 Kei Takasawa
Kei Takasawa Hirokazu Kanegane
Hirokazu Kanegane Kenichi Kashimada
Kenichi Kashimada Tomohiro Morio
Tomohiro Morio- 1Department of Pediatrics and Developmental Biology, Tokyo Medical and Dental University (TMDU), Tokyo, Japan
- 2Deparment of Child Health Development, Tokyo Medical and Dental University (TMDU), Tokyo, Japan
Inborn errors of immunity (IEI), caused by hereditary or genetic defects, are a group of more than 400 disorders, in which the immune system, including lymphocytes, neutrophils, macrophages, and complements, does not function properly. The endocrine system is frequently affected by IEI as an associated clinical feature and a complex network of glands which regulate many important body functions, including growth, reproduction, homeostasis, and energy regulation. Most endocrine disorders associated with IEI are hypofunction which would be treated with supplementation therapy, and early diagnosis and appropriate management are essential for favorable long-term outcomes in patients with IEI. In this review, we aimed to comprehensively summarize and discuss the current understanding on the clinical features and the pathophysiology of endocrine disorders in IEI. This review is composed with three parts. First, we discuss the two major pathophysiology of endocrinopathy in IEI, autoimmune response and direct effects of the responsible genes. Next, the details of each endocrinopathy, such as growth failure, hypothyroidism, hypoparathyroidism, adrenal insufficiency, diabetes mellitus (DM) are specified. We also illustrated potential endocrinopathy due to hematopoietic stem cell transplantation, including hypogonadism and adrenal insufficiency due to glucocorticoid therapy.
Introduction
Inborn errors of immunity (IEI), caused by hereditary or genetic defects, are a group of more than 400 disorders, in which the immune system, including lymphocytes, neutrophils, macrophages, and complements, does not function properly (1–4). While the clinical characteristics of each IEI differs, defects in the functions of the immune system generally lead to increased susceptibility to infection, which can be life-threatening or can cause permanent damage to various organs. Although the clinical symptoms of IEI are generally present at birth or in early childhood, patients can be affected with IEI at any age (1–4).
In addition to opportunistic infections, various clinical features are also involved in IEI. Autoimmunity and abnormal inflammation in the absence of apparent infection have often been observed clinically in association with IEI, and IEI has been linked to specific autoimmune/allergic complications at various frequencies (5). Furthermore, numerous causative genes of IEI are involved in the fundamental functions of cell differentiation and proliferation and are shared by various types of cells. Taken together, the clinical problems caused by the impaired molecular function of IEI genes are not limited to immunological systems (5).
Clinical complications of IEI can be classified into three groups according to the causes, the direct organ damages from opportunistic infections, chronic autoimmune/allergic inflammation, and impaired molecular functions of the causative genes (2, 5). Further, abnormal cell proliferation or carcinogenesis due to IEI could be form the pathogenesis of endocrinopathy. In any case, every organ can be affected by IEI, and for the clinical management of IEI, interventions for both opportunistic infections and clinical complications are essential.
The endocrine system is not an exception and is frequently affected by IEI as an associated clinical feature (Table 1) (12). The endocrine system is a complex network of glands that produce and release hormones which regulate many important body functions, including growth, reproduction, homeostasis, and energy regulation. During childhood, the endocrine system is essential for acquiring normal growth and secondary sexual characteristics (13), and pediatric endocrinopathy seriously affects long-term outcomes. On the other hand, most endocrine disorders associated with IEI are hypofunctions that can be treated with supplementation therapy. Therefore, early diagnosis and appropriate management are essential for favorable long-term outcomes in patients with IEI. In this review, we aim to summarize and discuss the current understanding of the clinical features and pathophysiology of endocrine disorders in IEI.
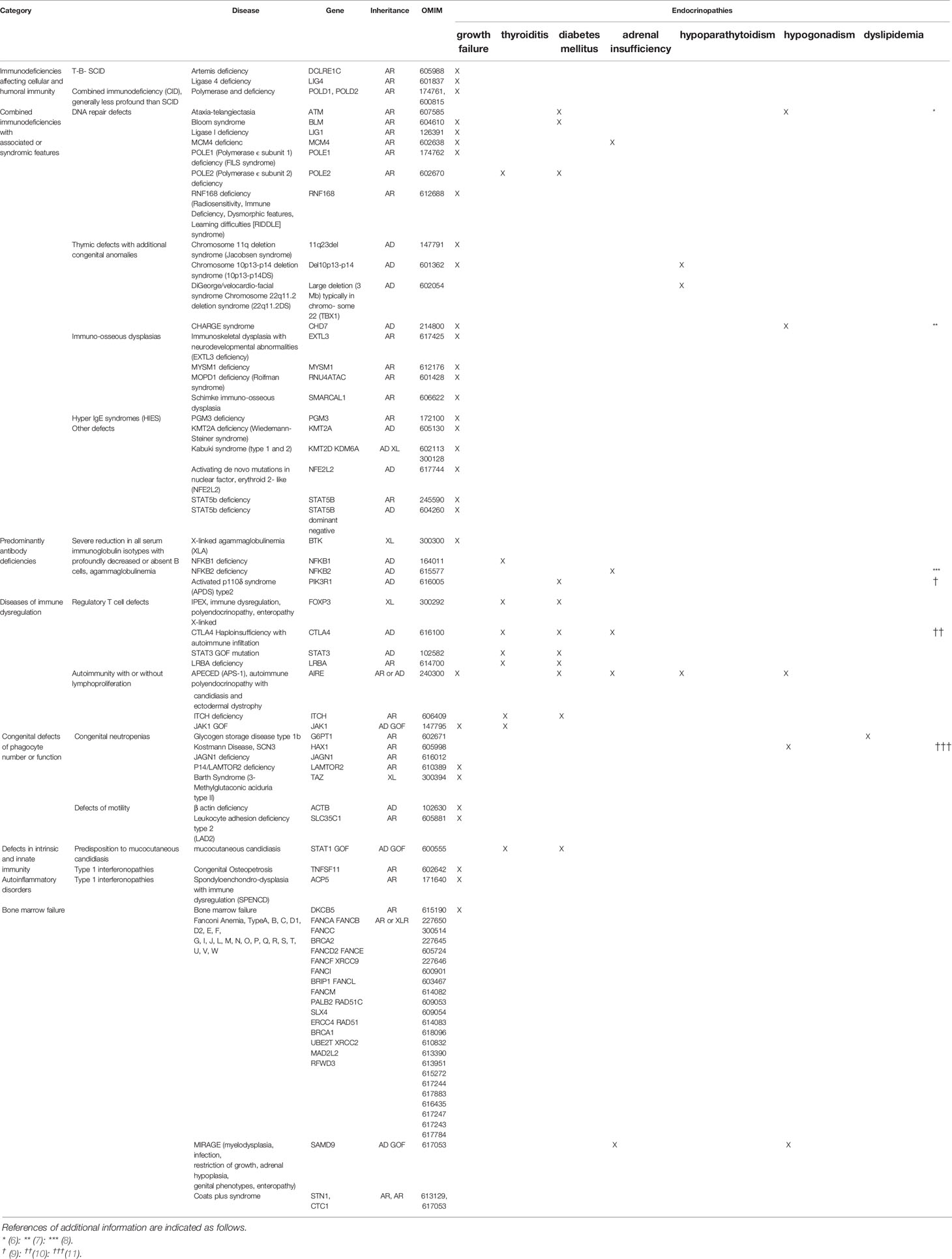
Table 1 Modified the classification of the International Union of Immunological Societies Expert Committee according to complications of endocrinopathy (2).
Pathophysiology of Endocrinopathy in IEI
Autoimmune
The immune system becomes self-tolerant through mechanisms called “tolerance”. Induction of tolerance is accomplished by education of both B and T cells, which occurs in both central (bone marrow and thymus) and peripheral (spleen and lymph nodes) lymphoid organs, and immune dysregulation causes autoimmune/allergic responses (14–17). Autoimmune diseases can be classified into systemic diseases, such as systemic lupus erythematosus, rheumatoid arthritis (RA), and systemic sclerosis, and organ-specific diseases, such as type 1 diabetes mellitus (T1DM) and autoimmune thyroiditis (17). Endocrine organs are the major targets of organ-specific autoimmune diseases, and most endocrinopathies in IEI are presumably mediated by autoimmunity (14–16).
The most common autoimmune endocrine disorders are autoimmune thyroid disorders and T1DM (2). Despite extremely rare conditions, hypophysitis, adrenalitis, ovarian failure, and hypoparathyroidism can occur due to an autoimmune response. Autoimmune endocrine disorders are characterized by associations with autoantibodies and/or autoreactive lymphocytes, which result from an interaction between environmental factors and genetic predisposition. For a genetic contribution to autoimmune endocrinopathies, epidemic studies of twins provide robust evidence that monozygotic twins have a higher concordance rate for disease than dizygotic twins, and familial accumulation is frequently observed. Most autoimmune endocrine disorders have been reported to be associated with specific major histocompatibility complex (MHC)/human leukocyte antigen (HLA) molecules.
Despite the long history of intensive investigations, the precise genetic mechanisms underlying autoimmune endocrine disorders are poorly understood. One of the possible explanations is the “autoimmune surveillance of hypersecreting mutants” (ASHM) hypothesis. The presence of autoreactive T cells whose nature is detecting self-antigens from hormone secretion pathway, dedicates to detect and to remove malfunctioned endocrine cells which potentially disrupt organismal homeostasis. The speculation looks persuasive, nevertheless, the speculation does not clarify the reason why certain endocrine organs, such as thyroid gland and beta cells, tend to be more affected.
A recent significant expansion of our understanding of monogenic IEI, which is associated with autoimmune endocrine disorders, would provide valuable insights into their pathophysiology.
