 Min-Hua Tseng1,2
Min-Hua Tseng1,2 Sung-Sen Yang3
Sung-Sen Yang3 Chih-Chien Sung3
Chih-Chien Sung3 Jhao-Jhuang Ding4
Jhao-Jhuang Ding4 Yu-Juei Hsu3Shih-Ming Chu5
Yu-Juei Hsu3Shih-Ming Chu5 Shih-Hua Lin3*
Shih-Hua Lin3*- 1Division of Nephrology, Department of Pediatrics, Chang Gung Memorial Hospital and Chang Gung University, Taoyuan, Taiwan
- 2Department of Pediatrics, Xiamen Chang Gung Hospital, Ximen, China
- 3Division of Nephrology, Department of Internal Medicine, Tri-Service General Hospital, National Defense Medical Center, Taipei, Taiwan
- 4Department of Pediatrics, Tri-Service General Hospital, National Defense Medical Center, Taipei, Taiwan
- 5Division of Neonatology, Department of Pediatrics, Chang Gung Memorial Hospital and Chang Gung University, Taoyuan, Taiwan
CNNM2 is primarily expressed in the brain and distal convoluted tubule (DCT) of the kidney. Mutations in CNNM2 have been reported to cause hypomagnesemia, seizure, and intellectual disability (HSMR) syndrome. However, the clinical and functional effect of CNNM2 mutations remains incompletely understood. We report our clinical encounter with a 1-year-old infant with HSMR features. Mutation screening for this trio family was performed using next-generation sequencing (NGS)-based whole exome sequencing (WES) with the identified mutation verified by Sanger sequencing. We identified a de novo heterozygous mutation c.G1439T (R480L) in the essential cystathionine β-synthase (CBS) domain of CNNM2 encoding CNNM2 (cyclin M2) without any other gene mutations related to hypomagnesemia. The amino acid involved in this missense mutation was conserved in different species. It was also found to be pathogenic based on the different software prediction models and ACGME criteria. In vitro studies revealed a higher expression of the CNNM2-R480L mutant protein compared to that of the wild-type CNNM2. Like the CNNM2-wild type, proper localization of CNNM2-R480L was shown on immunocytochemistry images. The Mg2+ efflux assay in murine DCT (mDCT) cells revealed a significant increase in intracellular Mg2+ green in CNNM2-R480L compared to that in CNNM2-WT. By using a simulation model, we illustrate that the R480L mutation impaired the interaction between CNNM2 and ATP-Mg2+. We propose that this novel R480L mutation in the CNNM2 gene led to impaired binding between Mg2+-ATP and CNNM2 and diminished Mg2+ efflux, manifesting clinically as refractory hypomagnesemia.
Introduction
Magnesium (Mg2+) is a pivotal cation and cofactor in maintaining cell function (Li et al., 2011; Jahnen-Dechent and Ketteler, 2012; de Baaij et al., 2015). In the gastrointestinal system, it is reabsorbed in the intestine and in the colon by fine-tuned control of an active transcellular Mg2+ channel, followed by extrusion to the blood via the CNNM4 Na+/Mg2+ exchanger. In the kidney, most of the filtered Mg2+ is reabsorbed paracellularly in the proximal tubule (PT) and thick-ascending loop of Henle (TALH) via different claudins (claudins 1 and 2 in the PT, and claudins 10, 14, 16, and 19 in TALH) (Stuiver et al., 2011; de Baaij et al., 2012; Curry and Yu, 2018; Ellison et al., 2021). Like the colon, the distal convoluted tubule (DCT) fine-tunes the reabsorption of tubular Mg2+ via the transcellular TRPM6 channel along with basolateral Mg2+ extrusion of Na+/Mg2+ exchanger. Inactivating mutations in claudins 16 and 19 in TALH are responsible for familial hypomagnesemia hypercalciuria nephrocalcinosis (FHHNC types I and II). Defective Mg2+ reabsorption in the DCT inevitably causes renal hypomagnesemia because there is no Mg2+ reabsorption in the downstream DCT. Gene mutations related to the regulation of Mg2+ transport in the DCT can cause renal hypomagnesemia. These genes include SLC12A3 encoding thiazide-sensitive NCC, TRMP6 encoding apical TRPM6 channel, HNF1B encoding HNF1β, PCBD1 encoding PCBD1, EGF encoding EGF, EGFR encoding EGFR, KCNJ10 encoding Kir4.1 (EAST syndrome), KCNA1 encoding Kv1.1, FXYD2 encoding γ-subunit of Na+-K+ ATPase, and CNNM2 encoding CNNM2 (cyclin M2) (Meij et al., 2003; Groenestege et al., 2007; Adalat et al., 2009; Glaudemans et al., 2009; Reichold et al., 2010; de Baaij et al., 2012; Ferrè et al., 2014; Sponder et al., 2016; Chen et al., 2021).
In 2011 and 2014, mutations in CNNM2 predominantly expressed in the DCT and brain were first reported to cause autosomal-dominant (majority) or recessive (minority) renal hypomagnesemia with seizure and intellectual disability (HSMR) (de Baaij et al., 2012; Chen et al., 2021; Franken et al., 2021; Accogli et al., 2019; Arjona and de Baaij, 2018; Bamhraz et al., 2021). In humans, the cyclin M (CNNM, also formerly known as ancient conserved domain protein, ACDP) family has four member proteins (CNNM1–4) sharing evolutionary homology. As shown in Figure 1, the CNNM2 structure consists of an extracellular domain, transmembrane domain (TMD) containing the domain of unknown function-21 (DFU21), Bateman domain or module containing two functionally essential cystathionine β-synthase (CBS) to bind Mg2+-ATP, and cytosolic cyclic-nucleotide-binding homology domain (CNBH). To date, only a few families with CNNM2 mutations have been reported (Arjona et al., 2014; Chen et al., 2021; Li et al., 2021). Although CNNM2 was once thought to be a basolateral Mg2+ transporter or Na+/Mg2+ exchanger, more recent evidence points to its function as an intracellular Mg2+ sensor, using a conformational change upon binding of Mg2+-ATP (Accogli et al., 2019). However, the precise mechanism by which CNNM2 mutation leads to a conformational change and impaired sensing of Mg2+-ATP to affect basolateral Mg2+ extrusion and/or apical Mg2+ reabsorption remains unknown.
