 Linyong Hu1
Linyong Hu1 Liangzhi Zhang1
Liangzhi Zhang1 Qi Li1
Qi Li1 Hongjin Liu1
Hongjin Liu1 Tianwei Xu1Na Zhao1
Tianwei Xu1Na Zhao1 Xueping Han1,2
Xueping Han1,2 Shixiao Xu1
Shixiao Xu1 Xinquan Zhao1
Xinquan Zhao1 Cunfang Zhang3*
Cunfang Zhang3*- 1Key Laboratory of Adaptation and Evolution of Plateau Biota, Northwest Institute of Plateau Biology, Chinese Academy of Sciences, Xining, China
- 2Technology Extension Service of Animal Husbandry of Qinghai, Xining, China
- 3State Key Laboratory of Plateau Ecology and Agriculture, Qinghai University, Xining, China
Copy number variation (CNV), an important source of genomic structural variation, can disturb genetic structure, dosage, regulation and expression, and is associated with phenotypic diversity and adaptation to local environments in mammals. In the present study, 24 resequencing datasets were used to characterize CNVs in three ecotypic populations of Tibetan sheep and assess CNVs related to domestication and adaptation in Qinghai-Tibetan Plateau. A total of 87,832 CNV events accounting for 0.3% of the sheep genome were detected. After merging the overlapping CNVs, 2777 CNV regions (CNVRs) were obtained, among which 1098 CNVRs were shared by the three populations. The average length of these CNVRs was more than 3 kb, and duplication events were more frequent than deletions. Functional analysis showed that the shared CNVRs were significantly enriched in 56 GO terms and 18 KEGG pathways that were mainly concerned with ABC transporters, olfactory transduction and oxygen transport. Moreover, 188 CNVRs overlapped with 97 quantitative trait loci (QTLs), such as growth and carcass QTLs, immunoglobulin QTLs, milk yield QTLs and fecal egg counts QTLs. PCDH15, APP and GRID2 overlapped with body weight QTLs. Furthermore, Vst analysis showed that RUNX1, LOC101104348, LOC105604082 and PAG11 were highly divergent between Highland-type Tibetan Sheep (HTS) and Valley-type Tibetan sheep (VTS), and RUNX1 and LOC101111988 were significantly differentiated between VTS and Oura-type Tibetan sheep (OTS). The duplication of RUNX1 may facilitate the hypoxia adaptation of OTS and HTS in Qinghai-Tibetan Plateau, which deserves further research in detail. In conclusion, for the first time, we represented the genome-wide distribution characteristics of CNVs in Tibetan sheep by resequencing, and provided a valuable genetic variation resource, which will facilitate the elucidation of the genetic basis underlying the distinct phenotypic traits and local adaptation of Tibetan sheep.
1 Introduction
Copy number variations (CNVs) and single nucleotide polymorphisms (SNPs), as significant genetic variations, play important roles in domestication and adaptation of animals and plants (Lye and Purugganan, 2019; Merot et al., 2020). Unlike SNPs, which refer to the substitution, deletion or insertion of just a single nucleotide for another, CNVs involve fragmental variation even larger than 1000 bp in size (Iafrate et al., 2004; Feuk et al., 2006). Therefore, it has been widely recognized that CNVs have the potential to markedly affect phenotypic traits of domestic animals (Salehian-Dehkordi et al., 2021).
In the past twenty years, a large number of CNVs have been revealed and identified by using array comparative genome hybridization (aCGH) chips and high-density SNP chips in domestic animals, such as cattle, sheep, goat, pig, horse, dog, chicken, turkey and duck (Bickhart and Liu, 2014; Strillacci et al., 2021). Meanwhile, numerous CNV-overlapping genes have been shown to be associated with coat color (Norris and Whan, 2008), growth, fertility and production (Zhou et al., 2016a; de Lemos et al., 2018a), immune response (Sasaki et al., 2016; Zhang et al., 2016; Chen J. et al., 2017; Chen L. et al., 2017; Xu et al., 2017), olfactory transduction (Zhou et al., 2016b; Upadhyay et al., 2017; de Lemos et al., 2018b; Mielczarek et al., 2018), molecular function (Letaief et al., 2017; Pierce et al., 2018), lipid metabolism (Gao et al., 2017; Xu et al., 2017; de Lemos et al., 2018b; Goyache et al., 2021) and environmental adaptability (Wang et al., 2016). Compared with aCGH and SNP chips, the sensitivity of which is mainly limited by the density of the probes, whole genome resequencing can be used to detect new and rare CNVs (Jenkins et al., 2016). Therefore, an increasing number of CNVs have been detected by using high-throughput sequencing. In terms of sheep, in addition to whole genome detection of new CNVs, the primary focus was uncovering the CNVs associated with economic traits. Yuan et al. found that 1855 CNVRs were associated with 166 quantitative trait loci (QTLs), including milk QTLs, carcass QTLs, and health-related QTLs in fine-wool sheep (Yuan et al., 2021). CNVR overlapping genes, such as SHE, PIGY, and BAG4, were reported to be correlated with body size in sheep (Jiang et al., 2019; Feng et al., 2020; Yang et al., 2021). The CNV-overlapping genes of BTG3, PTGS1 and PSPH were involved in fetal muscle development, prostaglandin (PG) synthesis, and bone color (Yang et al., 2018). Meanwhile, the correlation between CNVR-harboring genes and growth traits or phenotypic traits was also attractive. The agouti signaling protein (ASIP) gene duplication has been linked to typical white coat color (Norris and Whan, 2008). Distal-less homeobox 3 (DLX3) CNV is related to wool curling in Tan sheep (Ma et al., 2017).
Tibetan sheep (TS) is one of the three ancient coarse-wool sheep breeds in China and is predominantly distributed in the Qinghai-Tibetan Plateau (QTP). Even in nowadays, Tibetan sheep, together with yak, provide the Tibetan herders with the main food, fuel and clothing materials. Natural adaptation and artificial selection shaped the TS with the characteristics of adaptation to cold, food shortage, and hypoxia. TS can be divided into three ecotypic populations, namely, Highland-type Tibetan sheep (HTS), Valley-type Tibetan sheep (VTS) and Oura-type Tibetan sheep (OTS) (The photos of the three sheep population were shown in Supplementary Table S1). HTS is famous for its carpet wool production, with an average staple length of up to 25 cm. The proportion of dry dead wool in the OTS is relatively high but has remarkable meat performance. The appearance of VTS is similar to that of HTS but with a smaller body size and shorter staple length. Meanwhile, the VTS was mainly distributed in valley regions with relatively low altitudes (∼2000 m), in contrast to the HTS and OTS (both distributed above 3200 m).
It’s meaningful to elucidate the genomic distribution characteristics and potential functions of CNVs in TS. Nevertheless, there have been few reports on the analysis of CNVs in TS (Huang et al., 2021; Salehian-Dehkordi et al., 2021). Therefore, in the present study, we exploited a large number of CNVs in 24 individuals from the three TS populations using whole genome resequencing and identified CNVs associated with the molecular basis of phenotype differences, domestication and adaptation to QTPs.
