 Larisa V. Kovtonyuk
Larisa V. Kovtonyuk Kristin Fritsch
Kristin Fritsch Xiaomin Feng2
Xiaomin Feng2 Hitoshi Takizawa
Hitoshi Takizawa- 1Division of Hematology, University Hospital Zurich, University of Zurich, Zurich, Switzerland
- 2International Research Center for Medical Sciences, Kumamoto, Japan
All hematopoietic and immune cells are continuously generated by hematopoietic stem cells (HSCs) and hematopoietic progenitor cells (HPCs) through highly organized process of stepwise lineage commitment. In the steady state, HSCs are mostly quiescent, while HPCs are actively proliferating and contributing to daily hematopoiesis. In response to hematopoietic challenges, e.g., life-threatening blood loss, infection, and inflammation, HSCs can be activated to proliferate and engage in blood formation. The HSC activation induced by hematopoietic demand is mediated by direct or indirect sensing mechanisms involving pattern recognition receptors or cytokine/chemokine receptors. In contrast to the hematopoietic challenges with obvious clinical symptoms, how the aging process, which involves low-grade chronic inflammation, impacts hematopoiesis remains undefined. Herein, we summarize recent findings pertaining to functional alternations of hematopoiesis, HSCs, and the bone marrow (BM) microenvironment during the processes of aging and inflammation and highlight some common cellular and molecular changes during the processes that influence hematopoiesis and its cells of origin, HSCs and HPCs, as well as the BM microenvironment. We also discuss how age-dependent alterations of the immune system lead to subclinical inflammatory states and how inflammatory signaling might be involved in hematopoietic aging. Our aim is to present evidence supporting the concept of “Inflamm-Aging,” or inflammation-associated aging of hematopoiesis.
Introduction
Hematopoiesis
Hematopoiesis is an active, continuous process involving the production and consumption of mature blood cells that constitute the hemato-lymphoid system. It has been estimated that in a healthy individual (70 kg), approximately 5 × 1011 mature blood cells are produced daily throughout that individual’s lifetime (1). All blood cells arise from a small population of hematopoietic stem cells (HSCs) in the bone marrow (BM) that have two unique properties: self-renewing capacity, the ability to generate themselves, and multi-lineage differentiation capacity, the ability to produce all blood cell types, including red blood cells, platelets, myeloid lineage cells, such as monocytes and granulocytes, as well as lymphoid lineage cells, such as natural killer (NK) cells, B cells, and T cells. Since, in the steady state, most adult HSCs are in the G0 phase of cell cycle, i.e., they are quiescent and are estimated to turnover slowly on a monthly time scale (2–5), daily hematopoietic production is mainly sustained by highly proliferative downstream hematopoietic progenitor cells (HPCs) (6–8). Cellular behavior of HSCs, i.e., self-renewal, differentiation, and apoptosis, is tightly controlled by both cell-intrinsic factors, e.g., transcriptional regulatory networks and cellular metabolism, and cell-extrinsic factors, e.g., cytokines, chemokines, growth factors, metabolites, and exogenous pathogen-derived molecules (9–11). When mature hematopoietic cells are consumed and need to be replenished in response to hematopoietic challenges, HSCs can translate locally produced and/or systematically migrating external signals into hematopoiesis by increasing their own proliferation and differentiation (12, 13).
Hematopoietic Stem Cell Heterogeneity
Recent studies with single-cell transplantation and lineage tracing have revealed cellular heterogeneity within a functionally defined HSC population and have identified HSC subtypes with distinct lineage differentiation potentials (Figure 1A) (14–18). Myeloid-biased HSCs give rise to more myeloid lineage than lymphoid lineage cells, while lymphoid-biased HSCs favor lymphopoiesis over myelopoiesis, and balanced HSCs produce myeloid and lymphoid lineage cells equally (14, 16). It has also been indicated that there is a certain functional hierarchy within these lineage-biased HSCs (16, 19). Myeloid-biased HSCs defined as CD150highCD34− LKS have greater self-renewal potential than lymphoid-biased or balanced HSCs (CD150low/negativeCD34− LKS) and are able to replenish all types of HSC populations, indicating that myeloid-biased HSCs are higher in the HSC hierarchy than other HSCs. In addition, it was recently found that a subset of HSCs expressing von Willebrand factor efficiently gives rise to platelets and erythroid lineage cells earlier than other lineages, and this subset was therefore named platelet-biased HSCs. As the platelet-biased HSCs can generate myeloid-biased HSCs, they are suggested to be positioned at the apex of the hematopoietic hierarchy (17, 20).
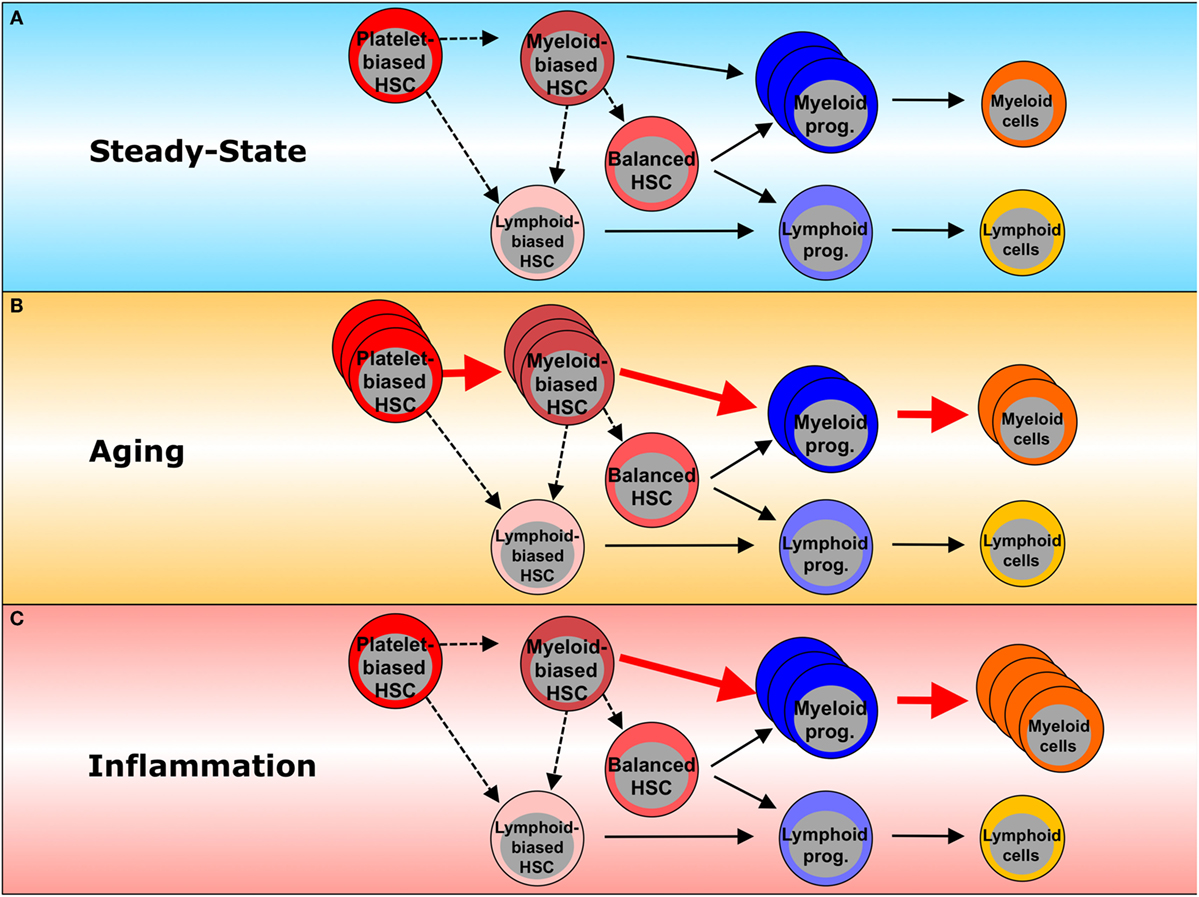
Figure 1. Inflammation- and aging-associated changes in hematopoiesis. (A) In steady state, platelet-biased HSCs are at the top of the hematopoietic hierarchy and are able to generate myeloid-biased and lymphoid-biased HSCs. In turn, myeloid-biased HSCs can generate both balanced- and lymphoid-biased HSCs, whereas lymphoid-biased HSCs do not generate their myeloid-biased counterparts. Platelet-biased HSCs have the potential to repopulate platelet populations faster than other HSC subsets. Myeloid-biased HSCs preferentially give rise to myeloid lineage cells through myeloid committed progenitors. Balanced HSCs make equal contributions to both myeloid and lymphoid lineages. Lymphoid-biased HSCs predominantly generate lymphoid over myeloid lineage cells through lymphoid-committed progenitors. Dashed lines represent the potential of one HSC subset to generate another HSC subset. Solid lines represent differentiation potential. (B) Inflammation enhances myeloid lineage production, including myeloid progenitors and mature myeloid cells, leading to myeloid bias in hematopoiesis. (C) During the processes of aging, myeloid-biased HSCs increase and produce more myeloid than lymphoid cells. Red arrows indicate the dominant differentiation pathway. Dashed lines represent a potential pathway. Solid lines represent the differentiation potential shown previously. The thickness of the lines reflects the relative contributions to each lineage commitment.
Cellular heterogeneity and lineage priming have been observed at the level of not only HSCs but also HPCs (6, 21–23). A subset of multipotent progenitors (MPPs), MPP2 (LKS Flt3−CD150+CD48+), preferentially gives rise to platelets, erythrocytes, and granulocyte-monocyte progenitors (GMPs), while MPP3 (LKS Flt3−CD150−CD48+) is primed to GMP, and to a lesser extent, the erythroid lineage with no potential for platelet generation. MPP4 (LKS Flt3+CD150−CD48+) is biased toward lymphoid lineage output. These MPP subsets are independent of one another and, as each is produced directly by HSCs, they lack the capacity to give rise to one another. However, it remains to be determined whether HSC subsets with distinct lineage outputs have any clonal relationship with MPP subsets.