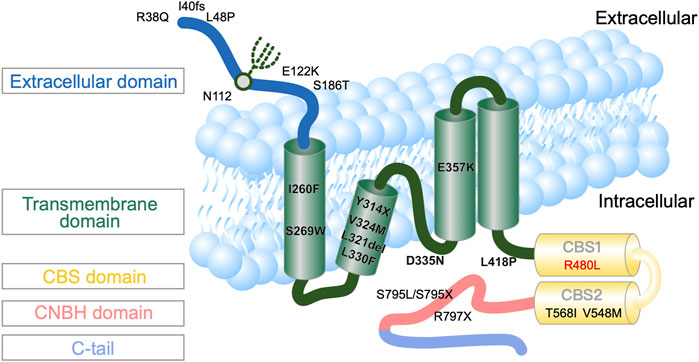
FIGURE 1. Topography of human CNNM2 with the position of the reported mutations (Stuiver et al., 2011). * indicates the mutation site and location in this patient.
We have encountered an infant with refractory hypomagnesemia and excessive renal Mg2+ wasting, seizure, and intellectual disability, consistent with the clinical description of HSMR syndrome. In this study, we aimed to identify the genetic mutation for her phenotype and to assess the functional impact of the identified mutation. Our results indicated that a de novo heterozygous mutation c.G1439T (R480L) in the CBS domain of the CNNM2 gene was identified in this proband. This missense CNNM2 R480L mutation was found to be pathogenic. In vitro studies revealed higher CNNM2-R480L protein expression with proper cellular localization on immunocytochemistry images. The impaired Mg2+ efflux with a significant increase in intracellular Mg2+ suggests that the CNNM2-R480L mutation blocks Mg2+ efflux. In our simulation model, this R480L mutation leads to an attenuated interaction between CNNM2 and ATP-Mg2+.
Materials and Methods
This study was approved by the ethics committee on human studies at Tri-Service General Hospital in Taiwan (IRB2-105-05-136). A trio family including the proband and her parents were enrolled. Written informed consent was obtained from the participants.
Index Case
The index case was a premature neonate who presented with generalized seizures at the age of 10 days. She was born preterm, gestational age 30 weeks, and was fed with breast and formula milk. No additives to feeding were noticed. The parents were non-consanguineous and no familial history of inherited disorders was reported. Her initial blood pressure was 90/45 mmHg. No anomalies were found on physical examination. Her laboratory characteristics were consistent with hypomagnesemia (0.8 mg/dl) with increased urine Mg2+ excretion (FEMg 6.5%), normokalemia, normocalcemia, and normocalciuria (Table 1). There was refractory hypomagnesemia with plasma Mg2+ ranging from 0.8 to 1.1 mg/dl despite high-dose oral and intravenous Mg2+ supplementation. At her 2 -year-old follow-up, she exhibited delayed neurodevelopment but decreased seizure frequency.
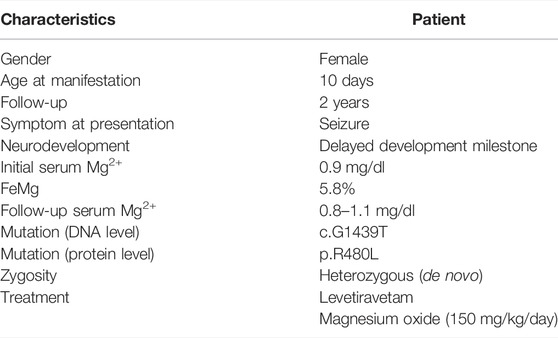
TABLE 1. Clinical characteristics.
Whole Exome Sequencing and Direct Sanger Sequencing
Genomic DNA was isolated from a peripheral venous blood sample. We performed exome capture using the Agilent Sure Select v6 and massively parallel sequencing using the HiSeq 4000 platform as previously reported (Li and Durbin, 2009; DePristo et al., 2011). Raw image analyses and base calling were performed using Illumina’s Pipeline with default parameters. Sequence data were aligned to the reference human genome using the Burrows–Wheeler Aligner (BWA) (Li and Durbin, 2009), and duplicate reads were removed using Picard tools. We used the Genome Analysis ToolKit (GATK) to perform the re-alignment and variation (SNP and InDel) detection (DePristo et al., 2011). Annovar was utilized to catalog the detected variations (Wang et al., 2010). Then, we filtered variations with a homo-polymer length >6 (and synonymous substitutions) or that were common (>2%) by dbSNP150 (http://www.ncbi.nlm.nih.gov/projects/SNP/), HapMap, the 1000 Genomes Project (http://www.1000 genomes.org), Exome Aggregation Consortium (ExAC) database, and the Genome Aggregation Database (gnomAD, https://gnomad.broadinstitute.org). Direct Sanger sequencing was performed for all patients and their parents to verify the genetic variants detected by WES. The data that support the findings of this study are available from the corresponding author upon reasonable request.
