 Concetta Scimone1,2
Concetta Scimone1,2 Luigi Donato1,2
Luigi Donato1,2 Concetta Alafaci1Francesca Granata1Carmela Rinaldi1Marcello Longo1
Concetta Alafaci1Francesca Granata1Carmela Rinaldi1Marcello Longo1 Rosalia D’Angelo1,2*
Rosalia D’Angelo1,2* Antonina Sidoti1,2
Antonina Sidoti1,2- 1Department of Biomedical and Dental Science and of Morphological and Functional Images, University of Messina, Messina, Italy
- 2Department of Vanguard Medicine and Therapies, Biomolecular Strategies and Neuroscience, I.E.ME.S.T., Palermo, Italy
Molecular signaling that leads to brain arteriovenous malformation (bAVM) is to date elusive and this is firstly due to the low frequency of familial cases. Conversely, sporadic bAVM is the most diffuse condition and represents the main source to characterize the genetic basis of the disease. Several studies were conducted in order to detect both germ-line and somatic mutations linked to bAVM development and, in this context, next generation sequencing technologies offer a pivotal resource for the amount of outputted information. We performed whole exome sequencing on a young boy affected by sporadic bAVM. Paired-end sequencing was conducted on an Illumina platform and filtered variants were validated by Sanger sequencing. We detected 20 likely gene-disrupting variants affecting as many loci. Of these variants, 11 are inherited novel variants and one is a de novo nonsense variant, affecting STK4 gene. Moreover, we also considered rare known variants affecting loci involved in vascular differentiation. In order to explain their possible involvement in bAVM pathogenesis, we analyzed molecular networks at Cytoscape platform. In this study we focus on some genetic point variations detected in a child affected by bAVM. Therefore, we suggest these novel affected loci as prioritized for further investigation on pathogenesis of bAVM lesions.
Introduction
Brain arteriovenous malformations (bAVM, OMIM #108010) are congenital malformations affecting microvessels. The absence of the capillary bed is peculiar of these lesions resulting in pathological direct shunt from arterioles to venules. Feeding arteries continue in an undifferentiated arteriovenous nidus that resolves in a single or more draining veins. At nidus, the vessels are enlarged and susceptible to rupture. Arterial and venous circulation are mixed within these lesions, therefore blood perfuses with a high pressure, increasing bleeding risk (Goldberg et al., 2018). Intracerebral hemorrhage (ICH) is the most common clinical manifestation appearing in about 50% of patients. The incidence of bAVM is approximately 0.01% and usually it develops sporadically (Osbun et al., 2017). Multiple lesions occur coexisting together with other vascular malformation syndromes, like hereditary hemorrhagic telangiectasia (HHT). HHT patients show multiple AVM affecting visceral organs and multiple telangiectasia at mucosal tissues. Mutations at genes involved in the TGF-βII transduction pathway cause different HHT phenotypes (Pawlikowska et al., 2018), (Karlsson and Cherif, 2018). Familial bAVM without HHT is extremely rare and only about 30 affected families were reported (van Beijnum et al., 2007). Although genetic causes are not yet elucidated, pedigree analysis confirmed an autosomal dominant pattern of inheritance with incomplete penetrance and variable expressivity. Three linkage studies were performed on 15 affected families and several chromosomal loci were mapped despite the exact genes are still unidentified (Oikawa et al., 2010). Mutational analyses were performed on ephrin family genes mapping on these regions but without success (Inoue et al., 2007). Recently, a novel missense mutation in ACVRL1 gene was reported as segregating within a family affected by hereditary bAVM without HHT (Yılmaz et al., 2017).
Expression studies performed on human lesion-derived specimens revealed alterations at several pathways controlling angiogenesis and angio-architecture maintenance, highlighting that lesions can arise due to impaired expression of ephrin proteins during early angiogenesis, following Hedgehog (Hh)-VEGF-Notch signaling activation (Lawson et al., 2002). This results in EFNB2 down-expression and loss of vein differentiation. Conversely, venous markers FLT4 and EPHB4 result expressed in arteries as consequence of notch signaling down-regulation (Lawson et al., 2001).
Next generation sequencing (NGS) technology, in the last years, is allowing to rapidly increase information regarding disease-causing loci. Here we present results of a whole exome sequencing (WES) performed on a young patient affected by sporadic bAVM. With support of bioinformatic tools and network analysis, likely gene-disrupting (LGD) variants at 20 loci, potentially linked to bAVM development, were detected.
Methods
Whole Exome Sequencing and Bioinformatic Analysis Pipeline
WES analysis was performed on a Caucasian child who manifested ICH due to a single AVM at the right cerebellar hemisphere. The lesion was totally resected following two surgeries and, to date, no recidivism is reported. However, he recently developed varicocele. His parents were unaffected despite the mother and the maternal grandmother showed varicose veins. The patient was classified as sporadic.
WES analysis was carried on DNA purified from whole peripheral blood by the QIAamp DNA Mini Kit (Qiagen). The extraction yield was assessed at NanoDrop spectrophotometer (Thermo Scientific). Exome capturing was performed by the SureSelectXT Human All Exon V6” (Agilent Genomics) kit. Library was set up in order to generate 100 bp length reads. Paired-ends sequencing was run on a Illumina HiSeq 2500 System.
The generated reads were subject to bioinformatic analysis, as described: quality check was performed by FastQC_Version_0.11.7 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc) tool. Reads presenting a Phred score ≥ 28 were selected, trimmed, and aligned to the reference human genome GRCh37 (hg19) by the Burrows-Wheeler Aligner (BWA) (Li and Durbin, 2009). Duplicate reads were removed by Picard toolkit Version_2.18.23 (Picard Toolkit.” 2019. Broad Institute, GitHub Repository. http://broadinstitute.github.io/picard/; Broad Institute). Genome Analysis Toolkit Version_4.1.3.0 (GATK) (https://software.broadinstitute.org/gatk/), ANNOVAR Version_2018Apr16 (Wang et al., 2010), and SnpEff Version_4.3T (Cingolani et al., 2012) were used for variant calling and variant annotation, respectively. Frequencies of detected variants were defined by the 1000 Genomes database.
Selection of Likely Gene-Disrupting Variants and Sanger Validation
LGD variants potentially involved in bAVM pathogenesis were strictly selected as follows. Firstly, all variants not reported in public human genome mutation database (HGMD®) or in SNP collection databases as dbSNP (https://www.ncbi.nlm.nih.gov/snp/), ExAC (http://exac.broadinstitute.org/), and Ensembl (https://www.ensembl.org/index.html) were considered. For variants already reported in literature, the first selective criterion was the minor allele frequency (MAF) reported in the 1000 Genome Project database and the cut-off value MAF < 0.01 (1%) was selected. Variants presenting MAF < 0.01% were further classified according to their functional class and missense, nonsense, frameshift, and splice site variants were considered. Within each group, we selected all those resulted potentially pathogenic at LRT (http://www.genetics.wustl.edu/jflab/lrt_query.html) (Chun et al., 2009), MutationTaster (http://www.mutationtaster.org) (Schwarz et al., 2014), and SIFT 4G (10.1038/nprot.2015.123) (Vaser et al., 2016) prediction tools.