Classically, autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED) and immunodeficiency, polyendocrinopathy, and enteropathy X-Linked syndrome (IPEX) exemplify monogenic autoimmune disorders associated with endocrine disorders (Figure 1). From the discovery of the AIRE gene in APECED (18, 19), defects in central tolerance with alteration of self-antigen expression levels in the thymus would be a cause of T1DM, and strictly controlled negative selection of T cells through tissue-specific self-antigen expression in the thymus is essential for preventing autoimmune endocrine disorders (20). Studies of another IEI associated with T1DM, IPEX syndrome have shown that the responsible gene, FOXP3 (21, 22), plays a critical role in the function of CD4+CD25+ regulatory T (Treg) cells (23, 24), and its defects cause loss of peripheral Tregs, leading to autoreactive T cell activation and proliferation (20, 23, 24).
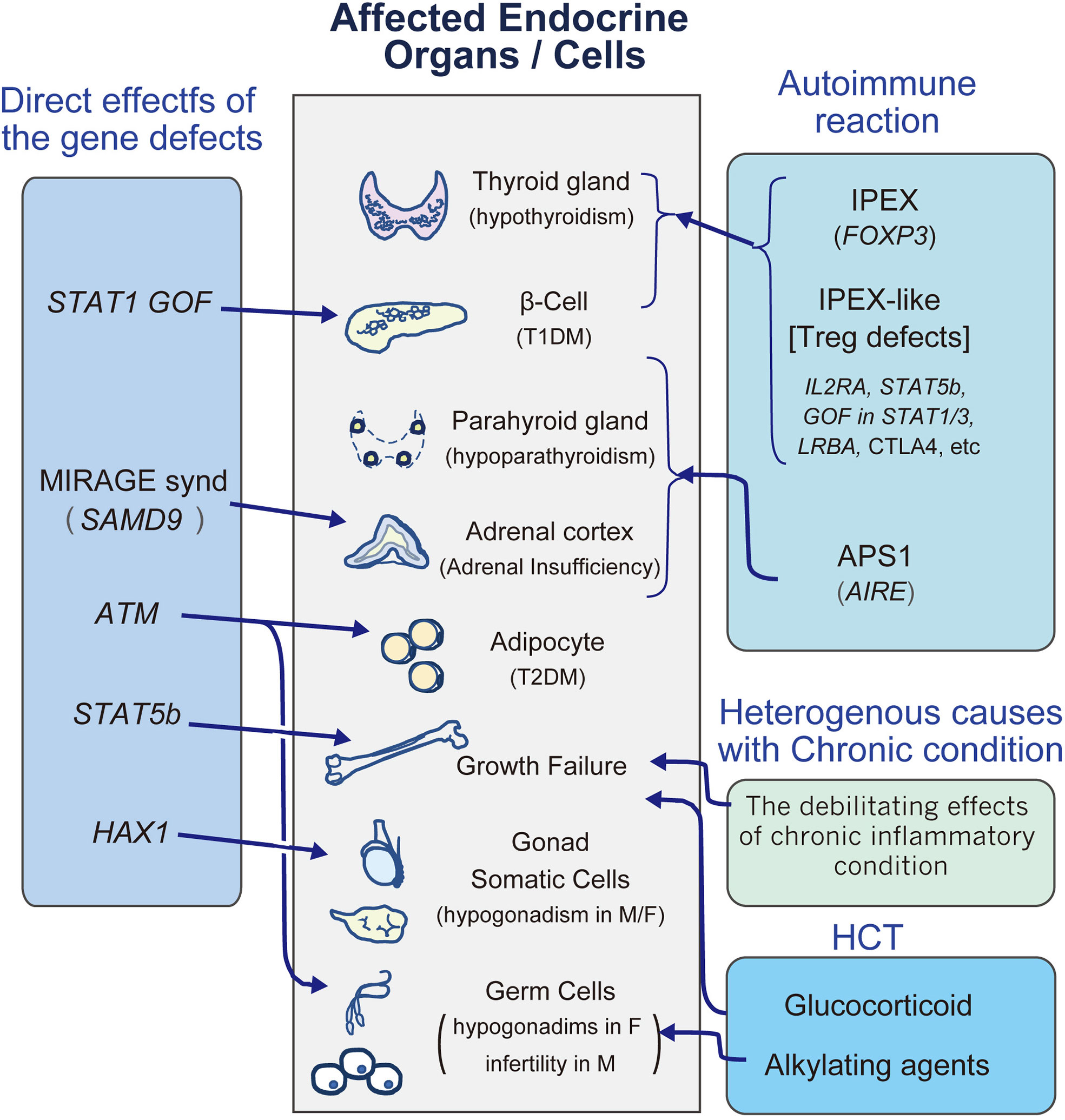
Figure 1 A diagram to review the major endocrinopathies in IEI. M, male; F, female; HCT, hematopoietic stem cell transplantation; T1DM, type1 diabetes mellitus; T2DM, type2 diabetes mellitus.
Recent studies have revealed that other molecules are involved in monogenic autoimmune disorders involving endocrinopathies. In cohorts of subjects with an IPEX-like phenotype, up to half do not exhibit pathogenic variant in FOXP3, and recent investigations revealed that a number of gene defects involved in Treg cell function cause an IPEX-like phenotype, such as IL2RA, STAT5b, GOF in STAT1/3, and LRBA (Figure 1) (25–32).
Additionally, several monogenic disorders in immune function have also been reported to be involved in T1DM. Immune checkpoint proteins play a central role in balancing effective immune responses to pathogens, as well as regulating autoimmune responses against self-tissues. CTLA4 is one of the components, and heterozygous deficiency of the protein gives rise to inappropriate polyclonal T-cell activation (10, 33, 34), leading to highly variable features of autoimmune responses, which may include endocrinopathies of adrenal insufficiency, T1DM, and thyroiditis (35). On the other hand, anti-CTLA-4 antibody therapy, which is a novel therapeutic approach to malignancy by targeting immune checkpoint proteins, is associated with serious immune-related adverse events, such as autoimmune hypophysitis (36, 37).
Endocrinopathy would be comorbidity of recently identified IEI. Inherited PD-1 deficiency and activated PI3Kδ syndrome (APDS) type 2, would associate with T1DM (38–40). Familial thyroiditis and T1DM are reported in patients with A20 (TNFAIP3) haploinsufficiency (41, 42).
Autoimmune endocrine disorders are progressive and are considered untreatable and irreversible. Generally, the treatment of endocrinopathy involves hormone supplementation instead of immunological therapy. However, the experience of endocrine disorders in IEI would provide valuable insights for developing novel therapeutic approaches for autoimmune endocrine disorders. Early hematopoietic cell transplantation (HCT) for IPEX appears to reverse the autoimmune complications, including T1DM, suggesting that Treg cells may be more tractable for the induction of tolerance and could be a possible target for the radical treatment of T1DM (9, 17, 43). Consistently, administration of ruxolitinib, an inhibitor of Janus kinase (JAK) 1 and 2 reversed hyperglycemia of T1DM in a case of STAT1 GOF (44).
Direct Effects of the Responsible Genes
Some endocrine disorders in IEI are thought to be caused by the direct influence of molecular defects (Figure 1).
STAT5b is a transcription factor that activates the transcription of IL-2Rα, FOXP3, and Bcl-2, and its deficiency exhibits combined immunodeficiencies with a reduced number of Treg cells and increased IgE (27, 45). The JAK2 and STAT pathways (STAT1, STAT3, STAT5A, and STAT5B) are also involved in the growth hormone (GH) receptor pathway, and we will discuss the details in C-2.
Kostmann disease is a type of severe congenital neutropenia (SCN) caused by HAX1 deficiency (46). HAX-1 is involved in the regulation of apoptosis and plays a role in myeloid differentiation. Cekic et al. investigated seven female patients with severe neutropenia due to the p.Trp44X variant in the HAX1 gene, and all female patients exhibited ovarian insufficiency. HAX1 mRNA is abundantly expressed in the testes and ovaries, and the authors speculated that the HAX1 gene plays an important role in ovarian development (11).
MIRAGE syndrome is a rare condition characterized by myelodysplasia, recurrent infection, restriction of growth, adrenal hypoplasia, genital phenotypes, and enteropathy. It is an autosomal dominant disorder typically caused by a de novo pathogenic variant of SAMD9/SAMD9L, whose gain of function variant increases the role of the molecule as a negative regulator of cellular proliferation by altering the endosome system (47, 48). This results in cytopenia, bone marrow failure, and immunodeficiency. In patients with MIRAGE syndrome, the adrenal glands are hypoplastic with severely disorganized structures, resulting in adrenal insufficiency. These findings are distinct from those of Addison’s disease caused by autoimmune inflammation (47–50).
ATM (ataxia telangiectasia, mutated) is essential for DNA repair, and its defects lead to ataxia telangiectasia (AT) with multisystem syndrome, including combined immunodeficiencies (51). Diabetes mellitus (DM) with severe insulin resistance is a major complication of AT. A study of AT mice model revealed that ATM regulates adipocyte differentiation, and in AT patients, adipocyte differentiation is disturbed, resembling lipodystrophy, which is one of the major causes of insulin resistance (52). ATM is also essential for germ cell meiosis. In mice, Atm deficiency results in severe meiotic disruption as early as leptonema of prophase I (53), resulting in germ cell depletion. Consistently, hypogonadism in AT is common only in female patients (6), because in contrast to testes, ovarian development is germ cell dependent.
STAT1 gain-of-function (GOF) variants impair the development of IL-17-producing T cells, resulting in mucocutaneous candidiasis (28, 31). It also causes autoimmune endocrinopathies, such as thyroiditis and T1DM. In a meta-analysis, autoimmune thyroiditis and T1DM were observed in 23.0% and 5% of STAT1 GOF patients, respectively (54). On the other hand, recent in vitro and in vivo studies have demonstrated an essential role for STAT1 in islet cell death. Stat1 ablation in T1DM model mice (NOD mice) ameliorated diabetes and insulitis, suggesting that in addition to autoimmune mechanisms, GOF of STAT1 would directly contribute to the pathogenesis of diabetes by inducing apoptosis of the islet beta cells (55).