2 Materials and methods
2.1 Animal sampling and genomic DNA sequencing
Twenty-four whole blood samples of Tibetan sheep were collected from their core distribution region in this study, including 8 highland-type sheep (HTS, Qilian County, 4 male and 4 female sheep, similar to the following two sheep groups), 8 valley-type sheep (VTS, Huangyuan County) and 8 outer-type sheep (OTS, Henan County). The samples of each population were randomly collected from more than three different groups and the information was seeked from the herdman to avoid the potential genetic relationship. The collected samples were stored in EDTA antifreezing tubes at -20°C. Genomic DNA was extracted with a QIAamp DNA Blood Mini Kits (Qiagen, Germany), and then the DNA integrity and concentration were checked with 1% agarose gel electrophoresis and Qubit® 3.0 Flurometer (Invitrogen, United States), respectively. A sequencing library was built with 0.2 μg genomic DNA from each sample using the NEB Next® UltraTM DNA Library Prep Kit (NEB, United States) following the manufacturer’s recommendations. Briefly, genomic DNA samples were fragmented by sonication to a size of ∼350 bp, and then DNA fragments were endpolished, A-tailed, and ligated with the full-length adapter, followed by further PCR amplification. The DNA libraries were sequenced using an Illumina HiSeq X-Ten platform (Illumina, United States), and 150 bp paired-end reads were generated and stored in FASTQ format. Paired reads with more than 10% unidentified nucleotides in either read, with low-quality bases (Phred quality value < 5) over 50%, and with more than 10 bp aligned to the adapter were removed by using Fastp (0.19.7) to obtain clean data.
2.2 CNV and CNVR detection and annotation
The following steps were required before CNV detection: 1) the clean reads of each sample were aligned against the reference genome of Ovis aries (Oar_v4.0) using BWA (Burrows–Wheeler Aligner) (Li and Durbin, 2009) and 2) alignment files were converted to BAM files using SAMtools software (Li et al., 2009). 3) SAMtools was also used to remove potential PCR duplications. If multiple read pairs have identical external coordinates, only the pair with the highest mapping quality is retained. CNVs were detected using CNVcaller with default parameters (-w: 800 bp, -l: 0.2, -u: 0.7, -g: 0.5) (Abyzov et al., 2011). The individual candidate CNV windows are defined using 2 criteria: 1) The normalized read depth (RD) must be significantly higher or lower than the normalized mean RD (deletions < 1–2 * STDEV; duplications > 1 + 2 * STDEV). 2) Considering that the normalized RD of heterozygous deletions and duplications should be approximately 0.5 and 1.5, respectively, an empirical standard for the normalized RD (deletions < 0.65; duplications > 1.35) must also be achieved. Low-frequency windows with low sequencing coverage were removed, and windows with an allele frequency ≥0.05 or at least 3 homozygous duplicated/deleted individuals were selected for further validation. If Pearson’s correlation index was significant at the p = 0.05 level by Student’s t test, these 2 adjacent non-overlapping windows were merged into 1 call. To avoid putative positive CNVRs, the population-level candidate CNVRs for each of the three populations and for TS as a whole was analyzed, respectively. CNV region definition, the distance between the 2 initial calls is less than 20% of their combined length, and the Pearson’s correlation index of the 2 CNVRs is significant at the p = 0.01 level. Furthermore, the distribution of these regions on the Ovis aries chromosome was analyzed by the karyoploteR package of Bioconductor (Gel and Serra, 2017). Functional annotation of CNVs was completed by ANNOVAR(Wang et al., 2010) and classified as intronic, exonic or intergenic.
2.3 Comparison with recent reports
To verify the reliability of our study, we compared the CNVR number and length of this study with three recently reports (Yuan et al., 2021; Huang et al., 2021; Salehian-Dehkordi et al., 2021). All these location information were annotated based on Oar_v4.0 reference genome.
2.4 Functional enrichment analysis of CNVR-harboring genes
CNVR-harboring genes were retrieved from the NCBI database by BioMart software (http://www.biomart.org/), and completely or partially (≥50%) overlapping genes were all reserved for later analysis. GO (Ashburner et al., 2000) and KEGG (Kanehisa and Goto, 2000) functional enrichment analyses were performed according to Huang et al. (Huang et al., 2009). The p value was calculated and subjected to FDR correction. The merged CNVRs were compared with QTLs in the animal QTL database (https://www.animalgenome.org/cgi-bin/QTLdb/OA/index), to further assess the CNVRs that were correlated with economic traits in three ecotypes of Tibetan sheep.
2.5 Sweep selective analysis of the CNVR
Tibetan sheep can be divided into three different ecotypes or populations according to their phenotypes and habitat environment. The three ecotypes have formed their own special characters under selection throughout long-term domestication and adaptation in the plateau. Therefore, the Vst (Redon et al., 2006) was calculated similar to population differentiation index Fst using the equation: Vst = (Vtotal–[Vpop1×Npop1+Vpop2×N pop2]/Ntotal)/Vtotal, where Vtotal is the total variance in LRRs (log-R ratios) of SNPs (within a defined CNVR) estimated among individuals of two populations, Vpop is the variance for each respective population, Npop is the sample size for each respective population, and Ntotal is the total sample size of the two population. Subsequently, the CNVRs with the top five Vst values were selected as candidate CNVRs, and functional enrichment analysis of these regions was performed.
2.6 qPCR and high depth resequencing validation
Quantitative real-time PCR was employed to validate the accuracy of CNVs detected in our study. 11 CNVR-harboring genes representing 5 deletion and 6 duplication types were selected. 9 sheep samples from the three TS populations were used for qPCR validation. The primers were designed using Primer 5 software based on our obtained genomic sequences, and DGAT1 gene was chosen as a reference gene (all primer information were shown in Supplementary Table S2 qPCR was performed using the TB Green PCR reagent kit (Takara Bio). Three replicates per sample and blank controls were required in the PCR. The 2−ΔΔCt method was used to calculate the copy number of the targeted genes [38–40].
3 Results
3.1 The landscape of copy number variation in Tibetan sheep
The total number of raw reads obtained for a single sheep varied between 20,759,076,000 (a highland-type sheep) and 24,338,034,900 (a valley-type sheep), and the high-quality data reached 516.242 GB, with an average of 21,510,086,113 bp per individual. The average depth was 7.10× ± 0.30×, and the coverage rate was 83.72% ± 1.63% at least 4× (Supplementary Table S1).
The CNVs were detected using CNVcaller as described by Wang et al. with default parameters (Wang et al., 2017). A total of 87,832 CNVs were detected in 24 samples (Figure 1). The number of duplication and deletion events was 21,830 and 7479 for HTS, 21,588 and 7079 for OTS, and 21,816 and 8040 for VTS, respectively. The average lengths were 1.96, 1.95 and 1.99 kb (Table 1, Supplementary Table S3). The frequency of duplication events was approximately 3 times higher in all three populations than that of deletions. Moreover, as shown in Figure 1A, more than 86.9% of CNVs were distributed within 0–2 kb, 11.3% were within 2–4 kb, and less than 2% were greater than 4 kb in size.
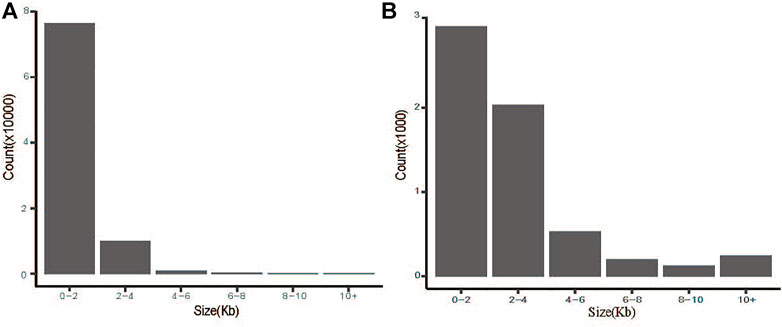
FIGURE 1. Size distribution of CNVs (A) and CNVRs (B) in Tibetan sheep.