Hematopoietic Aging
Hematopoietic Changes during Aging
Aging of the hematopoietic system is represented by functional declines in both the adaptive and the innate immune system, an immunosenescence that leads to high susceptibility to infections, low efficacy of vaccinations, and increased vulnerability to the development of autoimmunity and hematologic malignancies (24, 25). As shown in Figure 2, (a) B cell production decreases significantly with advancing age, i.e., the naïve B cell pool diminishes, while the memory B cell pool expands. Diversity of the B cell repertoire also decreases in association with lowered antibody affinity and impaired class switching. B cells are prone to produce auto-antibodies increasing the incidence of spontaneous autoimmunity (26, 27); (b) de novo T cell production also declines with aging partially due to thymic involution. CD8+ T cells undergo oligoclonal expansion and their repertoire is skewed toward previously encountered antigens, as niches for naïve T cells in peripheral lymphoid tissues become occupied by terminally differentiated cells (28); (c) NK cells show diminished cytotoxicity and cytokine secretion; (d) although myeloid cells increase in number, their functionality is decreased, e.g., neutrophils migrate less in response to stimuli, and macrophages have reduced phagocytic activity and decreased oxidative burst (29–31); and (e) erythropoiesis also declines in elderly people causing frequent anemia (32), while the thrombocytic lineage has not, to date, been reported to be significantly affected by aging.
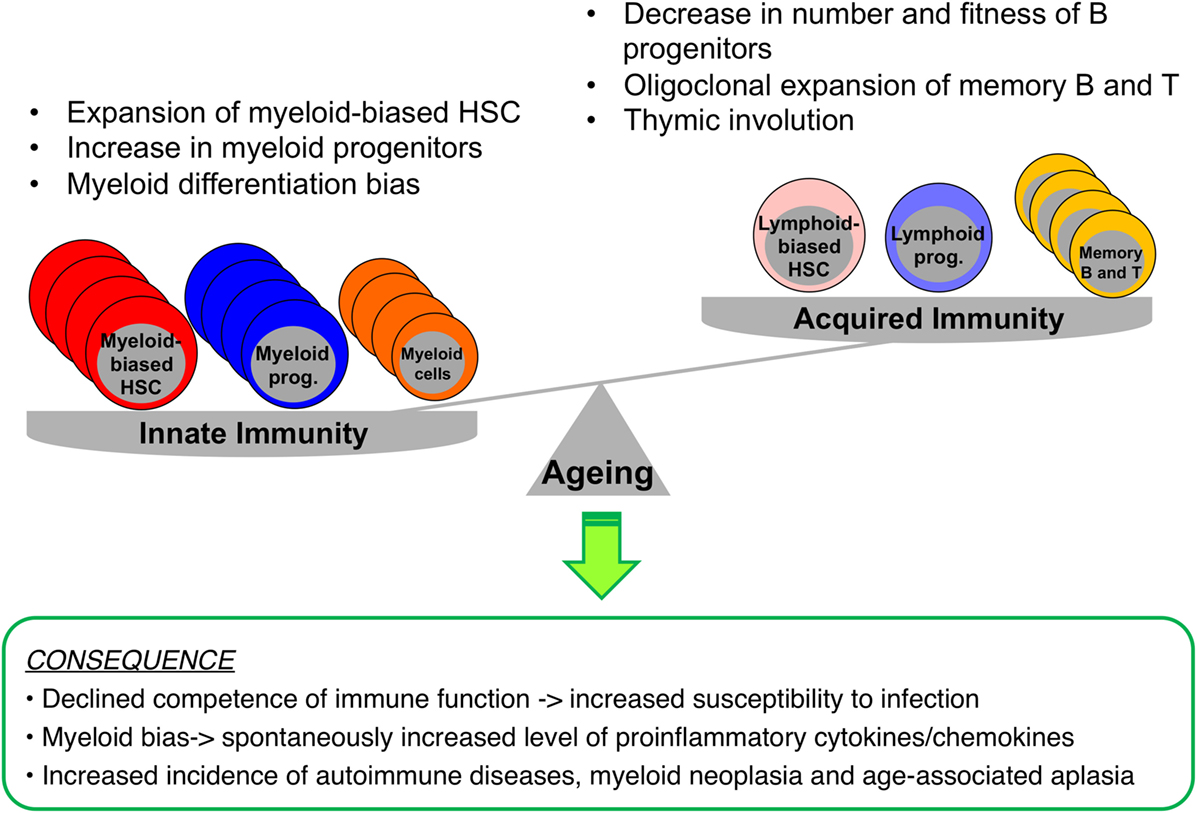
Figure 2. Impacts of aging on immunity and hematopoiesis. During aging, myelopoiesis results in the domination of hematopoiesis over lymphopoiesis due to increased numbers of myeloid-biased HSCs, myeloid progenitors, and myeloid cells, while the pool consisting of B and T cells shrinks. These hematopoietic changes result in increased dependence of the immune system on innate rather than acquired immunity, with an enhanced basal level of inflammation, increasing the risk of myeloid neoplasia or spontaneous anemia.
HSC Functional Alteration during Aging
Since multiple blood lineages change during the aging process, it is possible that hematopoietic aging is in part due to functional changes in early hematopoietic compartments that repopulate the affected lineages, including HSCs. Single-cell and limiting dilution transplantations have demonstrated the self-renewal capacity of HSCs to apparently be reduced on a per-cell basis during aging, as the frequency of phenotypically defined HSCs does not correlate with that of functionally defined HSCs in aged BM (19, 33–38). It was also shown that phenotypic HSCs (LKS CD34−Flt3−) upregulate CD150 expression (19, 37, 39), resulting in expansion of the myeloid-biased HSC population and the domination of this fraction over the entire aged HSC pool (39), with advancing age (Figure 1B). Consistent with the phenotypic characterization, hematopoietic repopulation after transplantation is biased toward myeloid cell production, and this change in differentiation potential persists over the course of serial transplantations, indicative of aging-associated cell-autonomous alterations in HSCs. Based on these observations, two possible theories for age-associated myeloid bias can be proposed: (a) clonal evolution within the aged HSC population, in which lymphoid-biased HSC clones turn into myeloid-biased or platelet-biased HSC clones via cell-intrinsic changes (40); (b) clonal composition shift, in which subsets of myeloid-biased or platelet-biased HSC clones dominate the entire HSC pool via clonal expansion and/or selection (16, 19, 34, 39, 41–43). Aging-associated myeloid lineage skewing may also involve disturbance in the composition of committed progenitors: aged mice show a decreased frequency of common lymphoid progenitors, while frequencies of GMPs are increased (37). These findings are accompanied by decreased B cell lymphopoiesis and diminished fitness of lymphoid progenitors, coinciding with altered receptor-associated kinase signaling (44). Moreover, the recent identification of myeloid-restricted progenitors with long-term repopulating capacity/self-renewal has raised new questions regarding the definition of HSCs (18, 45). Therefore, which level of the hematopoietic hierarchy is affected by aging remains uncertain.
The BM homing efficiency of aged HSCs is significantly reduced when transplanted intravenously into irradiated recipients (14), although similar mobilizing efficacies are observed in aged and young HSCs released into the circulation in response to granulocyte colony-stimulating factor (G-CSF) treatment (46). Transcriptome profiling of aged versus young HSCs has provided molecular insights into potential mechanisms of HSC aging (33, 47): aged HSCs show dysregulation of intracellular homeostasis, e.g., upregulated stress responses, increased pro-inflammatory signaling, protein misfolding, downregulated DNA repair machinery, and aberrant chromatin modification (19, 33, 37). Further investigations have demonstrated that aged HSCs accumulate more DNA damage possibly due to higher levels of intracellular reactive oxygen species (ROS) and naturally produced genotoxic metabolites (48–50), but interestingly, these cells are still able to efficiently repair the damage upon cell cycle induction (51). Other studies have indicated that accumulation of proliferative stress in aged HSCs causes inefficient DNA replication and transcriptional repression (52). Aged HSCs also exhibit activation of the mammalian target of rapamycin (mTOR) (53), autophagy-dependent survival (54), dysregulated DNA methylation, specifically at the site of genes controlling myeloid and lymphoid balancing (51), impaired histone modification (55), and disturbed cell polarity (56).
These characteristics of HSC aging can, in part, be experimentally recapitulated by increasing the proliferative history of HSCs or stressing them with multiple injections of myeloablative chemotherapeutic regimens (57), or by conducting serial transplantations (“experimental aging”) (34, 35). As this indicates that proliferative history might be associated with the aging process, several groups have compared the cycling activity of young versus aged HSCs. The results are, however, controversial: some data indicate that aged HSCs have increased cycling activity (36), whereas others suggest no difference in cell cycle status (33, 38), or more quiescent HSCs in aged as compared to young BM (4, 53). These seemingly discrepant results might be partially explained by differing immunophenotypic definitions of HSCs and/or different experimental approaches to measure cell cycle status [reviewed in Ref. (3)].