CAHRGE syndrome is a genetic syndrome with known pattern of features, coloboma, heart anomaly, choanal atresia, intellectual disability, genital and ear anomalies (56, 57). Heterozygous CHD7 gene deficiency causes most cases, and CHD7 protein regulates gene expression by chromatin remodeling (58). Immunodeficiency in CHARGE syndrome is rare, and occurs largely due to impairment in thymic development, and the severity of the immunodeficiency relates to the degree of thymic maldevelopment (7). The severest cases could be complete/near complete absence of T-cells and abnormal B-cell function with hypo-gammaglobulinemia. The syndrome is associated with impaired development of the pharyngeal arch structures, and in this regard, the clinical phenotypes overlap with 22q11.2 deletion syndrome (7). In addition to growth retardation, gonadotropin deficiency is one of the major clinical features of CHARGE syndrome. It causes micro penis in male and absent of pubertal development in both sexes (59).
Endocrine Disorders Associated With IEI
Multiorgan Endocrinopathy
APECED (APS-1)
APECED, also called autoimmune polyglandular syndrome type 1 (APS-1), is a rare condition with an approximate prevalence of 1:100,000 in US (60). and recessively inherited disorder caused by variants of the autoimmune regulator gene, AIRE. In addition to chronic mucocutaneous candidiasis (CMC) which is caused by the reduction of key cytokines for CMC, IL-22 and IL-17F (61), APECED is characterized by variable combinations of endocrine autoimmune diseases (62). AIRE consists of 14 exons spanning 11.9 kb of genomic DNA and encodes a 545-amino acid protein with a molecular weight of 58 kDa. AIRE plays an essential role in central tolerance induction by controlling the expression of tissue-specific self-antigens within the thymus. Disruption of AIRE function results in spontaneous autoimmunity in multiple organs with autoantibodies against organ specific antigens, such as TPO, Tg (thyroid gland), calcium sensing receptor (parathyroid gland), steroidogenic enzyme (adrenal cortex and gonad) and GAD/IA2/insulin (islet cells) (60).
In a classic large cohort study of 68 patients, the most common endocrinopathies were hypoparathyroidism and adrenal insufficiency (Addison’s disease), both of which were observed in more than 70% of patients (63). The onset of the two conditions occurs from early childhood and can be obvious at any age. Ovary is also would be a major target of autoimmune response, and approximately 40–70% of women with APECED will develop premature ovarian failure (63–65). On the other hand, T1DM and hypothyroidism affects less than 5-10% and 20-25% of the patients, respectively (65), and the age of onset is during adulthood than childhood.
In addition to endocrinopathy, all patients exhibit oral candidiasis and other problems, such as alopecia, vitiligo, and keratopathy, which affect the patients at various frequencies, and one of the possible causes is related to autoimmunity to interleukin (IL)-17 cytokines (66).
IPEX/IPEX Like Syndrome
IPEX syndrome is caused by loss of function of X linked gene, FOXP3 and characterized by immunodysregulation, polyendocrinopathy, enteropathy. The pathophysiology of IPEX syndrome is explained by defects for the development, survival, and/or function of Treg cells. In addition to “classic IPEX”, a group of patients with an IPEX- like phenotype without pathological variant in FOXP3 gene emerged, and several genes, such as IL2RA, STAT5b, GOF in STAT1/3, LRBA and CTLA4, have been identified as causative genes for IPEX-like phenotype. Recently, for categorizing the group of IPEX and IPEX like diseases, in which Treg deficiency caused by monogenic deficiency, “Tregopathies” is proposed (67).
Among the IPEX and IPEX- like patients, most common endocrinopathies are T1DM and thyroiditis. Additionally, few patients, less than 5%, develop adrenal insufficiency. In a cohort of 173 IPEX/IPEX like patients, endocrinopathy were seen at a similar frequency (IPEX 65% vs. IPEX-like 59%). On the other hand, there was almost a two-fold difference in the frequency of patients with T1DM in the IPEX cohort compared to the IPEX-like group (IPEX 49% vs. IPEX-like 28%) (32). Clinical thyroid disease was more prevalent in the IPEX-like group (IPEX 26% vs. IPEX-like 39%), and approximately more than 70% of cases with thyroiditis are associated with T1DM (68). Of IPEX like subjects, STAT5b and STAT1/3 GOF more likely evolve endocrinopathy with more than 90% of frequency (32).
Growth Failure
Endocrinologically, GH and thyroid hormone mainly contribute to linear growth, and deficiencies in these hormones lead to growth failure. However, other causes affect linear growth, e.g., undernutrition, rheumatologic inflammation, chronic renal failure, malignancy, respiratory failure, severe cardiac diseases, and genetic disease with primary effects on growth. Indeed, despite the risk of growth failure in IEI (69), GH deficiency or hypothyroidism is not documented in most IEI patients with growth failure (70). This suggests that the causes of growth failure in patients with IEI are heterogeneous.
Furthermore, glucocorticoids are strong growth suppressors, and the treatments for IEI would also cause growth failure. Few studies have evaluated treatments that affect linear growth in IEI. A retrospective study suggested that after HCT for IEI, linear growth was generally decreased rather than increased, and the glucocorticoid treatment duration for chronic graft-versus-host disease (cGVHD) was an independent risk factor for growth inhibition (71).
Among the causative genes for IEIome genes are directly involved in linear growth. The JAK-STAT signaling pathway plays an essential role in the immune system, and homozygous disruption of one of the components, STAT5b, leads to hyperimmunoglobulin E (IgE) syndrome (HIES) with low NK-cell numbers and modest T cell lymphopenia. STAT signals are also located downstream of the GH receptor (72), and a homozygous variant in STAT5b causes growth failure with GH insensitivity, i.e., normal serum GH levels with very low IGF-1 levels (45). Short stature in STAT5b defects is severe [-9.9 to -4.3SD), and GH therapy will not ameliorate growth failure. Although there are few data, IGF-1 therapy would be an option for short stature homozygous or heterozygous variants in STAT5b (73).
Although STAT3 is also involved in the GH signaling pathway, normal growth is maintained in the heterozygous loss of function of STAT3 in HIES because STAT3 may negatively regulate GH-induced STAT5b (74, 75). Indeed, the GOF variant of STAT3 inhibits linear growth in several hematologic autoimmune disorders (75).
Thyroiditis
The thyroid gland is a butterfly-shaped organ located at the base of the neck. It releases the thyroid hormones, T3 and T4, and its serum levels are strictly regulated by the TRH–TSH–thyroid axis without apparent circadian rhythm. Thyroid hormones regulate numerous body functions by controlling metabolism. During infancy and childhood, thyroid hormones also play essential roles in growth and brain development. Therefore, diagnosis and interventions for thyroid hypofunction (hypothyroidism) during childhood should not be delayed (76).
Except for congenital hypothyroidism and iodine deficiency, most cases of hypothyroidism are caused by autoimmune thyroiditis. Autoimmune thyroiditis is one of the most common endocrinopathies, with an estimated prevalence of 350 cases/100,000/year (95% confidence interval [CI]: 280–450) in women and 60 cases/100,000/year (95% CI: 30–120) in men (77). Most patients with thyroiditis exhibit no other immunological problems, such as systemic autoimmune diseases or immunodeficiencies (78). In contrast, patients with any problems in the immune system are at a high risk for thyroiditis. IEI is not an exception, and to date, a substantial number of IEI are reported to be associated with thyroiditis (17).
The clinical symptoms and signs of hypothyroidism are insidious and non-specific. Inactivity, general fatigue, and emotional symptoms linked to depression are major symptoms of hypothyroidism in adults (78). Except for growth retardation and developmental delay, clinical symptoms are reversible by LT4 supplementation therapy, and patients at high risk for hypothyroidism should be closely monitored. Regular examination of thyroid function (TSH, fT4, and fT3) is the most efficient approach (78). Frequent evaluation of neurological development and tracking of growth with a growth chart are essential for monitoring high-risk children. Autoantibodies for thyroid glands, such as TPO-Ab and Tg-Ab, would be excellent biomarkers for autoimmune thyroiditis, and euthyroid patients with TPO-Ab and/or Tg-Ab should be carefully followed up with regularly repeated thyroid function because of the risks for evolving hypothyroidism (77, 79).
Autoimmune thyroiditis occasionally cause hyperthyroidism, namely Graves’ disease. In this condition, autoantibodies that bind to the thyrotropin receptor (TSHR-Ab) stimulate thyroid gland in a TSH independent manner, leading to overproduction of thyroid hormone (80). The backgrounds of autoimmunity between hypothyroidism and hyperthyroidism are identical, and the patients with autoimmune thyroiditis are at high risk for developing hyperthyroidism. Many studies simply noted “thyroiditis”, which may evolve into either hyperthyroidism or hypothyroidism, and the epidemiological details of Graves’ diseases in IEI are not precisely clarified. The diagnosis of Graves’ disease can be confirmed by undetectable levels of TSH (< 0.3 mU/l), high serum free T4 and T3 levels, and the presence of TSH receptor antibodies (TRAb) (80). To date, associations with selective IgA deficiency, IPEX and Wiskott–Aldrich syndrome are reported (81–86). The clinical symptoms of hyperthyroidism are insidious as hypothyroidism, and regular examination of thyroid function for high risk group are essential.
Hypoparathyroidism
The parathyroid glands produce PTH and maintain serum calcium levels. The major function of PTH is to increase bone absorption, inhibit calcium excretion from the kidney, and activate 1αhydroxylase, which synthesizes active vitamin D3, 1-25(OH)2 Vit D3 (87). The serum calcium level is strictly regulated by PTH because of its essential role in numerous biological functions. When PTH secretion is insufficient as in hypoparathyroidism, hypocalcemia develops, which can cause various clinical problems, including tetany, arrhythmia, prolonged QT interval, hypotension, and neurological complications, such as seizures (87). Aside from postsurgical hypoparathyroidism, the major causes of hypoparathyroidism include abnormal parathyroid development and autoimmune-mediated destruction (87).