TABLE 1. Summary of CNVs and CNVRs identified in three Tibetan sheep populations.
A total of 2777 CNV regions (CNVRs) were obtained by merging overlapping CNVs in the three populations, and the numbers of duplication types in HTS, OTS and VTS were 1575, 1457 and 1484, respectively. Meanwhile, the numbers of deletion types were 521, 548 and 519. The duplication to deletion ratio of CNVRs is consistent with that of the CNVs. The average size of these CNVRs in the three populations was more than 3 kb, accounting for 0.3% of the sheep genome (Oar_v4.0, Table 1). The size distribution of all the CNVRs showed an L-shaped pattern, with 58.2% of the CNVRs located within 1–2 kb, 28.7% within 2–4 kb, 6.5% within 6–8 kb, and others greater than 8 kb in size (Figure 1B). Moreover, as shown by the Venn diagram, 1002 CNVRs were shared by all three populations: approximately 203 CNVRs were distributed uniquely in HTS, 201 CNVRs were distributed in VTS, and 183 CNVRs were distributed in OTS (Figure 2, Supplementary Table S4). The results indicated that these CNVRs were nonuniformly distributed across each chromosome (Figure 3A), and the number of CNVRs had a significant positive linear relationship with the corresponding chromosome size (R2 = 0.66, Figure 3B). More CNVRs were distributed on OARX (1180), OAR1 (162), OAR 3 and OAR 10 (138), and the fewest CNVRs were distributed on OAR 24 (11), as shown in Supplementary Figure S1 and Supplementary Table S5. Functional classification showed that 1839 CNVRs were located in intergenic regions, 812 were contained within introns, and the other 85 were located in exonic regions.
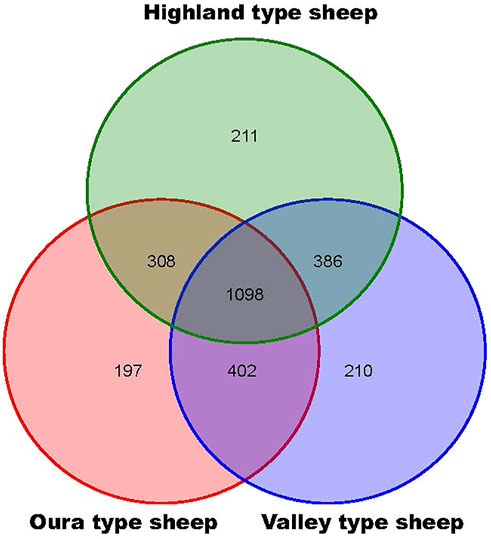
FIGURE 2. Venn diagram of CNVR numbers identified in three Tibetan sheep populations.
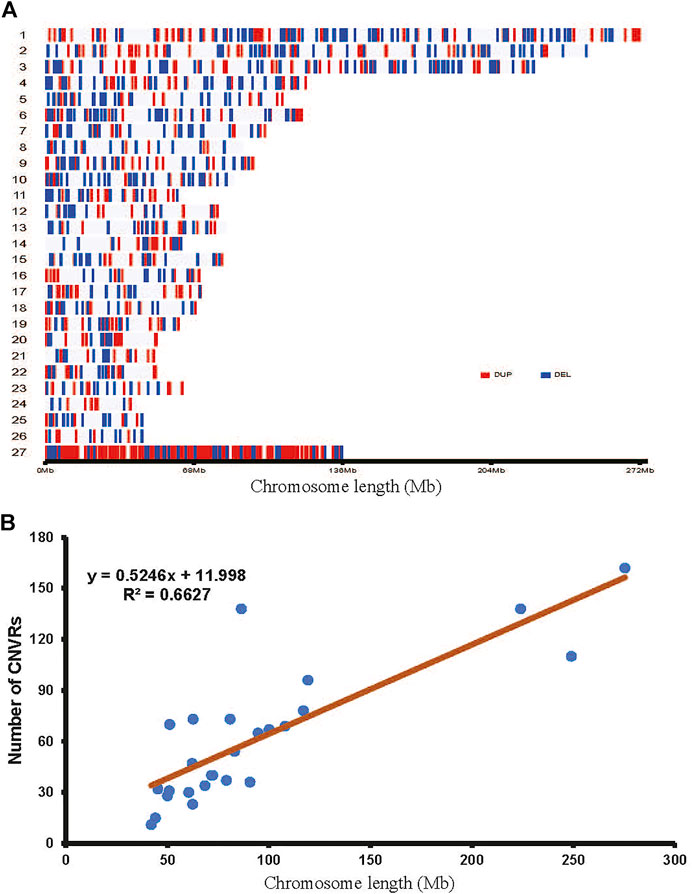
FIGURE 3. Genomic landscape of CNVRs in Tibetan sheep.
3.2 Comparison with recent reports on CNVs in sheep
The results of our study were compared with three recently reported studies on sheep CNVRs. As shown in Table 2, the CNVR count detected in sheep varied notably from 1217 to 24534. Accordingly, the CNVR count overlapping with this study varied from 145 to 421. The average CNVR length was several kb using resequencing platform, but increased to hundreds kb based on BeadChip platform.
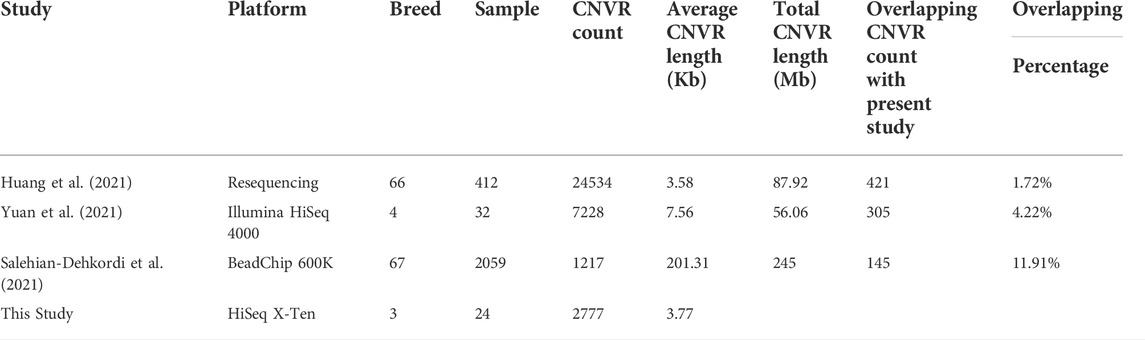
TABLE 2. Comparison of our study with three recent sheep CNV reports.
3.3 Functional annotation of CNVRs
Functional enrichment analysis was performed for all 2777 detected CNVRs. Among the CNVRs shared by the three populations of Tibetan sheep, 56 GO terms (p < 0.01) were enriched, including 23 biological processes, 8 cellular components and 25 molecular functions (Supplementary Table S6). These GO terms involved ion transport (GO:0006811, GO:0006812), sensory perception system (GO:0007600, GO:0050906, and GO:0050907), gas transport (GO:0015669), and pigmentation (GO:0032400, GO:0051875). KEGG pathway analysis showed that the shared CNVR-harboring genes were enriched in 18 pathways (p < 0.05, Supplementary Table S7), including disease defense (ko04612, ko05332, ko05330, ko05320 and ko04672), nutrition metabolism (ko02010, ko00020, and ko00910), hematopoietic cell lineage (ko04640), and ABC transporters (ko02010), among others. In particular, the CNVR-harboring genes specifically distributed in HTS mainly participated in pigmentation (GO:0043473, GO:0033059, GO0048753, GO:0051875, and GO:0032400) processes.
