 Ji-Ae Choi1,2,3*
Ji-Ae Choi1,2,3* Chang-Hwa Song1,2,3*
Chang-Hwa Song1,2,3*- 1Department of Medical Science, College of Medicine, Chungnam National University, Daejeon, South Korea
- 2Department of Microbiology, College of Medicine, Chungnam National University, Daejeon, South Korea
- 3Research Institute for Medical Sciences, College of Medicine, Chungnam National University, Daejeon, South Korea
The endoplasmic reticulum (ER) is the major organelle in the cell for protein folding and plays an important role in cellular functions. The unfolded protein response (UPR) is activated in response to misfolded or unfolded protein accumulation in the ER. However, the UPR successfully alleviates the ER stress. If UPR fails to restore ER homeostasis, apoptosis is induced. ER stress plays an important role in innate immune signaling in response to microorganisms. Dysregulation of UPR signaling contributes to the pathogenesis of a variety of infectious diseases. In this review, we summarize the contribution of ER stress to the innate immune response to invading microorganisms and its role in the pathogenesis of infectious diseases.
Introduction
The endoplasmic reticulum (ER) is crucial for maintaining cellular calcium homeostasis and for the production, processing, and transport of proteins and lipids (1). The rough ER working with membrane-bound ribosomes produces the protein and continues protein assembly, while the smooth ER synthesizes lipids, phospholipids, and steroids (2). The ER is sensitive to stresses that perturb the intracellular energy level, redox state, or calcium concentration. If the protein-folding function of the ER is reduced by stresses, unfolded proteins or misfolded proteins can accumulate. To prevent the resulting cytotoxicity, the unfolded protein response (UPR) is activated (1). ER-resident transmembrane proteins such as inositol-requiring enzyme 1 (IRE1), protein kinase R (PKR)-like ER kinase (PERK), and activating transcription factor 6 (ATF6) are implicated in activation of the UPR (3).
To maintain protein-folding homeostasis in the ER, mRNA translation is transiently attenuated through phosphorylation of eIF2α leading to ATF4 activation (2). IRE1α-dependent decay (RIDD) suppresses the load of new synthesized protein in ER through degradation of ER-localized mRNAs, ribosomal RNAs, and miRNAs (4, 5). Spliced X-box binding protein-1 (XBP-1) mRNA induced by activated IRE1α regulates the expression of numerous target genes including ER chaperones and ER-associated protein degradation (ERAD) components (6, 7). For instance, BiP is known to be an important factor in modulating the UPR to avoid apoptosis (4). ER stress regulates autophagy by modulating the release of calcium from the ER to cytosol and by modulating the activation of mTORC via PI3K/AKT or AMPK (8). Autophagy is also essential to maintain cellular homeostasis through degradation of dysfunctional components from the cells by using lysosomal degradation pathway (8, 9). The activation of three main molecules of UPR induces autophagy via regulation of ATG genes (8). The spliced XBP-1 directly binds to beclin-1 gene promotor region (10).
Another clearance mechanism to degrade the accumulation of misfolded proteins is the ERAD pathway. ERAD substrates recognized by ER chaperone are delivered to ERAD adaptors on ER membrane and then it is retro-translocated into the cytosol (11). The misfolded proteins undergo proteolytic degradation by the ubiquitin proteasome system to maintain the proteostasis (11). When the accumulation of misfolded proteins in the ER overwhelms the capacity of the ERAD system, ER stress, detected by the ER stress sensors IRE1, ATF6, and PERK, activates the UPR, but excessive ER stress may eventually lead to apoptosis (1, 12). Although the mechanism of UPR is well-known, it is not clear how the response regulates both apoptotic and adaptive pathways. A previous report suggested that instabilities of pro-survival and pro-apoptotic mRNAs and proteins mediate adaptation to ER stress (13). It was proposed that Death Receptor 5 (DR5) integrates dynamic UPR signals to control apoptosis in relation to ER stress (14).
In stressed cells, unfolded and misfolded proteins may accumulate in ER and ER-folding capacity is exceeded, causing apoptosis (15). PERK and eIF2α phosphorylation play an important role in protecting cells against the consequences of ER stress (16). Diverse stress stimuli-induced eIF2α dephosphorylation causes cells to die due to ER stress (12). Although C/EBP Homologous Protein (CHOP) is a well-known transcription factor induced by eIF2α phosphorylation, deregulated CHOP expression promotes apoptosis (17). CHOP promotes the expression of Bim, a pro-apoptotic protein, and decreases the expression of anti-apoptotic Bcl-2 (18). The GADD34, transcriptional target of CHOP, induces eIF2α dephosphorylation leading to restoration of protein translation (12). However, the overexpression of GADD34 can elicit apoptosis (12). Hyperactivated IRE1α cleaves and degrades precursor of miRNAs that normally repress translation of caspase-2 mRNA, and thus induces mitochondrial apoptotic pathway (19). Disruption of the cellular Ca2+ homeostasis induces calpain activation, which cleaves Bid and pro-caspase-12, and subsequently triggers caspase-3-dependent apoptosis (20).