For each functional class, variants showing low depth value were excluded. Subsequently, gene prioritization was established by enrichment analysis performed on Gene Ontology database, in relation to Biological Process annotations and Reactome pathways. Filtered LGD variants were validated by Sanger sequencing by using the BigDyeTerminator© v3.1 Cycle Sequencing Kit and run on a 3500 Genetic Analyzer (Applied Biosystems, Thermo Fisher Scientific), after amplification. Primer pairs and polymerase chain reaction conditions are provided as Supplementary Materials (SM1). Finally, physical and predicted interactions, co-expression, co-localization, and shared domain patterns of LGD gene sets were obtained by GeneMania Version_3.5. plugin 1 (Montojo et al., 2010) of Cytoscape Version_3.6.0 platform.
Screening of the Relatives
In order to detect possible de novo mutations and to establish segregation of selected variants within the family, the parents of the proband were genotyped by Sanger sequencing.
Allele Frequency Estimation
The frequencies of the novel variants were estimated in healthy population by restriction fragment length polymorphism (RFLP) analysis and Sanger sequencing. Enzymatic digestion was applied for variants in CLCN2, EFNA4, IGFBP7, TNXB, L2HGHD genes. Five units of specific enzyme were used to digest 0.5 µg of DNA. The restriction enzymes and the obtained fragments are listed in SM1. Variants detected in NAXE, TTLL11, TRMT10B, AOC3, STK4, SLIT2, and FLRT3 genes do not affect any restriction site, therefore these were screened by Sanger sequencing, as previously described.
Control group was made up by 106 and 94 Caucasian healthy males and females respectively, unrelated and randomly selected. The absence of lesions was confirmed by MRI investigation.
RNA Extraction, Quantitative Real-Time PCR, and Statistical Analysis
White blood cells (WBCs) were isolated from peripheral blood by Ficoll (Lympholyte® Cell Separation Media, Cedarlane). Total RNA was purified using TRIzol® reagent (Thermo Fisher Scientific), following manufacturer protocol. For each reaction, 1 µg of total RNA was retro-transcribed using the GeneAmp RNA PCR Core Kit® (Thermo Fisher Scientific) and applying the two-step protocol, as suggested in the datasheet. RT-PCR was conducted to evaluate the nonsense-mediated decay (NMD) as consequence of the nonsense mutation detected in STK4 gene. STK4 expression level in lymphocytes of the patient was compared with that observed in the negative control. The 2-years younger brother of the proband was considered as negative control. The absence of any STK4 nonsense mutation in the negative control was proven by Sanger sequencing. Equimolar quantities of sample/control complementary DNA (cDNA) were used for specific STK4 coding sequence amplification using the following primer pair: 5’-GAATACAGTGATAGGAACAC–3’ and 3’-CTCTTTACAAGACACTGTT-5’. The TaqMan® probe was designed downstream to the mutation site, in order to cover the exon-exon boundary. The probe had the following sequence 5’-VIC-CATCCAATGAGGGCAATCTTCATG–TAMRA-3’. For data normalization, human GAPDH housekeeping gene was used as endogenous reference. The probe for GAPDH was labeled with FAM as fluorophore and TAMRA as quencher. Multiplex TaqMan reactions were performed using the TaqMan® Master Mix (Thermo Fisher Scientific). For each reaction, 10 ng of cDNA were added to 3 µM of each primer, 5 µM of each probe and to the Master Mix. UltraPure™ DNase/RNase-Free Distilled Water was added up to reach 20 µl of total reaction volume. Quantitative RT-PCR reactions were conducted on a 7500 Real-Time PCR System (Applied Biosystems). RNA purification and RT-PCR reactions were trice replicated. Before multiplex RT-PCR reactions, singleplex reactions were conducted in order to evaluate the single threshold cycle (Ct) values of each amplicon. Different abundance of the STK4 cDNA was evaluated between the sample and the negative control. Relative quantification was determined by the 2-ΔΔCt method and was normalized against the expression of the human GAPDH gene. All data analyses were performed using the IBM SPSS 24 software for Macintosh (IBM Corp. Released 2013. IBM SPSS Statistics for Macintosh, Armonk, NY). A one-way analysis of variance (one-way ANOVA) was performed to compare between the sample groups. All p-values were Bonferroni’s corrected and considered significant if p < 0.05.
Protein Extraction and Western Blot Analysis
Proteins were extracted from WBCs, previously isolated from whole peripheral blood by Ficoll (Lympholyte® Cell Separation Media, Cedarlane). Radioimmunoprecipitation assay (RIPA) buffer supplied with protease inhibitor mix was used. Also in this case, the negative control considered was the younger brother of the proband and the samples were in parallel processed. Colorimetric protein quantification was performed by the Bradford protein assay. The protein amount of 30 µg was denatured, separated on a sodium dodecyl sulfate (SDS) (10%) 15% polyacrylamide gel and then blotted on a Immun-Blot® PVDF Membrane (Bio-Rad) by running for 2 h at 80 mA with transfer buffer (39 mM glycine, 48 mM Tris, 20% methanol) at 4°C. Membrane was stained in TBS 0.1%-Tween and blocked with 5% non-fat dry milk for 1 h at room temperature and then thrice washed in TBS–0.1% Tween. Overnight incubation with the primary anti-STK4 antibody (Genetex International Corporation, Hsinchu City, Taiwan, R.O.C.) was conducted in TBS-0.1%-Tween. Primary anti-STK4 antibody was 1:2,000 diluted. This step was followed by three washings in TBS–0.1% Tween and by the incubation with the secondary antibody peroxidase-conjugated goat anti-rabbit immunoglobulin G (Pierce, Rockford, IL, USA). Secondary antibody was diluted 1:20,000 and incubation was performed at room temperature for 1 h. In parallel, membrane for the β-actin immunodetection was processed for data normalization. Images were developed by the enhanced chemiluminescence system (ECL) (Amersham, Little Chalfont, UK) and digitally acquired (DiGit Blot Scanner with Image Studio 4.0 software, LI-Cor, Lincoln, NE, USA). Densitometric analysis was performed in order to calculate the STK4 protein amount of the sample compared to the control by Gel-Pro Analyzer v.4 software.
Ethical Statement
The study was approved by the local Ethical Committee (A.O.U. “G. Martino,” Messina, Italy) and it is compliant with Helsinki declaration.
All recruited subjects were fully informed ad written consent was obtained, also for the underage subjects.
Results
Whole Exome Sequencing and Bioinformatic Analysis
Sequencing run generated 86,816,620 reads. Phred quality score greater than 30 (Phred > 30) resulted for 92.83% of total reads. After duplicates discard, 69,592,810 were filtered and 99.09% mapped with hg19 RefSeq. Among these, 82.92% were on-target reads. Main reports of the sequence run, variant calling, and annotation results are provided as Supplementary Materials (SM2).
Selection of Likely Gene-Disrupting Variants and Sanger Validation
With exception of deep intronic variants, analysis outputted 63,171 variants. Of these, only 11,205 showed MAF < 0.01% and were clustered by functional class.
