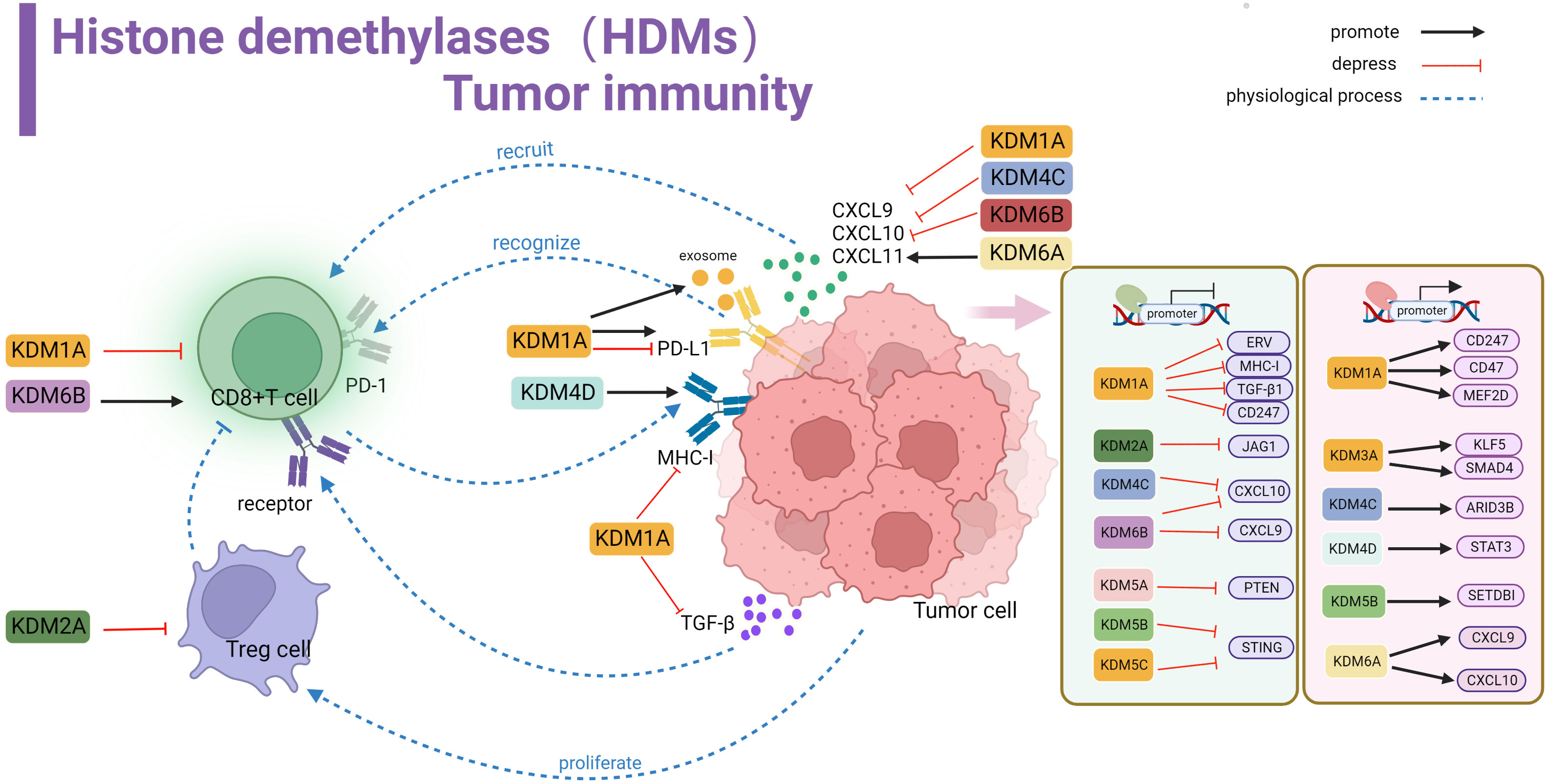
Figure 3 Histone demethylase(HDMs) involved in tumor immune summary. In tumor cells, KDM1A, KDM2A, KDM4C, KDM5A, KDM5B, KDM5C and KDM6B negatively regulate key genes and signaling pathways involved in stimulating T-cell anti-tumor immunity, including ERV, MHC-I, TGF-β, CD247, JAG1, CXCL10,9,STING and PTEN, affecting cellular KDM1A, KDM3A, KDM4C, KDM4D, KDM5B, KDM5A positively regulate related proteins involved in activating tumor surface antigens, including CD247,CD47 and other surface antigens, or by promoting MEF2D, KLFS, SMAD4, STAT3, ARID3B, SETDBI to promote or activate downstream KDM5A promotes CXCL9 and CXCL10 recruitment of T cells into the microenvironment. kdm1A inhibits TGF-β binding to T cell-associated receptors and suppresses MHC-I antigen expression. KDM1A promotes PD-L1 expression in exosomes. kdm1A and KDM6B affect T cell function. kdm2A alters the activity of regulatory T cells.
LSD1 and tumor immunity
Lysine-specific demethylase 1 (LSD1), also known as KDM1A, acts as an H3K4/9me eraser that binds to CoREST or nucleosome remodeling to repress gene transcription (131). LSD1 is highly expressed in most solid tumors, altering tumor immunogenicity and immune response by inhibiting or activating different signaling pathways. Shi et al. first discovered that inhibiting LSD1 can enhance endogenous transcription (EVR) expression, activate dsRNA stress and type I interferon activation, and improve the immunotherapy response of poorly immunogenic tumors (98). More importantly, LSD1 is inversely associated with CD8+ T cells in various tumors. In tumor cells, LSD1 largely affects the normal expression of MHC-I protein antigen by inhibiting the MHC-I encoding genes H2-D1 and H2-K2, which leads to the possibility that CD8+ T cells do not effectively recognize MHC-I prompting immune escape. The above mechanism has been observed in melanoma, breast cancer, and small-cell lung cancer (98–100)
Conclusions regarding the regulation of PD-L1 expression are inconsistent. In cervical cancer, LSD1 seems to be positively correlated with PD-L1 levels, in which H3K4me2 demethylation directly promoted the increase in PD-L1 expression (101). On the other hand, the demethylation of MEF2D in HCC indirectly promotes the PD-L1 expression, and this process is competitively inhibited by has-miR-329-3p (102). Moreover, in gastric cancer, LSD1 increases the level of PD-L1 found in exosomes and is transported to T-cell expression to inhibit tumor immunity (103). In contrast, LSD1 significantly suppresses the PD-L1 expression level in HNSCC (104). The surprising finding is that using the LSD1 inhibitor alone, despite its effective tumor suppression, the resulting exogenous TGF-1 binding to the CD8+ T cell surface receptors inhibits the cytotoxic effects (105), which may be one of the reasons why the clinical effects of LSD1 inhibitors are suboptimal. Alternatively, LSD1 performs an epigenetic program within CD8+ T cells. On the one hand, it inhibits the transcription of the progenitor phenotype gene TCF1, disrupting the progenitor cell population (106). On the other hand, eomesodermin (EOMES), a transcription factor associated with the regulation of T cell failure, promotes T cell dysfunction (107). These make T cell depletion fast and unsustained recovery, resulting in poor persistence of PD-1 blocking therapy. Current experimental data suggest that treatment with LSD1 inhibitors (ORY-1001, SP2509 or GSK2879552) in combination with PD-1/PD-L1 monoclonal antibodies enhances in vivo immunogenicity and has a long-term response (101, 104, 106).
