 Runjhun Mathur1,2
Runjhun Mathur1,2 Niraj Kumar Jha1,3,4
Niraj Kumar Jha1,3,4 Gaurav Saini5
Gaurav Saini5 Saurabh Kumar Jha1,4Sheo Prasad Shukla6Zita Filipejová7Kavindra Kumar Kesari8Danish Iqbal9,10Parma Nand1Vijay Jagdish Upadhye11
Saurabh Kumar Jha1,4Sheo Prasad Shukla6Zita Filipejová7Kavindra Kumar Kesari8Danish Iqbal9,10Parma Nand1Vijay Jagdish Upadhye11 Abhimanyu Kumar Jha1*Shubhadeep Roychoudhury12*Petr Slama13
Abhimanyu Kumar Jha1*Shubhadeep Roychoudhury12*Petr Slama13- 1Department of Biotechnology, School of Engineering and Technology, Sharda University, Greater Noida, India
- 2Dr. A.P.J Abdul Kalam Technical University, Lucknow, India
- 3Department of Biotechnology, School of Applied and Life Sciences (SALS), Uttaranchal University, Dehradun, India
- 4Department of Biotechnology Engineering and Food Technology, Chandigarh University, Mohali, India
- 5Department of Civil Engineering, Netaji Subhas University of Technology, Delhi, India
- 6Department of Civil Engineering, Rajkiya Engineering College, Banda, India
- 7Small Animal Clinic, University of Veterinary Sciences Brno, Brno, Czechia
- 8Department of Applied Physics, School of Science, Aalto University, Espoo, Finland
- 9Department of Medical Laboratory Sciences, College of Applied Medical Sciences, Majmaah University, Al Majma'ah, Saudi Arabia
- 10Health and Basic Sciences Research Center, Majmaah University, Al Majma'ah, Saudi Arabia
- 11Center of Research for Development (CR4D), Parul Institute of Applied Sciences (PIAS), Parul University, Vadodara, Gujarat
- 12Department of Life Science and Bioinformatics, Assam University, Silchar, India
- 13Department of Animal Morphology, Physiology, and Genetics, Faculty of AgriSciences, Mendel University in Brno, Brno, Czechia
Epigenetic modifications are inherited differences in cellular phenotypes, such as cell gene expression alterations, that occur during somatic cell divisions (also, in rare circumstances, in germ line transmission), but no alterations to the DNA sequence are involved. Histone alterations, polycomb/trithorax associated proteins, short non-coding or short RNAs, long non—coding RNAs (lncRNAs), & DNA methylation are just a few biological processes involved in epigenetic events. These various modifications are intricately linked. The transcriptional potential of genes is closely conditioned by epigenetic control, which is crucial in normal growth and development. Epigenetic mechanisms transmit genomic adaptation to an environment, resulting in a specific phenotype. The purpose of this systematic review is to glance at the roles of Estrogen signalling, polycomb/trithorax associated proteins, DNA methylation in breast cancer progression, as well as epigenetic mechanisms in breast cancer therapy, with an emphasis on functionality, regulatory factors, therapeutic value, and future challenges.
Introduction
The greatest serious hazard to women’s health in developed countries is breast cancer. Breast cancer is the most frequent cancer in women around the world (Bray et al., 2004; Chen et al., 2016) and distant metastasis is the major cause of poor survival (Gupta and Massague, 2006; Hanahan and Weinberg, 2011). The most common cause of death among breast cancer patients is metastasis. The molecular pathways that drive tumour cells to become metastatic have been thoroughly investigated, leading to major advances in prediction and treatment techniques. However, the significant high percentage of breast cancer related fatalities continues to be a major source of concern. As a result, elucidating novel metastasis-related molecular processes is critical for improving breast cancer therapy outcomes.
Since 2004, invasive breast cancer has been on the rise, in 2018, more than two million instances were reported around the world and more than 270,000 instances projected in the United States by 2020.
It is vital to completely understand the molecular pathways that enable breast cancer cell metastasis in order to create strategies to improve breast cancer patient survival and prognosis. Long non-coding RNAs (lncRNAs) have subsequently been revealed to play an important role promoting breast cancer metastasis through a variety of molecular pathways, albeit their exact functional characteristics have yet to be defined. Long noncoding RNAs (lncRNAs) have recently been linked to breast cancer metastasis in a number of studies (Bin et al., 2018; Klinge, 2018; Zhou et al., 2018; Arun and Spector, 2019; Tomar et al., 2020). Long noncoding RNAs are noncoding RNAs with a size of more than 200 nucleotides that have a role in a range of biological processes, particularly cancer cell invasion.
In transformed cells, epigenetic modifications include changes in DNA methylation, such as global hypomethylation or altered histone tail modification patterns, locus specific hypermethylation, as well as nucleosomal remodelling. DNA methylation is defined by Hinshelwood and Clark (Hinshelwood and Clark, 2008) (2008) as an enzyme-driven chemical modification to DNA sequence that happens most frequently at CpG dinucleotides among mammals.
DNA hypomethylation has been linked to gene reactivation and chromosomal instability, which can result in proto-oncogene overexpression, Imprinting loss, skewed or missing X-chromosomal inactivation, and increased recombination and mutation rates (De Smet et al., 2004). Gene suppression and genomic instability are connected to DNA hypermethylation as well as the suppression of tumour suppressor genes. In humans, PCDHB15 is a member of the cadherin superfamily of calcium-dependent cell-cell adhesion molecules that encodes for the PCDHB15 protein. CDH1 (also known as E-cadherin) and other cell adhesion molecules operate as epithelial-mesenchymal transition suppressors. In this CDH1 epigenetic silencing has been reported often in human cancer cases, including breast cancer (de Ruijter et al., 2020).
Another epigenetic process that can regulate gene expression by altering chromatin shape is post-translational histone tail modifications, which are linked to DNA methylation (Martin and Zhang, 2005; Baylin and Ohm, 2006). Furthermore, it has been demonstrated that several nucleosomal remodelling regulators are also engaged in DNA methylation and histone modification regulation (Esteller et al., 2000; Bird, 2002; Martin and Zhang, 2005; Baylin and Ohm, 2006). Understanding all of these epigenetic modifications and their role in breast carcinogenesis is critical for further advancements in breast cancer detection, prognosis, and treatment.
In the presence of the dinucleotide sequence CpG, DNA hypermethylation is a post-replication alteration that nearly exclusively affects cytosines’ pyrimidine ring (Pfeifer and Besaratinia, 2009). In mammalian genomes, the bulk of CpG dinucleotides (75%) are methylated. The quantity of 5-methylcytosine in 1% of all bases varied somewhat between tissue types. Repeating elements and transposons, which constitute roughly one-third of the human genome, include more than 90% of all methylation cytosines. Owing to the inherent carcinogenic potential of methylated cytosine residues, the proportion of Nucleotide bases in the genomes has been lowered over time, resulting in reduced number of CpGs than the quantitatively expected.
Cytosines that are methylated are more vulnerable to endogenous or exogenous mutagenesis mechanisms than other DNA bases, with CpG site mutation rates projected to be higher than other transitional mutations (Jones et al., 2008). Transitions from C to T at CpG dinucleotides account for almost a 1/3 of all known germ line and somatic and mutations, albeit the distribution varies depending on the tumour type (Lo and Sukumar, 2008). CpG islands are tiny DNA fragments (ranging in size from 200 base pairs to several kilo base pairs) found in 60% of all genes.
CpG islands that are ordinarily unmethylated in cancer cells may become methylated, potentially silencing critical genes such as tumour suppressor genes. At the same time, due to insufficient transcriptional regulation of typically silent genes like oncogenes or retrotransposons, CpG dinucleotides in other places can become unmethylated. DNA methylation silences tumour suppressor genes (TSGs) that govern tumor development, DNA repair genes, oestrogen receptor genes, or genes that regulate angiogenesis. Because transcription factors which interface with methylated DNA differ from those that interact with unmethylated DNA, DNA methylation has an impact on gene expression (Figure 1) Hypermethylation of promoter regions silences the gene, which is a critical step in carcinogenesis with substantial implications for cancer prevention.
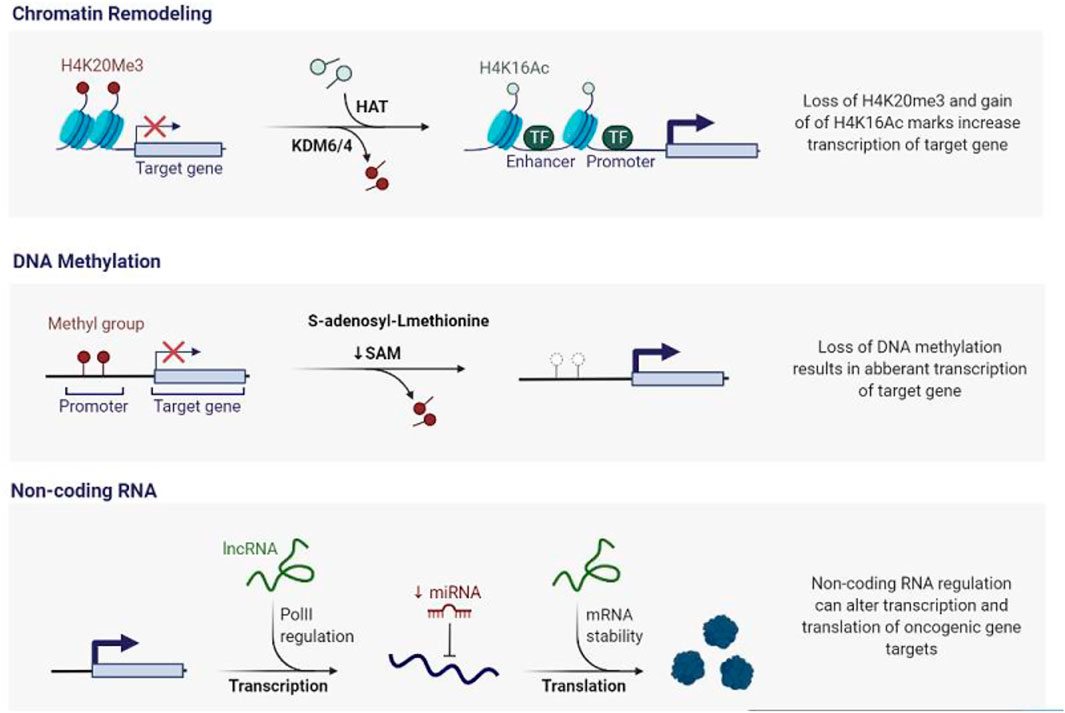
FIGURE 1. Epigenetic Deregulation in Cancer. A vast number of epigenetic modifiers are mutated or activated inappropriately during cancer genesis. Simultaneously, epigenetic alterations such as DNA methylation, histone modifications, and microRNAs cause aberrant gene expression, resulting in genomic instability.