Cell Culture, Construction of Plasmids, and Transfection
Murine distal convoluted tubular (mDCT) cells were cultured in a 1:1 mixture of Dulbecco’s modified Eagle’s medium with 1 g/L glucose, 1 mM sodium pyruvate, and Ham’s F-12 Nutrient Mix. Finally, 5% (v/v) fetal bovine serum,100 U/ml penicillin, and 0.1 mg/ml streptomycin were added to the growth medium. The cells were incubated at 37°C in a humidified 5% CO2 incubator. Human CNNM2 cDNA (NM_017649.5) was cloned into the pcDNA3.1 vector. The disease-causing mutation was obtained by a QuikChangeTM Site-Directed Mutagenesis Kit (Stratagene, San Diego, CA, United States of America). The primers used to introduce the mutations were R480L: forward primer 5′-GAGCGGCTACACCCTCATTCCAGTGTTTG-3′; reverse primer 5′-CAAACACTGGAATGAGGGTGTAGCCGCTC-3′, R480K: forward primer 5′-GGAGAGCGGCTACACCAAGATTCCAGTGTTTGAAGG-3′; reverse primer 5′-CCTTCAAACACTGGAATCTTGGTGTAGCCGCTCTCC-3′, V548M: forward primer 5′CTCACCTGGCTATCATGCAGCGGGTAAACAATG-3′; reverse primer 5′-CATTGTTTACCCGCTGCATGATAGCCAGGTGAG-3′, T568I: forward primer 5′-GAAGTTCTGGGAATCATCTTAGAAGATGTGATTG-3′; reverse primer 5′-CAATCACATCTTCTAAGATGACGATTCCCAGAACTTC-3′. The mDCT cells seeded in a six-well plate with 70–90% confluence were transfected by the indicated amount of plasmid DNA with a Lipofectamine 3000 Reagent (Thermo Fisher Scientific). The reported R480K, V548M, and T568I constructs were selected as negative controls. R480K constructed in CBS1 was selected as a charge-unchanged control. Two previously reported mutant constructs (V548M and T568I) in CBS2 were also performed.
Western Blotting
The mDCT cells were harvested for 24 h after transfection. The cell lysates were prepared in a RIPA lysis buffer with a protease inhibitor cocktail (Roche). Following the separation by centrifugation, the remaining protein lysates were denatured in an SDS sample reagent with 100 mM DTT for 30 min at 37°C and then analyzed by polyacrylamide-SDS mini-gels. The mDCT cells were transfected with a pcDN3.1 empty vector or disease-causing mutation and harvested after 24 h. The cell membrane fraction was subjected to the ProteoExtract native membrane protein extraction kit (Merck-Millipore), following the manufacturer’s description, and then analyzed by semiquantitative immunoblotting (Yamazaki et al., 2013; Chen et al., 2018). The immunoblots were detected with specific antibodies: self-generated CNNM2 (X1000), CNNM2 (X200 Cusabio), α-actin (X30000; Santa Cruz), Hsp70 (X2000; Enzo Life Science), and Na+-K+ ATPase (X1000; Santa Cruz).
Immunocytochemistry
The mDCT cells were seeded on a chamber slide (Millicell EZ slide) and transiently transfected with 0.5 μg of plasmid DNA. After 24 h , the cells were washed with PBS and fixed with 4% paraformaldehyde for 15 min. After PBS rinses, the cells were incubated for 1 h with 0.1 Triton X100 in PBS and then blocked with 1% BSA in PBST for another 30 min. Specific antibody CNNM2 (X200 Cusabio) was used for cell staining. After PBST rinses, cells were incubated with Alexa Fluor 488-conjugated goat anti-rabbit (X200 Invitrogen) for 1 h and stained with DAPI (5 ug/ml) for 5 min. The images were captured with a Leica DM2500 microscope.
Mg2+-Efflux Assay
The mDCT cells were cultured on a 96-black, clear bottom tissue culture plate (Corning), after transfection (24 h). The Mg2+ imaging of transfected cells was analyzed with Magnesium Green™ (Molecular Probes) as described previously (Yamazaki et al., 2013; Chen et al., 2018), with slight modifications. The cells were incubated with Mg2+ loading buffer including 2 μM Magnesium Green (Molecular Probes) at 37°C for 60 min. Then, the buffer was changed to buffer without Mg2+ (MgCl2 was replaced with 60 mM NaCl) and the fluorescent was recorded at 1-min intervals. The cells images were detected by ImageXpress Micro XLS (molecular devices) and fluorescence was measured using MetaXpress High content image acquisition and analysis software (Molecular Devices). The cell fluorescence was analyzed by the software setting for Cell Scoring.
Simulation of the Mutant Models
The resolved structures of the human CNNM-PRL complex (PDB code: 5LXQ) (Meij et al., 2003) were used as a template to generate the R480L mutant using the Built Mutants protocol (Biovia Discovery Studio 2019). The CHARMm force field was applied to the model followed by energy minimization using the smart algorithm in the calculation.
Statistical Analysis
The results were presented as mean ± standard deviation (SD) for continuous variables. Student’s t-test was used to compare differences between groups. When comparing the ratio of differences between groups, we used a ratio paired t-test with the Holm–Šídák method. A p-value less than 0.05 was considered statistically significant.
Results
Identification of Novel CNNM2 Mutation
To complete genetic diagnosis and counseling for this family, we pursued whole exome sequencing for the patient to identify the cause of pathogenesis. We obtained an average of 6.3G bases mapped to target exon regions with a mean depth of 81 times. About 98.18% of exons were covered at least 10 times. Overall, 53,172 single nucleotide variants and 6,224 small insertions or deletions were identified. Whole exome sequencing did not reveal mutations in SLC12A3, CLDN16, CLDN19, CLCNKB, and KCNJ10 but revealed one novel missense mutation (c.G1439T, p.R480L) located on the cystathionine-β-synthase (CBS) domain of CNNM2. Sanger sequencing for the patient and her parents confirmed this de novo heterozygous mutation in the CNNM2 gene (Figure 2A). R480 is a highly conserved amino acid in different animal species as shown in Figure 2B.
