 Fernando Díaz1*
Fernando Díaz1* André Luís A. Lima2Aline M. Nakamura2Fernanda Fernandes2
André Luís A. Lima2Aline M. Nakamura2Fernanda Fernandes2 Iderval Sobrinho Jr.2
Iderval Sobrinho Jr.2 Reinaldo A. de Brito2
Reinaldo A. de Brito2- 1Department of Entomology, University of Arizona, Tucson, AZ, United States
- 2Departamento de Genética e Evolução, Universidade Federal de São Carlos, São Carlos, Brazil
Introgression should no longer be considered as rare a phenomenon as once thought, since several studies have recently documented gene flow between closely related and radiating species. Here, we investigated evolutionary relationships among three closely related species of fruit flies of the Anastrepha fraterculus group (Anastrepha fraterculus, A. obliqua and A. sororcula). We sequenced a set of 20 genes and implemented a combined populational and phylogenetic inference with a model selection approach by an ABC framework in order to elucidate the demographic history of these species. The phylogenetic histories inferred from most genes showed a great deal of discordance and substantial shared polymorphic variation. The analysis of several population and speciation models reveal that this shared variation is better explained by introgression rather than convergence by parallel mutation or incomplete lineage sorting. Our results consistently showed these species evolving under an isolation with migration model experiencing a continuous and asymmetrical pattern of gene flow involving all species pairs, even though still showed a more closely related relationship between A. fraterculus and A. sororcula when compared with A. obliqua. This suggests that these species have been exchanging genes since they split from their common ancestor ∼2.6 MYA ago. We also found strong evidence for recent population expansion that appears to be consequence of anthropic activities affecting host crops of fruit flies. These findings point that the introgression here found may have been driven by genetic drift and not necessary by selection, which has implications for tracking and managing fruit flies.
Introduction
There is increased attention on the study of evolutionary relationships among recently diverged and radiating species (Beltran et al., 2002; Buckley et al., 2006; Maddison and Knowles, 2006; Carstens and Knowles, 2007; Mallet et al., 2007; Nunes et al., 2010; Martin et al., 2013; Cruickshank and Hahn, 2014; Meyer et al., 2016), particularly because they tend to be affected by incongruence among gene trees, as initially reviewed by Pamilo and Nei (1988) and Maddison (1997). This is often due to the stochasticity of the coalescent process, which is more likely the larger population sizes are relative to the divergence times (Pamilo and Nei, 1988), leading to incomplete lineage sorting (ILS). There are, though, other biological causes for disagreement among gene trees, such as introgressive hybridization, horizontal gene transfer and gene duplication (Maddison, 1997; Harrison and Larson, 2014), that complicate the correct identification of the underlying process involved in the incongruences. Introgression in particular has become an important issue in evolutionary biology, since the elucidation of mechanisms and effects of gene flow following speciation may help in the understanding of the nature of species boundaries (Cruickshank and Hahn, 2014; Harrison and Larson, 2014) as well as of species adaptation (Hedrick, 2013).
Differentiating between introgression and ILS is challenging because both processes can produce similar phylogenetic patterns (Holder et al., 2001), though several approaches have been recently proposed to formally test them (Beaumont et al., 2002; Hey and Nielsen, 2004; Buckley et al., 2006; Ané et al., 2007; Beerli and Palczewski, 2010; Larget et al., 2010; Meyer et al., 2016; Huang et al., 2017; Kamneva and Rosenberg, 2017). The relevance of introgression in speciation studies has been established in many taxa, several of them insects, including Drosophila species (Machado et al., 2002; Hey and Nielsen, 2004; Llopart et al., 2005; Bachtrog et al., 2006; Kulathinal et al., 2009; Nunes et al., 2010; Garrigan et al., 2012; Herrig et al., 2014; Beck et al., 2015), butterflies (Beltran et al., 2002; Mallet et al., 2007; Giraldo et al., 2008; Pardo-Diaz et al., 2012; Martin et al., 2013; Nadeau et al., 2014; Zhang et al., 2016) and mosquitoes (Marsden et al., 2011; Weetman et al., 2012; Clarkson et al., 2014; Norris et al., 2015). The study of introgression in other non-model species is more limited, even for pests in which the dynamics of introgression have a potential effect on adaptation to new environments that favor the formation of species complexes. That may be the case of the fruit fly Rhagoletis pomonella, where it has been suggested that introgression may be playing a relevant role during its speciation (Feder et al., 1999; Xie et al., 2007; Michel et al., 2010; Arcella et al., 2015).
