 Wendy Mao
Wendy Mao- Cell Biology, BioNTech Societas Europaea (SE), Gaithersburg, MD, United States
The antitumor potential of personalized immunotherapy, including adoptive T-cell therapy, has been shown in both preclinical and clinical studies. Combining cell therapy with targeted metabolic interventions can further enhance therapeutic outcomes in terms of magnitude and durability. The ability of a T cell receptor to recognize peptides derived from tumor neoantigens allows for a robust yet specific response against cancer cells while sparing healthy tissue. However, there exist challenges to adoptive T cell therapy such as a suppressive tumor milieu, the fitness and survival of transferred cells, and tumor escape, all of which can be targeted to further enhance the antitumor potential of T cell receptor-engineered T cell (TCR-T) therapy. Here, we explore current strategies involving metabolic reprogramming of both the tumor microenvironment and the cell product, which can lead to increased T cell proliferation, survival, and anti-tumor cytotoxicity. In addition, we highlight potential metabolic pathways and targets which can be leveraged to improve engraftment of transferred cells and obviate the need for lymphodepletion, while minimizing off-target effects. Metabolic signaling is delicately balanced, and we demonstrate the need for thoughtful and precise interventions that are tailored for the unique characteristics of each tumor. Through improved understanding of the interplay between immunometabolism, tumor resistance, and T cell signaling, we can improve current treatment regimens and open the door to potential synergistic combinations.
Introduction
Success of immunotherapy directed against cancer is predicated on the fact that tumor cells can be specifically targeted by various populations of immune cells, minimizing damage to healthy tissue while exclusively eliminating cancerous growth. Adoptive cellular therapy (ACT) for solid tumors has gained increased attention in recent years following its initial success in metastatic melanoma (1) and more recent promising results in lung (2) and pancreatic (3) cancers, demonstrating the potential for adoptively transferred tumor-antigen specific T cells to mediate an antitumor response. In this review, we will focus on TCR-T, which has already shown promising clinical efficacy in solid tumors (3–5). In this modality of ACT, T cell receptors (TCRs) targeting tumor antigens presented in the context of major histocompatibility (MHC) molecules are engineered onto isolated T cells and then infused into a patient, which will then mediate tumor regression (6) (Figure 1). Despite its successes, the field of ACT has faced many challenges, from inadequate immune infiltrate, a suppressive tumor microenvironment (TME), tumor heterogeneity and evolution, and exhausted effector cells, among others. Despite preclinical promise, many TCR-T therapies provide only limited benefit in clinic (7, 8), while in other contexts, tumors relapse after initial remission following treatment (9), suggesting that immune suppression and tumor evolution can dampen success of these therapies. Metabolic interventions hold promise for enhancing TCR-T survival, persistence, trafficking, and cytotoxicity against nutrient-devouring tumor cells. The TCR-T treatment process offers many opportunities for metabolic intervention, with the ability to precondition both T cells and the tumor separately prior to infusion to achieve the most benefit. These include treatment modalities that can be applied during engineering to attain better cell phenotypes, as well as systemic treatments pre-and post-infusion to condition the TME, each of which will be evaluated in subsequent sections, and are summarized in Figure 2. In this review, we will discuss methodologies of targeting metabolic pathways to not only increase the antitumor efficacy and durability of TCR-T, but also improve engineering and clinical protocols to further extend the capabilities of ACT.
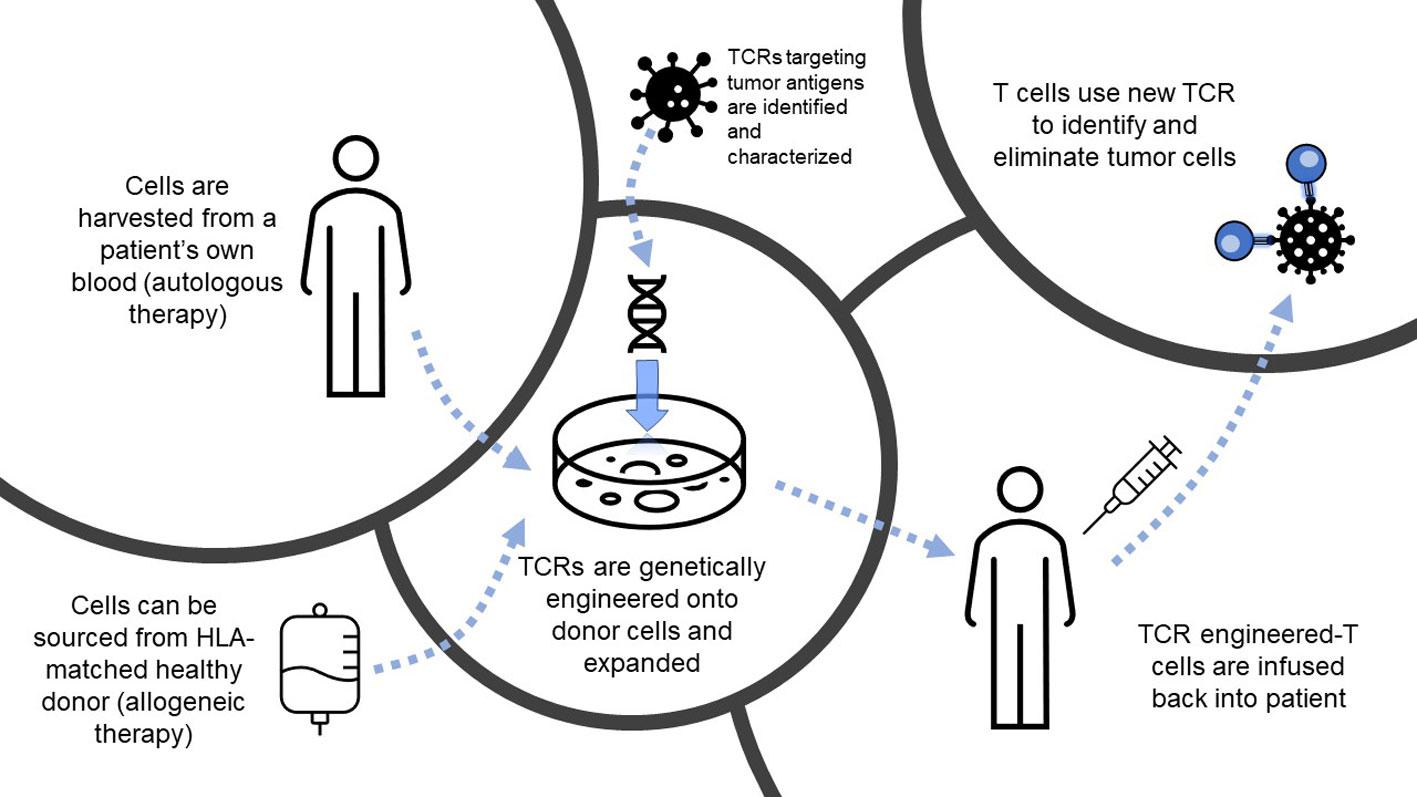
Figure 1 Overview of T cell receptor-engineered T cell (TCR-T) therapy. Depiction of the process of TCR-T, from autologous or allogeneic cell sourcing to engineering and infusion to target tumor cells. HLA, human leukocyte antigen; TCR, T cell receptor.
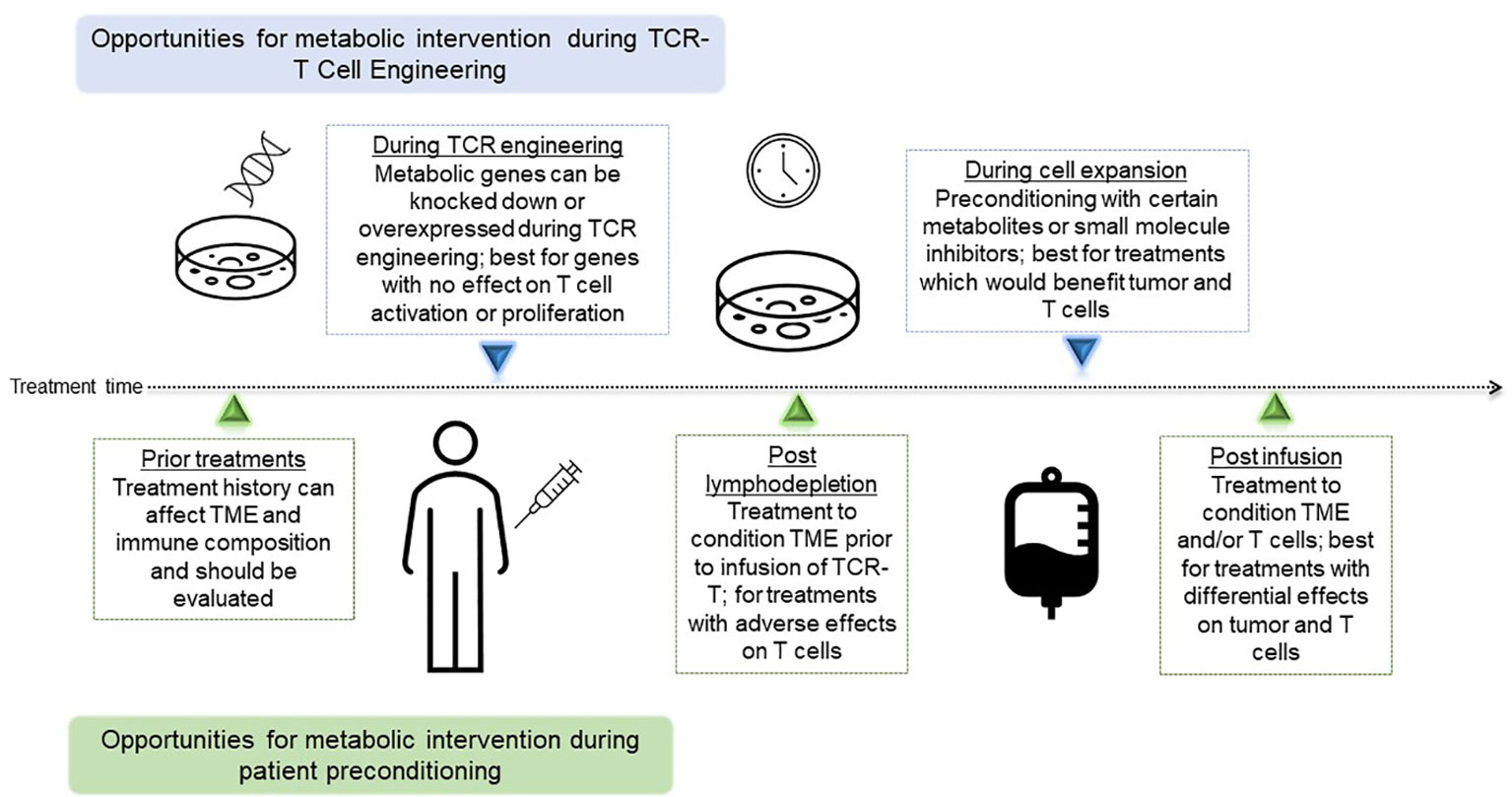
Figure 2 Timing of Opportunities for metabolic interventions during TCR-T. Depiction of metabolic interventions that can be deployed as part of the cell engineering and expansion process, or part of patient preconditioning and treatment. TCR-T, T cell receptor-engineered T cell; TCR, T cell receptor; TME, tumor microenvironment.