Aging-Associated Changes in BM Niche
Hematopoietic stem cell homeostasis is preserved in the BM microenvironment, the so-called HSC niche that supplies these cells with pivotal factors for their own maintenance (3, 58). Recent research on the BM niche has revealed a perivascular HSC niche comprised mesenchymal stromal cells (MSCs) and endothelial cells (ECs) as major cellular components, reflecting hierarchic HSC function and the effects exerted by aging (58, 59). MSCs are characterized by plastic adherence, high growth potential, and mesenchymal immunophenotypes, as well as differentiation into mesenchymal lineages, such as osteocytes, adipocytes, chondrocytes, fibroblasts, and epithelial cells (60, 61). Aged MSCs exhibit reduced clonogenic and proliferative capacity, and differentiation potentials are skewed toward adipogenesis at the expense of osteogenesis (62–64). These cells also show enlargement, telomere shortening, or p53/p21-mediated DNA damage accumulation, impaired DNA methylation or histone acetylation, and increased levels of ROS and nitric oxide (NO) (65–69). Although age-dependent mechanisms underlying adipogenesis-favoring MSC differentiation are not as yet fully understood, possible molecular changes have been reported, including activations of peroxisome proliferator-activated receptor gamma 2 and CCAAT/enhancer binding protein (70, 71). Adipogenesis enhancement in aged BM (72) might be linked to dysregulation of insulin growth factor signaling (27), changes in extracellular matrix composition, and decreased bone formation (73, 74). Since adipocytes are shown to negatively regulate HSC function and B-lymphopoiesis (75, 76), adipogenesis enhanced in aged BM might promote myelopoiesis over lymphopoiesis as well as impair HSC function. In fact, young HSCs in the aged environment reportedly tend to produce slightly more myeloid cells than in a young environment (37, 77).
Endothelial cells are another niche cell component that secrete HSC maintenance and retention factors, such as stem cell factor and CXC motif ligand (CXCL) 12 (58, 78). Aging involves decreases in CD31hiEmcnhi EC-associated osteoprogenitors (79), fewer PDGFRβ+/NG2+ perivascular cells, arterioles, and ECs, thereby resulting in reduced stem cell factor production (59). Activation of endothelial Notch signaling can reverse these age-dependent vascular niche alterations, without affecting aged HSC function. Additionally, vascular endothelial function declines with aging, due to reduced NO which in turn induces vasodilation, elevated oxidative stress causing genomic instability, and increased ROS levels associated with impaired proangiogenic functions of EC (80). As it has been suggested that NO production regulates CXCL12-mediated HSC mobilization (81), aging-related reductions of EC-derived NO and the enhancement of angiogenic function in the BM niche might be involved in aberrant HSC maintenance and/or retention in aged BM.
Hematopoietic Aging in Humans
While most of the data on aging of the hematopoietic system was obtained employing a mouse system, a few pioneering studies have indicated similar tendencies in the human hematopoietic system. HSCs containing fractions such as Lin−CD34+CD38− (29), Lin−CD34+CD38−CD90+CD45RA− (82), or Lin−CD34+CD10−CD123−CD45RA−CD90+ (83) increase with age. While GMPs appear to be retained at the same frequency (29, 82, 83), early B cell progenitors and CLPs decrease with advancing age (82, 83). Yet, the functionality and differentiation bias of HSCs remain unclear: one study, using xenograft mouse models, indicated no change in NSG-repopulating cell frequency and decreased myeloid lineage repopulation of aged HSCs (29), while another (82) showed a two-fold decreased engraftment with significant myeloid lineage dominance. Further molecular analyses indicated upregulations of myeloid and megakaryocyte-associated genes and downregulations of lymphoid differentiation genes (82, 83). These findings indicate major aging-associated changes in hematopoiesis to be conserved among species.
Inflammation in Hematopoiesis
Hematopoietic Responses to Inflammation
Inflammation is defined as a protective immune response, underlain by a variety of pathophysiological processes that are in part caused by infection and tissue injury/damage (84). There are many different types of endogenous and exogenous factors that can potentially induce local or systemic inflammation: mechanical stimuli (e.g., tissue damage, foreign objects), thermal stimuli (e.g., heat, cold), radiation (e.g., ultraviolet, ionizing, chemotherapeutic), chemical irritants (e.g., acids, alkalis, toxins), physical or psychological stress, autoimmunity (e.g., allergens, autoantigens), and pathogens (e.g., bacteria, viruses, fungi, protozoal parasites).
Inflammatory responses involving the hemato-immune system are exemplified by infection. The first line of defense against infection is often initiated by innate immunity: bacterial, viral, or fungal pathogens that break through the epithelial barrier will be recognized by pattern recognition receptors (PRRs) expressed on hematopoietic and non-hematopoietic cells, such as dendritic cells (DCs), macrophages, monocytes, and ECs (85). Upon ligation of PRR by pathogen-derived molecules, the innate immune cells secrete an array of inflammatory cytokines and chemokines, e.g., interleukin (IL)-1β, IL-6, CXCL-8, IL-12, and tumor necrosis factor (TNF)-α, as well as chemical mediators, e.g., leukotriene B4, prostaglandin E2 (PGE2), and histamine [reviewed in Ref. (86, 87)]. These biologically active factors attract immune effector cells from the circulation to the site of infection, and simultaneously increase vascular permeability to allow more immune effector cells to infiltrate and differentiate in the inflamed tissues. This process causes heat (calor), pain (dolor), redness (rubor), and swelling, which are referred to as hallmarks of inflammation (87).
Toll-like receptors (TLRs) belong to the PRR family and recognize microbial products derived from exogenous pathogen molecules, such as lipopolysaccharide (LPS) for TLR-4, lipopeptides for TLR-1, -2, and -6, bacterial and viral RNA and DNA for TLR-3, -7, -8, and -9, as well as possibly endogenous host molecules that are misfolded or modified, including heat shock proteins and fibronectin. Binding of the respective ligands to TLRs leads to cell proliferation, differentiation, and migration (88). For example, activation of TLR signaling on DCs is a key step necessary for their full maturation into antigen-presenting cells. When TLRs on DCs are ligated by the pathogen molecules encountered, DCs enhance the subcellular machineries that process and present pathogen-derived antigens on their cell surfaces, as well as triggering the production of pro-inflammatory cytokines/chemokines, e.g., macrophage inflammatory protein (MIP)-1α, MIP-1β, and the regulated on activation, normal T cell expressed and secreted (RANTES) chemokine. They also up/downregulate chemokine receptors, e.g., CCR1, CCR5, and CCR7, thereby directing their migration to secondary lymphoid organs, such as lymph nodes or the spleen, where they initiate a second-line defensive response by activating adaptive immunity [reviewed in Ref. (89)]. Naïve T cells are activated to proliferate and differentiate into functionally mature effector cells upon activation of two distinct signals through antigen-presenting cells expressing the major histocompatibility complex and co-stimulatory molecules. These effector T cells in turn migrate to the site of infection, and either activate other immune effector cells (macrophages, NK cells, neutrophils, eosinophils, mast cells, basophils) through cytokine secretion or kill the infected cells through release of cytolytic molecules. They also produce IL-4, IL-5, interferon (IFN)-γ, and tumor growth factor (TGF)-β to activate monoclonal B cells that recognize the specific antigen. This activation process facilitates clonal expansion and differentiation into plasma and memory B cells that secrete high-affinity antibodies, and therefore enhances subsequent humoral immune responses, e.g., neutralization, phagocytosis, and opsonization.
HSC Response to Infection
Since immune effector cells involved in both innate and adaptive immunity are short-lived except for the memory B and T cells maintained for life, they need to be replenished by the upstream HSCs and progenitor cells (HSPCs) in BM when consumed during inflammation (12). Given that HSCs tend to remain in a largely quiescent state, as mentioned above (5), understanding how these quiescent HSCs respond to inflammation and are activated to maintain hematopoietic homeostasis is becoming a major research focus. Recent findings have indicated that not only mature immune cells but also HSPCs are capable of responding to infection by directly sensing pathogen-associated molecule patterns through their respective PRR (4, 13, 90–92) (Figure 3). The activation of PRR in HSPCs leads to enhanced proliferation, increased mobilization from the BM, reduced self-renewal, and myelopoiesis-favoring differentiation (Figure 1C). Since HSPCs also express a broad spectrum of inflammatory cytokine/chemokine receptors (12, 93), they can detect milieu broad range of pro-inflammatory signals via their respective receptors, released systemically or locally by activated immune cells in response to infectious challenges, e.g., IFN-α/γ (94–98) (Figure 3). These two pathways are not mutually exclusive: G-CSF stimulation impairs HSC repopulating potential through upregulation of TLR-2 and -4 expressions and activation of the subsequent signaling in HSCs (99). When bound to TLR-2 and -4, HSPCs have been shown to have the capacity to secrete pro-inflammatory cytokines, e.g., IL-6, TGF-β, TNF-α, and granulocyte-macrophage colony-stimulating factor (GM-CSF), all of which activate their respective receptor signaling mechanisms and promote myelopoiesis in a paracrine or an autocrine fashion (13). The HSPC responses triggered by TLR activation appear to integrate the infectious signals into the process of hematopoiesis by recruiting themselves to the inflamed tissues, directing their differentiation, and reflecting the high demand toward myelopoiesis. A recent study identified stem cell-like megakaryocyte-committed progenitors as a novel inflammation responsive cell population remaining dormant and thereby show little contribution to megakaryopoiesis in the steady state, but in response to acute inflammation, these cells become metabolically active and rapidly produce platelets to replace those lost during inflammatory processes (45).
