 Jun Kido
Jun Kido Keishin Sugawara2
Keishin Sugawara2- 1Department of Pediatrics, Kumamoto University Hospital, Kumamoto, Japan
- 2Department of Pediatrics, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan
Ornithine transcarbamylase deficiency (OTCD) is an X-linked disorder. Several male patients with OTCD suffer from severe hyperammonemic crisis in the neonatal period, whereas others develop late-onset manifestations, including hyperammonemic coma. Females with heterozygous pathogenic variants in the OTC gene may develop a variety of clinical manifestations, ranging from asymptomatic conditions to severe hyperammonemic attacks, owing to skewed lyonization. We reported the variants of CPS1, ASS, ASL and OTC detected in the patients with urea cycle disorders through a nation-wide survey in Japan. In this study, we updated the variant data of OTC in Japanese patients and acquired information regarding genetic variants of OTC from patients with OTCD through an extensive literature review. The 523 variants included 386 substitution (330 missense, 53 nonsense, and 3 silent), eight deletion, two duplication, one deletion-insertion, 55 frame shift, two extension, and 69 no category (1 regulatory and 68 splice site error) mutations. We observed a genotype–phenotype relation between the onset time (neonatal onset or late onset), the severity, and genetic mutation in male OTCD patients because the level of deactivation of OTC significantly depends on the pathogenic OTC variants. In conclusion, genetic information about OTC may help to predict long-term outcomes and determine specific treatment strategies, such as liver transplantation, in patients with OTCD.
Introduction
Ornithine transcarbamylase (OTC; EC 2.1.3.3) is a mitochondrial enzyme that catalyzes the synthesis of citrulline from carbamoyl phosphate and ornithine during the urea cycle; inorganic phosphate is released as a by-product of the reaction. It is essential for the conversion of neurotoxic ammonia into non-toxic urea. In humans, OTC is exclusively expressed in the liver and small intestinal mucosa; however, it functions only in the liver during the urea cycle. The human OTC gene, which is 73 kb long and comprises 10 exons and nine introns (Hata et al., 1988), is located on the short arm of the X chromosome within band Xp21.1 (Lindgren et al., 1984). It encodes a precursor OTC protein that has a molecular weight of 39.9 kD and is composed of 354 amino acids. Upon entering the mitochondria, it undergoes post-transcriptional modification in which the 32 amino acid-long leader sequence is cleaved in two successive steps (Horwich et al., 1986). The mature OTC peptide has a molecular weight of 36.1 kD and is composed of 322 amino acids. The functional OTC holoenzyme is a homotrimer with a three-fold symmetry and three active sites, each of which is shared between two adjacent polypeptides (Shi et al., 1998).
The OTC deficiency (OTCD; MIM number: 311,250) is an X-linked disorder. Incidentally, the estimated frequency of OTCD is 1 per 80,000 births in Japan (Nagata et al., 1991), and recent studies indicate a prevalence of 1 per 62,000–77,000 births worldwide (Dionisi-Vici et al., 2002; Keskinen et al., 2008; Balasubramaniam et al., 2010; Summar et al., 2013). The OTCD phenotype is extremely heterogeneous. For instance, many male OTCD patients have severe hyperammonemic crisis in the neonatal stage, whereas others develop late-onset manifestations, including hyperammonemic coma (Kido et al., 2012; Kido et al., 2021a; Kido et al., 2021b). On the contrary, females with heterozygous pathogenic variants in the OTC gene may develop a variety of clinical manifestations, ranging from an asymptomatic condition to severe hyperammonemic attack, owing to the skewed lyonization phenomenon. Incidentally, the cloning of the human OTC gene has helped in the identification of mutations, most of which are “private” mutations (Yamaguchi et al., 2006). Majority of the mutation analysis may have been performed using PCR amplification of exons and flanking regions, followed by Sanger sequencing. In about 10%–15% of patients with clinically proven OTCD, no identifiable mutations have been detected in the routine molecular testing. In these patients, large deletions, duplications, and complex rearrangements associated with OTC or mutations in the promoter and enhancer region has been reported (Shchelochkov et al., 2009; Jang et al., 2018).
In the previous study (Kido et al., 2021d), we reported the variants of CPS1, ASS, ASL, and OTC detected in the patients with urea cycle disorders through a nation-wide survey in Japan and suggested that the onset time and severity in Japanese patients with OTCD can be estimated based on the type of OTC gene variant that they carry, thereby demonstrating a genotype–phenotype correlation in OTCD. In this study, we acquired information regarding 523 gene variants in patients with OTCD through a nationwide study in Japan and simultaneous literature review. Herein, we present our observations from the study and review. We also discuss the genotype–phenotype relationship and the clinical significance of these variants.
Material and methods
Previously, we had conducted nation-wide surveys on Japanese patients with urea cycle disorders (UCDs), such as OTCD, carbamoyl phosphate synthetase 1 deficiency, N-acetylglutamate synthase deficiency, argininosuccinate synthetase deficiency, argininosuccinate lyase deficiency, and arginase 1 deficiency (Kido et al., 2021c; 2021a; 2021b; 2021d). In the current survey, we acquired the clinical data of 128 patients with OTCD (73 males and 55 females), including genetic information of 62 of them (57 families). These patients were diagnosed and/or treated in different departments, including pediatrics, neonatology, endocrinology and metabolism, genetics, and transplant surgery, from 78 different hospitals between January 2000 and March 2018. Additionally, we acquired the clinical data of patients diagnosed with OTCD in our institution as well.
As part of the literature review, we surveyed the genetic information of OTCD patients available on PubMed (https://pubmed.ncbi.nlm.nih.gov) or Google Scholar (https://scholar.google.com) using the keywords “OTC mutation” and “OTCD mutation.” Moreover, we surveyed variants in the OTC gene by quoting exact words/phrases/statements from related papers (Yamaguchi et al., 2006; Caldovic et al., 2015; Choi et al., 2015). We also evaluated variants of OTCD patients reported in 112 papers.
Variant nomenclature followed the guidelines established by the Human Genome Variation Society (http://varnomen.hgvs.org/) (den Dunnen et al., 2016), and the variants were categorized by protein level descriptions. The public database ClinVar (http://www.ncbi.nlm.nih.gov/clinvar) (Landrum et al., 2020) was used for the classification of each variant. Bioinformatic tools, PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2) (Adzhubei et al., 2010) and SIFT (http://provean.jcvi.org/protein_batch_submit.php?species=human) (Choi et al., 2012) were used for predicting the potential impact of an amino acid alteration in missense mutations on the function of OTC.
Ethics statement
This study was approved by the ethical committee of the Faculty of Life Science, Kumamoto University (Ethics. No.1527). All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all patients or their legal guardians for being included in the study.
Results
We acquired information regarding 523 genetic variants of OTCD patients through additional nation-wide survey conducted in Japan, as well as through a review of the existing relevant literature. These variants in the OTC gene included 386 substitution (330 missense, 53 nonsense, and 3 silent), eight deletion, two duplication, one deletion-insertion, 55 frame shift, two extension, and 69 no category (1 regulatory, 68 splice site error) mutations (Table 1; Supplementary data 1–3). Table 1 and Supplementary data 1 depicts the onset time of the OTCD symptoms and the maximum blood ammonia concentrations for each variant of the OTCD patients.