Strategies for targeting tumor metabolism to improve T cell fitness
Tumor cells arise from transformations that fundamentally alter a cell’s proliferative and survival capacities. As such, the metabolic demands for cancer cells are different than those of a nonmalignant cell. In 1956, Warburg observed increased rates of glycolysis and lactate production in tumors, indicative of a preference for aerobic glycolysis over the more efficient oxidative phosphorylation pathway for generating ATP (10), a term later coined “The Warburg Effect”. Although less efficient than oxidative phosphorylation at ATP generation, the increased flux through aerobic glycolysis also generates metabolic intermediates necessary for supporting rapid proliferation, including glycerol and citrate for lipid biogenesis (11). The increased proliferative demands of tumor cells also increases rates of glutaminolysis to sustain anaplerosis, and this results in tumor metabolism of glutamine being higher than that of any other nonessential amino acid (12). The high utilization of these nutrients by tumor cells leads to competition for these resources with other cells, resulting in a glutamine restricted (13), glucose-poor, hypoxic, and acidic milieu (14, 15) which have been shown to suppress T cell infiltration and function (16–18), Figure 3. In addition, other immune subsets in the TME which are crucial to tumor-specific T cell activation, such as Dendritic Cells (DCs), are also adversely affected by the altered metabolic environment of the tumor (19, 20). Although the mechanism of immune cell suppression in the tumor is much more nuanced than simple nutrient depletion and competition, the unique milieu of the tumor requires that immune cells reshape metabolic flux to survive, which can then drastically affect their function (21). Targeting these nutrient pathways, summarized in Figure 3, may serve a dual purpose of not only reducing tumor growth, but also supporting the survival and antitumor efficacy of TCR-T.
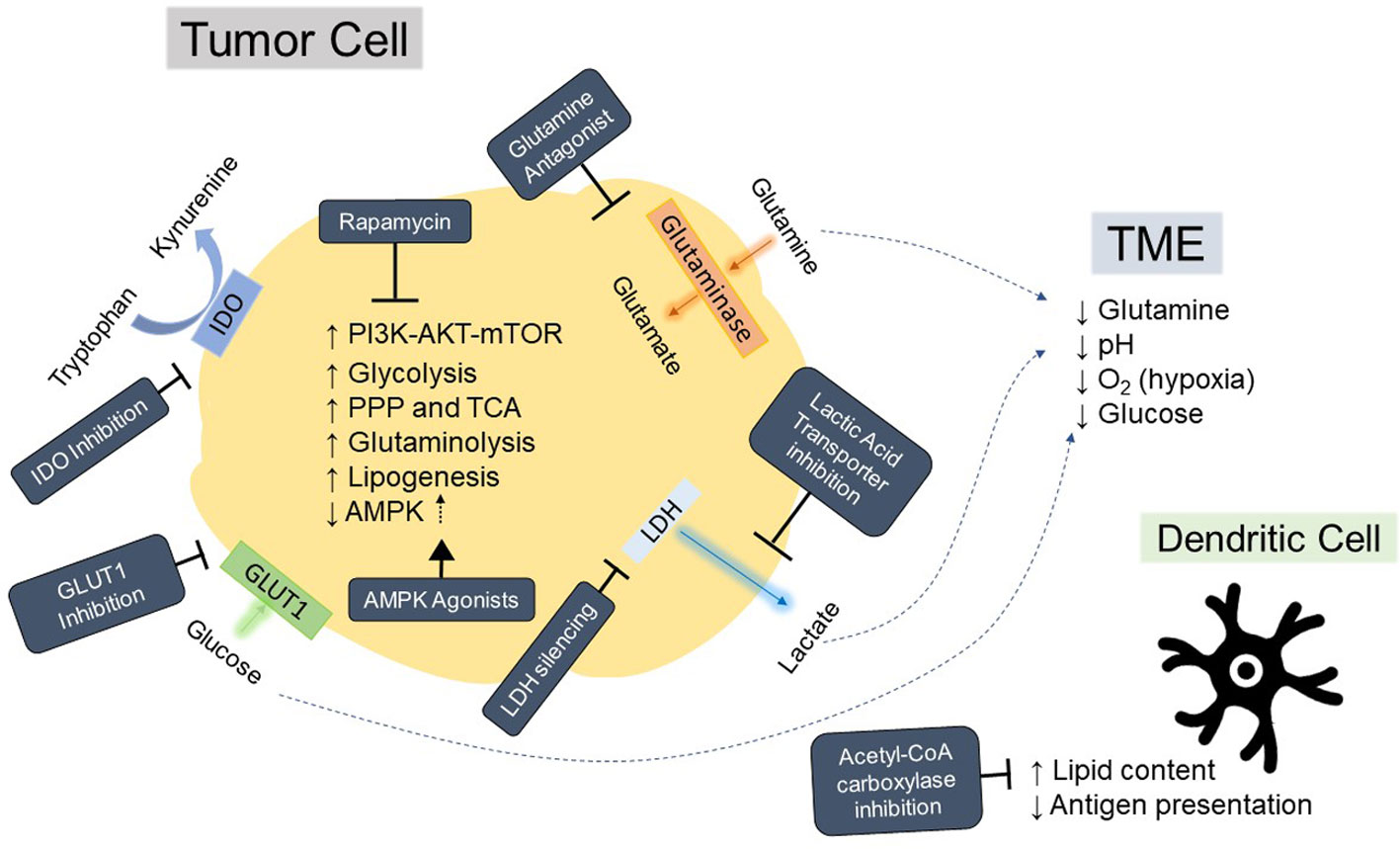
Figure 3 Hallmarks of tumor metabolism fueling immunosuppressive tumor microenvironment and strategies for modulation. Depiction of aspects of tumor metabolism that contribute to acidification, nutrient deprivation, and immunosuppression in the tumor microenvironment (TME) as well as general mechanisms for inhibiting these pathways. Strategies for inhibition or reversal of tumor phenotypes are in dark blue boxes with white text. AMPK, AMP-activated protein kinase; PI3K, Phosphoinositide 3 kinase; AKT, protein kinase B; mTOR, mammalian target of rapamycin; IDO, Indoleamine 2, 3-dioxygenase; LDH, lactate dehydrogenase; PPP, pentose phosphate pathway; TCA, the tricarboxylic acid cycle; O2, oxygen; GLUT1, Glucose transporter 1.
In addition to metabolite synthesis, cells normally integrate information regarding nutrient availability and stress to regulate survival and proliferation; unfortunately, tumor cells have co-opted many aspects of these signaling cascades, including ones involving Mammalian Target of Rapamycin (mTOR), in order to sustain rapid division (22). mTOR has long been thought of as a master regulator of cell growth and longevity in response to nutrients and growth factors, and exists in two functionally distinct complexes, mTOR Complex 1 (mTORC1) and mTOR Complex 2 (mTORC2) (23), each with differing drug sensitivities (24, 25). Aberrant expression of mTOR and associated pathway components are very common in solid tumors (26–28), and contributes to tumorigenesis and growth (29). In addition, mTOR also plays a role in T cell differentiation and function (30), making it an attractive target for enhancing antitumor immunity. Strategies for modulating mTOR signaling also can serve to enhance fitness and cytotoxicity of TCR-T while concomitantly reducing tumor cell growth (31).
Metabolic reshaping of the TME to enhance T cell fitness and function
The high level of glucose consumption by tumor cells provides not only fuel for glycolysis, but also increases flux through the Pentose Phosphate Pathway (PPP), which is consequently also elevated in malignant cells compared to normal cells (32). This pathway is crucial for generating intermediates for nucleic acid synthesis, nicotinamide-adenine dinucleotide phosphate (NADPH) for fatty acid synthesis (33), and generating glutathione to scavenge reactive oxidative species (ROS) (34), all of which are indispensable for rapidly dividing cells. Glucose-6-Phosphate Dehydrogenase (G6PD) is the rate limiting enzyme of the PPP, and its inhibition can induce autophagy (35) and senescence (36) in tumor cells, as well as inhibit their proliferation and metastasis (37). Interestingly, although T cells also proliferate rapidly upon activation, blockade of G6PD appears to generate superior CD8+ effector T cells which mediated a stronger tumor antigen-specific response, as well as increased proinflammatory cytokine secretion. In these T cells, the increased mitochondrial ROS is balanced by a concomitant increase in additional antioxidant enzymes to prevent oxidative damage (38). Taken together, these studies suggest that targeting the PPP through inhibition of G6PD can represent a “best of both worlds” situation in which tumor cells may not be as metabolically plastic as T cells due to their overreliance on specific metabolites, allowing T cells to leverage the shift in metabolic fuel to their benefit.
Evidence of plasticity in T cell metabolism has been demonstrated in the context of multiple metabolic pathways. Glutamine provides the fuel needed for the tricarboxylic acid (TCA) cycle, which in turn generates the intermediates needed for lipid, protein, and nucleic acid synthesis crucial for rapidly proliferating cells (39). Predictably, tumor cells rely heavily on this pathway for their survival, and indeed many tumors show evidence of being “addicted” to glutamine (40, 41). Treatment of tumor-bearing mice with glutamine antagonist JHU083, which impairs enzymes that require glutamine, induced not only durable antitumor effects, but also disrupted Warburg physiology by impairing glucose metabolism. Subsequently, glucose and glutamine concentrations both rose within the tumor, followed by a parallel decrease in hypoxia. T cells treated with glutamine antagonist showed increased markers of activation and memory, and demonstrated enhanced cytokine production upon restimulation. Interestingly, the T cells were found to be able to switch to utilizing acetate as a carbon source for the TCA cycle, a feat which the tumor cells were not able to replicate (42). In another study, the pharmacologic inhibition of glutamine transporter was able to impair tumor growth and also increase the concentration of glutamine in the tumor. However, as T cells adapted by upregulating a separate amino acid transporter, they were able to continue uptake of glutamine and were not affected by the drug – in addition to demonstrating enhanced activation and cytotoxic functions (43). Thus, although T cells and tumor cells rely on increased flux through many of the same pathways for survival and function, the superior metabolic plasticity of T cells can allow for metabolic interventions which reduce tumor fitness while enhancing T cell effector capabilities.
As part of Warburg physiology, large amounts of lactic acid are generated as byproducts of aerobic glycolysis, which is subsequently secreted from the tumor cells and contributes to the acidification of the tumor compartment (44). This increased concentration of lactic acid impairs cytotoxic function of both T cells and natural killer (NK) cells (45) by impairing their ability to secrete the lactic acid byproduct of their own increased glycolytic flux following activation, the secretion of which requires a concentration gradient (46). Treatment with Diclofenac, which inhibits the lactic acid transporters Monocarboxylate Transporter 1 and 4 (MCT1 and MCT4, respectively), enhanced the efficacy of checkpoint blockade immunotherapy through improved tumor control and cytokine secretion. In addition, T cells treated with diclofenac shifted their glucose flux to the TCA cycle and increased oxygen consumption, allowing them to retain their cytotoxic functions. However, it should be noted that proliferation of T cells was reduced following treatment with diclofenac (47), which could indicate that the timing of administration could be crucial in enhancing the efficacy of lactic-acid targeting therapies. Another paper also corroborated these findings through RNA interfering (RNAi)-nanoparticle mediated knockdown of lactate dehydrogenase (LDHA) in tumor cells, which also neutralized tumor pH and increased the infiltration and cytotoxicity of NK and T cells. Interestingly, the authors also conducted these experiments in immunocompromised mice and showed no effect on tumor growth, demonstrating that cytotoxic immune infiltrate was necessary for potentiating the antitumor effect of targeting lactic acid metabolism (48). Although targeting lactic acid metabolism may negatively affect proliferation of endogenous T cells, TCR-T can be administered after lactic acid transporter inhibition “preconditioning”, which could potentially render the tumor milieu more amenable to the survival and efficacy of the transferred cells.
