 Jae Hyun Park
Jae Hyun Park Jisook Moon
Jisook Moon- Department of Biotechnology, College of Life Science, CHA University, Seongnam, South Korea
Our previous paper showed that microRNAs (miRNAs) present within human placental or mesenchymal stem cell-derived extracellular vesicles (EVs) directly interacted with the RNA genome of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), inhibiting viral replication. In this paper, we analyzed whether these miRNAs could exert antiviral activity against other variants of SARS-CoV-2. We downloaded compete SARS-CoV-2 genome data submitted to the National Center for Biotechnology Information for each SARS-CoV-2 variant, aligned the data to the reference SARS-CoV-2 genome sequence, and then confirmed the presence of 3′ untranslated region (UTR) mutations. We identified one type of 3′ UTR mutation in the Alpha variant, four in the Beta variant, four in the Gamma variant, three in the Delta variant, and none in the Omicron variant. Our findings indicate that 3′ UTR mutations rarely occur as persistent mutations. Interestingly, we further confirmed that this phenomenon could suppress virus replication in the same manner as the previously discovered interaction of placental-EV-derived miRNA with 3′ UTRs of SARS-CoV-2. Because the 3′ UTR of the SARS-CoV-2 RNA genome has almost no mutations, it is expected to be an effective therapeutic target regardless of future variants. Thus, a therapeutic strategy targeting the 3′ UTR of SARS-CoV-2 is likely to be extremely valuable, and such an approach is also expected to be applied to all RNA-based virus therapeutics.
Introduction
As of April 2022, coronavirus 2019 (COVID-19) has infected >500 million people worldwide and has been reported as responsible for >6 million deaths. COVID-19 is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). This positive single-strand RNA virus has been identified as a variant of betacoronavirus. SARS-CoV-2 shows approximately 96% homology with bat coronavirus and 79.5% homology with SARS-CoV-1; it is the seventh human coronavirus discovered up to now (Zhou et al., 2020).
RNA viruses have a mutation rate 100–10,000 times higher than that of DNA viruses, and this high mutation rate is related to viral evolution as well as lethal mutagenesis (Duffy, 2018; Peck & Lauring, 2018). Thus, viruses with mutations related to viral replication, transformation, or immune system evasion have a competitive advantage over others, whereas mutations inappropriate for survival tend to be eliminated. In particular, the mutation of coronaviruses is slower than that of other RNA viruses, possibly because of a proofreading function. However, despite the low mutation rate of coronaviruses, novel variants have been reported (Callaway, 2020). A currently well-known SARS-CoV-2 mutation is the D614G spike protein mutation, which increases infectivity (Korber et al., 2020). According to William et al., whether mutations affecting the SARS-CoV-2 phenotype will break through infection- or vaccine-acquired immunity remains a point of contention; however, there is growing evidence that we should be prepared for mutations that can cause breakthrough infections (Harvey et al., 2021).
MicroRNAs (miRNAs) are small noncoding RNA molecules containing 18–25 bases that induce RNA degradation and translational suppression. miRNA can affect viral replication by direct interaction with the viral RNA genome or by acting on host mRNA (Trobaugh and Klimstra, 2017; Fani et al., 2018). For example, miRNAs can inhibit the replication of viruses, such as the human immunodeficiency virus 1, enterovirus 71, and hepatitis C virus (Jopling et al., 2005; Nathans et al., 2009; Zheng et al., 2013). In addition, approaches based on miRNA for SARS-CoV-2 have been recently reported (Alam and Lipovich, 2021; Fani et al., 2021; Hum et al., 2021).
Trobaugh et al. reported that eastern equine encephalitis virus replication was inhibited by host miR-142-3p, and the deletion of the miR-142-3p binding site on virus mutants was positively selected during virus replication. Furthermore, they suggested that there was an unknown mechanism at the corresponding binding site that was necessary for efficient virus replication (Trobaugh et al., 2014; Trobaugh and Klimstra, 2017).
