 An-Ko Chung1,2Ro-Ting Lin3
An-Ko Chung1,2Ro-Ting Lin3 Chun-Chieh Yeh4,5Chi-Ying Yang6Chang-Jiun Wu7
Chun-Chieh Yeh4,5Chi-Ying Yang6Chang-Jiun Wu7 Pei-Lung Chen1,8*Jaw-Town Lin9*
Pei-Lung Chen1,8*Jaw-Town Lin9*- 1Graduate Institute of Medical Genomics and Proteomics, National Taiwan University, Taipei, Taiwan
- 2Department of Internal Medicine, National Taiwan University College of Medicine, Taipei, Taiwan
- 3Department of Occupational Safety and Health, College of Public Health, China Medical University, Taichung, Taiwan
- 4School of Medicine, China Medical University, Taichung, Taiwan
- 5Department of Surgery, China Medical University Hospital, Taichung, Taiwan
- 6Department of Internal Medicine, Digestive Medicine Center, China Medical University Hospital, Taichung, Taiwan
- 7Department of Genomic Medicine, MD Anderson Cancer Center, University of Texas, Houston, TX, United States
- 8Department of Medical Genetics, National Taiwan University Hospital, Taipei, Taiwan
- 9Division of Gastroenterology and Hepatology, Department of Internal Medicine, E-Da Hospital, Kaohsiung, Taiwan
Identification of germline pathogenic variants in cancer patients is critical for treatment planning, genetic counseling, and health policymaking. However, previous estimates of the prevalence of germline etiology of pancreatic ductal adenocarcinoma (PDAC) were biased because they were based only on sequencing data of protein-coding regions of known PDAC candidate genes. To determine the percentage of patients with PDAC carrying germline pathogenic variants, we enrolled the inpatients from the digestive health clinics, hematology and oncology clinics, and surgical clinics of a single tertiary medical center in Taiwan for whole genome sequencing (WGS) analysis of genomic DNA. The virtual gene panel of 750 genes comprised PDAC candidate genes and those listed in the COSMIC Cancer Gene Census. The genetic variant types under investigation included single nucleotide substitutions, small indels, structural variants, and mobile element insertions (MEIs). In 8 of 24 (33.3%) patients with PDAC, we identified pathogenic/likely pathogenic variants, including single nucleotide substitutions and small indels in ATM, BRCA1, BRCA2, POLQ, SPINK1 and CASP8, as well as structural variants in CDC25C and USP44. We identified additional patients carrying variants that could potentially affect splicing. This cohort study demonstrates that an extensive analysis of the abundant information yielded by the WGS approach can uncover many pathogenic variants that could be missed by traditional panel-based or whole exome sequencing-based approaches. The percentage of patients with PDAC carrying germline variants might be much higher than previously expected.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) has an extremely poor prognosis. Approximately 10% of unselected patients with PDAC carried germline pathogenic variants (Yurgelun et al., 2019; Gardiner et al., 2022), with the rate being 30% among cases in populations with a strong family history of cancer and/or common founder variants (e.g., in Ashkenazi Jews) (Gardiner et al., 2022). These estimations of germline pathogenic variants related to PDAC might be biased because the number of candidate genes that had their coding regions tested is limited, and the approaches used were panel-based or whole exome sequencing (Yurgelun et al., 2019; Gardiner et al., 2022). This emphasizes a knowledge gap regarding the diagnostic rate of germline pathogenic variants in patients with PDAC using a comprehensive whole genome sequencing (WGS) approach. To ensure the quality of therapeutic planning, early genetic counseling for at-risk relatives, and health policymaking, precise data on the identification of germline pathogenic variants is critical.
Materials and methods
Subjects and sample collection
This retrospective cohort study recruited inpatients diagnosed with PDAC (C25.0–C25.9, based on ICD-10) in a tertiary medical center in Taiwan; details are available in our previous publication (Lin et al., 2022). We recruited 24 patients (men: 19; women: 5; median age at diagnosis: 56.7 years). Regarding lesion location, the head of the pancreas (C25.0) was the most common (13 patients), followed by the tail (C25.2; 7 patients), the body (C25.1; 3 patients), and both the head and body (1 patient). The study was approved by the Research Ethics Committee III of the China Medical University and Hospital (CMUH109-REC3-026). Genomic DNA was extracted from the participants’ peripheral blood mononuclear cells.