In addition to the metabolites mentioned previously, the anabolism/catabolism of other nutrients and amino acids can also drive immunosuppression in the TME and thus serve as potential targets for metabolic therapy. One example is the role of Indoleamine 2,3-dioxygenase (IDO), the first and rate-limiting step of tryptophan degradation as part of the kynurenine pathway, which fuels many essential biological processes including energy metabolism and generation of the co-factor Nicotinamide adenine dinucleotide (NAD+) (49). IDO is elevated in many tumor types and is correlated with poorer prognoses in solid tumors (50), and a higher kynurenine-to-tryptophan ratio is associated with shorter survival in patients with leukemia (51). One possible explanation for this association is that the tryptophan/kynurenine axis has been associated with multiple immunoregulatory effects – kynurenine can bind to the aryl hydrocarbon receptor (AHR), which skews T cells towards a immunosuppressive regulatory T cell (Treg) phenotype (52); additionally, it can induce exhaustion and immune checkpoint expression in T cells (53) and drive conversion of dendritic cells (DCs) towards a tolerogenic phenotype (54). In T cells specifically, IDO-mediated tryptophan deprivation was found to induce autophagy and inhibit both mTOR and protein kinase C theta (PKC-Θ) (55), which are crucial for T cell activation and proliferation (56). IDO inhibition in combination with a tumor vaccine was found to be able to convert suppressive Tregs into Th17-like cells in tumor draining lymph nodes of tumor-bearing mice, and this treatment both enhanced CD8 T cell activation and reduced tumor growth (57). Given their preclinical promise, IDO inhibitors which target IDO1 or the isoenzyme IDO2 have been and are currently being investigated in clinical trials. The ECHO-301/KEYNOTE-252 trial found that treatment with the IDO1 inhibitor Epacadostat did not improve overall survival or progression free survival in melanoma (58), which brings into question the utility of IDO inhibitors in a clinical setting. These disappointing results could be explained by several factors: the inability of Epacadostat to completely inhibit IDO1 even at the highest dose, the compensatory expression of other tryptophan-degrading enzymes such as IDO2, and patient cohort selection (59). Subsequent studies have focused on targeting other metabolites in the kynurenine pathway, including kynurenine itself. Treatment of tumors in vivo with an enhanced kynureninase to degrade kynurenine increased infiltration and proliferation of cytotoxic CD8s, resulting in improved tumor control in a variety of tumor types. Interestingly, the treatment had no effect on tumor growth in immunocompromised mice, suggesting that the antitumor mechanism of kynureninase is contingent upon the presence of immune cells (60). The example of IDO inhibitors demonstrates the importance of understanding compensatory mechanisms and possibility of targeting alternative components of the same pathway when investigating the effects of metabolic perturbations.
Increasing neoantigen presentation in the TME
Effective antitumor immune therapy is predicated on the function of dendritic cells (DCs), which uptake and present tumor antigen to activate naive T cells and induce their proliferation (61). DC ablation drastically reduces the activation of memory T cells - whose reactivation threshold is much lower and postulated to be DC-independent - in response to infections (62), thus suggesting that DCs play a role in maximizing the efficacy of T cell responses. In TCR-T, endogenous DCs can present tumor neoantigens to mobilize new cohorts of T cells of diverse TCR clonotypes, which is crucial to address epitope spread – especially since loss of target antigen is a common tumor mechanism of resistance in patients who relapse post-ACT (63). In the tumor, certain yet unknown tumor factors induce upregulation of scavenger receptors on DCs, increasing their uptake of lipoproteins and increasing their internal lipid content. These lipid-loaded DCs were shown to be unable to process and present antigen effectively, and were drastically impaired in their ability to activate T cells. Targeting fatty acid synthesis by impairing acetyl-CoA carboxylase, and thus forcing DCs to utilize exogenous lipids for triglyceride synthesis, reversed this effect (64). However, since proliferating T cells also increase their requirements for fatty acid synthesis to sustain division and inhibiting this pathway has been shown to be detrimental to cell expansion (65), systemic inhibition of acetyl-CoA carboxylase would have to be carefully timed to avoid interference with TCR-T.
A similar tale of functional balance can be told for the role of adenosine monophosphate-activated protein Kinase (AMPK) pathway in DC activation and T cell effector capabilities. AMPK is crucial to regulating energy balance within the cell, turning on ATP-producing catabolic pathways when cellular stress reduces the amount of ATP available (66). AMPK activation is crucial for protein kinase B (PKB; also known as AKT) activation in response to cellular stress (67), which in turn phosphorylates and inactivates Glycogen synthase kinase-3 beta (GSK3β), a negative regulator of cAMP response element-binding protein (CREB). This results in CREB-mediated enhancement of anti-inflammatory IL-10 transcription while reducing proinflammatory cytokine transcripts through reduction of Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling (68). Phosphorylation of the alpha subunit of AMPK (AMPKα) is required for AMPK activation (69). Knockdown of isoform 1 of this subunit, AMPKα1, in macrophages and DCs enhanced antigen presentation, decreased the production of the anti-inflammatory cytokine interleukin 10 (IL-10), and increased production of IL-17 as well as interferon gamma (IFN-y). This favored the skewing of activated helper T cells to T helper 1 (Th1) and 17 (Th17) phenotypes (70). In a tumor setting, these AMPKα1 deficient macrophages and dendritic cells would be poised to increase tumor antigen presentation, and induce a favorable proinflammatory phenotype of T cells. However, the effects of decreased AMPK in T cells remain less clear, but is necessary to consider if treatments targeting AMPK are to be administered concurrently with TCR-T. Although AMPKα1 deficient CD8 cells produced more proinflammatory IFN-y and IL-17A, they also had impaired viability compared to control cells when subjected to metabolic stress (71). This finding was corroborated by another study, in which the authors demonstrated that this requirement for AMPKα1 to drive T cell functionality was dependent on glucose concentration – that is, T cell impairment was observed in AMPKα1 deficient cells when glucose was scarce (72), as is the case in the tumor microenvironment. Interestingly, metformin, an AMPK agonist, has been shown to directly enhance antitumor function in tumor antigen specific T cells, increasing polyfunctionality and protecting T cells from apoptosis and exhaustion. The same effect was not noted in immunodeficient mice, suggesting that this antitumor effect was driven specifically by the presence of immune cells (73). Taken together, these studies indicate that the balance of AMPK signaling must be modulated very carefully; while increased signaling may lead to better immune cell survival and reduce flux through glycolytic pathway in cancer cells (74), it can also contribute to reducing the effects of cell stress signaling which can lead to tumor cell survival. Future treatment regimens may consider pretreating the T cells in TCR-T with an AMPK agonist to increase stress resistance, while utilizing tumor metabolomics to consider if AMPK is a viable target in each individual tumor setting.
Strategies for improving functionality and persistence of adoptively transferred T cells
In TCR-T, human T cells are engineered with a TCR of interest and then infused into the patient, where the hope is that those cells will traffic to the tumor and surrounding lymph nodes, setting up long-lived cytotoxic populations which will attack the tumor. In order for this therapy to be successful, the T cells must be able to survive in the TME, and thus are subject to many of the same pitfalls that plague immunotherapy in general – namely, exhaustion of effector cells, poor viability, and loss of effector function (75). The competition for glucose with tumor cells in the TME results in T cells with reduced capacity to produce IFN-y and decreased signaling through glycolysis and nutrient sensing pathways (76). Engagement of inhibitory immune checkpoint molecules such as Cytotoxic T-Lymphocyte Associated Protein 4 (CTLA-4) and Programmed Cell Death Protein 1 (PD-1) on T cells inhibits the glycolysis and amino acid metabolism which is crucial to effector differentiation and function (77). Hypoxia in the TME can upregulate the ligand for PD-1, Programmed death-ligand 1 (PD-L1) on tumors (78–81), as can glucose-depleted conditions (82), which engage inhibitory checkpoints on T cells and lead to their “exhaustion”. Even though the TME is known to be immunosuppressive and poor in nutrients, the survival and persistence of infiltrating immune cells is crucial to the sustained effectiveness of TCR-T. Already, blockade of CTLA-4, PD-1, and PD-L1 are being utilized to reinvigorate the dysfunction seen in exhausted T cells, and can restore effector T cell metabolic profiles (77) while dampening glycolytic flux in tumor cells (76). Tumor infiltration of T cells can also be influenced by nutrient changes in the TME, including hypoxia-mediated lactate accumulation which inhibits T cell migration (83), and aberrant lipid metabolism which negatively impacts function and migration of a variety of immune subsets (84). Here, we will summarize some strategies to “armor” T cells against metabolic challenges posed by the TME.
Targeting glucose metabolism to increase T cell functionality
Upon activation, T cells shift their metabolism from oxidative phosphorylation (OXPHOS) to glycolysis to meet energy requirements (85, 86), Figure 4. After the effector phase is completed, T cells yet again shift to rely on fatty acid metabolism for energy when they differentiate into memory T cells (Tmem) (87). Inhibition of glycolysis during antigen encounter drives T cells towards a memory phenotype (88) (Figure 4), but also reduces effector function (89). Glucose metabolites are also utilized in the PPP, which provides crucial intermediates for cell survival and growth, and scavenges ROS produced during cell proliferation. Inhibition of 6-phosphogluconate dehydrogenase (6PGD), the enzyme catalyzing the second step of the PPP, CD8+ T cells increased mitochondrial content and mitochondrial ROS production balanced by increased antioxidant production to protect against oxidative damage. Functionally, these cells increased IFN-y production and granzyme B, and showed superior effects against both tumors and infections, consistent with a T Effector Memory (TEM) phenotype. The cells also increased their expression of glucose-transporter 1 (GLUT1) to increase glucose uptake (38), which is crucial in a TME where there is tremendous competition for this crucial nutrient (76) (Figure 3). Blockade of 6PGD may also have the benefit of reprogramming immunosuppressive Tregs - which rely heavily on the antioxidant capacities of the PPP to balance the ROS produced by their fuel of choice – lipid oxidation - into Th1, 2, and 17 cells, which improved antitumor responses (90) (Figure 4).

Figure 4 Metabolic phenotypes of T cells and strategies for skewing towards memory phenotype. Depiction of changes in T cell metabolism upon activation, and highlighting differences in metabolism upon transition either to a memory phenotype or an exhausted effector cell. Strategies for improving memory phenotype skewing are also depicted in the blue box. 6PGD, 6-phosphogluconate dehydrogenase; PI3K, phosphoinositide 3 kinase; AKT, protein kinase B; mTOR, mammalian target of rapamycin; mTORC2, mTOR Complex 2.