In human malignancies and primary tumours, some tumour suppressor genes and other malignant genes have been discovered to be hypermethylated (Frigola et al., 2006). Cell -cycle control, DNA repair, cell death, cellular maintenance, and invasion are among their physiologic functions. Table 1 shows the genes which are most commonly methylation in breast cancer. Epigenetic cancer research has taken on a new dimension with the finding of long-range gene silence induced by epigenetic alterations (Stransky et al., 2006). Long-range epigenetic silencing appears to be ubiquitous during carcinogenesis, according to a recent study that revealed transcriptional dysregulation that can be regulated by epigenetic processes (Stransky et al., 2006).
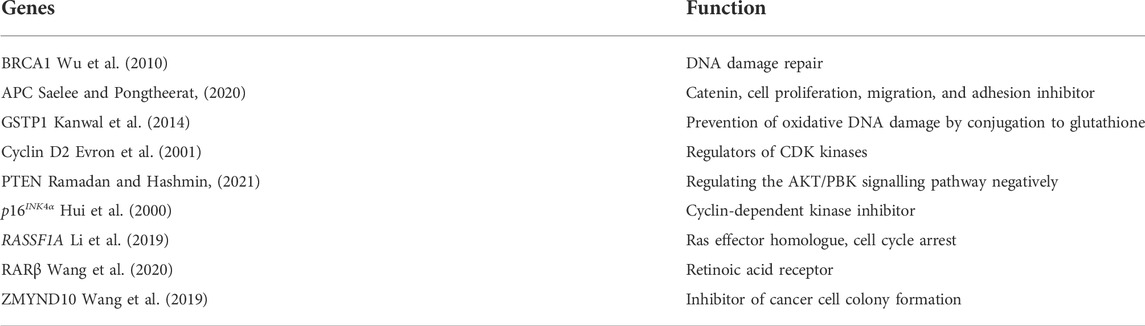
TABLE 1. Breast cancer genes that are hypermethylated.
Epigenetically regulated genes in breast cancer
Several research have sought to investigate the role of hypermethylation of TSG genes’ promoters in breast cancer, as well as the relationship between methylation of certain CGIs in TSGs and a variety of breast cancer clinical states. Table 1 lists the most important hypermethylated genes implicated in breast cancer functions so far. Methylation of these TSG promoters is linked to cancer cells losing all TSG protein products and developing a malignant phenotype. This DNA hypermethylation is a reversible signal, possibly due to the activity of Demethylase, which reverses the reaction of DNA methyltransferase and is a strong contender to be one of its key partners in shaping genome methylation patterns (Strahl and Allis, 2000; Ito et al., 2002). As a result, many recent research has focused on a novel strategy to cancer treatment that aims to block DNA hypermethylation and/or re-expression of silenced TSGs.
To create the transcriptional regulatory platform, DNA is packed into chromatin, a highly structured and dynamic protein–DNA complex. Histone modifications and composition interact with the binding of a variety of nonhistone proteins to control open (euchromatin) and closed (heterochromatin) chromatin states. The nucleosome is chromatin’s most basic component, wrapping 146 bp of DNA around an octamer made up of four core histones, an H3/H4 tetramer, and two H2A/H2B dimers (Strahl and Allis, 2000; Ito et al., 2002). The importance of local chromatin architecture in the regulation of gene expression is now widely acknowledged. Posttranslational changes to the N terminal tails of histones have a big impact on chromatin architecture.
Methylation, acetylation, phosphorylation, ubiquitination, sumoylation, ADP ribosylation, deamination, and proline isomerization are all covalent alterations to core histones (Schubeler et al., 2004; Shilatifard, 2006). The discovery of multiple histone modifications with varied functions in gene regulation aided the identification of a regulatory histone code, that defines at least partially overall transcriptional possibilities of a gene or genomic region (Xu et al., 2009). Altering the N terminus histone tail, which affects nucleosome density and positioning, enables this packed, inaccessible DNA accessible to DNA binding proteins during gene transcription initiation (Ito et al., 2002). Each histone modification acts as a chromatin organisation signal. Histone acetylation (hyperacetylation) is associated with increased transcriptional activity, whereas hypoacetylation (hypoacetylation) is associated with gene repression (Forsberg and Bresnick, 2001; Wade, 2001). Transcription related Protein (, p53, p73, E2F1, STAT1, GATA1, HMGB1, YY1, and NFkB etc), hormone response (GR, ER, and AR), nuclear transporter (Importin7), WNT signalling (catenin), DNA repair (Ku70) and heat shock/chaperone reaction (HSP90) are examples of HDAC substrates (Bolden et al., 2006; Kim et al., 2006).
For a long time, it was considered that methylation cytosines on DNA and deacetylated histones were two separate processes that could regulate chromatin structure and gene expression independently (Turner, 2000; Roth et al., 2001). HDACs have been linked to Epigenetic modifications by methyl group associated protein like MeCP2, which may read methylated sites on DNA and attract HDACs to them, or by HDACs directly interacting with DNA methyltransferases (Lo and Sukumar, 2008) (DNMTs). (Robertson et al., 2000; Bachman et al., 2001). Histone H3 lysine nine gets acetylated in functional chromatin regions, but when a genes is silenced, it becomes methylated, creating a binding domain for hetero-chromatin protein1 (HP1) (Litt et al., 2001; Peters et al., 2002). When serine 10 is phosphorylated, another epigenetic change, phosphorylation, prevents lysine nine from becoming methylated (Rea et al., 2000). PolyADP ribosylation is another alteration. PolyADP ribosylation has really been reported to affect chromatin structure through two methods, either covalently, by establishing short chains of Adenosine Diphosphate ribose polymers to histone proteins, or non-covalently, thereby attracting histones to the extended and branching polymers.
Epigenetic mechanism in breast cancer therapy
Diagnostic and prognostic methods based on epigenetics play an important role in precision medicine. Precision oncology benefits substantially from epigenetics-based diagnostic and prognostic techniques. Numerous DNA methylation diagnostic tests, in particular, are now being tested in clinics or are already in use. Precision oncology efforts to address dysregulated epigenetic pathways resulted in the development of epidrugs, or drugs that target epigenetic modulators. The FDA has approved only nine epidrugs, many more are in clinical studies for solid and hematological tumours (NCT01928576, NCT03179943), including antagonists of DNA methyltransferases (DNMTs) (NCT03164057, NCT02717884), EZH2, IDH and HDAC.The phase II trials (NCT00676663 and NCT00828854) exploring the epidrugs’ effectiveness when used in conjunction with normal treatment in oestrogen receptor positive (ER+) breast cancer are significant, reflecting recent developments in the knowledge of the epigenetic process. Due to ER expression, over 80% of all affected individuals are classified as ER+, and over 90 percent of all these patients have a 5 year cumulative rate of survival. Endocrine-based therapies such as ER-blockade, oestrogen synthesis inhibition, and selective ER degradation are used to treat most ER + cancers since ER is the primary oncogenic driver (Zardo et al., 2003; Thomas and Potter, 2013).
Long non coding RNAs
LncRNAs are RNA molecules having a length of more than 200 nucleotides but no apparent protein-coding function. Over 10,000 lncRNAs have been identified in the human transcriptome, with their genes located inter- or intra-genes in the genome. However, only a few have been thoroughly described. RNA polymerase II transcribes LncRNA genes, which then go through 5′ capping, splicing, 3′ cleavage, and polyadenylation. LncRNA loci are comparable to those of protein-coding genes at the chromatin level, although they frequently lack introns or have one or two. Splicing matures lncRNAs in the same way that it matures pre-mRNAs. n general, lncRNAs are found in the nucleus, although they have also been found in the cytoplasm and exosomes, and their expression levels are often lower than those of coding genes. Many investigations have found that their expression differs depending on the cell type24. In compared to protein-coding genes, lncRNAs are under low selected pressure, but their selective pressure is stronger than genomic repeat sequences. When comparing the sequences of lnRNAs from different species, brief highly conserved sequences can be found, demonstrating that they have preserved information about their cellular location and structure during evolution (Shang et al., 2000; Métivier et al., 2003; Moore and Proudfoot, 2009; Derrien et al., 2012; Rinn and Chang, 2012).
Following is a description of the lncRNAs H19, TINCR, MALAT, and NEAT1 DANCR, whose aberrant expression is linked to the growth and metastasis of BC.
H19 LncRNA
It has been established that BC development and dysregulated long non-coding RNA H19 (H19) expression are related (Yang et al., 2016; Hu et al., 2018). Over 70% of BC tumours, including ER+ and ER-, HER2+ and HER2-positive tumours, have highly expressed H19 (Yang et al., 2016; Wang et al., 2018). This lncRNA has greater expression in BC for a number of usual mutational polymorphisms as well (Vennin et al., 2017; Wang et al., 2018). Apoptosis suppression and cell proliferation promotion are two biological reactions in which the Akt signalling pathway is involved (Yang et al., 2016). The maternal allele encodes H19, a 2.3-kb lncRNA that is regarded as an oncogene in several malignancies. At the H19/IGF2 locus, a novel lncRNA called 91H is being produced in the H19 antisense direction. In breast cancer, the 91H lncRNA is in charge of preserving the genomic imprinting of the H19/IGF2 locus by preventing histone and DNA methylation on the maternal allele (Hu et al., 2018; Wang et al., 2018). E2F1 stimulates H19, which aids the G1-S transition in breast cancer cells (Vennin et al., 2017). Through the activation of Akt, the miR-675 produced by H19 downregulates the c-Cb1 and Cb1-b proteins and activates EGFR and c-Met to encourage cell growth (Berteaux et al., 2005; Zhang et al., 2017) discovered that overexpression of the lncRNA MEG3 inhibits cancer growth in a mouse model of breast cancer by inhibiting Akt signalling, in addition to causing cell cycle arrest in the G0/G1 phase. (Chen et al., 2017). demonstrated that lncRNA PTENP1 restricts the growth of breast cancer cells by downregulating the MAPK and AKT signalling pathways.