WGS and quality analysis
WGS was performed on the Illumina NovaSeq platform (Illumina, San Diego, CA, United States), with 2 × 150 bp paired-end reads to achieve 30 × coverage, followed by an adapter trimming and low-quality bases filtering with Phred quality scores greater than 30.
Germline variant detection and interpretation
Sequence analysis was conducted based on GATK Best Practice workflow (McKenna et al., 2010) (v4.2). Paired-end reads were aligned to the reference genome (GRCh38/hg38) using BWA-MEM(Li and Durbin, 2009) (v0.7.17). Variant calling was conducted using HaplotypeCaller. Variant quality score recalibrations were performed using VariantRecalibrator. Simple variants (e.g., single nucleotide variants or small indels) were then ready for annotation and automatic interpretation based on five classes (pathogenic, likely pathogenic, uncertain significance, likely benign, and benign) following the American College of Medical Genetics and Genomics guideline (Richards et al., 2015) using TAIGenomics software (https://www.taigenomics.com). Variants with high allele frequencies in the Genome Aggregation Database (gnomAD, https://gnomad.broad institute.org) or Taiwan Biobank (http://taiwanview.Twbiobank.Org.tw/index) databases were filtered out. Annotations of pathogenic and likely pathogenic variants were manually confirmed. Variants identified in the known PDAC genes or known cancer genes in DNA repair pathway (Supplementary Table S1) and the COSMIC Cancer Gene Census (CGC) (https://cancer.sanger.ac.uk/census) were selected (Supplementary Figure S1). We remove variants which are with read depth ≤10, genotype quality ≤20, or allele balance ≤0.2.
The potential of variants to affect splicing was explored using SpliceAI (Jaganathan et al., 2019). Variants with allele frequencies <1% and located in the candidate genes were tested. A score greater than 0.5 was considered positive.
Mobile element insertions (MEIs) were investigated using SCRAMble (Torene et al., 2020) (v1.0.2) and MELT (Gardner et al., 2017) (v2.2.2). All VCF files from SCRAMble and MELT were annotated using AnnotSV(Geoffroy et al., 2018) (v3.0.9).
Structural variants were examined using a combination of callers including Manta (Chen et al., 2016) (v1.6), Delly (Rausch et al., 2012) (v0.8.7), and SvABA (Wala et al., 2018) (v1.1.0). Structural variants detected by at least two callers were selected using SURVIVOR (Jeffares et al., 2017) (v1.0.7), followed by annotation using AnnotSV(Geoffroy et al., 2018) (v3.0.9).
Results
Between July 2020 and December 2020, a total of 24 participants fulfilled the inclusion criteria and had all biospecimens available (Supplementary Table S2).
Single nucleotide variants and small indels
To identify pathogenic variants, we first constructed a virtual panel of 750 genes, comprising known PDAC genes or known cancer genes in DNA repair pathway (Supplementary Table S1) and the COSMIC CGC Panel. From the list of single nucleotide variants and small indels identified in our cohort, we found six heterozygous pathogenic variants in six different patients. They include DNA damage response and DNA repair genes (ATM, BRCA1, BRCA2, and POLQ), pancreatitis gene (SPINK1) and cell apoptosis gene (CASP8; Table 1).