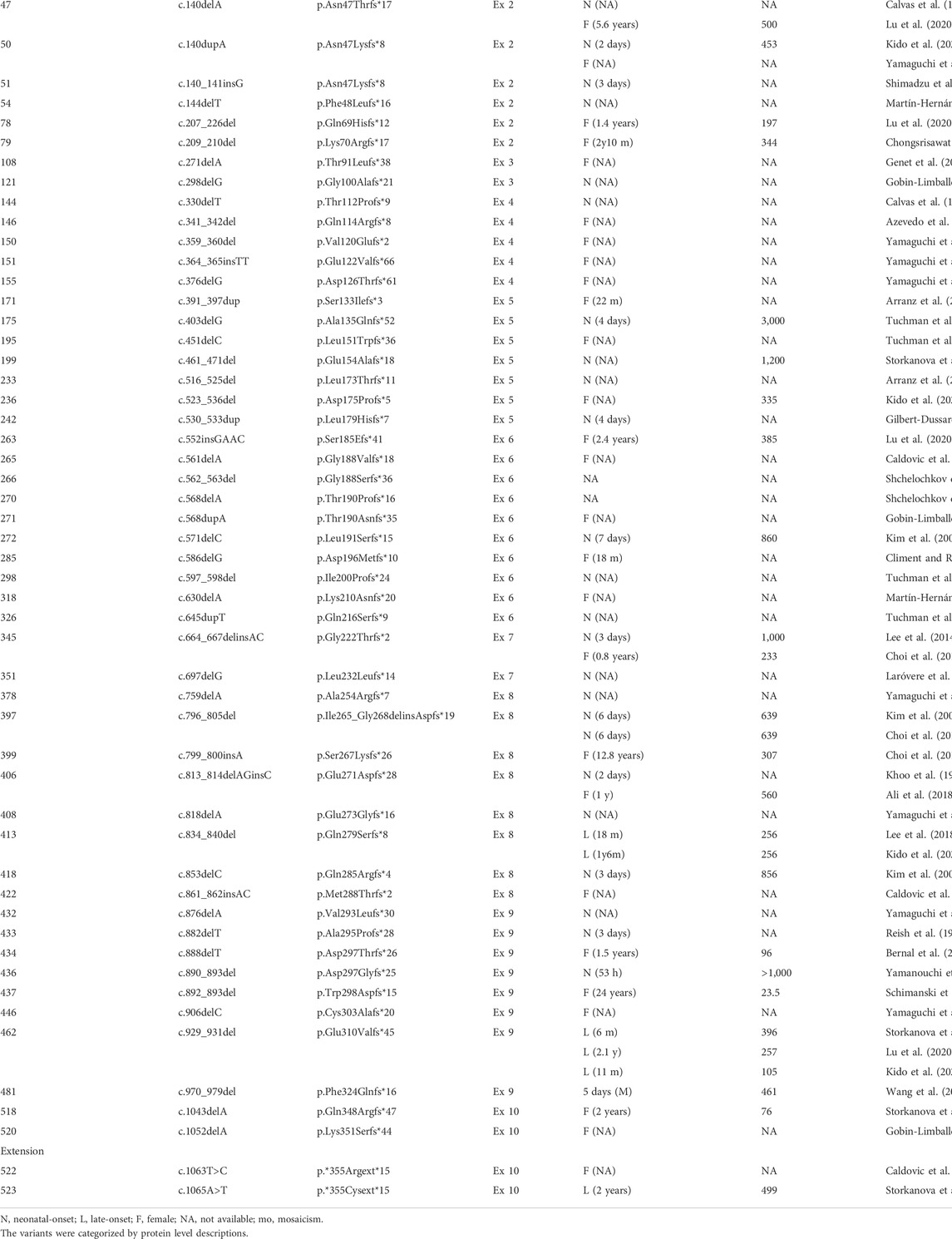
TABLE 1. Variants in the OTC gene and phenotype.
Among the missense variants, 108 variants have been identified in the male patients with neonatal onset of OTCD, while 81 variants have been identified in the male patients with late onset of OTCD. Eleven variants, namely the c.119G>A (p.Arg40His), c.304G>C (p.Ala102Pro), c.386G>A (p.Arg129His), c.481A>G (p.Asn161Asp), c.535C>T (p.Leu179Phe), c.540G>C (p.Gln180His), c.562G>C (p.Gly188Arg), c.725C>T (p.Thr242Ile), c.803T>C (p.Met268Thr), c.829C>T (p.Arg177Trp), and c.1028C>G (p.Thr343Arg), have been identified in case of both neonatal and late onset male OTCD patients. Additionally, the c.128T>C (p.Leu43Pro), c.530T>G (p.Leu177Arg), c.628A>C (p.Lys210Gln), and c.1025T>G (p.Leu342Pro) variants have been identified in female patients with neonatal onset of OTCD.
All nonsense variants detected in the male OTCD patients have been identified as the neonatal-onset type variant. Additionally, two silent variants, namely c.663G>A (p.Lys221=) and c.867G>A (p.Lys289=); three frame shift variants, specifically c.834_840delCCAGGCT (p.Gln279Serfs*8), c.929_931delAAG (p.Glu310Valfs*45), and c.1065A>T (p.*355Cysext*14); and two deletion/duplication variants, namely c.817_819delGAG (p.Glu273del) and c.928_930delGAA (p.Glu310del), have been identified in the late onset male OTCD patients.
The splicing-disrupting variants in introns 2, 3, 8, and 9 have been identified in case of both neonatal and late onset male OTCD patients. All splicing-disrupting variants in introns 5, 6, and 7 have been identified in the male patients with neonatal onset of OTCD. Moreover, although majority of the splicing-disrupting variants identified in introns 1 and 4 are specific for the male patients with neonatal onset of OTCD, only three variants, namely c.78-2A>G variant in intron 1 and c.386 + 1G>T, as well as c.386+4delT in intron 4, are specific for the late onset male OTCD patients.
Figure 1 demonstrates the amino acid substitutions in the OTC gene as detected in patients with OTCD. Incidentally, amino acid substitutions in exons 5 or 6 and in the α-helix or β-sheet structures are likely to result in neonatal onset of OTCD. Moreover, amino acid substitutions in positions 40, 52, 53, 59, 100, 102, 129, 158, 172, 176, 179, 180, 188, 191, 196, 220, 221, 225, 239, 242, 268, 269, 277, 289, 302,305,311, 337, 340, 343, and 345 are related to both neonatal and late onset OTCD patients.
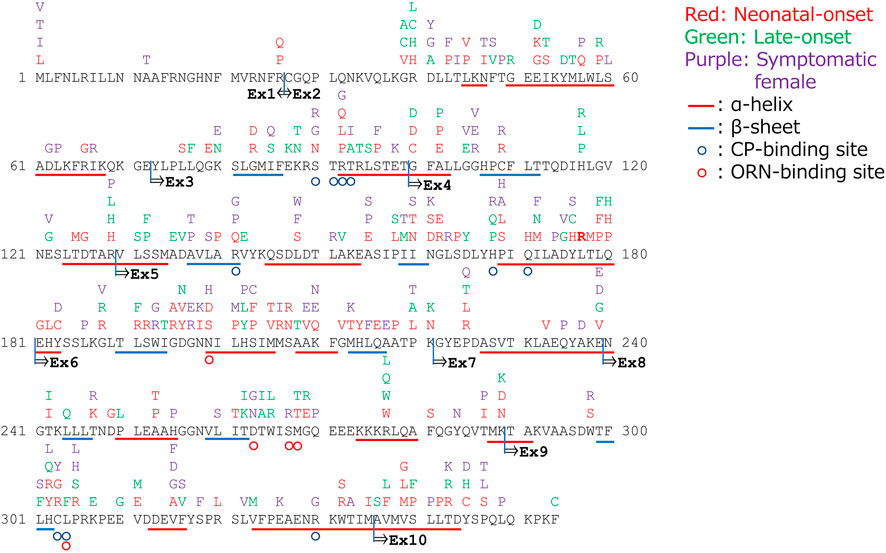
FIGURE 1. Amino acid substitutions in the gene encoding ornithine transcarbamylase (OTC) and their correlated phenotypes.
Discussion
In this study, we have suggested the genotype–phenotype correlations about the onset time of OTCD symptoms and the maximum blood ammonia levels, with respect to the identified variants of the OTC gene. While we could not demonstrate a linear relationship between genetic mutation, protein activity quantification, and clinical morbidity, we revealed the impact of the amino acid substitutions in OTC on the time of onset of the symptoms. According to Tuchman’s study, among the gene mutations leading to OTCD, majority (approximately 84%) are single-base substitutions, while small deletions or insertions and large deletions comprise a smaller proportion of the mutations (12% and 4%, respectively). The mutations are largely “private,” with recurrent mutations occurring mainly in CpG dinucleotides (Tuchman et al., 2002). Therefore, these are known as the mutation hotspots. Incidentally, a previous study indicated that majority of the mutations (80%) arise in the male germ cells (Tuchman et al., 1998). However, our survey demonstrated that the variants are equally likely to arise in any exon of the OTC gene.
The functional OTC holoenzyme is a homotrimeric protein, and each subunit contains an N-terminal domain that binds to carbamoyl phosphate and a C-terminal domain that binds to L-ornithine (Shi et al., 1998). Therefore, these domains are essential for the formation of the enzyme’s active site. Moreover, the α-helix and the β-sheet conformations are essential for retaining the structure of the functional enzyme. Hence, OTC variants that cannot retain the enzyme structure lead to the neonatal onset of OTCD, even if it is an amino acid substitution variant. Moreover, amino acids substitutions in the same position could lead to both neonatal and late onset of OTCD. The time of onset of disease symptoms and the disease severity may vary since the homotrimeric arrangement of the functional protein depends on the condition in the body. Splicing-disrupting mutations in the introns lead to heterogeneous variants, which, in turn, may be influenced by the condition in the body (Olga et al., 2020); hence, the OTC proteins synthesized are not all abnormal. Majority of the splicing-disrupting variants in intron 4 and all the splicing-disrupting variants in introns 5, 6, and 7 were associated with neonatal onset of OTCD. Although the number of exon sites removed in each splicing-disrupting variant was not evaluated, exons 5, 6, 7, and 8 were speculated to be essential for maintaining OTC function.