Here we consider the case of three closely related species of the Anastrepha fraterculus group, A. fraterculus, A. sororcula, and A. obliqua, which are some of the most important agricultural pests in South America, not only because they are widespread, but also because they inflict damage to a wide array of hosts (Aluja, 1994). These species encompass some of the most economically relevant in the genus, which is composed of 34 recognized species and includes some closely related species that show enough morphological similarities (Norrbom et al., 1999, 2012) to render species identification a difficult task. Despite their morphological lability, there are several differences in host preference, reproductive behavior (Aluja, 1994; Aluja et al., 1999; Sivinski et al., 1999; Juárez et al., 2015) and morphometry (Selivon and Perondini, 1998; Hernández-Ortiz et al., 2012, 2015; Perre et al., 2014, 2016) that suggest incipient speciation, so much so that A. fraterculus in particular has been considered a species complex, composed of several entities, three of them in Brazil (Hernández-Ortiz et al., 2004, 2015; Hendrichs et al., 2015; Vaníèková et al., 2015). Even though these studies have used data from a range of methods, including behavior, morphometrics, karyotype, isozymes, pheromone, cuticular hydrocarbons, mtDNA and reproductive studies, they lack a comprehensive sampling across the species’ range and fail to fully integrate molecular and phenotypic data, which complicates their use in species identification. An indication of the complex taxonomic questions in this genus is that single-gene molecular phylogenies based on mtDNA (McPheron et al., 1999; Smith-Caldas et al., 2001) or nuclear genes (Barr et al., 2005; Ruiz et al., 2007; Sarno et al., 2010; Gonçalves et al., 2013), and even using multiple genes (Mengual et al., 2017) have failed to identify species-specific fixed differences among the most closely related taxa, leading some to propose a multidisciplinary approach (Dias et al., 2016).
We took advantage of a recently developed cDNA library of reproductive female tissues of A. obliqua (Gonçalves et al., 2013), to select a set of 20 genes to study the evolutionary relationships and demographic history among three of the most widespread Anastrepha species in Brazil (Nascimento et al., 1993) using a multilocus analysis. For this, we integrated phylogenetic methods that account for ILS and introgression as well as compared several speciation models under an approximate Bayesian computation framework. Once we determined the interspecific relationships among species, we modeled their demographic history to better understand recent events, such as agricultural activities on temporal patterns of gene flow and population expansion. This helped us investigate the existence of evolutionary lineages in this group despite the occurrence of introgression, which has implications for tracking and managing fruit flies.
Materials and Methods
Sampling and Species Identification
Fruits of host–plants potentially infested with Anastrepha were collected from different regions in Brazil (Supplementary Table S1), and kept on vermiculite for ∼3 weeks in the laboratory until pupae were reared. After emergence and maturation of adults, specimens were identified using wing, ovipositor and other morphological markers following identification keys available on Norrbom et al. (2012) with the help of Drs. R. Zucchi and K. Uramoto. Specimens from the fraterculus group (particularly A. fraterculus, A. obliqua, and A. sororcula) were stored in 95% ethanol at -20°C until DNA extraction. Several localities have representatives of more than one Anastrepha species, though most are not from the same fruit (Supplementary Table S1). At least 20 individuals from each species were sampled across different locations in order to maximize their representation along their distribution in Brazil (Supplementary Table S1).
Despite strong evidence indicating that A. fraterculus in particular is a species complex (Vaníèková et al., 2015), the identification of these cryptic species requires large populational samples and an integrated taxonomy approach (Schutze et al., 2017) to enable morphometric differentiation, which seems to be the most effective way (Vaníèková et al., 2015), since egg chorion morphology and sex chromosomes (Selivon and Perondini, 2007; Vaníèková et al., 2015) are harder to get from field collected specimens. Because we still lack substantial information about species boundaries within this particular complex and which markers should be considered to identify individual specimens, we chose to, conservatively, maintain the use of A. fraterculus sensu latu in our analyses, particularly because most samples were directly collected from the field, several from small samples, preventing the use of an integrative approach for their identification.