Variants resulted potentially damaging by the three prediction tools were selected. The Gene Ontology annotation of biological processes and the reactome pathway analysis allowed to totally select 20 likely gene-disrupting (LGD) variants. In particular, eight are known single nucleotide variants (SNV) while 12 are novel, not yet reported in HGMD® or other variant collection databases. All selected variants were validated and confirmed by Sanger sequencing. Table 1 lists the 20 LGD variants selected as potentially involved in bAVM onset. Of these, 17 are missense, 2 are splicing variants, and only 1 is a nonsense variant (c.569G > A, p.Trp190Ter) affecting STK4 gene. Electropherograms were obtained in order to validate novel variants and are shown in Figure 1.
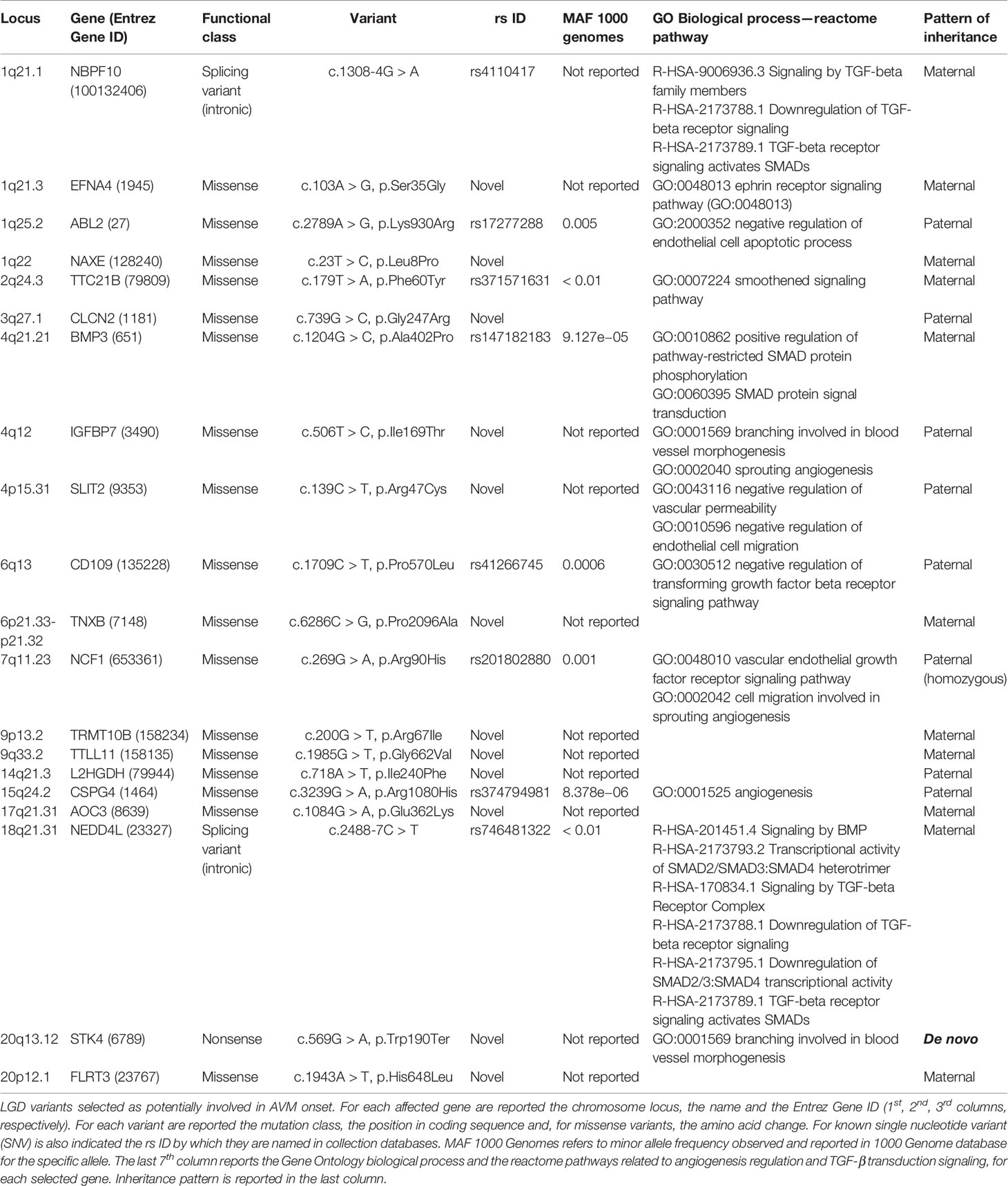
Table 1 Likely gene-disrupting (LGD) variants selected as potentially involved in arteriovenous malformation (AVM) onset.
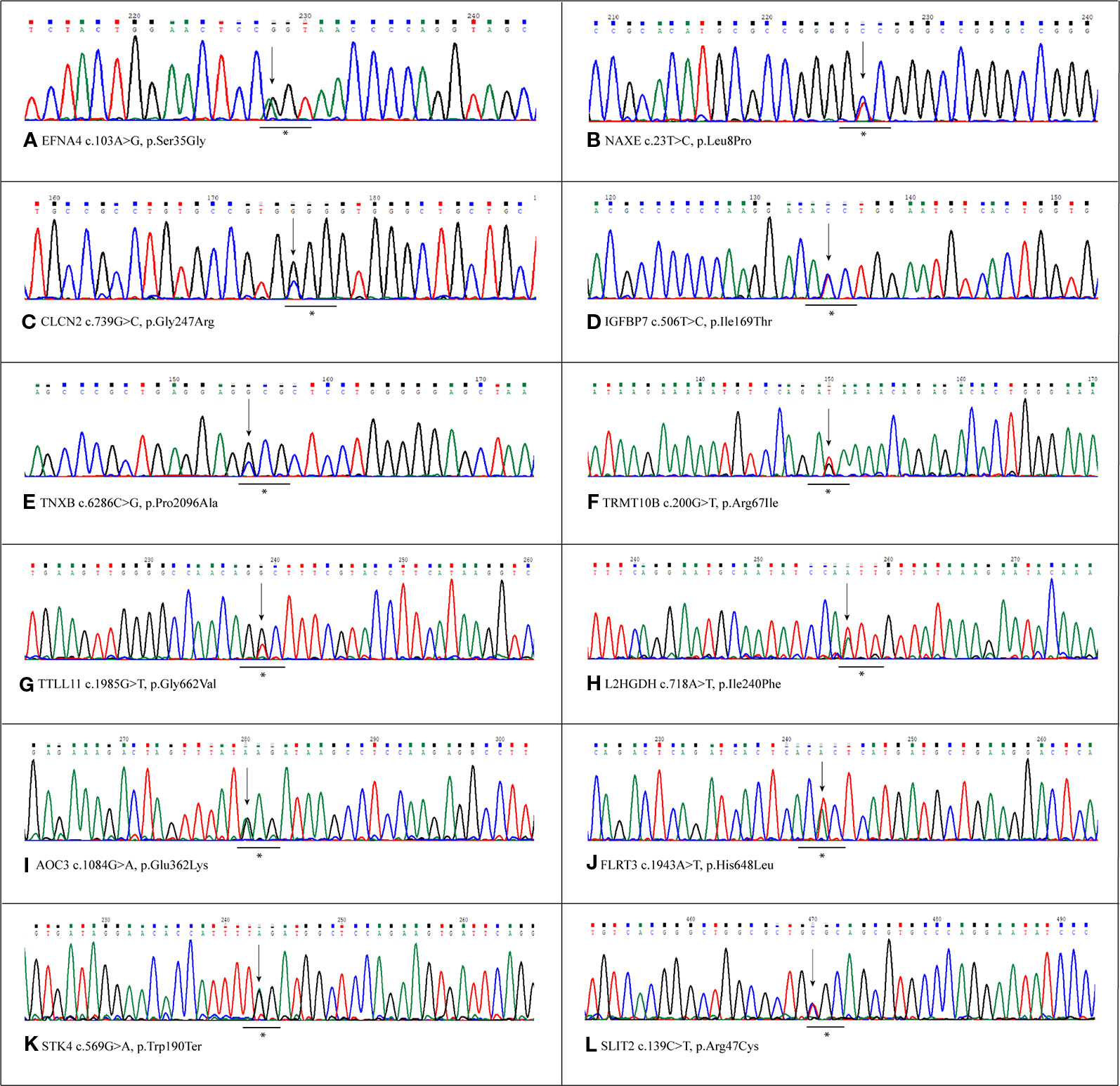
Figure 1 Sanger validation of novel variants. Electropherograms show the novel variants detected by whole exome sequencing (WES) and confirmed by Sanger sequencing. Each panel refers to the single variant described at the bottom of the same panel. The underlined asterisked triplets indicate the codons affected by mutations. All variants were detected in heterozygous condition. (A) Variant detected in EFNA4 gene, c.103A>G, p.Ser35Gly. (B) Variant detected in NAXe gene,c.23T>C, p.Leu8Pro. (C) Variant detected in CLCN2 gene, c.739C>G,p.Gly247Arg. (D) Variant detected in IGFBP7 gene, c.506T>C, p.Ile169Thr. (E) Variant detected in TNXB gene, c.286C>G, p.Pro2096Ala (F) Variant detected in TRMT10B gene, c.200G>T,p.Arg67Ile. (G) Variant detected in TTLL11 gene, c.1985G>T, p.Gly662Val. (H) Variant detected in L2HGDH gene, c.718A>T, p.Ile240Phe. (I) Variant detected in AOC3 gene, c.1084G>A, p.Glu362Lys (J) Variant detected in FLRT3 gene, c.1943A>T, p.His648Leu. (K) Variant detected in STK4 gene, c.569G>A, p.Trp190Ter. (L) Variant detected in SLIT2 gene, c.139C>T,p.Arg47Cys.