TINCR lncRNA
In 2018 (Liu et al., 2018), it was found that TINCR lncRNA (TINCR) influences how primary BC tumours develop and how they spread later. In a certain study of 24 patients, the qPCR technique identified greater TINCR BC expression compared to non-BC participants. Additionally, SP1-zinc finger transcriptional factor, which normally identifies the Guanine Cytosine -rich sequences in promoter regions, causes greater TINCR activity (Liu et al., 2018).
MALAT lncRNA
Multiple BC types have abnormal expression of the lncRNA MALAT (MALAT), and this abnormal expression is associated with metastasis and a poor prognosis (Jadaliha et al., 2016; Wu et al., 2019). Further evidence suggests that high concentrations of 17- oestradiol can impede this lncRNA activity (Zhao et al., 2014). In a fascinating study, individuals with early post-BC-resection fever had higher MALAT levels (Li et al., 2018), which was associated with a worse prognosis. Additionally, MALAT deletion in mouse 4T1 xenografts markedly reduced inflammation and the lung metastases that are frequently observed in BC (Li et al., 2018).
NEAT1 lncRNA
NEAT1 is a crucial oncogene in cancer and has a big impact on BC’s ability to induce EMT (Lu et al., 2016). In a sample of 179 BC patients, abnormal NEAT1 activity influenced chemoresistance and cancer cell stemness, and it has been expressed 6.86 times greater in BC patients than that in 192 controls (Shin et al., 2019).
Estrogen subtypes and ER signalling pathways
Estrogen promotes a variety of developmental processes in the body, involving reproductive maturity and bone growth, as well as energy balance via glycaemic control, intake rate, and thermoregulation. Estrogen also regulates mammary gland development through coordinating mitogenic and epigenetic processes. Several chemicals, as well as naturally occurring substances like polyphenols, which serve as a hypermethylation agent, can reverse the epigenetic silencing of tumour suppressor genes (Mathur and Jha, 2020). .Estrone, 17-Estradiol,Estriol, Estetrol (i.e, E1,E2,E3,E4) & Estrone-sulfate are the five major oestrogen subtypes (E1s). E1 and E2, the body’s two major estrogens, are reversibly transformed to E2, the physiologically active variety. Only E3 and E4 are identified throughout pregnancy, with E3 being the most prevalent. Because steroid sulfatases convert it to its active metabolite, E1 and E2, in situ, E1s is largely employed as an oestrogen reservoir (Mukherjee et al., 2017).
Epigenetic mechanism underlying Erα signalling
Epigenetic mechanisms are involved in ER signalling. In response to E2 stimulation, multitudes of ER co-regulators are transported to chromosomes in a synchronised way to ensure appropriate transcriptional and repressive activity at ER target sites. Regardless of the fact that every ER molecule usually stays on the chromatin for few moments at most, ER has been observed cycling on and off the chromatin for minutes and hours after E2 stimulation (Djebali et al., 2012; Johnson and O’Malley, 2012; Paakinaho et al., 2017; Wils and Bijlsma, 2018; Zhang et al., 2020). PRMTs, the SWI/SNF complex, P300/CBP & the Mediator complex, as well as the p160 family of proteins, are all significant epigenetic ER coactivators. Members of the p160 family the co—activators i.e., SRC-1, SRC-2, and SRC-3, bind to ER directly and act as a recruiting platform for other activating enzymes and proteins to be recruited by ER to change chromatin, including chromatin remodelling complexes Breast cancer messes up epigenetic mechanisms that are essential for mammary gland development. The mammary gland’s balance self-renewal, and tissue integrity is regulated by a variety of signalling cascades and chromatin moderators, and also hormonal factors. Embryonic, pubertal, & reproductive stages are all three stages in the development of the mammary gland. WNT and Hedgehog (HH) signalling pathways coordinate embryonic mammary gland development, whereas hormones control pubertal and reproductive stages (Suzuki et al., 2008).
The reactivation of various developmental pathways, which would be a common characteristic of many malignancies, is connected to the longevity of a mammary gland stem population of cells in cancer patients (Xiang et al., 2013). Derailment of important epigenetic mechanisms during breast development currently plays an essential role in the pathogenesis of breast cancer, according to research conducted in the last few years with the introduction of technical breakthroughs like as next-generation sequencing. This article discusses how the functional relationship between epigenetic alterations and developmental signalling cascades contributes to breast cancer.
WNT signalling epigenetic modification in ER + breast cancer
WNT signalling abnormalities have been associated to the onset and progression of a variety of cancers, including breast cancer. Breast cancer is aided by epigenetic suppression of WNT antagonist genes such as SFRP and DKK37. Chronic WNT signalling in breast cancer, which is linked to a poor prognosis, is caused by the methylation of these genes, which silences them (Bell et al., 2019). As a result of these changes, catenin stays constitutively active, resulting in enhanced stem cell replacement and division, which has been linked to disease resurgence (Serrano-Gomez et al., 2016). One of the constituent of the DKK family, i.e DKK3 had significantly more promoter methylation in tumours from individuals with lymph node metastases, advanced stage disease, or breast cancer samples with positive ER status. status of mammary cancer samples.
The link between both the WNT & ER signalling pathways, particularly via Polycomb protein EZH231, it has been suggested that DKK’s involvement of WNT signalling activity can relate forward into the ER dependent pathway (and vice versa) to strengthen survival and growth with DKK3 promoter methylation being associated with positive ER status.5-azacytidine and trichostatin A, for example, have been shown to restore DKK3 expression in vitro (Figure 2). In the clinic, however, attempts to re-establish ER expression with hypomethylating medications have failed, EMT influences the polarisation of mammary cells, milk flow patterns, particularly during pregnancy and during wound healing, cell movements are important. Mediated by ZEB1, SNAIL, and TWIST, among other transcription factors (TFs). SNAIL, for example, activates the Methyltransferase DNMT1 and inhibits CDH1 through DNA methylation. Furthermore, TGF-induced EMT modulates SNAIL transcription reactivation via the H3K27me3 demethylase KDM6B. Increased levels of SNAIL and KDM6B have been associated to cancer recurrence, metastases, and poor flatline survival in invasive breast carcinomas (Beatson, 1896; Liu et al., 2006; Saez-Ayala et al., 2013; Hanker et al., 2020). As a result, one can expect that targeting H3K27me3 demethylases in combination with DNA hypomethylating medicines, which are prospective treatment targets in other solid tumours such as castration-resistant prostate cancer, could reduce recurrence (Liu et al., 2001).
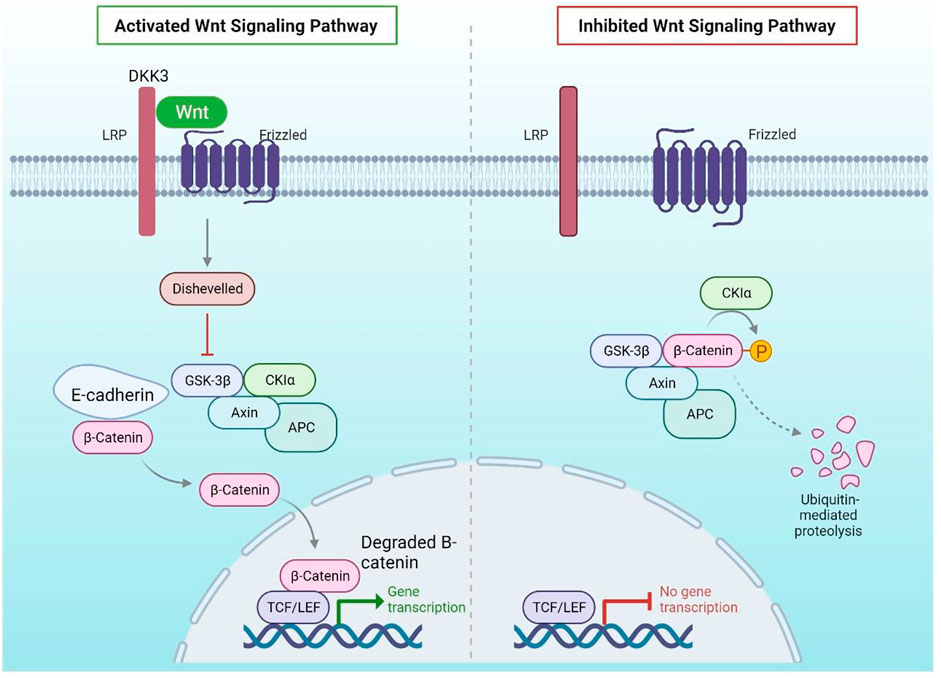
FIGURE 2. Wnt Signalling Pathway: DKK3 binds to LRP, a WNT pathway coactivator of Frizzled, in normal mammary epithelial cells, preventing the pathway from being activated in the presence of the WNT ligand. In the absence of WNT activation, E-Cadherin binds to cytoplasmic -catenin, which is destroyed by GSK3. The DKK3 promoter, on the other hand, is hypermethylated in breast cancer, resulting in its downregulation. LRP can coactivate Frizzled in the presence of the WNT ligand in the absence of DKK3, resulting in phosphorylation of DSH, which prevents GSK3 from degrading -catenin.