Molecular Procedures
DNA was extracted from individual flies by a slight modification of the method described by Chomczynski and Sacchi (1987) in order to maintain exoskeletons intact for future morphological analyses. Genes were selected from a cDNA library of reproductive tissues of A. obliqua (Gonçalves et al., 2013). For primer design (Supplementary Table S2), sequences were aligned to other Diptera available online (Supplementary Table S3) using the software ClustalW (Thompson et al., 1994) implemented in BioEdit (Hall, 1999) and the stability of their structures were checked using OligoAnalyzer 3.0. Only genes with consistent amplification across the three Anastrepha species (A. fraterculus, A. obliqua and A. sororcula) were considered (Supplementary Table S2). DNA samples were equimolarly pooled for each species in groups of 20 individuals (2N = 40), and then PCR amplifications were performed following manufacturer’s recommendations for Taq DNA polymerase kit (Fermentas Inc.) and 1/100th unit of Pfu polymerase (Cline et al., 1996) in a 26 μl final volume, involving 1 μl of pooled DNA and 0.2 μM of each primer. PCR amplifications involved 35 cycles of 94°C for 30 s followed by the annealing temperature of each gene for 30 s (Supplementary Table S2), 72°C extension for 1 min, and then a final extension at 72°C for 10 min. PCR products were purified by PEG 8000 precipitation (Lis and Schleif, 1975) and cloned using InsTAclone PCR Cloning Kit (Thermo Scientific) following manufacturer’s recommendations.
We have pooled samples in order to develop a time- and cost-effective molecular strategy that allowed us to sequence a large battery of loci required for coalescent simulations. Most analyses here considered to elucidate the demographic history of Anastrepha species are based on Bayesian genealogy sampler such as IMa2, as indicated below. Such methods are highly flexible in terms of number of estimated parameters, though such flexibility is limited by the number of loci considered, particularly for demographic parameters, since estimations that use data sets from 5 to 7 loci, which is standard in the field, has been demonstrated to generate false positives (Cruickshank and Hahn, 2014; Hey et al., 2015). For this reason, here we have decided to implement an alternative strategy to increase the number of loci sequenced in our multilocus sampling by pooling and posterior cloning of amplified pooled samples. Because we are aware of the potential issues associated when pooling samples for sequencing, we have designed a molecular framework in order to account for such issues and reduce their potential effects on the final conclusions of our population and phylogenetic analyses. The major issue when pooling samples is the indetermination of the haplotype structure, which we accounted for by performing molecular cloning of PCR products. This step allowed us to individualize every haplotype present in the pool in the form of multiple recombinant independent colonies. Then, a large enough number of these colonies were PCR amplified and their inserts purified and sequenced with M13 primers through the sequencing service of Macrogen Inc, Korea. Each colony was sampled and amplified with replication for sequencing (forward and reverse) in order to end up with a minimum of 20 colonies per species. Sequences obtained for each colony were inspected for incongruences and only colonies which showed no incongruences were retained. Sequences that differed from others by a single nucleotide were resequenced to confirm their haplotypes. We also accounted for possible duplicates by using a sampling size much lower than the pool size (k < 2N, 20 < 40) in order to minimize their probability, in such a way that we made it similar to the probability of getting duplicates from individual sequencing of diploid organisms. Both amplification steps were performed using a high fidelity Pfu polymerase to reduce PCR misincorporations (Cline et al., 1996). Finally, all alignments for each gene were analyzed using haplotype networks in order obtain the distribution of unique and duplicated haplotypes and such distributions were compared to expectations from the coalescent theory following Castelloe and Templeton (1994). With the mentioned framework, we were able to sequence a large set of 20 loci with the required definition of haplotype structure and minimal PCR misincorporations. The generated data set allowed proper site and haplotype-based parameter estimation since it retained the main fundamental assumptions for population analyses, such as a large number of neutrally independent loci with no within locus recombination.
Sequences were visually inspected for quality using Chromas v. 2.3.1 and aligned among themselves and to sequences of more distantly related taxa belonging to Tephritidae (Rhagoletis sp., Bactrocera sp., Ceratitis capitata) and Drosophilidae (Drosophila willistoni, Drosophila melanogaster, Drosophila grimshawi, Drosophila virilis, Drosophila mojavensis) available on GenBank (Supplementary Table S3). All sequences generated for Anastrepha species in this study are available on GenBank (Supplementary Table S4) The Tephritid species chosen are the most closely related species to Anastrepha for which there were available data for the genes here sequenced on GenBank at the time of this analysis, whereas Drosophilidae were used for some neutrality tests. All alignments were performed on inferred amino acid sequences using Clustal W and manually reconverted to DNA alignments.
