 B. Molloy1*†
B. Molloy1*† E. R. Jones1†
E. R. Jones1† N. D. Linhares1P. G. Buckley1
N. D. Linhares1P. G. Buckley1 T. R. Leahy2,3B. Lynch4,5
T. R. Leahy2,3B. Lynch4,5 I. Knerr5,6M. D. King4,5
I. Knerr5,6M. D. King4,5 K. M. Gorman4,5
K. M. Gorman4,5- 1Genuity Science, Dublin, Ireland
- 2Department of Paediatric Immunology, Children’s Health Ireland at Crumlin, Dublin, Ireland
- 3Department of Paediatrics, Trinity College, University of Dublin, Dublin, Ireland
- 4Department of Paediatric Neurology and Clinical Neurophysiology, Children’s Health Ireland at Temple Street, Dublin, Ireland
- 5School of Medicine and Medical Sciences, University College Dublin, Dublin, Ireland
- 6National Centre for Inherited Metabolic Disorders, Children’s Health Ireland at Temple Street, Dublin, Ireland
A uniparental disomy (UPD) screen using whole genome sequencing (WGS) data from 164 trios with rare disorders in the Irish population was performed to identify large runs of homozygosity of uniparental origin that may harbour deleterious recessive variants. Three instances of whole chromosome uniparental isodisomy (UPiD) were identified: one case of maternal isodisomy of chromosome 1 and two cases of paternal isodisomy of chromosome 2. We identified deleterious homozygous variants on isodisomic chromosomes in two probands: a novel p (Glu59ValfsTer20) variant in TMCO1, and a p (Pro222Leu) variant in PRKRA, respectively. The overall prevalence of whole chromosome UPiD in our cohort was 1 in 55 births, compared to 1 in ∼7,500 births in the general population, suggesting a higher frequency of UPiD in rare disease cohorts. As a distinct mechanism underlying homozygosity compared to biallelic inheritance, the identification of UPiD has important implications for family planning and cascade testing. Our study demonstrates that UPD screening may improve diagnostic yields by prioritising UPiD chromosomes during WGS analysis.
Introduction
Uniparental disomy (UPD) is generally a consequence of meiotic nondisjunction, where one or more chromosome pairs are of uniparental origin (Engel, 1980; Yamazawa et al., 2010; Del Gaudio et al., 2020). Nondisjunction events in meiosis I and II can lead to heterodisomy (UPhD), where both homologs of a chromosome are inherited from one parent, or isodisomy (UPiD), where two identical copies of one homolog are inherited from a single parent, respectively. This inheritance error may affect part or the whole chromosome.
A previous study on 214,915 trios estimated the prevalence of UPD (UPhD and UPiD) in the general population at 1 in ∼2,000 births, with unique per-chromosome rates that reflect the susceptibility of individual chromosomes to meiotic nondisjunction (Nakka et al., 2019). Independent assessments in clinical cohorts with diverse phenotypic indications, estimate the incidence of UPD to be between 1 in 176–500 births (King D. A. et al., 2014; Yauy et al., 2020; Scuffins et al., 2021). Whole chromosome UPiD was identified as the least common subtype of UPD, accounting for 27% of all uniparental disomy cases in 916,712 parent-child duos from the 23 and Me dataset, compared to 37% UPhD and 36% partial UPiD. The derived prevalence of UPiD is estimated to be 1 in ∼7,500 in the general population (Nakka et al., 2019).
Heterozygous deleterious variants on an affected chromosome in one parent may become homozygous in offspring as a consequence of partial or complete UPiD. Whole-genome sequencing (WGS) data is a powerful resource for investigating UPD inheritance errors in patients with suspected rare Mendelian disorders (Bis et al., 2017).
Here, we describe an identity-by-descent UPD screen of WGS data from 164 parent-child trios with rare disorders. Rare variant analysis was performed to identify deleterious variants on affected chromosomes. We show that applying in silico methods for the screening of UPD events in every patient with a suspected Mendelian disorder focuses WGS analysis and can improve diagnostic yields.