It is well-known that ER stress is associated with the pathogenesis of various diseases such as obesity, diabetes, cancer, neurodegenerative disorders, inflammatory diseases, and infectious diseases. However, we have only vague ideas of how ER stress is involved in the pathogenesis of infectious diseases. Recently, many scientists are trying to unveil the implication of ER stress in infectious diseases. Thus, a better understanding of the regulatory mechanisms of ER stress will be important in the development of new therapeutics to treat refractory infectious diseases.
The UPR in Immunity
ER stress induces an inflammatory response by activating UPR transcription factors and plays an important role in the pathogenesis of inflammatory and autoimmune diseases, such as obesity, diabetes, atherosclerosis, myositis, and inflammatory bowel disease (21). The UPR has important roles in the development of immune cells because UPR regulates immune cell differentiation, activation, and cytokine production (21). The immunostimulant lipopolysaccharide (LPS) induces inflammatory cytokine production and activates the transcription of ER chaperone genes, including spliced XBP-1, BiP, ATF4, and CHOP (21, 22). Spliced XBP1s in response to toll-like receptor (TLR) is necessary for macrophages to produce proinflammatory cytokines (23). It has been known that the IRE1α-XBP-1 pathway activates production of TNF and IL-6 in macrophages of cystic fibrosis patients (24). Additionally, the IRE1α/TRAF2 pathway-mediated NOD1 and NOD2 signaling provides ER-stress-induced inflammation (25). Many studies have shown that the role of ER stress is associated with immune cell differentiation, activation, and cytokine production.
A recent report suggests that XBP-1 is important for the differentiation of Th17 cells (26). Interestingly, it is known that the IRE1–XBP-1 pathway is activated by acute infection and is required for T cell differentiation (27). Another role of the IRE1–XBP-1 pathway is to support the functions of plasma cells and the survival of dendritic cells (28, 29). UPR is implicated in host immunity due to its involvement in calcium signaling, glycosylation, lipid metabolism, and oxidative protein folding (30). Activation of the T-cell receptor, the B-cell receptor, the Fc-γ receptor, and various cytokine receptors causes calcium efflux from the ER through inositol 1,4,5-trisphosphate receptor (IP3R) (30). The resulting increased intracellular calcium concentration activates cellular signaling molecules to promote T-cell activation, maturation of myeloid cells, and cellular differentiation, adhesion, and death (31). The increased levels of ROS triggered by ER stress activate not only proinflammatory signals but also inflammasome formation, suggesting that ER stress exerts immunogenic effects (32).
The transcription factor nuclear factor kappaB (NF-κB) regulates the immune response of the host. ER-stress-mediated activation of NF-κB may modulate the production of cytokines (21). The IRE1α-TRAF2–IKK interaction is known to activate NF-κB, which in turn leads to the production of TNF-α (33). IRE1α induces inflammation by activating JNK (34, 35). The interaction between ER stress and MAPKs (JNK, p38, and ERK) may contribute to inflammatory responses. Therefore, the UPR plays a critical role in regulating the immune response.
Relationship of ER Stress With Pathogens
In mammalian cells, the UPR is triggered by three ER-stress sensor proteins, IRE1, PERK, and ATF6, to restore ER homeostasis (3). Infection by the majority of known pathogens activates the UPR. Modulation of the functions of the ER by pathogens can result in their survival/replication or clearance because ER stress is associated with autophagy or apoptosis. Although little is known about the role of the ER-stress response in the pathogenesis of viral and bacterial infection, the regulation of ER stress might be important in intractable infectious diseases.
Bacterial Infection and ER Stress
Bacterial virulence factors are involved in UPR activation. LPS from Gram-negative bacteria binds to TLR4 that is delivered by Glucose-regulated protein 94 (Grp94) (36). The expression of Grp94 (an HSP90-like protein specialized for protein folding and quality control in the ER) is increased by LPS stimulation (36). The cytotoxin subtilase, produced by Shiga-toxigenic Escherichia coli, cleaves BiP, resulting in DNA fragmentation and UPR-mediated apoptosis (37). Similarly, the Shiga toxin produced by the enteric pathogens Shigella dysenteriae serotype 1 and enterohemorrhagic E. coli increases ER-stress-mediated apoptosis by inducing release of Ca2+ from the ER to the cytosol and upregulating PERK-CHOP-mediated DR5 (38, 39). Subunit A of unfolded cholera toxin (CT) retro-translocates through the ER membrane to the cytoplasm, where it directly binds to the ER-luminal domain of IRE1α (40). Streptolysin O and streptolysin S of group A Streptococcus (GAS) induce the production of ATF4, which upregulates the expression of proliferation-related genes of GAS (41). ER stress is also important for biofilm formation, microcolony aggregation, distribution, and spread of GAS during infection of soft tissues (42). The pore-forming toxin listeriolysin O (LLO) produced by Listeria monocytogenes induces the three axes of the UPR before cell entry (43). Therefore, diverse bacterial taxa induce the UPR by secreting toxins (Figure 1A).