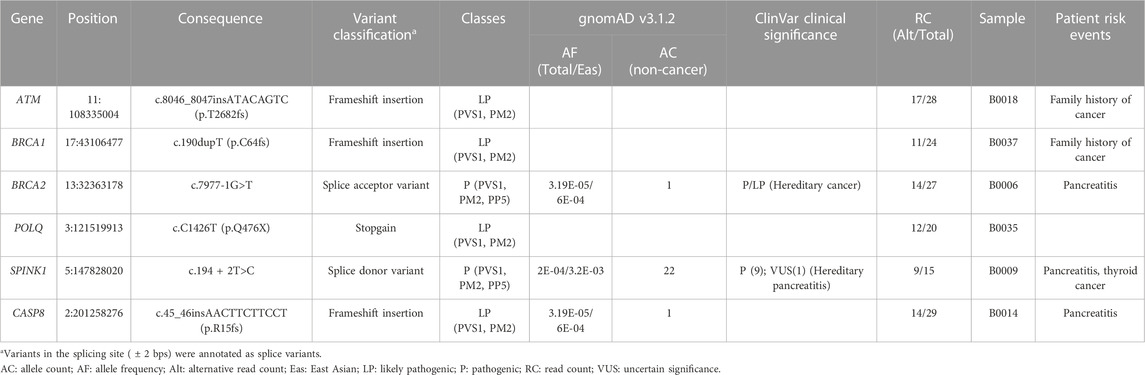
TABLE 1. Disease-causing germline variants or small indels identified in PDAC patients.
All variants disrupted at least one protein domain in these genes, suggesting that variants lead to loss of function (Figure 1A). Specifically, the patient carrying the SPINK1 (c.194 + 2T>C) variant had a history of chronic pancreatitis (Table 1). Two variants in ATM and BRCA1 were identified in patients with a family history of cancer (Table 1).
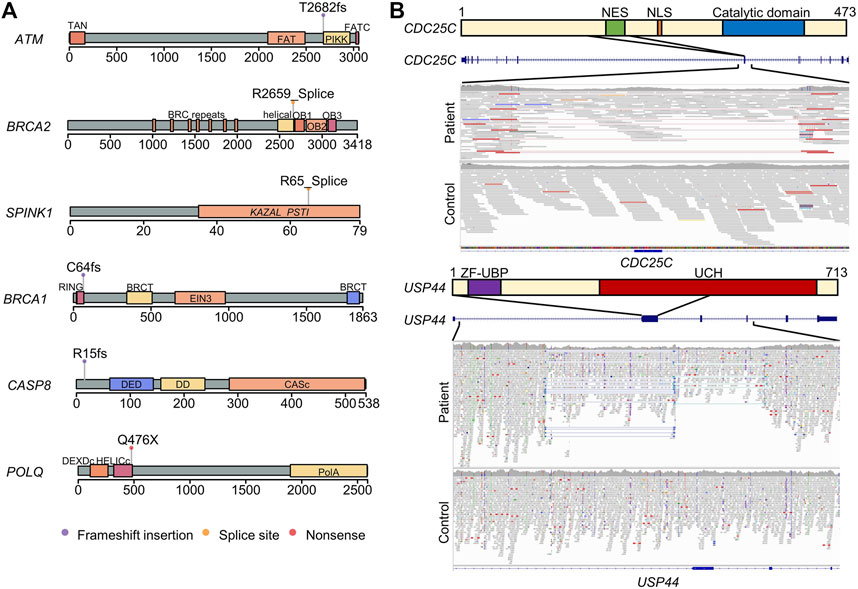
FIGURE 1. Schematic diagram of the pathogenic germline variants and structural variants identified in patients with pancreatic ductal adenocarcinoma. (A) Protein structures and domains were generated using maftools package. The lollipops represent the positions of pathogenic germline variants. (B) Deletion in the region containing nuclear export signal in CDC25C. The upper panel represented the CDC25C protein structure and domain. The middle panel is a screenshot of UCSC Genome Browser in the region of CDC25C gene. The lower panel is a screenshot of integrative genomics viewer of the deletion region. The paired reads with unexpected insert-size are visualized in the link line with red color. NES, nuclear export signal. NLS, nuclear localization sequence. A combined deletion and inversion in the second exon of USP44. The read depth decreased in the patient compared to the control, and paired-end inversion reads are visualized with light and dark blue colors.