JmjC family and tumor immunity
KDM2
KDM2 is mainly responsible for the demethylation of the H3 lysine 36(H3K36) residues, and its family members include KDM2A and KDM2B (132). In glioma, LncRNA HOXA-AS2 promotes KDM2A expression by binding to miR-302a, thus recruiting H3K4me3 to demethylate JAG1 and promoting the proliferation and immune tolerance of regulatory T cells (108). In addition, KDM2A may promote immune body suppression Fumarate as an important metabolite may antagonize inhibitory histones and promote immune regulation (133, 134). In conclusion, KDM2 serves as a considerable therapeutic target.
KDM3
KDM3 is mainly composed of KDM3A, KDM3B, and KDM3C, which can specifically catalyze the demethylation of histone H3K9me1/2 (135). Using CRISPR screening in a mouse model of pancreatic cancer, KDM3A was found to be an epigenetic modulator of the response to immunotherapy. KDM3A mainly affects the KLF5 and SMAD4 transcription factor activity, regulates the epidermal growth factor receptor (EFFR) expression, and affects the T cell infiltration and the infiltration of dendritic cell DC (109). This suggests that KDM3A is closely related to the composition of the immune microenvironment. Therefore, eliminating KDM3A could help overcome immunotherapy resistance and enhance sensitivity to therapeutic effects, thereby creating a microenvironment for T-cell inflammation.
KDM4
The KDM4 protein family is composed of (KDM4A-C) and KDM4D, and several studies have found them to be overexpressed in cancer and to have the ability to malignant tumor growth (136). Notably, while maintaining tumor growth, they simultaneously suppress the activity of some pathways to interfere with normal immunosuppression. In HNSCC, the knockdown of KDM4A led to the activation of both types I IFN interferon signaling and DNA replication stress signal cGAS-STING, along with the significant upregulation of CXCL9, CXCL10, and CXCL11, and significantly increases the effect of the combined PD-1 blocking treatment (110). KDM4B is also recommended as a clinical prognostic marker and is closely associated with immune cell infiltration and immune checkpoint molecular expression (111). In colon cancer cell culture, KDM4B elevates HOXC4 expression by driving H3K27me3 demethylation to induce the expression of PD-L1, and exogenous miR-15a was able to prevent tumor escape events from occurring (112). Moreover, KDM4C is negatively associated with CD8+ T cells in lung cancer; transcription sequencing found that KDM4C mainly downregulates the transcript level of CXCL10 and inhibits T cell recruitment to tumors and killing (113). KDM4C is also involved in the regulation of PD-L1 expression, and the main mechanism is the transcriptional activation of the Notch gene and PD-L1 through ARID3B recruitment to regulate chromatin structure, whereas KDM4D promotes PD-L1 expression through the SP-1/STAT3/IRF1 signaling pathway, assisting the immune escape of in colorectal cancer (114, 115).
KDM5
The KDM5 protein family, including KDM5A-C and KDM5D, is responsible for removing histone H3 lysine 4 dimethylation and trimethylation (H3K4me2 and H3K4me3) (116). It is an attractive target in cancer therapy. Several prospective raw letter analyses have shown that KDM5 is closely associated with regulaing immune infiltration and expressing immune-related molecules, and is considered a prospective candidate for epigenetic anti-tumor therapy (117–119). In clinical treatment, some patients have low tumor cell PD-L1 abundance, so they cannot respond well to ICB. One study showed that increased KDM5A gene expression or protein abundance, promoting PD-L1 upregulation to accommodate the PD-1 treatment response, is a valuable clinical response tag (137). In melanoma, high expression of KDM5B can recruit the H3K9 methyltransferase SETDB1 to interact in the suppression of endogenous retrotransposable elements and block subsequent RNA and DNA sensing pathways as well as type I interferon responses, resulting in the inability of the organism to respond positively to tumor rejection and immune responses (40). A similar mechanism has been found in breast cancer. The STING promoter is directly transcriptionally repressed by KDM5B and KDM5C, disrupting the cGAS/STING pathway signaling and failing to activate a robust interferon response (120). Using KDM5 inhibitors reversed the normal transmission of this signaling pathway. It has also been suggested that combining of immunotherapy and KDM5 inhibitors could maximize the anti-tumor immune response, thus representing a potential therapeutic modality of interest.
KDM6
The KDM6 subfamily consists of three distinct members, i.e., KDM6A (also called UTX), KDM6B (also called JMJD3), and KDM6C (also called UTY), capable of removing di-and trimethylated H3K27, thereby activating or repressing target gene transcription (121). Its Function is highly dependent on the specific of the cell type pathological environment (122). The molecular basis of KDM6 in tumors is still in its infancy, and only a few studies have addressed this issue. Yet, several studies have shown a high correlation between its mutations and tumor immunity. A functional screen for lysine demethylase in HCC showed that KDM6A is closely associated with immune infiltration (123). In bladder cancer and its subtypes, KDM6A is a more frequently mutated gene, that negatively regulates the signaling pathways of the immune system and suppresses tumor immunity (124–126). In medulloblastoma, KDM6A activates the expression of Th1-type chemokines and promotes cell migration (127). Moreover, KDM6B inhibit CXCL9 and CXCL10 expression in colon cancer and exerts an anti-tumor immune effects (26). In contrast, the effect of KDM6B is positively regulated for CD8+ T cells. KDM6B can promote the differentiation of mature CD8+ T cells by demethylating the expression of GZMB and FasL (128). Inhibition of KDM6B resulted in reduced of toxicity-related genes in CD8+ T cells (129). Little experimental support exists for the specific mechanism of KDM6B in tumor progression and immune cell infiltration. However, available pan-cancer analyses suggest that KDM6B expression is associated with TMB, MSI and immune cell infiltration, and influences the response to immunotherapy and clinical outcome (130).