Polycomb complexes and HH signalling
Epigenetic changes in breast cancer impair HH signalling, which is an important developmental pathway. Cancer progression is driven by increased ligand-dependent pathway activation and unregulated cell division (Early Breast Cancer Trialists’ Collaborative Group, 2005). Whenever the promoters of the SHH, HH Ligand, or its subsequent receptors, PTCH, is hypomethylated, the pathway is activated more ligand-dependently, resulting in uncontrolled cell division and cancer progression (Early Breast Cancer Trialists’ Collaborative Group, 2005). HH signalling also helps to promote normal and tumorigenic breast stem cells by boosting the production of PCGF4 (BMI1), a constituent of the PRC1 complex (Jeselsohn et al., 2015). Breast cancer stem cells have already been related to hormonal therapy resistance; however, it is unclear whether the appearance of stem cell like features in resistant cells is attributable to the multiplication of pre-existing highly specialised tumour cells or to epigenetic modifications that promote dynamic reprogramming (Jeselsohn et al., 2015).
According to the findings, combining medicines that directly block HH signalling with epigenetic modifiers like DNMTs to recover HH antagonistic control could alter cancer stem cell viability and differentiation. It has previously been reported on the utilisation of two-step approaches that combine several classes of medicines to trigger a process known as targeted phenotypic flipping to treat resilient melanoma cells to lineage-specific therapy (Razavi et al., 2018). WNT and Hedgehog signalling pathways are required for the development of embryonic mammary glands, and their activation must be carefully coordinated spatially and temporally (SHH). In healthy mammary epithelial cells, DKK3 binds to LRP, a Frizzled WNT pathway coactivator, preventing the route from becoming activated in the vicinity of the WNT ligand. E-Cadherin attaches to cytoplasmic -catenin in the lack of WNT activation, which is eliminated by GSK3.
The oncogenic E2-ER axis is the focus of endocrine treatment. The very first-time steroid hormonal signalling being linked to breast tumor progression when both ovaries were surgically removed from patients with breast cancer, resulting in tumour regression and pave the way for endocrine therapy (Hanker et al., 2020). Treatments that decrease estrogen synthesis as well as techniques that specifically target the estrogen receptor are used in hormonal therapy, which is really the benchmark for ER + breast cancer (ER). Selective oestrogen receptor degraders (SERDs), Selected Oestrogen Receptor Modulators (SERMs), and Aromatase Inhibitors are the three categories (AIs) (Fanning et al., 2016). In addition, next-generation ER targeting medications are presently being investigated in ER+/HER2 metastatic melanoma as adjuvant therapy or in combination with other therapy (Helleman et al., 2008).
Mechanisms Of endocrine therapy resistance, as well as possible alternatives
Considering the fact that the endocrine therapy is effective in the treatment of ER + breast cancer patients, resistance develops in around 25% of early-stage patients and virtually all metastatic patients, results in a poor clinical prognosis (Liu et al., 2006; Serrano-Gomez et al., 2016; Bell et al., 2019); (Jansen et al., 2005; Generali et al., 2006; Pontiggia et al., 2009). Resistance to endocrine therapy has been classified as either intrinsic or acquired. Patients with breast cancer frequently experience clonally distinct progression as a result of the selection of genetic changes under treatment (Folgiero et al., 2008).
Resistance mechanisms associated with the tumor microenvironment and the host
The tumour microenvironment’s role as a regulator of these pathways and contribution to endocrine responsiveness has recently been established. This theory has been supported by studies including gene expression studies and biomarkers linked to hormonal therapy outcomes (Encarnacion et al., 1993; Brinkman et al., 2010) as well as the more complex in vitro or in vivo existing experimental systems (Gutierrez et al., 2005). Endocrine resistance is linked to stromal cells (endothelial, fibroblasts and immune cells), structural features of the microenvironment and soluble substances (e.g., interleukins and growth factors) as well as other micro-environmental variables including hypoxia and acidity (Lopez-Tarruella and Schiff, 2007).
The role of tumorigenic cell pathways in modulating these microenvironmental and extracellular stimulus has previously been characterised (Munzone et al., 2006; Xia et al., 2006), inferring potential signalling components (e.g., SRC Kinase/integrin/FAK) that might be targeted to overcome endocrine resistance (Osborne et al., 2003; Shi et al., 2009). In addition, the list of other host genome–associated variables influencing endocrine sensitivity is rising as a consequence of new pharmacogenomic and high-throughput research.
Resistance mechanisms associated with tumors
However, as previously indicated, the most of pathways that may play a role in endocrine resistance originates in tumour cells. These pathways can be split into three categories, each of which has components and mechanisms that overlap.
The two different types of ER regulator are ER and ER coregulators. The first group includes the ER, with coregulators, as well as other factors that alter the normal ER activity and modify receptor functions in relation to endocrine therapy. In refractory endocrine cancers, reduction of ER synthesis (i.e., the ER isoform) culminates in an endocrine-insensitive phenotype, that is rare (Lavinsky et al., 1998; Shou et al., 2004; Musgrove and Sutherland, 2009). Treatments that block growth factor receptors pathways that are known for down regulation, ER can enhance ER expression and endocrine sensitivities both in experimental and clinical situations (Schiff et al., 2004; Levin and Pietras, 2008; Santen et al., 2009) Reduced endocrine responsiveness has also been associated to the expression of various ER splicing variation, specifically the recently found minor variance ER36 (Musgrove and Sutherland, 2009) and oestrogen-related receptors. Furthermore, statistics suggest that ER coregulators, whether negative (corepressors) or positive (coactivators), have a role in defining endocrine sensitivity and resistance by influencing the balance of agonistic vs. antagonistic SERM activity. Both in clinical and experimental contexts, dysregulation of a ER co-activator AIB1 (also abbreviated as SRC3 or NCoA3) has been associated to tamoxifen resilience (Span et al., 2003; Butt et al., 2005), while reduced expression of the co-repressor NCoR has been detected in tamoxifen-resistance experimental malignancies (Arpino et al., 2008). The ER as well as its coregulators are heavily influenced by posttranslational modifications. Growth factor receptor [e.g., FGFR (fibro-blast growth factor receptor, EGFR/HER2, and IGF1-R ) and other cellular and stress-related kinases [e.g.p42/44, JNK , AKT, and PKA (protein kinase A and p38 MAPKs ), PAK1] regulate several posttranslational modifications (p21-activated kinase). Ubiquitination, Methylation, Phosphorylation, and other posttranslational modifications of ER and its co-regulators have been identified to alter ER activity and susceptibility to various endocrine therapies (Chakraborty et al., 2010) Outside of the nucleus, ER interfaces with cytoplasmic and membrane signalling complexes to activate and regulate a variety of growth factor receptors as well as other cell - signalling cascades (Kern et al., 1994; Morgan et al., 2009; Shi et al., 2009; Spoerke et al., 2016).
Cell cycle signalling molecules
Molecules associated in cellular and biological responsiveness to endocrine therapy, such as cell growth inhibition and apoptosis induction, are included in endocrine resistance–related pathways. The majority of information on the participation of these pathways comes from preclinical investigations. Positive cell-cycle regulators, notably those directing G1 phase progression, and also negative cell cycle regulator, have both been demonstrated to disrupt and decrease endocrine therapy’s antiproliferative action, tends to result in resistance (Fribbens et al., 2016). Endocrine resistance is caused by overexpression of positive cell-cycle regulators MYC & cyclins E1 and D1, which activate cyclin-dependent kinases (CDKs) for G1 phase or reduce the inhibitory effects of negative cell-cycle regulators (p21 & p27) (Gates et al., 2018).
Growth factor receptor pathways
In the case that the ER system is effectively inhibited, the third set of regulatory mechanisms in endocrine resistance would comprise those that can provide alternate proliferation and migration inputs to tumours. Importantly, through bi-directional interactions and control of the ER, these mechanisms—such as growth factors and other cellular-kinase pathways—might be able to offset the inhibitory activity of endocrine therapy. Many of these pathways, on the otherhand, it might develop into ER-independent drivers of tumour development and survival, making patients susceptible to all kinds of endocrine therapy, either early or later on. It has been suggested that fibroblast growth factor (FGF), insulin/IGF1 receptors HER, tyrosine kinase receptors, and vascular endothelial growth-factor (VEGF) receptors are all involved (Harrod et al., 2017; Jeselsohn et al., 2018; Morel et al., 2020).
Alteration Of ESR1 And Genes Involved In Estrogen-Mediated Signalling.
The tumour cell’s reliance on ER for growth and survival is targeted by endocrine treatment. As a result, bypassing pharmacological inhibition relies on the accumulation of changes in the ER and its downstream targets. The main mechanism of resistance in most cases is ligand independent ER reactivation (Oronsky et al., 2014). Constitutive ER activation can be mediated by mutations in the ESR1 gene (which codes for ER) and is a major driver of acquired resistance. The majority of ER mutations occur in the LBD at two neighbouring amino acids: tyrosine at position 537 transformed to asparagine, cysteine, or serine (ERY537 N/C/S) and aspartic acid at position 538 altered to glycine (ERD538G). From a structural standpoint, these changes remain stable ER in an agonists configuration, resulting in constitutively active state (Oronsky et al., 2014). ESR1 mutations are detected in less than 1% of original tumours, however they are reported in 20–40% of tumours after endocrine therapy and have been associated to poor AI & tamoxifen efficacy (O’Neil et al., 2017; Zucchetti et al., 2019; Xu et al., 2020). The nearly complete detection of ESR1 alterations in hematologic malignancies after the AI therapy shows that under the constraints of endocrine treatment, uncommon, resistant clones can be selected.
Several studies have focused on establishing new therapeutic strategies for tumor tissues with ESR1 mutations in recent years. Continuous ER signalling encourages hormone independent development and thus is linked to a distinct transcription network involved in signaling pathways and metastasis as a result of this process (Fukumoto et al., 2018). Activating kinases, epigenetic modifying enzymes and ER co-regulators, are required for the development of ESR1 mutants (Fukumoto et al., 2018). .As a consequence, they could be employed to treat ESR1 mutant malignancies in the preclinical stage. Another type of genetic mutation discovered in metastatic ER + breast cancer is ESR1 gene fusion events, which are likely to represent novel resistance drivers. As a result of ESR1 chromosomal rearrangement occurrences, the ER’s LBD is replaced by another protein.