Methods
Samples and DNA isolation
The Research Ethics Committee of Children’s Health Ireland (CHI) at Temple Street approved the study protocol (Study number: 16.032). Informed consent was obtained according to current ethical and legal guidelines. Probands with undiagnosed rare diseases and both parents were recruited in CHI at Temple Street. The probands had a wide variety of indications for genetic testing. Genomic DNA was isolated at Genuity Science, Ireland, from whole peripheral blood using Qiagen’s Flexigene precipitation chemistry on FlexSTAR PLUS automated isolation instrument (AutoGen Inc., Holliston, MA, United States), according to the manufacturer’s protocol.
Whole-genome sequencing (WGS)
Whole-genome sequencing was performed using Illumina TruSEQ DNA PCR-free whole-genome sequencing with 2 × 150 bp pair-end sequencing reads on a NovaSeq-6000 (Illumina Inc, San Diego, CA, United States), following the manufacturer’s protocol. The minimum mean sequencing coverage was 30x, with a minimum of 95% of target bases covered at least ten times. Using an automated Sentieon (Freed et al., 2017) based pipeline, raw sequencing FASTQ data were aligned to the GRCh38/hg38 reference genome build via Burrows-Wheeler aligner (BWA) and variants were called using mathematical models of the Best Practices Genome Analysis Toolkit (GATK).
UPD screen
For each trio, GVCF files containing WGS data were merged and joint genotyped using GATK (Poplin et al., 2017). Biallelic variants with a minor allele frequency of
WGS rare variant analysis
VCF and BAM files from the trios were uploaded to Genuity Science Clinical Sequence Analyzer (CSA®). All data was stored according to genomic position in the Genuity Science Genomically Ordered Relational Database (GORdb) to facilitate rapid access by the CSA® (version 4.18) user interface and Sequence Miner visualisation software, which were used for variant analysis. Variants were annotated for functional effect by Variant Effect Prediction (VEP-Ensembl, Version 96.2) and annotated with allele frequencies from publicly available large population databases such as Single Nucleotide Polymorphism database (dbSNP) and Genome Aggregation Database (GnomAD) (Karczewski et al., 2020), as well as an internal database of Irish controls. The analysis was limited to a subset of genes based on case-specific HPO terms, gene panels and OMIM classifications. Only rare variants (Minor Allele Frequency <0.03) classified as moderate or high impact by VEP were analysed. Alamut Visual software (version 2.12, Interactive Biosoftware) was used to analyse the impact of candidate variants.
Sanger sequencing
Sanger sequencing was performed using the BigDye® Direct Cycle Sequencing Kit in combination with the BigDye XTerminator™ Purification Kit (Applied Biosystems, Foster City, CA, EUA) and the Applied Biosystems SeqStudio™ Genetic Analyzer. Sequencing data was analysed using the software GeneStudio™ Professional Edition Version 2.2.0.0 (GeneStudio, Inc.).
Results
Our UPD screen of 164 trios revealed three instances of whole chromosome UPiD: one case of maternal isodisomy of chromosome 1 (P1) and two cases of paternal isodisomy of chromosome 2 (P2 and P3). Uniparental disomy was detected by isolating large chromosomal regions where homozygous genotypes in the proband could not be ascribed to biparental inheritance (Figure 1A). No partial UPD events were found in our dataset (with a 5 Mb limit of resolution). Heterozygosity across each chromosome was assessed to differentiate between UPiD and UPhD (Figure 1B). Heterozygosity across chromosome 1 (P1) and chromosome 2 (P2 and P3) approached zero, indicative of UPiD. Some other probands exhibited low heterozygosity across their chromosomes which was due to recent consanguinity in their family tree. Conversely, one proband exhibited high levels of heterozygosity on chromosome 21. This proband had Down Syndrome.