Polymorphism and Genetic Structure
In order to quantify the genetic diversity for each species, we estimated a set of descriptive parameters using the software DNAsp v5 (Librado and Rozas, 2009), including the number of variable sites (S), number of haplotypes (h), haplotype diversity (Hd), nucleotide diversity (π) and the average number of nucleotide differences (θW). Sampled genes encode for proteins that perform different metabolic functions and are therefore likely to be under different selective constrains. We performed a set of neutrality tests including Tajima’s D (Tajima, 1989), Fu and Li’s D and F tests (Fu and Li, 1993) and Fu’s FS statistic (Fu, 1997) using the software DNAsp. A comparison among these tests was used for inferring any deviation from neutral expectations, population structure or drastic changes in population sizes (recent bottlenecks or population expansion). All genes were assessed for recombination events with GENECONV (Sawyer, 1989) and RDP methods (Martin and Rybicki, 2000) both implemented in RDP v4 (Martin et al., 2015), as well as the GARD test (a genetic algorithm for recombination detection) (Kosakovsky-Pond et al., 2006a,b) implemented in the package HyPhy. Furthermore, the sequences were checked for substitution saturation using the software DAMBE v5 (Xia, 2013).
Divergence among species was estimated by comparing the number of synonymous (Ks) and non-synonymous (Ka) changes per site and Ka/Ks ratio using the software DNAsp v5 (Librado and Rozas, 2009). The genetic structure among species was estimated through pairwise ΦST, an analogue version of the Wright’s fixation index FST (Wright, 1951; Weir and Cockerham, 1984) which is estimated by an analysis of molecular variance (AMOVA), taking into account information on the genetic distances among haplotypes as well as their frequencies following Excoffier et al. (1992). Significance was based on 10,000 permutations in the software Arlequin version 3.5.2 (Excoffier and Lischer, 2010).
Given the close relationship among Anastrepha species in the fraterculus group, and considering previous results from different genes (Ruiz et al., 2007; Sarno et al., 2010; Gonçalves et al., 2013), we expected to find significant levels of shared polymorphism among them. This shared variation could be the result of retained ancestral polymorphisms, when variation present in the ancestral species persists after speciation, as well as introgressive hybridization following speciation (Holder et al., 2001; Harrison and Larson, 2014). We tested the hypothesis for introgression by contrasting phylogenetic inferences as well as evaluating speciation and demographic models (as explained below). However, part of the shared variation may have arisen as a result of convergence by parallel mutation among species (Clark, 1997). We quantified numbers of shared polymorphisms and fixed differences between each species pair, and then assessed how much polymorphic variation could be expected by parallel mutation under the assumption that mutations occur randomly and independently with equal probability at all sites following Clark (1997) and Kliman et al. (2000).
Phylogenetic Analyses
We investigated phylogenetic relationships individually for each locus using Bayesian inference in MrBayes (Huelsenbeck and Ronquist, 2001) after testing for the best evolutionary model inferred in the software jModelTest v0.1.1 (Posada, 2008). Four independent runs were used from different starting points by a Metropolis-coupled Markov Chain Monte Carlo analysis, one cold and three incrementally heated (heating parameter = 0.2) for 10 million generations, sampling every 400th tree. All runs were checked for convergence with the standard deviation of split frequencies being less than 0.01. Parameter estimates were then analyzed in Tracer (Drummond and Rambaut, 2007) to ensure that these had reached stable values with adequate mixing and ESSs above 200.
Because we did not expect reciprocal monophyly for the species here studied, or even a general agreement on the topology of most genes here investigated, we implemented a multilocus inference of the species tree. For this, we performed a comparative phylogenetic analysis using ∗BEAST (Bayesian Evolutionary Analysis Sampling Trees) (Drummond and Rambaut, 2007), which accounts for stochasticity in the coalescent process, and BUCKy (Bayesian Untangling of Concordance Knots, Bayesian concordance analysis) (Larget et al., 2010), a method that accounts for discordance among gene trees without making any assumption about the cause of discordance (Ané et al., 2007). The comparison between these methods was used for inferring causes of discordance among gene trees. The gene-trees approach implemented in ∗BEAST assumes that discordance among gene trees is only the result of stochastic coalescence of gene lineages within a species phylogeny. ∗BEAST was run for 200 million generations, sampling every 1000 generations. A maximum clade credibility tree was generated using the program Tree Annotator v.1.6.2 provided in the BEAST package, with a burn-in of 10%. Statistical parameters were analyzed in Tracer (Drummond and Rambaut, 2007) to assure that these had reached stable values with adequate mixing and ESS above 200.