Although the correlation between the conservation of miRNA binding sites in the viral 3′ untranslated region (UTR) and the life cycle of the virus is unclear, the secondary or tertiary structure of the 3′ UTR in RNA viruses is a necessary control element for RNA replication (Williams et al., 1999). In fact, Koyama et al., who analyzed 10,022 SARS-CoV-2 genomes that had been uploaded to databases by April 2020, reported two types of 3′ UTR mutations: 131 mutations of 29742G→T and 115 mutations of 29870C→A (Koyama et al., 2020). This extremely low mutation rate indicates that the SARS-CoV-2 3′ UTR region is much more stable (i.e., less susceptible to mutations) than other regions.
In a previous study, we studied the antiviral effect of miRNAs based on the SARS-CoV-2 3′ UTR mutation stability and the potential interaction between miRNAs and the viral RNA genome. More specifically, we selected five miRNAs (miR-92a-3p, miR-26a-5p, miR-23a-3p, miR-103a-3p, and miR-181a-5p) present in placental extracellular vesicles (EVs) that were predicted to interact with the RNA genome of SARS-CoV-2. We also experimentally verified that the miRNAs bound to the complementary 3′ UTR of SARS-CoV-2 and inhibited viral replication (Figure 1A) (Park et al., 2021). Moreover, five miRNAs not only blocked SARS-CoV-2 replication but also regulated stress-induced inflammatory environment. Here, we further investigated whether the 3′ UTR of the SARS-CoV-2 binding site for EV miRNAs is conserved for various currently occurring variants using sequencing information submitted to the National Center for Biotechnology Information (NCBI) and predicted the potential effects of EV miRNAs on future variants.
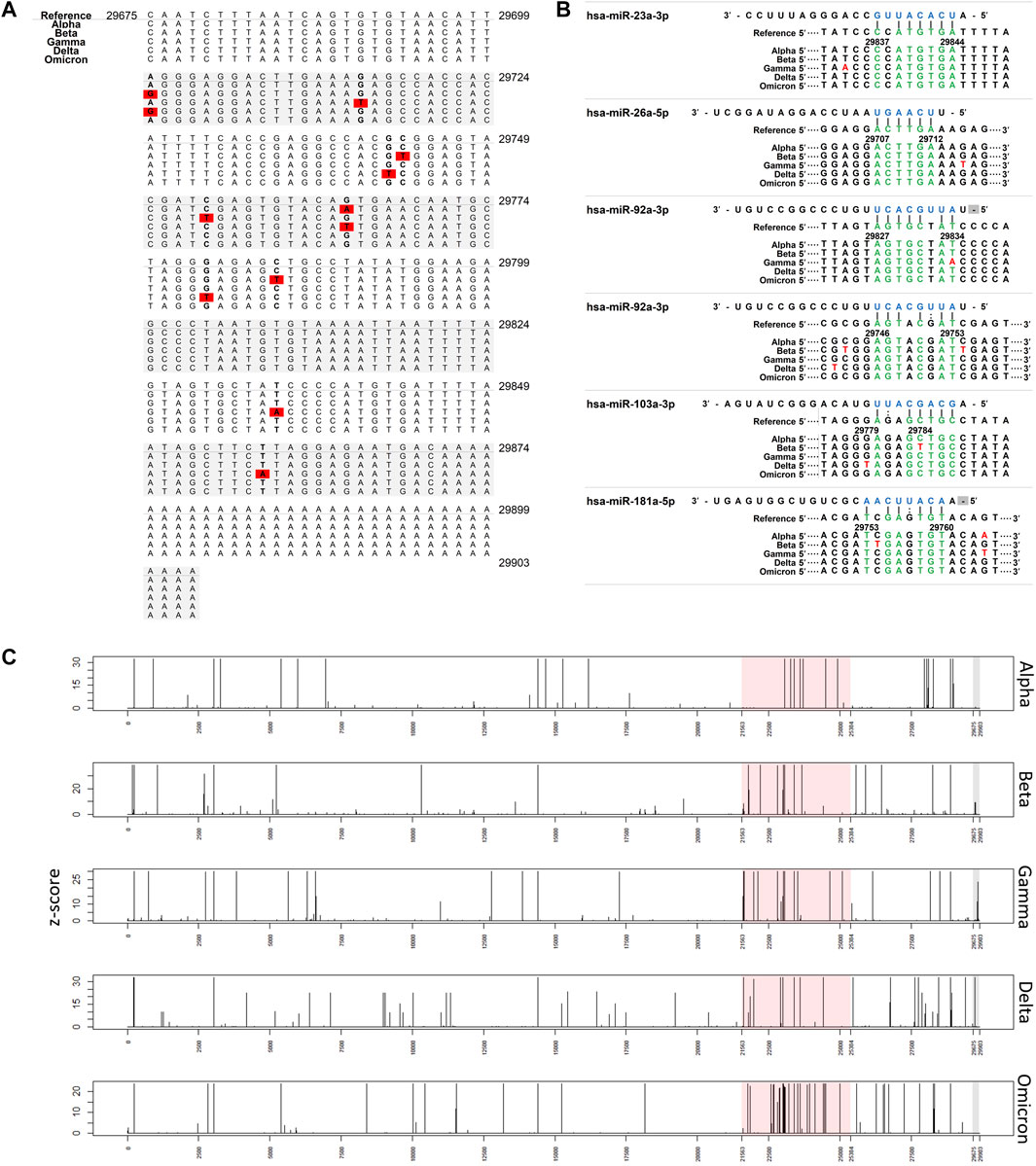