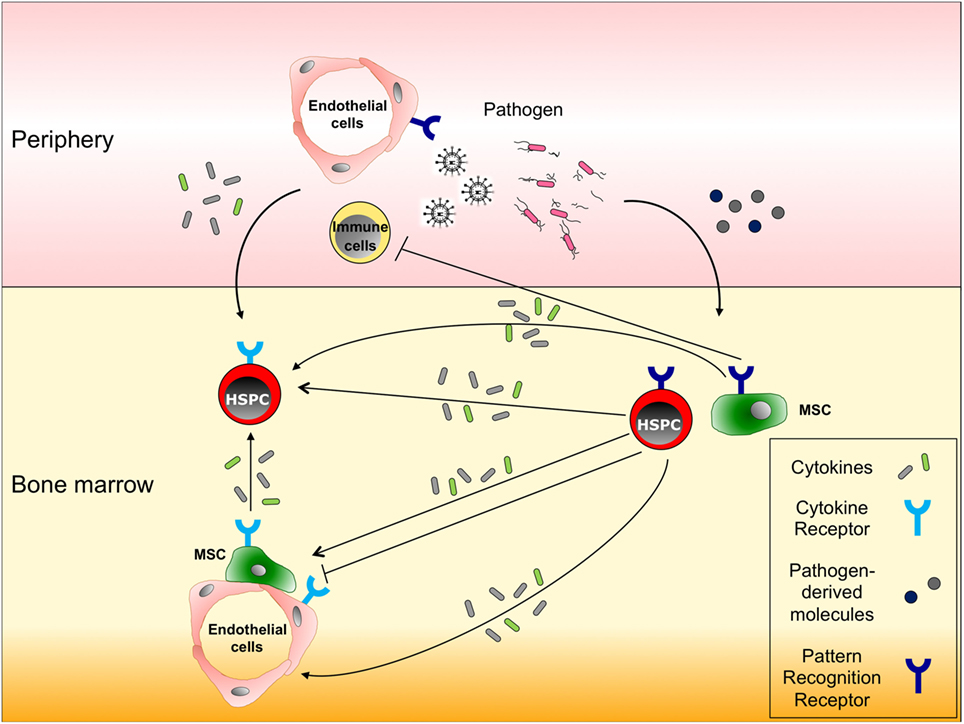
Figure 3. Responses of hematopoietic stem and progenitor cells (HSPCs), and non-hematopoietic bone marrow cells to infection. HSPCs and non-hematopoietic cells in bone marrow (BM), such as mesenchymal stromal cells (MSCs) and endothelial cells, express both cytokine and pattern recognition receptors (PRRs) on their surfaces. In response to infections in peripheral tissues, (a) immune cells or endothelial cells secrete pro-inflammatory cytokines that migrate to the BM and stimulate the respective receptors expressed on HSPCs, thereby inducing their proliferation, migration and/or differentiation; (b) alternatively, migrating cytokines also can act on MSCs or endothelial cells to enhance their pro-inflammatory cytokine production; and (c) some pathogen-derived molecules reach the BM and activate HSPCs directly through PRRs or indirectly through pro-inflammatory cytokines produced by PRR expressing MSCs or HSPCs.
BM Niche Response to Infection
MSCs can influence both innate and acquired immunity through cell–cell contact or secretion of soluble factors, e.g., TNF, IL-10, IL-6, and PGE2 (100), which inhibit T cell function, DC maturation, and the activation and proliferation of both B and NK cells. MSCs have also been shown to express several TLRs, the activation of which controls inflammatory cytokine production critical for their immunosuppressive function (101, 102). TLR activation in MSCs reportedly not only influences their functions, e.g., differentiation, proliferation, migration, immunomodulation, and bone regeneration (103–105), but also regulates HSPC proliferation and differentiation toward myeloid development, as well as monocyte egress (106, 107) (Figure 3). Moreover, it has been shown that pro-inflammatory cytokines control the productions of other cytokines/chemokines by MSCs. IFN-γ alone or in combination with TNF or IL-1 induces the production of nitric oxide synthase or PGE2 in MSCs, thereby inhibiting T or NK cell activation, respectively (108). The secretion of IFN-γ by cytotoxic CD8+ T cells also leads indirectly to an activation of HPCs by promoting the IL-6 production in MSCs (109). During early hematopoietic regulation, G-CSF suppresses the production of CXCL12 from BM MSCs and mobilizes HSC into circulating blood (110).
ECs also express multiple PRRs, and when activated, regulate various immune responses acting against infection [reviewed in Ref. (111)] (Figure 3). In response to TLR4 activation, ECs produce G-CSF that contributes to rapid neutrophil production in the BM (112, 113), and at the same time, induces neutrophil recruitment to infectious sites (114, 115). Pro-inflammatory cytokine signaling influences EC function through nuclear factor κB (NFκB) activity. It has also been shown that stimulation of ECs in the BM with TNF-α and LPS expands the HPC population through modulation of Notch signals (66). When stimulated with IL-1β and TNF-α, ECs in the BM are induced to produce GM-CSF, which leads to the recruitment of neutrophils and expansion of HPCs in the BM (116, 117). Interestingly, it was also shown that through MSC-EC interaction, MSCs upregulate IL-6 production modulating the responses of ECs to inflammatory cytokines (118). The results obtained in these studies suggest that ECs in the BM play important roles in hematopoietic regulation during inflammation.
Taken together, these findings highlight previously unappreciated HSPC responses to infection via TLR-mediated direct or cytokine-mediated indirect sensing mechanisms. Inflammatory responses play beneficial roles in the activation and replenishment of hemato-immune system components and thereby contribute to controlling infection. However, if the inflammatory response is not discontinued in a timely manner, instead being unnecessarily sustained even after infection has resolved, it might ultimately have detrimental effects, e.g., tissue damage, chronic diseases, and even cancer. In fact, uncontrolled persistent inflammatory signaling is known to promote the development of chronic diseases, e.g., rheumatoid arthritis, inflammatory bowel disease, asthma, and aplasia. Recent studies have also demonstrated that sustained IFN-α/γ activation during chronic infection impairs HSC function and can ultimately lead to BM failure (94, 119). Chronic TNF-α signaling is associated with myelodysplastic syndrome and BM failure (120). Therefore, inflammatory responses must stop when no longer needed. The appropriate timing of this cessation is critical.
Inflammation of the Hematopoietic System in Humans
Not only murine but also human HSCs have been shown to express TLRs (121, 122). The stimulation of agonists for TLR2, TLR7, and TLR8 in vitro induces cytokine production, e.g., IL-1b, IL-6, IL-8, TNF-α, and GM-CSF, as well as cell differentiation of the myeloid lineage (122, 123). In vitro TLR9 binding by CpG DNA reportedly upregulates IL-8 expression via activation of ERK1/2 and p38, both mitogen-activated protein kinases, but not of NFκB (121). Nevertheless, in contrast to murine HSCs, quiescent human HSCs appear to be fully resistant to infection with both intracellular bacteria, such as Listeria monocytogenes and Salmonella enterica, and extracellular bacteria, including Yersinia enterocolitica (124). In contrast, when human CD34+ HSPCs were cultured with Escherichia coli, they showed upregulated production of pro-inflammatory cytokines such as IL-1, IL-6, IL-8, and TNF through NFκB activation, as do human CD34+-derived granulocyte–macrophage lineage cells (125). Regarding the HSPC response to inflammatory cytokines, the exposure of human CD34+ HSPCs to IFN-γ produces drastic transcriptional changes in genes involved in pro-apoptotic processes, immune responses, and myeloid proliferation that results in an increased number of viable cells (126, 127). While some transcriptional changes are specific to HSPCs, others, e.g., cell growth and signal transduction, generally occur in stromal cells incubated with IFN-γ (127). In contrast, in vitro stimulation with IFN-γ and TNF severely compromised the ability of HSPCs to undergo multi-lineage reconstitution in xenografted mice (128, 129). Future studies are required to unravel the details of the different HSPC responses to inflammatory cues and the underlying molecular mechanisms.
Aging-Associated Inflammation: “Inflamm-Aging”
There are similarities between hematopoietic alterations during inflammation and those that occur with aging. In response to aging and bacterial infection, myelopoiesis becomes dominant over lymphopoiesis in relation to immunosenescence (36, 38, 130, 131). Most notably, B-lymphopoiesis is impaired due to a decreased level of E47, a transcription factor essential for B cell development, in aged (132) and LPS-treated mice (130). The aging-associated myeloid dominance and/or adipogenesis in BM might be triggered by increased basal levels of pro-inflammatory cytokines even in the absence of infection. Indeed, levels of circulating pro-inflammatory cytokines, such as IL-6, TNF-α, IL-1Rα, and C-reactive protein, are reportedly upregulated in healthy elderly populations (25, 133–138). These observations allow us to hypothesize that “Inflamm-Aging” represents a subclinical grade of chronic inflammation possibly contributing to the initiation and/or acceleration of hematopoietic aging.
Aging-associated HSC alterations, including reduced self-renewal and myelopoiesis-favored differentiation, are very similar to the functional changes occurring in HSCs exposed to chronic inflammatory stimuli: the HSC pool is shifted to an increased proportion of CD150high HSCs that predominantly produce myeloid lineage cells, as mentioned earlier (19, 39). This was also the case when mice were given a daily low dose of LPS for 1 month, suggesting exogenous stimulation to be involved in HSC aging (130). Recent studies have indicated that accumulation of myeloid-skewed HSCs with aging is mediated by activated signaling of TLR4 (130), P-selectin (33), NFκB (33), the RANTES-mTOR pathway (53, 77), and TGF-β (19). Since numerous pro-inflammatory cytokines are known to be produced by myeloid cell lineages or adipocytes, there might be a positive feedback mechanism by which, when aging processes begin, HSCs with myeloid-skewed differentiation gradually accumulate in BM via proliferative signals. As a consequence, these cells give rise to more myeloid cells that are prone to spontaneously produce pro-inflammatory cytokines, thereby further advancing myeloid dominance (Figure 2). In parallel, adipogenic MSC differentiation during aging also contributes to the promotion of inflammatory cytokine production and enhanced myelopoiesis. Moreover, inflammatory conditions foster the production and release of ROS in hematopoietic cells, a genotoxic reagent known to damage DNA, and might cause genetic ablations in adjacent cells, such as MSCs and ECs in BM (48). Intracellular ROS production has indeed been shown to be induced and to cause DNA damage accumulation in HSCs during viral infections and aging (49, 119), although another study detected no DNA damage in aged HSCs (52).