Endocrine-resistant breast cancer is caused by epigenetic factors
According to a whole-genome sequencing study, epigenetic factors are among the most commonly changed factors in human malignancies. The most frequent genetic modifications in many types of cancer are inactivating mutations as well as the loss of SNF/SWI subunits. In breast cancer, ARID1A impacts breast luminal lineage adherence and sensitivity to endocrine treatment.
Patients’ poor response to SERDs suggests that ARID1A loss-of-function mutations are more frequent in endocrine-resistant metastatic situations, implying that they can also cause endocrine resistance.
ARID1A deficiency affects chromatin accessibility and transcription factor binding, as well as the binding of ER and FOXA1 to chromatin, all of which influence luminal cell destiny.
According to Xu et al., long-term ER suppression could result in the generation of individuals with ARID1A inactivating mutation, promoting a luminal-to-basal phenotypic transition (Bitler et al., 2015). In the clinic, ER + tumours cured with endocrine therapy, reduce ER expression, by becoming resistant to hormone therapy. The increasing prevalence of ARID1A mutation in endocrine resistant breast cancer, including its prevalence in other cancers, highlights the need of treating ARID1A mutant tumours with targeted therapeutic strategies (Morel et al., 2020).
One of the therapy paradigms examined in ARID1A mutant cancers is synthesised lethality, which relates to the lethal consequence of simultaneous alteration of two genes which, when separately disrupted, do not impact cell viability (Morel et al., 2020). For example EZH2 suppression and ARID1A mutations are synthetically fatal in ovarian cancer, and HDAC2 inhibition amplifies this effect. In ARID1A defective cells, HDAC2 is attracted to EZH2/ARID1A co-target genes including such PIK3IP1, a PI3K/AKT signalling inhibitor, leading in incorrect stimulation of this mitogenic system (Shiino et al., 2016). These two processes, ARID1A loss of function and enhanced PI3K/AKT signalling, are typically detected in endocrine-resistant breast cancer cells (Shiino et al., 2016). As a consequence, one can believe that targets EZH2 in breast cancer patients with ARID1A mutations might be a promising treatment option.
Epigenetic avenues in the endocrine therapy
In the realm of endocrine therapy, there are a variety of epigenetic options. Despite the fact that endocrine and molecularly targeted have been shown to cure the great majority of breast cancers, they have failed to target a tiny percentage of the population, leading to recurrence and therapeutic resistance. Compounding variables like tumour genetic instability enables tumours to adapt to a range of stresses, including the selective pressure produced by therapeutic drugs, which we addressed previously. As a result, precise patient classification and personalised treatment approaches would be required to reduce the significant morbidity and mortality of individuals with ER + metastatic breast cancer (Shiino et al., 2016).
Tumorigenesis and medication resistance are both influenced by epigenetic instability. Epidrugs have primarily been used to treat haematological malignancies, with limited efficacy in solid tumors. The failure to treat solid tumours, on the other side, can be traced back to a one size fits all approach. Epigenetic reprogramming’s plasticity enhances cancerous cells’ overall fitness, making individualised cancer treatment much more challenging. According to multiple preclinical and clinical studies, epidrugs have synergistic benefits with a number of therapeutic methods, including immunotherapy, radiation, and endocrine therapy. One of the most notable areas of current drug discovery operations is the development of small compounds that target chromatin regulators (Shiino et al., 2016).
The majority of the studies focused on epigenetic changes that occur when cancer progresses and resistance develops. Small molecule HDACi blockers (vorinostat and entinostat ) and also DNA hypomethylating drugs (decitabine & 5-azacytidine) have been explored as re-sensitizing strategies to endocrine therapy in ER + preclinical models. The modes of action of DNMT inhibitors (DNMTs) have been proposed as de-methylation of tumour suppressor genes and an unique viral mimicking mechanism. Furthermore, in endocrine-resistant breast cancer, epigenetic dysregulation is a prevalent occurrence. For example, in roughly 20% of patients that continue through tamoxifen treatment, promoter hyper-methylation of ESR1 causes loss in ER expression (Shiino et al., 2016). In ER human breast cancer cells, letrozole (AI) and entinostat (HDACi) and can re-establish ER & aromatase expression, resulting in growth suppression.
Conclusion
New findings, as with all parts of science, produce new questions, and some of the most important unanswered questions like. Is it possible to use the dynamic nature of epigenetic modifications to develop short-term treatment techniques to avoid selection toward a resistant phenotype, or are epidrugs’ underlying processes contributing to resistance formation is possible to follow disease progression and therapy response using epigenetic markers. Endocrine therapy has been proven to be an important treatment option for hormone-responsive breast cancers. However, there is still an urgent need to develop strategies to combat the phenotype of resistance that appears to be unavoidable. Recent epidrug breakthroughs attest to the developing new era of epigenetic-based treatments for screening and treating a variety of disorders, including breast cancer.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Acknowledgments
The authors would like to acknowledge the support from their respective institutes throughout the review writing process.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Arpino, G., Wiechmann, L., Osborne, C. K., and Schiff, R. (2008). Crosstalk between the estrogen receptor and theHER tyrosine kinase receptor family: Molecular mechanism and clinical implications for endocrine therapy resistance. Endocr. Rev. 29, 217–233. doi:10.1210/er.2006-0045
Arun, G., and Spector, D. L. (2019). MALAT1 long non-coding RNA and breast cancer. RNA Biol. 16, 860–863. doi:10.1080/15476286.2019.1592072
Bachman, K. E., Rountree, M. R., and Baylin, S. B. (2001). Dnmt3a and Dnmt3b are transcriptional repressors that exhibit unique localization properties to heterochromatin. J. Biol. Chem. 276, 32282–32287. doi:10.1074/jbc.m104661200
Baylin, S. B., and Ohm, J. E. (2006). Epigenetic gene silencing in cancer-mechanism for early oncogenic pathway addiction? Nat. Rev. Cancer 6, 107–116. doi:10.1038/nrc1799
Beatson, G. T. (1896). On the treatment of inoperable cases of carcinoma of the mamma: Suggestions for a new method of treatment, with illustrative cases. Trans. Med. Chir. Soc. Edinb. 15, 153–179.
Bell, C. C., Fennell, K. A., Chan, Y. C., Rambow, F., Yeung, M. M., Vassiliadis, D., et al. (2019). Targeting enhancer switching overcomes non-genetic drug resistance in acute myeloid leukaemia. Nat. Commun. 10, 2723. doi:10.1038/s41467-019-10652-9
Berteaux, N., Lottin, S., Monte, D., Pinte, S., Quatannens, B., Coll, J., et al. (2005). H19 mRNA-like noncoding RNA promotes breast cancer cell proliferation through positive control by E2F1. J. Biol. Chem. 280 (33), 29625–29636. doi:10.1074/jbc.M504033200
Bin, X., Hongjian, Y., Xiping, Z., Bo, C., Shifeng, Y., and Binbin, T. (2018). Research progresses in roles of LncRNA and its relationships with breast cancer. Cancer Cell. Int. 18, 179. doi:10.1186/s12935-018-0674-0
Bird, A. (2002). DNA methylation patterns and epigenetic memory. Genes. Dev. 16, 6–21. doi:10.1101/gad.947102
Bitler, B. G., Aird, K. M., Garipov, A., Li, H., Amatangelo, M., Kossenkov, A. V., et al. (2015). Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat. Med. 21, 231–238. doi:10.1038/nm.3799
Bolden, J. E., Peart, M. J., and Johnstone, R. W. (2006). Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 5, 769–784. doi:10.1038/nrd2133
Bray, F., McCarron, P., and Parkin, D. M. (2004). The changing global patterns of female breast cancer incidence and mortality. Breast Cancer Res. 6, 229–239. doi:10.1186/bcr932
Brinkman, A., de Jong, D., Tuinman, S., Azaouagh, N., van Agthoven, T., and Dorssers, L. C. J. (2010). The substrate domain of BCAR1 is essential for antiestrogen-resistant proliferation of human breast cancer cells. Breast Cancer Res. Treat. 120, 401–408. doi:10.1007/s10549-009-0403-4
Butt, A. J., McNeil, C. M., Musgrove, E. A., and Sutherland, R. L. (2005). Downstream targets of growth factor and oestrogen signaling and endocrine resistance. Endocrine-Related Cancer. 12, S47–S59. doi:10.1677/erc.1.00993
Chakraborty, A. K., Welsh, A., and Digiovanna, M. P. (2010). Co-targeting the insulin-like growth factor I receptor enhances growth-inhibitory and proapoptotic effects of antiestrogens in human breast cancer cell lines. Breast Cancer Res. Treat. 120, 327–335. doi:10.1007/s10549-009-0382-5
Chen, S., Wang, Y., Zhang, J. H., Xia, Q. J., Sun, Q., Li, Z. K., et al. (2017). Long non-coding RNA PTENP1 inhibits proliferation and migration of breast cancer cells via AKT and MAPK signaling pathways. Oncol. Lett. 14 (4), 4659–4662. doi:10.3892/ol.2017.6823
Chen, W., Zheng, R., Baade, P. D., Zhang, S., Zeng, H., Bray, F., et al. (2016). Cancer statistics in China, 2015. CA A Cancer J. Clin. 66, 115–132. doi:10.3322/caac.21338
de Ruijter, T. C., van der Heide, F., Smits, K. M., Aarts, M. J., van Engeland, M., and Heijnen, V. C. G. (2020). Prognostic DNA methylation markers for hormone receptor breast cancer: A systematic review. Breast Cancer Res. 22, 13. doi:10.1186/s13058-020-1250-9
De Smet, C., Loriot, A., and Boon, T. (2004). Promoter-dependent mechanism leading to selective hypomethylation within the 5′ region of gene MAGE-A1 in tumor cells. Mol. Cell. Biol. 24, 4781–4790. doi:10.1128/MCB.24.11.4781-4790.2004
Derrien, T., Johnson, R., Bussotti, G., Tanzer, A., Djebali, S., Tilgner, H., et al. (2012). The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 22, 1775–1789. doi:10.1101/gr.132159.111
Djebali, S., Lagarde, J., Kapranov, P., Lacroix, V., Borel, C., Mudge, J. M., et al. (2012). Evidence for transcript networks composed of chimeric RNAs in human cells. PLoS One 7, e28213. doi:10.1371/journal.pone.0028213
Early Breast Cancer Trialists’ Collaborative Group (2005). Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15- year survival: Overview of the randomised trials. Lancet 365, 1687–1717.