22q11.2 deletion syndrome (also known as DiGeorge syndrome) is the most common microdeletion syndrome in humans, with an incidence of 1:4000 live births. Its clinical manifestations are highly variable, including congenital heart disease, palatal abnormalities, immune deficiency due to thymic hypoplasia with fewer T cells, characteristic facial features, hearing impairment, and hypoparathyroidism. The cause of hypoparathyroidism in 22q11.2 deletion syndrome is congenital defects, and the onset of hypocalcemia occurs mainly during the neonatal period. However, recent analysis revealed that autoimmune responses are involved in the pathogenesis of hypoparathyroidism, and hypocalcemia may occur at any age (88). These findings suggest that routine screening of serum calcium levels is essential for the long-term follow-up of the syndrome.
Chromosome 10p13-p14 deletion syndrome (10p13-p14DS) was originally discovered in patients with clinical phenotypes resembling 22q11.2 deletion syndrome. The syndrome also has a wide variety of clinical symptoms, including immunodeficiency and hypoparathyroidism (89, 90). Although the etiology of hypoparathyroidism in 10p13-p14 deletion syndrome is thought to be congenital defects, the involvement of autoimmune responses can be considered as 22q11.2 deletion syndrome. Further accumulation of cases with long-term follow-up of hypoparathyroidism would provide valuable insights for hypoparathyroidism mediated by autoimmune mechanisms in 10p13-p14 deletion syndrome.
The clinical features of APECED including hypopara-thyroidism are discussed in C-1.
Adrenal Insufficiency
Adrenal insufficiency is a disorder that occurs when the adrenal glands are not capable of producing sufficient adrenal hormones, mainly cortisol. Cortisol protects the body from physical stress and is essential for life. Therefore, adrenal insufficiency can be life-threatening, requiring prompt and appropriate diagnosis. However, the diagnosis of adrenal insufficiency is challenging. First, the common symptoms are non-specific, such as fatigue, muscle weakness, loss of appetite, weight loss, and abdominal pain. Second, one-spot blood sampling is usually insufficient for the endocrinological diagnosis, and precise studies, including ACTH stimulation test or circadian rhythm, are required. Thus, identifying patients who are at a high risk for adrenal insufficiency is crucial (91).
The production of cortisol is strictly regulated by the hypothalamus–pituitary–adrenal (HPA) axis. Except for iatrogenic adrenal insufficiency due to glucocorticoid therapy, major causes of adrenal insufficiency are deficiency of ACTH secretion from the pituitary gland (secondary adrenal insufficiency) and organ failure of the adrenal gland (primary adrenal insufficiency) (91, 92).
Most causes of ACTH deficiency are anatomical defects of the pituitary gland, in which the production of multiple pituitary hormones is impaired. Thus, isolated ACTH deficiency is rare (93). Recently, loss-of-function variants of NFKB2 have been shown to cause adrenal insufficiency due to isolated ACTH deficiency (8, 94, 95).
NFKB2 belongs to the NF-kB family, which is a group of evolutionarily conserved transcription factors involved in organ development, immune systems, and oncogenesis. Impaired function of NF-kB transcription factors or their regulators has been reported to be associated with primary immunodeficiency and autoimmunity (96). Classically, the condition characterized by a combination of common variable immunodeficiency (CVID) and ACTH insufficiency is called DAVID syndrome. ACTH deficiency occurred in 44% of pathologic variant carriers with a median age at onset of 8.2 years (SD ± 4.2, median 7.0, range 4–15 years) and is difficult to predict based on genetic alterations. The age at onset of ACTH deficiency suggests that the condition is acquired rather than congenital, and ACTH deficiency is presumed to be caused by T cell-mediated autoimmune diseases (8).
Except for congenital conditions, primary adrenal insufficiency is mainly caused by an autoimmune response. Isolated adrenal insufficiency accounts for up to 40% of patients with autoimmune primary adrenal insufficiency, and one or more organ-specific autoimmune endocrinopathies, such as autoimmune thyroid disease, T1DM, and premature ovarian insufficiency, can be associated with adrenal insufficiency. 21-Hydroxylase is a major autoantigen in autoimmune primary adrenal insufficiency (97), and the etiologies of primary adrenal insufficiency can be screened for using 21-hydroxylase antibodies (98). Of numerous genes that cause autoimmune adrenal insufficiency, AIRE is a representative gene that is associated with IEI (92). A recent cohort study of 691 patients with autoimmune adrenal insufficiency reported that variants of the CTLA4 gene would increase susceptibility to adrenal insufficiency (99).
Recently another IEI gene has been identified to cause primary adrenal insufficiency. MCM4 (mini chromosome maintenance deficient 4 homolog) is part of a hetero-hexameric helicase complex which is important for DNA replication and genome integrity, and pathological variations were identified in an Irish travelling community manifesting with familial adrenal insufficiency (100, 101). Adrenal insufficiency is progressive and onset was usually in childhood following a period of normal adrenal function. ACTH resistance without mineralocorticoid deficiency is the characteristic of the condition. Although NK cell function is disrupted in the syndrome, the precise mechanisms of adrenal insufficiency are remained to be clarified (102).
Adrenal hypoplasia due to SAMD9 is discussed in B-2.
Diabetes Mellitus
DM is a condition of chronic hyperglycemia due to the dysregulation of glucose homeostasis. Insulin is the only hormone that decreases blood sugar, and the pathophysiology of diabetes can be classified into two main groups: insulin deficiency and increased insulin resistance. The major pathogenesis of insulin deficiency is an autoimmune response. A targeted immune response by both T and B cells leads to the destruction of insulin-producing β-cells in the islets of the pancreas. This condition is called T1DM, and some IEI are frequently associated with T1DM, such as APS type 1 (AIRE), IPEX (FOXP3), CTLA4, LRBA, and GOF of STAT1 (22, 31, 35, 63, 103).
One distinctive clinical feature of T1DM associated with IEI is age at onset. Generally, an average age of onset of childhood-onset T1DM is approximately 9 ~ 10 years (104), and onset of T1DM occurs after six months of age. Accordingly, diabetes that develops before six months of age is classified as neonatal diabetes, in which other pathophysiologies, such as congenital malfunction of β-cells, are involved (105). However, several studies have revealed that T1DM associated with IEI frequently develops before six months of age (43, 103). In such patients, T1DM would be the first clinical symptom, and subsequently, immunodeficiency becomes clinically obvious. Accordingly, for T1DM patients whose onset is before six months of age, intensive genetic analysis, including IEI genes, is required.
Some IEI due to DNA repair defects, such as AT and Bloom syndrome, are reported to be associated with DM (106). In such IEI, insulin synthesis is generally maintained, and extremely increased insulin resistance has been suggested to contribute to the development of DM. In AT, lipodystrophy has been suggested as a cause of DM (52), and possibly, in Bloom syndrome, similar mechanisms would be involved, although precise analyses have not yet been reported.
A recent prospective analysis of 39 patients with AT showed a significant increase in HbA1c and fasting glucose levels with age (107). OGTT has good sensitivity for insulin resistance, and 7/39 (17.9%) patients had diabetes. In AT, metformin did not always lead to sufficient glycemic control (107).
Endocrine and Metabolic Disorders Caused by HCT for IEI
Impaired Reproductive Function Caused by HCT
Most IEI are due to defects in genes that are essential for hematopoietic cells to function normally, and supportive therapies, such as immunoglobulin G (IgG) infusions and antibiotics, are occasionally unsatisfactory for long-term outcomes. The only curative approach is the replacement of impaired cells by healthy donor hematopoietic stem cells and allogeneic HCT (allo HCT) (108–110). One of the clinical implications of HCT is the long-term complications that cause chronic and irreversible damage to various organs (111), including the endocrine glands. The major potential causes for long-term complications are conditioning regimens with cytotoxic agents and therapies for GVHD, including glucocorticoids (71).
The purpose of conditioning therapies is to ablate immune cells to allow stable engraftment of donor-derived HSCs. Classically, antineoplastic alkylating agents are used as key drugs in conditioning regimens for HCT. Although recent technical advances in HCT have enabled the reduction of the intensity of the regimen, alkylating agents are still widely used as the central drug (112, 113).
One of the common adverse effects of alkylating agents is germ cell toxicity. The clinical phenotypes of germ cell defects differ between female and male patients. In the ovaries, follicular structures, including oocytes, are essential for the differentiation and maintenance of steroidogenic ovarian cells, granulosa cells, and theca cells. Oocyte depletion due to alkylating agents causes hypogonadism with loss of estrogen and progesterone. In contrast, Leydig cells, which are steroidogenic cells in the testes, are viable in the absence of germ cells. Accordingly, normal androgen synthesis does not indicate the viability of germ cells in the testes, i.e., even after developing secondary sexual characteristics, future fertility of the male subjects is not warranted in IEI patients after HCT.
In contrast to malignant diseases, the conditioning regimen for IEI is generally less toxic, and the complications of HCT for patients with IEI would be different from that of malignant diseases, requiring careful evaluation of the toxicity of alkylating agents. However, evaluating the adverse effects of alkylating agents is not straightforward in IEI. First, the majority of severe IEI cases requiring HCT are male patients because of X-linked genetic diseases. Second, in contrast to hypogonadism, screening for isolated infertility in male patients is difficult, especially during childhood.