Neonatal onset of OTCD leads to severe symptoms, and a majority of these patients suffer from hyperammonemia attacks resulting in a maximum blood ammonia concentration of ≥360 μmol/L at the time of onset (Kido et al., 2018; 2021b; 2021a). Such hyperammonemia attacks could damage the brain significantly and lead to poor neurodevelopmental outcomes in patients with OTCD (Kido et al., 2012; 2021a; 2021b).
Family members of OTCD patients, males as well as females, may also develop symptoms of OTCD, such as hyperammonemia attacks. Incidentally, if a male child is born to a female who has a family history of OTCD and possesses a known neonatal onset type variant, then immediate intervention will be necessary after birth to prevent a hyperammonemia attack that may cause blood ammonia levels to rise above 360 μmol/L. In fact, if the maximum blood ammonia levels can be controlled within 360 μmol/L during the first as well as subsequent hyperammonemia attacks, then these patients with neonatal onset OTCD are likely to acquire normal neurodevelopmental outcomes. Moreover, if the maximum ammonia concentrations could be controlled within 360 μmol/L in patients with neonatal onset OTCD, then early liver transplantation may help to achieve a stable overall health condition as well as proper neurodevelopmental outcomes (Kido et al., 2018; 2021a). Such patients may live a life with normal social activity.
There is a degree of genotype–phenotype correlation in male OTCD patients because the level of deactivation of OTC depends extensively on the pathogenic OTC variants. Therefore, the information about the OTC variants discussed in this study may help to develop early intervention strategies for patients who possess variants associated with neonatal onset OTCD; early liver transplantation should be considered as an optional therapy for such patients. Other notable OTC therapeutic options include gene and exon skipping therapy that may become available for clinical application in the near future (Supplementary data 4) (Balestra et al., 2020; Baruteau et al., 2021; and Wang et al., 2012 and, 2022a).
In future, it is important to establish a new medical system that will be able to provide a better prognosis by referring to the patient’s genetic information and intervening at an appropriate time. Moreover, we should consider the need of more comprehensive prenatal genetic testing system for OTC gene because the current prenatal genetic testing of OTC is applied to known mutations in the families with OTC gene mutation or OTCD patients in each institution in Japan. These will help to develop subsequent treatment strategies, including liver transplantation, which may help to save the patients’ lives.
In conclusion, we investigated the impact of OTCD variants on clinical aspects of Japanese patients through an additional nationwide study and an extensive literature review. Genetic information about OTC variations may help to predict long-term outcomes of the OTCD patients, as well as determine specific treatment strategies, such as liver transplantation. In particular, such genetic information is beneficial for performing prenatal diagnosis and designing intervention strategies for neonates born to females possessing the neonate onset variants.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The studies involving human participants were reviewed and approved by the ethical committee of the Faculty of Life Science, Kumamoto University (Ethics No. 1527). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
JK and KN designed the research; JK, KS, TS, and SM contributed to practicing medicine, DNA analysis, and data collection from the OTCD patients; J.K and KS verified and analyzed the data and performed the literature review; JK wrote the manuscript; and JK and KN supervised the research. All authors have read and approved the final manuscript for submission. All authors have agreed to be personally accountable for their own contributions and answer any questions related to the accuracy or integrity of any part of the work.
Funding
This work was supported in part by a Health and Labor Sciences Research Grant for Research on Rare and Intractable Diseases from the Ministry of Health, Labor and Welfare, Japan (grant number JPMH20FC1025), a Grant-in-Aid for Practical Research Project for Rare/Intractable Diseases from the Japan Agency for Medical Research and Development (AMED; grant numbers JP19ek0109276, JP21ek0109482), and a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology, Japan (Japan Society for the Promotion of Science [JSPS] KAKENHI; grant number JP20K08207). The funders had no role in designing the study, collecting and analyzing the data, preparation of the manuscript, or the decision to publish.
Acknowledgments
We are grateful to all 731 institutions that participated in the present study, and in particular, to the 110 institutions that kindly provided useful clinical information regarding patients with UCDs. Additionally, we would like to thank Dr. Tomohiro Horita, Dr. Kiyotaka Kosugiyama, Dr. Atsuko Noguchi, Dr. Chikahiko Numakura, Dr. Yutaka Suzuki, Dr. Masayoshi Nagao, Dr. Hiroshi Kobayashi, Dr. Masahisa Kobayashi, Dr. Manabu Abe, Dr. Keiji Tsuchiya, Dr. Mirai Hattori, Dr. Seiichi Shimizu, Dr. Masahiro Takeda, Dr. Yoshihiro Hirata, Dr. Hajime Uchida, Dr. Mureo Kasahara, Dr. Reiko Horikawa, Dr. Yoichi Wada, Dr. Narutaka Mochizuki, Dr. Kei Murayama, Dr. Tomoko Lee, Dr. Hiroshi Mochizuki, Dr. Yoriko Watanabe, Dr. Yusuke Fujisawa, Dr. Kenichi Kinjo, Dr. Tomotaka Kono, Dr. Asako Tajima, Dr. Masaru Shimura, Dr. Tomoyo Itonaga, Dr. Masaki Kanazawa, Dr. Atsushi Iwabuchi, Dr. Jiro Kagawa, Dr. Keiko Ichimoto, Dr. Akira Otake, Dr. Kaoru Hagita, Dr. Tatsuya Suzuki, Dr. Yasuhiko Ago, Dr. Yoko Nakajima, Dr. Akihiro Tanemura, Dr. Yoshinori Satomura, Dr. Toko Shibuya, Dr. Tohru Yorifuji, Dr. Jun Mori, Dr. Mari Hasegawa, Dr. Takenori Suga, Dr. Mahoko Furujo, Dr. Reina Ogata, Dr. Nobuhiko Koga, Dr. Fusako Sasaki, Dr. Toshihiko Kakiuchi, Dr. Nanae Kawano, Dr. Toshihiko Nonaka, Dr. Kenji Nakamura, Dr. Kazuyuki Yotsumata, Dr. Yasutsugu Chinen, Dr. Hiromi Nyuzuki, Dr. Hiroshi Yoshida, Dr. Hiroyuki Iijima, Dr. Tetsuya Ito, Dr. Shinichi Hirose, Dr. Kaori Fukui, Dr. Kanako Kojima-Ishii, Dr. Yuichi Mushimoto, Dr. Shinobu Yoshida, Dr. Mika Ishige, and Dr. Norio Sakai for providing medical information regarding patients with UCDs. We are also extremely grateful to Ms. Naomi Yano and Ms. Yuri Ikita for their assistance during the survey analysis.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.952467/full#supplementary-material
References
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249. doi:10.1038/nmeth0410-248
Ali, E. Z., Zakaria, Y., Mohd Radzi, M. A., Ngu, L. H., and Jusoh, S. A. (2018). Mutation study of Malaysian patients with ornithine transcarbamylase deficiency: Clinical, molecular, and bioinformatics analyses of two novel missense mutations of the OTC gene. BioMed Res. Int. 2018, 1–15. doi:10.1155/2018/4320831
Arranz, J. A., Riudor, E., Marco-Marín, C., and Rubio, V. (2007). Estimation of the total number of disease-causing mutations in ornithine transcarbamylase (OTC) deficiency. Value of the OTC structure in predicting a mutation pathogenic potential. J Inher Metab Disea 30, 217–226. doi:10.1007/s10545-007-0429-x
Azevedo, L., Soares, P. A., Quental, R., Vilarinho, L., Teles, E. L., Martins, E., et al. (2006). Mutational spectrum and linkage disequilibrium patterns at the ornithine transcarbamylase gene (OTC). Ann. Hum. Genet. 70, 797–801. doi:10.1111/j.1469-1809.2006.00283.x
Azevedo, L., Vilarinho, L., Leão Teles, E., and Amorim, A. (2002). Ornithine transcarbamylase deficiency: A novel splice site mutation in a family with meiotic recombination and a new useful SNP for diagnosis. Mol. Genet. Metabolism 76, 68–70. doi:10.1016/S1096-7192(02)00013-6
Balasubramaniam, S., Rudduck, C., Bennetts, B., Peters, G., Wilcken, B., and Ellaway, C. (2010). Contiguous gene deletion syndrome in a female with ornithine transcarbamylase deficiency. Mol. Genet. Metabolism 99, 34–41. doi:10.1016/j.ymgme.2009.08.007
Balestra, D., Ferrarese, M., Lombardi, S., Ziliotto, N., Branchini, A., Petersen, N., et al. (2020). An exon-specific small nuclear U1 RNA (ExSpeU1) improves hepatic OTC expression in a splicing-defective spf/ash mouse model of ornithine transcarbamylase deficiency. Ijms 21, 8735. doi:10.3390/ijms21228735
Bartholomew, D. W., and McClellan, J. (1998). A novel missense mutation in the human ornithine transcarbamylase gene. Hum. Mutat. 12, 220.