Variants with potential to affect splicing
We found six heterozygous variants predicted to alter splicing; two in DNA repair-associated genes (BRCA1 and BAP1) and four in tumor suppressor genes (ARHGEF10L, ELL, MYH9, and NCOR2) (Supplementary Table S3). Due to a lack of mRNA/cDNA data to confirm the change in splicing patterns, they were not counted as pathogenic variants in this study.
Structural variants or MEIs
We observed one in-frame deletion of the seventh exon (amino acids 154–205) of CDC25C, resulting in the loss of the nuclear export signal (amino acids 190–199) but preserving the catalytic domain and nuclear localization signal; the deletion may cause the accumulation of CDC25C in the nucleus and promote a cell cycle without cellular localization control (Figure 1B).
We found a complex structural variant, indicating an inversion and deletion event, interrupting USP44 (Figure 1B). USP44 is a recently discovered tumor suppressor gene implicated in PDAC (Yang et al., 2019).
MEIs may interrupt gene function, but we did not find any suspected MEIs in any gene in our PDAC virtual panel.
Discussion
We identified pathogenic germline variants in eight out of 24 (33.3%) patients with PDAC in Taiwan. This high rate may be partially explained by the WGS approach to examine a comprehensive list of 750 genes, diverse genetic variant types, and sequence information within non-coding regions. Our cohort was not considered to be of younger age (range, 36.1–82.5 years) or have a stronger family history (Lin et al., 2022).
Among the eight variants, the pathogenicity prediction (Richards et al., 2015) and gene-disease correlation of five variants or small indels in ATM, BRCA1, BRCA2, POLQ, and SPINK1 were unequivocal. The pathogenicity predictions of the frameshift insertions in CASP8 was also convincing. Two of them (BRCA2 c.7977-1G>T; SPINK1 c.194 + 2T>C) are listed in the ClinVar database. The BRCA2 variant was consistently reported to be pathogenic; for the SPINK1 variant, there were conflicting interpretations of pathogenicity, with nine identifying it as pathogenic and one as uncertain significance. These two variants had both been reported as pathogenic variants in published pancreatic cancer studies (Chian et al., 2021; Yin et al., 2022). Although the other variants in BRCA1, ATM, and POLQ were not previously reported and were identified as novel variants in this current study, pathogenic variants in these genes have also been reported in pancreatic cancer and therefore these genes have been known to cause PDAC (Earl et al., 2020; Mizukami et al., 2020). Inactivation and somatic mutation of CASP8 are reported in various cancers (Mandal et al., 2020), and a recent report found a colorectal cancer patient with somatic loss of heterozygosity in CASP8 (Choi et al., 2021). In addition, the level of caspase 8, which is encoded by CASP8, was reduced in pancreatic cancer according to a previous study (Jakubowska et al., 2016). The two structural variants were also predicted to have major effects on CDC25C and USP44, respectively, and while the first CDC25C has not been related to PDAC, the latter has recently begun to be linked to PDAC (Yang et al., 2019). Therefore, CASP8 and CDC25C are suggested to be novel pathogenic genes of PDAC, and this study supports the potential of USP44 to cause PDAC.
We also identified six deep intronic variants predicted to alter splicing in DNA repair-associated genes (BRCA1 and BAP1) or tumor suppressor genes (ARHGEF10L, ELL, MYH9, and NCOR2; Supplementary Table S3). They might be disease-causing variants related to PDAC, but we could not confirm this due to a lack of data. We did not identify disease-causing MEIs in our sample.
This study had a modest sample size and lacked confirmatory experiments to determine the disease-causing roles of the identified variants, leaving space for future investigation. Comprehensive and thorough genomic analyses in large cohorts are needed to support our finding that the prevalence of PDAC patients carrying pathogenic germline variants might be higher than previously estimated. Overall, our results demonstrate the potential for the WGS-based approach to uncover pathogenic genes/variants that could be missed by traditional panel-based or WES-based approaches. Although CASP8, CDC25C, and USP44 have been undervalued, they were shown to be plausible PDAC genes. The percentage of patients with PDAC carrying germline etiology (33.3% in this study) might be much higher than previously expected.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: PRJNA947736.