3.4 QTLs overlapping with identified CNVRs
To further reveal the CNVRs associated with sheep economic traits and confirm their hereditary effects, the detected CNVRs were compared with QTLs in the sheep QTL database. We found 188 CNVRs overlapping with 97 quantitative trait loci (QTLs), including milk production and quality (61 CNVRs), fecal egg counts (56 CNVRs), tail fat deposition (21 CNVRs), immunoglobulin A level (20 CNVRs), bone development (14 CNVRs), growth and carcass traits (36 CNVRs) (Supplementary Table S8). Some CNVR-harboring genes related to slaughter performance were uncovered, such as PCDH15 (protocadherin related 15) gene located in body weight (slaughter) QTL (95871), carcass bone percentage QTL (95870), hot carcass weight QTL 95872) and muscle weight in carcass QTL (95853); APP (Amyloid precursor protein) gene located in muscle weight in carcass QTL 95851) and total muscle area QTL (95852). Meanwhile, the ASIP, LOC101111988 and LOC105606907 genes located in the tail fat deposition QTL (127012) were also detected.
3.5 CNVRs diverging among populations
The Vst statistic was used to analyze the population differentiation of CNVRs among HTS, VTS and OTS. This method is similar to Fst in estimating the population-specific selective pressure at the gene level but uses the protein-coding genes annotated by CNVR. The average values of Vst across all of the detected responding CNVRs were 0.1061 for ‘HTS vs. OTS’, 0.099 for ‘HTS vs. VTS’, and 0.096 for ‘VTS vs. OTS’ (Supplementary Table S9). As shown in Figure 4 and Supplementarty Table S9, the divergent CNVRs were distributed unevenly on the chromosome. Eighteen CNVR overlapping genes or loci, including RUNX family transcription factor 1 (RUNX1), glutamate receptor 4 (GRIA4), LOC101104348, LOC105604082, pregnancy-associated glycoproteins 11 (PAG11) and LOC106990378, were the top 1% of genes that showed significant divergence between HTS and VTS (Figure 4A). There were 15 overlapping genes or loci, including RAPGEF1, MED27, LOC105615522, RUNX1, LOC101113153, LOC105607734 and LOC101111988 (located upstream of ASIP), among others, which were the top 1% genes highly differentiated between the VTS and OTS (Figure 4C; Table 3). Among them, only RUNX1, LOC101104348, LOC105604082, PAG11 and LOC101111988 were located in exonic or intronic regions. Unfortunately, few significantly divergent genes located in exonic or intronic regions were detected between HTS and OTS. Notably, RUNX1 was detected in both the ‘HTS vs. VTS’ pairs and ‘VTS vs. OTS’ pairs. The CNV within RUNX1 was duplication type with the length of 2400 bp and located in the intronic region (Table 3). It is worth noting that ASIP, playing a vital role in coat color, also showed significant variation between ‘VTS vs. OTS’ pairs and ‘HTS vs. OTS’ pairs.
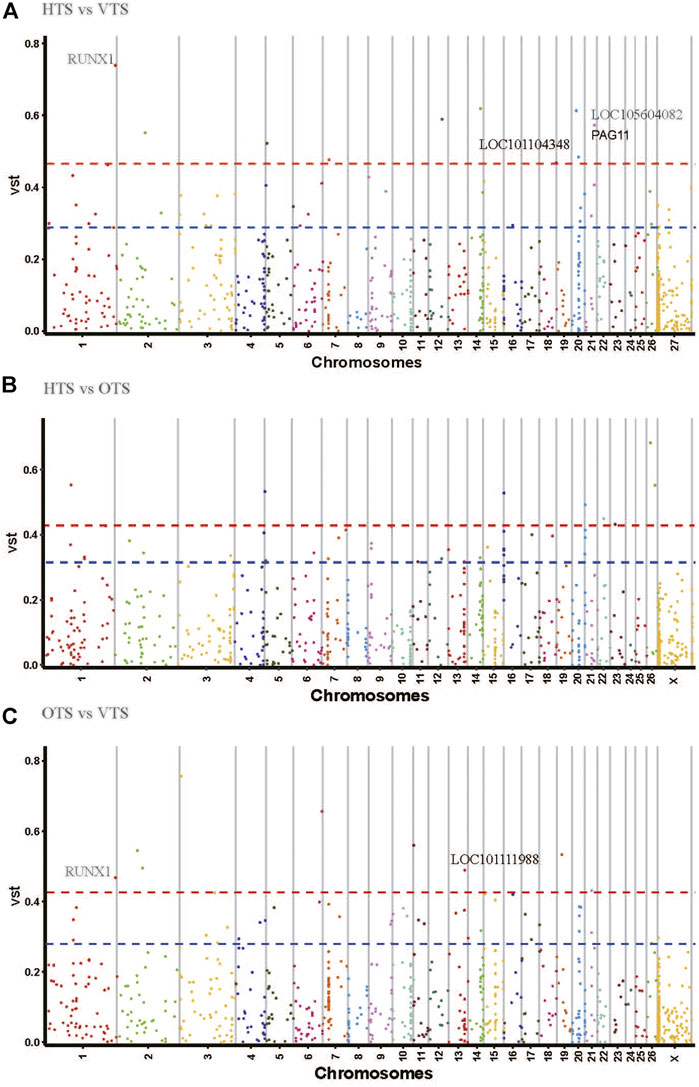
FIGURE 4. Genome wide Vst value plots for CNVRs in three population pairs.

TABLE 3. CNVR-harboring genes showing high divergence in two population pair s.
3.6 CNV validation by qPCR
qPCR was used to verify the accuracy of CNVR predictions. 11 CNVRs including 5 deletions and 6 duplications were selected according to their Vst value (Supplementary Table S9). qPCR results showed that the five deletions (LOC101111526, PLCB1, GUCY1A2, GRIA4, and TMEM144) and six duplications (LOC105602432, RUNX1, PAG11, LOC101113153, and ASIP) were all consistent with the results based on whole genome sequencing prediction (Supplementary Figure S2).
4 Discussion
To date, there are many reports regarding the detection CNVs of sheep using aCGH, SNP array and genome resequencing (Fontanesi et al., 2011a; Jenkins et al., 2016; Di Gerlando et al., 2022). In the present study, we obtained 2777 CNVRs, including 1965 duplications and 812 deletions. The detected CNVR counts is higher than 619 reported by Yang et al. (Yang et al., 2018), comparable to 2394, 3488, and 4301 reported by Guo et al. (Guo et al., 2018), and Cheng et al. (Cheng et al., 2020), but far less than 24,534 claimed by Huang et al. (Huang et al., 2021). The CNVRs accounted for ∼0.3% of the sheep reference genome in our research. This coverage ratio is comparable with previous reports (Ma et al., 2015; Cheng et al., 2020) but lower than the 10% reported by Salehian-Dehkordi et al. (Salehian-Dehkordi et al., 2021). Consistent with the other reports, the number of CNVRs had a significant positive linear relationship with the corresponding chromosome size (Yuan et al., 2021). Meanwhile, the counts of overlapping CNVRs ranged from 145 to 421, which was comparable with Yuan et al. (Yuan et al., 2021). aCGH arrays and SNP chips were all constructed based on limited known probes. The limited probe resolution restricts the top ceiling of the CNV number that can be identified. Besides, they can’t be used to identify new CNVs especially for less studied native breed (Jenkins et al., 2016). Due to the difference in CNV detection platforms, the detected CNVs vary widely in different studies. In terms of the CNVs captured by aCGH and SNP chips, the number usually ranges from dozens to hundreds, and the average length is approximately 100–300 kb (Liu et al., 2013; Ma et al., 2015; Wu et al., 2015; Jenkins et al., 2016; Zhu et al., 2016). In addition to the detection platform and algorithm, the tested breed and individual size used were other considerable factors that results in the CNVRs counts obtained (Sudmant et al., 2015; Yuan et al., 2021; Huang et al., 2021; Prunier et al., 2022). In addition, more deletion evens than duplication were detected in this research, which is similar to Huang, et al. (Huang et al., 2021). But this result is contrary to Yuan et al. (Yuan et al., 2021), that’s may due to CNVcaller were more sensitive in identifying duplication (Wang et al., 2017). Moreover, it may be the characteristic of Tibetan sheep.