Baruteau, J., Cunningham, S. C., Yilmaz, B. S., Perocheau, D. P., Eaglestone, S., Burke, D., et al. (2021). Safety and efficacy of an engineered hepatotropic AAV gene therapy for ornithine transcarbamylase deficiency in cynomolgus monkeys. Mol. Ther. - Methods & Clin. Dev. 23, 135–146. doi:10.1016/j.omtm.2021.09.005
Ben-Ari, Z., Dalal, A., Morry, A., Pitlik, S., Zinger, P., Cohen, J., et al. (2010). Adult-onset ornithine transcarbamylase (OTC) deficiency unmasked by the Atkins' diet. J. Hepatology 52, 292–295. doi:10.1016/j.jhep.2009.11.014
Bernal, A. C., Tubio, M. C., Crespo, C., and Eiroa, H. D. (2021). Clinical and genetic characterization and biochemical correlation at presentation in 48 patients diagnosed with urea cycle disorders at the hospital juan P garrahan, Argentina. J. Inborn Errors Metab. Screen. 9, e20200026. doi:10.1590/2326-4594-jiems-2020-0026
Bijarnia-Mahay, S., Häberle, J., Jalan, A. B., Puri, R. D., Kohli, S., Kudalkar, K., et al. (2018). Urea cycle disorders in India: Clinical course, biochemical and genetic investigations, and prenatal testing. Orphanet J. Rare Dis. 13, 174. doi:10.1186/s13023-018-0908-1
Bisanzi, S., Morrone, A., Donati, M. A., Pasquini, E., Spada, M., Strisciuglio, P., et al. (2002). Genetic analysis in nine unrelated Italian patients affected by OTC deficiency: Detection of novel mutations in the OTC gene. Mol. Genet. Metabolism 76, 137–144. doi:10.1016/S1096-7192(02)00028-8
Caldovic, L., Abdikarim, I., Narain, S., Tuchman, M., and Morizono, H. (2015). Genotype-phenotype correlations in ornithine transcarbamylase deficiency: A mutation update. J. Genet. Genomics 42, 181–194. doi:10.1016/j.jgg.2015.04.003
Calvas, P., Ségues, B., Rozet, J. M., Rabier, D., Bonnefond, J. P., and Munnich, A. (1998). Novel intragenic deletions and point mutations of the ornithine transcarbamylase gene in congenital hyperammonemia. Hum. Mutat. 11, S81–S84. doi:10.1002/humu.1380110128
Carstens, R. P., Fenton, W. A., and Rosenberg, L. R. (1991). Identification of RNA splicing errors resulting in human ornithine transcarbamylase deficiency. Am. J. Hum. Genet. 48, 1105–1114. Available at: http://www.ncbi.nlm.nih.gov/pubmed/2035531.
Cavicchi, C., Donati, M. A., Parini, R., Rigoldi, M., Bernardi, M., Orfei, F., et al. (2014). Sudden unexpected fatal encephalopathy in adults with OTC gene mutations-Clues for early diagnosis and timely treatment. Orphanet J. Rare Dis. 9, 1–10. doi:10.1186/s13023-014-0105-9
Choi, J. H., Lee, B. H., Kim, J. H., Kim, G. H., Kim, Y. M., Cho, J., et al. (2015). Clinical outcomes and the mutation spectrum of the OTC gene in patients with ornithine transcarbamylase deficiency. J. Hum. Genet. 60, 501–507. doi:10.1038/jhg.2015.54
Choi, Y., Sims, G. E., Murphy, S., Miller, J. R., and Chan, A. P. (2012). Predicting the functional effect of amino acid substitutions and indels. PLoS One 7, e46688. doi:10.1371/journal.pone.0046688
Chongsrisawat, V., Damrongphol, P., Ittiwut, C., Ittiwut, R., Suphapeetiporn, K., and Shotelersuk, V. (2018). The phenotypic and mutational spectrum of Thai female patients with ornithine transcarbamylase deficiency. Gene 679, 377–381. doi:10.1016/j.gene.2018.09.026
Climent, C., García-Pérez, M. A., Sanjurjo, P., Ruiz-Sanz, J. I., Vilaseca, M. A., Pineda, M., et al. (1999). Identification of a cytogenetic deletion and of four novel mutations (Q69X, I172F, G188V, G197R) affecting the gene for ornithine transcarbamylase (OTC) in Spanish patients with OTC deficiency. Hum. Mutat. 14, 352–353. doi:10.1002/(SICI)1098-1004(199910)14:4<352:AID-HUMU15>3.0.CO;2-D
Climent, C., and Rubio, V. (2002). Identification of seven novel missense mutations, two splice-site mutations, two microdeletions and a polymorphic amino acid substitution in the gene for ornithine transcarbamylase (OTC) in patients with OTC deficiency. Hum. Mutat. 19, 185–186. doi:10.1002/humu.9011
Daijo, K., Kawaoka, T., Nakahara, T., Nagaoki, Y., Tsuge, M., Hiramatsu, A., et al. (2017). Late-onset ornithine transcarbamylase deficiency associated with hyperammonemia. Clin. J. Gastroenterol. 10, 383–387. doi:10.1007/s12328-017-0753-0
den Dunnen, J. T., Dalgleish, R., Maglott, D. R., Hart, R. K., Greenblatt, M. S., McGowan-Jordan, J., et al. (2016). HGVS recommendations for the description of sequence variants: 2016 update. Hum. Mutat. 37, 564–569. doi:10.1002/humu.22981
Diggelen, O. P., Zaremba, J., He, W., Keulemans, J. L. M., Boer, A. M., Reuser, A. J. J., et al. (2008). Asymptomatic and late-onset ornithine transcarbamylase (OTC) deficiency in males of a five-generation family, caused by an A208T mutation. Clin. Genet. 50, 310–316. doi:10.1111/j.1399-0004.1996.tb02380.x
Dionisi-Vici, C., Rizzo, C., Burlina, A. B., Caruso, U., Sabetta, G., Uziel, G., et al. (2002). Inborn errors of metabolism in the Italian pediatric population: A national retrospective survey. J. Pediatr. 140, 321–329. doi:10.1067/mpd.2002.122394
Engel, K., Nuoffer, J.-M., Mühlhausen, C., Klaus, V., Largiadèr, C. R., Tsiakas, K., et al. (2008). Analysis of mRNA transcripts improves the success rate of molecular genetic testing in OTC deficiency. Mol. Genet. Metabolism 94, 292–297. doi:10.1016/j.ymgme.2008.03.009
Ensenauer, R., Tuchman, M., El-Youssef, M., Kotagal, S., Ishitani, M. B., Matern, D., et al. (2005). Management and outcome of neonatal-onset ornithine transcarbamylase deficiency following liver transplantation at 60 days of life. Mol. Genet. Metabolism 84, 363–366. doi:10.1016/j.ymgme.2004.12.011
Fantur, M., Karall, D., Scholl-Buergi, S., Häberle, J., Rauchenzauner, M., and Fruehwirth, M. (2013). Recurrent somnolence in a 17-month-old infant: Late-onset ornithine transcarbamylase (OTC) deficiency due to the novel hemizygous mutation c.535C > T (p.Leu179Phe). Eur. J. Paediatr. Neurology 17, 112–115. doi:10.1016/j.ejpn.2012.05.007
Feldmann, D., Rozet, J. M., Pelet, A., Hentzen, D., Briand, P., Hubert, P., et al. (1992). Site specific screening for point mutations in ornithine transcarbamylase deficiency. J. Med. Genet. 29, 471–475. Available at: http://www.ncbi.nlm.nih.gov/pubmed/1353535.