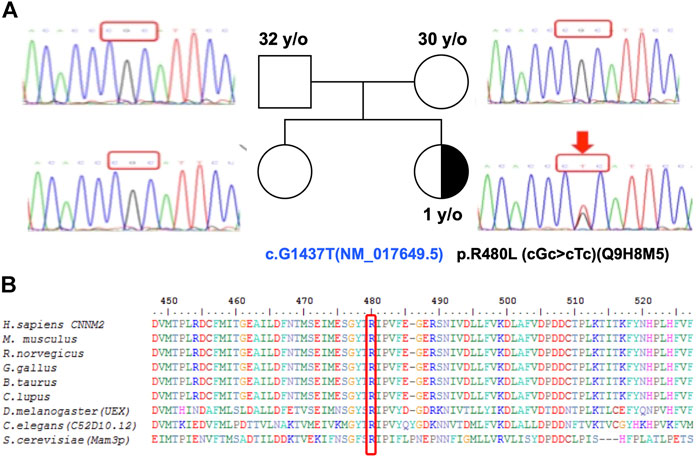
FIGURE 2. A multiple sequence alignment shows R480 as a highly conserved residue. (A) Pedigree showing the index patient and her parents with heterozygous CNNM2 mutation with a half-filled black symbol. (B) The R480 of CNNM2 is a highly conserved amino acid (Stuiver et al., 2011). The sequence accession IDs are H. sapiens: Q9H8M5, M. musculus: Q3TWN3, R. norvegicus: Q5U2P1, G. gallus: K7CZV7, B. taurus: A0A3Q1LRB9, C. lupus: E2RJ19, D. melanogaster (UEX): A0A0B7P9G0, C. elegans (C52D10.12): A3QM97, and S. cerevisiae (Mam3p): N1NX85.
In vitro Expression of Novel CNNM2 Mutants
The total and membrane expressions of wild-type CNNM2 (amino acids 1–875) and mutant CNNM2 (R480L, R480K, V548M, and T568I) proteins were examined after transient transfection of CNNM2 in mDCT cell lines. As shown in Figure 3 and Supplementary Table S1, the membrane expression was increased in CNNM2-R480L and CNNM2-R480K but not in CNNM2-V548M and CNNM2-T568I. Actin and HSp70 were used as the loading control and ubiquitination, respectively. There was no difference in the expression of actin and Hsp70 between cells with wild-type and all CNNM2 mutants.
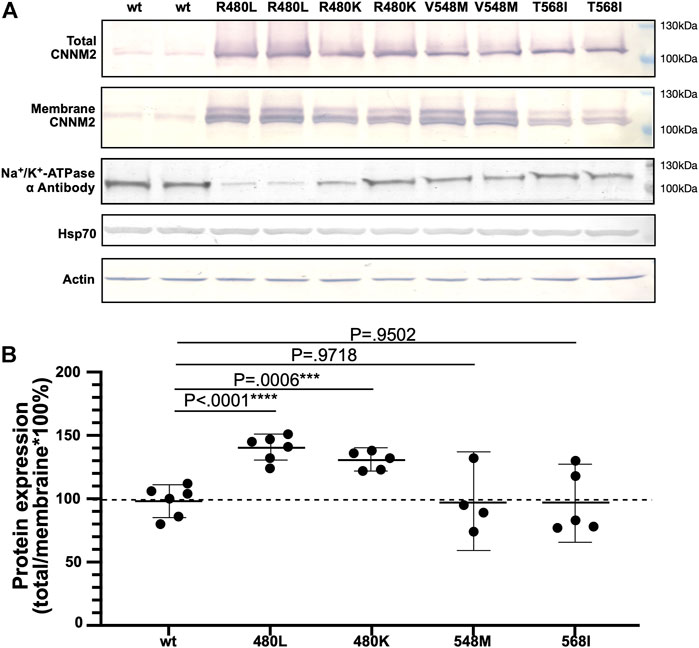
FIGURE 3. Expression of the mutant CNNM2. (A) mDCT cells were transfected with pcDNA3.1.HA.CNNM2-wild type, pcDNA3.1.HA.CNNM2-R480L, pcDNA3.1.HA.CNNM2-R480L, pcDNA3.1.HA.CNNM2-V548M, and pcDNA3.1.HA.CNNM2-T568I expression constructs. Cells extracts were subjected to western blot analysis and probed with antibodies against CNNM2. Antibodies to α-actin and Hsp70 were for loading control of mDCT cells. (B) Results are representative of three independent experiments and show increased membrane expression of CNNM2-R480L and CNNM2-R480K.
Immunocytochemistry Microscopy
The immunocytochemistry images of mDCT cells with anti-nuclei (blue) and anti-CNNM2 (green) demonstrated that CNNM2-wild type, CNNM2-R480L, and other negative controls (CNNM2-V548M and CNNM2-T568I) were properly localized adjacent to the cell membrane (Figure 4). Of note, the expression of CNNM2-R480L was higher than that of the CNNM2-wild type.
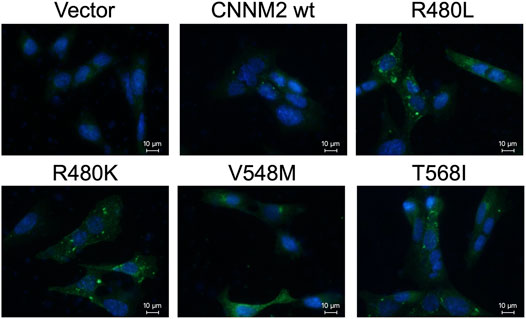
FIGURE 4. Immunocytochemistry microscopy. Images of transiently transfected murine distal convoluted tubular cells (mDCT) with wild-type and mutant constructs. Cells were immunostained with antibodies against the CNNM2 (in green). Nuclei were counterstained with DAPI (in blue). This demonstrates that the expressions of mutant CNNM2 are higher than those of wild-type CNNM2.