Regarding affected loci, role of EFNA4, IGFBP7, SLIT2, and STK4 in vessel maturation was already demonstrated. However there are no pertinent GO annotations for the eight loci NAXE, CLCN2, TNXB, TRMT10B, TTLL11, L2HGDH, AOC3, and FLRT3. In order to obtain any functional relation among these and the other loci reported in Table 1, network analysis by GeneMANIA plugin of Cytoscape platform was performed. We obtained two different networks, showed in Figure 2. Details on nodes and edges are provided as Supplementary Materials (SM3). The eight previous novel loci were also given as input together with the other NBPF10, BMP3, CD109, NEDD4L, known to be involved in TGF-β transduction pathway, and TGFB2 and ACVRL1, since it resulted mutated in the patient affected by sporadic bAVM (Figure 2A). Network showed in Figure 2B was obtained giving as input the same eight loci and all those involved in angiogenesis, as ABL2, EFNA4, TTC21B, IGFBP7, SLIT2, NCF1, CSPG4, STK4.
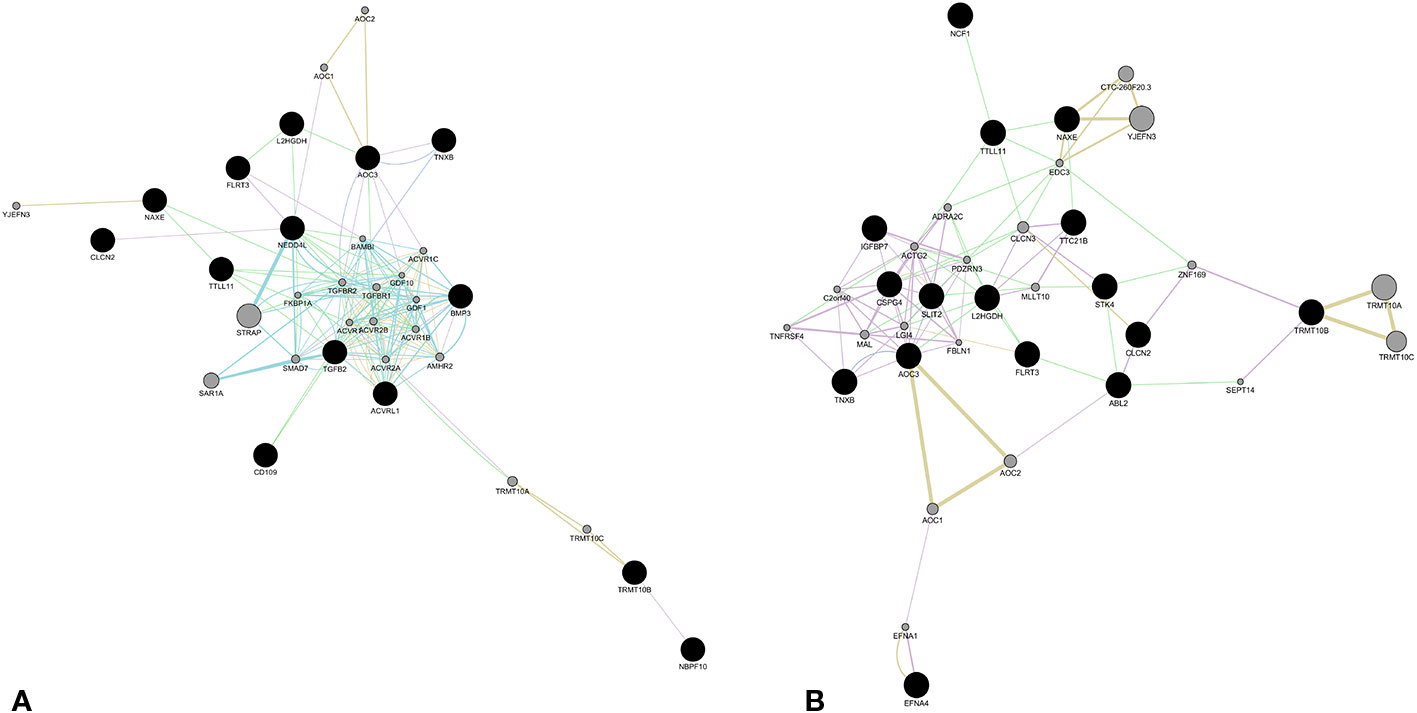
Figure 2 Molecular network obtained by GeneMania plugin (Cytoscape). The network shows patterns of co-expression (violet), co-localization (blue), genetic interaction (green), pathway (light blue), predicted interaction (orange), and shared proteins domains (yellow), obtained from merger of genes affecting by likely gene-disrupting (LGD) variants. The eight novel loci were merged with those involved in TGFβ signaling (A) and those involved in angiogenesis and vessel differentiation (B). Color of each edge is in relation of the kind of interaction. Black nodes represent input genes. Gray nodes resulted from analysis. Details about nodes and edges as well as annotations are available in Tables 1–5, supplied in Supplementary Materials SM3.