A recent report suggested that lower doses of alkylating agents for a reduced intensity conditioning regimen (RIC) would reduce the toxicity, including hypogonadism. A long-term follow-up study of 43 pediatric patients with non-malignant disease after HCT conditioned by RIC regimen revealed that all three postpubertal female patients before HCT resumed normal menstrual cycles post-HCT, and apparent hypogonadism were observed only one (5%) patient (112). A similar outcome was obtained from another cohort conditioned by fludarabine and melphalan based RIC (114). On the other hand, another report suggested that even RIC regimen would be a risk for hypogonadism in female patients (71). Treosulfan is an alkylating agent and increasingly used in recent pediatric practice because of less systemic toxicity than busulphan. By assessing bio markers for potential fertility, such as Anti-Müllerian hormone (AMH) and inhibin B, a treosulfan-based regimen confers a more favorable outcomes for gonadal reserve than fludarabine and melphalan or busulphan/cyclophosphamide based in both sexes (115).
With regard to the gonadal toxicity of alkylating agents, we should be aware that there is no uniformly “safe” or “toxic” alkylating agent dose threshold. Despite a cohort based on malignant diseases in which higher doses of alkylating agents are generally administered, a study of 214 cancer survivors found that despite a negative correlation between cyclophosphamide equivalent dose and sperm concentration, a firm threshold dose could not be established because azoospermia was observed even at the lowest dose (116).
Another issue for reproduction in IEI patients is age at HCT. Most patients with IEI receive HCT before the reproductive age (108, 112), resulting in limited options for the preservation of fertility before HCT, such as cryopreservation of gonadal tissues. However, at present, cryopreservation of gonadal tissues is considered an experimental technique, and this procedure may only be utilized for carefully selected patients as an experimental protocol (117). Further accumulation of these cases is necessary to elucidate the germ cell toxicity of RIC.
Endocrine and Metabolic Disorders Caused by Prolonged Glucocorticoid Therapy
Long-term administration of glucocorticoids for GVHD after HCT or autoimmune response can cause various clinical problems, such as iatrogenic Cushing’s syndrome. The major adverse effects in the metabolic and endocrine systems include glucose intolerance, truncal obesity, and osteoporosis. For these adverse effects, careful and regular monitoring is required (118). Although the clinical presentation of iatrogenic Cushing’s syndrome in the pediatric population is similar to that in adults, there are some child-specific features, such as growth retardation. For the management of pediatric patients treated with systemic glucocorticoid therapy, frequent evaluation of linear growth and body weight change by plotting a growth chart is essential (119).
In addition to Cushing’s syndrome, patients treated with glucocorticoids are at high risk for adrenal insufficiency. Anti-inflammatory glucocorticoid treatment for IEI suppresses ACTH and CRH production from the pituitary gland and hypothalamus, respectively, decreasing endogenous cortisol synthesis (118). Subsequently, prolonged ACTH suppression leads to adrenal gland atrophy. Atrophic adrenal glands are not capable of promptly and properly synthesizing sufficient endogenous cortisol by abrupt cessation or rapid withdrawal of glucocorticoids, resulting in symptoms of adrenal insufficiency (118).
Furthermore, patients treated with low to medium doses of glucocorticoids are also at high risk for potentially life-threatening acute adrenal insufficiency. The low to medium dose of glucocorticoids suppresses the HPA axis, leading to adrenal atrophy. However, such a dose of glucocorticoid might not sufficiently tolerate physical stresses, such as fever, major injuries, seizure, and frequent vomiting, in which the required volume of glucocorticoid is dramatically increased (118). Physicians should always pay attention to adrenal insufficiency in patients treated with glucocorticoids.
Conclusion remarks
In summary, we comprehensively reviewed and discussed the current understanding on the clinical features and the pathophysiology of endocrine disorders in IEI. We discussed the two major pathophysiology of endocrinopathy in IEI, autoimmune response and direct effects of the responsible genes, the details of each endocrinopathy, and potential endocrinopathy due to hematopoietic stem cell transplantation. Our expanding understanding on the molecular mechanisms of IEI have shed light on the new aspects of pathophysiology of endocrinopathies. Greater appreciation of these issues will provide valuable insights of the mechanisms underpinning endocrinopathies.
Author Contributions
All authors conceptualized, planned, wrote, and approved the manuscript.
Funding
The manuscript is partially funded by a Japanese nonprofit organization (NPO), Heart-link working project.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to extend my sincere thanks to Kosuke Imai (Tokyo Medical and Dental University) for valuable suggestions and comments.
References
1. Bousfiha A, Jeddane L, Picard C, Ailal F, Bobby Gaspar H, Al-Herz W, et al. The 2017 IUIS Phenotypic Classification for Primary Immunodeficiencies. J Clin Immunol (2018) 38(1):129–43. doi: 10.1007/s10875-017-0465-8
2. Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human Inborn Errors of Immunity: 2019 Update on the Classification From the International Union of Immunological Societies Expert Committee. J Clin Immunol (2020) 40(1):24–64. doi: 10.1007/s10875-019-00737-x
3. Chinn IK, Chan AY, Chen K, Chou J, Dorsey MJ, Hajjar J, et al. Diagnostic Interpretation of Genetic Studies in Patients With Primary Immunodeficiency Diseases: A Working Group Report of the Primary Immunodeficiency Diseases Committee of the American Academy of Allergy, Asthma & Immunology. J Allergy Clin Immunol (2020) 145(1):46–69. doi: 10.1016/j.jaci.2019.09.009
4. Yamashita M, Inoue K, Okano T, Morio T. Inborn Errors of Immunity-Recent Advances in Research on the Pathogenesis. Inflamm Regen (2021) 41(1):9. doi: 10.1186/s41232-021-00159-6
5. Walter JE, Ayala IA, Milojevic D. Autoimmunity as a Continuum in Primary Immunodeficiency. Curr Opin Pediatr (2019) 31(6):851–62. doi: 10.1097/MOP.0000000000000833
6. Nissenkorn A, Levy-Shraga Y, Banet-Levi Y, Lahad A, Sarouk I, Modan-Moses D. Endocrine Abnormalities in Ataxia Telangiectasia: Findings From a National Cohort. Pediatr Res (2016) 79(6):889–94. doi: 10.1038/pr.2016.19
7. Mehr S, Hsu P, Campbell D. Immunodeficiency in CHARGE Syndrome. Am J Med Genet C Semin Med Genet (2017) 175(4):516–23. doi: 10.1002/ajmg.c.31594
8. Klemann C, Camacho-Ordonez N, Yang L, Eskandarian Z, Rojas-Restrepo JL, Frede N, et al. Clinical and Immunological Phenotype of Patients With Primary Immunodeficiency Due to Damaging Mutations in NFKB2. Front Immunol (2019) 10:297. doi: 10.3389/fimmu.2019.00297
9. Yamauchi T, Takasawa K, Kamiya T, Kirino S, Gau M, Inoue K, et al. Hematopoietic Stem Cell Transplantation Recovers Insulin Deficiency in Type 1 Diabetes Mellitus Associated With IPEX Syndrome. Pediatr Diabetes (2019) 20(7):1035–40. doi: 10.1111/pedi.12895
10. Verma N, Burns SO, Walker LSK, Sansom DM. Immune Deficiency and Autoimmunity in Patients With CTLA-4 (CD152) Mutations. Clin Exp Immunol (2017) 190(1):1–7. doi: 10.1111/cei.12997
11. Cekic S, Saglam H, Gorukmez O, Yakut T, Tarim O, Kilic SS. Delayed Puberty and Gonadal Failure in Patients With HAX1 Mutation. J Clin Immunol (2017) 37(6):524–8. doi: 10.1007/s10875-017-0412-8
12. Cheng MH, Anderson MS. Insights Into Type 1 Diabetes From the Autoimmune Polyendocrine Syndromes. Curr Opin Endocrinol Diabetes Obes (2013) 20(4):271–8. doi: 10.1097/MED.0b013e32836313eb
13. Hage C, Gan HW, Ibba A, Patti G, Dattani M, Loche S, et al. Advances in Differential Diagnosis and Management of Growth Hormone Deficiency in Children. Nat Rev Endocrinol (2021) 17. doi: 10.1038/s41574-021-00539-5