FIGURE 1. Interaction of miRNAs on SARS-CoV-2 3′ UTRs. (A) Nucleotide change in 3′ UTR of SARS-CoV-2 Red: nucleotide changes (associated with Table 2). (B) miRNA binding site in 3′ UTR of SARS-CoV-2. Nucleotide changes observed more than 1% were presented. (C) Plot showing the frequency of nucleotide changes in the SARS-CoV-2 genome. The frequency was scaled by the z-score within each variant. Red shading indicates spike glycoprotein coding region and grey shading indicates the 3′ UTR region.
Materials and Methods
Severe Acute Respiratory Syndrome Coronavirus 2 Complete Sequence Data
We analyzed sequence data selected by the World Health Organization for five variants of concern (Alpha, Beta, Delta, Gamma, and Omicron) as of December 2021. The SARS-CoV-2 reference genome (NC_045512.2) and complete genome sequence data of SARS-CoV-2 were downloaded from the NCBI and NCBI Virus databases, respectively. The sequence data were selected using the following filters: txid2697049; nucleotide completeness: complete; and collection dates: ∼2021-12-17.
Each variant was sorted according to the Pango lineage classification option of NCBI Virus. The Pango lineages used for the search were B.1.1.7, B.1.351, P.1, B.1.617.2, and BA.1 for the Alpha, Beta, Delta, Gama, and Omicron variants, respectively (Table 1).
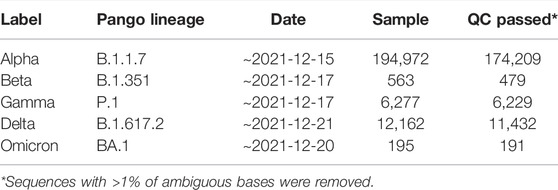
TABLE 1. SARS-CoV-2 sample information.
Sequencing Data Alignment and Analysis
We performed multiple sequence alignment against the SARS-CoV-2 reference genome using MAFFT software version 7.487 (Nakamura et al., 2018), which can rapidly calculate full-length multiple sequence alignments of closely-related viral genomes. Sequences with >1% of ambiguous bases were removed. For 3′ UTR region analysis, we extracted the sequences aligned between 29,675 and 29,903 in the SARS-CoV-2 reference genome (range of the 3′ UTR).