Screening of the Relatives
Genotyping of the parents of the young patient revealed that all detected variants were inherited with the exception of the novel nonsense c.569G > A (p.Trp190Ter) affecting STK4 locus. This variant was not detected in any parents. Patterns of inheritance for each variant are reported in Table 1.
Allele Frequency Estimation
The frequencies of the novel variants were estimated on a cohort of 200 healthy subjects, heterogeneous for sex and age. None of the controls carried the variants, therefore they can be considered mutations (data not shown).
Real Time-PCR, Gene Expression, and Protein Detection
STK4 expression level was measured on both c.569G > A and wild-type WBCs. The mature wild-type messenger RNA (mRNA) consists of 11 exons and the substituted nucleotide is comprised in the 6th. It was predicted to create a premature stop codon leading to the synthesis of a 190 amino acid truncated protein against the 487 wild-type one. Therefore the hypothesis of NMD was evaluated as mechanism involved in premature degradation of mRNA molecule transcribed by the mutated allele (Lindeboom et al., 2019). This hypothesis was assayed and confirmed by RT-PCR (Figure 3A). The results are reported as logarithmic values and highlight the reduction of about 20% of the STK4 transcript in the c.596G > A WBCs, compared to the wild-type cells. The ANOVA test was applied to determine statistical significance of the data showing a significative p-value = 2.455E−50. Results are shown as the mean values of the three replicates.
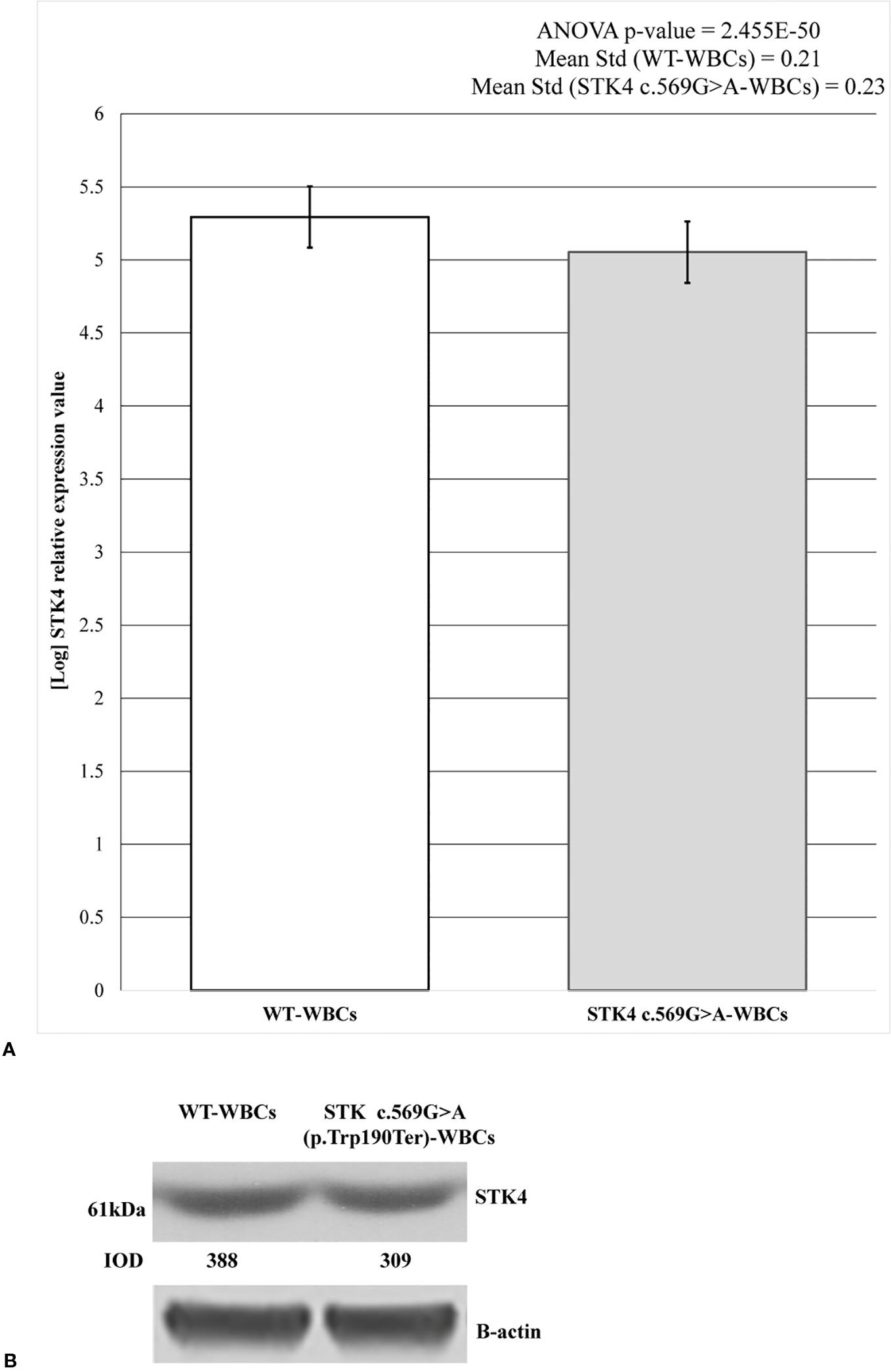
Figure 3 STK4 expression values and protein detection in control white blood cells (WBCs) and c.569G > A (p.Trp190Ter) WBCs. (A) The histogram shows the logarithmic STK4 relative expression values coming from real-time (RT)-PCR experiment, compared between wild-type and mutated sample groups. As reported, ANOVA test resulted significant (p-value = 2.455E−50). The presence of STK4 c.569G > A variant determines an expression reduction of about 20%, compared to the wild-type control. Reported values represent the mean of the three replicates. (B) Protein detection by western blot analysis showing a reduction of the STK4 protein synthesis in the heterozygous c.569G > A (p.Trp190Ter) mutation carrier. The integrated optical intensity (IOD) values are reported.
These results were also confirmed by western blot analysis. As predicted, the synthesis of a truncated 21.7 kDa protein results following the c.569G > A (p.Trp190Ter) nonsense mutation (https://web.expasy.org/compute_pi/), compared to the 52.335 kDa wild-type one. The 61 kDa product results from anti-STK4-STK4 complex. As expected, in the c.569G > A heterozygous mutant only the wild-type protein was observed (Figure 3B) and this resulted reduced by about the 20% when compared to the wild-type condition, as shown by the densitometric analysis. Taken together, these data highlight that the mutant allele is prematurely degraded probably by the NMD.