14. Farmer JR, Ong MS, Barmettler S, Yonker LM, Fuleihan R, Sullivan KE, et al. Common Variable Immunodeficiency Non-Infectious Disease Endotypes Redefined Using Unbiased Network Clustering in Large Electronic Datasets. Front Immunol (2017) 8:1740. doi: 10.3389/fimmu.2017.01740
15. Ochs HD, Oukka M, Torgerson TR. TH17 Cells and Regulatory T Cells in Primary Immunodeficiency Diseases. J Allergy Clin Immunol (2009) 123(5):977–983; quiz 984-975. doi: 10.1016/j.jaci.2009.03.030
16. Goyal R, Bulua AC, Nikolov NP, Schwartzberg PL, Siegel RM. Rheumatologic and Autoimmune Manifestations of Primary Immunodeficiency Disorders. Curr Opin Rheumatol (2009) 21(1):78–84. doi: 10.1097/BOR.0b013e32831cb939
17. Amaya-Uribe L, Rojas M, Azizi G, Anaya JM, Gershwin ME. Primary Immunodeficiency and Autoimmunity: A Comprehensive Review. J Autoimmun (2019) 99:52–72. doi: 10.1016/j.jaut.2019.01.011
18. Nagamine K, Peterson P, Scott HS, Kudoh J, Minoshima S, Heino M, et al. Positional Cloning of the APECED Gene. Nat Genet (1997) 17(4):393–8. doi: 10.1038/ng1297-393
19. Finnish-German AC. An Autoimmune Disease, APECED, Caused by Mutations in a Novel Gene Featuring Two PHD-Type Zinc-Finger Domains. Nat Genet (1997) 17(4):399–403. doi: 10.1038/ng1297-399
20. Husebye ES, Anderson MS. Autoimmune Polyendocrine Syndromes: Clues to Type 1 Diabetes Pathogenesis. Immunity (2010) 32(4):479–87. doi: 10.1016/j.immuni.2010.03.016
21. Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, et al. X-Linked Neonatal Diabetes Mellitus, Enteropathy and Endocrinopathy Syndrome is the Human Equivalent of Mouse Scurfy. Nat Genet (2001) 27(1):18–20. doi: 10.1038/83707
22. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked Syndrome (IPEX) is Caused by Mutations of FOXP3. Nat Genet (2001) 27(1):20–1. doi: 10.1038/83713
23. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 Programs the Development and Function of CD4+CD25+ Regulatory T Cells. Nat Immunol (2003) 4(4):330–6. doi: 10.1038/ni904
24. Hori S, Nomura T, Sakaguchi S. Control of Regulatory T Cell Development by the Transcription Factor Foxp3. Science (2003) 299(5609):1057–61. doi: 10.1126/science.1079490
25. Caudy AA, Reddy ST, Chatila T, Atkinson JP, Verbsky JW. CD25 Deficiency Causes an Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked-Like Syndrome, and Defective IL-10 Expression From CD4 Lymphocytes. J Allergy Clin Immunol (2007) 119(2):482–7. doi: 10.1016/j.jaci.2006.10.007
26. Lowe CE, Cooper JD, Brusko T, Walker NM, Smyth DJ, Bailey R, et al. Large-Scale Genetic Fine Mapping and Genotype-Phenotype Associations Implicate Polymorphism in the IL2RA Region in Type 1 Diabetes. Nat Genet (2007) 39(9):1074–82. doi: 10.1038/ng2102
27. Cohen AC, Nadeau KC, Tu W, Hwa V, Dionis K, Bezrodnik L, et al. Cutting Edge: Decreased Accumulation and Regulatory Function of CD4+ CD25(high) T Cells in Human STAT5b Deficiency. J Immunol (2006) 177(5):2770–4. doi: 10.4049/jimmunol.177.5.2770
28. Liu L, Okada S, Kong XF, Kreins AY, Cypowyj S, Abhyankar A, et al. Gain-Of-Function Human STAT1 Mutations Impair IL-17 Immunity and Underlie Chronic Mucocutaneous Candidiasis. J Exp Med (2011) 208(8):1635–48. doi: 10.1084/jem.20110958
29. Charbonnier LM, Janssen E, Chou J, Ohsumi TK, Keles S, Hsu JT, et al. Regulatory T-Cell Deficiency and Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked-Like Disorder Caused by Loss-of-Function Mutations in LRBA. J Allergy Clin Immunol (2015) 135(1):217–27. doi: 10.1016/j.jaci.2014.10.019
30. Haapaniemi EM, Kaustio M, Rajala HL, van Adrichem AJ, Kainulainen L, Glumoff V, et al. Autoimmunity, Hypogammaglobulinemia, Lymphoproliferation, and Mycobacterial Disease in Patients With Activating Mutations in STAT3. Blood (2015) 125(4):639–48. doi: 10.1182/blood-2014-04-570101
31. Uzel G, Sampaio EP, Lawrence MG, Hsu AP, Hackett M, Dorsey MJ, et al. Dominant Gain-of-Function STAT1 Mutations in FOXP3 Wild-Type Immune Dysregulation-Polyendocrinopathy-Enteropathy-X-Linked-Like Syndrome. J Allergy Clin Immunol (2013) 131(6):1611–23. doi: 10.1016/j.jaci.2012.11.054
32. Gambineri E, Ciullini Mannurita S, Hagin D, Vignoli M, Anover-Sombke S, DeBoer S, et al. Clinical, Immunological, and Molecular Heterogeneity of 173 Patients With the Phenotype of Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked (IPEX) Syndrome. Front Immunol (2018) 9:2411. doi: 10.3389/fimmu.2018.02411
33. Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, et al. Immune Dysregulation in Human Subjects With Heterozygous Germline Mutations in CTLA4. Science (2014) 345(6204):1623–7. doi: 10.1126/science.1255904
34. Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, et al. Autosomal Dominant Immune Dysregulation Syndrome in Humans With CTLA4 Mutations. Nat Med (2014) 20(12):1410–6. doi: 10.1038/nm.3746
35. Nistico L, Buzzetti R, Pritchard LE, van der Auwera B, Giovannini C, Bosi E, et al. The CTLA-4 Gene Region of Chromosome 2q33 is Linked to, and Associated With, Type 1 Diabetes. Belgian Diabetes Registry. Hum Mol Genet (1996) 5(7):1075–80. doi: 10.1093/hmg/5.7.1075
36. Albarel F, Gaudy C, Castinetti F, Carre T, Morange I, Conte-Devolx B, et al. Long-Term Follow-Up of Ipilimumab-Induced Hypophysitis, a Common Adverse Event of the Anti-CTLA-4 Antibody in Melanoma. Eur J Endocrinol (2015) 172(2):195–204. doi: 10.1530/EJE-14-0845
37. Blansfield JA, Beck KE, Tran K, Yang JC, Hughes MS, Kammula US, et al. Cytotoxic T-Lymphocyte-Associated Antigen-4 Blockage can Induce Autoimmune Hypophysitis in Patients With Metastatic Melanoma and Renal Cancer. J Immunother (2005) 28(6):593–8. doi: 10.1097/01.cji.0000178913.41256.06
38. Ogishi M, Yang R, Aytekin C, Langlais D, Bourgey M, Khan T, et al. Inherited PD-1 Deficiency Underlies Tuberculosis and Autoimmunity in a Child. Nat Med (2021) 27. doi: 10.1038/s41591-021-01388-5
39. Deau MC, Heurtier L, Frange P, Suarez F, Bole-Feysot C, Nitschke P, et al. A Human Immunodeficiency Caused by Mutations in the PIK3R1 Gene. J Clin Invest (2014) 124(9):3923–8. doi: 10.1172/JCI75746
40. Nakagawa R, Takasawa K, Yeh TW, Imai K, Kashimada K, Morio T. Type 1 Diabetes Mellitus Associated With Activated Phosphatidylinositol 3-Kinase Delta Syndrome, Type 2. J Diabetes (2018) 10(5):421–2. doi: 10.1111/1753-0407.12638
41. Hori T, Ohnishi H, Kadowaki T, Kawamoto N, Matsumoto H, Ohara O, et al. Autosomal Dominant Hashimoto’s Thyroiditis With a Mutation in TNFAIP3. Clin Pediatr Endocrinol (2019) 28(3):91–6. doi: 10.1297/cpe.28.91
42. Kadowaki S, Hashimoto K, Nishimura T, Kashimada K, Kadowaki T, Kawamoto N, et al. Functional Analysis of Novel A20 Variants in Patients With Atypical Inflammatory Diseases. Arthritis Res Ther (2021) 23(1):52. doi: 10.1186/s13075-021-02434-w
43. Baud O, Goulet O, Canioni D, Le Deist F, Radford I, Rieu D, et al. Treatment of the Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked Syndrome (IPEX) by Allogeneic Bone Marrow Transplantation. N Engl J Med (2001) 344(23):1758–62. doi: 10.1056/NEJM200106073442304
44. Chaimowitz NS, Ebenezer SJ, Hanson IC, Anderson M, Forbes LR. STAT1 Gain of Function, Type 1 Diabetes, and Reversal With JAK Inhibition. N Engl J Med (2020) 383(15):1494–6. doi: 10.1056/NEJMc2022226
45. Hwa V. STAT5B Deficiency: Impacts on Human Growth and Immunity. Growth Horm IGF Res (2016) 28:16–20. doi: 10.1016/j.ghir.2015.12.006
46. Klein C, Grudzien M, Appaswamy G, Germeshausen M, Sandrock I, Schaffer AA, et al. HAX1 Deficiency Causes Autosomal Recessive Severe Congenital Neutropenia (Kostmann Disease). Nat Genet (2007) 39(1):86–92. doi: 10.1038/ng1940
47. Narumi S, Amano N, Ishii T, Katsumata N, Muroya K, Adachi M, et al. SAMD9 Mutations Cause a Novel Multisystem Disorder, MIRAGE Syndrome, and are Associated With Loss of Chromosome 7. Nat Genet (2016) 48(7):792–7. doi: 10.1038/ng.3569
48. Buonocore F, Kuhnen P, Suntharalingham JP, Del Valle I, Digweed M, Stachelscheid H, et al. Somatic Mutations and Progressive Monosomy Modify SAMD9-Related Phenotypes in Humans. J Clin Invest (2017) 127(5):1700–13. doi: 10.1172/JCI91913