Prediction of MiRNA-Viral 3′ Untranslated Region Interaction
We used the PITA tool (Kertesz et al., 2007) to investigate and predict the miRNA binding sites in the 3′ UTR of SARS-CoV-2, using the SARS-CoV-2 complete genome sequence (NC_045512.2), from which the 3′ UTR sequence was also extracted.
Graphics
Plots were drawn using R programming language. The frequency of nucleotide changes in the SARS-CoV-2 genome was scaled by the z-score.
Results
We analyzed the following sequence data obtained from the NCBI database: 194,972 Alpha variants of SARS-CoV-2 (Pango lineage B.1.1.7), 563 Beta variants (B.1.351), 6277 Gamma variants (P.1), 12,162 Delta variants (B.1.617.2), and 195 Omicron variants (BA.1) (Table 1). Sequences with >1% of ambiguous bases were removed. We aligned the sequencing data obtained from NCBI to the SARS-CoV-2 reference genome (accession number: NC_045512.2) using the MAFFT Tool (Nakamura et al., 2018), and through this, the position where the mutation occurred for each variant was determined. We first investigated nucleotide changes at the 3′ UTR of SARS-CoV-2. Although we found 348 cases of nucleotide changes in the Alpha variant, 17 in the Beta variant, 86 in the Gamma variant, 138 in the Delta variant, and 3 in the Omicron variant. The frequency of nucleotide changes in the 3′ UTR was very low in five variants. We further investigated the nucleotide changes with a frequency of >1% in all samples for each variant, and the following nucleotide changes were identified: 29764G→A in the Alpha variant; 29754C→T, 29743C→T, 29700A→G, and 29784C→T in the Beta variant; 29834T→A, 29858T→A, 29764G→T, and 29715G→T in the Gamma variant; and 29742G→T, 29779G→T, and 29700A→G in the Delta variant (Figure 1A and Table 2). Table 2 shows nucleotide changes that were observed in more than 1%. We identified that nucleotide changes in the 3′ UTR had a lower frequency of mutation compared to the spike glycoprotein coding region currently undergoing rapid mutation. In the Omicron variant, one case