Discussion
Brain AVM is a rare disease usually observed as sporadic condition. Despite the genetic bases of the disease are not yet well elucidated, more evidences indicate possible common pathways as cause of sporadic, hereditary AVM and HHT onset (Walcott et al., 2018). Familial AVM without HHT is an extremely unusual condition, therefore many genetic studies are madeon sporadic patients. Here we reported the main data collected by WES analysis performed on a child affected by sporadic AVM with the aim to increase the very little knowledge regarding genetic and molecular etiology of the disease. Among all detected variants, we firstly focused on the novel ones and then considered those presenting MAF < 0.01 and affecting genes whom GO enrichment revealed any possible involvement in TGF-βII transduction pathway or in angiogenesis regulation. Based on functional class, 1 nonsense, 2 splice affecting, and 17 missense mutations have been considered (Table 1). Selected genes were clustered in three groups. The first includes all genes involved in TGF-β/SMAD transduction pathway, as NBPF10, BMP3, CD109, NEDD4L genes (Lei et al., 2017; Yamakawa et al., 2018). All detected variants were already reported in ExAC database although no clinical significance was to date assigned.
The second group comprises genes that regulate angiogenetic process as well as arterial and vein differentiation. These genes are ABL2, EFNA4, TTC21B, IGFBP7, SLIT2, NCF1, CSPG4, STK4 (Amyere et al., 2017; Errede et al., 2018; Romano et al., 2018; Tumelty et al., 2018). Among these, EFNA4, IGFBP7, and SLIT2 genes are affected by novel missense mutations while STK4 carries a novel nonsense mutation leading to a 190 amino acid truncated protein against 487 amino acid wild-type one.
In the third cluster are grouped all genes affected by novel variants that showed no annotation with TGF-βII and angiogenetic pathways, and these are NAXE, CLCN2, TNXB, TRMT10B, TTLL11, L2HGDH, AOC3, FLRT3. Knowledge about these genes are really few and several of them are not yet reported in HGMD database, being not associated to any pathological phenotypes. Briefly, a recent study showed involvement of NAXE in vascular remodeling via modulation of cholesterol metabolism and γ-secretases, proving the attenuation of Notch signaling and the enhance of angiogenesis following NAXE depletion (Mao et al., 2017).
TNXB contributes to angiogenesis and the decrease of TGFβ1 expression level in TNXB knockout mice was reported as modulated by VEGF-A. TNXB acts by encouraging neovascularization and by suppressing anti-angiogenic VEGF-B mRNA expression in vivo (Sumioka et al., 2018). Moreover, it is mutated in a small cohort of patients affected by Ehlers-Danlos syndromes, a heterogeneous group of rare monogenic conditions characterized by vascular and generalized connective tissue fragility (Malfait, 2018).
AOC3 encodes for the amino oxidase enzyme also expressed in cerebral venules and in blood-brain barrier capillaries (https://discovery.lifemapsc.com/in-vivo-development/brain/blood-brain-barrier/endothelial-cells). Associated phenotypes include different vascular impairments (Vijayan and Reddy, 2016).
FLRT3 encodes for the fibronectin leucine rich transmembrane protein 3, an extracellular adhesion molecule, implicated in several cellular processes as axon guidance, pericardium, and retinal vasculature development.
Based on current knowledge, the other loci here considered are not involved in angiogenesis or in vascular differentiation.
However, among all selected LGD variants, we found the de novo c.569G > A (p.Trp190Ter) nonsense mutation affecting STK4 gene that leads to a truncated protein and the our hypothesis is that this variant could mainly be involved in the development of the bAVM phenotype. This is due to different reasons. Firstly, it is a de novo mutation harbored only by the young patient and this could be justify the onset of the sporadic condition.
Homozygous mutations in STK4 are linked to the autosomal recessive primary immunodeficiency (Abdollahpour et al., 2012). The nonsense mutation here we reported was detected in heterozygous condition and this can be well accepted if we consider the dominant pattern of inheritance of bAVM. Moreover, RT-PCR and western blot data showed a statistically significant reduction both of the STK4 transcript and of the protein in mutated sample, when compared to the wild-type one. Gene Ontology annotations for biological processes report STK4 involved in “branching involved in blood vessel morphogenesis” (GO:0001569), defined as the process of coordinated growth and sprouting of blood vessels giving rise to the organized vascular system. In particular, recent evidence suggests that hypoxia-induced reactive oxygen species (ROS) biosynthesis from mitochondria could be a major upstream regulator of Stk4 activation in endothelial cells. Activated stk4 promotes planar and perpendicular vascular branching for regulating tip endothelial cells polarity and sprouting angiogenesis (Kim et al., 2019). Moreover, it was recently shown that STK4 acts as pro-apoptotic factor in damaged venous endothelial cells by the MIEF1 pathway. In particular, the study was conducted on human umbilical vein endothelial cells (HUVECs) treated with tumor necrosis factor α, in order to mimicking lower limb chronic venous disorders. Chronic venous disorders, indeed, also include telangiectasias (Lu et al., 2019). STK4 was firstly identified as a member of Hippo signal pathway that regulates cell proliferation, growth, differentiation, and apoptosis (Varelas and Wrana, 2012). A crosstalk between the Hippo pathway and the BMP/TGFβ signaling was demonstrated in endothelial cells and this finding could elucidate a potential role of Hippo pathway in bAVM and HHT pathogenesis (Young et al., 2015).
In addition, it is well known that human brain AVMs over-express COUP-TFII, PROX1, and other genes, also known to regulate key aspects of angiogenesis and lymphangiogenesis. These genes have similar expression patterns and a subset of them significantly correlated with the presence of preoperative edema and acute hemorrhage (Shoemakerl et al., 2014). STK4 is not included among these genes, but homozygous nonsense mutation in STK4 cause primary immunodeficiency syndrome, given its central role in immunity (Abdollahpour et al., 2012).
Based on these evidence, we think that consequences on angiogenesis of this loss of function mutation must be further validated in order to confirm our hypothesis of its role in the development of bAVM.
However, also a pathogenic model based on interaction among low-penetrance loci could be considered. In this context, the other genes we selected could act as modifier genes. This idea comes from the observation of their expression profile. In particular, TGFβ, BMP, and IGF family members are highly expressed at embryo stage particularly in mesoderm, by which originate vasculature, connective tissue, skeleton, and urinary tract. All considered variants affect genes associated to connective tissue, glomerulus, and skeleton impairment (Demirdas et al., 2017; Emlet et al., 2017; Schock et al., 2017; Wunnenburger et al., 2017). However, a critical point regards the clinical significance of variants detected by the analysis. Although some of these have already been reported in variant collection databases, no information is available about the phenotypes of the variant carriers. In this context, a valid example is given by rs147182183 (c.1204G > C, p.Ala402Pro) affecting BMP3 locus: it is reported in SNP collection databases with the very low allele frequency equal to 9.127e−05 in European non-Finnish population. However, it is unknown if it was found in healthy subjects or in affected patients, also asymptomatic. BMP3, indeed, is a relevant candidate locus. Moreover, high variant rate in genes involved in BMP/TGF-β and VEGF/VEGFR pathways was detected in a cohort of 100 patients affected by sporadic bAVM (Wang et al., 2018).