Mg2+-Efflux Assays
To evaluate the impact of R480L on the function of CNNM2, we examined the CNNM2-dependent Mg2+ efflux in a cellular assay with wild-type and mutant CNNM2. The mDCT cells were transfected with the indicated constructs treated with Mg2+ Green and then subjected to Mg2+ depletion. As shown in Figure 5 and Supplementary Table S2, intracellular Mg2+ Green was significantly higher in CNNM2-R480L compared to wild type and other CNNM2 mutants, with p-values < 0.05 from min 1 to min 5. This finding indicates that the R480L mutation located in the CBS1 domain of CNNM2 blocks Mg2+ efflux activity.
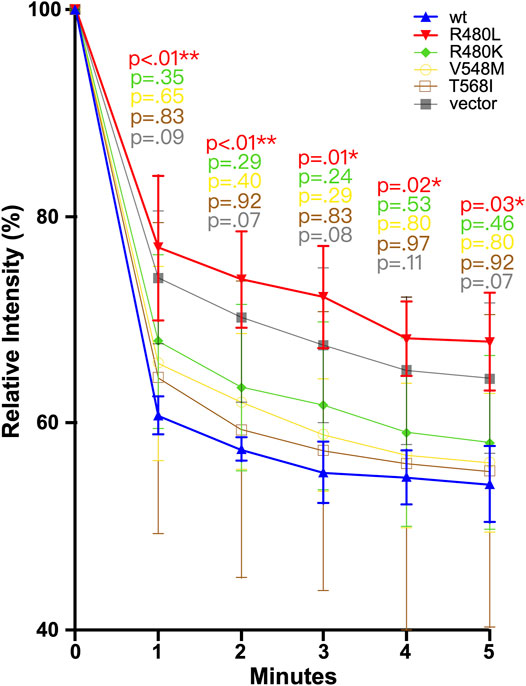
FIGURE 5. Magnesium efflux assay. mDCT cells transfected with the indicated constructs were loaded with Mg2+ green and then subjected to the Mg depletion condition. R480L had significantly reduced Mg2+ efflux than other mutants and wild type. In comparison with wild-type and mutant, a p-value was calculated. A p-value <0.05 is considered statistically significant. *p<0.05, **p < 0.01.
Simulation Model of the CNNM2 Mutant
The interaction from the side chain of Arg480 with the γ-phosphate of ATP was lost after being replaced by leucine (Figure 6). The mutation of arginine to leucine led to the reduction of the binding energy of Mg2+-ATP in the CBS module of CNNM2 by approximately 352 kcal/mol, which suggested that this mutation may cause significant impairment in the binding ability with Mg2+-ATP.
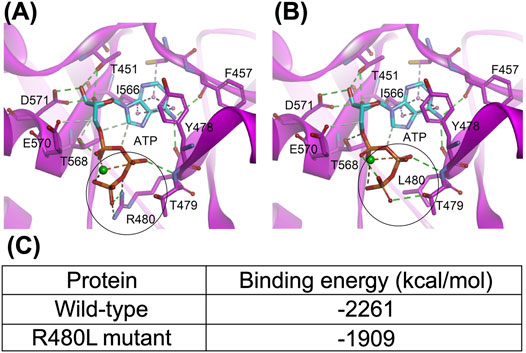
FIGURE 6. Structure of the CNNM-PRL complex (PDB code: 5LXQ). (A) and (B), the wild-type and R480L mutant CBS module of CNNM2, respectively. The dashed lines represent the hydrogen bond (green), electrostatic (orange), and hydrophobic (pink) interactions. The metal ion is shown as a green sphere. The black circle highlights the mutation site; (C) indicates that this mutation in the CBS module results in significant impairment of the binding ability with MgATP.
Discussion
In this study, we have identified a novel and de novo heterozygous mutation c.G1439T (R480L) in the CBS domain of the CNNM2 gene in a trio family with typical HSMR. In vitro studies showed that this CNNM2-R480L had a higher expression level than the CNNM2-wild type and proper localization to the plasma membrane. The Mg2+ efflux assay in mDCT cells revealed the blockade of intracellular Mg2+ efflux under Mg2+ depletion. The simulation model also predicted the attenuated interaction of this mutant protein with Mg2+-ATP.
To date, nearly 20mutations including ours in CNNM2 have been reported responsible for HSMR (Stuiver et al., 2011; de Baaij et al., 2012; Arjona and de Baaij, 2018; Accogli et al., 2019; Bamhraz et al., 2021; Franken et al., 2021). Autosomal-dominant inheritance is the most common type with the majority of mutations being de novo and missense as shown in this patient (Stuiver et al., 2011; de Baaij et al., 2012; Accogli et al., 2019; García-Castaño et al., 2020; Chen et al., 2021; Franken et al., 2021). The clinical features including profound hypomagnesemia, epilepsy, and neurodevelopmental delay in our proband were in line with phenotypes reported by a previous large cohort study (Franken et al., 2021).) Epilepsy might be a result of disturbed brain development rather than impaired Mg2+ efflux. Arjona et al. had shown that knockdown of CNNM2 isoforms in zebrafish resulted in disturbed brain development including neurodevelopmental impairments (Arjona et al., 2014). Although the correlation between phenotype and genotype was poor, patients with homozygous mutation usually have more severe forms of structural brain abnormalities (Stuiver et al., 2011; Accogli et al., 2019; Franken et al., 2021).