Preconditioning regimens to increase T cell survival in the TME
In previous sections, we have explored the evidence for T cell metabolic plasticity which allows them to adjust flux through various pathways to adapt to environmental cues. This plasticity can not only be leveraged to favor survival of T cells over tumor cells, but also to induce metabolic changes which prepare T cells for the nutrient challenges they will face in the TME. Although glucose has been shown to be essential for T cell effector function, transient glucose restriction (TGR) in activated cells can help enhance antitumor function. In a mouse model of lymphoma, adoptively transferred effector CD8s preconditioned in low glucose conditions were able to mediate complete clearance of tumor, with an enhanced effector phenotype and increased number in circulation. TGR T cells redirected their utilization of glucose carbons to the TCA cycle rather than glycolysis, resulting in a drastic increase in ATP generation, and were better poised to handle oxidative stress due to the sustained synthesis of the reactive oxidative species (ROS) scavenger glutathione – two characteristics which allow these T cells to better survive in the TME compared to cells which have not undergone preconditioning. Metabolomic interrogation of these TGR effector cells showed that these cells were able to enhance their glucose uptake following re-exposure to glucose and increase flux through the PPP (91), both of which can boost effector function and improve ability to compete for nutrients.
In a similar vein, glutamine restriction can also yield comparable results to glucose restriction. T cells cultured in glutamine depleted conditions or treated with inhibitors of glutamine metabolism were shown to increase survival and promote tumor clearance. Phenotyping of the tumor infiltrate showed that glutamine restricted T cells displayed lower levels of exhaustion markers and increased proliferation, cytokine production, and markers associated with long-lived memory populations. Metabolically, these cells showed increased mitochondrial spare respiratory capacity, reduced mitochondrial ROS, and increased glycolytic flux, indicating that glutamine restriction can skew cells metabolically towards a memory-like phenotype with greater self-renewal and antitumor capabilities (92). Just as in the case of glucose restriction, glutamine restriction can rewire T cells to utilize available nutrients more effectively while also increasing oxidative stress tolerance, giving preconditioned effector cells an edge over their non-pretreated counterparts in the TME.
During glycolysis, the generation of pyruvate from phosphoenolpyruvate (PEP) by pyruvate kinase generates one molecule of ATP (93). Knockdown or pharmacological inhibition of PEP-generating enzymes T cells led to a decrease in calcium flux upon activation in T cells, which showed diminished cytokine production and effector capacities. In T cells, overexpression of phosphoenolpyruvate carboxykinase 1 (PCK1), which can generate PEP from the TCA cycle intermediate oxaloacetate, improved function in both CD4s and CD8s both in terms of measured calcium flux and antitumor effects. Interestingly, overexpression of PCK1 only increased intracellular PEP in low glucose conditions (94), making it a promising modification for increasing efficacy in the glucose-depleted TME while preventing aberrant metabolism in the periphery. This suggests that despite low levels of crucial nutrients in the TME, strategies for increasing expression of intermediates in key pathways can obviate some of the effects of nutrient deprivation in T cells.
Cell therapies comprising greater populations of memory cells compared to terminally differentiated effectors can yield greater antitumor efficacy and long-term durability (95). Other studies have found that a naïve/stem cell memory population resulted in reduced cytokine release syndrome and improved antitumor efficacy compared to bulk unselected T cell infusion, which comprises mostly terminally differentiated effectors; however, stem cell memory T cells are extremely rare in circulation and their expansion requires additional engineering steps; additionally, the initial cytotoxic functions of these cells are not as robust as bulk effectors (96). Both effector/central memory cells and naïve/stem cell memory populations exhibit the ability to expand and differentiate, making them very valuable for cell therapy. One of the hallmarks of a memory T cell is their ability to respond rapidly and robustly to antigen rechallenge. Critical to this rapid recall response is carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase (CAD), the rate-limiting enzyme of the de novo pyrimidine synthesis pathway. Inhibition of CAD decreases available pre-rRNA, which limits the ribosomal biogenesis required for synthesizing new proteins to support proliferation and cytokine production. Overexpression of CAD was shown to enhance proliferation and improve cytokine production upon rechallenge (97). This suggests that methods of increasing flux through the de novo pyrimidine synthesis pathway ex vivo could yield an improved cohort of memory cells for infusion (Figure 4), with enhanced antitumor and proliferative capacities, potentially ameliorating the need for repeated infusions.
Alternate pathways through which T cells generate fuel in glucose-depleted conditions can also identify specific targets for preconditioning regimens to further enhance T cell function. T cells can utilize acetate as an alternative to glucose in glucose-poor environments, and ex vivo exposure to acetate can increase histone acetylation and chromatin accessibility to epigenetically remodel exhausted T cells into active effectors again. These cells show increased cytokine production and promote tumor clearance (98). Inosine can also be utilized by T cells as an alternative to glucose, and treatment with inosine after adoptive transfer also enhances antitumor effector function in tumor models that are unable to metabolize inosine (99). Although it could be speculated that inosine preconditioning would also enhance T cells’ ability to utilize this alternative fuel source in a tumor setting, studies still need to be completed to test this hypothesis. Additionally, the ability of some tumors to also metabolize inosine (100, 101) and thus compete with T cells for this fuel source would also limit its utility.
Strategies for navigating the complexities of mTOR signaling to increase T cell fitness
During T cell activation and differentiation, mTOR signaling through mTORC1 and mTORC2 have differing effects on T cell fate and phenotype. Constitutive mTORC1 activation in T cells generates highly potent effector CD8s, but at the cost of inhibiting transition to a memory phenotype (102, 103). However, although inhibition of mTORC1 led to generation of long-lived memory cells which exhibited poorer effector function, these cells also failed to display a hallmark of memory cells – ability to generate a recall response upon rechallenge. Interestingly, in the T cells where constitutive mTORC1 signaling decreased memory formation, administration of rapamycin – an mTORC1 inhibitor - was able to re-enable the formation of memory recall responses (102). A similar study also showed that rapamycin treatment during initial activation and expansion favored the generation of memory precursors, while treatment during the contraction phase favored the differentiation of memory cells (104). The importance of precisely timing this inhibition is further supported by the observation that mTORC1 signaling is inexorably linked to glucose uptake and glycolysis, and treatment with rapamycin early upon activation both impairs glucose metabolism and decreases perforin production (105). These studies suggest that the inhibition of mTORC1 can be leveraged to achieve both a potent effector response and generation of long-lived memory through the carefully timed targeting of mTORC1 (Figure 5).
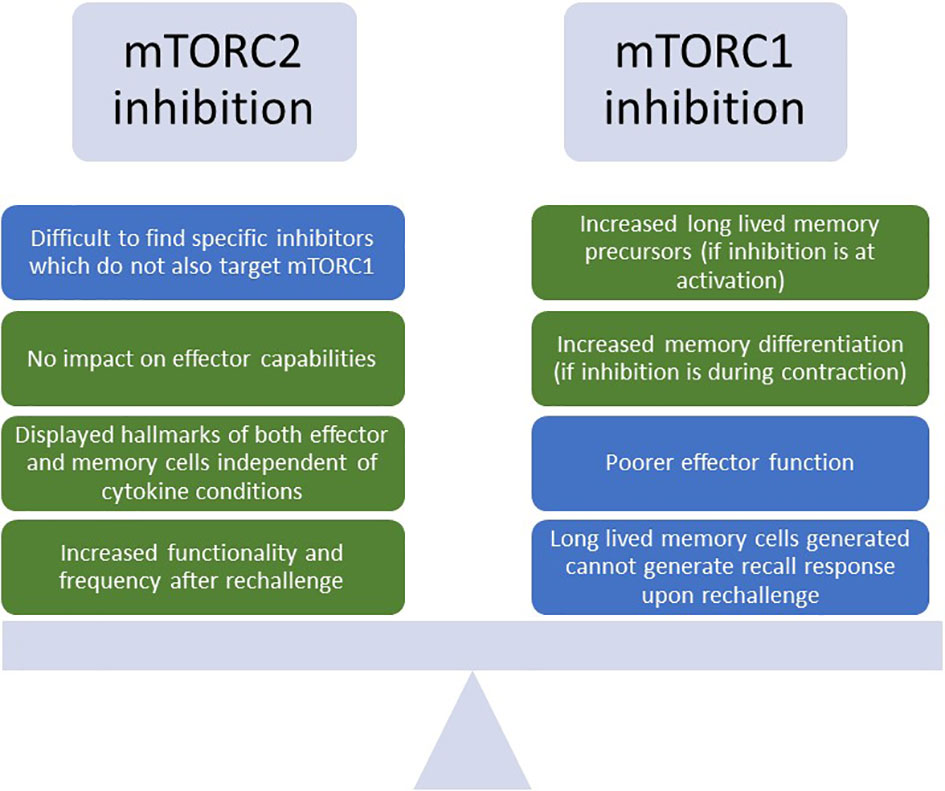
Figure 5 Benefits and drawbacks of inhibiting mTORC1 or mTORC2 to enhance T cell phenotypes for TCR-T. Depiction of the challenges and advantages of inhibiting either mTORC1 (mTOR Complex 1) or mTORC2 (mTOR Complex 2) for T cell enhancement. Green boxes indicate benefits, and blue boxes indicate challenges. mTOR, mammalian target of rapamycin.
Due to the complexity of targeting mTORC1 to enhance potent and long-lived T cell responses, mTORC2 has also emerged as a potential target. T-cell specific knockdown of Rapamycin-insensitive companion of mTOR (Rictor), a binding partner of mTORC2 crucial to its activation, was able to generate robust T cell responses similar to wild type controls upon vaccination, indicating that deficiencies in mTORC2 did not affect effector capabilities. Interestingly, upon rechallenge, these cells displayed increased markers of memory, robust cytokine production, and were present at a higher frequency in the spleen. This is possibly due to the fact that mTORC2 prevents activation and nuclear translocation of Forkhead Box O1 (FOXO1), a transcription factor which is responsible for upregulation of memory-associated transcripts like L-selectin (CD62L) and C-C Motif Chemokine Receptor 7 (CCR7) (106). Additionally, they displayed hallmarks of both effector and memory T cells, with increased glycolytic flux and enhanced spare respiratory capacity (SRC) for long-term survival, regardless of whether the culture media contained cytokines favoring effector or memory formation (102). This study suggests that inhibition of mTORC2 can lead to the generation of T cells bearing favorable characteristics of both potent effectors and long-lived memory, enhancing their survival in the TME while also decreasing their dependence on exogenous cytokines for maintenance of phenotype. Thus, targeting of mTORC2 could be a viable alternative to mTORC1 inhibition in generating cells endowed with both intact effector functions and memory-like persistence, with greater metabolic resilience to the challenges posed by the TME; however, a challenge remains in identifying specific mTORC2 inhibitors which do not target mTORC1 as well (107) (Figure 5).