Conclusions and outlook
In the past decade, human cancer prevention and treatment have entered a new era with the emergence of immunotherapy. In the process of gradually understanding the potential mechanism of tumor cell occurrence and development, to the mechanism of killing malignant cells and avoiding the effect of the immune system, researchers have also developed corresponding therapeutic drugs for clinical practice, including immune checkpoint inhibitors, epigenetic targeted drugs, etc. Nevertheless, the low response rate and immune resistance in practical clinical applications led to identification of so-called “cold tumor”.
The concentrated research on histone methylation modifying enzymes in epigenetics advances our new understanding of “cold tumors” in human cancer, and builds the bridge between tumor cells and immune cells, promoting a deeper understanding of the complexity and diversity of the tumor immune microenvironment. Current studies on the involvement of histone methylase and demethylase in anti-tumor immunity mainly includes (1): regulation of tumor immunogenic antigen expression; (2) their influence on the activation of immune-related pathways; (3) regulation of expression of chemokines/cytokines and induced immune-related factors; (4) regulation of immune cells, including immune cell activation, immune cell depletion and functional remodeling, and immune memory. The above regulatory mechanisms provide a more comprehensive picture of the facilitative/suppressive immune microenvironment shaped by aberrant histone methylation modifications at the transcriptional and translational levels. Furthermore, the contribution of histone methylation modifications for tumor immune escape mechanism, immunotherapy tolerance mechanism, and immune stress has brought new perspectives and approaches for solving the “cold tumor” dilemma.
The above studies are still in their infancy but provide a solid theoretical basis for future preclinical and clinical development of combination therapies using epigenetic modulators and immunotherapeutic agents and show great potential. This will be a new therapeutic paradigm targeting improved and enhanced immune efficacy. We expect that based on the rapid development of immunogenomics, immunoproteomics, and immunobioinformatics, the complex structures in the tumor immune microenvironment will be revealed more comprehensively in the future. Together with the development of research on immune features in preclinical tumor models, this will greatly improve our understanding of the role of histone methylation in the immune microenvironment, facilitating clinical translation and the construction of precise therapeutic systems. Therefore, the development of this field is an important breakthrough to improve the efficacy of immunotherapy for the benefit of more patients. Based on the current research, we still need further studies to explore the role of histone methylation mutations in the regulation of immune resistance in different types of tumors. Meanwhile, the combination of single cell sequencing and spatial transcriptome sequencing will fully reveal the importance of histone methyl esterases in the tumor microenvironment, providing finer evidence to support the mechanism of epigenetic involvement in immune regulation. In addition, experimental models of combining multiple histone methylation modulators with immunotherapeutic agents will be developed, and rational and less toxic optimization protocols will be sought to advance clinical practice.
In conclusion, understanding the regulatory mechanisms of histone methylation modifying enzymes will improve immunotherapy.
Author contributions
YZ and JC collected relevant literature, prepared data, and drafted the manuscript. HL and RM participated in the design of this review. RH, FF and XX participated in the summary and drawing of tables and pictures. XL and JD made strict revisions to the manuscript. All authors read and approve the final draft.
Funding
This work was supported in part by the National Natural Science Foundation of China (81360366,81302169); Guizhou Science and Technology Plan Project (Foundation (2019) 1198, Foundation Foundation [2020] 1Z064).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Li B, Chan HL, Chen P. Immune checkpoint inhibitors: Basics and challenges. Curr Med Chem (2019) 26(17):3009–25. doi: 10.2174/0929867324666170804143706
2. Abreu TR, Fonseca NA, Gonçalves N, Moreira JN. Current challenges and emerging opportunities of CAR-T cell therapies. J Control Release. (2020) 319:246–61. doi: 10.1038/s41423-020-0488-6
3. Yarchoan M, Johnson BA 3rd, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer. (2017) 17(9):569. doi: 10.1038/nrc.2017.74
4. Haanen JB, Robert C. Immune checkpoint inhibitors. Prog Tumor Res (2015) 42:55–66. doi: 10.1159/000437178
5. Pansy K, Uhl B, Krstic J, Szmyra M, Fechter K, Santiso A, et al. Immune regulatory processes of the tumor microenvironment under malignant conditions. Int J Mol Sci (2021) 22(24):13311. doi: 10.3390/ijms222413311
6. Fiala O, Šorejs O, Šustr J, Fínek J. Side effects and efficacy of immunotherapy. nežádoucí účinky a efekt imunoterapie. Klin Onkol. (2020) 33(1):8–10. doi: 10.14735/amko20208
7. van den Bulk J, Verdegaal EM, de Miranda NF. Cancer immunotherapy: broadening the scope of targetable tumours. Open Biol (2018) 8(6):180037. doi: 10.1098/rsob.180037
8. Schoenfeld AJ, Hellmann MD. Acquired resistance to immune checkpoint inhibitors. Cancer Cell (2020) 37(4):443–55. doi: 10.1016/j.ccell.2020.03.017
9. Filipp FV. Crosstalk between epigenetics and metabolism-yin and yang of histone demethylases and methyltransferases in cancer. Brief Funct Genomics (2017) 16(6):320–5. doi: 10.1093/bfgp/elx001
10. Tian X, Zhang S, Liu HM, Zhang YB, Blair CA, Mercola D, et al. Histone lysine-specific methyltransferases and demethylases in carcinogenesis: New targets for cancer therapy and prevention. Curr Cancer Drug Targets. (2013) 13(5):558–79. doi: 10.2174/1568009611313050007
11. Tost J, Gay S, Firestein G. Epigenetics of the immune system and alterations in inflammation and autoimmunity. Epigenomics. (2017) 9(4):371–3. doi: 10.2217/epi-2017-0026