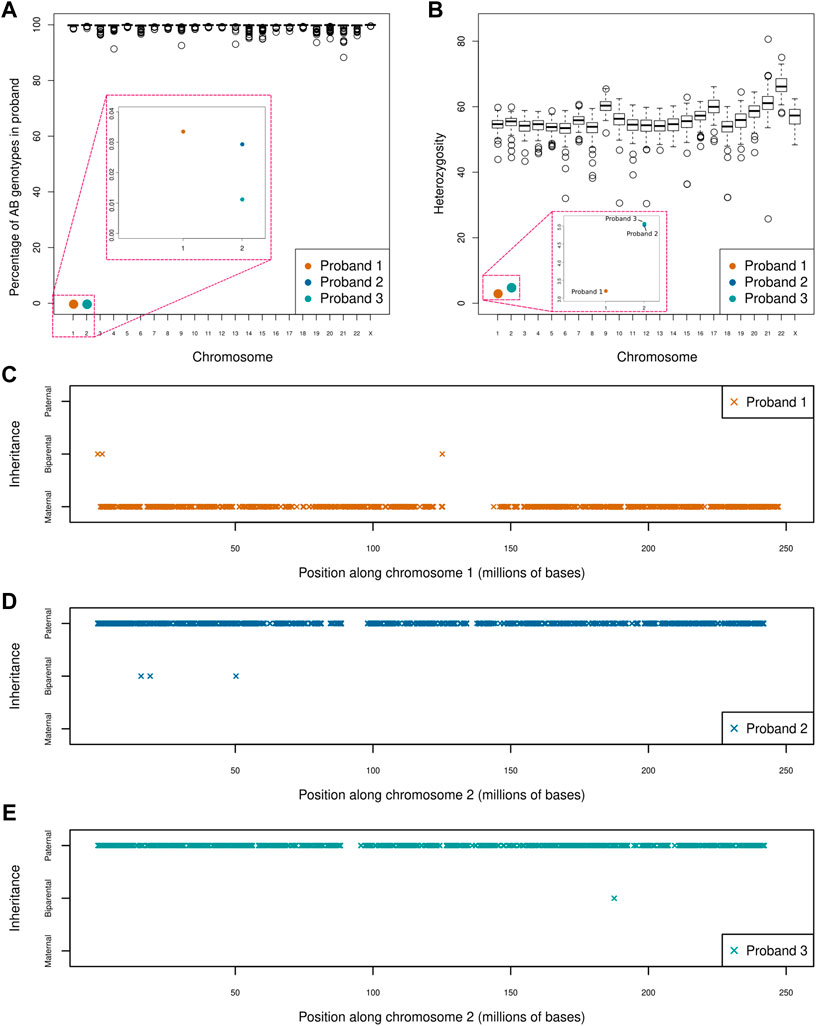
FIGURE 1. Uniparental disomy screen of 164 parent-child trios (A) Boxplot shows the percentage of AB genotypes in each child (represented by circles) when one parent has an AA genotype and the other parent has a BB genotype across each chromosome. The boxes in red indicate a zoom of that region of the plot for ease of visualisation. Three clear outliers can be seen affecting chromosome 1 (P1) and chromosome 2 (P2 and P3). Large regions of apparent Mendelian error across single chromosomes (e.g., in chromosomes 4, 9, and 14) were identified. An assessment of coverage across these regions confirmed these events to be a consequence of inherited or de novo deletions rather than partial UPD events. One of the large deletions had previously been identified by chromosomal microarray (B) Boxplot shows the percentage heterozygosity across each chromosome for every proband (C–E) Shows the direction and extent of disomy in P1 (C), P2 (D) and P3 (E). Proband 1 shows maternal isodisomy of chromosome 1 with 99.97% of genotypes matching that of the mother, while proband 2 and 3 exhibits paternal isodisomy of chromosome 2 with 99.99 and 99.97% of genotypes matching those of the father, respectively. This indicates that all three UPiD events affect the whole chromosome (these small deviations from 100% were attributed to sequencing and genotype calling errors). Only genotypes where the parents were homozygous for different alleles were used in this analysis.
Analysis of WGS data from all three probands prioritised rare homozygous variants in the isodisomic chromosomes which were not homozygous in the parents’ samples. Only variants in genes associated with the probands’ phenotypes were analysed (see phenotype summary on Table 1 and detailed clinical history in Supplementary Data). A homozygous frameshift variant at position chr1:165,759,558 c.176_177del p. (Glu59ValfsTer20) (NM_019026.6) in the Transmembrane and coiled-coil domains protein 1 (TMCO1) gene (MIM: 614123) was identified in P1. The genome of P1 was sequenced to a mean coverage of 30.35x. The genotype position had 0x coverage while the position 3’ of the deletion on the forward strand had 25x coverage and a genotype call ratio of 1. The variant is absent from gnomAD (Karczewski et al., 2020). Bi-allelic loss-of-function variants in TMCO1 are associated with Craniofacial dysmorphism, skeletal anomalies, and intellectual disability syndrome (CFSMR; MIM: 213980). The clinical description of P1 is consistent with the spectrum of the condition (Table 1; Supplementary Data).