each of 29742G→T, 29772T→C, and 29818A→T mutations located in the 3′ UTR of SARS-CoV-2 were found, but in all three cases, the frequency was <1% of the total samples (n = 191). It must be taken into account that in the case of the Omicron variant, the sample size was small as of December 2021, so continuous monitoring and analysis are required. All nucleotide changes that we identified are presented in Supplementary Table S1.
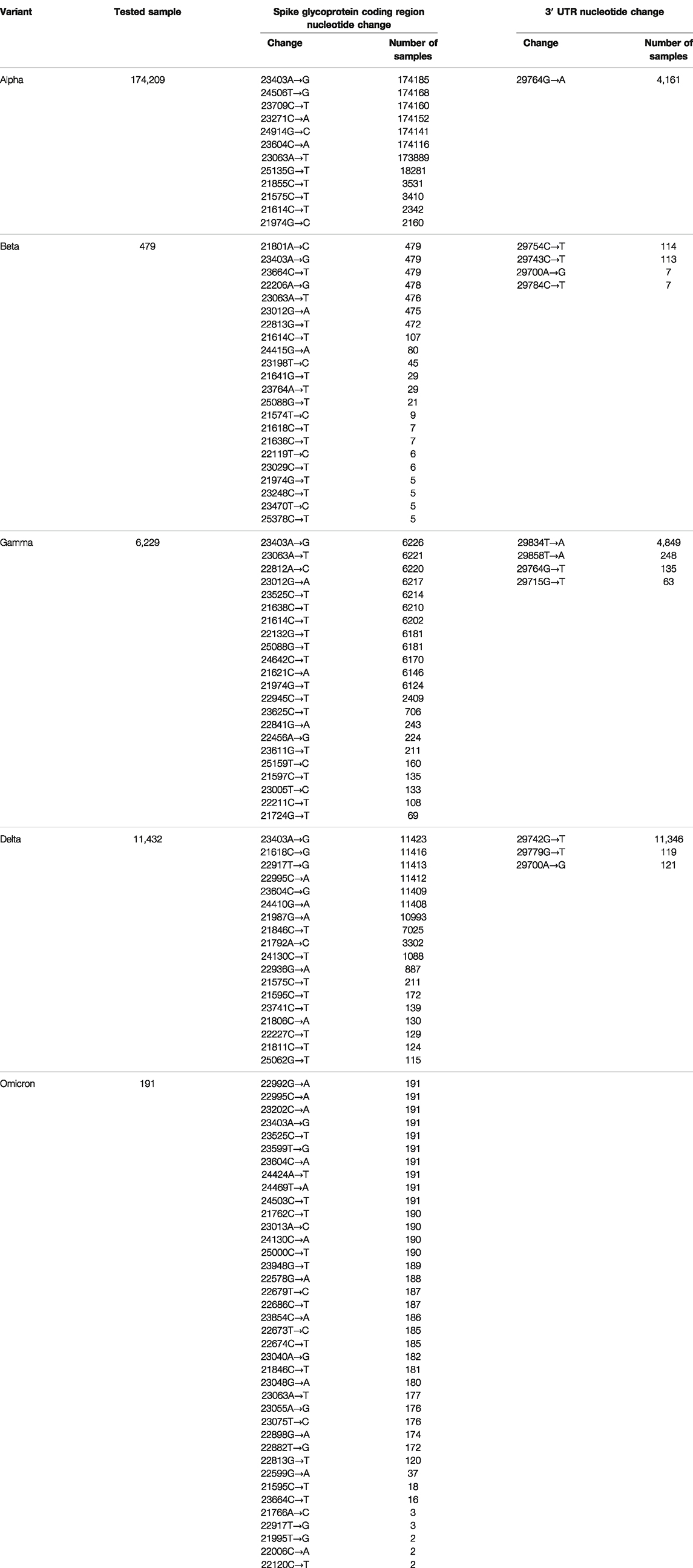
TABLE 2. Number of nucleotide changes in the 3′ UTR of SARS-CoV-2.
We used a miRNA binding prediction tool to further investigate how these 3′ UTR mutations affect miRNA binding (Table 3). Table 3 shows thermodynamic energy required for binding (kcal/mol). Lower energy indicates stronger binding prediction. In a recent study from our laboratory (Park et al., 2021), five miRNAs (hsa-miR-103a-3p, hsa-miR-181a-5p, hsa-miR-23a-5p, hsa-miR-26a-5p, and hsa-miR-92a-3p) were predicted and experimentally verified to interact with mutant 3′ UTR sites of SARS-CoV-2 reference sequences (Figure 1B). Most of the interactions between these miRNAs and the mutation sites on 3′ UTRs of the SARS-CoV-2 genome did not change their binding energy when compared with interactions between the reference 3′ UTRs of the SARS-CoV-2 genome. However, the binding of hsa-miR-103a-3p in the Beta variant and hsa-miR-92a-3p in the Gamma variant to the 29784C→T and 29834T→A mutations, respectively, of the 3′ UTR of SARS-CoV-2 genome was not predicted. Notably, the binding affinity to hsa-miR-181a-5p was increased compared with the reference sequence in the 29754C→T mutation in the Beta variant. These findings suggest that the miRNA binding site can disappear as the result of specific 3′ UTR mutations of SARS-CoV-2, consequently diminishing most of the related miRNA affinity. However, in certain miRNAs, the binding affinity is predicted to increase despite the site change due to mutation, suggesting that efficacy may vary depending on miRNA characteristics (Table 3).