12. Li B, Severson E, Pignon JC, Zhao H, Li T, Novak J, et al. Comprehensive analyses of tumor immunity: Implications for cancer immunotherapy. Genome Biol (2016) 17(1):174. doi: 10.1186/s13059-016-1028-7
13. Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, et al. Tumor cell-intrinsic factors underlie heterogeneity of immune cell infiltration and response to immunotherapy. Immunity. (2018) 49(1):178–193.e7. doi: 10.1016/j.immuni.2018.06.006
14. Jenuwein T, Allis CD. Translating the histone code. Science. (2001) 293(5532):1074–80. doi: 10.1126/science.1063127
15. Peterson CL, Laniel MA. Histones and histone modifications. Curr Biol (2004) 14(14):R546–51. doi: 10.1016/j.cub.2004.07.007
16. Schneider R, Bannister AJ, Kouzarides T. Unsafe SETs: Histone lysine methyltransferases and cancer. Trends Biochem Sci (2002) 27(8):396–402. doi: 10.1016/s0968-0004(02)02141-2
17. Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer. (2013) 13(1):37–50. doi: 10.1038/nrc3409
18. Moore KE, Gozani O. An unexpected journey: Lysine methylation across the proteome. Biochim Biophys Acta (2014) 1839(12):1395–403. doi: 10.1016/j.bbagrm.2014.02.008
19. Shen C, Vakoc CR. Gain-of-function mutation of chromatin regulators as a tumorigenic mechanism and an opportunity for therapeutic intervention. Curr Opin Oncol (2015) 27(1):57–63. doi: 10.1097/CCO.0000000000000151
20. Ennishi D, Takata K, Béguelin W, Duns G, Mottok A, Farinha P, et al. Molecular and genetic characterization of MHC deficiency identifies EZH2 as therapeutic target for enhancing immune recognition. Cancer Discovery (2019) 9(4):546–63. doi: 10.1158/2159-8290.CD-18-1090
21. Burr ML, Sparbier CE, Chan KL, Chan YC, Kersbergen A, Lam EYN, et al. An evolutionarily conserved function of polycomb silences the MHC class I antigen presentation pathway and enables immune evasion in cancer. Cancer Cell (2019) 36(4):385–401.e8. doi: 10.1016/j.ccell.2019.08.008
22. Morel KL, Sheahan AV, Burkhart DL, Baca SC, Boufaied N, Liu Y, et al. EZH2 inhibition activates a dsRNA-STING-interferon stress axis that potentiates response to PD-1 checkpoint blockade in prostate cancer. Nat Cancer. (2021) 2(4):444–56. doi: 10.1038/s43018-021-00185-w
23. Xiao G, Jin LL, Liu CQ, Wang YC, Meng YM, Zhou ZG, et al. EZH2 negatively regulates PD-L1 expression in hepatocellular carcinoma. J Immunother Cancer. (2019) 7(1):300. doi: 10.1186/s40425-019-0784-9
24. Yin H, Wang Y, Wu Y, Zhang X, Zhang X, Liu J, et al. EZH2-mediated epigenetic silencing of miR-29/miR-30 targets LOXL4 and contributes to tumorigenesis, metastasis, and immune microenvironment remodeling in breast cancer. Theranostics. (2020) 10(19):8494–512. doi: 10.7150/thno.44849
25. Yin Y, Qiu S, Li X, Huang B, Xu Y, Peng Y. EZH2 suppression in glioblastoma shifts microglia toward M1 phenotype in tumor microenvironment. J Neuroinflammation. (2017) 14(1):220. doi: 10.1186/s12974-017-0993-4
26. Nagarsheth N, Peng D, Kryczek I, Wu K, Li W, Zhao E, et al. PRC2 epigenetically silences Th1-type chemokines to suppress effector T-cell trafficking in colon cancer. Cancer Res (2016) 76(2):275–82. doi: 10.1158/0008-5472.CAN-15-1938
27. Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature. (2015) 527(7577):249–53. doi: 10.1038/nature15520
28. Li J, Wang W, Zhang Y, Cieślik M, Guo J, Tan M, et al. Epigenetic driver mutations in ARID1A shape cancer immune phenotype and immunotherapy. J Clin Invest. (2020) 130(5):2712–26. doi: 10.1016/j.celrep.2018.05.050
29. Wang D, Quiros J, Mahuron K, Pai CC, Ranzani V, Young A, et al. Targeting EZH2 reprograms intratumoral regulatory T cells to enhance cancer immunity. Cell Rep (2018) 23(11):3262–74. doi: 10.1016/j.celrep.2018.05.050
30. Zhao E, Maj T, Kryczek I, Li W, Wu K, Zhao L, et al. Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat Immunol (2016) 17(1):95–103. doi: 10.1038/ni.3313
31. Weber EW, Parker KR, Sotillo E, Lynn RC, Anbunathan H, Lattin J, et al. Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science. (2021) 372(6537):eaba1786. doi: 10.1126/science.aba1786
32. Kato S, Weng QY, Insco ML, Chen KY, Muralidhar S, Pozniak J, et al. Gain-of-Function genetic alterations of G9a drive oncogenesis. Cancer Discovery (2020) 10(7):980–97. doi: 10.1158/2159-8290.CD-19-0532
33. Seier JA, Reinhardt J, Saraf K, Ng SS, Layer JP, Corvino D, et al. Druggable epigenetic suppression of interferon-induced chemokine expression linked to MYCN amplification in neuroblastoma. J Immunother Cancer. (2021) 9(5):e001335. doi: 10.1136/jitc-2020-001335
34. Spiliopoulou P, Spear S, Mirza H, Garner I, McGarry L, Grundland-Freile F, et al. Dual G9A/EZH2 inhibition stimulates antitumor immune response in ovarian high-grade serous carcinoma. Mol Cancer Ther (2022) 21(4):522–34. doi: 10.1158/1078-0432.CCR-20-3463
35. Kelly GM, Al-Ejeh F, McCuaig R, Casciello F, Ahmad Kamal N, Ferguson B, et al. G9a inhibition enhances checkpoint inhibitor blockade response in melanoma. Clin Cancer Res (2021) 27(9):2624–35. doi: 10.1158/1078-0432.CCR-20-3463
36. Paschall AV, Yang D, Lu C, Choi JH, Li X, Liu F, et al. H3K9 trimethylation silences fas expression to confer colon carcinoma immune escape and 5-fluorouracil chemoresistance. J Immunol (2015) 195(4):1868–82. doi: 10.4049/jimmunol.1402243
37. Xia S, Wu J, Zhou W, Zhang M, Zhao K, Liu J, et al. SLC7A2 deficiency promotes hepatocellular carcinoma progression by enhancing recruitment of myeloid-derived suppressors cells. Cell Death Dis (2021) 12(6):570. doi: 10.1038/s41419-021-03853-y
38. Griffin GK, Wu J, Iracheta-Vellve A, Patti JC, Hsu J, Davis T, et al. Epigenetic silencing by SETDB1 suppresses tumour intrinsic immunogenicity. Nature. (2021) 595(7866):309–14. doi: 10.1038/s41586-021-03520-4
39. Lin J, Guo D, Liu H, Zhou W, Wang C, Müller I, et al. The SETDB1-TRIM28 complex suppresses antitumor immunity. Cancer Immunol Res (2021) 9(12):1413–24. doi: 10.1158/2326-6066.CIR-21-0754
40. Zhang SM, Cai WL, Liu X, Thakral D, Luo J, Chan LH, et al. KDM5B promotes immune evasion by recruiting SETDB1 to silence retroelements. Nature. (2021) 598(7882):682–7. doi: 10.1038/s41586-021-03994-2