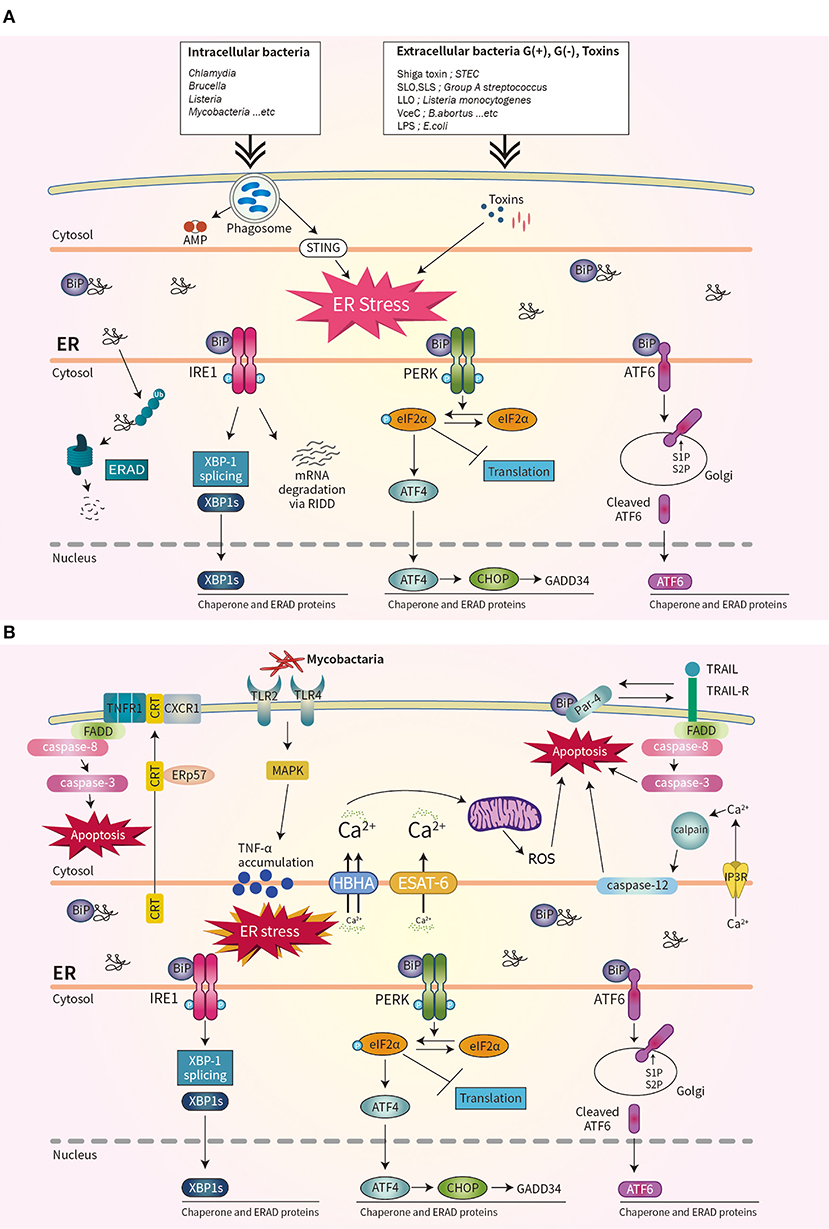
Figure 1. Schematic overview of unfolded protein response (UPR) signaling during bacterial infection. (A) Three ER stress sensors–IRE1, PERK, and ATF6–are activated when the accumulation of misfolded protein aggregates promotes recruitment of BiP. Bacterial infection and toxins activate the UPR. (B) During mycobacterial infection, co-translocated calreticulin and ERp57 form a complex with TNFR1 and CXCR1 in the plasma membrane, leading to apoptosis, and suppression of intracellular Mtb. The interaction of Par-4 and BiP leads to apoptosis by inducing Mtb-mediated ER stress and activating the FADD/caspase-8/-3 pathway. The mycobacterial antigens HBHA and ESAT-6 affect the ER membrane and induce the release of Ca2+ from the ER to mitochondria, leading to the production of reactive oxygen species and apoptosis.
The obligate intracellular pathogen Chlamydia induces the UPR by upregulating BiP (44, 45). Chlamydia infection also induces TLR4/IRE1-mediated activation of PKR, which enhances IFN-β production (46). Brucella abortus localizes to the ER by transforming its fine reticular pattern into a thicker tubular structure (47). Also, VceC of B. abortus directly binds BiP and selectively activates the IRE1–XBP-1 pathway, which increases the IL-6 level (47). Enhancement of the UPR by co-stimulation with IFN-β promotes the replication of B. abortus in host cells (48). Thus, modulating the UPR may be useful for treating brucellosis or chlamydia infection (Figure 1A).