The importance of testing the fit to a multispecies coalescent model has been demonstrated recently (Buckley et al., 2006; Reid et al., 2014) since species trees estimated by ∗BEAST are good phylogeny estimations only when these assumptions are not violated. Since introgressive hybridization is likely to be one of the causes for discordance among gene trees, we implemented formal tests in order to reject Incomplete Lineage Sorting (ILS) as the only cause for discordance among gene trees. First, we ran BUCKy using all genes in order to estimate the Primary Concordance Tree (PCT) and the Population Tree (PT) using gene trees obtained from MrBayes. Analyses were run for 4 million generations with four different heating chains after a 200,000-generation burn-in. Prior values for the parameter α were estimated from the level of discordance among gene trees using the R script ‘prior_standalone.r’1. Hypotheses for introgressive hybridization were tested by comparing the PCT vs. PT trees. These two trees are expected to be in agreement when ILS is responsible for the discordance among most of the gene trees. A lack of agreement is therefore evidence for introgressive hybridization or other biological causes (Larget et al., 2010).
When ILS was not rejected as the only cause for disagreement in gene trees, we tested the fit to the multispecies coalescent model for each individual loci using the R package P2C2M (Posterior Predictive Checks of Coalescent Models) (Gruenstaeudl et al., 2015). This package implements a posterior predictive simulation using gene and species trees generated by ∗BEAST. For this, we used the software script ‘BEAUTiAutomator.py’ implemented in the R package in order to set up the XML input file for ∗BEAST, instead of the software BEAUTi implemented in BEAST. Using this approach we were able to identify genes showing poor coalescent likelihood using the statistic LCWT (likelihood of the coalescent waiting times) (Reid et al., 2014) as well as genes showing deep coalescence with the statistic NDC (number of deep coalescences) (Maddison, 1997). Both ∗BEAST and BUCKy were run twice, with and without genes showing poor fit to coalescent assumptions.
ABC Approach
The occurrence of introgression is a clear violation of the coalescent assumptions, which complicates the interpretation of phylogenetic analyses among Anastrepha species. We used an Approximate Bayesian Computation (ABC) framework (Tavare et al., 1997; Beaumont et al., 2002) to investigate whether the patterns of genetic variation could be explained in the absence of gene flow as well as test the topology of relationship among Anastrepha species. We compared the three possible topology models among these species considering two speciation models (Figure 1), a strict isolation scenario (SI model) as well as the isolation with migration model (IM model) using the ABCtoolbox 2.0 package (Wegmann et al., 2010). We evaluated such models by a hierarchical procedure, comparing the IM against SI model within each possible topology. Then, the topology models were compared in the presence of migration within an IM speciation model (Figure 1).
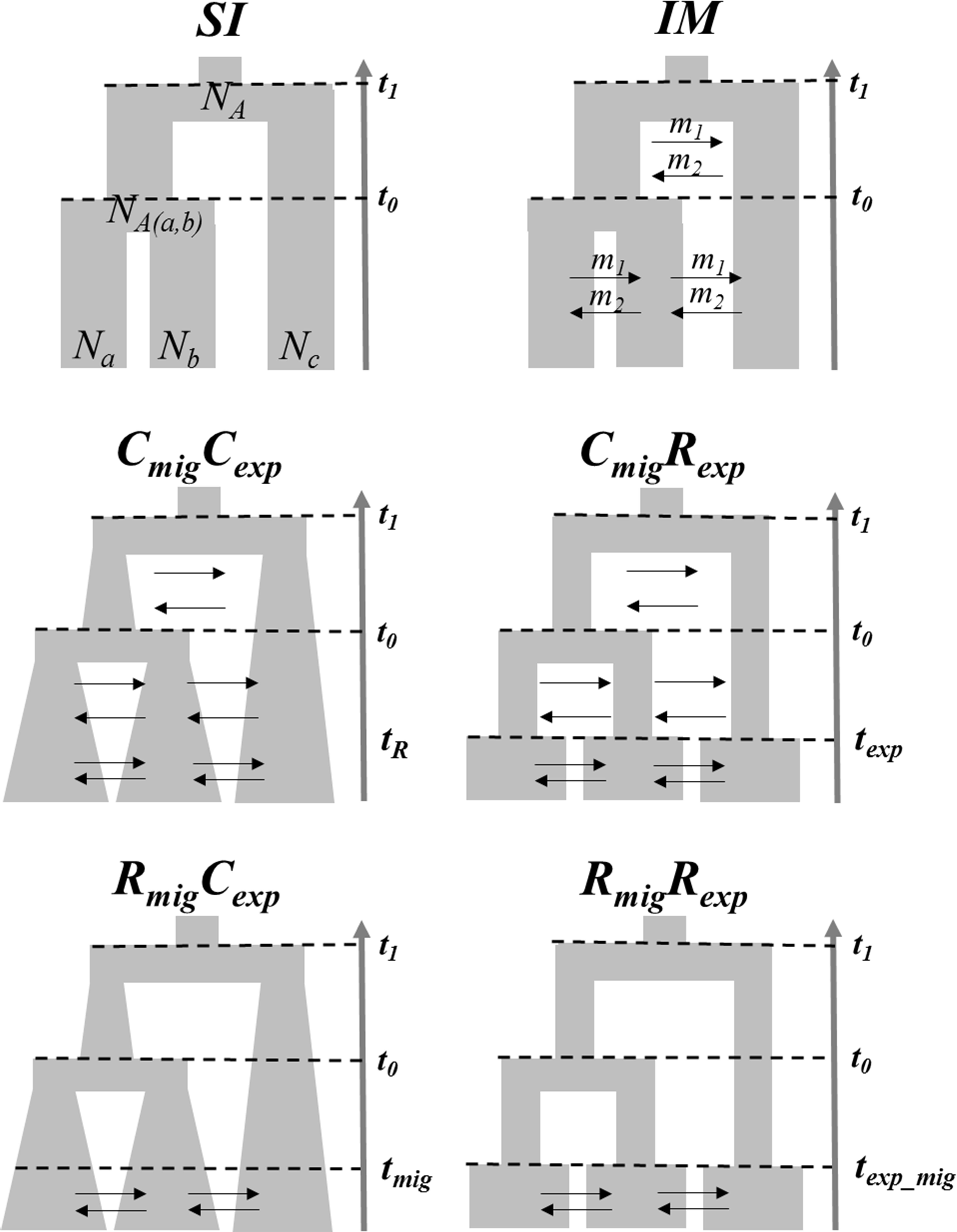