Discussion
Since numerous inflammatory factors are increased in aged hematopoietic tissues, and inflammation- and aging-associated hematopoietic changes share common cellular and molecular alterations, it is reasonable to speculate that low-grade inflammation might be involved in hematopoietic aging with reduced fitness of both adaptive and innate immune cells. Given that some hematopoietic phenotypes during inflammation and aging arise from functional alterations in HSPCs, as discussed above (19, 34, 39), it would be worthwhile to elucidate the underlying common mechanisms. Future research could yield meaningful insights into cell-intrinsic changes in HSPC quantity and quality, e.g., how aspects of HSPC population dynamics such as functional heterogeneity and population size change, whether all subsets of HSCs with a distinct lineage output respond equally to inflammatory stimuli or only the minor fraction is responsive, how the self-renewal and differentiation capacities of HSC are altered on a per-cell basis, and molecular changes in cellular signaling, such as alterations in cellular metabolism, transcriptional networks, epigenetic modifications, and genomic instability (Figure 4). It is also essential to understand to what extent inflamm-aging-associated cell-extrinsic factors influence HSPC biology, including signals derived from the BM niche, tissue damage/repair, infection, obesity, or the microbiome. In addition, the fundamental task that remains is identification of the factor(s) initially triggering the process of hematopoietic inflamm-aging. Inflammation- or aging-related external stimuli appear to force quiescent HSCs to proliferate and impair their self-renewal and differentiation capacities, as suggested by evidence that HSC cycling in response to chemotherapy administration or hematopoietic stress accelerates the manifestation of aging phenotypes (119). These data suggest that the central features of HSCs aging might be attributable to accumulation of a proliferative history that is closely associated with perturbed self-renewal and differentiation.
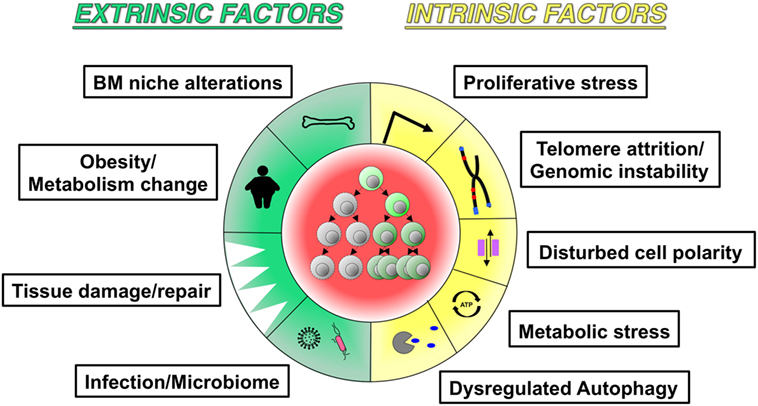
Figure 4. Potential hallmarks of inflamm-aging. The schematic figure highlights similarities between aging and inflammation-associated changes in hematopoiesis and hematopoietic stem cells: (a) extrinsic factors include bone marrow niche alterations, metabolic changes, tissue damage/repair, and infection/microbiome; (b) intrinsic factors include proliferative stress, telomere attrition/genomic instability, dysregulated autophagy, metabolic stress, and dysregulated autophagy.
The incidence of myeloid leukemia increases with aging (139). It was recently demonstrated that age-related clonal hematopoiesis with somatic mutations is a risk factor for hematopoietic malignancies (140–144). Inflammation has long been regarded as a driving force for cancer development in many tissues (145). Indeed, high resolutional sequencing of human hematologic malignancies have revealed somatic mutations in different inflammatory signaling genes, such as NFκB, myeloid differentiation primary response gene 88, TLR4, and TNF receptor-associated factors, all of which result in hyperproliferation or prolonged survival of tumor cells (146). These findings imply a critical role of inflammatory signaling as a driving force for leukemogenesis. It would be interesting to investigate how chronically sustained inflammatory stimuli are involved in the acquisition of genetic ablations in pre-leukemic clones and the roles of clonal evolution in the development of hematologic malignancies. It may also merit mention that no mutations in inflammatory signaling cascades have been found in myeloid malignancies, raising the possibility of a protective mechanism functioning to prevent malignant myeloid transformation of HSCs.
Inflammation and aging have thus far been seen as two independent pathophysiological processes. However, a growing body of evidence has highlighted biological changes in hematopoiesis and HSCs that are common to both inflammation and aging. Thus, it is likely that sustained inflammatory stimuli contribute to hematopoietic aging and possibly leukemogenesis, supporting the inflamm-aging concept. Since inflammation and aging might both be involved in increased risk for leukemogenesis, eliminating unwanted inflamm-aging factors is a potential approach to preserving both HSC and immune functions, and thereby preventing a functional decline in hematopoiesis and the emergence of malignant clones. Future investigation is required to better characterize hematopoietic inflamm-aging processes at the tissue, cellular, and molecular levels.
Author Contributions
LVK, KF, and XF wrote the manuscript and prepared the figures. MGM and HT wrote and critically revised the manuscript. All the authors approved the final version of this manuscript for submission.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer DL and handling editor declared their shared affiliation, and the handling editor states that the process nevertheless met the standards of a fair and objective review.
Funding
This work was supported by the Swiss National Science Foundation (310030_146528/1), the Promedica Foundation (Chur, Switzerland), and the Clinical Research Priority Program of the University of Zurich to MGM, as well as by the JSPS KAKENHI (15H01519), the Takeda Science Foundation, and the KANAE Foundation for the Promotion of Medical Science to HT.
References
1. Fliedner TM, Graessle D, Paulsen C, Reimers K. Structure and function of bone marrow hemopoiesis: mechanisms of response to ionizing radiation exposure. Cancer Biother Radiopharm (2002) 17:405–26. doi:10.1089/108497802760363204
2. Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell (2005) 121:1109–21. doi:10.1016/j.cell.2005.05.026
3. Nakamura-Ishizu A, Takizawa H, Suda T. The analysis, roles and regulation of quiescence in hematopoietic stem cells. Development (2014) 141:4656–66. doi:10.1242/dev.106575
4. Takizawa H, Regoes RR, Boddupalli CS, Bonhoeffer S, Manz MG. Dynamic variation in cycling of hematopoietic stem cells in steady state and inflammation. J Exp Med (2011) 208:273–84. doi:10.1084/jem.20101643
5. Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell (2008) 135:1118–29. doi:10.1016/j.cell.2008.10.048
6. Busch K, Klapproth K, Barile M, Flossdorf M, Holland-Letz T, Schlenner SM, et al. Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature (2015) 518:542–6. doi:10.1038/nature14242
7. Passegué E, Wagers AJ, Giuriato S, Anderson WC, Weissman IL. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J Exp Med (2005) 202:1599–611. doi:10.1084/jem.20050967
8. Sun J, Ramos A, Chapman B, Johnnidis JB, Le L, Ho Y-J, et al. Clonal dynamics of native haematopoiesis. Nature (2014) 514:322–7. doi:10.1038/nature13824
9. Chow A, Lucas D, Hidalgo A, Méndez-Ferrer S, Hashimoto D, Scheiermann C, et al. Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J Exp Med (2011) 208:261–71. doi:10.1084/jem.20101688
10. Kaushansky K. Lineage-specific hematopoietic growth factors. N Engl J Med (2006) 354:2034–45. doi:10.1056/NEJMra052706
11. Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell (2008) 132:631–44. doi:10.1016/j.cell.2008.01.025
12. Takizawa H, Boettcher S, Manz MG. Demand-adapted regulation of early hematopoiesis in infection and inflammation. Blood (2012) 119:2991–3002. doi:10.1182/blood-2011-12-380113
13. Zhao JL, Ma C, O’Connell RM, Mehta A, DiLoreto R, Heath JR, et al. Conversion of danger signals into cytokine signals by hematopoietic stem and progenitor cells for regulation of stress-induced hematopoiesis. Cell Stem Cell (2014) 14:445–59. doi:10.1016/j.stem.2014.01.007
14. Dykstra B, Kent D, Bowie M, McCaffrey L, Hamilton M, Lyons K, et al. Long-term propagation of distinct hematopoietic differentiation programs in vivo. Cell Stem Cell (2007) 1:218–29. doi:10.1016/j.stem.2007.05.015
15. Morikawa S, Mabuchi Y, Kubota Y, Nagai Y, Niibe K, Hiratsu E, et al. Prospective identification, isolation, and systemic transplantation of multipotent mesenchymal stem cells in murine bone marrow. J Exp Med (2009) 206:2483–96. doi:10.1084/jem.20091046
16. Morita Y, Ema H, Nakauchi H. Heterogeneity and hierarchy within the most primitive hematopoietic stem cell compartment. J Exp Med (2010) 207:1173–82. doi:10.1084/jem.20091318
17. Sanjuan-Pla A, Macaulay IC, Jensen CT, Woll PS, Luis TC, Mead A, et al. Platelet-biased stem cells reside at the apex of the haematopoietic stem-cell hierarchy. Nature (2013) 502:232–6. doi:10.1038/nature12495
18. Yamamoto R, Morita Y, Ooehara J, Hamanaka S, Onodera M, Rudolph KL, et al. Clonal analysis unveils self-renewing lineage-restricted progenitors generated directly from hematopoietic stem cells. Cell (2013) 154:1112–26. doi:10.1016/j.cell.2013.08.007
19. Challen GA, Boles NC, Chambers SM, Goodell MA. Distinct hematopoietic stem cell subtypes are differentially regulated by TGF-beta1. Cell Stem Cell (2010) 6:265–78. doi:10.1016/j.stem.2010.02.002
20. Grover A, Sanjuan-Pla A, Thongjuea S, Carrelha J, Giustacchini A, Gambardella A, et al. Single-cell RNA sequencing reveals molecular and functional platelet bias of aged haematopoietic stem cells. Nat Commun (2016) 7:11075. doi:10.1038/ncomms11075