Personalized strategies for assessing T cell metabolic vulnerabilities
In recent years, advances in sequencing and analysis technologies have made it possible to conduct large-scale screens at a reduced cost, bringing truly personalized medicine within reach. As mentioned previously, different tumors will have varying metabolic vulnerabilities, and T cells from individual patients will also display similar diversity in sensitivities. As such, it may be prudent to leverage in vitro pharmacological or genetic screens to determine optimal treatment regimens for personalized metabolic medicine. A recent study described insights gained from such a screening process, where T-cell and tumor cells were placed in coculture in the presence of 240 different pharmacological perturbations to assess which compounds influenced T cell activation. Not only were they able to gain understandings about vulnerabilities of T cells and their underlying mechanisms, but they were also able to identify compounds in which T cells were differentially affected compared to tumor cells. For example, in the context of a B16-OVA tumor with OT-1 T cells, glutathione peroxidase 4 (GPX4) inhibition induced ferroptosis in CD8+ T cells while leaving tumor cells mostly unaffected, and this sensitivity was attributed to acyl-CoA synthetase long-chain family member 4 (ACSL4) (108), one of the key regulators of ferroptosis (109). One could imagine a scenario in which patient T cells could be initially screened against either tumor organoids or cell lines to identify metabolic enhancements which would benefit T cell survival over that of tumors, or to detect susceptibilities. Through performing personalized tumor-T cell screens, T cells for TCR-T can be engineered to reduce their metabolic vulnerabilities, and concurrent pharmacological interventions to target specific tumor vulnerabilities can be identified.
Strategies for improving TCR-T screening and engineering
The fundamental principle behind TCR-T is that tumor-specific TCRs can be engineered onto a bulk population of T cells, and this population can then be redirected to target the tumor while sparing healthy tissue (110). However, this means that TCR discovery is also of high importance – TCRs must not only be specific for the tumor antigen, but also induce cytotoxicity and cytokine production in T cells once engaged. TCR signal strength has been correlated with T cell functional state, with high-strength interactions inducing inhibitory receptors and acquiring a molecular profile associated with dysfunction (111), suggesting that selecting TCRs with higher affinity may reduce efficacy of the final cell product. As such, methods for identifying TCRs with optimal affinity and influence on T cell phenotype can be very time consuming. These methods can include assessing individual TCR affinity, functional avidity, and conducting cytotoxicity assays to ensure that engineered TCRs have a high enough affinity to detect tumor targets, but not so elevated that it adversely affects their function (112). Understanding how metabolism can influence T cell sensitivity to antigen can decrease TCR screening time, and perhaps increase the number of TCRs that enter the final stages of preclinical evaluation to broaden the targetable patient population.
Another challenge in TCR-T has been optimizing the phenotype of the TCR-engineered T cell population throughout the expansion process to yield long-lived cells which will persist and carry out effector functions in their new niche. It has been found that a central memory phenotype of adoptively transferred T cells was optimal for obtaining the best antitumor responses in patients (113); however, a diverse starting population of T cells poses a challenge as to how to induce this profile for therapy. Previously, we have examined how metabolism plays a crucial role in T cell differentiation into memory and effector subsets, and this knowledge can also be applied to the engineering process to enhance efficacy of the final TCR-T product.
Fine tuning peptide sensitivity of TCRs
T cell sensitivity to their cognate peptide/major histocompatibility molecule (MHC) appear to be governed by both intrinsic and induced factors – there is an intrinsic affinity of a TCR for its ligand, but also induced dynamic TCR clustering (114), both of which modulate the strength of activation. As such, a single T cell clone has the ability to differentiate into both high avidity and low avidity effectors, suggesting that the sensitivity of a T cell can be modulated at the time of activation and is not dependent on the TCR alone (115). After this period of plasticity, T cells develop an activation threshold “set point”, whereupon encounters with sub-threshold or supra-threshold levels of antigen result in apoptosis (116). This poses many issues for TCR therapy, from the initial screening needed for identifying optimal TCRs based on functional avidity, to the engineering process for clinical products. However, understanding the mechanism through which T cells are able to adjust their sensitivity can allow for enhanced cell therapy products and even new applications.
Upon activation, naïve T cells typically switch their metabolism from primarily oxidative phosphorylation to glycolysis (117). However, upon strong peptide stimulation, flux through both OXPHOS and glycolysis were increased. This increased OXPHOS upregulated the transcription factor NFATC1 leading to an increased production of IL-4, which signaled in an autocrine manner to reduce functional avidity of high-peptide-dose stimulated cells. IL-4 neutralizing antibodies were able to block this effect and restore sensitivity (118). Physiologically, the production of IL-4 and its autocrine signaling in cells that have encountered high peptide stimulation can serve as a mechanism for inducing tolerance in the periphery. This has implications for TCR-T, where TCR discovery efforts may be hindered by the activation “set point” of cells used to screen for reactivity. Of course, recent advancements in screening capabilities using phage display or yeast libraries remove the need for using T cells or APCs for initial TCR screening, which allows for identification of putative antigen-reactive TCRs without being affected by T cell or APC phenotypes; however, these systems may not be optimal for low-affinity interactions (119), and T cell-based systems are still needed to validate TCRs for functional relevance. In addition to the complexities of TCR discovery, when it comes to manufacturing the cell product, the activation threshold of different T cells in the starting population may be different even when bearing the same TCR, affecting the sensitivity of the final product (118). It could be postulated that monitoring and adjusting IL-4 levels could help alleviate some of these concerns. Additionally, the fine-tuning of activation threshold can be seen as a blessing, in which T cells could theoretically be able to discern between increased levels of antigen on tumor cells versus low physiological levels of antigen on normal tissue. Due to the challenges of identifying TCRs against antigens specific to the tumor, being able to modulate sensitivity of T cells to target overexpressed tumor antigens such as HER-2 (120) would greatly expand TCR-T treatment to more indications.
Optimizing expansion culture conditions to improve T cell functionality
Cell manufacturing for TCR-T incorporates large-scale cell culture capable of expanding TCR-engineered T cells in preparation for clinical infusion. Multiple factors other than the TCR itself have been shown to influence the final cell product, and composition of media in which cells are expanded can play a large role in their antitumor capabilities (121). It has been shown that expanded T cells in vitro have a different metabolic profile than cells activated in physiologic conditions, with the former acquiring more of a Warburg-like metabolism and the latter displaying higher rates of oxidative metabolism (122). The differentially active metabolic and signaling pathways between cultured and physiologically conditioned cells could raise concerns regarding the fitness of these cultured cells once placed into a physiologic setting, such as patient infusion. A recent study exploring the impact of several different media formulations on cell therapy products found that exposure to ascites from ovarian cancer patients reduced IFN-y and tumor necrosis factor alpha (TNF-a) production by T cells regardless of expansion condition. However, there were differences in the extent to which ascites exposure reduced viability, cytokine production, and mitochondrial activity, suggesting that different media formulations with varying levels of nutrients can affect T cell resilience in TME-like conditions (123). Additionally, supplementing with interleukin-7 and -15 in culture during T cell expansion can promote a central memory phenotype and robust antitumor function (124); IL-7 has been found to induce expression of glycerol channel aquaporin 9 (AQP9) on memory T cells, which imports glycerol for fatty acid esterification and triglyceride synthesis required for survival (125). Additional studies have shown the synergistic potential of IL-21 with IL-15 in enhancing cell function and antitumor effects (126, 127); T cells cultured in IL-21 media increased fatty acid oxidation over glycolysis, skewed towards a central memory phenotype, and displayed reduced exhaustion markers (128). Thus, cytokine supplementation in media can have profound effects on T cell metabolism and phenotype, and can be utilized to generate cell populations with enhanced self-renewal and differentiation capabilities. To date, there is no standardized expansion media condition for cell therapy production across the field. Optimizing expansion conditions by adjusting the levels of key metabolites can generate cell products which persist better in the TME and retain more of their antitumor function under immunosuppressive conditions.
Discussion
Harnessing the immune system for attacking tumors is an area of active investigation, and TCR-T is a promising treatment modality which endow T cells with new specificity against tumor antigens. This has the potential to treat even advanced disease; indeed, a recently published case report highlighted the ability of Kirsten rat sarcoma virus (Kras) G12D mutation-targeting TCR-T to mediate regression of metastatic pancreatic cancer, with the response ongoing 6 months post-treatment (3). To ensure more cases of tumor regression like this one, there needs to be a deeper understanding of how infused T cells interact with the immunosuppressive TME, which has an altered nutrient and metabolite profile. Metabolism plays a key role in T cell fitness and effector capacity, and certain alterations in nutrient conditions can abrogate activation altogether. However, these metabolites also play a role in tumor cell survival, and finding the precise metabolic targets which enhance T cell function at the expense of the tumor can lead to new treatments given alongside TCR-T to improve efficacy. In this review, we highlight several methods of targeting both tumor and T cell metabolism to improve antitumor response. However, one of the largest challenges in the immune-oncology sphere is tumor heterogeneity, and there is no guarantee that metabolic vulnerabilities will be consistent even within the same tumor, let alone across indications. Personalized screening can help to identify unique susceptibilities specific to tumors and patient T cells; however, it will need to keep in mind the limitations set by tumor evolution and heterogeneity. In addition, metabolism is a matter of balance, and as seen with the example of mTOR targeting, perturbations must be timed precisely to be able to yield the desired phenotype. While the plasticity of T cells allows them to acclimate to different nutrient conditions, metabolic interventions during a T cell response raises the possibility of skewing towards immunosuppressive populations such as Tregs, as is the case with mTORC1 inhibition in certain conditions (129). New methods of three dimensional automated coculture screening can more closely mimic dynamics of T cell activation and help identify the effects of individual metabolic perturbations on T cell function and phenotype and identify potential targets for enhancement (130), and can be applied for personalized medicine as well. Further studies into the temporal relationship between metabolism, trajectory of activation, and in vivo phenotype will shed further light on potential treatment regimens. Due to the nature of T cell engineering and expansion for cell therapy, there is a unique window of opportunity to modulate the phenotypic profile of T cells prior to infusion, in addition to post-infusion systemic treatment. Additionally, most metabolic enhancements, such as nutrient preconditioning, are very cost-effective and simple to implement – without the need for lengthy regulatory approvals that accompany genetic engineering.
Future TCR-T approaches will seek to shorten engineering time, improve cell function and phenotype, and broaden the applicability of therapy to more patients. Although initial trials with TCR-T utilized a patient’s own cells for engineering (autologous cell therapy), this requires that engineering and expansion be completed for each individual, which is resource and time intensive. There has recently been a push towards “off the shelf” allogeneic products, whereby healthy donor cells can be engineered to form a bank of cells which can then be infused into any HLA-matched patient, reducing time and engineering costs; additionally, the availability of large numbers of these cells allows for repeat dosing (131, 132). However, as with any transplant scenario, there is concern for graft-versus-host disease (GVHD), and much effort has been put into preventing transferred cells from attacking healthy host tissue due to alloreactivity. In addition to current engineering controls to test TCRs for alloreactivity (133, 134) and suppress endogenous TCR expression (135, 136), infusing cells with a memory phenotype, which is favored for cell therapy, can reduce the chances of GVHD occurrence (137–139). Above, we have discussed multiple methods through which to influence T cell metabolism to generate memory cells which are more resilient and polyfunctional, many of which can be easily translated into current engineering protocols. Metabolic interventions represent a promising treatment modality to augment the efficacy of TCR-T, leading to a more robust cell product that is more resilient to the immunosuppressive effects of the TME and demonstrate superior antitumor efficacy.