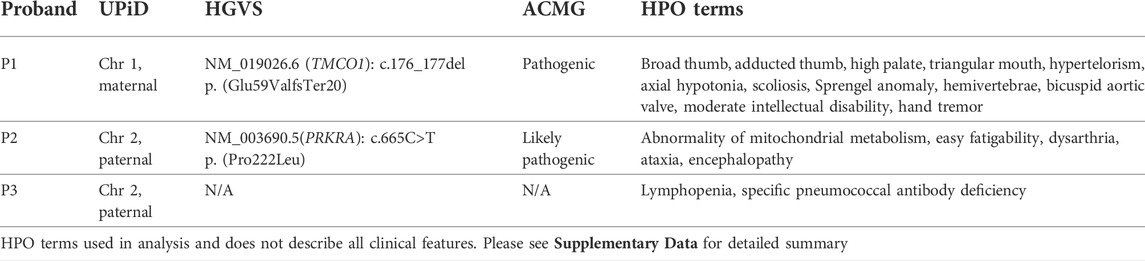
TABLE 1. Summary of clinical features and analysis results. Phenotype column shows HPO terms used in analysis.
Analysis of P2 identified a previously reported homozygous missense variant; chr2:178436264G>A c.665C>T p. (Pro222Leu) (NM_003690.5) in the Protein kinase, interferon-inducible double-stranded RNA-dependent activator (PRKRA) gene. The genome of P2 was sequenced to a mean coverage of 41.79x. The genotype position had 48x coverage and a genotype call ratio of 1. The p. (Pro222Leu) missense variant is a known variant associated with a dystonia-parkinsonism phenotype (MIM: 612067). The variant allele frequency is 0.00009905 on gnomAD v2.1.1, with no homozygotes reported. It has been reported in dbSNP (rs121434410) and classified as pathogenic in ClinVar (VCV000006346.4).
No homozygous pathogenic variants were identified in P3. The variants identified in P1 and P2 were confirmed by Sanger sequencing. A review of P1 and P2 at a multidisciplinary meeting confirmed clinical correlation with findings. Both variants were classified according to ACMG guidelines (Richards et al., 2015) (Table 1).
Discussion
We applied a WGS screening approach to identify UPD events in 164 probands from an undiagnosed cohort. Chromosomes with whole chromosome isodisomy were prioritised in the WGS analysis for deleterious variation. We identified three independent events, two of which rendered parental heterozygous pathogenic variants homozygous in probands (P1 and P2). The maternal UPiD event on chromosome 1 unmasked a novel frameshift variant in the TMCO1 gene, predicted to produce two null allele, associated with CFSMR (MIM:213980). The paternal UPiD event on chromosome 2 in P2 uncovered a recurrent missense variant in the PRKRA gene reported in Dystonia 16 (MIM: 612067).
Recent studies provide estimates on the incidence of UPD in clinical cohorts with diverse phenotypic indications, ranging from 1 in 176–500 (King D. A. et al., 2014; Yauy et al., 2020; Scuffins et al., 2021), compared to 1 in 2,000 in the general population (Nakka et al., 2019) One study found that approximately half of UPD events were considered diagnostic or indirectly diagnostic (Scuffins et al., 2021). Clinically significant homozygosity as a consequence of UPD accounted for 1699 diagnoses, six of which localised to chromosome 1 (Scuffins et al., 2021). The incidence of UPD in our cohort was approximately 1 in 55 births which is higher than previously reported (King D. A. et al., 2014; Nakka et al., 2019; Yauy et al., 2020; Scuffins et al., 2021). It should be noted that we did not screen for mosaic uniparental disomy events, which could yet increase the incidence of UPD further. Whole chromosome UPiD is the rarest type of UPD encountered in the general population (1 in ∼7,500) compared to UPhD (1 in ∼5,500) and partial UPiD (1 in ∼5,500) (Nakka et al., 2019). However, in our cohort of 164 probands, it was the only type of UPD identified. Whole chromosome isodisomy may present a greater clinical risk, as the probability of unmasking rare and damaging recessive variants increases in proportion to the quantity of DNA affected. It is therefore not surprising that an undiagnosed cohort could have a higher proportion of complete isodisomy events.