Ethics statement
The studies involving human participants were reviewed and approved by Research Ethics Committee III of the China Medical University and Hospital (CMUH109-REC3-026). The patients/participants provided their written informed consent to participate in this study.
Author contributions
A-KC: data curation; formal analysis; investigation; writing—original draft; writing—review and editing. R-TL: data curation; formal analysis; investigation; resources; writing—review and editing. C-CY: data curation; investigation; resources; writing—review and editing. C-YY: data curation; investigation; resources; writing—review and editing. C-JW: data curation; investigation; resources; writing—review and editing. P-LC: conceptualization; writing—original draft; writing—review and editing; supervision. J-TL: conceptualization; resources; writing—review and editing.
Funding
This work was funded by the National Science and Technology Council, Taiwan (109-2327-B-039-001, 110-2314-B-650-012-MY3, and 110-2314-B-039-027-MY3 to J-TL; 111-2314-B-039-020-MY2 and 110-2314-B-039-058 to R-TL; 108-2314-B-002-069-MY3 to P-LC) and the Taipei Institute of Pathology (TIP-111-006 to R-TL).
Acknowledgments
We would like to acknowledge Dr. Pei-Ling Tsou for helpful discussion and the editors at Editage for their English language editing. We thank the National Center for High-performance Computing (NCHC) of National Applied Research Laboratories (NARLabs) in Taiwan for providing computational and storage resources.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1172365/full#supplementary-material
References
Chen, X., Schulz-Trieglaff, O., Shaw, R., Barnes, B., Schlesinger, F., Kallberg, M., et al. (2016). Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32, 1220–1222. doi:10.1093/bioinformatics/btv710
Chian, J., Sinha, S., Qin, Z., and Wang, S. M. (2021). BRCA1 and BRCA2 variation in Taiwanese general population and the cancer cohort. Front. Mol. Biosci. 8, 685174. doi:10.3389/fmolb.2021.685174
Choi, Y. Y., Shin, S. J., Lee, J. E., Madlensky, L., Lee, S. T., Park, J. S., et al. (2021). Prevalence of cancer susceptibility variants in patients with multiple Lynch syndrome related cancers. Sci. Rep. 11, 14807. doi:10.1038/s41598-021-94292-4
Earl, J., Galindo-Pumarino, C., Encinas, J., Barreto, E., Castillo, M. E., Pachon, V., et al. (2020). A comprehensive analysis of candidate genes in familial pancreatic cancer families reveals a high frequency of potentially pathogenic germline variants. EBioMedicine 53, 102675. doi:10.1016/j.ebiom.2020.102675
Gardiner, A., Kidd, J., Elias, M. C., Young, K., Mabey, B., Taherian, N., et al. (2022). Pancreatic ductal carcinoma risk associated with hereditary cancer-risk genes. J. Natl. Cancer Inst. 114, 996–1002. doi:10.1093/jnci/djac069
Gardner, E. J., Lam, V. K., Harris, D. N., Chuang, N. T., Scott, E. C., Pittard, W. S., et al. (2017). The mobile element locator tool (MELT): Population-scale mobile element discovery and biology. Genome Res. 27, 1916–1929. doi:10.1101/gr.218032.116
Geoffroy, V., Herenger, Y., Kress, A., Stoetzel, C., Piton, A., Dollfus, H., et al. (2018). AnnotSV: An integrated tool for structural variations annotation. Bioinformatics 34, 3572–3574. doi:10.1093/bioinformatics/bty304
Jaganathan, K., Kyriazopoulou Panagiotopoulou, S., Mcrae, J. F., Darbandi, S. F., Knowles, D., Li, Y. I., et al. (2019). Predicting splicing from primary sequence with deep learning. Cell 176, 535–548. doi:10.1016/j.cell.2018.12.015