Galloway, P. J., MacPhee, G. B., Alea, P., and Robinson, P. H. (2000). Severe hyperammonaemia in a previously healthy teenager. Ann. Clin. Biochem. 37, 727–728. doi:10.1258/0004563001899807
García-Pérez, M. A., Sanjurjo, P., and Rubio, V. (1995b). Demonstration of the spf-ash mutation in Spanish patients with ornithine transcarbamylase deficiency of moderate severity. Hum. Genet. 95, 183–186. doi:10.1007/BF00209398
García-Pérez, M. A., Sanjurjo, P. S., Briones, M. J., García-Muñoz, V., and Rubio, V. (1995a). A splicing mutation, a nonsense mutation (Y167X) and two missense mutations (I159T and A209V) in Spanish patients with ornithine transcarbamylase deficiency. Hum. Genet. 96, 549–551. doi:10.1007/BF00197410
Genet, S., Cranston, T., and Middleton-Price, H. R. (2000). Mutation detection in 65 families with a possible diagnosis of ornithine carbamoyltransferase deficiency including 14 novel mutations. J. Inherit. Metab. Dis. 23, 669–676. doi:10.1023/A:1005614409241
Gilbert-Dussardier, B., Segues, B., Rozet, J.-M., Rabier, D., Calvas, P., de Lumley, L., et al. (1996). Partial duplication [dup. TCAC (178)] and novel point mutations (T125M, G188R, A209V, and H302L) of the ornithine transcarbamylase gene in congenital hyperammonemia. Hum. Mutat. 8, 74–76. doi:10.1002/(SICI)1098-1004(1996)8:1<74:AID-HUMU11>3.0.CO;2-O
Giorgi, M., Morrone, A., Donati, M. A., Ciani, F., Bardelli, T., Biasucci, G., et al. (2000). Lymphocyte mRNA analysis of the ornithine transcarbamylase gene in Italian OTCD male patients and manifesting carriers: Identification of novel mutations. Hum. Mutat. 15(4):380–381. doi:10.1002/(SICI)1098-1004(200004)15:4<380:AID-HUMU12>3.0.CO;2-Q
Gobin-Limballe, S., Ottolenghi, C., Reyal, F., Arnoux, J. B., Magen, M., Simon, M., et al. (2021). OTC deficiency in females: Phenotype-genotype correlation based on a 130-family cohort. J Inher Metab Disea 44, 1235–1247. doi:10.1002/jimd.12404
Grompe, M., Caskey, C. T., and Fenwick, R. G. (1991). Improved molecular diagnostics for ornithine transcarbamylase deficiency. Am. J. Hum. Genet. 48, 212–222. Available at: http://www.ncbi.nlm.nih.gov/pubmed/1671317.
Grompe, M., Muzny, D. M., and Caskey, C. T. (1989). Scanning detection of mutations in human ornithine transcarbamoylase by chemical mismatch cleavage. Proc. Natl. Acad. Sci. U.S.A. 86, 5888–5892. doi:10.1073/pnas.86.15.5888
Gyato, K., Wray, J., Huang, Z. J., Yudkoff, M., and Batshaw, M. L. (2004). Metabolic and neuropsychological phenotype in women heterozygous for ornithine transcarbamylase deficiency. Ann. Neurol. 55, 80–86. doi:10.1002/ana.10794
Hata, A., Matsuura, T., Setoyama, C., Shimada, K., Yokoi, T., Akaboshi, I., et al. (1991). A novel missense mutation in exon 8 of the ornithine transcarbamylase gene in two unrelated male patients with mild ornithine transcarbamylase deficiency. Hum. Genet. 87, 28–32. doi:10.1007/BF01213087
Hata, A., Setoyama, C., Shimada, K., Takeda, E., Kuroda, Y., Akaboshi, I., et al. (1989). Ornithine transcarbamylase deficiency resulting from a C-to-T substitution in exon 5 of the ornithine transcarbamylase gene. Am. J. Hum. Genet. 45, 123–127. Available at: http://www.ncbi.nlm.nih.gov/pubmed/2741942.
Hata, A., Tsuzuki, T., Shimada, K., Takiguchi, M., Mori, M., and Matsuda, I. (1988). Structure of the human ornithine transcarbamylase Gene1. J. Biochem. 103, 302–308. doi:10.1093/oxfordjournals.jbchem.a122265
Horwich, A. L., Kalousek, F., Fenton, W. A., Pollock, R. A., and Rosenberg, L. E. (1986). Targeting of pre-ornithine transcarbamylase to mitochondria: Definition of critical regions and residues in the leader peptide. Cell 44, 451–459. doi:10.1016/0092-8674(86)90466-6
Hoshide, R., Matsuura, T., Komaki, S., Koike, E., Ueno, I., and Matsuda, I. (1993). Specificity of PCR-SSCP for detection of the mutant ornithine transcarbamylase (OTC) gene in patients with OTC deficiency. J Inher Metab Disea 16, 857–862. doi:10.1007/BF00714278
Hoshide, R., Matsuura, T., Sagara, Y., Kubo, T., Shimadzu, M., Endo, F., et al. (1996). Prenatal monitoring in a family at high risk for ornithine transcarbamylase (OTC) deficiency: A new mutation of an A-to-C transversion in position +4 of intron 1 of the OTC gene that is likely to abolish enzyme activity. Am. J. Med. Genet. 64, 459–464. doi:10.1002/(SICI)1096-8628(19960823)64:3<459:AID-AJMG3>3.0.CO;2-K
Hübler, A., Seidel, J., Patzer, L., Bellstedt, K., and Schramm, D. (2001). Interdisziplinäre Behandlung des frühmanifesten Ornithintranscarbamylase(OTC)-Mangels1 - zwei Patientenberichte und Literaturübersicht -. Z. Geburtshilfe Neonatol. 205, 236–241. doi:10.1055/s-2001-19056
Hwu, W. L., Huang, Y. T., Chien, Y. H., Yeh, H. Y., Lu, F., Chou, S. P., et al. (2003a). Gene symbol: OTC. Disease: Ornithine carbamoyltransferase deficiency. Hum. Genet. 113, 365–366. doi:10.1007/s00439-003-0994-5
Hwu, W. L., Huang, Y. T., Chien, Y. H., Yeh, H. Y., Lu, F., Chou, S. P., et al. (2003b). Gene symbol: OTC. Disease: Ornithine carbamoyltransferase deficiency. Hum. Genet. 113, 365–368. doi:10.1007/s00439-003-0995-4
Jamroz, E., Paprocka, J., Sokół, M., Popowska, E., and Ciara, E. (2013). Magnetic resonance spectroscopy and molecular studies in ornithine transcarbamylase deficiency novel mutation c.802A>G in exon 8 (p.Met268Val)Met268Val). Neurol. i Neurochir. Pol. 47, 283–289. doi:10.5114/ninp.2013.35488
Jang, Y. J., LaBella, A. L., Feeney, T. P., Braverman, N., Tuchman, M., Morizono, H., et al. (2018). Disease-causing mutations in the promoter and enhancer of the ornithine transcarbamylase gene. Hum. Mutat. 39, 527–536. doi:10.1002/humu.23394
Keskinen, P., Siitonen, A., and Salo, M. (2008). Hereditary urea cycle diseases in Finland. Acta Paediatr. Int. J. Paediatr. 97, 1412–1419. doi:10.1111/j.1651-2227.2008.00923.x
Khoo, A. S. B., Balraj, P., Rachedi, A., Chin, C. N., and Volpi, L. (1999). A novel complex mutation of the OTC (ornithine transcarbamylase) gene in a Malaysian pedigree. Hum. Mutat. 14, 448. doi:10.1002/(SICI)1098-1004(199911)14:5<448:AID-HUMU14>3.0.CO;2-T
Kido, J., Matsumoto, S., Häberle, J., Inomata, Y., Kasahara, M., Sakamoto, S., et al. (2021a). Role of liver transplantation in urea cycle disorders: Report from a nationwide study in Japan. J Inher Metab Disea 44, 1311–1322. doi:10.1002/jimd.12415
Kido, J., Matsumoto, S., Häberle, J., Nakajima, Y., Wada, Y., Mochizuki, N., et al. (2021b). Long-term outcome of urea cycle disorders: Report from a nationwide study in Japan. J Inher Metab Disea 44, 826–837. doi:10.1002/jimd.12384
Kido, J., Matsumoto, S., Ito, T., Hirose, S., Fukui, K., Kojima-Ishii, K., et al. (2021c). Physical, cognitive, and social status of patients with urea cycle disorders in Japan. Mol. Genet. Metabolism Rep. 27, 100724. doi:10.1016/j.ymgmr.2021.100724
Kido, J., Matsumoto, S., Mitsubuchi, H., Endo, F., and Nakamura, K. (2018). Early liver transplantation in neonatal-onset and moderate urea cycle disorders may lead to normal neurodevelopment. Metab. Brain Dis. 33, 1517–1523. doi:10.1007/s11011-018-0259-6