49. Nagamachi A, Matsui H, Asou H, Ozaki Y, Aki D, Kanai A, et al. Haploinsufficiency of SAMD9L, an Endosome Fusion Facilitator, Causes Myeloid Malignancies in Mice Mimicking Human Diseases With Monosomy 7. Cancer Cell (2013) 24(3):305–17. doi: 10.1016/j.ccr.2013.08.011
50. Wong JC, Bryant V, Lamprecht T, Ma J, Walsh M, Schwartz J, et al. Germline SAMD9 and SAMD9L Mutations are Associated With Extensive Genetic Evolution and Diverse Hematologic Outcomes. JCI Insight (2018) 3(14):1–11. doi: 10.1172/jci.insight.121086
51. Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, et al. A Single Ataxia Telangiectasia Gene With a Product Similar to PI-3 Kinase. Science (1995) 268(5218):1749–53. doi: 10.1126/science.7792600
52. Takagi M, Uno H, Nishi R, Sugimoto M, Hasegawa S, Piao J, et al. ATM Regulates Adipocyte Differentiation and Contributes to Glucose Homeostasis. Cell Rep (2015) 10(6):957–67. doi: 10.1016/j.celrep.2015.01.027
53. Barlow C, Liyanage M, Moens PB, Tarsounas M, Nagashima K, Brown K, et al. Atm Deficiency Results in Severe Meiotic Disruption as Early as Leptonema of Prophase I. Development (1998) 125(20):4007–17. doi: 10.1242/dev.125.20.4007
54. Zhang W, Chen X, Gao G, Xing S, Zhou L, Tang X, et al. Clinical Relevance of Gain- and Loss-Of-Function Germline Mutations in STAT1: A Systematic Review. Front Immunol (2021) 12:654406. doi: 10.3389/fimmu.2021.654406
55. Kim S, Kim HS, Chung KW, Oh SH, Yun JW, Im SH, et al. Essential Role for Signal Transducer and Activator of Transcription-1 in Pancreatic Beta-Cell Death and Autoimmune Type 1 Diabetes of Nonobese Diabetic Mice. Diabetes (2007) 56(10):2561–8. doi: 10.2337/db06-1372
56. Hall BD. Choanal Atresia and Associated Multiple Anomalies. J Pediatr (1979) 95(3):395–8. doi: 10.1016/S0022-3476(79)80513-2
57. Hittner HM, Hirsch NJ, Kreh GM, Rudolph AJ. Colobomatous Microphthalmia, Heart Disease, Hearing Loss, and Mental Retardation–a Syndrome. J Pediatr Ophthalmol Strabismus (1979) 16(2):122–8. doi: 10.3928/0191-3913-19790301-10
58. Lalani SR, Safiullah AM, Fernbach SD, Harutyunyan KG, Thaller C, Peterson LE, et al. Spectrum of CHD7 Mutations in 110 Individuals With CHARGE Syndrome and Genotype-Phenotype Correlation. Am J Hum Genet (2006) 78(2):303–14. doi: 10.1086/500273
59. Pinto G, Abadie V, Mesnage R, Blustajn J, Cabrol S, Amiel J, et al. CHARGE Syndrome Includes Hypogonadotropic Hypogonadism and Abnormal Olfactory Bulb Development. J Clin Endocrinol Metab (2005) 90(10):5621–6. doi: 10.1210/jc.2004-2474
60. Dittmar M, Kahaly GJ. Polyglandular Autoimmune Syndromes: Immunogenetics and Long-Term Follow-Up. J Clin Endocrinol Metab (2003) 88(7):2983–92. doi: 10.1210/jc.2002-021845
61. Kisand K, Boe Wolff AS, Podkrajsek KT, Tserel L, Link M, Kisand KV, et al. Chronic Mucocutaneous Candidiasis in APECED or Thymoma Patients Correlates With Autoimmunity to Th17-Associated Cytokines. J Exp Med (2010) 207(2):299–308. doi: 10.1084/jem.20091669
62. Heino M, Peterson P, Kudoh J, Shimizu N, Antonarakis SE, Scott HS, et al. APECED Mutations in the Autoimmune Regulator (AIRE) Gene. Hum Mutat (2001) 18(3):205–11. doi: 10.1002/humu.1176
63. Ahonen P, Myllarniemi S, Sipila I, Perheentupa J. Clinical Variation of Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy (APECED) in a Series of 68 Patients. N Engl J Med (1990) 322(26):1829–36. doi: 10.1056/NEJM199006283222601
64. Szeliga A, Calik-Ksepka A, Maciejewska-Jeske M, Grymowicz M, Smolarczyk K, Kostrzak A, et al. Autoimmune Diseases in Patients With Premature Ovarian Insufficiency-Our Current State of Knowledge. Int J Mol Sci (2021) 22(5):1–11. doi: 10.3390/ijms22052594
65. Constantine GM, Lionakis MS. Lessons From Primary Immunodeficiencies: Autoimmune Regulator and Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy. Immunol Rev (2019) 287(1):103–20. doi: 10.1111/imr.12714
66. Puel A, Doffinger R, Natividad A, Chrabieh M, Barcenas-Morales G, Picard C, et al. Autoantibodies Against IL-17a, IL-17F, and IL-22 in Patients With Chronic Mucocutaneous Candidiasis and Autoimmune Polyendocrine Syndrome Type I. J Exp Med (2010) 207(2):291–7. doi: 10.1084/jem.20091983
67. Cepika AM, Sato Y, Liu JM, Uyeda MJ, Bacchetta R, Roncarolo MG. Tregopathies: Monogenic Diseases Resulting in Regulatory T-Cell Deficiency. J Allergy Clin Immunol (2018) 142(6):1679–95. doi: 10.1016/j.jaci.2018.10.026
68. Park JH, Lee KH, Jeon B, Ochs HD, Lee JS, Gee HY, et al. Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked (IPEX) Syndrome: A Systematic Review. Autoimmun Rev (2020) 19(6):102526. doi: 10.1016/j.autrev.2020.102526
69. Westermann-Clark E, Meehan CA, Meyer AK, Dasso JF, Amre D, Ellison M, et al. Primary Immunodeficiency in Children With Autoimmune Cytopenias: Retrospective 154-Patient Cohort. Front Immunol (2021) 12:649182. doi: 10.3389/fimmu.2021.649182
71. Sutani A, Miyakawa Y, Tsuji-Hosokawa A, Nomura R, Nakagawa R, Nakajima K, et al. Gonadal Failure Among Female Patients After Hematopoietic Stem Cell Transplantation for non-Malignant Diseases. Clin Pediatr Endocrinol (2019) 28(4):105–12. doi: 10.1297/cpe.28.105
72. Herrington J, Smit LS, Schwartz J, Carter-Su C. The Role of STAT Proteins in Growth Hormone Signaling. Oncogene (2000) 19(21):2585–97. doi: 10.1038/sj.onc.1203526
73. Wit JM, Joustra SD, Losekoot M, van Duyvenvoorde HA, de Bruin C. Differential Diagnosis of the Short IGF-I-Deficient Child With Apparently Normal Growth Hormone Secretion. Horm Res Paediatr (2021) 94(3-4):81–104. doi: 10.1159/000516407
74. Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, et al. Dominant-Negative Mutations in the DNA-Binding Domain of STAT3 Cause Hyper-IgE Syndrome. Nature (2007) 448(7157):1058–62. doi: 10.1038/nature06096
75. Flanagan SE, Haapaniemi E, Russell MA, Caswell R, Allen HL, De Franco E, et al. Activating Germline Mutations in STAT3 Cause Early-Onset Multi-Organ Autoimmune Disease. Nat Genet (2014) 46(8):812–4. doi: 10.1038/ng.3040
76. Peters C, van Trotsenburg ASP, Schoenmakers N. DIAGNOSIS OF ENDOCRINE DISEASE: Congenital Hypothyroidism: Update and Perspectives. Eur J Endocrinol (2018) 179(6):R297–317. doi: 10.1530/EJE-18-0383
77. Vanderpump MP, Tunbridge WM, French JM, Appleton D, Bates D, Clark F, et al. The Incidence of Thyroid Disorders in the Community: A Twenty-Year Follow-Up of the Whickham Survey. Clin Endocrinol (Oxf) (1995) 43(1):55–68. doi: 10.1111/j.1365-2265.1995.tb01894.x
78. Ragusa F, Fallahi P, Elia G, Gonnella D, Paparo SR, Giusti C, et al. Hashimotos’ Thyroiditis: Epidemiology, Pathogenesis, Clinic and Therapy. Best Pract Res Clin Endocrinol Metab (2019) 33(6):101367. doi: 10.1016/j.beem.2019.101367
79. Huber G, Staub JJ, Meier C, Mitrache C, Guglielmetti M, Huber P, et al. Prospective Study of the Spontaneous Course of Subclinical Hypothyroidism: Prognostic Value of Thyrotropin, Thyroid Reserve, and Thyroid Antibodies. J Clin Endocrinol Metab (2002) 87(7):3221–6. doi: 10.1210/jcem.87.7.8678
80. Leger J, Kaguelidou F, Alberti C, Carel JC. Graves’ Disease in Children. Best Pract Res Clin Endocrinol Metab (2014) 28(2):233–43. doi: 10.1016/j.beem.2013.08.008
81. Bacchetta R, Barzaghi F, Roncarolo MG. From IPEX Syndrome to FOXP3 Mutation: A Lesson on Immune Dysregulation. Ann N Y Acad Sci (2018) 1417(1):5–22. doi: 10.1111/nyas.13011
82. Limbach M, Saare M, Tserel L, Kisand K, Eglit T, Sauer S, et al. Epigenetic Profiling in CD4+ and CD8+ T Cells From Graves’ Disease Patients Reveals Changes in Genes Associated With T Cell Receptor Signaling. J Autoimmun (2016) 67:46–56. doi: 10.1016/j.jaut.2015.09.006
83. Mohammadi J, Ramanujam R, Jarefors S, Rezaei N, Aghamohammadi A, Gregersen PK, et al. IgA Deficiency and the MHC: Assessment of Relative Risk and Microheterogeneity Within the HLA A1 B8, DR3 (8.1) Haplotype. J Clin Immunol (2010) 30(1):138–43. doi: 10.1007/s10875-009-9336-2
84. Singh K, Chang C, Gershwin ME. IgA Deficiency and Autoimmunity. Autoimmun Rev (2014) 13(2):163–77. doi: 10.1016/j.autrev.2013.10.005
85. Yin X, Latif R, Tomer Y, Davies TF. Thyroid Epigenetics: X Chromosome Inactivation in Patients With Autoimmune Thyroid Disease. Ann N Y Acad Sci (2007) 1110:193–200. doi: 10.1196/annals.1423.021