Jakubowska, K., Guzinska-Ustymowicz, K., Famulski, W., Cepowicz, D., Jagodzinska, D., and Pryczynicz, A. (2016). Reduced expression of caspase-8 and cleaved caspase-3 in pancreatic ductal adenocarcinoma cells. Oncol. Lett. 11, 1879–1884. doi:10.3892/ol.2016.4125
Jeffares, D. C., Jolly, C., Hoti, M., Speed, D., Shaw, L., Rallis, C., et al. (2017). Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat. Commun. 8, 14061. doi:10.1038/ncomms14061
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi:10.1093/bioinformatics/btp324
Lin, R. T., Chen, P. L., Yang, C. Y., Yeh, C. C., Lin, C. C., Huang, W. H., et al. (2022). Risk factors related to age at diagnosis of pancreatic cancer: A retrospective cohort pilot study. BMC Gastroenterol. 22, 243. doi:10.1186/s12876-022-02325-7
Mandal, R., Barron, J. C., Kostova, I., Becker, S., and Strebhardt, K. (2020). Caspase-8: The double-edged sword. Biochim. Biophys. Acta Rev. Cancer 1873, 188357. doi:10.1016/j.bbcan.2020.188357
Mckenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi:10.1101/gr.107524.110
Mizukami, K., Iwasaki, Y., Kawakami, E., Hirata, M., Kamatani, Y., Matsuda, K., et al. (2020). Genetic characterization of pancreatic cancer patients and prediction of carrier status of germline pathogenic variants in cancer-predisposing genes. EBioMedicine 60, 103033. doi:10.1016/j.ebiom.2020.103033
Rausch, T., Zichner, T., Schlattl, A., Stutz, A. M., Benes, V., and Korbel, J. O. (2012). Delly: Structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28, i333–i339. doi:10.1093/bioinformatics/bts378
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular Pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Torene, R. I., Galens, K., Liu, S., Arvai, K., Borroto, C., Scuffins, J., et al. (2020). Mobile element insertion detection in 89,874 clinical exomes. Genet. Med. 22, 974–978. doi:10.1038/s41436-020-0749-x
Wala, J. A., Bandopadhayay, P., Greenwald, N. F., O'rourke, R., Sharpe, T., Stewart, C., et al. (2018). SvABA: Genome-wide detection of structural variants and indels by local assembly. Genome Res. 28, 581–591. doi:10.1101/gr.221028.117
Yang, C., Zhu, S., Yang, H., Deng, S., Fan, P., Li, M., et al. (2019). USP44 suppresses pancreatic cancer progression and overcomes gemcitabine resistance by deubiquitinating FBP1. Am. J. Cancer Res. 9, 1722–1733.
Yin, L., Wei, J., Lu, Z., Huang, S., Gao, H., Chen, J., et al. (2022). Prevalence of germline sequence variations among patients with pancreatic cancer in China. JAMA Netw. Open 5, e2148721. doi:10.1001/jamanetworkopen.2021.48721
Yurgelun, M. B., Chittenden, A. B., Morales-Oyarvide, V., Rubinson, D. A., Dunne, R. F., Kozak, M. M., et al. (2019). Germline cancer susceptibility gene variants, somatic second hits, and survival outcomes in patients with resected pancreatic cancer. Genet. Med. 21, 213–223. doi:10.1038/s41436-018-0009-5
Keywords: pancreatic ductal adenocarcinoma (PADC), whole genome sequencing (WGS), germline genetic testing, structural variant (SV), cancer genetic
Citation: Chung A-K, Lin R-T, Yeh C-C, Yang C-Y, Wu C-J, Chen P-L and Lin J-T (2023) Diagnostic rate of germline pathogenic variants in pancreatic ductal adenocarcinoma patients using whole genome sequencing. Front. Genet. 14:1172365. doi: 10.3389/fgene.2023.1172365
Received: 23 February 2023; Accepted: 25 April 2023;
Published: 10 May 2023.
Edited by:
Salih Ibrahem, University of Kirkuk, IraqReviewed by:
Chao Ling, Peking Union Medical College Hospital (CAMS), ChinaVignesh Ravichandran, Memorial Sloan Kettering Cancer Center, United States
Copyright © 2023 Chung, Lin, Yeh, Yang, Wu, Chen and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pei-Lung Chen, paylong@ntu.edu.tw; Jaw-Town Lin, jawtown@gmail.com