FIGURE 1. Speciation models among the three Anastrepha species compared by the ABC approach. A model under a strict isolation (SI) was first compared with an isolation with migration model (IM). These two speciation models were compared in the presence of the three alternative models of topology. t0 is the number of generations since the first speciation event (backwards in time) while t1 corresponds to the second speciation event. Na, Nb and Nc are effective populations sizes for each species. NA(a,b) is the ancestral population size of a and b while NA is the ancestral population size of the common ancestor for the three species. Bidirectional migration rates m1 and m2 are the proportion of migrants per generation. Once the more likely topology was confirmed under an IM model, four models were further compared using MIGRATE and IMa2 results as priors by a second ABC analysis in order to test for temporal patterns of migration and population expansion (continuous vs recent). A model with constant migration and population expansion CmigCexp, a model with constant migration and recent population expansion CmigRexp, a model with recent migration and constant population expansion RmigCexp, and a model with recent migration and population expansion RmigRexp. The parameters texp, tmig and texp_mig represent the time (3,000 generations) since the species experienced population expansion, migration or both after a period of strict isolation.
Once the most likely speciation and topology models were confirmed (See results), we ran MIGRATE software version 3.5.2 (Beerli and Palczewski, 2010) in order to compare specific models for the direction of migration. Then, the IMa2 software (Hey and Nielsen, 2004) was run under an IM scenario in order to compute population parameters of population sizes for each species (recent and ancestral) as well as migration rates and coalescent times between species (see below). Because we detected migration among these species as well as evidence for population expansion (see Results), MIGRATE and IMa2 results were then used as priors for a second ABC analysis to investigate whether the introgression detected among these fruit flies and their population expansion have been favored by the recent burst of agriculture activities. Given the fact that by the turn of the 1700, Brazil underwent a population burst from mining and agriculture in the midlands (Furtado, 1959), which would involve ∼3,000 generations of fruit flies, we evaluated four demographic models comparing temporal patterns of gene flow and population expansion (Figure 1). (i) A model with continuous gene flow and population expansion (CmigCexp); (ii) a model with continuous gene flow and recent population expansion (CmigRexp); (iii) a model with recent gene flow and constant population expansion (RmigCexp), and (iv) a model with recent gene flow and population expansion (RmigRexp) (Figure 1). Again, these models were compared with a hierarchical procedure in a pairwise comparison between the CmigCexp against each of the non-contiguous models, and then, the best two models were compared in order to select the most likely model.