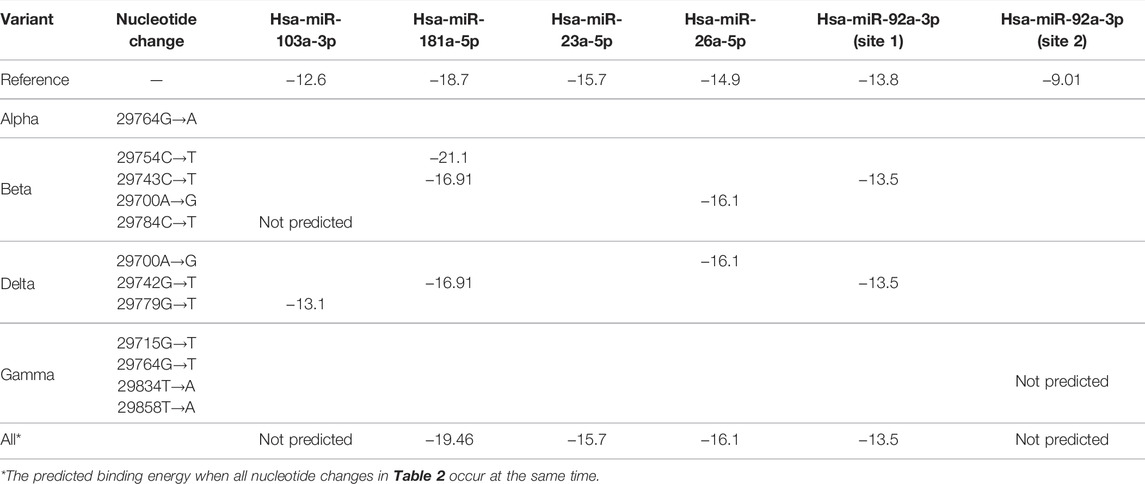
TABLE 3. Binding energy prediction in 3′ UTRs of SARS-CoV-2.
Subsequently, to determine whether the miRNA binding site in the 3′ UTR is conserved among variants, we investigated the frequency of mutations in the human miRNA binding site in the SARS-CoV-2 3′ UTR (Figure 1B). The colored spot in Figure 1B shows the site where many nucleotide changes have been detected. In the Alpha variant, mutations were found within the miRNA binding site, but the frequency was extremely low (<1%). Furthermore, hardly any mutations in the hsa-miR-23a-3p binding site were found in the five variants. This suggests that hsa-miR-23a-3p can be used as a therapeutic agent regardless of the SARS-CoV-2 variant.
Subsequently, to measure how often nucleotide changes occur in the 3′ UTR, we investigated the frequency of nucleotide changes in the whole SARS-CoV-2 genome to determine the relative frequency of 3′ UTR nucleotide changes (Figure 1C). Mutations in 3′ UTR were relatively minor when compared with the frequency of nucleotide changes in the whole genome. In particular, nucleotide changes in the 3′ UTR region were detected less frequently than those in the spike glycoprotein coding region. Major mutations in the 3′ UTR were only found in the Beta, Gamma, and Delta variants.
Discussion
We found a major UTR mutation in the 3′ UTR in the SARS-CoV-2 Gamma and Delta variants. In the Gamma variant, the 29834T→A mutation accounted for 78% of the analyzed cases, and in the Delta variant, the 29742G→T mutation accounted for 99% of the analyzed cases. The effect of these 3′ UTR mutations on the replication and infectivity of the Gamma and Delta variants is unknown. We speculated that they would not affect the binding affinity of the five miRNAs (miR-92a-3p, miR-26a-5p, miR-23a-3p, miR-103a-3p, and miR-181a-5p) because we previously identified antiviral effects when these five miRNAs bound to the SARS-CoV-2 3′ UTR region (Figure 1B).