Truncating genetic variants in patients with CFSMR are hypothesized to abrogate the normal protein function of TMCO1. All pathogenic variants identified to date are consistent with this hypothesis (Xin et al., 2010; Caglayan et al., 2013; Tender and Ferreira, 2018; Sharkia et al., 2019). TMCO1 is an important regulator of many cellular processes including transcription, cell death, and proliferation, acting on the maintenance of endoplasmic reticulum (ER) Ca2+ homeostasis by preventing overfilling of ER stores with Ca2+ ions (Wang et al., 2016). This is the first report of TMCO1 and UPD, an ever-growing list of disorders associated with UPD (King J. E. et al., 2014).
The homozygous variant p. (Pro222Leu) in P2 was first identified in six individuals from two consanguineous families (Camargos et al., 2008). Dystonia 16 (MIM: 612067) is a generalized dystonia, with predominant oromandibular, craniofacial and axial involvement. PACT, the protein counterpart of PRKRA, activates PKR, a regulator of how cells respond to viral infections, ER stress, growth factor deprivation, and oxidative stress. The p. (Pro222Leu) variant produces an altered kinetic response to ER-stress. It is thought to foster stronger homo-dimer interactions with itself and with the RNA-activated protein kinase, PKR, leading to an aberrant stress response with a concomitant increase in apoptosis (Vaughn et al., 2015; Burnett et al., 2020). While the age-of-onset for P2 (3 years) is slightly younger than previous reports (average 8 years (range: 4–14 years) (Lange et al., 2021), the phenotype is otherwise typical of Dystonia 16, with predominant dysarthria. The encephalopathic illness (Supplementary Data) may have precipitated earlier onset of presentation (Masnada et al., 2021).
Attributing homozygosity to an inheritance error rather than parental carriers has important implications for family planning and cascade testing. The identification of a homozygous variant would often imply a recurrence risk of 25%. Establishing the source of homozygosity facilitates accurate genetic counselling (recurrence risk is negligible) and avoids the incorrect presumption of non-paternity (King J. E. et al., 2014).
The UPiD event in P3 may represent a benign and incidental finding with no pathological impact. It may equally represent a clinically significant event, but at this time no candidate gene was identified. As the curation of novel disease-gene associations is a continuing effort we stress the importance of periodic reanalysis for P3 (Costain et al., 2018; James et al., 2020).
In summary, our data supports UPD screening as a method of clinical value in the analysis of rare disorders. The incidence of UPiD in our cohort was approximately 1 in 55 births which is higher than previously reported incidences in the general population (Nakka et al., 2019) This may represent an overestimation, or it could suggest that isodisomic events present greater clinical risk. While isodisomy in many instances is benign, the coincidence of isodisomy and a severe congenital disorder should be investigated as potentially causal.
Data availability statement
The datasets presented in this article are not readily available because they relate to paediatric rare disease patients under the care of Children’s Health Ireland. Requests to access the datasets should be directed to Children’s Health Ireland.
Ethics statement
The studies involving human participants were reviewed and approved by The Research Ethics Committee of Children’s Health Ireland (CHI) at Temple Street (Study number: 16.032). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
BM, EJ, NL, and PB designed the study and analysed the genomic data. TL, BL, IK, MK, and KG provided clinical input.
Funding
This research was supported by the Genuity Science.
Acknowledgments
We thank all the clinicians and families who participated in this study. We thank the lab team at Genuity Science for generating the data, including Tim Chambers, Stephanie O’Brien and Claire Garvin who performed Sanger sequencing. We thank Aoife Coughlan, Stephanie Roe, Emma Gillen, Jackie Dolan and Anne Jones for their support, Javier Núñez, Raony Guimaraes, Jóhann Haukur Sigurðsson, Hildur Ólafsdóttir, Árni Einarsson, Heiðdís Rut Hreinsdóttir and Valdimar Ragnarsson for their bioinformatic/technical support, Emma Clarke for her clinical assistance and James O’Byrne for reviewing the manuscript.