Bacterial infections are frequently caused in chronic diseases such as obesity, type 1 and type 2 diabetes (T2D), and atherosclerosis (49, 50). The reason that altered immune functions in these chronic diseases are observed is because the immune system can be affected by chronic stress (15, 50). It has been known that different ER stress sensors are activated during bacterial infection (11). Therefore, understanding the diverse roles of ER stress sensors during bacterial infection might be effective to treat chronic diseases in the future.
Mycobacterial Infection and ER Stress
Mycobacterium tuberculosis (Mtb) is the causative agent of tuberculosis (TB), does not produce toxins, and grows very slowly. However, Mtb infection induces ER stress in host cells. Mycobacterial ESAT-6 and HBHA induce ER stress by promoting ROS production, by disrupting intracellular calcium homeostasis (51, 52). ER-stress-induced apoptosis suppresses the intracellular growth of Mtb by activating caspase-12 in the outer membrane of the ER (52). Phosphorylation of eIF2α is reported to be an important component of the ER stress response that modulates the intracellular survival of Mtb (Figure 1B) (53).
Interestingly, Mtb-induced ER chaperones contribute to the translocation of CRT or Par-4 to the plasma membrane of macrophages, leading to apoptosis and suppression of Mtb growth (54, 55). Also, mycobacterial infection induces an ER-stress response due to accumulation of misfolded or unfolded TNF-α in the ER; this indicates that Mtb-mediated overproduction of proinflammatory cytokines induces ER stress in macrophages (Figure 1B) (56). Moreover, activation of the RIDD pathway suppresses the intracellular growth of mycobacteria (57). Therefore, investigation of the regulatory mechanism of ER stress during mycobacterial infection might suggest new therapeutic targets for multidrug-resistant TB.
Viral Infection and ER Stress
Viruses modulate host defense mechanisms to escape the host immune response. Viruses may interact with the host UPR to maintain an environment favorable for establishment of persistent infection. Indeed, viral infection can disturb ER stress (58). ER stress and the UPR are reported not to protect against infection by reovirus and hepatitis B virus but, rather, promote their replication (59, 60).
The PERK pathway is important for host antiviral defense (61). The PERK-mediated phosphorylation of eIF2α may be responsible for regulating viral replication (62). Similarly, regulation of the phosphorylation of eIF2α is important for the survival of enveloped viruses, such as herpes simplex virus (HSV) (63). HSV reduces the level of MHC-I by promoting its ER-associated degradation (ERAD), leading to suppression of the immune response (64). Infection by human immunodeficiency virus type 1 induces the degradation of CD4 by ERAD (64). RIDD degrades mRNAs to reduce ER load and alleviates ER stress (9). Although it is unclear whether RIDD activation is beneficial for all viral infectivity, RIDD is closely associated with viral RNA synthesis (4, 9). This is likely because virus replication requires large quantities of membrane proteins and lipids, which are produced in the ER (Figure 2).
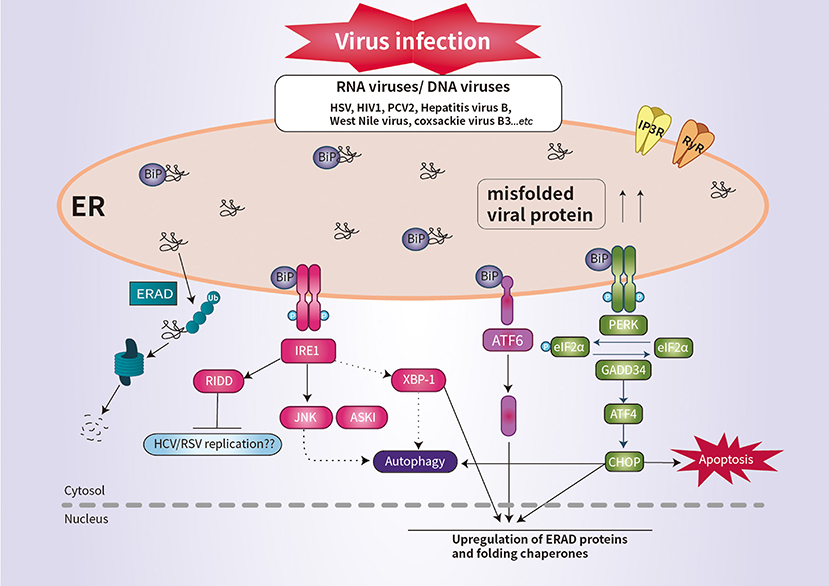
Figure 2. Schematic overview of UPR signaling during viral infection. Viral infection induces ER stress and the UPR, which promotes cell survival by inhibiting apoptosis. Some viral infection induces ER-stress-mediated apoptosis by promoting the synthesis of CHOP. IRE1 activates RIDD to promote the degradation of ER-localized mRNAs. The activation of RIDD may enhance viral protein synthesis. The interaction between the UPR pathways and the autophagic response is implicated in the pathogenesis of viral infection.