Kido, J., Matsumoto, S., Sugawara, K., Sawada, T., and Nakamura, K. (2021d). Variants associated with urea cycle disorders in Japanese patients: Nationwide study and literature review. Am. J Med Genet. Pt A 185, 2026–2036. doi:10.1002/ajmg.a.62199
Kido, J., Nakamura, K., Mitsubuchi, H., Ohura, T., Takayanagi, M., Matsuo, M., et al. (2012). Long-term outcome and intervention of urea cycle disorders in Japan. J. Inherit. Metab. Dis. 35, 777–785. doi:10.1007/s10545-011-9427-0
Kim, G. H., Choi, J. H., Lee, H. H., Park, S., Kim, S. S., and Yoo, H. W. (2006). Identification of novel mutations in the human ornithine transcarbamylase (OTC) gene of Korean patients with OTC deficiency and transient expression of the mutant proteins in vitro. Hum. Mutat. 27, 1159. doi:10.1002/humu.9465
Kogo, T., Satoh, Y., Kanazawa, M., Yamamoto, S., Takayanagi, M., Ohtake, A., et al. (1998). Expression analysis of two mutant human ornithine transcarbamylases in COS-7 cells. J. Hum. Genet. 43, 54–58. doi:10.1007/s100380050037
Komaki, S., Matsuura, T., Oyanagi, K., Hoshide, R., Kiwaki, K., Endo, F., et al. (1997). Familial lethal inheritance of a mutated paternal gene in females causing X-linked ornithine transcarbamylase (OTC) deficiency. Am. J. Med. Genet. 69, 177–181. doi:10.1002/(SICI)1096-8628(19970317)69:2<177:AID-AJMG12>3.0.CO;2-I
Kumar, R. D., Burrage, L. C., Bartos, J., Ali, S., Schmitt, E., Nagamani, S. C. S., et al. (2021). A deep intronic variant is a common cause of OTC deficiency in individuals with previously negative genetic testing. Mol. Genet. Metabolism Rep. 26, 100706. doi:10.1016/j.ymgmr.2020.100706
Landrum, M. J., Chitipiralla, S., Brown, G. R., Chen, C., Gu, B., Hart, J., et al. (2020). ClinVar: Improvements to accessing data. Nucleic Acids Res. 48, D835–D844. doi:10.1093/nar/gkz972
Laróvere, L. E., Silvera Ruiz, S. M., Arranz, J. A., and Dodelson de Kremer, R. (2018). Mutation spectrum and genotype-phenotype correlation in a cohort of Argentine patients with ornithine transcarbamylase deficiency: A single-center experience. J. Inborn Errors Metabolism Screen. 6, 232640981881317. doi:10.1177/2326409818813177
Lee, J. H., Kim, G. H., Yoo, H. W., and Cheon, C. K. (2014). OTC gene in ornithine transcarbamylase deficiency: Clinical course and mutational spectrum in seven Korean patients. Pediatr. Neurol. 51, 354–359. doi:10.1016/j.pediatrneurol.2014.03.029
Lee, T., Misaki, M., Shimomura, H., Tanaka, Y., Yoshida, S., Murayama, K., et al. (2018). Late-onset ornithine transcarbamylase deficiency caused by a somatic mosaic mutation. Hum. Genome Var. 5, 22. doi:10.1038/s41439-018-0022-x
Lee, T., Yoshii, K., Yoshida, S., Suga, T., Nakamura, K., Sasai, H., et al. (2020). Retrospective evaluations revealed pre-symptomatic citrulline concentrations measured by newborn screening were significantly low in late-onset ornithine transcarbamylase deficiency patients. Clin. Chim. Acta 510, 633–637. doi:10.1016/j.cca.2020.08.027
Leibundgut, E., Liechti-Gallati, S., Colombo, J. P., and Wermuth, B. (1995). Ornithine transcarbamylase deficiency: New sites with increased probability of mutation. Hum. Genet. 95, 191–196. doi:10.1007/BF00209400
Leibundgut, E., Liechti-Gallati, S., Colombo, J. P., and Wermuth, B. (1997). Ornithine transcarbamylase deficiency: Ten new mutations and high proportion of de novo mutations in heterozygous females. Hum. Mutat. 9, 409–411. doi:10.1002/(SICI)1098-1004(1997)9:5<409:AID-HUMU5>3.0.CO;2-Z
Leibundgut, E., Wermuth, B., Colombo, J.-P., and Liechti-Gallati, S. (1996b). Ornithine transcarbamylase deficiency: Characterization of gene mutations and polymorphisms. Hum. Mutat. 8, 333–339. doi:10.1002/(SICI)1098-1004(1996)8:4<333:AID-HUMU6>3.0.CO;2-8
Li, S., Cai, Y., Shi, C., Liu, M., Liu, B., Lin, L., et al. (2018). Gene mutation analysis and prenatal diagnosis of the ornithine transcarbamylase (OTC) gene in two families with ornithine transcarbamylase deficiency. Med. Sci. Monit. 24, 7431–7437. doi:10.12659/MSM.911295
Lin, H. Y., Lin, H. Y., and Lin, S. P. (2010). Novel human pathological mutations. Hum. Genet. 127, 463–490. doi:10.1007/s00439-010-0788-5
Lindgren, V., de Martinville, B., Horwich, A. L., Rosenberg, L. E., and Francke, U. (1984). Human ornithine transcarbamylase locus mapped to band Xp21.1 near the Duchenne muscular dystrophy locus. Science 226, 698–700. doi:10.1126/science.6494904
Liu, F., Bao, L., Liang, R., Zhao, X., Li, Z., Du, Z., et al. (2021). Identification of rare variants causing urea cycle disorders: A clinical, genetic, and biophysical study. J. Cell. Mol. Med. 25, 4099–4109. doi:10.1111/jcmm.16379
Lu, D., Han, F., Qiu, W., Zhang, H., Ye, J., Liang, L., et al. (2020). Clinical and molecular characteristics of 69 Chinese patients with ornithine transcarbamylase deficiency. Orphanet J. Rare Dis. 15, 340. doi:10.1186/s13023-020-01606-2
Luksan, O., Jirsa, M., Eberova, J., Minks, J., Treslova, H., Bouckova, M., et al. (2010). Disruption of OTC promoter-enhancer interaction in a patient with symptoms of ornithine carbamoyltransferase deficiency. Hum. Mutat. 31, E1294–E1303. doi:10.1002/humu.21215
Maddalena, A., Spence, J. E., O'Brien, W. E., and Nussbaum, R. L. (1988). Characterization of point mutations in the same arginine codon in three unrelated patients with ornithine transcarbamylase deficiency. J. Clin. Invest. 82, 1353–1358. doi:10.1172/JCI113738
Martín-Hernández, E., Aldámiz-Echevarría, L., Castejón-Ponce, E., Pedrón-Giner, C., Couce, M. L., Serrano-Nieto, J., et al. (2014). Urea cycle disorders in Spain: An observational, cross-sectional and multicentric study of 104 cases. Orphanet J. Rare Dis. 9, 187. doi:10.1186/s13023-014-0187-4
Matsuda, I., and Tanase, S. (1997). The ornithine transcarbamylase (OTC) gene: Mutations in 50 Japanese families with OTC deficiency. Am. J. Med. Genet. 71, 378–383. doi:10.1002/(SICI)1096-8628(19970905)71:4<378:AID-AJMG2>3.0.CO;2-Q
Matsuura, T., Hoshide, R., Fukushima, M., Sakiyama, T., Owada, M., and Matsuda, I. (1993). Prenatal monitoring of ornithine transcarbamoylase deficiency in two families by DNA analysis. J Inher Metab Disea 16, 31–38. doi:10.1007/BF00711312
Matsuura, T., Hoshide, R., Kiwaki, K., Komaki, S., Koike, E., Endo, F., et al. (1994). Four newly identified ornithine transcarbamylase (OTC) mutations (D126G, R129H, I172M and W332X) in Japanese male patients with early-onset OTC deficiency. Hum. Mutat. 3, 402–406. doi:10.1002/humu.1380030415
Matsuura, T., Hoshide, R., Komaki, S., Kiwaki, K., Endo, F., Nakamura, S., et al. (1995). Identification of two new aberrant splicings in the ornithine carbamoyltransferase (OCT) gene in two patients with early and late onset OCT deficiency. J Inher Metab Disea 18, 273–282. doi:10.1007/BF00710415
Matsuura, T., and Matsuda, I. (1998). Ornithine transcarbamylase deficiency (OTCD). Ryoikibetsu Shokogun Shirizu, 170–174. Available at: http://www.ncbi.nlm.nih.gov/pubmed/9590019.