Conflict of interest
Authors BM, EJ, NL, and PB were employed by the company Genuity Science.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.945296/full#supplementary-material
References
Bis, D. M., Schüle, R., Reichbauer, J., Synofzik, M., Rattay, T. W., Soehn, A., et al. (2017). Uniparental disomy determined by whole-exome sequencing in a spectrum of rare motoneuron diseases and ataxias. Mol. Genet. Genomic Med. 5, 280–286. doi:10.1002/mgg3.285
Burnett, S. B., Vaughn, L. S., Sharma, N., Kulkarni, R., and Patel, R. C. (2020). Dystonia 16 (DYT16) mutations in PACT cause dysregulated PKR activation and eIF2α signaling leading to a compromised stress response. Neurobiol. Dis. 146, 105135. doi:10.1016/j.nbd.2020.105135
Caglayan, A. O., Per, H., Akgumus, G., Gumus, H., Baranoski, J., Canpolat, M., et al. (2013). Whole-exome sequencing identified a patient with TMCO1 defect syndrome and expands the phenotic spectrum. Clin. Genet. 84, 394–395. doi:10.1111/cge.12088
Camargos, S., Scholz, S., Simón-Sánchez, J., Paisán-Ruiz, C., Lewis, P., Hernandez, D., et al. (2008). DYT16, a novel young-onset dystonia-parkinsonism disorder: Identification of a segregating mutation in the stress-response protein PRKRA. Lancet. Neurol. 7, 207–215. doi:10.1016/S1474-4422(08)70022-X
Costain, G., Jobling, R., Walker, S., Reuter, M. S., Snell, M., Bowdin, S., et al. (2018). Periodic reanalysis of whole-genome sequencing data enhances the diagnostic advantage over standard clinical genetic testing. Eur. J. Hum. Genet. 26, 740–744. doi:10.1038/s41431-018-0114-6
Del Gaudio, D., Shinawi, M., Astbury, C., Tayeh, M. K., Deak, K. L., and Raca, G. (2020). Diagnostic testing for uniparental disomy: A points to consider statement from the American College of medical genetics and genomics (ACMG). Genet. Med. 22, 1133–1141. doi:10.1038/s41436-020-0782-9
Engel, E. (1980). A new genetic concept: Uniparental disomy and its potential effect, isodisomy. Am. J. Med. Genet. 6, 137–143. doi:10.1002/ajmg.1320060207
Freed, D., Aldana, R., Weber, J. A., and Edwards, J. S. (2017). The Sentieon Genomics Tools - a fast and accurate solution to variant calling from next-generation sequence data (preprint). Bioinformatics. doi:10.1101/115717
James, K. N., Clark, M. M., Camp, B., Kint, C., Schols, P., Batalov, S., et al. (2020). Partially automated whole-genome sequencing reanalysis of previously undiagnosed pediatric patients can efficiently yield new diagnoses. NPJ Genom. Med. 5, 33. doi:10.1038/s41525-020-00140-1
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. doi:10.1038/s41586-020-2308-7
King, D. A., Fitzgerald, T. W., Miller, R., Canham, N., Clayton-Smith, J., Johnson, D., et al. (2014). A novel method for detecting uniparental disomy from trio genotypes identifies a significant excess in children with developmental disorders. Genome Res. 24, 673–687. doi:10.1101/gr.160465.113
King, J. E., Dexter, A., Gadi, I., Zvereff, V., Martin, M., Bloom, M., et al. (2014). Maternal uniparental isodisomy causing autosomal recessive GM1 gangliosidosis: A clinical report. J. Genet. Couns. 23, 734–741. doi:10.1007/s10897-014-9720-9
Lange, L. M., Junker, J., Loens, S., Baumann, H., Olschewski, L., Schaake, S., et al. (2021). Genotype-phenotype relations for isolated dystonia genes: MDSGene systematic review. Mov. Disord. 36, 1086–1103. doi:10.1002/mds.28485