CHOP plays an important role in suppressing infection of host cells by RNA viruses (9). For example, porcine circovirus type 2 (PCV2) triggers the eIF2α-ATF4–CHOP pathway and activation of caspases (65). West Nile virus and coxsackie virus B3 induce ER-stress-mediated apoptosis by promoting the synthesis of CHOP (66, 67). Interestingly, IRE1 is reportedly essential for induction of autophagy during infection with infectious bronchitis virus (Figure 2) (68). Thus, further investigation of the ER stress response would enhance our understanding of the pathogenesis of viral infection.
Conclusion and Future Perspectives
Knowledge of the mechanisms by which viruses modulate the UPR has advanced more than that of bacteria. The interaction between pathogenic bacteria and ER stress is under active investigation, but the role of ER stress in the pathogenesis of infectious diseases is unclear. How ER stress modulates bacterial survival and how bacteria modulate ER stress to promote their replication need to be studied. ER stress is not only associated with autophagy but also with the immune response to pathogens. Targeting ER stress and the UPR with small molecules is emerging as a promising therapy for treatment of various diseases such as neurodegeneration, cancer, metabolic diseases, stroke, and heart disease (69). Therefore, studies of the regulatory mechanisms of ER stress during pathogenic infection are warranted. The results of such efforts are likely to lead to the development of novel host-derived therapeutics for infection by multidrug-resistant bacteria or emerging viruses.
Author Contributions
J-AC and C-HS contributed to the writing of the manuscript, proofreading, editing, and figure preparation.
Funding
This work was supported by the research fund of Chungnam National University and by the Brain Korea 21 PLUS Project for Medical Science, Chungnam National University College of Medicine. The funders had no role in the study design, data collection, and analysis decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. (2006) 7:880–5. doi: 10.1038/sj.embor.7400779
2. Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. (2010) 140:900–17. doi: 10.1016/j.cell.2010.02.034
3. Bravo R, Parra V, Gatica D, Rodriguez AE, Torrealba N, Paredes F, et al. Endoplasmic reticulum and the unfolded protein response: dynamics and metabolic integration. Int Rev Cell Mol Biol. (2013) 301:215–90. doi: 10.1016/B978-0-12-407704-1.00005-1
4. Maurel M, Chevet E, Tavernier J, Gerlo S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem Sci. (2014) 39:245–54. doi: 10.1016/j.tibs.2014.02.008
5. Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. (2006) 313:104–7. doi: 10.1126/science.1129631
6. Uemura A, Oku M, Mori K, Yoshida H. Unconventional splicing of XBP1 mRNA occurs in the cytoplasm during the mammalian unfolded protein response. J Cell Sci. (2009) 122:2877–86. doi: 10.1242/jcs.040584
7. Acosta-Alvear D, Zhou Y, Blais A, Tsikitis M, Lents NH, Arias C, et al. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol Cell. (2007) 27:53–66. doi: 10.1016/j.molcel.2007.06.011
8. Rashid HO, Yadav RK, Kim HR, Chae HJ. ER stress: autophagy induction, inhibition and selection. Autophagy. (2015) 11:1956–77. doi: 10.1080/15548627.2015.1091141
9. Jheng JR, Ho JY, Horng JT. ER stress, autophagy, and RNA viruses. Front Microbiol. (2014) 5:388. doi: 10.3389/fmicb.2014.00388
10. Vidal RL, Figueroa A, Court FA, Thielen P, Molina C, Wirth C, et al. Targeting the UPR transcription factor XBP1 protects against Huntington's disease through the regulation of FoxO1 and autophagy. Hum Mol Genet. (2012) 21:2245–62. doi: 10.1093/hmg/dds040
11. Celli J, Tsolis RM. Bacteria, the ER and the unfolded protein response: friends or foes? Nat Rev Microbiol. (2015) 13:71. doi: 10.1038/nrmicro3393
12. Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. (2011) 13:184–90. doi: 10.1038/ncb0311-184
13. Rutkowski DT, Arnold SM, Miller CN, Wu J, Li J, Gunnison KM, et al. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. (2006) 4:e374. doi: 10.1371/journal.pbio.0040374