Among selected LGD variants and according to literature data, clearly NBPF10, EFNA4, NAXE, TTC21B, BMP3, IGFBP7, SLIT2, CD109, TNXB, CSPG4, AOC3, NEDD4L, STK4, and FLRT3 could be valid candidate loci to consider in a more wide cohort of patients. In particular, STK4 resulted affected by a de novo loss of function mutation. Limits of this study are indeed the single-case considered and the lack of functional data about the effects of these variants on vascular development. Genotyping of a larger cohort of patients will allow to more finely select mutations or genes for functional validations.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
The study was approved by the local Ethical Committee A.O.U. “G. Martino”, Messina. The study involves human participants and was performed according to Helsinki declaration. All involved subjects were fully informed and written consent was obtained.
Author Contributions
CS wrote the manuscript and conducted laboratory investigation. LD performed bioinformatic analysis. CA and FG recruited the patient and formulated the diagnosis. CR contributed to laboratory investigation. ML provided at financial support. RD’A conceived the study and corrected the manuscript. AS coordinated the research team.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
All researchers want to thank the members of the family to have agreed and collaborate to achieve this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00146/full#supplementary-material
Supplementary Table 1 | The table lists the nodes represented in Figure 2A. For each node are reported the node IDs in the different databases (Ensembl, Entrez, RefSeq. UniProt). Node type refers to the origin of the node within the network: query nodes were given as input while result nodes were inferred by the analysis.
Supplementary Table 2 | The table explains the details of the network edges reported in Figure 2A. For each edge, the nature of the link (co-expression, co-localization, genetic interaction, shared protein domains) and the source (network column) are also provided. The other columns show the statistical parameters derived from network assembly.
Supplementary Table 3 | Edge table of Figure 2B. For each edge, the nature of the link (co-expression, co-localization, genetic interaction, shared protein domains) and the source (network column) are also provided. The other columns show the statistical parameters derived from network assembly.
Supplementary Table 4 | Annotation table of nodes represented in Figure 2. Nodes are grouped based on GeneOntology annotation.
References
Abdollahpour, H., Appaswamy, G., Kotlarz, D., Diestelhorst, J., Beier, R., Schäffer, A. A., et al. (2012). The phenotype of human STK4 deficiency. Blood 119, 3450–3457. doi: 10.1182/blood-2011-09-378158
Amyere, M., Revencu, N., Helaers, R., Pairet, E., Baselga, E., Cordisco, M., et al. (2017). Germline loss-of-function mutations in EPHB4 cause a second form of Capillary malformation-Arteriovenous malformation (CM-AVM2) deregulating RAS-MAPK signaling. Circulation 136, 1037–1048. doi: 10.1161/CIRCULATIONAHA.116.026886
Chun, S., Fay, J. C. (2009). Identification of deleterious mutations within three human genomes. Genome Res. 19, 1553–1561. doi: 10.1101/gr.092619.109
Cingolani, P., Platts, A., Wang, l., Coon, M., Nguyen, T., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92. doi: 10.4161/fly.19695
Demirdas, S., Dulfer, E., Robert, L., Kempers, M., van Beek, D., Micha, D., et al. (2017). Recognizing the tenascin-X deficient type of Ehlers-Danlos syndrome: a cross-sectional study in 17 patients. Clin. Genet. 91, 411–425. doi: 10.1111/cge.12853
Emlet, D. R., Pastor-Soler, N., Marciszyn, A., Wen, X., Gomez, H., Humphries, W. H., 4th, et al. (2017). Insulin-like growth factor binding protein 7 and tissue inhibitor of metalloproteinases-2: differential expression and secretion in human kidney tubule cells. Am. J. Physiol. Renal Physiol. 312, F284–F296. doi: 10.1152/ajprenal.00271.2016
Errede, M., Mangieri, D., Longo, G., Girolamo, F., de Trizio, I., Vimercati, A., et al. (2018). Tunneling nanotubes evoke pericyte/endothelial communication during normal and tumoral angiogenesis. Fluids Barriers CNS 15, 28. doi: 10.1186/s12987-018-0114-5
Goldberg, J., Raabe, A., Bervini, D. (2018). Natural history of brain arteriovenous malformations: systematic review. J. Neurosurg. Sci. 62, 437–443. doi: 10.23736/S0390-5616.18.04452-1
Inoue, S., Liu, W., Inoue, K., Mineharu, Y., Takenaka, K., Yamakawa, H., et al. (2007). Combination of linkage and association studies for brain arteriovenous malformation. Stroke 38, 1368–1370. doi: 10.1161/01.STR.0000260094.03782.59
Karlsson, T., Cherif, H. (2018). Mutations in the ENG, ACVRL1, and SMAD4 genes and clinical manifestations of hereditary haemorrhagic telangiectasia: experience from the Center for Osler’s Disease, Uppsala University Hospital. Ups. J. Med. Sci. 123, 153–157. doi: 10.1080/03009734.2018.1483452
Kim, Y. H., Choi, J., Yang, M. J., Hong, S. P., Lee, C., Kubota, Y., et al. (2019). A MST1–FOXO1 cascade establishes endothelial tip cell polarity and facilitates sprouting angiogenesis. Nat. Commun. 10, 838. doi: 10.1038/s41467-019-08773-2
Lawson, N. D., Scheer, N., Pham, V. N., Kim, C. H., Chitnis, A. B., Campos-Ortega, J. A., et al. (2001). Notch signaling is required for arterial-venous differentiation during embryonic vascular development. Development 128, 3675–3683.