41. Guo E, Xiao R, Wu Y, Lu F, Liu C, Yang B, et al. WEE1 inhibition induces anti-tumor immunity by activating ERV and the dsRNA pathway. J Exp Med (2022) 219(1):e20210789. doi: 10.1084/jem.20210789
42. Cuellar TL, Herzner AM, Zhang X, Goyal Y, Watanabe C, Friedman BA, et al. Silencing of retrotransposons by SETDB1 inhibits the interferon response in acute myeloid leukemia. J Cell Biol (2017) 216(11):3535–49. doi: 10.1083/jcb.201612160
43. Zhang L, Tian S, Zhao M, Yang T, Quan S, Song L, et al. SUV39H1-mediated DNMT1 is involved in the epigenetic regulation of Smad3 in cervical cancer. Anticancer Agents Med Chem (2021) 21(6):756–65. doi: 10.2174/1871520620666200721110016
44. Pace L, Goudot C, Zueva E, Gueguen P, Burgdorf N, Waterfall JJ, et al. The epigenetic control of stemness in CD8+ T cell fate commitment. Science. (2018) 359(6372):177–86. doi: 10.1126/science.aah6499
45. Wakabayashi Y, Tamiya T, Takada I, Fukaya T, Sugiyama Y, Inoue N, et al. Histone 3 lysine 9 (H3K9) methyltransferase recruitment to the interleukin-2 (IL-2) promoter is a mechanism of suppression of IL-2 transcription by the transforming growth factor-β-Smad pathway. J Biol Chem (2011) 286(41):35456–65. doi: 10.1074/jbc.M111.236794
46. Lu M, Zhao B, Liu M, Wu L, Li Y, Zhai Y, et al. Pan-cancer analysis of SETD2 mutation and its association with the efficacy of immunotherapy. NPJ Precis Oncol (2021) 5(1):51. doi: 10.1038/s41698-021-00193-0
47. Li K, Liu J, Wu L, Xiao Y, Li J, Du H, et al. Genomic correlates of programmed cell death ligand 1 (PD-L1) expression in Chinese lung adenocarcinoma patients. Cancer Cell Int (2022) 22(1):138. doi: 10.1186/s12935-022-02488-z
48. Liu W, Ren D, Xiong W, Jin X, Zhu L. A novel FBW7/NFAT1 axis regulates cancer immunity in sunitinib-resistant renal cancer by inducing PD-L1 expression. J Exp Clin Cancer Res (2022) 41(1):38. doi: 10.1186/s13046-022-02253-0
49. Lu C, Paschall AV, Shi H, Savage N, Waller JL, Sabbatini ME, et al. The MLL1-H3K4me3 axis-mediated PD-L1 expression and pancreatic cancer immune evasion. J Natl Cancer Inst (2017) 109(6):djw283. doi: 10.1093/jnci/djw283
50. Xu H, Liang XL, Liu XG, Chen NP. The landscape of PD-L1 expression and somatic mutations in hepatocellular carcinoma. J Gastrointest Oncol (2021) 12(3):1132–40. doi: 10.21037/jgo-21-251
51. Shi Y, Lei Y, Liu L, Zhang S, Wang W, Zhao J, et al. Integration of comprehensive genomic profiling, tumor mutational burden, and PD-L1 expression to identify novel biomarkers of immunotherapy in non-small cell lung cancer. Cancer Med (2021) 10(7):2216–31. doi: 10.1002/cam4.3649
52. Lv J, Zhu Y, Ji A, Zhang Q, Liao G. Mining TCGA database for tumor mutation burden and their clinical significance in bladder cancer. Biosci Rep (2020) 40(4):BSR20194337. doi: 10.1042/BSR20194337
53. Ji Q, Cai Y, Shrestha SM, Shen D, Zhao W, Shi R. Construction and validation of an immune-related gene prognostic index for esophageal squamous cell carcinoma. BioMed Res Int (2021) 2021:7430315. doi: 10.1155/2021/7430315
54. Salem ME, Puccini A, Xiu J, Raghavan D, Lenz HJ, Korn WM, et al. Comparative molecular analyses of esophageal squamous cell carcinoma, esophageal adenocarcinoma, and gastric adenocarcinoma. Oncologist. (2018) 23(11):1319–27. doi: 10.1634/theoncologist.2018-0143
55. Isshiki Y, Melnick A. Epigenetic mechanisms of therapy resistance in diffuse Large b cell lymphoma (DLBCL). Curr Cancer Drug Targets. (2021) 21(4):274–82. doi: 10.2174/1568009620666210106122750
56. Farah CS. Molecular landscape of head and neck cancer and implications for therapy. Ann Transl Med (2021) 9(10):915. doi: 10.21037/atm-20-6264
57. Oreskovic E, Wheeler EC, Mengwasser KE, Fujimura E, Martin TD, Tothova Z, et al. Genetic analysis of cancer drivers reveals cohesin and CTCF as suppressors of PD-L1. Proc Natl Acad Sci U S A. (2022) 119(7):e2120540119. doi: 10.1073/pnas.2120540119
58. Kwesi-Maliepaard EM, Aslam MA, Alemdehy MF, van den Brand T, McLean C, Vlaming H, et al. The histone methyltransferase DOT1L prevents antigen-independent differentiation and safeguards epigenetic identity of CD8+ T cells. Proc Natl Acad Sci U S A. (2020) 117(34):20706–16. doi: 10.1073/pnas.1920372117
59. Kagoya Y, Nakatsugawa M, Saso K, Guo T, Anczurowski M, Wang CH, et al. DOT1L inhibition attenuates graft-versus-host disease by allogeneic T cells in adoptive immunotherapy models. Nat Commun (2018) 9(1):1915. doi: 10.1038/s41467-018-04262-0
60. Sun D, Wang W, Guo F, Pitter MR, Du W, Wei S, et al. DOT1L affects colorectal carcinogenesis via altering T cell subsets and oncogenic pathway. Oncoimmunology. (2022) 11(1):2052640. doi: 10.1080/2162402X.2022.2052640
61. Sellers WR, Loda M. The EZH2 polycomb transcriptional repressor–a marker or mover of metastatic prostate cancer? Cancer Cell (2002) 2(5):349–50. doi: 10.1016/s1535-6108(02)00187-3
63. Booth CAG, Barkas N, Neo WH, Boukarabila H, Soilleux EJ, Giotopoulos G, et al. Ezh2 and Runx1 mutations collaborate to initiate lympho-myeloid leukemia in early thymic progenitors. Cancer Cell (2018) 33(2):274–291.e8. doi: 10.1016/j.ccell.2018.01.006
64. Cao H, Li L, Yang D, Zeng L, Yewei X, Yu B, et al. Recent progress in histone methyltransferase (G9a) inhibitors as anticancer agents. Eur J Med Chem (2019) 179:537–46. doi: 10.1016/j.ejmech.2019.06.072
65. Jan S, Dar MI, Wani R, Sandey J, Mushtaq I, Lateef S, et al. Targeting EHMT2/ G9a for cancer therapy: Progress and perspective. Eur J Pharmacol (2021) 893:173827. doi: 10.1016/j.ejphar.2020.173827
66. Karanth AV, Maniswami RR, Prashanth S, Govindaraj H, Padmavathy R, Jegatheesan SK, et al. Emerging role of SETDB1 as a therapeutic target. Expert Opin Ther Targets. (2017) 21(3):319–31. doi: 10.1080/14728222.2017.1279604
67. Weirich S, Khella MS, Jeltsch A. Structure, activity and function of the Suv39h1 and Suv39h2 protein lysine methyltransferases. Life (Basel). (2021) 11(7):703. doi: 10.3390/life11070703
68. Li W. Histone methyltransferase SETD2 in lymphoid malignancy. Gallamini M, Juweid M, eds. Lymphoma. Brisbane (AU): Exon Publications. (2021).