Author contributions
WM, conceptualization, writing‐original draft, and figure design.
Acknowledgments
The author would like to acknowledge the research team at BioNTech for their support of this work.
Conflict of interest
The author is an employee of BioNTech USA.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. a preliminary report. N Engl J Med (1988) 319(25):1676–80. doi: 10.1056/NEJM198812223192527
2. Xia Y, Tian X, Wang J, Qiao D, Liu X, Xiao L, et al. Treatment of metastatic non-small cell lung cancer with NY-ESO-1 specific TCR engineered-T cells in a phase I clinical trial: A case report. Oncol Lett (2018) 16(6):6998–7007. doi: 10.3892/ol.2018.9534
3. Leidner R, Sanjuan Silva N, Huang H, Sprott D, Zheng C, Shih YP, et al. Neoantigen T-cell receptor gene therapy in pancreatic cancer. N Engl J Med (2022) 386(22):2112–9. doi: 10.1056/NEJMoa2119662
4. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science (2006) 314(5796):126–9. doi: 10.1126/science.1129003
5. Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol (2011) 29(7):917–24. doi: 10.1200/JCO.2010.32.2537
6. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science (2015) 348(6230):62–8. doi: 10.1126/science.aaa4967
7. Hong DS, Butler MO, Pachynski RK, Sullivan R, Kebriaei P, Boross-Harmer S, et al. Phase 1 clinical trial evaluating the safety and anti-tumor activity of ADP-A2M10 SPEAR T-cells in patients with MAGE-A10+ head and neck, melanoma, or urothelial tumors. Front Oncol (2022) 12:818679. doi: 10.3389/fonc.2022.818679
8. Shablak A, Hawkins RE, Rothwell DG, Elkord E. T Cell-based immunotherapy of metastatic renal cell carcinoma: modest success and future perspective. Clin Cancer Res (2009) 15(21):6503–10. doi: 10.1158/1078-0432.CCR-09-1605
9. Klebanoff CA. T-Cell receptor gene therapy clinically targeting a TP53 public neoantigen. Cancer Immunol Res (2022) 10(8):919. doi: 10.1158/2326-6066.CIR-22-0386
10. Warburg O. On the origin of cancer cells. Science (1956) 123(3191):309–14. doi: 10.1126/science.123.3191.309
11. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the warburg effect: the metabolic requirements of cell proliferation. Science (2009) 324(5930):1029–33. doi: 10.1126/science.1160809
12. Medina MA. Glutamine and cancer. J Nutr (2001) 131(9 Suppl):2539S–42S; discussion 50S-1S. doi: 10.1093/jn/131.9.2539S
13. Roberts E, Simonsen DG, Tanaka KK, Tanaka T. Free amino acids in growing and regressing ascites cell tumors: host resistance and chemical agents. Cancer Res (1956) 16(10 Part 1):970–8.
14. Ngwa VM, Edwards DN, Philip M, Chen J. Microenvironmental metabolism regulates antitumor immunity. Cancer Res (2019) 79(16):4003–8. doi: 10.1158/0008-5472.CAN-19-0617
15. Bellone M, Calcinotto A, Filipazzi P, De Milito A, Fais S, Rivoltini L. The acidity of the tumor microenvironment is a mechanism of immune escape that can be overcome by proton pump inhibitors. Oncoimmunology (2013) 2(1):e22058. doi: 10.4161/onci.22058
16. Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol (2010) 185(2):1037–44. doi: 10.4049/jimmunol.0903586
17. Wu H, Estrella V, Beatty M, Abrahams D, El-Kenawi A, Russell S, et al. T-Cells produce acidic niches in lymph nodes to suppress their own effector functions. Nat Commun (2020) 11(1):4113. doi: 10.1038/s41467-020-17756-7
18. Bannoud N, Dalotto-Moreno T, Kindgard L, Garcia PA, Blidner AG, Marino KV, et al. Hypoxia supports differentiation of terminally exhausted CD8 T cells. Front Immunol (2021) 12:660944. doi: 10.3389/fimmu.2021.660944
19. Raychaudhuri D, Bhattacharya R, Sinha BP, Liu CSC, Ghosh AR, Rahaman O, et al. Lactate induces pro-tumor reprogramming in intratumoral plasmacytoid dendritic cells. Front Immunol (2019) 10:1878. doi: 10.3389/fimmu.2019.01878
20. de Lima Thomaz L, Peron G, Oliveira J, da Rosa LC, Thome R, Verinaud L. The impact of metabolic reprogramming on dendritic cell function. Int Immunopharmacol (2018) 63:84–93. doi: 10.1016/j.intimp.2018.07.031
21. Reinfeld BI, Madden MZ, Wolf MM, Chytil A, Bader JE, Patterson AR, et al. Cell-programmed nutrient partitioning in the tumour microenvironment. Nature (2021) 593(7858):282–8. doi: 10.1038/s41586-021-03442-1
22. Kim LC, Cook RS, Chen J. mTORC1 and mTORC2 in cancer and the tumor microenvironment. Oncogene (2017) 36(16):2191–201. doi: 10.1038/onc.2016.363
23. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell (2012) 149(2):274–93. doi: 10.1016/j.cell.2012.03.017
24. Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol (2004) 14(14):1296–302. doi: 10.1016/j.cub.2004.06.054
25. Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol (2004) 6(11):1122–8. doi: 10.1038/ncb1183
26. Populo H, Lopes JM, Soares P. The mTOR signalling pathway in human cancer. Int J Mol Sci (2012) 13(2):1886–918. doi: 10.3390/ijms13021886
27. Sato T, Nakashima A, Guo L, Coffman K, Tamanoi F. Single amino-acid changes that confer constitutive activation of mTOR are discovered in human cancer. Oncogene (2010) 29(18):2746–52. doi: 10.1038/onc.2010.28
28. Tian T, Li X, Zhang J. mTOR signaling in cancer and mTOR inhibitors in solid tumor targeting therapy. Int J Mol Sci (2019) 20(3):755. doi: 10.3390/ijms20030755
29. Ciuffreda L, Di Sanza C, Incani UC, Milella M. The mTOR pathway: a new target in cancer therapy. Curr Cancer Drug Targets (2010) 10(5):484–95. doi: 10.2174/156800910791517172
30. Pollizzi KN, Powell JD. Regulation of T cells by mTOR: the known knowns and the known unknowns. Trends Immunol (2015) 36(1):13–20. doi: 10.1016/j.it.2014.11.005
31. El Hage A, Dormond O. Combining mTOR inhibitors and T cell-based immunotherapies in cancer treatment. Cancers (Basel) (2021) 13(6):1359. doi: 10.3390/cancers13061359
32. Jin L, Zhou Y. Crucial role of the pentose phosphate pathway in malignant tumors. Oncol Lett (2019) 17(5):4213–21. doi: 10.3892/ol.2019.10112
33. Patra KC, Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci (2014) 39(8):347–54. doi: 10.1016/j.tibs.2014.06.005
34. Kuehne A, Emmert H, Soehle J, Winnefeld M, Fischer F, Wenck H, et al. Acute activation of oxidative pentose phosphate pathway as first-line response to oxidative stress in human skin cells. Mol Cell (2015) 59(3):359–71. doi: 10.1016/j.molcel.2015.06.017
35. Mele L, la Noce M, Paino F, Regad T, Wagner S, Liccardo D, et al. Glucose-6-phosphate dehydrogenase blockade potentiates tyrosine kinase inhibitor effect on breast cancer cells through autophagy perturbation. J Exp Clin Cancer Res (2019) 38(1):160. doi: 10.1186/s13046-019-1164-5
36. Sukhatme VP, Chan B. Glycolytic cancer cells lacking 6-phosphogluconate dehydrogenase metabolize glucose to induce senescence. FEBS Lett (2012) 586(16):2389–95. doi: 10.1016/j.febslet.2012.05.052
37. Mele L, Paino F, Papaccio F, Regad T, Boocock D, Stiuso P, et al. A new inhibitor of glucose-6-phosphate dehydrogenase blocks pentose phosphate pathway and suppresses malignant proliferation and metastasis in vivo. Cell Death Dis (2018) 9(5):572. doi: 10.1038/s41419-018-0635-5
38. Daneshmandi S, Cassel T, Lin P, Higashi RM, Wulf GM, Boussiotis VA, et al. Blockade of 6-phosphogluconate dehydrogenase generates CD8(+) effector T cells with enhanced anti-tumor function. Cell Rep (2021) 34(10):108831. doi: 10.1016/j.celrep.2021.108831
39. Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer (2016) 16(10):619–34. doi: 10.1038/nrc.2016.71
40. Zhang J, Pavlova NN, Thompson CB. Cancer cell metabolism: the essential role of the nonessential amino acid, glutamine. EMBO J (2017) 36(10):1302–15. doi: 10.15252/embj.201696151
41. Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci (2010) 35(8):427–33. doi: 10.1016/j.tibs.2010.05.003
42. Leone RD, Zhao L, Englert JM, Sun IM, Oh MH, Sun IH, et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science (2019) 366(6468):1013–21. doi: 10.1126/science.aav2588
43. Edwards DN, Ngwa VM, Raybuck AL, Wang S, Hwang Y, Kim LC, et al. Selective glutamine metabolism inhibition in tumor cells improves antitumor T lymphocyte activity in triple-negative breast cancer. J Clin Invest (2021) 131(4):e140100. doi: 10.1172/JCI140100
44. Jiang B. Aerobic glycolysis and high level of lactate in cancer metabolism and microenvironment. Genes Dis (2017) 4(1):25–7. doi: 10.1016/j.gendis.2017.02.003
45. Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab (2016) 24(5):657–71. doi: 10.1016/j.cmet.2016.08.011
46. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood (2007) 109(9):3812–9. doi: 10.1182/blood-2006-07-035972
47. Renner K, Bruss C, Schnell A, Koehl G, Becker HM, Fante M, et al. Restricting glycolysis preserves T cell effector functions and augments checkpoint therapy. Cell Rep (2019) 29(1):135–50 e9. doi: 10.1016/j.celrep.2019.08.068
48. Zhang YX, Zhao YY, Shen J, Sun X, Liu Y, Liu H, et al. Nanoenabled modulation of acidic tumor microenvironment reverses anergy of infiltrating T cells and potentiates anti-PD-1 therapy. Nano Lett (2019) 19(5):2774–83. doi: 10.1021/acs.nanolett.8b04296
49. Moffett JR, Namboodiri MA. Tryptophan and the immune response. Immunol Cell Biol (2003) 81(4):247–65. doi: 10.1046/j.1440-1711.2003.t01-1-01177.x
50. Moon YW, Hajjar J, Hwu P, Naing A. Targeting the indoleamine 2,3-dioxygenase pathway in cancer. J Immunother Cancer (2015) 3:51. doi: 10.1186/s40425-015-0094-9
51. Corm S, Berthon C, Imbenotte M, Biggio V, Lhermitte M, Dupont C, et al. Indoleamine 2,3-dioxygenase activity of acute myeloid leukemia cells can be measured from patients' sera by HPLC and is inducible by IFN-gamma. Leuk Res (2009) 33(3):490–4. doi: 10.1016/j.leukres.2008.06.014
52. Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol (2010) 185(6):3190–8. doi: 10.4049/jimmunol.0903670
53. Wu D, Zhu Y. Role of kynurenine in promoting the generation of exhausted CD8(+) T cells in colorectal cancer. Am J Transl Res (2021) 13(3):1535–47.