R480L mutation involved a highly conserved residue in the CBS domain of CNNM2 protein and was pathogenic based on a higher score of pathogenicity by different software prediction and ACME criteria (Richards et al., 2015). We first examined in vitro studies for this R480L mutant. A higher CNNM2-R480L protein expression with proper cellular localization on immunocytochemistry images was found. In contrast to the previous findings, the membranous expressions of mutant CNNM2-R480L were higher than those of the CNNM2-wild type (Franken et al., 2021). The exact mechanism of increased expression of CNNM2-R480L on the plasma membrane remained to be elucidated. We speculate that it is secondary to a compensatory effect of impaired function of mutant or different unique mutations in the CBS domain. We confirmed that the R480L mutation did not alter protein trafficking or membrane localization. Both arginine (R) and lysine (K) are basic amino acids with similar physiochemical properties. The difference between the membranous expression of R480L and R480K was minimal in this study. However, the charged R containing the guanidino head group, as opposed to the amino head group in the charged K, is solvated by more oxygen atoms and can form more H-bonds. However, the charged R containing guanidino head group interacts with individual polar molecules more weakly due to its delocalized charge (Meloni et al., 2020). Furthermore, the positively charged residues R480 in CNNM2 plus the threonine residue of T568 are essential for stabilizing a phosphate moiety (Corral-Rodríguez et al., 2014).
CNNM2 contains an extracellular N-terminal domain, a transmembrane domain of the unknown function (DUF21), and a large cytosolic region including a cystathionine-β-synthase (CBS) domain and a putative cyclic nucleotide-binding homology (CNBH) domain (Chen et al., 2020). The mutation R480L is located in the CBS domain, and this Arg480 has been reported to interact with Glu570 to form a salt bridge and with the acidic aspartate residue D583 for strengthening CBS dimerization (Corral-Rodríguez et al., 2014; Chen et al., 2020). Previous studies have demonstrated the importance of CBS dimerization for binding Mg2+-ATP by the mutation of hydrophobic residues lining the interface of dimerization (Baykov et al., 2011; Chen et al., 2020). In line with this finding, our simulation model predicts that R480L alters the binding force between CNNM2 and Mg2+-ATP. Therefore, R480L might diminish its binding with Mg2+-ATP by impairment of the formation of CBS dimerization (Figure 7).
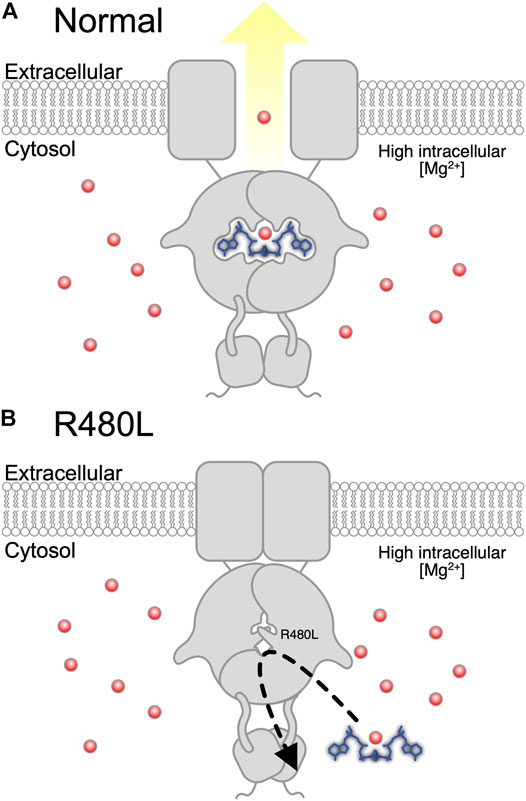
FIGURE 7. Proposed pathogenesis of hypomagnesemia due to R480L of CNNM2. (A) Normally, Mg2+ is effluxed after Mg2+-ATP binds to wild-type CNNM2 under magnesium depletion. (B) Mutation in CNNM2 yields a decreased binding force between CNNM2 and Mg2+-ATP and subsequent hypomagnesemia under magnesium depletion.
We conducted a cellular Mg2+ efflux assay to evaluate the effect of R480L on CNNM2 transport activity. Consistent with previous studies, R480L impaired the Mg2+ efflux activity in the presence of significantly high intracellular Mg2+ under Mg2+ depletion. It has been shown that Mg2+-ATP binding is required for cellular Mg2+ efflux since mutations that abolished Mg2+-ATP binding prevent Mg2+ efflux (Arjona and de Baaij, 2018). Of note, our simulation model predicts the reduction of the binding energy of Mg2+-ATP in the CBS1 of CNNM2 by R480L mutation. However, we did not find a significant impairment of Mg2+ efflux in CNNM2 T568I in CBS2, unlike a previous report (Corral-Rodríguez et al., 2014; Hirata et al., 2014). This discrepancy might be due to the following reasons. First, the cell line (mDCT cell) we utilized was different from that (HEK293) used by the previous study (Hirata et al., 2014). Second, we presented the relative intensity of intracellular Mg2+-green by calculating the mean fluorescence of all cells over 300 gray levels. Hirata et al. presented the relative intensity as the mean fluorescence of 10 cells. In fact, we found that the relative intensity of CNNM2 T568I on the Mg2+ efflux assay was higher than the wild type, although the difference was not statistically significant. Altogether, our study demonstrated that this R480L mutation resulted in the diminished binding with Mg2+-ATP and consequently led to impairment of Mg2+ efflux. This defective Mg2+ efflux might account for the hypomagnesemia in this patient with CNNM2 R480L mutation.
There were a few limitations in our study. First, we did not provide direct evidence for the R480L mutation in causing impairment of CBS dimerization by the crystal structure. Second, isothermal titration calorimetry was not performed to confirm the diminishment of the interaction of Mg2+-ATP and the CBS-pair. However, the simulation model showed the reduction of the binding energy of ATP-Mg2+ in the CBS module of CNNM2 by R480L mutation.