McCullough, B. A., Yudkoff, M., Batshaw, M. L., Wilson, J. M., Raper, S. E., and Tuchman, M. (2000). Genotype spectrum of ornithine transcarbamylase deficiency: Correlation with the clinical and biochemical phenotype. Am. J. Med. Genet. 93, 313–319. doi:10.1002/1096-8628(20000814)93:4<313:AID-AJMG11>3.0.CO;2-M
Meng, L., Jiang, T., Qin, L., Ma, D., Chen, Y., Han, S., et al. (2013). Molecular diagnosis of OTC gene mutation in a Chinese family with ornithine transcarbamylase deficiency. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 30, 195–198. doi:10.3760/cma.j.issn.1003-9406.2013.04.016
Mohamed, S., Hamad, M. H., Kondkar, A. A., and Abu-Amero, K. K. (2015). A novel mutation in ornithine transcarbamylase gene causing mild intermittent hyperammonemia. Smj 36, 1229–1232. doi:10.15537/smj.2015.10.12127
Mukhtar, A., Dabbous, H., El Sayed, R., Aboulfetouh, F., Bahaa, M., Abdelaal, A., et al. (2013). A novel mutation of the ornithine transcarbamylase gene leading to fatal hyperammonemia in a liver transplant recipient. Am. J. Transplant. 13, 1084–1087. doi:10.1111/ajt.12146
Nagata, N., Matsuda, I., and Oyanagi, K. (1991). Estimated frequency of urea cycle enzymopathies in Japan. Am. J. Med. Genet. 39, 228–229. doi:10.1002/ajmg.1320390226
Nguyen, H. H., Khanh Nguyen, N., Dung Vu, C., Thu Huong Nguyen, T., and Nguyen, N. L. (2020). Late-onset ornithine transcarbamylase deficiency and variable phenotypes in Vietnamese females with OTC mutations. Front. Pediatr. 8, 321. doi:10.3389/fped.2020.00321
Nishiyori, A., Yoshino, M., Kato, H., Matsuura, T., Hoshide, R., Matsuda, I., et al. (1997). The R40H mutation in a late onset type of human ornithine transcarbamylase deficiency in male patients. Hum. Genet. 99, 171–176. doi:10.1007/s004390050333
Nishiyori, A., Yoshino, M., Tananari, Y., Matsuura, T., Hoshide, R., Mastuda, I., et al. (1998). Y55D mutation in ornithine transcarbamylase associated with late-onset hyperammonemia in a male. Hum. Mutat. 11, S131–S133. doi:10.1002/humu.1380110144
Ogino, W., Takeshima, Y., Nishiyama, A., Okizuka, Y., Yagi, M., Tsuneishi, S., et al. (2007). Mutation analysis of the ornithine transcarbamylase (OTC) gene in five Japanese OTC deficiency patients revealed two known and three novel mutations including a deep intronic mutation. Kobe J. Med. Sci. 53, 229–240. Available at: http://www.ncbi.nlm.nih.gov/pubmed/18204299.
Olga, S., Natalia, S., Igor, B., Alena, C., Ekaterina, Z., Oksana, R., et al. (2020). A novel splice site mutation in OTC gene of a female with ornithine transcarbamylase deficiency and her asymptomatic mosaic father. J. Genet. 99, 29. doi:10.1007/s12041-020-1189-8
Oppliger Leibundgut, E., Wermuth, B., Colombo, J. P., and Liechti-Gallati, S. (1996a). Identification of four novel splice site mutations in the ornithine transcarbamylase gene. Hum. Genet. 97, 209–213. doi:10.1007/BF02265267
Popowska, E., Ciara, E., Rokicki, D., and Pronicka, E. (1999). Three novel and one recurrent ornithine carbamoyltransferase gene mutations in Polish patients. J. Inherit. Metab. Dis. 22, 92–93. doi:10.1023/A:1005476021549
Qin, L., Wang, J., Tian, X., Yu, H., Truong, C., Mitchell, J. J., et al. (2016). Detection and quantification of mosaic mutations in disease genes by next-generation sequencing. J. Mol. Diagnostics 18, 446–453. doi:10.1016/j.jmoldx.2016.01.002
Ramanathan, M., Uppalapu, S., and Patel, N. M. (2017). Hiding in plain sight: A case of ornithine transcarbamylase deficiency unmasked post-liver transplantation. Am. J. Transpl. 17, 1405–1408. doi:10.1111/ajt.14174
Rapp, B., Häberle, J., Linnebank, M., Wermuth, B., Marquardt, T., Harms, E., et al. (2001). Genetic analysis of carbamoylphosphate synthetase I and ornithine transcarbamylase deficiency using fibroblasts. Eur. J. Pediatr. 160, 283–287. doi:10.1007/s004310100725
Reish, O., Plante, R. J., and Tuchman, M. (1993). Four new mutations in the ornithine transcarbamylase gene. Biochem. Med. Metabolic Biol. 50, 169–175. doi:10.1006/bmmb.1993.1058
Rivera-Barahona, A., Sánchez-Alcudia, R., Viecelli, H. M., Rüfenacht, V., Pérez, B., Ugarte, M., et al. (2015). Functional characterization of the spf/ash splicing variation in OTC deficiency of mice and man. PLoS One 10, e0122966. doi:10.1371/journal.pone.0122966
Schimanski, U., Krieger, D., Horn, M., Stremmel, W., Wermuth, B., and Theilmann, L. (1996). A novel two-nucleotide deletion in the ornithine transcarbamylase gene causing fatal hyperammonia in early pregnancy. Hepatology 24, 1413–1415. doi:10.1053/jhep.1996.v24.pm000893817210.1002/hep.510240618
Schultz, R. E. H., and Salo, M. K. (2000). Under recognition of late onset ornithine transcarbamylase deficiency. Arch. Dis. Child. 82, 390–391. doi:10.1136/adc.82.5.390
Ségues, B., Veber, P. S., Rabier, D., Calvas, P., Saudubray, J. M., Gilbert-Dussardier, B., et al. (1996). A 3-base pair in-frame deletion in exon 8 (delGlu272/273) of the ornithine transcarbamylase gene in late-onset hyperammonemic coma. Hum. Mutat. 8, 373–374. doi:10.1002/(SICI)1098-1004)8:4<373:AID-HUMU13>3.0.CO;2-#10.1002/(SICI)1098-1004(1996)8:4<373:AID-HUMU13>3.0.CO;2-#
Shao, Y., Jiang, M., Lin, Y., Mei, H., Zhang, W., Cai, Y., et al. (2017). Clinical and mutation analysis of 24 Chinese patients with ornithine transcarbamylase deficiency. Clin. Genet. 92, 318–322. doi:10.1111/cge.13004
Shchelochkov, O. A., Li, F. Y., Geraghty, M. T., Gallagher, R. C., Van Hove, J. L., Lichter-Konecki, U., et al. (2009). High-frequency detection of deletions and variable rearrangements at the ornithine transcarbamylase (OTC) locus by oligonucleotide array CGH. Mol. Genet. Metabolism 96, 97–105. doi:10.1016/j.ymgme.2008.11.167
Shi, D., Morizono, H., Ha, Y., Aoyagi, M., Tuchman, M., and Allewell, N. M. (1998). 1.85-Å resolution crystal structure of human ornithine transcarbamoylase complexed withN-Phosphonacetyl-l-ornithine. J. Biol. Chem. 273, 34247–34254. doi:10.1074/jbc.273.51.34247
Shimadzu, M., Matsumoto, H., Matsuura, T., Kobayashi, K., Komaki, S., Kiwaki, K., et al. (1998). Ten novel mutations of the ornithine transcarbamylase (OTC) gene in OTC deficiency. Hum. Mutat. 11, S5–S7. doi:10.1002/humu.1380110103
Staudt, M., Wermuth, B., Freisinger, P., Hässler, A., and Pontz, B. F. (1998). Symptomatic ornithine carbamoyltransferase deficiency (point mutation H202P) with normal in vitro activity. J. Inherit. Metab. Dis. 21, 71–72. doi:10.1023/A:1005315531630
Storkanova, G., Vlaskova, H., Chuzhanova, N., Zeman, J., Stranecky, V., Majer, F., et al. (2013). Ornithine carbamoyltransferase deficiency: Molecular characterization of 29 families. Clin. Genet. 84, 552–559. doi:10.1111/cge.12085