Encarnacion, C. A., Ciocca, D. R., McGuire, W. L., Clark, G. M., Fuqua, S. A., and Osborne, C. K. (1993). Measurement of steroid hormone receptors in breast cancer patients on tamoxifen. Breast Cancer Res. Treat. 26, 237–246. doi:10.1007/BF00665801
Esteller, M., Silva, J. M., Dominguez, G., Bonilla, F., Matias-Guiu, X., Lerma, E., et al. (2000). Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J. Natl. Cancer Inst. 92, 564–569. doi:10.1093/jnci/92.7.564
Evron, E., Umbricht, C. B., Korz, D., Raman, V., Loeb, D. M., Niranjan, B., et al. (2001). Loss of cyclin D2 expression in the majority of breast cancers is associated with promoter hypermethylation. Cancer Res. 61 (6), 2782–2787. 1 TUMOR BIOLOGY| MARCH 01 2001.;
Fanning, S. W., Mayne, C. G., Dharmarajan, V., Carlson, K. E., Martin, T. A., Novick, S. J., et al. (2016). Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. Elife 5, e12792. doi:10.7554/eLife.12792
Folgiero, V., Avetrani, P., Bon, G., Di Carlo, S. E., Fabi, A., Nistico, C., et al. (2008). Induction of ErbB-3 expression by alpha6beta4 integrin contributes to tamoxifen resistance in ERbeta1-negative breast carcinomas. PLoS One 3, e1592. doi:10.1371/journal.pone.0001592
Forsberg, E. C., and Bresnick, E. H. (2001). Histone acetylation beyond promoters: Long-range acetylation patterns in the chromatin world. Bioessays. 23, 820–830. doi:10.1002/bies.1117
Fribbens, C., O'Leary, B., Kilburn, L., Hrebien, S., Garcia-Murillas, I., Beaney, M., et al. (2016). Plasma ESR1 mutations and the treatment of estrogen receptor-positive advanced breast cancer. J. Clin. Oncol. 34, 2961–2968. doi:10.1200/JCO.2016.67.3061
Frigola, J., Song, J., Stirzaker, C., Hinshelwood, R. A., Peinado, M. A., and Clark, S. J. (2006). Epigenetic remodeling in colorectal cancer results in coordinate gene suppression across an entire chromosome band. Nat. Genet. 38, 540–549. doi:10.1038/ng1781
Fukumoto, T., Park, P. H., Wu, S., Fatkhutdinov, N., Karakashev, S., Nacarelli, T., et al. (2018). Repurposing Pan-HDAC inhibitors for ARID1A-mutated ovarian cancer. Cell. Rep. 22, 3393–3400. doi:10.1016/j.celrep.2018.03.019
Gates, L. A., Gu, G., Chen, Y., Rohira, A. D., Lei, J. T., Hamilton, R. A., et al. (2018). Proteomic profiling identifies key coactivators utilized by mutant ERα proteins as potential new therapeutic targets. Oncogene 37, 4581–4598. doi:10.1038/s41388-018-0284-2
Generali, D., Berruti, A., Brizzi, M. P., Campo, L., Bonardi, S., Wigfield, S., et al. (2006). Hypoxia-inducible factor-1alpha expression predicts a poor response to primary chemoendocrine therapy and disease-free survival in primary human breast cancer. Clin. Cancer Res. 12, 4562–4568. doi:10.1158/1078-0432.CCR-05-2690
Gupta, G. P., and Massague, J. (2006). Cancer metastasis: Building a framework. Cell. 127, 679–695. doi:10.1016/j.cell.2006.11.001
Gutierrez, M. C., Detre, S., Johnston, S., Mohsin, S. K., Shou, J., Allred, D. C., et al. (2005). Molecular changes in tamoxifen-resistant breast cancer:relationship between estrogen receptor, HER-2, and p38 mitogen-activated protein kinase. J. Clin. Oncol. 23, 2469–2476. doi:10.1200/JCO.2005.01.172
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: The next generation. Cell. 144, 646–674. doi:10.1016/j.cell.2011.02.013
Hanker, A. B., Sudhan, D. R., and Arteaga, C. L. (2020). Overcoming endocrine resistance in breast cancer. Cancer Cell. 37, 496–513. doi:10.1016/j.ccell.2020.03.009
Harrod, A., Fulton, J., Nguyen, V. T. M., Periyasamy, M., Ramos-Garcia, L., Lai, C. F., et al. (2017). Genomic modelling of the ESR1 Y537S mutation for evaluating function and new therapeutic approaches for metastatic breast cancer. Oncogene 36, 2286–2296. doi:10.1038/onc.2016.382
Helleman, J., JansenMP, , Ruigrok-Ritstier, K., van Staveren, I. L., Look, M. P., Meijer-van Gelder, M. E., et al. (2008). Association of an extracellular matrix gene cluster with breast cancer prognosis and endocrine therapy response. Clin. Cancer Res. 14, 5555–5564. doi:10.1158/1078-0432.CCR-08-0555
Hinshelwood, R. A., and Clark, S. J. (2008). Breast cancer epigenetics: Normal human mammary epithelial cells as a model system. J. Mol. Med. 86, 1315–1328. doi:10.1007/s00109-008-0386-3
Hu, Q., Yin, J., Zeng, A., Jin, X., Zhang, Z., Yan, W., et al. (2018). H19 functions as a competing endogenous RNA to regulate EMT by sponging miR-130a-3p in glioma. Cell. Physiol. biochem. 50 (1), 233–245. doi:10.1159/000494002
Hui, R., Macmillan, R. D., Kenny, F. S., Musgrove, E. A., Blamey, R. W., Nicholson, R. I., et al. (2000). INK4a gene expression and methylation in primary breast cancer: Overexpression of p16INK4a messenger RNA is a marker of poor prognosis. Clin. Cancer Res. 6 (7), 2777–2787.
Ito, K., Barnes, P. J., and Adcock, I. M. (2002). Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin- 1beta-induced histone H4 acetylation on lysines 8 and 12. Mol. Cell. Biol. 20, 6891–6903. doi:10.1128/MCB.20.18.6891-6903.2000
Jadaliha, M., Zong, X., Malakar, P., Ray, T., Singh, D. K., Freier, S. M., et al. (2016). Functional and prognostic significance of long non-coding RNA MALAT1 as a metastasis driver in ER negative lymph node negative breast cancer. Oncotarget 7, 40418–40436. doi:10.18632/oncotarget.9622
Jansen, M. P., Foekens, J. A., van Staveren, I. L., Dirkzwager-Kiel, M. M., Ritstier, K., Look, M. P., et al. (2005). Molecular classification of tamoxifen-resistant breast carcinomas by gene expression profiling. J. Clin. Oncol. 23, 732–740. doi:10.1200/JCO.2005.05.145
Jeselsohn, R., Bergholz, J. S., Pun, M., Cornwell, M., Liu, W., Nardone, A., et al. (2018). Allele-specific chromatin recruitment and therapeutic vulnerabilities of ESR1 activating mutations. Cancer Cell. 33, 173–186.e5. doi:10.1016/j.ccell.2018.01.004
Jeselsohn, R., Buchwalter, G., De Angelis, C., Brown, M., and Schiff, R. (2015). ESR1 mutations—A mechanism for acquired endocrine resistance in breast cancer. Nat. Rev. Clin. Oncol. 12, 573–583. doi:10.1038/nrclinonc.2015.117
Johnson, A. B., and O’Malley, B. W. (2012). Steroid receptor coactivators 1, 2, and 3: Critical regulators of nuclear receptor activity and steroid receptor modulator (SRM)-based cancer therapy. Mol. Cell. Endocrinol. 348, 430–439. doi:10.1016/j.mce.2011.04.021
Jones, S., Zhang, X., Parsons, D. W., Lin, J. C., Leary, R. J., Angenendt, P., et al. (2008). Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321, 1801–1806. doi:10.1126/science.1164368
Kanwal, R., Pandey, M., Natarajan, B., MacLennan, G. T., Fu, P., Lee E Ponsky, M. D., et al. (2014). Protection against oxidative DNA damage and stress in human prostate by glutathione S-transferase P1. Mol. Carcinog. 53 (1), 8–18. doi:10.1002/mc.21939
Kern, F. G., McLeskey, S. W., Zhang, L., Kurebayashi, J., Liu, Y., Ding, I. Y., et al. (1994). Transfected MCF-7 cells as a model for breast-cancer progression. Breast Cancer Res. Treat. 31, 153–165. doi:10.1007/BF00666149
Kim, T. Y., Bang, Y. J., and Robertson, K. D. (2006). Histone deacetylase inhibitors for cancer therapy. Epigenetics 1, 15–24. doi:10.4161/epi.1.1.2644
Klinge, C. M. (2018). Non-coding RNAs in breast cancer: Intracellular and intercellular Communication. Noncoding. RNA 4, 40. doi:10.3390/ncrna4040040
Lavinsky, R. M., Jepsen, K., Heinzel, T., Torchia, J., Mullen, T. M., Schiff, R., et al. (1998). Diverse signaling pathways modulate nuclear receptor recruitment of N-CoR and SMRT complexes. Proc. Natl. Acad. Sci. U. S. A. 95, 2920–2925. doi:10.1073/pnas.95.6.2920
Levin, E. R., and Pietras, R. J. (2008). Estrogen receptors outside the nucleus in breast cancer. Breast Cancer Res. Treat. 108, 351–361. doi:10.1007/s10549-007-9618-4
Li, M., Wang, C., Yu, B., Zhang, X., Shi, F., and Liu, X. (2019). Diagnostic value of RASSF1A methylation for breast cancer: A meta-analysis. Biosci. Rep. 39 (6). doi:10.1042/bsr20190923
Li, Z., Xu, L., Liu, Y., Fu, S., Tu, J., Hu, Y., et al. (2018). LncRNA MALAT1 promotes relapse of breast cancer patients with postoperative fever. Am. J. Transl. Res. 10, 3186–3197.