Masnada, S., Martinelli, D., Correa-Vela, M., Agolini, E., Baide-Mairena, H., Marcé-Grau, A., et al. (2021). PRKRA-related disorders: Bilateral striatal degeneration in addition to DYT16 spectrum. Mov. Disord. 36, 1038–1040. doi:10.1002/mds.28492
Nakka, P., Pattillo Smith, S., O’Donnell-Luria, A. H., McManus, K. F., Mountain, J. L., Ramachandran, S., et al. (2019). Characterization of prevalence and Health consequences of uniparental disomy in four million individuals from the general population. Am. J. Hum. Genet. 105, 921–932. doi:10.1016/j.ajhg.2019.09.016
Poplin, R., Ruano-Rubio, V., DePristo, M. A., Fennell, T. J., Carneiro, M. O., Van der Auwera, G. A., et al. (2017). Scaling accurate genetic variant discovery to tens of thousands of samples. Genomics. doi:10.1101/201178
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). ACMG laboratory quality assurance CommitteeStandards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Scuffins, J., Keller-Ramey, J., Dyer, L., Douglas, G., Torene, R., Gainullin, V., et al. (2021). Uniparental disomy in a population of 32, 067 clinical exome trios. Genet. Med. 23, 1101–1107. doi:10.1038/s41436-020-01092-8
Sharkia, R., Zalan, A., Jabareen-Masri, A., Hengel, H., Schöls, L., Kessel, A., et al. (2019). A novel biallelic loss-of-function mutation in TMCO1 gene confirming and expanding the phenotype spectrum of cerebro-facio-thoracic dysplasia. Am. J. Med. Genet. A 179, 1338–1345. doi:10.1002/ajmg.a.61168
Tender, J. A. F., and Ferreira, C. R. (2018). Cerebro-facio-thoracic dysplasia (Pascual-Castroviejo syndrome): Identification of a novel mutation, use of facial recognition analysis, and review of the literature. Transl. Sci. Rare Dis. 3, 37–43. doi:10.3233/TRD-180022
Vaughn, L. S., Bragg, D. C., Sharma, N., Camargos, S., Cardoso, F., and Patel, R. C. (2015). Altered activation of protein kinase PKR and enhanced apoptosis in dystonia cells carrying a mutation in PKR activator protein PACT. J. Biol. Chem. 290, 22543–22557. doi:10.1074/jbc.M115.669408
Wang, Q.-C., Zheng, Q., Tan, H., Zhang, B., Li, X., Yang, Y., et al. (2016). TMCO1 is an ER Ca(2+) load-activated Ca(2+) channel. Cell 165, 1454–1466. doi:10.1016/j.cell.2016.04.051
Xin, B., Puffenberger, E. G., Turben, S., Tan, H., Zhou, A., and Wang, H. (2010). Homozygous frameshift mutation in TMCO1 causes a syndrome with craniofacial dysmorphism, skeletal anomalies, and mental retardation. Proc. Natl. Acad. Sci. U. S. A. 107, 258–263. doi:10.1073/pnas.0908457107
Yamazawa, K., Ogata, T., and Ferguson-Smith, A. C. (2010). Uniparental disomy and human disease: An overview. Am. J. Med. Genet. C Semin. Med. Genet. 154C, 329–334. doi:10.1002/ajmg.c.30270
Keywords: genetic screen, rare disease, genomics, uniparental, pathogenic
Citation: Molloy B, Jones ER, Linhares ND, Buckley PG, Leahy TR, Lynch B, Knerr I, King MD and Gorman KM (2022) Uniparental disomy screen of Irish rare disorder cohort unmasks homozygous variants of clinical significance in the TMCO1 and PRKRA genes. Front. Genet. 13:945296. doi: 10.3389/fgene.2022.945296
Received: 16 May 2022; Accepted: 23 August 2022;
Published: 14 September 2022.
Edited by:
Ben Pode-Shakked, Sheba Medical Center, IsraelReviewed by:
Hongzheng Dai, Baylor College of Medicine, United StatesPriyanka Nakka, Regeneron Genetic Center, United States
Copyright © 2022 Molloy, Jones, Linhares, Buckley, Leahy, Lynch, Knerr, King and Gorman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: B. Molloy, ben.molloy@genuitysci.com
†These authors have contributed equally to this work