21. Cabezas-Wallscheid N, Klimmeck D, Hansson J, Lipka DB, Reyes A, Wang Q, et al. Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA methylome analysis. Cell Stem Cell (2014) 15:507–22. doi:10.1016/j.stem.2014.07.005
22. Oguro H, Ding L, Morrison SJ. SLAM family markers resolve functionally distinct subpopulations of hematopoietic stem cells and multipotent progenitors. Cell Stem Cell (2013) 13:102–16. doi:10.1016/j.stem.2013.05.014
23. Pietras EM, Reynaud D, Kang Y-A, Carlin D, Calero-Nieto FJ, Leavitt AD, et al. Functionally distinct subsets of lineage-biased multipotent progenitors control blood production in normal and regenerative conditions. Cell Stem Cell (2015) 17:35–46. doi:10.1016/j.stem.2015.05.003
24. Dorshkind K, Montecino-Rodriguez E, Signer RAJ. The ageing immune system: is it ever too old to become young again? Nat Rev Immunol (2009) 9:57–62. doi:10.1038/nri2471
25. Haq K, McElhaney JE. Ageing and respiratory infections: the airway of ageing. Immunol Lett (2014) 162:323–8. doi:10.1016/j.imlet.2014.06.009
26. Frasca D, Landin AM, Riley RL, Blomberg BB. Mechanisms for decreased function of B cells in aged mice and humans. J Immunol (2008) 180:2741–6. doi:10.4049/jimmunol.180.5.2741
27. Linton PJ, Dorshkind K. Age-related changes in lymphocyte development and function. Nat Immunol (2004) 5:133–9. doi:10.1038/ni1033
28. Akbar AN, Fletcher JM. Memory T cell homeostasis and senescence during aging. Curr Opin Immunol (2005) 17:480–5. doi:10.1016/j.coi.2005.07.019
29. Kuranda K, Vargaftig J, de la Rochere P, Dosquet C, Charron D, Bardin F, et al. Age-related changes in human hematopoietic stem/progenitor cells. Aging Cell (2011) 10:542–6. doi:10.1111/j.1474-9726.2011.00675.x
30. Ogata K, Yokose N, Tamura H, An E, Nakamura K, Dan K, et al. Natural killer cells in the late decades of human life. Clin Immunol Immunopathol (1997) 84:269–75. doi:10.1006/clin.1997.4401
31. Plowden J, Renshaw-Hoelscher M, Engleman C, Katz J, Sambhara S. Innate immunity in aging: impact on macrophage function. Aging Cell (2004) 3:161–7. doi:10.1111/j.1474-9728.2004.00102.x
33. Chambers SM, Shaw CA, Gatza C, Fisk CJ, Donehower LA, Goodell MA. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol (2007) 5:e201. doi:10.1371/journal.pbio.0050201
34. Dykstra B, Olthof S, Schreuder J, Ritsema M, de Haan G. Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J Exp Med (2011) 208:2691–703. doi:10.1084/jem.20111490
35. Harrison DE, Astle CM. Loss of stem cell repopulating ability upon transplantation. Effects of donor age, cell number, and transplantation procedure. J Exp Med (1982) 156:1767–79. doi:10.1084/jem.156.6.1767
36. Morrison SJ, Wandycz AM, Akashi K, Globerson A, Weissman IL. The aging of hematopoietic stem cells. Nat Med (1996) 2:1011–6. doi:10.1038/nm1296-1282
37. Rossi DJ, Bryder D, Zahn JM, Ahlenius H, Sonu R, Wagers AJ, et al. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc Natl Acad Sci U S A (2005) 102:9194–9. doi:10.1073/pnas.0503280102
38. Sudo K, Ema H, Morita Y, Nakauchi H. Age-associated characteristics of murine hematopoietic stem cells. J Exp Med (2000) 192:1273–80. doi:10.1084/jem.192.9.1273
39. Beerman I, Bhattacharya D, Zandi S, Sigvardsson M, Weissman IL, Bryder D, et al. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc Natl Acad Sci U S A (2010) 107:5465–70. doi:10.1073/pnas.1000834107
40. Waterstrat A, Van Zant G. Effects of aging on hematopoietic stem and progenitor cells. Curr Opin Immunol (2009) 21:408–13. doi:10.1016/j.coi.2009.05.002
41. Cho RH, Sieburg HB, Muller-Sieburg CE. A new mechanism for the aging of hematopoietic stem cells: aging changes the clonal composition of the stem cell compartment but not individual stem cells. Blood (2008) 111:5553–61. doi:10.1182/blood-2007-11-123547
42. Goto M. Inflammaging (inflammation + aging): a driving force for human aging based on an evolutionarily antagonistic pleiotropy theory? Biosci Trends (2008) 2:218–30.
43. Muller-Sieburg CE, Sieburg HB. Clonal diversity of the stem cell compartment. Curr Opin Hematol (2006) 13:243–8. doi:10.1097/01.moh.0000231421.00407.65
44. Henry CJ, Marusyk A, Zaberezhnyy V, Adane B, DeGregori J. Declining lymphoid progenitor fitness promotes aging-associated leukemogenesis. Proc Natl Acad Sci U S A (2010) 107:21713–8. doi:10.1073/pnas.1005486107
45. Haas S, Hansson J, Klimmeck D, Loeffler D, Velten L, Uckelmann H, et al. Inflammation-induced emergency megakaryopoiesis driven by hematopoietic stem cell-like megakaryocyte progenitors. Cell Stem Cell (2015) 17:422–34. doi:10.1016/j.stem.2015.07.007
46. Verovskaya E, Broekhuis MJC, Zwart E, Weersing E, Ritsema M, Bosman LJ, et al. Asymmetry in skeletal distribution of mouse hematopoietic stem cell clones and their equilibration by mobilizing cytokines. J Exp Med (2014) 211:487–97. doi:10.1084/jem.20131804
47. Tremaroli V, Bäckhed F. Functional interactions between the gut microbiota and host metabolism. Nature (2012) 489:242–9. doi:10.1038/nature11552
48. Ito K, Hirao A, Arai F, Takubo K, Matsuoka S, Miyamoto K, et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med (2006) 12:446–51. doi:10.1038/nm1388
49. Rossi DJ, Bryder D, Seita J, Nussenzweig A, Hoeijmakers J, Weissman IL. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature (2007) 447:725–9. doi:10.1038/nature05862
50. Rübe CE, Fricke A, Widmann TA, Fürst T, Madry H, Pfreundschuh M, et al. Accumulation of DNA damage in hematopoietic stem and progenitor cells during human aging. PLoS One (2011) 6:e17487. doi:10.1371/journal.pone.0017487
51. Beerman I, Seita J, Inlay MA, Weissman IL, Rossi DJ. Quiescent hematopoietic stem cells accumulate DNA damage during aging that is repaired upon entry into cell cycle. Cell Stem Cell (2014) 15:37–50. doi:10.1016/j.stem.2014.04.016
52. Flach J, Bakker ST, Mohrin M, Conroy PC, Pietras EM, Reynaud D, et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature (2014) 512:198–202. doi:10.1038/nature13619
53. Chen C, Liu Y, Liu Y, Zheng P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci Signal (2009) 2:ra75. doi:10.1126/scisignal.2000559
54. Warr MR, Binnewies M, Flach J, Reynaud D, Garg T, Malhotra R, et al. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature (2013) 494:323–7. doi:10.1038/nature11895
55. Sun D, Luo M, Jeong M, Rodriguez B, Xia Z, Hannah R, et al. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell (2014) 14:673–88. doi:10.1016/j.stem.2014.03.002
56. Florian MC, Dörr K, Niebel A, Daria D, Schrezenmeier H, Rojewski M, et al. Cdc42 activity regulates hematopoietic stem cell aging and rejuvenation. Cell Stem Cell (2012) 10:520–30. doi:10.1016/j.stem.2012.04.007
57. Beerman I, Bock C, Garrison BS, Smith ZD, Gu H, Meissner A, et al. Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging. Cell Stem Cell (2013) 12:413–25. doi:10.1016/j.stem.2013.01.017
58. Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature (2014) 505:327–34. doi:10.1038/nature12984
59. Kusumbe AP, Ramasamy SK, Itkin T, Mäe MA, Langen UH, Betsholtz C, et al. Age-dependent modulation of vascular niches for haematopoietic stem cells. Nature (2016) 532:380–4. doi:10.1038/nature17638
60. Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy (2006) 8:315–7. doi:10.1080/14653240600855905
61. Liu Z-J, Zhuge Y, Velazquez OC. Trafficking and differentiation of mesenchymal stem cells. J Cell Biochem (2009) 106:984–91. doi:10.1002/jcb.22091
62. Singh L, Brennan TA, Russell E, Kim J-H, Chen Q, Brad Johnson F, et al. Aging alters bone-fat reciprocity by shifting in vivo mesenchymal precursor cell fate towards an adipogenic lineage. Bone (2016) 85:29–36. doi:10.1016/j.bone.2016.01.014
63. Tuljapurkar SR, McGuire TR, Brusnahan SK, Jackson JD, Garvin KL, Kessinger MA, et al. Changes in human bone marrow fat content associated with changes in hematopoietic stem cell numbers and cytokine levels with aging. J Anat (2011) 219:574–81. doi:10.1111/j.1469-7580.2011.01423.x
64. Walenda T, Bork S, Horn P, Wein F, Saffrich R, Diehlmann A, et al. Co-culture with mesenchymal stromal cells increases proliferation and maintenance of haematopoietic progenitor cells. J Cell Mol Med (2010) 14:337–50. doi:10.1111/j.1582-4934.2009.00776.x
65. Behrens A, van Deursen JM, Rudolph KL, Schumacher B. Impact of genomic damage and ageing on stem cell function. Nat Cell Biol (2014) 16:201–7. doi:10.1038/ncb2928
66. Fernandez L, Rodriguez S, Huang H, Chora A, Fernandes J, Mumaw C, et al. Tumor necrosis factor-alpha and endothelial cells modulate Notch signaling in the bone marrow microenvironment during inflammation. Exp Hematol (2008) 36:545–58. doi:10.1016/j.exphem.2007.12.012
67. Kornicka K, Marycz K, Tomaszewski KA, Marędziak M, Śmieszek A. The effect of age on osteogenic and adipogenic differentiation potential of human adipose derived stromal stem cells (hASCs) and the impact of stress factors in the course of the differentiation process. Oxid Med Cell Longev (2015) 2015:309169. doi:10.1155/2015/309169
68. Zhang D-Y, Wang H-J, Tan Y-Z. Wnt/β-catenin signaling induces the aging of mesenchymal stem cells through the DNA damage response and the p53/p21 pathway. PLoS One (2011) 6:e21397. doi:10.1371/journal.pone.0021397
69. Zheng Y, He L, Wan Y, Song J. H3K9me-enhanced DNA hypermethylation of the p16INK4a gene: an epigenetic signature for spontaneous transformation of rat mesenchymal stem cells. Stem Cells Dev (2013) 22:256–67. doi:10.1089/scd.2012.0172