Litt, M. D., Simpson, M., Gaszner, M., Allis, C. D., and Felsenfeld, G. (2001). Correlation between histone lysine methylation and developmental changes at the chicken beta-globin locus. Science 293, 2453–2455. doi:10.1126/science.1064413
Liu, H., Lee, E. S., Deb Los Reyes, A., Zapf, J. W., and Jordan, V. C. (2001). Silencing and reactivation of the selective estrogen receptor modulator-estrogen receptor alpha complex. Cancer Res. 61, 3632–3639.
Liu, S., Dontu, G., Mantle, I. D., Patel, S., Ahn, N. s., Jackson, K. W., et al. (2006). Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 66, 6063–6071. doi:10.1158/0008-5472.CAN-06-0054
Liu, Y., Du, Y., Hu, X., Zhao, L., and Xia, W. (2018). Up-regulation of CeRNA TINCR by SP1 contributes to tumorigenesis in breast cancer. BMC Cancer 18, 367. doi:10.1186/s12885-018-4255-3
Lo, P. K., and Sukumar, S. (2008). Epigenomics and breast cancer. Pharmacogenomics 12, 1879–1902. doi:10.2217/14622416.9.12.1879
Lopez-Tarruella, S., and Schiff, R. (2007). The dynamics of estrogen receptor status in breast cancer: Reshaping the paradigm. Clin. Cancer Res. 13, 6921–6925. doi:10.1158/1078-0432.CCR-07-1399
Lu, Y., Li, T., Wei, G., Liu, L., Chen, Q., Xu, L., et al. (2016). The long non-coding RNA NEAT1 regulates epithelial to mesenchymal transition and radioresistance in through MiR-204/ZEB1 Axis in nasopharyngeal carcinoma. Tumour Biol. 37, 11733–11741. doi:10.1007/s13277-015-4773-4
Martin, C., and Zhang, Y. (2005). The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell. Biol. 6, 838–849. doi:10.1038/nrm1761
Mathur, R., and Jha, A. K. (2020). Reversal of hypermethylation and reactivation of tumor suppressor genes due to natural compounds in breast cancer cells.
Métivier, R., Penot, G., Hubner, M. R., Reid, G., Brand, H., Kos, M., et al. (2003). Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell. 115, 751–763. doi:10.1016/s0092-8674(03)00934-6
Moore, M. J., and Proudfoot, N. J. (2009). Pre-mRNA processing reaches back to transcription and ahead to translation. Cell. 136, 688–700. doi:10.1016/j.cell.2009.02.001
Morel, D., Jeffery, D., Aspeslagh, S., Almouzni, G., and Postel-Vinay, S. (2020). Combining epigenetic drugs with other therapies for solid tumours—Past lessons and future promise. Nat. Rev. Clin. Oncol. 17, 91–107. doi:10.1038/s41571-019-0267-4
Morgan, L., Gee, J., Pumford, S., Farrow, L., Finlay, P., Robertson, J., et al. (2009). Elevated Src kinase activity attenuates tamoxifen response in vitro and is associated with poor prognosis clinically. Cancer Biol. Ther. 8, 1550–1558. doi:10.4161/cbt.8.16.8954
Mukherjee, N., Calviello, L., Hirsekorn, A., de Pretis, S., Pelizzola, M., and Ohler, U. (2017). Integrative classification of human coding and noncoding genes through RNA metabolism profiles. Nat. Struct. Mol. Biol. 24, 86–96. doi:10.1038/nsmb.3325
Munzone, E., Curigliano, G., Rocca, A., Bonizzi, G., Renne, G., Goldhirsch, A., et al. (2006). Reverting estrogen-receptor-negative phenotype in HER-2-overexpressing advanced breast cancer patients exposed to trastuzumab plus chemotherapy. Breast Cancer Res. 8, R4. doi:10.1186/bcr1366
Musgrove, E. A., and Sutherland, R. L. (2009). Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer 9, 631–643. doi:10.1038/nrc2713
O’Neil, N. J., Bailey, M. L., and Hieter, P. (2017). Synthetic lethality and cancer. Nat. Rev. Genet. 18, 613–623. doi:10.1038/nrg.2017.47
Oronsky, B., Oronsky, N., Knox, S., Fanger, G., and Scicinski, J. (2014). Episensitization: Therapeutic tumor resensitization by epigenetic agents: A review and reassessment. Anticancer. Agents Med. Chem. 14, 1121–1127. doi:10.2174/1871520614666140418144610
Osborne, C. K., Bardou, V., Hopp, T. A., Chamness, G. C., Hilsenbeck, S. G., Fuqua, S. A. W., et al. (2003). Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J. Natl. Cancer Inst. 95, 353–361. doi:10.1093/jnci/95.5.353
Paakinaho, V., Presman, D. M., Ball, D. A., Johnson, T. A., Schiltz, R. L., Levitt, P., et al. (2017). Single-molecule analysis of steroid receptor and cofactor action in living cells. Nat. Commun. 8, 15896. doi:10.1038/ncomms15896
Peters, A. H., Mermoud, J. E., Carroll, D. O., Pagani, M., Schweizer, D., Brockdorff, N., et al. (2002). Histone H3 lysine 9 methylation is an epigenetic imprint of facultative heterochromatin. Nat. Genet. 30, 77–80. doi:10.1038/ng789
Pfeifer, G. P., and Besaratinia, A. (2009). Mutational spectra of human cancer. Hum. Genet. 125, 493–506. doi:10.1007/s00439-009-0657-2
Pontiggia, O., Rodriguez, V., Fabris, V., Raffo, D., Bumaschny, V., Fiszman, G., et al. (2009). Establishment of an in vitro estrogen-dependent mouse mammary tumor model: A new tool to understand estrogen responsiveness and development of tamoxifen resistance in the context of stromal-epithelial interactions. Breast Cancer Res. Treat. 116, 247–255. doi:10.1007/s10549-008-0113-3
Ramadan, A., and Hashim, M., Amr Abouzid & Menha Swellam Clinical impact of PTEN methylation status as a prognostic marker for breast cancer J. Genet. Eng. Biotechnol. volume 19, Article number: 66 (2021)
Razavi, P., Chang, M. T., Xu, G., Bandlamudi, C., Ross, D. S., Vasan, N., et al. (2018). The genomic landscape of endocrine-resistant advanced breast cancers. Cancer Cell. 34, 427–438.e6. doi:10.1016/j.ccell.2018.08.008
Rea, S., Eisenhaber, F., O’Carroll, D., Strahl, B. D., Sun, Z. W., Schmid, M., et al. (2000). Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406, 593–599. doi:10.1038/35020506
Rinn, J. L., and Chang, H. Y. (2012). Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 81, 145–166. doi:10.1146/annurev-biochem-051410-092902
Robertson, K. D., Ait-Si-Ali, S., Yokochi, T., Wade, P. A., Jones, P. L., and Wolffe, A. P. (2000). DNMT1 forms a complex with Rb, E2F1, and HDAC1 and represses transcription from E2F-responsive promoters. Nat. Genet. 25, 338–342. doi:10.1038/77124
Roth, S. Y., Denu, J. M., and Allis, C. D. (2001). Histone acetyltransferases. Annu. Rev. Biochem. 70, 81–120. doi:10.1146/annurev.biochem.70.1.81
Saelee, P., and Pongtheerat, T. (2020). APC promoter hypermethylation as a prognostic marker in breast cancer patients. Asian pac. J. Cancer Prev. 21 (12), 3627–3632. doi:10.31557/apjcp.2020.21.12.3627
Saez-Ayala, M., Montenegro, M. F., Sanchez-Del-Campo, L., Fernandez-Perez, M. P., Chazarra, S., Freter, R., et al. (2013). Directed phenotype switching as an effective antimelanoma strategy. Cancer Cell. 24, 105–119. doi:10.1016/j.ccr.2013.05.009
Santen, R. J., Fan, P., Zhang, Z., Bao, Y., Song, R. X. D., and Yue, W. (2009). Estrogen signals via an extranuclear pathway involving IGF-1R and EGFR in tamoxifen-sensitive and -resistant breast cancer cells. Steroids 74, 586–594. doi:10.1016/j.steroids.2008.11.020
Schiff, R., Massarweh, S. A., Shou, J., Bharwani, L., Mohsin, S. K., and Osborne, C. K. (2004). Cross-talk between estrogen receptor and growth factor pathways as a molecular target for overcoming endocrine resistance. Clin. Cancer Res. 10, 331S–36S. doi:10.1158/1078-0432.ccr-031212
Schubeler, D., macAlpine, D. M., Scalzo, D., Wirbelauer, C., Kooperberg, C., van Leeuwen, F., et al. (2004). The histone modification pattern of active genes revealed through genome-wide chromatin analysis of a higher eukaryote. Genes. Dev. 518, 1263–1271. doi:10.1101/gad.1198204
Serrano-Gomez, S. J., Maziveyi, M., and Alahari, S. K. (2016). Regulation of epithelial mesenchymal transition through epigenetic and post-translational modifications. Mol. Cancer 15, 18. doi:10.1186/s12943-016-0502-x
Shang, Y., Hu, X., DiRenzo, J., Lazar, M. A., and Brown, M. (2000). Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell. 103, 843–852. doi:10.1016/s0092-8674(00)00188-4
Shi, L., Dong, B., Li, Z., Lu, Y., Ouyang, T., Li, J., et al. (2009). Expression of ER-{alpha}36, a novel variant of estrogen receptor {alpha}, and resistance to tamoxifen treatment in breast cancer. J. Clin. Oncol. 27, 3423–3429. doi:10.1200/JCO.2008.17.2254