Meanwhile, the 29784C→T and 29834T→A mutations in the SARS-CoV-2 3′ UTR region of the Beta and Gamma variants were predicted to inhibit the binding affinity of hsa-miR-103a-3p and hsa-miR-92a-3p, respectively. These are sites of attachment for the seed regions of miRNAs, which play an essential role in miRNA-RNA interaction. Analysis of the match of these seed regions using the miRNA binding prediction tool revealed 3′ UTR mismatches. Although hsa-miR-92a-3p was not predicted to bind at position 29834 of the SARS-CoV-2 3′ UTR, it was predicted to bind to another site in the SARS-CoV-2 3′ UTR (Table 3), and this site was unaffected by the 29834T→A 3′ UTR mutation. Additionally, because the other binding site has a higher binding affinity with hsa-miR-92a-3p than the site at position 29834, it plays a more prominent role in the interaction between hsa-miR-92a-3p and the SARS-CoV-2 3′ UTR than the binding site at position 29834. In a previous study, we observed that a combination treatment of miRNAs was more effective against the 3′ UTR of SARS-CoV-2, suggesting that there is a synergistic effect on the miRNAs. We suspect that the synergistic effect on the miRNAs, which may be attributed to different binding sites.
Although the antiviral efficacy of miRNAs against SARS-CoV-2 has been predicted despite mutations that have occurred thus far, there are some miRNAs whose binding affinity is predicted to change due to mutation. Regarding the binding of hsa-miR-181-a-5p, the 29743C→T mutation of the Beta variant and the 29742G→T mutation of the Delta variant are expected to slightly decrease the binding affinity. In contrast, the binding energy of hsa-miR-181-a-5p is predicted to be stronger in the 29754C→T mutation of the Beta variant. Thus, changes in the binding affinity of miRNAs according to variants will be a major consideration when establishing an antiviral treatment strategy using miRNAs for COVID-19 in the future.
It was revealed that the binding sites of the five miRNAs that we previously reported as showing an antiviral effect (Park et al., 2021) were mostly conserved in other variants, even in newly occurring SARS-CoV-2 variants. These findings predict that EV- or miRNA-based antiviral therapy will be effective against other major variants of SARS-CoV-2 that are likely to continue to occur in the future.
The current study did not investigate whether the reason why these five miRNA binding sites are mostly conserved across variants is to preserve the 3′ UTR function or whether there is an unknown mechanism, as Trobaugh et al. suggested (Trobaugh et al., 2014; Trobaugh and Klimstra, 2017). Although it is necessary to experimentally verify the antiviral efficacy of a therapeutic strategy targeting the 3′ UTR of SARS-CoV-2, our evidence convincingly shows the potential of miRNAs as the most appropriate antiviral treatment to counter the emergence of virus variants, which is the most significant problem.
Data Availability Statement
Publicly available datasets were analyzed in this study. This data can be found here: https://www.ncbi.nlm.nih.gov/labs/virus/
Author Contributions
JM contributed to the conception of the study. JP performed the computational analysis. JM and JP wrote the manuscript. All authors approved the submitted version.
Funding
This work was supported by the Bio and Medical Technology Development Program of the NRF funded by the Korean government, MSIP (NRF-2019M3A9H1103765).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.893141/full#supplementary-material
Reference
Callaway, E. (2020). The Coronavirus Is Mutating - Does it Matter? Nature 585 (7824), 174–177. doi:10.1038/d41586-020-02544-6