70. Shockley KR, Lazarenko OP, Czernik PJ, Rosen CJ, Churchill GA, Lecka-Czernik B. PPARgamma2 nuclear receptor controls multiple regulatory pathways of osteoblast differentiation from marrow mesenchymal stem cells. J Cell Biochem (2009) 106:232–46. doi:10.1002/jcb.21994
71. Takeshita S, Fumoto T, Naoe Y, Ikeda K. Age-related marrow adipogenesis is linked to increased expression of RANKL. J Biol Chem (2014) 289:16699–710. doi:10.1074/jbc.M114.547919
72. Justesen J, Stenderup K, Ebbesen EN, Mosekilde L, Steiniche T, Kassem M. Adipocyte tissue volume in bone marrow is increased with aging and in patients with osteoporosis. Biogerontology (2001) 2:165–71. doi:10.1023/A:1011513223894
73. Bellantuono I, Aldahmash A, Kassem M. Aging of marrow stromal (skeletal) stem cells and their contribution to age-related bone loss. Biochim Biophys Acta (2009) 1792:364–70. doi:10.1016/j.bbadis.2009.01.008
74. Wagner W, Bork S, Horn P, Krunic D, Walenda T, Diehlmann A, et al. Aging and replicative senescence have related effects on human stem and progenitor cells. PLoS One (2009) 4:e5846. doi:10.1371/journal.pone.0005846
75. Kennedy DE, Knight KL. Inhibition of B lymphopoiesis by adipocytes and IL-1-producing myeloid-derived suppressor cells. J Immunol (2015) 195:2666–74. doi:10.4049/jimmunol.1500957
76. Naveiras O, Nardi V, Wenzel PL, Hauschka PV, Fahey F, Daley GQ. Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature (2009) 460:259–63. doi:10.1038/nature08099
77. Ergen AV, Boles NC, Goodell MA. Rantes/Ccl5 influences hematopoietic stem cell subtypes and causes myeloid skewing. Blood (2012) 119:2500–9. doi:10.1182/blood-2011-11-391730
78. Nombela-Arrieta C, Pivarnik G, Winkel B, Canty KJ, Harley B, Mahoney JE, et al. Quantitative imaging of haematopoietic stem and progenitor cell localization and hypoxic status in the bone marrow microenvironment. Nat Cell Biol (2013) 15:533–43. doi:10.1038/ncb2730
79. Kusumbe AP, Ramasamy SK, Adams RH. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature (2014) 507:323–8. doi:10.1038/nature13145
80. Groleau J, Dussault S, Haddad P, Turgeon J, Ménard C, Chan JS, et al. Essential role of copper-zinc superoxide dismutase for ischemia-induced neovascularization via modulation of bone marrow-derived endothelial progenitor cells. Arterioscler Thromb Vasc Biol (2010) 30:2173–81. doi:10.1161/ATVBAHA.110.212530
81. Gur-Cohen S, Itkin T, Chakrabarty S, Graf C, Kollet O, Ludin A, et al. PAR1 signaling regulates the retention and recruitment of EPCR-expressing bone marrow hematopoietic stem cells. Nat Med (2015) 21:1307–17. doi:10.1038/nm.3960
82. Pang WW, Price EA, Sahoo D, Beerman I, Maloney WJ, Rossi DJ, et al. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc Natl Acad Sci U S A (2011) 108:20012–7. doi:10.1073/pnas.1116110108
83. Rundberg Nilsson A, Soneji S, Adolfsson S, Bryder D, Pronk CJ. Human and murine hematopoietic stem cell aging is associated with functional impairments and intrinsic megakaryocytic/erythroid bias. PLoS One (2016) 11:e0158369. doi:10.1371/journal.pone.0158369
84. Chen GY, Núñez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol (2010) 10:826–37. doi:10.1038/nri2873
85. Heine H, Lien E. Toll-like receptors and their function in innate and adaptive immunity. Int Arch Allergy Immunol (2003) 130:180–92. doi:10.1159/000069517
86. Lawrence T, Willoughby DA, Gilroy DW. Anti-inflammatory lipid mediators and insights into the resolution of inflammation. Nat Rev Immunol (2002) 2:787–95. doi:10.1038/nri915
87. Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol (2008) 8:349–61. doi:10.1038/nri2294
88. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol (2010) 11:373–84. doi:10.1038/ni.1863
89. Sallusto F, Lanzavecchia A. Understanding dendritic cell and T-lymphocyte traffic through the analysis of chemokine receptor expression. Immunol Rev (2000) 177:134–40. doi:10.1034/j.1600-065X.2000.17717.x
90. Burberry A, Zeng MY, Ding L, Wicks I, Inohara N, Morrison SJ, et al. Infection mobilizes hematopoietic stem cells through cooperative NOD-like receptor and Toll-like receptor signaling. Cell Host Microbe (2014) 15:779–91. doi:10.1016/j.chom.2014.05.004
91. Massberg S, Schaerli P, Knezevic-Maramica I, Köllnberger M, Tubo N, Moseman EA, et al. Immunosurveillance by hematopoietic progenitor cells trafficking through blood, lymph, and peripheral tissues. Cell (2007) 131:994–1008. doi:10.1016/j.cell.2007.09.047
92. Nagai Y, Garrett KP, Ohta S, Bahrun U, Kouro T, Akira S, et al. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity (2006) 24:801–12. doi:10.1016/j.immuni.2006.04.008
93. Kovtonyuk LV, Manz MG, Takizawa H. Enhanced thrombopoietin but not G-CSF receptor stimulation induces self-renewing hematopoietic stem cell divisions in vivo. Blood (2016) 127:3175–9. doi:10.1182/blood-2015-09-669929
94. Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature (2010) 465:793–7. doi:10.1038/nature09135
95. Essers MAG, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature (2009) 458:904–8. doi:10.1038/nature07815
96. Pietras EM, Lakshminarasimhan R, Techner J-M, Fong S, Flach J, Binnewies M, et al. Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. J Exp Med (2014) 211:245–62. doi:10.1084/jem.20131043
97. Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S, et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat Cell Biol (2016) 18:607–18. doi:10.1038/ncb3346
98. Sato T, Onai N, Yoshihara H, Arai F, Suda T, Ohteki T. Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon-dependent exhaustion. Nat Med (2009) 15:696–700. doi:10.1038/nm.1973
99. Schuettpelz LG, Borgerding JN, Christopher MJ, Gopalan PK, Romine MP, Herman AC, et al. G-CSF regulates hematopoietic stem cell activity, in part, through activation of Toll-like receptor signaling. Leukemia (2014) 28:1851–60. doi:10.1038/leu.2014.68
100. Keating A. Mesenchymal stromal cells: new directions. Cell Stem Cell (2012) 10:709–16. doi:10.1016/j.stem.2012.05.015
101. Opitz CA, Litzenburger UM, Lutz C, Lanz TV, Tritschler I, Köppel A, et al. Toll-like receptor engagement enhances the immunosuppressive properties of human bone marrow-derived mesenchymal stem cells by inducing indoleamine-2,3-dioxygenase-1 via interferon-beta and protein kinase R. Stem Cells (2009) 27:909–19. doi:10.1002/stem.7
102. Sioud M, Mobergslien A, Boudabous A, Fløisand Y. Evidence for the involvement of galectin-3 in mesenchymal stem cell suppression of allogeneic T-cell proliferation. Scand J Immunol (2010) 71:267–74. doi:10.1111/j.1365-3083.2010.02378.x
103. Bunnell BA, Betancourt AM, Sullivan DE. New concepts on the immune modulation mediated by mesenchymal stem cells. Stem Cell Res Ther (2010) 1:34. doi:10.1186/scrt34
104. Martino MM, Maruyama K, Kuhn GA, Satoh T, Takeuchi O, Müller R, et al. Inhibition of IL-1R1/MyD88 signalling promotes mesenchymal stem cell-driven tissue regeneration. Nat Commun (2016) 7:11051. doi:10.1038/ncomms11051
105. Zhang W, Huang S, Huang X, Li J, Ye P, Xu J, et al. Regulation of human mesenchymal stem cell differentiation by TREM-2. Hum Immunol (2015) 77:476–82. doi:10.1016/j.humimm.2015.06.005
106. Shi C, Jia T, Méndez-Ferrer S, Hohl TM, Serbina NV, Lipuma L, et al. Bone marrow mesenchymal stem and progenitor cells induce monocyte emigration in response to circulating toll-like receptor ligands. Immunity (2011) 34:590–601. doi:10.1016/j.immuni.2011.02.016
107. Ziegler P, Boettcher S, Takizawa H, Manz MG, Brümmendorf TH. LPS-stimulated human bone marrow stroma cells support myeloid cell development and progenitor cell maintenance. Ann Hematol (2016) 95:173–8. doi:10.1007/s00277-015-2550-5
108. Ren G, Zhang L, Zhao X, Xu G, Zhang Y, Roberts AI, et al. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell (2008) 2:141–50. doi:10.1016/j.stem.2007.11.014
109. Schürch CM, Riether C, Ochsenbein AF. Cytotoxic CD8+ T cells stimulate hematopoietic progenitors by promoting cytokine release from bone marrow mesenchymal stromal cells. Cell Stem Cell (2014) 14:460–72. doi:10.1016/j.stem.2014.01.002
110. Schajnovitz A, Itkin T, D’Uva G, Kalinkovich A, Golan K, Ludin A, et al. CXCL12 secretion by bone marrow stromal cells is dependent on cell contact and mediated by connexin-43 and connexin-45 gap junctions. Nat Immunol (2011) 12:391–8. doi:10.1038/ni.2017
111. Manz MG, Boettcher S. Emergency granulopoiesis. Nat Rev Immunol (2014) 14:302–14. doi:10.1038/nri3660
112. Boettcher S, Ziegler P, Schmid MA, Takizawa H, van Rooijen N, Kopf M, et al. Cutting edge: LPS-induced emergency myelopoiesis depends on TLR4-expressing nonhematopoietic cells. J Immunol (2012) 188:5824–8. doi:10.4049/jimmunol.1103253
113. Boettcher S, Gerosa RC, Radpour R, Bauer J, Ampenberger F, Heikenwalder M, et al. Endothelial cells translate pathogen signals into G-CSF-driven emergency granulopoiesis. Blood (2014) 124:1393–403. doi:10.1182/blood-2014-04-570762
114. Andonegui G, Bonder CS, Green F, Mullaly SC, Zbytnuik L, Raharjo E, et al. Endothelium-derived Toll-like receptor-4 is the key molecule in LPS-induced neutrophil sequestration into lungs. J Clin Invest (2003) 111:1011–20. doi:10.1172/JCI16510
115. Andonegui G, Zhou H, Bullard D, Kelly MM, Mullaly SC, McDonald B, et al. Mice that exclusively express TLR4 on endothelial cells can efficiently clear a lethal systemic Gram-negative bacterial infection. J Clin Invest (2009) 119:1921–30.