The GO enrichment analysis showed that the detected CNVRs harboring genes shared by the three populations were significantly enriched in ion transport, sensory perception system, gas transport and pigmentation. Among them, sensory perception was also significantly enriched in yaks (Qiu et al., 2012; Hui et al., 2019) and other sheep breeds (Ma et al., 2015; Yuan et al., 2021). And this was even observed in Rangifer tarandus caribou, a wild boreal ruminant (Prunier et al., 2022). The three TS populations live in alpine grasslands where the weather conditions are much harsher. As yak, TS faced the similar living conditions especially the lack of herbage in cold season. The well-developed sensory perception system was important to improve their ability to acquire food and avoid noxious weeds (Qiu et al., 2012). In the KEGG pathway analysis, it is noteworthy that the CNVR-harboring genes shared by the three populations were significantly enriched in nutrition metabolism, ABC transporters, disease defense and hematopoietic cell lineage. The enrichment of nutrient metabolism terms, including nitrogen metabolism, citrate cycle, and bile secretion, was important for the digestion of nutrients in TS, especially in the cold season. Similar results showing the enrichment of CNVR-harboring genes in ABC transporters were also reported in humans (Sun et al., 2014; Dron et al., 2018), cattle (Liu et al., 2011; Lee et al., 2013; Upadhyay et al., 2017) and goats (Guan et al., 2020). In mammals, ABC transporters can carry a broad array of endogenous metabolites, such as amino acids, peptides, and sugars, across lipid membranes, which facilitate the absorption and utilization of these nutrients. Overall, the number of enriched CNVR overlapping genes associated with forage intake and consumption may be helpful for their adaptation to the local environment (Allen et al., 2009).
Notably, the olfactory transduction pathway was significantly enriched specifically in OTSs. Enrichment of the olfactory transduction pathway has been reported in cattle Zhang et al., 2015; Gao et al., 2017; Lemos et al., 2018; Mielczarek et al., 2018), yaks (Guang-Xin et al., 2020), sheep (Jiasen et al., 2013; Liu et al., 2013; Yuan et al., 2021) and goats (Guan et al., 2020). It has been revealed to influence food consumption (Ma, 2008) and as a factor to assess feed efficiency and performance in crossbred beef cattle (Abo-Ismail et al., 2014) and residual feed intake in pigs (Do et al., 2014). Meanwhile, more CNVR harboring genes were enriched in amino acid and VFA metabolism pathways in the OTS. In regards to the much better meat performance of OTS than HTS and VTS (Zhao, 2010). More CNVR harboring genes enriched in the olfactory transduction pathway, together with protein and energy metabolism pathways, may explain the better production performance in OTS.
QTLs, which contain genetic variants affecting the economic traits of domestic animals, can be used to select candidate CNV-overlapping genes that affect phenotypes in sheep (Hu et al., 2022). After integrating CNVs into QTLs, we found 188 CNVRs overlapping with 97 sheep QTL regions in this study. Many CNVs overlapping genes, such as PCDH15, APP and GRID2, are located in growth and carcass QTL regions. The PCDH15 gene was identified to be associated with the concentration of the neurotransmitter glutamate (Glu) in cattle (Chen et al., 2020). Zheng et al. indicated that homozygous APP deficiency leads to a 15%–20% reduction in body weight (Hui et al., 1995). An et al. reported that adipocyte-specific and mitochondria-targeted APP-overexpressing mice display increased body mass (An et al., 2019). GRID2 was also identified as being associated with body weight in rats (Keele et al., 2018). So, these identified CNV harboring genes provide candidate molecular associated markers for future sheep breeding.
Selective sweeping can reveal putative regions that undergo environmental and artificial selection during local adaptation and domestication. To screen the critical CNVR significantly divergent between different populations, the pairwise Vst value was estimated (Chen et al., 2020; Salehian-Dehkordi H, 2021; Yuan et al., 2021). Here, the five CNVR harboring genes RUNX1, LOC101104348, LOC105604082, PAG11 and LOC101111988 showed significant pairwise differentiation among HTS, OTS and VTS. RUNX1 has been reported overlapping with a CNV in Chaka sheep (Cheng et al., 2020). It is well known that RUNX1 plays a crucial role in hematopoiesis, leukemogenesis and neural development. Lin et al. reported that GWAS hit SNPs associated with colostrum albumin concentration were enriched in RUNX1 in Chinese Holsteins, and these mutations might initiate the hyperactivation of inflammatory and innate immunity (Lin et al., 2020). In pigs, the GWAS hit SNP within RUNX1 is associated with the mean corpuscular volume level (Lee et al., 2016). Moreover, HIF-1α facilitates RUNX1 transcriptional activity under hypoxic conditions and triggers hematopoietic stem cell differentiation, which ultimately improves oxygen transport to peripheral tissues (Lee et al., 2017). As we know, Chaka sheep is a cultivated breed mainly with Tibetan sheep as crossbreeding ewes and adapted to a low oxygen environment for a long time (Zhao, 2010). Hence, we speculated that CNVR-harboring RUNX1 has undergone strict selection pressure in Tibetan and Chaka sheep, possibly helpful for their adaptation to a low oxygen environment at high altitudes. PAG11 is a pregnancy-associated glycoprotein that is expressed in the trophoblast of the ruminant placenta and influences embryo growth and survival (Shorten et al., 2018). In contrast to VTS, HTS always faces shortage of forage and cold environments in their late pregnancy stage. We thought the divergence of CNVR-harboring PAG11 between HTS and VTS may be related to their different habitat conditions, especially in the pregnancy stage. Notably, CNVR-harboring LOC101111988, located upstream of ASIP, was divergent between VTS vs. OTS (Vst = 0.489) and HTS vs. OTS (Vst = 0.284). The ASIP gene has been widely studied in mammals for its effect on animal coat color. Individuals with normal or duplication alleles of the ASIP gene are generally white or gray coat color, but individuals with normal or single deletion alleles in ASIP almost entirely have solid-black coat color in sheep (Norris and Whan, 2008; Fontanesi et al., 2011b; Han et al., 2015). In our study, the duplication of the LOC101111988 in VTS and HTS might account for their mainly white coat color. Meanwhile, the deletion in LOC101111988 might be the basis for the primary brown covering color, especially in the neck in OTS.
5 Conclusion
In this study, the whole genome characteristics were described in three ecotypic populations of Tibetan sheep. A total of 87,832 CNV events and 2777 CNVRs covering 0.3% of the sheep genome were captured. A few CNVR-harboring genes, such as PCDH15, APP, RUNX1, PAG11, and ASIP, were uncovered and may be associated with body weight, environmental adaptation, fertility, and coat color. Above all, our results provide a valuable genome-wide variation resource in Tibetan sheep for the elucidation of the genetic basis underlying the distinct phenotypic traits and local adaptation of Tibetan sheep.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://ngdc.cncb.ac.cn/, CRA005573.