69. Yuan H, Han Y, Wang X, Li N, Liu Q, Yin Y, et al. SETD2 restricts prostate cancer metastasis by integrating EZH2 and AMPK signaling pathways. Cancer Cell (2020) 38(3):350–365.e7. doi: 10.1016/j.ccell.2020.05.022
70. Rao RC, Dou Y. Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat Rev Cancer. (2015) 15(6):334–46. doi: 10.1038/nrc3929
71. Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell (2011) 20(1):66–78. doi: 10.1016/j.ccr.2011.06.010
72. Cha B, Jho EH. Protein arginine methyltransferases (PRMTs) as therapeutic targets. Expert Opin Ther Targets. (2012) 16(7):651–64. doi: 10.1517/14728222.2012.688030
73. Hou J, Wang Y, Shi L, Chen Y, Xu C, Saeedi A, et al. Integrating genome-wide CRISPR immune screen with multi-omic clinical data reveals distinct classes of tumor intrinsic immune regulators. J Immunother Cancer. (2021) 9(2):e001819. doi: 10.1136/jitc-2020-001819
74. Schonfeld M, Zhao J, Komatz A, Weinman SA, Tikhanovich I. The polymorphism rs975484 in the protein arginine methyltransferase 1 gene modulates expression of immune checkpoint genes in hepatocellular carcinoma. J Biol Chem (2020) 295(20):7126–37. doi: 10.1074/jbc.RA120.013401
75. Zheng NN, Zhou M, Sun F, Huai MX, Zhang Y, Qu CY, et al. Combining protein arginine methyltransferase inhibitor and anti-programmed death-ligand-1 inhibits pancreatic cancer progression. World J Gastroenterol (2020) 26(26):3737–49. doi: 10.3748/wjg.v26.i26.3737
76. Zhao J, O'Neil M, Vittal A, Weinman SA, Tikhanovich I. PRMT1-dependent macrophage IL-6 production is required for alcohol-induced HCC progression. Gene Expr. (2019) 19(2):137–50. doi: 10.3727/105221618X15372014086197
77. Liu K, Ma J, Ao J, Mu L, Wang Y, Qian Y, et al. The oncogenic role and immune infiltration for CARM1 identified by pancancer analysis. J Oncol (2021) 2021:2986444. doi: 10.1155/2021/2986444
78. Lin J, Liu H, Fukumoto T, Zundell J, Yan Q, Tang CA, et al. Targeting the IRE1α/XBP1s pathway suppresses CARM1-expressing ovarian cancer. Nat Commun (2021) 12(1):5321. doi: 10.1038/s41467-021-25684-3
79. Kim EJ, Liu P, Zhang S, Donahue K, Wang Y, Schehr JL, et al. BAF155 methylation drives metastasis by hijacking super-enhancers and subverting anti-tumor immunity. Nucleic Acids Res (2021) 49(21):12211–33. doi: 10.1093/nar/gkab1122
80. Zhang LX, Gao J, Long X, Zhang PF, Yang X, Zhu SQ, et al. The circular RNA circHMGB2 drives immunosuppression and anti-PD-1 resistance in lung adenocarcinomas and squamous cell carcinomas via the miR-181a-5p/CARM1 axis. Mol Cancer. (2022) 21(1):110. doi: 10.1186/s12943-022-01586-w
81. Zhang Z, Guo Z, Xu X, Cao D, Yang H, Li Y, et al. Structure-based discovery of potent CARM1 inhibitors for solid tumor and cancer immunology therapy. J Med Chem (2021) 64(22):16650–74. doi: 10.1021/acs.jmedchem.1c01308
82. Kumar S, Zeng Z, Bagati A, Tay RE, Sanz LA, Hartono SR, et al. CARM1 inhibition enables immunotherapy of resistant tumors by dual action on tumor cells and T cells. Cancer Discovery (2021) 11(8):2050–71. doi: 10.1158/2159-8290.CD-20-1144
83. Kim H, Kim H, Feng Y, Li Y, Tamiya H, Tocci S, et al. PRMT5 control of cGAS/STING and NLRC5 pathways defines melanoma response to antitumor immunity. Sci Transl Med (2020) 12(551):eaaz5683. doi: 10.1126/scitranslmed.aaz5683
84. Luo Y, Gao Y, Liu W, Yang Y, Jiang J, Wang Y, et al. Myelocytomatosis-protein arginine n-methyltransferase 5 axis defines the tumorigenesis and immune response in hepatocellular carcinoma. Hepatology. (2021) 74(4):1932–51. doi: 10.1002/hep.31864
85. Hu R, Zhou B, Chen Z, Chen S, Chen N, Shen L, et al. PRMT5 inhibition promotes PD-L1 expression and immuno-resistance in lung cancer. Front Immunol (2022) 12:722188. doi: 10.3389/fimmu.2021.722188
86. Jiang Y, Yuan Y, Chen M, Li S, Bai J, Zhang Y, et al. PRMT5 disruption drives antitumor immunity in cervical cancer by reprogramming T cell-mediated response and regulating PD-L1 expression. Theranostics. (2021) 11(18):9162–76. doi: 10.7150/thno.59605
87. Zheng Y, Chen Z, Zhou B, Chen S, Han L, Chen N, et al. PRMT5 deficiency enforces the transcriptional and epigenetic programs of Klrg1+CD8+ terminal effector T cells and promotes cancer development. J Immunol (2022) 208(2):501–13. doi: 10.4049/jimmunol.2100523
88. Strobl CD, Schaffer S, Haug T, Völkl S, Peter K, Singer K, et al. Selective PRMT5 inhibitors suppress human CD8+ T cells by upregulation of p53 and impairment of the AKT pathway similar to the tumor metabolite MTA. Mol Cancer Ther (2020) 19(2):409–19. doi: 10.1158/1535-7163.MCT-19-0189
89. Nagai Y, Ji MQ, Zhu F, Xiao Y, Tanaka Y, Kambayashi T, et al. PRMT5 associates with the FOXP3 homomer and when disabled enhances targeted p185erbB2/neu tumor immunotherapy. Front Immunol (2019) 10:174. doi: 10.3389/fimmu.2019.00174
90. Tang J, Frankel A, Cook RJ, Kim S, Paik WK, Williams KR, et al. PRMT1 is the predominant type I protein arginine methyltransferase in mammalian cells. J Biol Chem (2000) 275(11):7723–30. doi: 10.1074/jbc.275.11.7723
91. Chen D, Ma H, Hong H, Koh SS, Huang SM, Schurter BT, et al. Regulation of transcription by a protein methyltransferase. Science. (1999) 284(5423):2174–7. doi: 10.1126/science.284.5423.2174