86. Struja T, Kutz A, Fischli S, Meier C, Mueller B, Recher M, et al. Is Graves’ Disease a Primary Immunodeficiency? New Immunological Perspectives on an Endocrine Disease. BMC Med (2017) 15(1):174. doi: 10.1186/s12916-017-0939-9
87. Mannstadt M, Bilezikian JP, Thakker RV, Hannan FM, Clarke BL, Rejnmark L, et al. Hypoparathyroidism. Nat Rev Dis Primers (2017) 3:17080. doi: 10.1038/nrdp.2017.55
88. Lima K, Abrahamsen TG, Wolff AB, Husebye E, Alimohammadi M, Kampe O, et al. Hypoparathyroidism and Autoimmunity in the 22q11.2 Deletion Syndrome. Eur J Endocrinol (2011) 165(2):345–52. doi: 10.1530/EJE-10-1206
89. Daw SC, Taylor C, Kraman M, Call K, Mao J, Schuffenhauer S, et al. A Common Region of 10p Deleted in DiGeorge and Velocardiofacial Syndromes. Nat Genet (1996) 13(4):458–60. doi: 10.1038/ng0896-458
90. Hasegawa T, Hasegawa Y, Aso T, Koto S, Nagai T, Tsuchiya Y, et al. HDR Syndrome (Hypoparathyroidism, Sensorineural Deafness, Renal Dysplasia) Associated With Del(10)(P13). Am J Med Genet (1997) 73(4):416–8. doi: 10.1002/(SICI)1096-8628(19971231)73:4<416::AID-AJMG9>3.0.CO;2-L
91. Rushworth RL, Torpy DJ, Falhammar H. Adrenal Crisis. N Engl J Med (2019) 381(9):852–61. doi: 10.1056/NEJMra1807486
92. Husebye ES, Pearce SH, Krone NP, Kampe O. Adrenal Insufficiency. Lancet (2021) 397(10274):613–29. doi: 10.1016/S0140-6736(21)00136-7
93. Zaloga GP, Marik P. Hypothalamic-Pituitary-Adrenal Insufficiency. Crit Care Clin (2001) 17(1):25–41. doi: 10.1016/S0749-0704(05)70150-0
94. Chen K, Coonrod EM, Kumanovics A, Franks ZF, Durtschi JD, Margraf RL, et al. Germline Mutations in NFKB2 Implicate the Noncanonical NF-kappaB Pathway in the Pathogenesis of Common Variable Immunodeficiency. Am J Hum Genet (2013) 93(5):812–24. doi: 10.1016/j.ajhg.2013.09.009
95. Lee CE, Fulcher DA, Whittle B, Chand R, Fewings N, Field M, et al. Autosomal-Dominant B-Cell Deficiency With Alopecia Due to a Mutation in NFKB2 That Results in Nonprocessable P100. Blood (2014) 124(19):2964–72. doi: 10.1182/blood-2014-06-578542
96. Gilmore TD. Introduction to NF-Kappab: Players, Pathways, Perspectives. Oncogene (2006) 25(51):6680–4. doi: 10.1038/sj.onc.1209954
97. Winqvist O, Karlsson FA, Kampe O. 21-Hydroxylase, a Major Autoantigen in Idiopathic Addison’s Disease. Lancet (1992) 339(8809):1559–62. doi: 10.1016/0140-6736(92)91829-W
98. Bornstein SR, Allolio B, Arlt W, Barthel A, Don-Wauchope A, Hammer GD, et al. Diagnosis and Treatment of Primary Adrenal Insufficiency: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab (2016) 101(2):364–89. doi: 10.1210/jc.2015-1710
99. Wolff AS, Mitchell AL, Cordell HJ, Short A, Skinningsrud B, Ollier W, et al. CTLA-4 as a Genetic Determinant in Autoimmune Addison’s Disease. Genes Immun (2015) 16(6):430–6. doi: 10.1038/gene.2015.27
100. Gineau L, Cognet C, Kara N, Lach FP, Dunne J, Veturi U, et al. Partial MCM4 Deficiency in Patients With Growth Retardation, Adrenal Insufficiency, and Natural Killer Cell Deficiency. J Clin Invest (2012) 122(3):821–32. doi: 10.1172/JCI61014
101. Hughes CR, Guasti L, Meimaridou E, Chuang CH, Schimenti JC, King PJ, et al. MCM4 Mutation Causes Adrenal Failure, Short Stature, and Natural Killer Cell Deficiency in Humans. J Clin Invest (2012) 122(3):814–20. doi: 10.1172/JCI60224
102. Fluck CE. Mechanisms in Endocrinology: Update on Pathogenesis of Primary Adrenal Insufficiency: Beyond Steroid Enzyme Deficiency and Autoimmune Adrenal Destruction. Eur J Endocrinol (2017) 177(3):R99–111. doi: 10.1530/EJE-17-0128
103. Johnson MB, De Franco E, Lango Allen H, Al Senani A, Elbarbary N, Siklar Z, et al. Recessively Inherited LRBA Mutations Cause Autoimmunity Presenting as Neonatal Diabetes. Diabetes (2017) 66(8):2316–22. doi: 10.2337/db17-0040
104. Svensson M, Nystrom L, Schon S, Dahlquist G. Age at Onset of Childhood-Onset Type 1 Diabetes and the Development of End-Stage Renal Disease: A Nationwide Population-Based Study. Diabetes Care (2006) 29(3):538–42. doi: 10.2337/diacare.29.03.06.dc05-1531
105. Beltrand J, Busiah K, Vaivre-Douret L, Fauret AL, Berdugo M, Cave H, et al. Neonatal Diabetes Mellitus. Front Pediatr (2020) 8:540718. doi: 10.3389/fped.2020.540718
106. Mori S, Kondo N, Motoyoshi F, Yamaguchi S, Kaneko H, Orii T. Diabetes Mellitus in a Young Man With Bloom’s Syndrome. Clin Genet (1990) 38(5):387–90. doi: 10.1111/j.1399-0004.1990.tb03601.x
107. Donath H, Hess U, Kieslich M, Theis M, Ohlenschlager U, Schubert R, et al. Diabetes in Patients With Ataxia Telangiectasia: A National Cohort Study. Front Pediatr (2020) 8:317. doi: 10.3389/fped.2020.00317
108. Miyamoto S, Umeda K, Kurata M, Nishimura A, Yanagimachi M, Ishimura M, et al. Hematopoietic Cell Transplantation for Severe Combined Immunodeficiency Patients: A Japanese Retrospective Study. J Clin Immunol (2021). doi: 10.1007/s10875-021-01112-5
109. Sullivan KM, Parkman R, Walters MC. Bone Marrow Transplantation for Non-Malignant Disease. Hematol Am Soc Hematol Educ Program (2000) 319–38. doi: 10.1182/asheducation.V2000.1.319.319
110. Ono S, Takeshita K, Kiridoshi Y, Kato M, Kamiya T, Hoshino A, et al. Hematopoietic Cell Transplantation Rescues Inflammatory Bowel Disease and Dysbiosis of Gut Microbiota in XIAP Deficiency. J Allergy Clin Immunol Pract (2021) 9. doi: 10.1016/j.jaip.2021.05.045
111. Kahn JM, Brazauskas R, Tecca HR, Bo-Subait S, Buchbinder D, Battiwala M, et al. Subsequent Neoplasms and Late Mortality in Children Undergoing Allogeneic Transplantation for Nonmalignant Diseases. Blood Adv (2020) 4(9):2084–94. doi: 10.1182/bloodadvances.2019000839
112. Madden LM, Hayashi RJ, Chan KW, Pulsipher MA, Douglas D, Hale GA, et al. Long-Term Follow-Up After Reduced-Intensity Conditioning and Stem Cell Transplantation for Childhood Nonmalignant Disorders. Biol Blood Marrow Transplant (2016) 22(8):1467–72. doi: 10.1016/j.bbmt.2016.04.025
113. Rao K, Amrolia PJ, Jones A, Cale CM, Naik P, King D, et al. Improved Survival After Unrelated Donor Bone Marrow Transplantation in Children With Primary Immunodeficiency Using a Reduced-Intensity Conditioning Regimen. Blood (2005) 105(2):879–85. doi: 10.1182/blood-2004-03-0960
114. Fujino H, Ishida H, Iguchi A, Onuma M, Kato K, Shimizu M, et al. High Rates of Ovarian Function Preservation After Hematopoietic Cell Transplantation With Melphalan-Based Reduced Intensity Conditioning for Pediatric Acute Leukemia: An Analysis From the Japan Association of Childhood Leukemia Study (JACLS). Int J Hematol (2019) 109(5):578–83. doi: 10.1007/s12185-019-02627-9
115. Leiper A, Houwing M, Davies EG, Rao K, Burns S, Morris E, et al. Anti-Mullerian Hormone and Inhibin B After Stem Cell Transplant in Childhood: A Comparison of Myeloablative, Reduced Intensity and Treosulfan-Based Chemotherapy Regimens. Bone Marrow Transplant (2020) 55(10):1985–95. doi: 10.1038/s41409-020-0866-9
116. Green DM, Liu W, Kutteh WH, Ke RW, Shelton KC, Sklar CA, et al. Cumulative Alkylating Agent Exposure and Semen Parameters in Adult Survivors of Childhood Cancer: A Report From the St Jude Lifetime Cohort Study. Lancet Oncol (2014) 15(11):1215–23. doi: 10.1016/S1470-2045(14)70408-5
117. Rivas Leonel EC, Lucci CM, Amorim CA. Cryopreservation of Human Ovarian Tissue: A Review. Transfus Med Hemother (2019) 46(3):173–81. doi: 10.1159/000499054
118. Hopkins RL, Leinung MC. Exogenous Cushing’s Syndrome and Glucocorticoid Withdrawal. Endocrinol Metab Clin North Am (2005) 34(2):371–84, ix. doi: 10.1016/j.ecl.2005.01.013
Keywords: endocrinopathy, thyroiditis, diabetes mellitus, growth failure, inborn errors of immunity (IEIs), HCT
Citation: Takasawa K, Kanegane H, Kashimada K and Morio T (2021) Endocrinopathies in Inborn Errors of Immunity. Front. Immunol. 12:786241. doi: 10.3389/fimmu.2021.786241
Received: 30 September 2021; Accepted: 01 November 2021;
Published: 23 November 2021.
Edited by:
Satoshi Okada, Hiroshima University, JapanReviewed by:
Hidenori Ohnishi, Gifu University, JapanBergithe Eikeland Oftedal, University of Bergen, Norway
Copyright © 2021 Takasawa, Kanegane, Kashimada and Morio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kenichi Kashimada, kkashimada.ped@tmd.ac.jp