14. Lu M, Lawrence DA, Marsters S, Acosta-Alvear D, Kimmig P, Mendez AS, et al. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science. (2014) 345:98–101. doi: 10.1126/science.1254312
15. Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. (2008) 454:455–62. doi: 10.1038/nature07203
16. Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, et al. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell. (2001) 7:1165–76. doi: 10.1016/S1097-2765(01)00265-9
17. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. (2007) 8:519–29. doi: 10.1038/nrm2199
18. Puthalakath H, O'Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. (2007) 129:1337–49. doi: 10.1016/j.cell.2007.04.027
19. Upton JP, Wang L, Han D, Wang ES, Huskey NE, Lim L, et al. IRE1α cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science. (2012) 338:818–22. doi: 10.1126/science.1226191
20. Nakagawa T, Yuan J. Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J Cell Biol. (2000) 150:887–94. doi: 10.1083/jcb.150.4.887
21. Bettigole SE, Glimcher LH. Endoplasmic reticulum stress in immunity. Annu Rev Immunol. (2015) 33:107–38. doi: 10.1146/annurev-immunol-032414-112116
22. Endo M, Oyadomari S, Suga M, Mori M, Gotoh T. The ER stress pathway involving CHOP is activated in the lungs of LPS-treated mice. J Biochem. (2005) 138:501–7. doi: 10.1093/jb/mvi143
23. Martinon F, Chen X, Lee AH, Glimcher LH. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol. (2010) 11:411–8. doi: 10.1038/ni.1857
24. Lara-Reyna S, Scambler T, Holbrook J, Wong C, Jarosz-Griffiths HH, Martinon F, et al. Metabolic reprograming of cystic fibrosis macrophages via the IRE1α arm of the unfolded protein response results in exacerbated inflammation. Front Immunol. (2019) 10:1789. doi: 10.3389/fimmu.2019.01789
25. Keestra-Gounder AM, Byndloss MX, Seyffert N, Young BM, Chavez-Arroyo A, Tsai AY, et al. NOD1 and NOD2 signalling links ER stress with inflammation. Nature. (2016) 532:394–7. doi: 10.1038/nature17631
26. Brucklacher-Waldert V, Ferreira C, Stebegg M, Fesneau O, Innocentin S, Marie JC, et al. Cellular stress in the context of an inflammatory environment supports TGF-beta-independent T Helper-17 differentiation. Cell Rep. (2017) 19:2357–70. doi: 10.1016/j.celrep.2017.05.052
27. Kamimura D, Bevan MJ. Endoplasmic reticulum stress regulator XBP-1 contributes to effector CD8+ T cell differentiation during acute infection. J Immunol. (2008) 181:5433–41. doi: 10.4049/jimmunol.181.8.5433
28. Iwawaki T, Akai R, Kohno K. IRE1α disruption causes histological abnormality of exocrine tissues, increase of blood glucose level, and decrease of serum immunoglobulin level. PLoS ONE. (2010) 5:e13052. doi: 10.1371/journal.pone.0013052
29. Osorio F, Tavernier SJ, Hoffmann E, Saeys Y, Martens L, Vetters J, et al. The unfolded-protein-response sensor IRE-1α regulates the function of CD8α+ dendritic cells. Nat Immunol. (2014) 15:248–57. doi: 10.1038/ni.2808
30. Feske S, Okamura H, Hogan PG, Rao A. Ca2+/calcineurin signalling in cells of the immune system. Biochem Biophys Res Commun. (2003) 311:1117–32. doi: 10.1016/j.bbrc.2003.09.174
31. Vig M, Kinet JP. Calcium signaling in immune cells. Nat Immunol. (2009) 10:21–7. doi: 10.1038/ni.f.220
32. Menu P, Mayor A, Zhou R, Tardivel A, Ichijo H, Mori K, et al. ER stress activates the NLRP3 inflammasome via an UPR-independent pathway. Cell Death Dis. (2012) 3:e261. doi: 10.1038/cddis.2011.132
33. Hu P, Han Z, Couvillon AD, Kaufman RJ, Exton JH. Autocrine tumor necrosis factor α links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1α-mediated NF-kappaB activation and down-regulation of TRAF2 expression. Mol Cell Biol. (2006) 26:3071–84. doi: 10.1128/MCB.26.8.3071-3084.2006
34. Kaser A, Lee AH, Franke A, Glickman JN, Zeissig S, Tilg H, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. (2008) 134:743–56. doi: 10.1016/j.cell.2008.07.021
35. Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. (2000) 287:664–6. doi: 10.1126/science.287.5453.664
36. Coope A, Milanski M, Arruda AP, Ignacio-Souza LM, Saad MJ, Anhe GF, et al. Chaperone insufficiency links TLR4 protein signaling to endoplasmic reticulum stress. J Biol Chem. (2012) 287:15580–9. doi: 10.1074/jbc.M111.315218