Conclusion
We identified a novel and de novo heterozygous R480L mutation in the CBS domain of the CNNM2 gene in a trio family with severe HSMR. This highly expressed CNNM2-R480L properly localizes to the plasma membrane but impairs Mg2+ efflux likely through the attenuated interaction with Mg2+-ATP, resulting in the clinical manifestation of refractory hypomagnesemia.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at https://www.ncbi.nlm.nih.gov/bioproject, accession number: PRJNA851908.
Ethics Statement
The studies involving human participants were reviewed and approved by the Tri-Service General Hospital in Taiwan (IRB2-105-05-136). Written informed consent to participate in this study was provided by the participant's legal guardian/next of kin. The animal study was reviewed and approved by Tri-Service General Hospital in Taiwan (IRB2-105-05-136). Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
All authors provided contributions to study conception and design, acquisition of data or analysis and interpretation of data, drafting the article or revising it critically for important intellectual content, and final approval of the version to be published. Here are the most important contributions of each author: M-HT, S-SY, and CS designed the study. Data were collected by J-JD and SC. Analysis was carried out by S-SY, CS, J-JD, and Y-JH. S-HL takes final responsibility for this article.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.875013/full#supplementary-material
References
Accogli, A., Scala, M., Calcagno, A., Napoli, F., Di Iorgi, N., Arrigo, S., et al. (2019). CNNM2 Homozygous Mutations Cause Severe Refractory Hypomagnesemia, Epileptic Encephalopathy and Brain Malformations. Eur. J. Med. Genet. 62, 198–203. doi:10.1016/j.ejmg.2018.07.014
Adalat, S., Woolf, A. S., Johnstone, K. A., Wirsing, A., Harries, L. W., Long, D. A., et al. (2009). HNF1BMutations Associate with Hypomagnesemia and Renal Magnesium Wasting. J. Am. Soc. Nephrol. 20, 1123–1131. doi:10.1681/ASN.2008060633
Arjona, F. J., and de Baaij, J. H. F. (2018). CrossTalk Opposing View: CNNM Proteins Are Not Na+/Mg2+ Exchangers but Mg2+ Transport Regulators Playing a Central Role in Transepithelial Mg2+ (Re)absorption. J. Physiol. 596, 747–750. doi:10.1113/JP275249
Arjona, F. J., de Baaij, J. H. F., Schlingmann, K. P., Lameris, A. L. L., van Wijk, E., Flik, G., et al. (2014). CNNM2 Mutations Cause Impaired Brain Development and Seizures in Patients with Hypomagnesemia. PLoS Genet. 10, e1004267. doi:10.1371/journal.pgen.1004267
Bamhraz, A. A., Franken, G. A. C., de Baaij, J. H. F., Rodrigues, A., Grady, R., Deveau, S., et al. (2021). Diagnostic Dilemma in an Adolescent Girl with an Eating Disorder, Intellectual Disability, and Hypomagnesemia. Nephron 145, 717–720. doi:10.1159/000518173
Baykov, A. A., Tuominen, H. K., and Lahti, R. (2011). The CBS Domain: A Protein Module with an Emerging Prominent Role in Regulation. ACS Chem. Biol. 6, 1156–1163. doi:10.1021/cb200231c
Chen, Y. S., Kozlov, G., Fakih, R., Funato, Y., Miki, H., and Gehring, K. (2018). The Cyclic Nucleotide-Binding Homology Domain of the Integral Membrane Protein CNNM Mediates Dimerization and Is Required for Mg2+ Efflux Activity. J. Biol. Chem. 293, 19998–20007. doi:10.1074/jbc.RA118.005672
Chen, Y. S., Kozlov, G., Fakih, R., Yang, M., Zhang, Z., Kovrigin, E. L., et al. (2020). Mg2+-ATP Sensing in CNNM, a Putative Magnesium Transporter. Structure 28, 324–335.e4. doi:10.1016/j.str.2019.11.016
Chen, Y. S., Kozlov, G., Moeller, B. E., Rohaim, A., Fakih, R., Roux, B., et al. (2021). Crystal Structure of an Archaeal CorB Magnesium Transporter. Nat. Commun. 12, 4028. doi:10.1038/s41467-021-24282-7
Corral-Rodríguez, M. Á., Stuiver, M., Abascal-Palacios, G., Diercks, T., Oyenarte, I., Ereño-Orbea, J., et al. (2014). Nucleotide Binding Triggers a Conformational Change of the CBS Module of the Magnesium Transporter CNNM2 from a Twisted towards a Flat Structure. Biochem. J. 464, 23–34. doi:10.1042/BJ20140409
Curry, J. N., and Yu, A. S. L. (2018). Magnesium Handling in the Kidney. Adv. Chronic Kidney Dis. 25, 236–243. doi:10.1053/j.ackd.2018.01.003
de Baaij, J. H. F., Hoenderop, J. G. J., and Bindels, R. J. M. (2015). Magnesium in Man: Implications for Health and Disease. Physiol. Rev. 95, 1–46. doi:10.1152/physrev.00012.2014
de Baaij, J. H. F., Stuiver, M., Meij, I. C., Lainez, S., Kopplin, K., Venselaar, H., et al. (2012). Membrane Topology and Intracellular Processing of Cyclin M2 (CNNM2). J. Biol. Chem. 287, 13644–13655. doi:10.1074/jbc.M112.342204
DePristo, M. A., Banks, E., Poplin, R., Garimella, K. V., Maguire, J. R., Hartl, C., et al. (2011). A Framework for Variation Discovery and Genotyping Using Next-Generation DNA Sequencing Data. Nat. Genet. 43, 491–498. doi:10.1038/ng.806