All models were run using a likelihood-free ABC–MCMC method (Wegmann et al., 2009) and the program Fastsimcoal 2 (Excoffier and Foll, 2011) was used to simulate 10,000,00 samples with a proposal range of φ = 1 and tolerance δ = 0.1. The MCMC sampling was previously calibrated based on 10,000 simulated samples under a standard mode and partial least squares (PLS) components were extracted using the specific R script of the ABCtoolbox (Wegmann et al., 2010) from the summary statistics in order to calculate distances in the Markov Chain. The observed summary statistics were chosen based on the potential information for differentiating the models here evaluated. We used 31 statistics calculated by the software Arlsumstat, a modified version of Arlequin 3.5.2 (Excoffier and Lischer, 2010), in order to summarize the genetic information of sequences at all loci in the three species. For each species, we computed the number of segregating sites, number of haplotypes, number of nucleotide differences, Tajima’s D (Tajima, 1989) and Fu’s FS statistic (Fu, 1997). For each pair of species, we computed deviations between the mentioned summary statistics as well as ΦST and the average number of pairwise differences. The 5,000 closest simulated samples were retained and compared with the observed summary statistics using a regression adjustment to a general linear model (GLM) (Leuenberger and Wegmann, 2010). Statistics were checked for redundancy and highly correlated statistics were pruned. The model selection was performed following pairwise comparisons based on the calculation of Bayes-Factors (BF) and the corresponding posterior probability given the model. The robustness of each comparison was also estimated based on 2,000 pseudo-observed data sets simulated in each alternative model.
Migration Direction Among Species
Because we found evidence for introgression (see Results), the software MIGRATE version 3.5.2 was run to test migration models among the three species by coupled-MCMC simulations (Beerli and Palczewski, 2010). Ten replicates were used per run with a burn-in of 100 thousand steps followed by 100 million steps sampled every 100th. Each replicate was run under a static heating approach implementing four incrementally heated chains. Given that we used a set of 20 different nuclear genes, we accounted for different evolutionary rates using the option of relative mutation rates estimated from the data. Analyses were performed in two steps: first, eight migration models were tested including different possibilities for pairwise comparisons between species, as well as the panmictic model. This included three migration models for one parameter (migration involving only two species and no migration among the others), three models for two parameters (migration involving three species but only two pairwise possibilities) and the full migration model involving three species and all pairwise possibilities. Marginal likelihoods and Bayes factors were estimated to calculate the probability for each model. Because Marginal likelihoods became stable much faster than convergence, we ran these tests using increments of 10 million steps until the marginal likelihoods stabilized. Then, the model with the highest probability according to the Bayes factors was run again in order to improve the convergence of migration parameters.
Isolation With Migration Model (IM)
Once we confirmed the most likely topology among Anastrepha species under an IM speciation model, we estimated demographic history parameters using the software IMa2 (Hey and Nielsen, 2004, 2007; Hey, 2010a), including the effective population sizes of the ancestral (θA1 and θA2) and the descendent populations (θ1, θ2, and θ3), bidirectional migration rates between population pairs (m1 and m2) and the divergence times (t0 and t1). We used the IMa2 program to simulate gene genealogies using a Markov Chain Monte Carlo (MCMC) approach to obtain posterior distributions of population demographic parameters (Nielsen and Wakeley, 2001; Hey and Nielsen, 2004). The IM model assumes that one panmitic ancestral population is first divided into two descendant populations and then one of these two populations divides again into another two populations, which may experience gene flow after each split (Wakeley and Hey, 1997; Hey, 2006, 2010a,b; Hey and Nielsen, 2007). The assumptions of neutrality and non-recombination for each locus were previously tested as described above, and then several preliminary runs were performed in order to test for different combinations of heating terms and number of MCMC chains until the overall MCMC simulation ended up with high swaps between adjacent chains (∼0.6–0.8) and good mixing. Finally, we run 100 Markov chains (a = 0.99 and b = 0.80) under the HKY model (Hasegawa et al., 1985). Prior values were established following recommendations in the IMa2 manual. The program was run indefinitely using the M mode for over 3,000,000 steps of burn-in until it reached good mixing based on the Effective Sample Sizes (ESS) and parameter trends during burn-in. Then, the program was run using the L mode for additional steps specifying details for the IM model and number of parameters to estimate. Because all parameters estimated by IMa2 are scaled by mutation rates, we estimated the mutation rate per year for each locus based on the split time reported for Tephritidae (∼36 million years) (Beverley and Wilson, 1984; Norrbom, 1994; Han and Ro, 2016) and a generation time of 0.11 years for Anastrepha (Celedonio-Hurtado et al., 1988; Joachim-Bravo et al., 2003) in order to convert the estimated parameters into demographic scales.