37. Morinaga N, Yahiro K, Matsuura G, Moss J, Noda M. Subtilase cytotoxin, produced by Shiga-toxigenic Escherichia coli, transiently inhibits protein synthesis of Vero cells via degradation of BiP and induces cell cycle arrest at G1 by downregulation of cyclin D1. Cell Microbiol. (2008) 10:921–9. doi: 10.1111/j.1462-5822.2007.01094.x
38. Lee SY, Lee MS, Cherla RP, Tesh VL. Shiga toxin 1 induces apoptosis through the endoplasmic reticulum stress response in human monocytic cells. Cell Microbiol. (2008) 10:770–80. doi: 10.1111/j.1462-5822.2007.01083.x
39. Lee MS, Cherla RP, Leyva-Illades D, Tesh VL. Bcl-2 regulates the onset of shiga toxin 1-induced apoptosis in THP-1 cells. Infect Immun. (2009) 77:5233–44. doi: 10.1128/IAI.00665-09
40. Wernick NL, De Luca H, Kam WR, Lencer WI. N-terminal extension of the cholera toxin A1-chain causes rapid degradation after retrotranslocation from endoplasmic reticulum to cytosol. J Biol Chem. (2010) 285:6145–52. doi: 10.1074/jbc.M109.062067
41. Baruch M, Belotserkovsky I, Hertzog BB, Ravins M, Dov E, McIver KS, et al. An extracellular bacterial pathogen modulates host metabolism to regulate its own sensing and proliferation. Cell. (2014) 156:97–108. doi: 10.1016/j.cell.2013.12.007
42. Vajjala A, Biswas D, Tay WH, Hanski E, Kline KA. Streptolysin-induced endoplasmic reticulum stress promotes group A Streptococcal host-associated biofilm formation and necrotising fasciitis. Cell Microbiol. (2019) 21:e12956. doi: 10.1111/cmi.12956
43. Pillich H, Loose M, Zimmer KP, Chakraborty T. Activation of the unfolded protein response by Listeria monocytogenes. Cell Microbiol. (2012) 14:949–64. doi: 10.1111/j.1462-5822.2012.01769.x
44. Shima K, Klinger M, Schutze S, Kaufhold I, Solbach W, Reiling N, et al. The role of endoplasmic reticulum-related BiP/GRP78 in interferon gamma-induced persistent Chlamydia pneumoniae infection. Cell Microbiol. (2015) 17:923–34. doi: 10.1111/cmi.12416
45. George Z, Omosun Y, Azenabor AA, Partin J, Joseph K, Ellerson D, et al. The roles of unfolded protein response pathways in chlamydia pathogenesis. J Infect Dis. (2017) 215:456–65. doi: 10.1093/infdis/jiw569
46. Webster SJ, Ellis L, O'Brien LM, Tyrrell B, Fitzmaurice TJ, Elder MJ, et al. IRE1α mediates PKR activation in response to Chlamydia trachomatis infection. Microbes Infect. (2016) 18:472–83. doi: 10.1016/j.micinf.2016.03.010
47. de Jong MF, Starr T, Winter MG, den Hartigh AB, Child R, Knodler LA, et al. Sensing of bacterial type IV secretion via the unfolded protein response. MBio. (2013) 4:e00418–12. doi: 10.1128/mBio.00418-12
48. Guimaraes ES, Gomes MTR, Campos PC, Mansur DS, Dos Santos AA, Harms J, et al. Brucella abortus cyclic dinucleotides trigger STING-dependent unfolded protein response that favors bacterial replication. J Immunol. (2019) 202:2671–81. doi: 10.4049/jimmunol.1801233
49. Gan YH. Host susceptibility factors to bacterial infections in type 2 diabetes. PLoS Pathog. (2013) 9:e1003794. doi: 10.1371/journal.ppat.1003794
50. Gomez JA, Rutkowski DT. Experimental reconstitution of chronic ER stress in the liver reveals feedback suppression of BiP mRNA expression. Elife. (2016) 5:e20390. doi: 10.7554/eLife.20390
51. Choi HH, Shin DM, Kang G, Kim KH, Park JB, Hur GM, et al. Endoplasmic reticulum stress response is involved in Mycobacterium tuberculosis protein ESAT-6-mediated apoptosis. FEBS Lett. (2010) 584:2445–54. doi: 10.1016/j.febslet.2010.04.050
52. Choi JA, Lim YJ, Cho SN, Lee JH, Jeong JA, Kim EJ, et al. Mycobacterial HBHA induces endoplasmic reticulum stress-mediated apoptosis through the generation of reactive oxygen species and cytosolic Ca2+ in murine macrophage RAW 264.7 cells. Cell Death Dis. (2013) 4:e957. doi: 10.1038/cddis.2013.489
53. Lim YJ, Choi JA, Choi HH, Cho SN, Kim HJ, Jo EK, et al. Endoplasmic reticulum stress pathway-mediated apoptosis in macrophages contributes to the survival of Mycobacterium tuberculosis. PLoS ONE. (2011) 6:e28531. doi: 10.1371/journal.pone.0028531