54. Belladonna ML, Grohmann U, Guidetti P, Volpi C, Bianchi R, Fioretti MC, et al. Kynurenine pathway enzymes in dendritic cells initiate tolerogenesis in the absence of functional IDO. J Immunol (2006) 177(1):130–7. doi: 10.4049/jimmunol.177.1.130
55. Metz R, Rust S, Duhadaway JB, Mautino MR, Munn DH, Vahanian NN, et al. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: A novel IDO effector pathway targeted by d-1-methyl-tryptophan. Oncoimmunology (2012) 1(9):1460–8. doi: 10.4161/onci.21716
56. Isakov N, Altman A. Protein kinase c(theta) in T cell activation. Annu Rev Immunol (2002) 20:761–94. doi: 10.1146/annurev.immunol.20.100301.064807
57. Sharma MD, Hou DY, Liu Y, Koni PA, Metz R, Chandler P, et al. Indoleamine 2,3-dioxygenase controls conversion of Foxp3+ tregs to TH17-like cells in tumor-draining lymph nodes. Blood (2009) 113(24):6102–11. doi: 10.1182/blood-2008-12-195354
58. Long GV, Dummer R, Hamid O, Gajewski TF, Caglevic C, Dalle S, et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol (2019) 20(8):1083–97. doi: 10.1016/S1470-2045(19)30274-8
59. Eynde B, Baren Nv, Baurain J-F. Is there a clinical future for IDO1 inhibitors after the failure of epacadostat in melanoma? Annu Rev Cancer Biol (2020) 4(1):241–56. doi: 10.1146/annurev-cancerbio-030419-033635
60. Triplett TA, Garrison KC, Marshall N, Donkor M, Blazeck J, Lamb C, et al. Reversal of indoleamine 2,3-dioxygenase-mediated cancer immune suppression by systemic kynurenine depletion with a therapeutic enzyme. Nat Biotechnol (2018) 36(8):758–64. doi: 10.1038/nbt.4180
61. Gardner A, de Mingo Pulido A, Ruffell B. Dendritic cells and their role in immunotherapy. Front Immunol (2020) 11:924. doi: 10.3389/fimmu.2020.00924
62. Zammit DJ, Cauley LS, Pham QM, Lefrancois L. Dendritic cells maximize the memory CD8 T cell response to infection. Immunity (2005) 22(5):561–70. doi: 10.1016/j.immuni.2005.03.005
63. Hubbe ML, Jaehger DE, Andresen TL, Andersen MH. Leveraging endogenous dendritic cells to enhance the therapeutic efficacy of adoptive T-cell therapy and checkpoint blockade. Front Immunol (2020) 11:578349. doi: 10.3389/fimmu.2020.578349
64. Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, Tyurin VA, et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med (2010) 16(8):880–6. doi: 10.1038/nm.2172
65. Lee J, Walsh MC, Hoehn KL, James DE, Wherry EJ, Choi Y. Regulator of fatty acid metabolism, acetyl coenzyme a carboxylase 1, controls T cell immunity. J Immunol (2014) 192(7):3190–9. doi: 10.4049/jimmunol.1302985
66. Hardie DG, Carling D. The AMP-activated protein kinase–fuel gauge of the mammalian cell? Eur J Biochem (1997) 246(2):259–73. doi: 10.1111/j.1432-1033.1997.00259.x
67. Han F, Li CF, Cai Z, Zhang X, Jin G, Zhang WN, et al. The critical role of AMPK in driving akt activation under stress, tumorigenesis and drug resistance. Nat Commun (2018) 9(1):4728. doi: 10.1038/s41467-018-07188-9
68. Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol (2005) 6(8):777–84. doi: 10.1038/ni1221
69. Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, et al. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem (1996) 271(44):27879–87. doi: 10.1074/jbc.271.44.27879
70. Carroll KC, Viollet B, Suttles J. AMPKalpha1 deficiency amplifies proinflammatory myeloid APC activity and CD40 signaling. J Leukoc Biol (2013) 94(6):1113–21. doi: 10.1189/jlb.0313157
71. MacIver NJ, Blagih J, Saucillo DC, Tonelli L, Griss T, Rathmell JC, et al. The liver kinase B1 is a central regulator of T cell development, activation, and metabolism. J Immunol (2011) 187(8):4187–98. doi: 10.4049/jimmunol.1100367
72. Blagih J, Coulombe F, Vincent EE, Dupuy F, Galicia-Vazquez G, Yurchenko E, et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity (2015) 42(1):41–54. doi: 10.1016/j.immuni.2014.12.030
73. Eikawa S, Nishida M, Mizukami S, Yamazaki C, Nakayama E, Udono H. Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proc Natl Acad Sci U S A (2015) 112(6):1809–14. doi: 10.1073/pnas.1417636112
74. Hu L, Zeng Z, Xia Q, Liu Z, Feng X, Chen J, et al. Metformin attenuates hepatoma cell proliferation by decreasing glycolytic flux through the HIF-1alpha/PFKFB3/PFK1 pathway. Life Sci (2019) 239:116966. doi: 10.1016/j.lfs.2019.116966
75. Liu Y, Yan X, Zhang F, Zhang X, Tang F, Han Z, et al. TCR-T immunotherapy: The challenges and solutions. Front Oncol (2021) 11:794183. doi: 10.3389/fonc.2021.794183
76. Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell (2015) 162(6):1229–41. doi: 10.1016/j.cell.2015.08.016
77. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun (2015) 6:6692. doi: 10.1038/ncomms7692
78. van Duijn A, Willemsen KJ, van Uden NOP, Hoyng L, Erades S, Koster J, et al. A secondary role for hypoxia and HIF1 in the regulation of (IFNgamma-induced) PD-L1 expression in melanoma. Cancer Immunol Immunother (2022) 71(3):529–40. doi: 10.1007/s00262-021-03007-1
79. Messai Y, Gad S, Noman MZ, Le Teuff G, Couve S, Janji B, et al. Renal cell carcinoma programmed death-ligand 1, a new direct target of hypoxia-inducible factor-2 alpha, is regulated by von hippel-lindau gene mutation status. Eur Urol (2016) 70(4):623–32. doi: 10.1016/j.eururo.2015.11.029
80. Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res (2014) 74(3):665–74. doi: 10.1158/0008-5472.CAN-13-0992
81. Ruf M, Moch H, Schraml P. PD-L1 expression is regulated by hypoxia inducible factor in clear cell renal cell carcinoma. Int J Cancer (2016) 139(2):396–403. doi: 10.1002/ijc.30077
82. Yu Y, Liang Y, Li D, Wang L, Liang Z, Chen Y, et al. Glucose metabolism involved in PD-L1-mediated immune escape in the malignant kidney tumour microenvironment. Cell Death Discovery (2021) 7(1):15. doi: 10.1038/s41420-021-00401-7
83. Haas R, Smith J, Rocher-Ros V, Nadkarni S, Montero-Melendez T, D'Acquisto F, et al. Lactate regulates metabolic and pro-inflammatory circuits in control of T cell migration and effector functions. PloS Biol (2015) 13(7):e1002202. doi: 10.1371/journal.pbio.1002202
84. Guak H, Krawczyk CM. Implications of cellular metabolism for immune cell migration. Immunology (2020) 161(3):200–8. doi: 10.1111/imm.13260
85. Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science (2013) 342(6155):1242454. doi: 10.1126/science.1242454
86. Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab (2014) 20(1):61–72. doi: 10.1016/j.cmet.2014.05.004
87. O'Sullivan D, van der Windt GJ, Huang SC, Curtis JD, Chang CH, Buck MD, et al. Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity (2014) 41(1):75–88. doi: 10.1016/j.immuni.2014.06.005
88. Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest (2013) 123(10):4479–88. doi: 10.1172/JCI69589
89. Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O'Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell (2013) 153(6):1239–51. doi: 10.1016/j.cell.2013.05.016
90. Daneshmandi S, Cassel T, Higashi RM, Fan TW, Seth P. 6-phosphogluconate dehydrogenase (6PGD), a key checkpoint in reprogramming of regulatory T cells metabolism and function. Elife (2021) 10:e67476. doi: 10.7554/eLife.67476
91. Klein Geltink RI, Edwards-Hicks J, Apostolova P, O'Sullivan D, Sanin DE, Patterson AE, et al. Metabolic conditioning of CD8(+) effector T cells for adoptive cell therapy. Nat Metab (2020) 2(8):703–16. doi: 10.1038/s42255-020-0256-z
92. Nabe S, Yamada T, Suzuki J, Toriyama K, Yasuoka T, Kuwahara M, et al. Reinforce the antitumor activity of CD8(+) T cells via glutamine restriction. Cancer Sci (2018) 109(12):3737–50. doi: 10.1111/cas.13827
93. Bar-Even A, Flamholz A, Noor E, Milo R. Rethinking glycolysis: on the biochemical logic of metabolic pathways. Nat Chem Biol (2012) 8(6):509–17. doi: 10.1038/nchembio.971
94. Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell (2015) 162(6):1217–28. doi: 10.1016/j.cell.2015.08.012
95. Blaeschke F, Stenger D, Kaeuferle T, Willier S, Lotfi R, Kaiser AD, et al. Induction of a central memory and stem cell memory phenotype in functionally active CD4(+) and CD8(+) CAR T cells produced in an automated good manufacturing practice system for the treatment of CD19(+) acute lymphoblastic leukemia. Cancer Immunol Immunother (2018) 67(7):1053–66. doi: 10.1007/s00262-018-2155-7
96. Arcangeli S, Bove C, Mezzanotte C, Camisa B, Falcone L, Manfredi F, et al. CAR T cell manufacturing from naive/stem memory T lymphocytes enhances antitumor responses while curtailing cytokine release syndrome. J Clin Invest (2022) 132(12):e150807. doi: 10.1172/JCI150807
97. Claiborne MD, Sengupta S, Zhao L, Arwood ML, Sun IM, Wen J, et al. Persistent CAD activity in memory CD8(+) T cells supports rRNA synthesis and ribosomal biogenesis required at rechallenge. Sci Immunol (2022) 7(71):eabh4271. doi: 10.1126/sciimmunol.abh4271
98. Qiu J, Villa M, Sanin DE, Buck MD, O'Sullivan D, Ching R, et al. Acetate promotes T cell effector function during glucose restriction. Cell Rep (2019) 27(7):2063–74 e5. doi: 10.1016/j.celrep.2019.04.022
99. Wang T, Gnanaprakasam JNR, Chen X, Kang S, Xu X, Sun H, et al. Inosine is an alternative carbon source for CD8(+)-t-cell function under glucose restriction. Nat Metab (2020) 2(7):635–47. doi: 10.1038/s42255-020-0219-4