92. Stopa N, Krebs JE, Shechter D. The PRMT5 arginine methyltransferase: Many roles in development, cancer and beyond. Cell Mol Life Sci (2015) 72(11):2041–59. doi: 10.1007/s00018-015-1847-9
93. Pedersen MT, Helin K. Histone demethylases in development and disease. Trends Cell Biol (2010) 20(11):662–71. doi: 10.1016/j.tcb.2010.08.011
94. Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. (2004) 119(7):941–53. doi: 10.1016/j.cell.2004.12.012
95. Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature. (2006) 439(7078):811–6. doi: 10.1038/nature04433
96. Arifuzzaman S, Khatun MR, Khatun R. Emerging of lysine demethylases (KDMs): From pathophysiological insights to novel therapeutic opportunities. BioMed Pharmacother. (2020) 129:110392. doi: 10.1016/j.biopha.2020.110392
97. Yang J, Hu Y, Zhang B, Liang X, Li X. The JMJD family histone demethylases in crosstalk between inflammation and cancer. Front Immunol (2022) 13:881396. doi: 10.3389/fimmu.2022.881396
98. Sheng W, LaFleur MW, Nguyen TH, Chen S, Chakravarthy A, Conway JR, et al. LSD1 ablation stimulates anti-tumor immunity and enables checkpoint blockade. Cell. (2018) 174(3):549–563.e19. doi: 10.1016/j.cell.2018.05.052
99. Zhou Z, van der Jeught K, Fang Y, Yu T, Li Y, Ao Z, et al. An organoid-based screen for epigenetic inhibitors that stimulate antigen presentation and potentiate T-cell-mediated cytotoxicity. Nat BioMed Eng. (2021) 5(11):1320–35. doi: 10.1038/s41551-021-00805-x
100. Nguyen EM, Taniguchi H, Chan JM, Zhan YA, Chen X, Qiu J, et al. Targeting lysine-specific demethylase 1 rescues major histocompatibility complex class I antigen presentation and overcomes programmed death-ligand 1 blockade resistance in SCLC. J Thorac Oncol (2022) 17(8):1014–31. doi: 10.1016/j.jtho.2022.05.014
101. Xu S, Wang X, Yang Y, Li Y, Wu S. LSD1 silencing contributes to enhanced efficacy of anti-CD47/PD-L1 immunotherapy in cervical cancer. Cell Death Dis (2021) 12(4):282. doi: 10.1038/s41419-021-03556-4
102. Wang Y, Cao K. KDM1A promotes immunosuppression in hepatocellular carcinoma by regulating PD-L1 through demethylating MEF2D. J Immunol Res (2021) 2021:9965099. doi: 10.1155/2021/9965099
103. Shen DD, Pang JR, Bi YP, Zhao LF, Li YR, Zhao LJ, et al. LSD1 deletion decreases exosomal PD-L1 and restores T-cell response in gastric cancer. Mol Cancer. (2022) 21(1):75. doi: 10.1186/s12943-022-01557-1
104. Han Y, Xu S, Ye W, Wang Y, Zhang X, Deng J, et al. Targeting LSD1 suppresses stem cell-like properties and sensitizes head and neck squamous cell carcinoma to PD-1 blockade. Cell Death Dis (2021) 12(11):993. doi: 10.1038/s41419-021-04297-0
105. Sheng W, Liu Y, Chakraborty D, Debo B, Shi Y. Simultaneous inhibition of LSD1 and TGFβ enables eradication of poorly immunogenic tumors with anti-PD-1 treatment. Cancer Discovery (2021) 11(8):1970–81. doi: 10.1158/2159-8290.CD-20-0017
106. Liu Y, Debo B, Li M, Shi Z, Sheng W, Shi Y. LSD1 inhibition sustains T cell invigoration with a durable response to PD-1 blockade. Nat Commun (2021) 12(1):6831. doi: 10.1038/s41467-021-27179-7
107. Tu WJ, McCuaig RD, Tan AHY, Hardy K, Seddiki N, Ali S, et al. Targeting nuclear LSD1 to reprogram cancer cells and reinvigorate exhausted T cells via a novel LSD1-EOMES switch. Front Immunol (2020) 11:1228. doi: 10.3389/fimmu.2020.01228
108. Zhong C, Tao B, Li X, Xiang W, Peng L, Peng T, et al. HOXA-AS2 contributes to regulatory T cell proliferation and immune tolerance in glioma through the miR-302a/KDM2A/JAG1 axis. Cell Death Dis (2022) 13(2):160. doi: 10.1038/s41419-021-04471-4
109. Li J, Yuan S, Norgard RJ, Yan F, Sun YH, Kim IK, et al. Epigenetic and transcriptional control of the epidermal growth factor receptor regulates the tumor immune microenvironment in pancreatic cancer. Cancer Discovery (2021) 11(3):736–53. doi: 10.1158/2159-8290.CD-20-0519
110. Zhang W, Liu W, Jia L, Chen D, Chang I, Lake M, et al. Targeting KDM4A epigenetically activates tumor-cell-intrinsic immunity by inducing DNA replication stress. Mol Cell (2021) 81(10):2148–2165.e9. doi: 10.1016/j.molcel.2021.02.038
111. Zhang M, Liu Y, Hou S, Wang Y, Wang C, Yin Y, et al. KDM4B, a potential prognostic biomarker revealed by large-scale public databases and clinical samples in uterine corpus endometrial carcinoma. Mol Omics. (2022) 18(6):506–19. doi: 10.1039/d1mo00287b
112. Liu L, Yu T, Jin Y, Mai W, Zhou J, Zhao C. MicroRNA-15a carried by mesenchymal stem cell-derived extracellular vesicles inhibits the immune evasion of colorectal cancer cells by regulating the KDM4B/HOXC4/PD-L1 axis. Front Cell Dev Biol (2021) 9:629893. doi: 10.3389/fcell.2021.629893
113. Jie X, Chen Y, Zhao Y, Yang X, Xu Y, Wang J, et al. Targeting KDM4C enhances CD8+ T cell mediated antitumor immunity by activating chemokine CXCL10 transcription in lung cancer. J Immunother Cancer. (2022) 10(2):e003716. doi: 10.1136/jitc-2021-003716
114. Liao TT, Lin CC, Jiang JK, Yang SH, Teng HW, Yang MH. Harnessing stemness and PD-L1 expression by AT-rich interaction domain-containing protein 3B in colorectal cancer. Theranostics. (2020) 10(14):6095–112. doi: 10.7150/thno.44147