54. Jo SH, Choi JA, Lim YJ, Lee J, Cho SN, Oh SM, et al. Calreticulin modulates the intracellular survival of mycobacteria by regulating ER-stress-mediated apoptosis. Oncotarget. (2017) 8:58686–98. doi: 10.18632/oncotarget.17419
55. Han JY, Lim YJ, Choi JA, Lee JH, Jo SH, Oh SM, et al. The role of prostate apoptosis response-4 (Par-4) in Mycobacterium tuberculosis infected macrophages. Sci Rep. (2016) 6:32079. doi: 10.1038/srep32079
56. Oh SM, Lim YJ, Choi JA, Lee J, Cho SN, Go D, et al. TNF-α-mediated ER stress causes elimination of Mycobacterium fortuitum reservoirs by macrophage apoptosis. FASEB J. (2018) 32:3993–4003. doi: 10.1096/fj.201701407R
57. Go D, Lee J, Choi JA, Cho SN, Kim SH, Son SH, et al. Reactive oxygen species-mediated endoplasmic reticulum stress response induces apoptosis of M. avium-infected macrophages by activating IRE1α-regulated IRE1-dependent decay (RIDD) pathway. Cell Microbiol. (2019) 21:e13094. doi: 10.1111/cmi.13094
58. Chan SW. Unfolded protein response in hepatitis C virus infection. Front Microbiol. (2014) 5:233. doi: 10.3389/fmicb.2014.00233
59. Smith JA, Schmechel SC, Raghavan A, Abelson M, Reilly C, Katze MG, et al. Reovirus induces and benefits from an integrated cellular stress response. J Virol. (2006) 80:2019–33. doi: 10.1128/JVI.80.4.2019-2033.2006
60. Huang ZM, Tan T, Yoshida H, Mori K, Ma Y, Yen TS. Activation of hepatitis B virus S promoter by a cell type-restricted IRE1-dependent pathway induced by endoplasmic reticulum stress. Mol Cell Biol. (2005) 25:7522–33. doi: 10.1128/MCB.25.17.7522-7533.2005
61. Baltzis D, Qu LK, Papadopoulou S, Blais JD, Bell JC, Sonenberg N, et al. Resistance to vesicular stomatitis virus infection requires a functional cross talk between the eukaryotic translation initiation factor 2α kinases PERK and PKR. J Virol. (2004) 78:12747–61. doi: 10.1128/JVI.78.23.12747-12761.2004
62. Perkins DJ, Barber GN. Defects in translational regulation mediated by the α subunit of eukaryotic initiation factor 2 inhibit antiviral activity and facilitate the malignant transformation of human fibroblasts. Mol Cell Biol. (2004) 24:2025–40. doi: 10.1128/MCB.24.5.2025-2040.2004
63. Mulvey M, Arias C, Mohr I. Maintenance of endoplasmic reticulum (ER) homeostasis in herpes simplex virus type 1-infected cells through the association of a viral glycoprotein with PERK, a cellular ER stress sensor. J Virol. (2007) 81:3377–90. doi: 10.1128/JVI.02191-06
64. Byun H, Gou Y, Zook A, Lozano MM, Dudley JP. ERAD and how viruses exploit it. Front Microbiol. (2014) 5:330. doi: 10.3389/fmicb.2014.00330
65. Zhou Y, Qi B, Gu Y, Xu F, Du H, Li X, et al. Porcine circovirus 2 deploys PERK pathway and GRP78 for its enhanced replication in PK-15 cells. Viruses. (2016) 8:E56. doi: 10.3390/v8020056
66. Zhang HM, Ye X, Su Y, Yuan J, Liu Z, Stein DA, et al. Coxsackievirus B3 infection activates the unfolded protein response and induces apoptosis through downregulation of p58IPK and activation of CHOP and SREBP1. J Virol. (2010) 84:8446–59. doi: 10.1128/JVI.01416-09
67. Medigeshi GR, Lancaster AM, Hirsch AJ, Briese T, Lipkin WI, Defilippis V, et al. West Nile virus infection activates the unfolded protein response, leading to CHOP induction and apoptosis. J Virol. (2007) 81:10849–60. doi: 10.1128/JVI.01151-07
68. Fung TS, Liu DX. The ER stress sensor IRE1 and MAP kinase ERK modulate autophagy induction in cells infected with coronavirus infectious bronchitis virus. Virology. (2019) 533:34–44. doi: 10.1016/j.virol.2019.05.002
Keywords: ER stress, infection, infectious disease, UPR (unfolded protein response), bacteria, viruses, pathogen, apoptosis
Citation: Choi J-A and Song C-H (2020) Insights Into the Role of Endoplasmic Reticulum Stress in Infectious Diseases. Front. Immunol. 10:3147. doi: 10.3389/fimmu.2019.03147
Received: 23 August 2019; Accepted: 27 December 2019;
Published: 31 January 2020.
Edited by:
Cláudia Pereira, University of Coimbra, PortugalReviewed by:
Guillaume Thibault, Nanyang Technological University, SingaporePatrick Lajoie, University of Western Ontario, Canada
Copyright © 2020 Choi and Song. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ji-Ae Choi, jiae9035@gmail.com; Chang-Hwa Song, songch@cnu.ac.kr