Ethics statement
The animal study was reviewed and approved by Ethical Committee of Northwest Institute of Plateau Biology, CAS. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author contributions
LH, SX, XZ and CZ initiated the study and designed the project. LH, HL, TX, NZ and XH collected the samples. LH, LZ, QL, and CZ performed the bioinformatics and experimental analysis. LH, QL and CZ summarized the results. LH and CZ wrote the manuscript. All authors read and approved the final manuscript.
Funding
This study was supported in part by The Second Tibetan Plateau Scientific Expedition and Research (STEP) program (Grant No. 2019QZKK0302), Key R & D Project of Qinghai Province (2020-NK-166), and Basic Applied Study of Qinghai Province (2014-ZJ-710).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.971464/full#supplementary-material
References
Abo-Ismail, M. K., Voort, G. V., Squires, J. J., Swanson, K. C., Mandell, I. B., Liao, X. P., et al. (2014). Single nucleotide polymorphisms for feed efficiency and performance in crossbred beef cattle. BMC Genet. 15, 14. doi:10.1186/1471-2156-15-14
Abyzov, A., Urban, A. E., Snyder, M., and Gerstein, M. (2011). CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 21 (6), 974–984. doi:10.1101/gr.114876.110
Allen, M. S., Bradford, B. J., and Oba, M. (2009). Board Invited Review: The hepatic oxidation theory of the control of feed intake and its application to ruminants. J. Anim. Sci. 87 (10), 3317–3334. doi:10.2527/jas.2009-1779
An, Y. A., Crewe, C., Asterholm, I. W., Sun, K., Chen, S., Zhang, F., et al. (2019). Dysregulation of amyloid precursor protein impairs adipose tissue mitochondrial function and promotes obesity. Nat. Metab. 1 (12), 1243–1257. doi:10.1038/s42255-019-0149-1
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat. Genet. 25 (1), 25–29. doi:10.1038/75556
Bickhart, D. M., and Liu, G. E. (2014). The challenges and importance of structural variation detection in livestock. Front. Genet. 5, 37. doi:10.3389/fgene.2014.00037
Chen, J., Lee, M. O., and Womack, J. E. (2017a). Genetic variation and gene conversions within the bovine NK-lysin gene family. Anim. Genet. 48 (2), 225–227. doi:10.1111/age.12521
Chen, L., Chamberlain, A. J., Reich, C. M., Daetwyler, H. D., and Hayes, B. J. (2017b). Detection and validation of structural variations in bovine whole-genome sequence data. Genet. Sel. Evol. 49 (1), 13. doi:10.1186/s12711-017-0286-5
Chen, Q., Qu, K., Ma, Z., Zhan, J., Lei, C., Shen, J., et al. (2020). Genome-wide association study identifies genomic loci associated with neurotransmitter concentration in cattle. Front. Genet. 11, 139. doi:10.3389/fgene.2020.00139
Cheng, J., Cao, X., Hanif, Q., Pi, L., Hu, L., Huang, Y., et al. (2020). Integrating genome-wide CNVs into QTLs and high confidence GWAScore regions identified positional candidates for sheep economic traits. Front. Genet. 11, 569. doi:10.3389/fgene.2020.00569
de Lemos, M. V. A., Peripolli, E., Berton, M. P., Braga Feitosa, F. L., Olivieri, B. F., Stafuzza, N. B., et al. (2018a). Association study between copy number variation and beef fatty acid profile of Nellore cattle. J. Appl. Genet. 59 (2), 203–223. doi:10.1007/s13353-018-0436-7
de Lemos, M. V. A., Peripolli, E., Berton, M. P., Feitosa, F. L. B., Olivieri, B. F., Stafuzza, N. B., et al. (2018b). Association study between copy number variation and beef fatty acid profile of Nellore cattle. J. Appl. Genet. 59 (2), 203–223. doi:10.1007/s13353-018-0436-7
Di Gerlando, R., Mastrangelo, S., Tolone, M., Rizzuto, I., Sutera, A. M., Moscarelli, A., et al. (2022). Identification of copy number variations and genetic diversity in Italian insular sheep breeds. Animals. 12 (2), 217. doi:10.3390/ani12020217
Do, D. N., Strathe, A. B., Ostersen, T., Pant, S. D., and Kadarmideen, H. N. (2014). Genome-wide association and pathway analysis of feed efficiency in pigs reveal candidate genes and pathways for residual feed intake. Front. Genet. 5, 307. doi:10.3389/fgene.2014.00307
Dron, J. S., Wang, J., Berberich, A. J., Iacocca, M. A., Cao, H., Yang, P., et al. (2018). Large-scale deletions of the ABCA1 gene in patients with hypoalphalipoproteinemia. J. Lipid Res. 59 (8), 1529–1535. doi:10.1194/jlr.P086280
Feng, Z., Li, X., Cheng, J., Jiang, R., Huang, R., Wang, D., et al. (2020). Copy number variation of the PIGY gene in sheep and its association analysis with growth traits. Animals. 10 (4), 688. doi:10.3390/ani10040688
Feuk, L., Carson, A. R., and Scherer, S. W. (2006). Structural variation in the human genome. Nat. Rev. Genet. 7 (2), 85–97. doi:10.1038/nrg1767
Fontanesi, L., Beretti, F., Martelli, P. L., Colombo, M., Dall'Olio, S., Occidente, M., et al. (2011a). A first comparative map of copy number variations in the sheep genome. Genomics 97 (3), 158–165. doi:10.1016/j.ygeno.2010.11.005
Fontanesi, L., Dall'Olio, S., Beretti, F., Portolano, B., and Russo, V. (2011b). Coat colours in the Massese sheep breed are associated with mutations in the agouti signalling protein (ASIP) and melanocortin 1 receptor (MC1R) genes. Animal 5 (1), 8–17. doi:10.1017/s1751731110001382
Gao, Y., Jiang, J., Yang, S., Hou, Y., Liu, G. E., Zhang, S., et al. (2017). CNV discovery for milk composition traits in dairy cattle using whole genome resequencing. BMC Genomics 18 (1), 265. doi:10.1186/s12864-017-3636-3
Gel, B., and Serra, E. (2017). karyoploteR: an R/Bioconductor package to plot customizable genomes displaying arbitrary data. Bioinformatics 33 (19), 3088–3090. doi:10.1093/bioinformatics/btx346
Goyache, F., Fernandez, I., Tapsoba, A. S. R., Traore, A., Menendez-Arias, N. A., and Alvarez, I. (2021). Functional characterization of copy number variations regions in djallonke sheep. J. Animal Breed. Genet. 138 (5), 600–612. doi:10.1111/jbg.12542
Guan, D., Martínez, A., Castelló, A., Landi, V., Amills, M., Fernandez-Alvarez, J., et al. (2020). A genome-wide analysis of copy number variation in Murciano-Granadina goats. Genet. Sel. Evol. 52 (1), 44. doi:10.1186/s12711-020-00564-4
Guang-Xin, E., Yang, B. G., Zhu, Y. B., Duang, X. H., An, T. W., Luo, X. L., et al. (2020). Genome-wide selective sweep analysis of the high-altitude adaptability of yaks by using the copy number variant. 3 Biotech. 10 (6), 259. doi:10.1007/s13205-020-02254-w