Duffy, S. (2018). Why Are RNA Virus Mutation Rates So Damn High? PLoS Biol. 16 (8), e3000003. doi:10.1371/journal.pbio.3000003
Fani, M., Zandi, M., Rezayi, M., Khodadad, N., Langari, H., and Amiri, I. (2019). The Role of microRNAs in the Viral Infections. Cpd 24 (39), 4659–4667. doi:10.2174/1381612825666190110161034
Harvey, W. T., Carabelli, A. M., Jackson, B., Gupta, R. K., Thomson, E. C., Harrison, E. M., et al. (2021). SARS-CoV-2 Variants, Spike Mutations and Immune Escape. Nat. Rev. Microbiol. 19 (7), 409–424. doi:10.1038/s41579-021-00573-0
Jopling, C. L., Yi, M., Lancaster, A. M., Lemon, S. M., and Sarnow, P. (2005). Modulation of Hepatitis C Virus RNA Abundance by a Liver-specific MicroRNA. Science 309 (5740), 1577–1581. doi:10.1126/science.1113329
Kertesz, M., Iovino, N., Unnerstall, U., Gaul, U., and Segal, E. (2007). The Role of Site Accessibility in microRNA Target Recognition. Nat. Genet. 39 (10), 1278–1284. doi:10.1038/ng2135
Korber, B., Fischer, W. M., Gnanakaran, S., Yoon, H., Theiler, J., Abfalterer, W., et al. (2020). Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell. 182 (4), 812–827. doi:10.1016/j.cell.2020.06.043
Koyama, T., Platt, D., and Parida, L. (2020). Variant Analysis of SARS-CoV-2 Genomes. Bull. World Health Organ. 98 (7), 495–504. doi:10.2471/BLT.20.253591
Nakamura, T., Yamada, K. D., Tomii, K., and Katoh, K. (2018). Parallelization of MAFFT for Large-Scale Multiple Sequence Alignments. Bioinforma. Oxf. Engl. 34 (14), 2490–2492. doi:10.1093/bioinformatics/bty121
Nathans, R., Chu, C.-y., Serquina, A. K., Lu, C.-C., Cao, H., and Rana, T. M. (2009). Cellular MicroRNA and P Bodies Modulate Host-HIV-1 Interactions. Mol. Cell. 34 (6), 696–709. doi:10.1016/j.molcel.2009.06.003
Park, J. H., Choi, Y., Lim, C.-W., Park, J.-M., Yu, S.-H., Kim, Y., et al. (2021). Potential Therapeutic Effect of Micrornas in Extracellular Vesicles from Mesenchymal Stem Cells against SARS-CoV-2. Cells 10 (9), 2393. doi:10.3390/cells10092393
Peck, K. M., and Lauring, A. S. (2018). Complexities of Viral Mutation Rates. J. Virol. 92 (14), 17. doi:10.1128/JVI.01031-17
Trobaugh, D. W., Gardner, C. L., Sun, C., Haddow, A. D., Wang, E., Chapnik, E., et al. (2014). RNA Viruses Can Hijack Vertebrate microRNAs to Suppress Innate Immunity. Nature 506 (7487), 245–248. doi:10.1038/nature12869
Trobaugh, D. W., and Klimstra, W. B. (2017). MicroRNA Regulation of RNA Virus Replication and Pathogenesis. Trends Mol. Med. 23 (1), 80–93. doi:10.1016/j.molmed.2016.11.003
Williams, G. D., Chang, R.-Y., and Brian, D. A. (1999). A Phylogenetically Conserved Hairpin-type 3′ Untranslated Region Pseudoknot Functions in Coronavirus RNA Replication. J. Virol. 73 (10), 8349–8355. doi:10.1128/JVI.73.10.8349-8355.1999
Zheng, Z., Ke, X., Wang, M., He, S., Li, Q., Zheng, C., et al. (2013). Human MicroRNA Hsa-miR-296-5p Suppresses Enterovirus 71 Replication by Targeting the Viral Genome. J. Virol. 87 (10), 5645–5656. doi:10.1128/JVI.02655-12
Keywords: SARS-CoV-2, COVID-19, extracellular vesicles (EVs), virus variants, miRNA
Citation: Park JH and Moon J (2022) Conserved 3′ UTR of Severe Acute Respiratory Syndrome Coronavirus 2: Potential Therapeutic Targets. Front. Genet. 13:893141. doi: 10.3389/fgene.2022.893141
Received: 10 March 2022; Accepted: 09 June 2022;
Published: 30 June 2022.
Edited by:
Graziano Pesole, University of Bari Aldo Moro, ItalyReviewed by:
Igor Jurak, University of Rijeka, CroatiaBobo Mok, The University of Hong Kong, Hong Kong SAR, China
Copyright © 2022 Park and Moon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jisook Moon, jmoon@cha.ac.kr