Shiino, S., Kinoshita, T., Yoshida, M., Jimbo, K., Asaga, S., Takayama, S., et al. (2016). Prognostic impact of discordance in hormone receptor status between primary and recurrent sites in patients with recurrent breast cancer. Clin. Breast Cancer 16, e133–e140. doi:10.1016/j.clbc.2016.05.014
Shilatifard, A. (2006). Chromatin modifications by methylation and ubiquitination: Implications in the regulation of gene expression. Annu. Rev. Biochem. 75, 243–269. doi:10.1146/annurev.biochem.75.103004.142422
Shin, V. Y., Chen, J., Cheuk, I. W. Y., Siu, M. T., Ho, C. W., Wang, X., et al. (2019). Long non-coding RNA NEAT1 confers oncogenic role in triple-negative breast cancer through modulating chemoresistance and cancer stemness. Cell. Death Dis. 10, 270–310. doi:10.1038/s41419-019-1513-5
Shou, J., Massarweh, S., Osborne, C. K., Wakeling, A. E., Ali, S., Weiss, H., et al. (2004). Mechanisms of tamoxifen resistance: Increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J. Natl. Cancer Inst. 96, 926–935. doi:10.1093/jnci/djh166
Span, P. N., Tjan-Heijnen, V. C., Manders, P., Beex, L. V. A. M., and Sweep, C. G. J. (2003). Cyclin-E is a strong predictor of endocrine therapy failure in human breast cancer. Oncogene 22, 4898–4904. doi:10.1038/sj.onc.1206818
Spoerke, J. M., Gendreau, S., Walter, K., Qiu, J., Wilson, T. R., Savage, H., et al. (2016). Heterogeneity and clinical significance of ESR1 mutations in ER-positive metastatic breast cancer patients receiving fulvestrant. Nat. Commun. 7, 11579. doi:10.1038/ncomms11579
Strahl, B. D., and Allis, C. D. (2000). The language of covalent histone modifications. Nature 403, 41–45. doi:10.1038/47412
Stransky, N., Vallot, C., Reyal, F., Bernard-Pierrot, I., de Medina, S. G., Segraves, R., et al. (2006). Regional copy number-independent deregulation of transcription in cancer. Nat. Genet. 38, 1386–1396. doi:10.1038/ng1923
Suzuki, H., Toyota, M., Carraway, H., Caraway, H., Gabrielson, E., Ohmura, T., et al. (2008). Frequent epigenetic inactivation of Wnt antagonist genes in breast cancer. Br. J. Cancer 98, 1147–1156. doi:10.1038/sj.bjc.6604259
Thomas, M. P., and Potter, B. V. (2013). The structural biology of oestrogen metabolism. J. Steroid Biochem. Mol. Biol. 137, 27–49. doi:10.1016/j.jsbmb.2012.12.014
Tomar, D., Yadav, A. S., Kumar, D., Bhadauriya, G., and Kundu, G. C. (2020). Non-coding RNAs as potential therapeutic targets in breast cancer. Biochim. Biophys. Acta. Gene Regul. Mech. 1863, 194378. doi:10.1016/j.bbagrm.2019.04.005
Turner, B. M. (2000). Histone acetylation and an epigenetic code. Bioessays 22, 836–845. doi:10.1002/1521-1878(200009)22:9<836::aid-bies9>3.0.co;2-x
Vennin, C., Spruyt, N., Robin, Y. M., Chassat, T., Le Bourhis, X., and Adriaenssens, E. (2017). The long non-coding RNA 91 H increases aggressive phenotype of breast cancer cells and up-regulates H19/IGF2 expression through epigenetic modifications. Cancer Lett. 385, 198–206. doi:10.1016/j.canlet.2016.10.023
Wade, P. A. (2001). Transcriptional control at regulatory checkpoints by histone deacetylases: Molecular connections between cancer and chromatin. Hum. Mol. Genet. 10, 693–698. doi:10.1093/hmg/10.7.693
Wang, M., Han, D., Yuan, Z., Hu, H., Zhao, Z., Yang, R., et al. (2018). Long non-coding RNA H19 confers 5-Fu resistance in colorectal cancer by promoting SIRT1-mediated autophagy. Cell. Death Dis. 9 (12), 1149. doi:10.1038/s41419-018-1187-4
Wang, X., Liu, Y., Sun, H., Ge, A., Li, D., Fu, J., et al. (2020). DNA methylation in RARβ gene as a mediator of the association between healthy lifestyle and breast cancer: A case-control study. Cancer Manag. Res. 12, 4677–4684. doi:10.2147/CMAR.S244606
Wang, Yan, Dan, Liangying, Li, Qianqian, Li, Lili, Lan, Zhong, Shao, Bianfei, et al. Putti, Xiaoqian He, Yixiao Feng, Yong Lin & Tingxiu Xiang ZMYND10, an epigenetically regulated tumor suppressor, exerts tumor-suppressive functions via miR145-5p/NEDD9 axis in breast cancer Clin. Epigenetics volume 11, Article number: 184 (2019)
Wils, L. J., and Bijlsma, M. F. (2018). Epigenetic regulation of the Hedgehog and Wnt pathways in cancer. Crit. Rev. Oncol. Hematol. 121, 23–44. doi:10.1016/j.critrevonc.2017.11.013
Wu, Jiaxue, Lu, Lin-Yu, and Yu, Xiaochun (2010). The role of BRCA1 in DNA damage response.Protein Cell. 1 (2), 117–123. doi:10.1007/s13238-010-0010-5
Wu, Y., Shao, A., Wang, L., Hu, K., Yu, C., Pan, C., et al. (2019). The role of LncRNAs in the distant metastasis of breast cancer. Front. Oncol. 9, 407. doi:10.3389/fonc.2019.00407
Xia, W., Bacus, S., Hegde, P., Husain, I., Strum, J., Liu, L., et al. (2006). A model of acquired autoresistance to a potent ErbB2 tyrosine kinase inhibitor and a therapeutic strategy to prevent its onset in breast cancer. Proc. Natl. Acad. Sci. U. S. A. 103, 7795–7800. doi:10.1073/pnas.0602468103
Xiang, T., Li, L., Yin, X., Zhong, L., Peng, W., Qiu, Z., et al. (2013). Epigenetic silencing of the WNT antagonist Dickkopf 3 disrupts normal Wnt/β-catenin signalling and apoptosis regulation in breast cancer cells. J. Cell. Mol. Med. 17, 1236–1246. doi:10.1111/jcmm.12099
Xu, D., Bai, J., Duan, Q., Costa, M., and Dai, W. (2009). Covalent modifications of histones during mitosis and meiosis. Cell. Cycle 8, 3688–3694. doi:10.4161/cc.8.22.9908
Xu, G., Chhangawala, S., Cocco, E., Razavi, P., Cai, Y., Otto, J. E., et al. (2020). ARID1A determines luminal identity and therapeutic response in estrogen-receptor-positive breast cancer. Nat. Genet. 52, 198–207. doi:10.1038/s41588-019-0554-0
Yang, S. X., Polley, E., and Lipkowitz, S. (2016). New insights on PI3K/AKT pathway alterations and clinical outcomes in breast cancer. Cancer Treat. Rev. 45, 87–96. doi:10.1016/j.ctrv.2016.03.004
Zardo, G., Reale, A., De Matteis, G., Buontempo, S., and Caiafa, P. (2003). A role for poly (ADP-ribosyl)ation in DNA methylation. Biochem. Cell. Biol. 81, 197–208. doi:10.1139/o03-050
Zhang, C. Y., Yu, M. S., Li, X., Zhang, Z., Han, C. R., and Yan, B. (2017). Overexpression of long non-coding RNA MEG3 suppresses breast cancer cell proliferation, invasion, and angiogenesis through AKT pathway. Tumour Biol. 39 (6), 1010428317701311. doi:10.1177/1010428317701311
Zhang, Y., Chan, H. L., Garcia-Martinez, L., Karl, D. L., Weich, N., Slingerland, J. M., et al. (2020). Estrogen induces dynamic ERα and RING1B recruitment to control gene and enhancer activities in luminal breast cancer. Sci. Adv. 6, eaaz7249. doi:10.1126/sciadv.aaz7249
Zhao, Z., Chen, C., Liu, Y., and Wu, C. (2014). 17β-Estradiol treatment inhibits breast cell proliferation, migration and invasion by decreasing MALAT-1 RNA level. Biochem. Biophys. Res. Commun. 445, 388–393. doi:10.1016/j.bbrc.2014.02.006
Zhou, S., He, Y., Yang, S., Hu, J., Zhang, Q., Chen, W., et al. (2018). The regulatory roles of lncRNAs in the process of breast cancer invasion and metastasis. Biosci. Rep. 38, BSR20180772. doi:10.1042/BSR20180772
Keywords: cancer, breast, epigenetics, estrogen, therapy
Citation: Mathur R, Jha NK, Saini G, Jha SK, Shukla SP, Filipejová Z, Kesari KK, Iqbal D, Nand P, Upadhye VJ, Jha AK, Roychoudhury S and Slama P (2022) Epigenetic factors in breast cancer therapy. Front. Genet. 13:886487. doi: 10.3389/fgene.2022.886487
Received: 28 February 2022; Accepted: 20 July 2022;
Published: 23 September 2022.
Edited by:
César López-Camarillo, Universidad Autónoma de la Ciudad de México, MexicoReviewed by:
Dilek Cansu Gurer, Izmir Institute of Technology, TurkeyAmir Yunus, Universiti Sains Malaysia (USM), Malaysia
Copyright © 2022 Mathur, Jha, Saini, Jha, Shukla, Filipejová, Kesari, Iqbal, Nand, Upadhye, Jha, Roychoudhury and Slama. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Abhimanyu Kumar Jha, abhimanyujha630@gmail.com; Shubhadeep Roychoudhury, shubhadeep1@gmail.com