Lawson, N. D., Vogel, A. M., Weinstein, B. M. (2002). Sonic hedgehog and vascular endothelial growth factor act upstream of the Notch pathway during arterial endothelial differentiation. Dev. Cell. 3, 127–136. doi: 10.1016/S1534-5807(02)00198-3
Lei, D., Jin, X., Wen, L., Dai, H., Ye, Z., Wang, G. (2017). Bmp3 is required for integrity of blood brain barrier by promoting pericyte coverage in Zebrafish embryos. Curr. Mol. Med. 17, 298–303. doi: 10.2174/1566524017666171106114234
Li, H., Durbin, R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Lindeboom, R. G. H., Vermeulen, M., Lehner, B., Supek, F. (2019). The impact of nonsense-mediated mRNA decay on genetic disease, gene editing and cancer immunotherapy. Nat. Genet. 51, 1645–1651. doi: 10.1038/s41588-019-0517-5
Lu, H., Liu, X., Guo, W. (2019). Melatonin attenuates inflammation-related venous endothelial cells apoptosis through modulating the MST1-MIEF1 pathway. J. Cell Physiol. 234, 23675–23684. doi: 10.1002/jcp.28935
Malfait, F. (2018). Vascular aspects of the Ehlers-Danlos Syndromes. Matrix Biol. 71–72. doi: 10.1016/j.matbio.2018.04.013
Mao, R., Meng, S., Gu, Q., Araujo-Gutierrez, R., Kumar, S., Yan, Q. (2017). AIBP limits angiogenesis through γ-secretase-mediated upregulation of notch signaling. Circ Res. 120, 1727–1739. doi: 10.1161/CIRCRESAHA.116.309754
Montojo, J., Zuberi, K., Rodriguez, H., Kazi, F., Wright, G., Donaldson, S. L., et al. (2010). GeneMANIA Cytoscape plugin: fast gene function predictions on the desktop. Bioinformatics 26, 2927–2928. doi: 10.1093/bioinformatics/btq562
Oikawa, M., Kuniba, H., Kondoh, T., Kinoshita, A., Nagayasu, T., Niikawa, N., et al. (2010). Familial brain arteriovenous malformation maps to 5p13-q14, 15q11-q13 or 18p11: linkage analysis with clipped fingernail DNA on high-density SNP array. Eur. J. Med. Genet. 53, 244–249. doi: 10.1016/j.ejmg.2010.06.007
Osbun, J. W., Reynolds, M. R., Barrow, D. L. (2017). Arteriovenous malformations: epidemiology, clinical presentation, and diagnostic evaluation. Handb. Clin. Neurol. 143, 25–29. doi: 10.1016/B978-0-444-63640-9.00003-5
Pawlikowska, L., Nelson, J., Guo, D. E., McCulloch, C. E., Lawton, M. T., Kim, H., et al. (2018). Brain vscular malformation consortium HHT Investigator Group. Association of common candidate variants with vascular malformations and intracranial hemorrhage in hereditary hemorrhagic telangiectasia. Mol. Genet. Genom. Med. 6, 350–356. doi: 10.1002/mgg3.377
Romano, E., Manetti, M., Rosa, I., Fioretto, B. S., Ibba-Manneschi, L., Matucci-Cerinic, M., et al. (2018). Slit2/Robo4 axis may contribute to endothelial cell dysfunction and angiogenesis disturbance in systemic sclerosis. Ann. Rheumatol. Dis. 77, 1665–1674. doi: 10.1136/annrheumdis-2018-213239
Schock, E. N., Struve, J. N., Chang, C. F., Williams, T. J., Snedeker, J., Attia, A. C., et al. (2017). A tissue-specific role for intraflagellar transport genes during craniofacial development. PloS One 12, e0174206. doi: 10.1371/journal.pone.0174206
Schwarz, J. M., Cooper, D. N., Schuelke, M., Seelow, D. (2014). MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods 11, 361–362. doi: 10.1038/nmeth.2890
Shoemakerl, L. D., Fuentes, L. F., Santiago, S. M., Allen, B. M., Cook, D. J., Steinberg, G. K., et al. (2014). Human brain arteriovenous malformations express lymphatic-associated genes. Ann. Clin. Transl. Neur. 1, 982–995. doi: 10.1002/acn3.142
Sumioka, T., Iwanishi, H., Okada, Y., Nidegawa, Y., Miyajima, M., Matsumoto, K. I., et al. (2018). Loss of tenascin X gene function impairs injury-induced stromal angiogenesis in mouse corneas. J. Cell. Mol. Med. 22, 948–956. doi: 10.1111/jcmm.13397
Tumelty, K. E., Higginson-Scott, N., Fan, X., Bajaj, P., Knowlton, K. M., Shamashkin, M., et al. (2018). Identification of direct negative cross-talk between the SLIT2 and bone morphogenetic protein-Gremlin signaling pathways. J. Biol. Chem. 293, 3039–3055. doi: 10.1074/jbc.M117.804021
van Beijnum, J., van der Worp, H. B., Schippers, H. M., van Nieuwenhuizen, O., Kappelle, L. J., Rinkel, G. J., et al. (2007). Familial occurrence of brain arteriovenous malformations: a systematic review. J. Neurol. Neurosurg. Psychiatry 78, 1213–1217. doi: 10.1136/jnnp.2006.112227
Varelas, X., Wrana, J. L. (2012). Coordinating developmental signaling: novel roles for the Hippo pathway. Trends Cell Biol. 22, 88–96. doi: 10.1016/j.tcb.2011.10.002
Vaser, R., Adusumalli, S., Ngak Leng, S., Sikic, M., Ng, P. C. (2016). SIFT missense predictions for genomes. Nat. Protoc. 11, 1–9. doi: 10.1038/nprot.2015.123
Vijayan, M., Reddy, P. H. (2016). Stroke, vascular dementia, and Alzheimer’s disease: molecular links. J. Alzheimers Dis. 54, 427–443. doi: 10.3233/JAD-160527
Walcott, B. P., Winkler, E. A., Zhou, S., Birk, H., Guo, D., Koch, M. J., et al. (2018). Identification of a rare BMP pathway mutation in a non-syndromic human brain arteriovenous malformation via exome sequencing. Hum. Genome Var. 5, 18001. doi: 10.1038/hgv.2018.1
Wang, K., Li, M., Hakonarson, H. (2010). ANNOVAR: Functional annotation of genetic variants from next-generation sequencing data. Nucleic Acids Res. 38, e164. doi: 10.1093/nar/gkq603
Wang, K., Zhao, S., Liu, B., Zhang, Q., Li, Y., Liu, J., et al. (2018). Perturbations of BMP/TGF-β and VEGF/VEGFR signalling pathways in non-syndromic sporadic brain arteriovenous malformations (BAVM). J. Med. Genet. 55, 675–684. doi: 10.1136/jmedgenet-2017-105224
Wunnenburger, S., Schultheiss, U. T., Walz, G., Hausknecht, B., Ekici, A. B., Kronenberg, F., et al (2017). Associations between genetic risk variants for kidney diseases and kidney disease etiology. Sci. Rep. 7, 13944. doi: 10.1038/s41598-017-13356-6
Yılmaz, B., Toktaş, Z. O., Akakın, A., Işık, S., Bilguvar, K., Kılıç, T., et al. (2017). Familial occurrence of brain arteriovenous malformation: a novel ACVRL1 mutation detected by whole exome sequencing. J. Neurosurg. 126, 1879–1883. doi: 10.3171/2016.6.JNS16665
Yamakawa, D., Jia, W., Kidoya, H., Hosojima, S., Torigata, M., Zhang, L., et al. (2018). Visualization of proliferative vascular Endothelial cells in tumors in vivo by imaging their partner of Sld5-1 promoter activity. Am. J. Pathol. 188, 1300–1314. doi: 10.1016/j.ajpath.2018.01.015
Keywords: brain arteriovenous malformations, pathogenic variants, exome, molecular signaling, vascular differentiation
Citation: Scimone C, Donato L, Alafaci C, Granata F, Rinaldi C, Longo M, D’Angelo R and Sidoti A (2020) High-Throughput Sequencing to Detect Novel Likely Gene-Disrupting Variants in Pathogenesis of Sporadic Brain Arteriovenous Malformations. Front. Genet. 11:146. doi: 10.3389/fgene.2020.00146
Received: 15 March 2019; Accepted: 07 February 2020;
Published: 28 February 2020.
Edited by:
Jordi Pérez-Tur, Superior Council of Scientific Investigations (CSIC), SpainReviewed by:
Alessio Branchini, University of Ferrara, ItalySteven D. Chang, Stanford University, United States
Copyright © 2020 Scimone, Donato, Alafaci, Granata, Rinaldi, Longo, D’Angelo and Sidoti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rosalia D’Angelo, rdangelo@unime.it