Guo, J., Tao, H., Li, P., Li, L., Zhong, T., Wang, L., et al. (2018). Whole-genome sequencing reveals selection signatures associated with important traits in six goat breeds. Sci. Rep. 8 (1), 10405. doi:10.1038/s41598-018-28719-w
Han, J. L., Yang, M., Yue, Y. J., Guo, T. T., Liu, J. B., Niu, C. E., et al. (2015). Analysis of agouti signaling protein (ASIP) gene polymorphisms and association with coat color in Tibetan sheep (Ovis aries). Genet. Mol. Res. 14 (1), 1200–1209. doi:10.4238/2015.February.6.22
Hu, Z.-L., Park, C. A., and Reecy, J. M. (2022). Bringing the animal QTLdb and CorrDB into the future: Meeting new challenges and providing updated services. Nucleic Acids Res. 50 (D1), D956–D961. doi:10.1093/nar/gkab1116
Huang, D. W., Sherman, B. T., and Lempicki, R. A. (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4 (1), 44–57. doi:10.1038/nprot.2008.211
Huang, Y., Li, Y., Wang, X., Yu, J., Cai, Y., Zheng, Z., et al. (2021). An atlas of CNV maps in cattle, goat and sheep. Sci. China. Life Sci. 64 (10), 1747–1764. doi:10.1007/s11427-020-1850-x
Hui, W., Chai, Z., Dan, H., Ji, Q., Zhong, J., Zhang, C., et al. (2019). A global analysis of CNVs in diverse yak populations using whole-genome resequencing. BMC Genomics 20 (1), 61. doi:10.1186/s12864-019-5451-5
Hui, Z., Jiang, M., Trumbauer, M. E., Sirinathsinghji, D., Ploeg, L., Smith, D. W., et al. (1995). beta-Amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell 81 (4), 525–531. doi:10.1016/0092-8674(95)90073-x
Iafrate, A. J., Feuk, L., Rivera, M. N., Listewnik, M. L., Donahoe, P. K., Qi, Y., et al. (2004). Detection of large-scale variation in the human genome. Nat. Genet. 36 (9), 949–951. doi:10.1038/ng1416
Jenkins, G. M., Goddard, M. E., Black, M. A., Brauning, R., Auvray, B., Dodds, K. G., et al. (2016). Copy number variants in the sheep genome detected using multiple approaches. BMC Genomics 17, 441. doi:10.1186/s12864-016-2754-7
Jiang, R., Cheng, J., Cao, X.-K., Ma, Y.-L., Chaogetu, B., Huang, Y.-Z., et al. (2019). Copy number variation of the SHE gene in sheep and its association with economic traits. Animals. 9 (8), 531. doi:10.3390/ani9080531
Jiasen, L., Li, Z., Lingyang, X., Ren, H., Lu, J., Zhang, X., et al. (2013). Analysis of copy number variations in the sheep genome using 50K SNP BeadChip array. BMC Genomics 14 (1), 229. doi:10.1186/1471-2164-14-229
Kanehisa, M., and Goto, S. (2000). KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28 (1), 27–30. doi:10.1093/nar/28.1.27
Keele, G. R., Prokop, J. W., He, H., Holl, K., Littrell, J., Deal, A., et al. (2018). Genetic fine-mapping and identification of candidate genes and variants for adiposity traits in outbred rats. Obesity 26 (1), 213–222. doi:10.1002/oby.22075
Lee, J.-B., Yoo, C.-K., Park, H.-B., Cho, I.-C., and Lim, H.-T. (2016). Association of the single nucleotide polymorphisms in RUNX1, DYRK1A, and KCNJ15 with blood related traits in pigs. Asian-Australas. J. Anim. Sci. 29 (12), 1675–1681. doi:10.5713/ajas.16.0348
Lee, K. T., Chung, W. H., Lee, S. Y., Choi, J. W., Kim, J., Lim, D., et al. (2013). Whole-genome resequencing of Hanwoo (Korean cattle) and insight into regions of homozygosity. BMC Genomics 14, 519. doi:10.1186/1471-2164-14-519
Lee, S. H., Manandhar, S., and Lee, Y. M. (2017). Roles of RUNX in hypoxia-induced responses and angiogenesis. Adv. Exp. Med. Biol. 962, 449–469. doi:10.1007/978-981-10-3233-2_27
Lemos, M. D., Peripolli, E., Berton, M. P., Feitosa, F., Olivieri, B. F., Stafuzza, N. B., et al. (2018). Association study between copy number variation and beef fatty acid profile of Nellore cattle. J. Appl. Genet. 59 (2), 203–223. doi:10.1007/s13353-018-0436-7
Letaief, R., Rebours, E., Grohs, C., Meersseman, C., Fritz, S., Trouilh, L., et al. (2017). Identification of copy number variation in French dairy and beef breeds using next-generation sequencing. Genet. Sel. Evol. 49 (1), 77. doi:10.1186/s12711-017-0352-z
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25 (14), 1754–1760. doi:10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25 (16), 2078–2079. doi:10.1093/bioinformatics/btp352
Lin, S., Wan, Z., Zhang, J., Xu, L., and Sun, D. (2020). Genome-wide association studies for the concentration of albumin in colostrum and serum in Chinese holstein. Animals. 10 (12), 2211. doi:10.3390/ani10122211
Liu, G. E., Brown, T., Hebert, D. A., Cardone, M. F., Hou, Y., Choudhary, R. K., et al. (2011). Initial analysis of copy number variations in cattle selected for resistance or susceptibility to intestinal nematodes. Mamm. Genome 22 (1-2), 111–121. doi:10.1007/s00335-010-9308-0
Liu, J., Zhang, L., Xu, L., Ren, H., Lu, J., Zhang, X., et al. (2013). Analysis of copy number variations in the sheep genome using 50K SNP BeadChip array. BMC Genomics 14, 229. doi:10.1186/1471-2164-14-229
Lye, Z. N., and Purugganan, M. D. (2019). Copy number variation in domestication. Trends Plant Sci. 24 (4), 352–365. doi:10.1016/j.tplants.2019.01.003
Ma, M. (2008). Encoding olfactory signals via multiple chemosensory systems. Crit. Rev. Biochem. Mol. Biol. 42 (6), 463–480. doi:10.1080/10409230701693359
Ma, Q., Liu, X., Pan, J., Ma, L., Ma, Y., He, X., et al. (2017). Genome-wide detection of copy number variation in Chinese indigenous sheep using an ovine high-density 600 K SNP array. Sci. Rep. 7 (1), 912. doi:10.1038/s41598-017-00847-9
Ma, Y., Zhang, Q., Lu, Z., Zhao, X., and Zhang, Y. (2015). Analysis of copy number variations by SNP50 BeadChip array in Chinese sheep. Genomics 106 (5), 295–300. doi:10.1016/j.ygeno.2015.08.001
Merot, C., Oomen, R. A., Tigano, A., and Wellenreuther, M. (2020). A roadmap for understanding the evolutionary significance of structural genomic variation. Trends Ecol. Evol. 35 (7), 561–572. doi:10.1016/j.tree.2020.03.002
Mielczarek, M., Fraszczak, M., Nicolazzi, E., Williams, J. L., and Szyda, J. (2018). Landscape of copy number variations in Bos taurus: Individual - and inter-breed variability. BMC Genomics 19 (1), 410. doi:10.1186/s12864-018-4815-6
Norris, B. J., and Whan, V. A. (2008). A gene duplication affecting expression of the ovine ASIP gene is responsible for white and black sheep. Genome Res. 18 (8), 1282–1293. doi:10.1101/gr.072090.107