116. Broudy VC, Kaushansky K, Segal GM, Harlan JM, Adamson JW. Tumor necrosis factor type alpha stimulates human endothelial cells to produce granulocyte/macrophage colony-stimulating factor. Proc Natl Acad Sci U S A (1986) 83:7467–71. doi:10.1073/pnas.83.19.7467
117. Sieff CA, Tsai S, Faller DV. Interleukin 1 induces cultured human endothelial cell production of granulocyte-macrophage colony-stimulating factor. J Clin Invest (1987) 79:48–51. doi:10.1172/JCI112806
118. Luu NT, McGettrick HM, Buckley CD, Newsome PN, Rainger GE, Frampton J, et al. Crosstalk between mesenchymal stem cells and endothelial cells leads to downregulation of cytokine-induced leukocyte recruitment. Stem Cells (2013) 31:2690–702. doi:10.1002/stem.1511
119. Walter D, Lier A, Geiselhart A, Thalheimer FB, Huntscha S, Sobotta MC, et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature (2015) 520:549–52. doi:10.1038/nature14131
120. Kitagawa M, Saito I, Kuwata T, Yoshida S, Yamaguchi S, Takahashi M, et al. Overexpression of tumor necrosis factor (TNF)-alpha and interferon (IFN)-gamma by bone marrow cells from patients with myelodysplastic syndromes. Leukemia (1997) 11:2049–54. doi:10.1038/sj.leu.2400844
121. Kim JM, Kim NI, Oh Y-K, Kim Y-J, Youn J, Ahn M-J. CpG oligodeoxynucleotides induce IL-8 expression in CD34+ cells via mitogen-activated protein kinase-dependent and NF-kappaB-independent pathways. Int Immunol (2005) 17:1525–31. doi:10.1093/intimm/dxh345
122. Sioud M, Fløisand Y, Forfang L, Lund-Johansen F. Signaling through toll-like receptor 7/8 induces the differentiation of human bone marrow CD34+ progenitor cells along the myeloid lineage. J Mol Biol (2006) 364:945–54. doi:10.1016/j.jmb.2006.09.054
123. Sioud M, Fløisand Y. TLR agonists induce the differentiation of human bone marrow CD34+ progenitors into CD11c+ CD80/86+ DC capable of inducing a Th1-type response. Eur J Immunol (2007) 37:2834–46. doi:10.1002/eji.200737112
124. Kolb-Mäurer A, Wilhelm M, Weissinger F, Bröcker E-B, Goebel W. Interaction of human hematopoietic stem cells with bacterial pathogens. Blood (2002) 100:3703–9. doi:10.1182/blood-2002-03-0898
125. Kim JM, Oh Y-K, Kim Y-J, Youn J, Ahn M-J. Escherichia coli up-regulates proinflammatory cytokine expression in granulocyte/macrophage lineages of CD34 stem cells via p50 homodimeric NF-kappaB. Clin Exp Immunol (2004) 137:341–50. doi:10.1111/j.1365-2249.2004.02542.x
126. Caux C, Moreau I, Saeland S, Banchereau J. Interferon-gamma enhances factor-dependent myeloid proliferation of human CD34+ hematopoietic progenitor cells. Blood (1992) 79:2628–35.
127. Zeng W, Miyazato A, Chen G, Kajigaya S, Young NS, Maciejewski JP. Interferon-gamma-induced gene expression in CD34 cells: identification of pathologic cytokine-specific signature profiles. Blood (2006) 107:167–75. doi:10.1182/blood-2005-05-1884
128. Dybedal I, Bryder D, Fossum A, Rusten LS, Jacobsen SE. Tumor necrosis factor (TNF)-mediated activation of the p55 TNF receptor negatively regulates maintenance of cycling reconstituting human hematopoietic stem cells. Blood (2001) 98:1782–91. doi:10.1182/blood.V98.6.1782
129. Yang L, Dybedal I, Bryder D, Nilsson L, Sitnicka E, Sasaki Y, et al. IFN-gamma negatively modulates self-renewal of repopulating human hemopoietic stem cells. J Immunol (2005) 174:752–7. doi:10.4049/jimmunol.174.2.752
130. Esplin BL, Shimazu T, Welner RS, Garrett KP, Nie L, Zhang Q, et al. Chronic exposure to a TLR ligand injures hematopoietic stem cells. J Immunol (2011) 186:5367–75. doi:10.4049/jimmunol.1003438
131. Kim M, Moon H-B, Spangrude GJ. Major age-related changes of mouse hematopoietic stem/progenitor cells. Ann N Y Acad Sci (2003) 996:195–208. doi:10.1111/j.1749-6632.2003.tb03247.x
132. Van der Put E, Frasca D, King AM, Blomberg BB, Riley RL. Decreased E47 in senescent B cell precursors is stage specific and regulated posttranslationally by protein turnover. J Immunol (2004) 173:818–27. doi:10.4049/jimmunol.173.2.818
133. Baylis D, Bartlett DB, Patel HP, Roberts HC. Understanding how we age: insights into inflammaging. Longev Healthspan (2013) 2:8. doi:10.1186/2046-2395-2-8
134. Bruunsgaard H, Andersen-Ranberg K, Jeune B, Pedersen AN, Skinhøj P, Pedersen BK. A high plasma concentration of TNF-alpha is associated with dementia in centenarians. J Gerontol A Biol Sci Med Sci (1999) 54:M357–64. doi:10.1093/gerona/54.7.M357
135. Cohen HJ, Pieper CF, Harris T, Rao KM, Currie MS. The association of plasma IL-6 levels with functional disability in community-dwelling elderly. J Gerontol A Biol Sci Med Sci (1997) 52:M201–8. doi:10.1093/gerona/52A.4.M201
136. Emerging Risk Factors Collaboration, Sarwar N, Gao P, Seshasai SRK, Gobin R, Kaptoge S, et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: a collaborative meta-analysis of 102 prospective studies. Lancet (2010) 375:2215–22. doi:10.1016/S0140-6736(10)60484-9
137. Ferrucci L, Corsi A, Lauretani F, Bandinelli S, Bartali B, Taub DD, et al. The origins of age-related proinflammatory state. Blood (2005) 105:2294–9. doi:10.1182/blood-2004-07-2599
138. Hasegawa Y, Sawada M, Ozaki N, Inagaki T, Suzumura A. Increased soluble tumor necrosis factor receptor levels in the serum of elderly people. Gerontology (2000) 46:185–8. doi:10.1159/000022157
139. Neukirchen J, Schoonen WM, Strupp C, Gattermann N, Aul C, Haas R, et al. Incidence and prevalence of myelodysplastic syndromes: data from the Düsseldorf MDS-registry. Leuk Res (2011) 35:1591–6. doi:10.1016/j.leukres.2011.06.001
140. Busque L, Patel JP, Figueroa ME, Vasanthakumar A, Provost S, Hamilou Z, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet (2012) 44:1179–81. doi:10.1038/ng.2413
141. Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med (2014) 371:2477–87. doi:10.1056/NEJMoa1409405
142. Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med (2014) 371:2488–98. doi:10.1056/NEJMoa1408617
143. Laurie CC, Laurie CA, Rice K, Doheny KF, Zelnick LR, McHugh CP, et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat Genet (2012) 44:642–50. doi:10.1038/ng.2271
144. Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med (2014) 20:1472–8. doi:10.1038/nm.3733
145. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi:10.1016/j.cell.2011.02.013
Keywords: ageing, inflammation, hematopoietic stem cell, niche, cytokine, pathogen recognition receptor
Citation: Kovtonyuk LV, Fritsch K, Feng X, Manz MG and Takizawa H (2016) Inflamm-Aging of Hematopoiesis, Hematopoietic Stem Cells, and the Bone Marrow Microenvironment. Front. Immunol. 7:502. doi: 10.3389/fimmu.2016.00502
Received: 30 June 2016; Accepted: 27 October 2016;
Published: 14 November 2016
Edited by:
Laura Schuettpelz, Washington University in St. Louis, USAReviewed by:
Katherine C. MacNamara, Albany Medical College, USADan Link, Washington University in St. Louis, USA
Copyright: © 2016 Kovtonyuk, Fritsch, Feng, Manz and Takizawa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hitoshi Takizawa, htakizawa@kumamoto-u.ac.jp
†Larisa V. Kovtonyuk and Kristin Fritsch contributed equally.