100. Wieczorek P, Balut-Wieczorek M, Jasinski M, Szablonski W, Antczak A. Inosine monophosphate dehydrogenase 2 as a marker of aggressive and advanced prostate cancer. Cent Eur J Urol (2018) 71(4):399–403. doi: 10.5173/ceju.2018.1696
101. Duan S, Huang W, Liu X, Liu X, Chen N, Xu Q, et al. IMPDH2 promotes colorectal cancer progression through activation of the PI3K/AKT/mTOR and PI3K/AKT/FOXO1 signaling pathways. J Exp Clin Cancer Res (2018) 37(1):304. doi: 10.1186/s13046-018-0980-3
102. Pollizzi KN, Patel CH, Sun IH, Oh MH, Waickman AT, Wen J, et al. mTORC1 and mTORC2 selectively regulate CD8(+) T cell differentiation. J Clin Invest (2015) 125(5):2090–108. doi: 10.1172/JCI77746
103. Sowell R, Rogozinska M, Marzo A. Signaling through Tsc2-mTOR regulates memory CD8 T cell differentiation. (LYM4P.755). J Immunol (2014) 192(1 Supplement):65.12–2. Available at: https://www.jimmunol.org/content/192/1_Supplement/65.12
104. Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, et al. mTOR regulates memory CD8 T-cell differentiation. Nature (2009) 460(7251):108–12. doi: 10.1038/nature08155
105. Finlay DK, Rosenzweig E, Sinclair LV, Feijoo-Carnero C, Hukelmann JL, Rolf J, et al. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J Exp Med (2012) 209(13):2441–53. doi: 10.1084/jem.20112607
106. Kerdiles YM, Beisner DR, Tinoco R, Dejean AS, Castrillon DH, DePinho RA, et al. Foxo1 links homing and survival of naive T cells by regulating l-selectin, CCR7 and interleukin 7 receptor. Nat Immunol (2009) 10(2):176–84. doi: 10.1038/ni.1689
107. Murray ER, Cameron AJM. Towards specific inhibition of mTORC2. Aging (Albany NY) (2017) 9(12):2461–2. doi: 10.18632/aging.101346
108. Drijvers JM, Gillis JE, Muijlwijk T, Nguyen TH, Gaudiano EF, Harris IS, et al. Pharmacologic screening identifies metabolic vulnerabilities of CD8(+) T cells. Cancer Immunol Res (2021) 9(2):184–99. doi: 10.1158/2326-6066.CIR-20-0384
109. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol (2017) 13(1):91–8. doi: 10.1038/nchembio.2239
110. Xu Y, Morales AJ, Cargill MJ, Towlerton AMH, Coffey DG, Warren EH, et al. Preclinical development of T-cell receptor-engineered T-cell therapy targeting the 5T4 tumor antigen on renal cell carcinoma. Cancer Immunol Immunother (2019) 68(12):1979–93. doi: 10.1007/s00262-019-02419-4
111. Shakiba M, Zumbo P, Espinosa-Carrasco G, Menocal L, Dundar F, Carson SE, et al. TCR signal strength defines distinct mechanisms of T cell dysfunction and cancer evasion. J Exp Med (2022) 219(2):e20201966. doi: 10.1084/jem.20201966
112. Campillo-Davo D, Flumens D, Lion E. The quest for the best: How TCR affinity, avidity, and functional avidity affect TCR-engineered T-cell antitumor responses. Cells (2020) 9(7):1720. doi: 10.3390/cells9071720
113. Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med (2018) 24(5):563–71. doi: 10.1038/s41591-018-0010-1
114. Drake DR 3rd, Braciale TJ. Cutting edge: lipid raft integrity affects the efficiency of MHC class I tetramer binding and cell surface TCR arrangement on CD8+ T cells. J Immunol (2001) 166(12):7009–13. doi: 10.4049/jimmunol.166.12.7009
115. Kroger CJ, Alexander-Miller MA. Cutting edge: CD8+ T cell clones possess the potential to differentiate into both high- and low-avidity effector cells. J Immunol (2007) 179(2):748–51. doi: 10.4049/jimmunol.179.2.748
116. Alexander-Miller MA, Leggatt GR, Sarin A, Berzofsky JA. Role of antigen, CD8, and cytotoxic T lymphocyte (CTL) avidity in high dose antigen induction of apoptosis of effector CTL. J Exp Med (1996) 184(2):485–92. doi: 10.1084/jem.184.2.485
117. Roos D, Loos JA. Changes in the carbohydrate metabolism of mitogenically stimulated human peripheral lymphocytes. II. relative importance of glycolysis and oxidative phosphorylation on phytohaemagglutinin stimulation. Exp Cell Res (1973) 77(1):127–35. doi: 10.1016/0014-4827(73)90561-2
118. Crofts KF, Holbrook BC, Soto-Pantoja DR, Ornelles DA, Alexander-Miller MA. TCR dependent metabolic programming regulates autocrine IL-4 production resulting in self-tuning of the CD8(+) T cell activation setpoint. Front Immunol (2020) 11:540. doi: 10.3389/fimmu.2020.00540
119. Wang L, Lan X. Rapid screening of TCR-pMHC interactions by the YAMTAD system. Cell Discovery (2022) 8(1):30. doi: 10.1038/s41421-022-00386-2
120. Gutierrez C, Schiff R. HER2: biology, detection, and clinical implications. Arch Pathol Lab Med (2011) 135(1):55–62. doi: 10.5858/2010-0454-RAR.1
121. Stock S, Schmitt M, Sellner L. Optimizing manufacturing protocols of chimeric antigen receptor T cells for improved anticancer immunotherapy. Int J Mol Sci (2019) 20(24):6223. doi: 10.3390/ijms20246223
122. Ma EH, Verway MJ, Johnson RM, Roy DG, Steadman M, Hayes S, et al. Metabolic profiling using stable isotope tracing reveals distinct patterns of glucose utilization by physiologically activated CD8(+) T cells. Immunity (2019) 51(5):856–70 e5. doi: 10.1016/j.immuni.2019.09.003
123. MacPherson S, Keyes S, Kilgour MK, Smazynski J, Chan V, Sudderth J, et al. Clinically relevant T cell expansion media activate distinct metabolic programs uncoupled from cellular function. Mol Ther Methods Clin Dev (2022) 24:380–93. doi: 10.1016/j.omtm.2022.02.004
124. Cha E, Graham L, Manjili MH, Bear HD. IL-7 + IL-15 are superior to IL-2 for the ex vivo expansion of 4T1 mammary carcinoma-specific T cells with greater efficacy against tumors in vivo. Breast Cancer Res Treat (2010) 122(2):359–69. doi: 10.1007/s10549-009-0573-0
125. Cui G, Staron MM, Gray SM, Ho PC, Amezquita RA, Wu J, et al. IL-7-Induced glycerol transport and TAG synthesis promotes memory CD8+ T cell longevity. Cell (2015) 161(4):750–61. doi: 10.1016/j.cell.2015.03.021
126. Zeng R, Spolski R, Finkelstein SE, Oh S, Kovanen PE, Hinrichs CS, et al. Synergy of IL-21 and IL-15 in regulating CD8+ T cell expansion and function. J Exp Med (2005) 201(1):139–48. doi: 10.1084/jem.20041057
127. Li Y, Cong Y, Jia M, He Q, Zhong H, Zhao Y, et al. Targeting IL-21 to tumor-reactive T cells enhances memory T cell responses and anti-PD-1 antibody therapy. Nat Commun (2021) 12(1):951. doi: 10.1038/s41467-021-21241-0
128. Loschinski R, Bottcher M, Stoll A, Bruns H, Mackensen A, Mougiakakos D. IL-21 modulates memory and exhaustion phenotype of T-cells in a fatty acid oxidation-dependent manner. Oncotarget (2018) 9(17):13125–38. doi: 10.18632/oncotarget.24442
129. Chapman NM, Chi H. mTOR signaling, tregs and immune modulation. Immunotherapy (2014) 6(12):1295–311. doi: 10.2217/imt.14.84
130. Slaats J, Dieteren CE, Wagena E, Wolf L, Raaijmakers TK, van der Laak JA, et al. Metabolic screening of cytotoxic T-cell effector function reveals the role of CRAC channels in regulating lethal hit delivery. Cancer Immunol Res (2021) 9(8):926–38. doi: 10.1158/2326-6066.CIR-20-0741
131. Martinez Bedoya D, Dutoit V, Migliorini D. Allogeneic CAR T cells: An alternative to overcome challenges of CAR T cell therapy in glioblastoma. Front Immunol (2021) 12:640082. doi: 10.3389/fimmu.2021.640082
132. Zhao J, Lin Q, Song Y, Liu D. Universal CARs, universal T cells, and universal CAR T cells. J Hematol Oncol (2018) 11(1):132. doi: 10.1186/s13045-018-0677-2
133. Poncette L, Chen X, Lorenz FK, Blankenstein T. Effective NY-ESO-1-specific MHC II-restricted T cell receptors from antigen-negative hosts enhance tumor regression. J Clin Invest (2019) 129(1):324–35. doi: 10.1172/JCI120391
134. Sanderson JP, Crowley DJ, Wiedermann GE, Quinn LL, Crossland KL, Tunbridge HM, et al. Preclinical evaluation of an affinity-enhanced MAGE-A4-specific T-cell receptor for adoptive T-cell therapy. Oncoimmunology (2020) 9(1):1682381. doi: 10.1080/2162402X.2019.1682381
135. Stenger D, Stief TA, Kaeuferle T, Willier S, Rataj F, Schober K, et al. Endogenous TCR promotes in vivo persistence of CD19-CAR-T cells compared to a CRISPR/Cas9-mediated TCR knockout CAR. Blood (2020) 136(12):1407–18. doi: 10.1182/blood.2020005185
136. Thomas S, Stauss HJ, Morris EC. Molecular immunology lessons from therapeutic T-cell receptor gene transfer. Immunology (2010) 129(2):170–7. doi: 10.1111/j.1365-2567.2009.03227.x
137. Chen BJ, Deoliveira D, Cui X, Le NT, Son J, Whitesides JF, et al. Inability of memory T cells to induce graft-versus-host disease is a result of an abortive alloresponse. Blood (2007) 109(7):3115–23. doi: 10.1182/blood-2006-04-016410
138. Anderson BE, McNiff J, Yan J, Doyle H, Mamula M, Shlomchik MJ, et al. Memory CD4+ T cells do not induce graft-versus-host disease. J Clin Invest (2003) 112(1):101–8. doi: 10.1172/JCI17601
Keywords: metabolism, T-cell receptor therapy, adoptive cell therapy, tumor microenvironment, cancer immunology and immunotherapy, T cell fitness
Citation: Mao W (2022) Overcoming current challenges to T-cell receptor therapy via metabolic targeting to increase antitumor efficacy, durability, and tolerability. Front. Immunol. 13:1056622. doi: 10.3389/fimmu.2022.1056622
Received: 29 September 2022; Accepted: 31 October 2022;
Published: 21 November 2022.
Edited by:
Laura A. Solt, University of Florida, United StatesReviewed by:
Anna Schurich, Department of Immunobiology, Faculty of Life Sciences & Medicine, King’s College London, United KingdomScott Weber, Brigham Young University, United States
Copyright © 2022 Mao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wendy Mao, wendy.mao@biontech.us