115. Chen Q, Zhuang S, Hong Y, Yang L, Guo P, Mo P, et al. Demethylase JMJD2D induces PD-L1 expression to promote colorectal cancer immune escape by enhancing IFNGR1-STAT3-IRF1 signaling. Oncogene. (2022) 41(10):1421–33. doi: 10.1038/s41388-021-02173-x
116. Rasmussen PB, Staller P. The KDM5 family of histone demethylases as targets in oncology drug discovery. Epigenomics. (2014) 6(3):277–86. doi: 10.2217/epi.14.14
117. Hao F. Systemic profiling of KDM5 subfamily signature in non-Small-Cell lung cancer. Int J Gen Med (2021) 14:7259–75. doi: 10.2147/IJGM.S329733
118. Duan Y, Du Y, Gu Z, Zheng X, Wang C. Expression, prognostic value, and functional mechanism of the KDM5 family in pancreatic cancer. Front Cell Dev Biol (2022) 10:887385. doi: 10.3389/fcell.2022.887385
119. Chen XJ, Ren AQ, Zheng L, Zheng ED. Predictive value of KDM5C alterations for immune checkpoint inhibitors treatment outcomes in patients with cancer. Front Immunol (2021) 12:664847. doi: 10.3389/fimmu.2021.664847
120. Wu L, Cao J, Cai WL, Lang SM, Horton JR, Jansen DJ, et al. KDM5 histone demethylases repress immune response via suppression of STING. PLoS Biol (2018) 16(8):e2006134. doi: 10.1371/journal.pbio.2006134
121. Chen S, Shi Y. A new horizon for epigenetic medicine? Cell Res (2013) 23(3):326–8. doi: 10.1038/cr.2012.136
122. Hua C, Chen J, Li S, Zhou J, Fu J, Sun W, et al. KDM6 demethylases and their roles in human cancers. Front Oncol (2021) 11:779918. doi: 10.3389/fonc.2021.779918
123. Qu LH, Fang Q, Yin T, Yi HM, Mei GB, Hong ZZ, et al. Comprehensive analyses of prognostic biomarkers and immune infiltrates among histone lysine demethylases (KDMs) in hepatocellular carcinoma. Cancer Immunol Immunother. (2022) 71(10):2449–67. doi: 10.1186/s12885-021-08372-9
124. Chen X, Lin X, Pang G, Deng J, Xie Q, Zhang Z. Significance of KDM6A mutation in bladder cancer immune escape. BMC Cancer (2021) 21(1):635. doi: 10.1186/s12885-021-08372-9
125. Kardos J, Chai S, Mose LE, Selitsky SR, Krishnan B, Saito R, et al. Claudin-low bladder tumors are immune infiltrated and actively immune suppressed. JCI Insight (2016) 1(3):e85902. doi: 10.1172/jci.insight.85902
126. Zhu G, Pei L, Li Y, Gou X. EP300 mutation is associated with tumor mutation burden and promotes antitumor immunity in bladder cancer patients. Aging (Albany NY). (2020) 12(3):2132–41. doi: 10.18632/aging.102728
127. Yi J, Shi X, Xuan Z, Wu J. Histone demethylase UTX/KDM6A enhances tumor immune cell recruitment, promotes differentiation and suppresses medulloblastoma. Cancer Lett (2021) 499:188–200. doi: 10.1016/j.canlet.2020.11.031)
128. Zhang H, Hu Y, Liu D, Liu Z, Xie N, Liu S, et al. The histone demethylase Kdm6b regulates the maturation and cytotoxicity of TCRαβ+CD8αα+ intestinal intraepithelial lymphocytes. Cell Death Differ (2022) 29(7):1349–63. doi: 10.1038/s41418-021-00921-w
129. Xu T, Schutte A, Jimenez L, Gonçalves ANA, Keller A, Pipkin ME, et al. Kdm6b regulates the generation of effector CD8+ T cells by inducing chromatin accessibility in effector-associated genes. J Immunol (2021) 206(9):2170–83. doi: 10.4049/jimmunol.2001459
130. Ding JT, Yu XT, He JH, Chen DZ, Guo F. A pan-cancer analysis revealing the dual roles of lysine (K)-specific demethylase 6B in tumorigenesis and immunity. Front Genet (2022) 13:912003. doi: 10.3389/fgene.2022.912003
131. Hino S, Kohrogi K, Nakao M. Histone demethylase LSD1 controls the phenotypic plasticity of cancer cells. Cancer Sci (2016) 107(9):1187–92. doi: 10.1111/cas.13004
132. Liu L, Liu J, Lin Q. Histone demethylase KDM2A: Biological functions and clinical values (Review). Exp Ther Med (2021) 22(1):723. doi: 10.3892/etm.2021.10155
133. Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, et al. Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev (2012) 26(12):1326–38. doi: 10.1101/gad.191056.112
134. Kornberg MD, Bhargava P, Kim PM, Putluri V, Snowman AM, Putluri N, et al. Dimethyl fumarate targets GAPDH and aerobic glycolysis to modulate immunity. Science. (2018) 360(6387):449–53. doi: 10.1126/science.aan466
135. Sui Y, Gu R, Janknecht R. Crucial functions of the JMJD1/KDM3 epigenetic regulators in cancer. Mol Cancer Res (2021) 19(1):3–13. doi: 10.1158/1541-7786.MCR-20-0404
136. Berry WL, Janknecht R. KDM4/JMJD2 histone demethylases: epigenetic regulators in cancer cells. Cancer Res (2013) 73(10):2936–42. doi: 10.1158/0008-5472.CAN-12-4300
Keywords: histone methylation, epigenetic modification, tumor, antitumor immunity, immunotherapy
Citation: Zhang Y, Chen J, Liu H, Mi R, Huang R, Li X, Fan F, Xie X and Ding J (2023) The role of histone methylase and demethylase in antitumor immunity: A new direction for immunotherapy. Front. Immunol. 13:1099892. doi: 10.3389/fimmu.2022.1099892
Received: 16 November 2022; Accepted: 23 December 2022;
Published: 11 January 2023.
Edited by:
Catherine Sautes-Fridman, U1138 Centre de Recherche des Cordeliers (CRC)(INSERM), FranceReviewed by:
Limin Xia, Huazhong University of Science and Technology, ChinaMatias I. Hepp, Universidad Católica de la Santísima Concepción, Chile
Copyright © 2023 Zhang, Chen, Liu, Mi, Huang, Li, Fan, Xie and Ding. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jie Ding, dingjiexy@126.com
†These authors have contributed equally to this work and share first authorship