Pierce, M. D., Dzama, K., and Muchadeyi, F. C. (2018). Genetic diversity of seven cattle breeds inferred using copy number variations. Front. Genet. 9, 163. doi:10.3389/fgene.2018.00163
Prunier, J., Carrier, A., Gilbert, I., Poisson, W., Albert, V., Taillon, J., et al. (2022). CNVs with adaptive potential in Rangifer tarandus: genome architecture and new annotated assembly. Life Sci. Alliance 5 (3), e202101207. doi:10.26508/lsa.202101207
Qiu, Q. Z., Zhang, G., Ma, T., Qian, W., Wang, J., Ye, Z., et al. (2012). The yak genome and adaptation to life at high altitude. Nat. Genet. 44 (8), 946–949. The yak genome and adaptation to life at high altitude. doi:10.1038/ng.2343
Redon, R., Ishikawa, S., Fitch, K. R., Feuk, L., Perry, G. H., Andrews, T. D., et al. (2006). Global variation in copy number in the human genome. Nature 444 (7118), 444–454. doi:10.1038/nature05329
Salehian-Dehkordi, H., Xu, Y.-X., Xu, S.-S., Li, X., Luo, L.-Y., Liu, Y.-J., et al. (2021). Genome-wide detection of copy number variations and their association with distinct phenotypes in the world's sheep. Front. Genet. 12, 670582. doi:10.3389/fgene.2021.670582
Sasaki, S., Watanabe, T., Nishimura, S., and Sugimoto, Y. (2016). Genome-wide identification of copy number variation using high-density single-nucleotide polymorphism array in Japanese Black cattle. BMC Genet. 17, 26. doi:10.1186/s12863-016-0335-z
Shorten, P. R., Ledgard, A. M., Donnison, M., Pfeffer, P. L., Mcdonald, R. M., and Berg, D. K. (2018). A mathematical model of the interaction between bovine blastocyst developmental stage and progesterone-stimulated uterine factors on differential embryonic development observed on Day 15 of gestation. J. Dairy Sci. 101 (1), 736–751. doi:10.3168/jds.2017-12845
Strillacci, M. G., Marelli, S. P., Milanesi, R., Zaniboni, L., Punturiero, C., and Cerolini, S. (2021). Copy number variants in four Italian Turkey breeds. Animals. 11 (2), 391. doi:10.3390/ani11020391
Sudmant, P. H., Mallick, S., Nelson, B. J., Hormozdiari, F., Krumm, N., Huddleston, J., et al. (2015). Global diversity, population stratification, and selection of human copy-number variation. Science 349 (6253), aab3761. doi:10.1126/science.aab3761
Sun, Y., Shi, N., Lu, H., Zhang, J., Ma, Y., Qiao, Y., et al. (2014). ABCC4 copy number variation is associated with susceptibility to esophageal squamous cell carcinoma. Carcinogenesis 35 (9), 1941–1950. doi:10.1093/carcin/bgu043
Upadhyay, M., da Silva, V. H., Megens, H. J., Visker, M., Ajmone-Marsan, P., Balteanu, V. A., et al. (2017). Distribution and functionality of copy number variation across European cattle populations. Front. Genet. 8, 108. doi:10.3389/fgene.2017.00108
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38 (16), e164. doi:10.1093/nar/gkq603
Wang, M. D., Dzama, K., Rees, D. J. G., and Muchadeyi, F. C. (2016). Tropically adapted cattle of Africa: Perspectives on potential role of copy number variations. Anim. Genet. 47 (2), 154–164. doi:10.1111/age.12391
Wang, X., Zheng, Z., Cai, Y., Chen, T., Li, C., Fu, W., et al. (2017). CNVcaller: Highly efficient and widely applicable software for detecting copy number variations in large populations. Gigascience 6 (12), 1–12. doi:10.1093/gigascience/gix115
Wu, Kai-Feng, Zhou, Huan-Min, Zhang, Dong, Wang, S. Y., Xing, Y. P., Cao, J. W., et al. (2015). Genome-wide analysis of copy number variations in Chinese sheep using array comparative genomic hybridization. Small Ruminant Res. 128, 19–26. doi:10.1016/j.smallrumres.2015.04.014
Xu, Y., Jiang, Y., Shi, T., Cai, H., Lan, X., Zhao, X., et al. (2017). Whole-genome sequencing reveals mutational landscape underlying phenotypic differences between two widespread Chinese cattle breeds. PLoS One 12 (8), e0183921. doi:10.1371/journal.pone.0183921
Yang, L., Xu, L., Zhou, Y., Liu, M., Wang, L., Kijas, J. W., et al. (2018). Diversity of copy number variation in a worldwide population of sheep. Genomics 110 (3), 143–148. doi:10.1016/j.ygeno.2017.09.005
Yang, Z., Cao, X., Ma, Y., Cheng, J., Song, C., Jiang, R., et al. (2021). Novel copy number variation of the BAG4 gene is associated with growth traits in three Chinese sheep populations. Anim. Biotechnol. 32 (4), 461–469. doi:10.1080/10495398.2020.1719124
Yuan, C., Lu, Z., Guo, T., Yue, Y., Wang, X., Wang, T., et al. (2021). A global analysis of CNVs in Chinese indigenous fine-wool sheep populations using whole-genome resequencing. BMC Genomics 22 (1), 78. doi:10.1186/s12864-021-07387-7
Zhang, Q., Ma, Y., Wang, X., Zhang, Y., and Zhao, X. (2015). Identification of copy number variations in Qinchuan cattle using BovineHD Genotyping Beadchip array. Mol. Genet. Genomics 290 (1), 319–327. doi:10.1007/s00438-014-0923-4
Zhang, X., Wang, K., Wang, L., Yang, Y., Ni, Z., Xie, X., et al. (2016). Genome-wide patterns of copy number variation in the Chinese yak genome. BMC Genomics 17, 379. doi:10.1186/s12864-016-2702-6
Zhou, Y., Utsunomiya, Y. T., Xu, L., Hay, E. H. A., Bickhart, D. M., Alexandre, P. A., et al. (2016a). Genome-wide CNV analysis reveals variants associated with growth traits in Bos indicus. BMC Genomics 17, 419. doi:10.1186/s12864-016-2461-4
Zhou, Y., Utsunomiya, Y. T., Xu, L., Hay el, H. A., Bickhart, D. M., Sonstegard, T. S., et al. (2016b). Comparative analyses across cattle genders and breeds reveal the pitfalls caused by false positive and lineage-differential copy number variations. Sci. Rep. 6, 29219. doi:10.1038/srep29219
Keywords: copy number variations, Tibetan sheep, whole genome resequencing, adaptation, domestication
Citation: Hu L, Zhang L, Li Q, Liu H, Xu T, Zhao N, Han X, Xu S, Zhao X and Zhang C (2022) Genome-wide analysis of CNVs in three populations of Tibetan sheep using whole-genome resequencing. Front. Genet. 13:971464. doi: 10.3389/fgene.2022.971464
Received: 17 June 2022; Accepted: 23 August 2022;
Published: 07 September 2022.
Edited by:
Wendy Mercedes Rauw, Spanish National Research Council (CSIC), SpainReviewed by:
Annelin Molotsi, Stellenbosch University, South AfricaHosein Salehian Dehkordi, Chinese Academy of Sciences, China
Copyright © 2022 Hu, Zhang, Li, Liu, Xu, Zhao, Han, Xu, Zhao and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cunfang Zhang, zcf_1023@126.com