Strautnleks, S., and Malcolm, S. (1993). Novel mutation affecting a splice site in exon 4 of the ornithine carbamoyl transferase gene. Hum. Mol. Genet. 2, 1963–1964. doi:10.1093/hmg/2.11.1963
Summar, M. L., Koelker, S., Freedenberg, D., Le Mons, C., Haberle, J., Lee, H. S., et al. (2013). The incidence of urea cycle disorders. Mol. Genet. Metabolism 110, 179–180. doi:10.1016/j.ymgme.2013.07.008
Takanashi, J., Kurihara, A., Tomita, M., Kanazawa, M., Yamamoto, S., Morita, F., et al. (2002). Distinctly abnormal brain metabolism in late-onset ornithine transcarbamylase deficiency. Neurology 59, 210–214. doi:10.1212/WNL.59.2.210
Tanaka, A., Wada, T., Maruyama, M., Tanaka, A., Takikawa, H., and Komatsu, Y. (2005). Hyperammonemia-induced encephalopathy due to ornithine transcarbamylase deficiency in an adult woman: Identification of novel missense mutations. J. Gastroenterol. 40, 106–107. doi:10.1007/s00535-004-1502-y
Tsai, M. Y., Holzknecht, R. A., and Tuchman, M. (1993). Single-strand conformational polymorphism and direct sequencing applied to carrier testing in families with ornithine transcarbamylase deficiency. Hum. Genet. 91, 321–325. doi:10.1007/BF00217350
Tuchman, M., Holzknecht, R. A., Gueron, A. B., Berry, S. A., and Tsai, M. Y. (1992). Six new mutations in the ornithine transcarbamylase gene detected by single-strand conformational polymorphism. Pediatr. Res. 32, 600–604. doi:10.1203/00006450-199211000-00024
Tuchman, M., Jaleel, N., Morizono, H., Sheehy, L., and Lynch, M. G. (2002). Mutations and polymorphisms in the human ornithine transcarbamylase gene. Hum. Mutat. 19, 93–107. doi:10.1002/humu.10035
Tuchman, M., Morizono, H., Rajagopal, B. S., Plante, R. J., and Allewell, N. M. (1997). Identification of 'private' mutations in patients with ornithine transcarbamylase deficiency. J. Inherit. Metab. Dis. 20, 525–527. doi:10.1023/A:1005301513465
Tuchman, M., Morizono, H., Rajagopal, B. S., Plante, R. J., and Allewell, N. M. (1998). The biochemical and molecular spectrum of ornithine transcarbamylase deficiency. J. Inherit. Metab. Dis. 21, 40–58. doi:10.1023/A:1005353407220
Tuchman, M., Morizono, H., Reish, O., Yuan, X., and Allewell, N. M. (1995). The molecular basis of ornithine transcarbamylase deficiency: Modelling the human enzyme and the effects of mutations. J. Med. Genet. 32, 680–688. doi:10.1136/jmg.32.9.680
Tuchman, M., Plante, R. J., Giguère, Y., and Lemieux, B. (1994). The ornithine transcarbamylase gene: New "Private" mutations in four patients and study of a polymorphism. Hum. Mutat. 3, 318–320. doi:10.1002/humu.1380030325
Ueta, A., Sumi, S., Kidouchi, K., Ito, T., Ban, K., Hamajima, N., et al. (2001). Intra-day variations in urinary pyrimidines in ornithine carbamoyltransferase deficiency and healthy individuals. Clin. Chim. Acta 308, 187–189. doi:10.1016/S0009-8981(01)00472-7
Valik, D., Sedova, Z., Starha, J., Zeman, J., Hruba, E., and Dvorakova, L. (2004). Acute hyperammonaemic encephalopathy in a female newborn caused by a novel, de novo mutation in the ornithine transcarbamylase gene. Acta Paediatr. 93, 710–711. doi:10.1111/j.1651-2227.2004.tb03002.x
Vella, S., Steiner, F., Schlumbom, V., Zurbrügg, R., Wiesmann, U. N., Schaffner, T., et al. (1997). Mutation of ornithine transcarbamylase (H136R) in a girl with severe intermittent orotic aciduria but normal enzyme activity. J. Inherit. Metab. Dis. 20, 517–524. doi:10.1023/A:1005397329395
Wang, L., Morizono, H., Lin, J., Bell, P., Jones, D., McMenamin, D., et al. (2012). Preclinical evaluation of a clinical candidate AAV8 vector for ornithine transcarbamylase (OTC) deficiency reveals functional enzyme from each persisting vector genome. Mol. Genet. Metabolism 105, 203–211. doi:10.1016/j.ymgme.2011.10.020
Wang, L. P., Luo, H. Z., Song, M., Yang, Z. Z., Yang, F., Cao, Y. T., et al. (2022b). Hemizygous deletion in the OTC gene results in ornithine transcarbamylase deficiency: A case report. Wjcc 10, 1417–1422. doi:10.12998/wjcc.v10.i4.1417
Wang, L., Warzecha, C. C., Kistner, A., Chichester, J. A., Bell, P., Buza, E. L., et al. (2022a). Prednisolone reduces the interferon response to AAV in cynomolgus macaques and may increase liver gene expression. Mol. Ther. Methods Clin. Dev. 24, 292–305. doi:10.1016/j.omtm.2022.01.007
Wang, Y., Liu, X., Wu, H., Liu, H., Wang, C., and He, X. (2014). Analysis of clinical features, metabolic profiling and gene mutations of patients with ornithine transcarbamylase deficiency. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 31, 148–151. doi:10.3760/cma.j.issn.1003-9406.2014.02.005
Wu, B., Wang, Y. Z., Cao, L. R., Zhang, R. P., Fang, Y. L., Zhang, Y. Q., et al. (2018). Case Report A novel mutation in ornithine transcarbamylase gene identified from a Chinese child with ornithine transcarbamylase deficiency. Int. J. Clin. Exp. Med. 11, 6344–6350. Available at: www.ijcem.com/(Accessed December 2, 2021).
Yamaguchi, S., Brailey, L. L., Morizono, H., Bale, A. E., and Tuchman, M. (2006). Mutations and polymorphisms in the human ornithine transcarbamylase (OTC) gene. Hum. Mutat. 27, 626–632. doi:10.1002/humu.20339
Yamanouchi, H., Yokoo, H., Yuhara, Y., Maruyama, K., Sasaki, A., Hirato, J., et al. (2002). An autopsy case of ornithine transcarbamylase deficiency. Brain Dev. 24, 91–94. doi:10.1016/S0387-7604(01)00408-9
Yoo, H. W., Kim, G. H., and Lee, D. H. (1996). Identification of new mutations in the ornithine transcarbamylase (OTC) gene in Korean families. J Inher Metab Disea 19, 31–42. doi:10.1007/BF01799346
Zheng, Z., Lin, Y., Lin, W., Zhu, L., Jiang, M., Wang, W., et al. (2020). Clinical and genetic analysis of five Chinese patients with urea cycle disorders. Mol. Genet. Genomic Med. 8, e1301. doi:10.1002/mgg3.1301
Zhou, Q., Huang, H., Ma, L., and Zhu, T. (2020). The application of next-generation sequencing (NGS) in neonatal-onset urea cycle disorders (UCDs): Clinical course, metabolomic profiling, and genetic findings in nine Chinese hyperammonemia patients. BioMed Res. Int. 2020, 1–11. doi:10.1155/2020/5690915
Keywords: ornithine transcarbamylase deficiency, X-linked disorder, hyperammonemia, late onset OTCD, neonatal onset OTCD
Citation: Kido J, Sugawara K, Sawada T, Matsumoto S and Nakamura K (2022) Pathogenic variants of ornithine transcarbamylase deficiency: Nation-wide study in Japan and literature review. Front. Genet. 13:952467. doi: 10.3389/fgene.2022.952467
Received: 25 May 2022; Accepted: 25 August 2022;
Published: 11 October 2022.
Edited by:
Sunita Bijarnia-Mahay, Sir Ganga Ram Hospital, IndiaReviewed by:
Wei Qu, Capital Medical University, ChinaPetr O. Ilyinskii, Selecta Biosciences, United States
Copyright © 2022 Kido, Sugawara, Sawada, Matsumoto and Nakamura. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Kido, kidojun@kuh.kumamoto-u.ac.jp