Ellison, D. H., Maeoka, Y., and McCormick, J. A. (2021). Molecular Mechanisms of Renal Magnesium Reabsorption. J. Am. Soc. Nephrol. 32, 2125–2136. doi:10.1681/ASN.2021010042
Ferrè, S., de Baaij, J. H. F., Ferreira, P., Germann, R., de Klerk, J. B. C., Lavrijsen, M., et al. (2014). Mutations in PCBD1 Cause Hypomagnesemia and Renal Magnesium Wasting. J. Am. Soc. Nephrol. 25, 574–586. doi:10.1681/ASN.2013040337
Franken, G. A. C., Müller, D., Mignot, C., Keren, B., Lévy, J., Tabet, A. C., et al. (2021). The Phenotypic and Genetic Spectrum of Patients with Heterozygous Mutations in Cyclin M2 (CNNM2). Hum. Mutat. 42, 473–486. doi:10.1002/humu.24182
García-Castaño, A., Madariaga, L., Antón-Gamero, M., Mejia, N., Ponce, J., Gómez-Conde, S., et al. (2020). Novel Variant in the CNNM2 Gene Associated with Dominant Hypomagnesemia. PLoS ONE 15, e0239965. doi:10.1371/journal.pone.0239965
Glaudemans, B., van der Wijst, J., Scola, R. H., Lorenzoni, P. J., Heister, A., van der Kemp, A. W., et al. (2009). A Missense Mutation in the Kv1.1 Voltage-Gated Potassium Channel-Encoding Gene KCNA1 Is Linked to Human Autosomal Dominant Hypomagnesemia. J. Clin. Invest. 119, 936–942. doi:10.1172/JCI36948
Groenestege, W. M. T., Thébault, S., van der Wijst, J., van den Berg, D., Janssen, R., Tejpar, S., et al. (2007). Impaired Basolateral Sorting of Pro-EGF Causes Isolated Recessive Renal Hypomagnesemia. J. Clin. Invest. 117, 2260–2267. doi:10.1172/JCI31680
Hirata, Y., Funato, Y., Takano, Y., and Miki, H. (2014). Mg2+-dependent Interactions of ATP with the Cystathionine-β-Synthase (CBS) Domains of a Magnesium Transporter. J. Biol. Chem. 289, 14731–14739. doi:10.1074/jbc.M114.551176
Jahnen-Dechent, W., and Ketteler, M. (2012). Magnesium Basics. Clin. Kidney J. 5, i3–i14. doi:10.1093/ndtplus/sfr163
Li, F.-Y., Chaigne-Delalande, B., Kanellopoulou, C., Davis, J. C., Matthews, H. F., Douek, D. C., et al. (2011). Second Messenger Role for Mg2+ Revealed by Human T-Cell Immunodeficiency. Nature 475, 471–476. doi:10.1038/nature10246
Li, H., and Durbin, R. (2009). Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 25, 1754–1760. doi:10.1093/bioinformatics/btp69810.1093/bioinformatics/btp324
Li, X., Bao, S., Wang, W., Shi, X., Hu, Y., Li, F., et al. (2021). Case Report: CNNM2 Mutations Cause Damaged Brain Development and Intractable Epilepsy in a Patient without Hypomagnesemia. Front. Genet. 12, 705734. doi:10.3389/fgene.2021.705734
Meij, I. C., Koenderink, J. B., Jong, J. C., Pont, J. J. H. H. M., Monnens, L. A. H., Heuvel, L. P. W. J., et al. (2003). Dominant Isolated Renal Magnesium Loss Is Caused by Misrouting of the Na+,K+-ATPase γ-Subunit. Ann. N. Y. Acad. Sci. 986, 437–443. doi:10.1111/j.1749-6632.2003.tb07226.x
Meloni, B. P., Mastaglia, F. L., and Knuckey, N. W. (2020). Cationic Arginine-Rich Peptides (CARPs): A Novel Class of Neuroprotective Agents with a Multimodal Mechanism of Action. Front. Neurol. 11, 108. doi:10.3389/fneur.2020.00108
Reichold, M., Zdebik, A. A., Lieberer, E., Rapedius, M., Schmidt, K., Bandulik, S., et al. (2010). KCNJ10 Gene Mutations Causing EAST Syndrome (Epilepsy, Ataxia, Sensorineural Deafness, and Tubulopathy) Disrupt Channel Function. Proc. Natl. Acad. Sci. U.S.A. 107, 14490–14495. doi:10.1073/pnas.1003072107
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Sponder, G., Mastrototaro, L., Kurth, K., Merolle, L., Zhang, Z., Abdulhanan, N., et al. (2016). Human CNNM2 Is Not a Mg2+ Transporter Per Se. Pflugers Arch. - Eur. J. Physiol. 468, 1223–1240. doi:10.1007/s00424-016-1816-7
Stuiver, M., Lainez, S., Will, C., Terryn, S., Günzel, D., Debaix, H., et al. (2011). CNNM2, Encoding a Basolateral Protein Required for Renal Mg2+ Handling, Is Mutated in Dominant Hypomagnesemia. Am. J. Hum. Genet. 88, 333–343. doi:10.1016/j.ajhg.2011.02.005
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 38, e164. doi:10.1093/nar/gkq603
Keywords: CNNM2, hypomagnesemia, seizure, HSMR syndrome, renal magnesium wasting
Citation: Tseng M-H, Yang S-S, Sung C-C, Ding J-J, Hsu Y-J, Chu S-M and Lin S-H (2022) Novel CNNM2 Mutation Responsible for Autosomal-Dominant Hypomagnesemia With Seizure. Front. Genet. 13:875013. doi: 10.3389/fgene.2022.875013
Received: 19 February 2022; Accepted: 27 April 2022;
Published: 29 June 2022.
Edited by:
Ping Liang, Brock University, CanadaReviewed by:
Kai Hu, Central South University, ChinaAn-Ping Chen, Cytovia Therapeutics, United States
Copyright © 2022 Tseng, Yang, Sung, Ding, Hsu, Chu and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shih-Hua Lin, l521116@ndmctsgh.edu.tw