Results
Polymorphism and Genetic Structure Among Species
We obtained an average of 55 sequences for each gene across all species. These amplicons had an average of 384 sites after excluding introns for the 20 genes analyzed (Table 1). Between 14 and 119 polymorphic sites and 16 and 59 different haplotypes were detected on each gene. Even though we did not amplify individualized specimens, we sampled from a large enough pool per species (2N = 40) to make it more likely that sequences represent different DNA copies rather than a second amplification of the same DNA. The large number of individual haplotypes for the great majority of genes here studied, and almost absence of duplicated haplotypes was further evidence of that. In addition, haplotype networks for all genes here analyzed showed that singletons (unique haplotypes) are mostly located in tips (data not shown, available upon request) while more frequent haplotypes (with two or more copies) are generally located in network interiors, as expected by the coalescent theory (Castelloe and Templeton, 1994), suggesting that amplification was not biased by individual haplotypes from our pooling that would significantly interfere with our analyses. We failed to detect any evidence of recombination in any of the genes sampled. Most haplotypes had very low frequencies, being therefore restricted to a single species, which led to high haplotype diversity values (Table 1). All species showed similar nucleotide and haplotype diversity values (Table 1). Tajima’s D and Fu and Li’s D and F neutrality tests were not significant for most genes within individual species (Supplementary Table S5), which may be further evidence that there were no major biases introduced by the analysis of pooled data. On the other hand, Fu’s FS statistic was significantly negative for most genes within species (Supplementary Table S5), a pattern expected for population expansions.
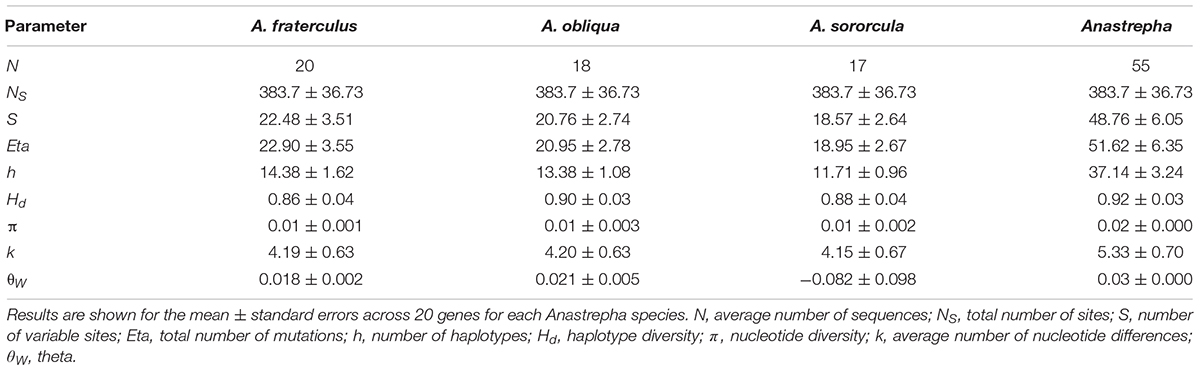
TABLE 1. Descriptive parameters for haplotype and nucleotide diversity.
The average divergence assessed by comparing the number of synonymous (Ks) and non-synonymous (Ka) substitutions suggested that the nuclear genes here investigated are not particularly conserved or subjected to heterogenic patterns of divergence driven by selection (Table 2). The genetic structure assessed by ΦST analyses show significant population differentiation among the three Anastrepha species here studied. Most loci showed significant genetic structure both overall as well as in most pairwise comparisons here performed, which showed wide amplitude of ΦST values ranging between 0 and 0.86 (Table 2).
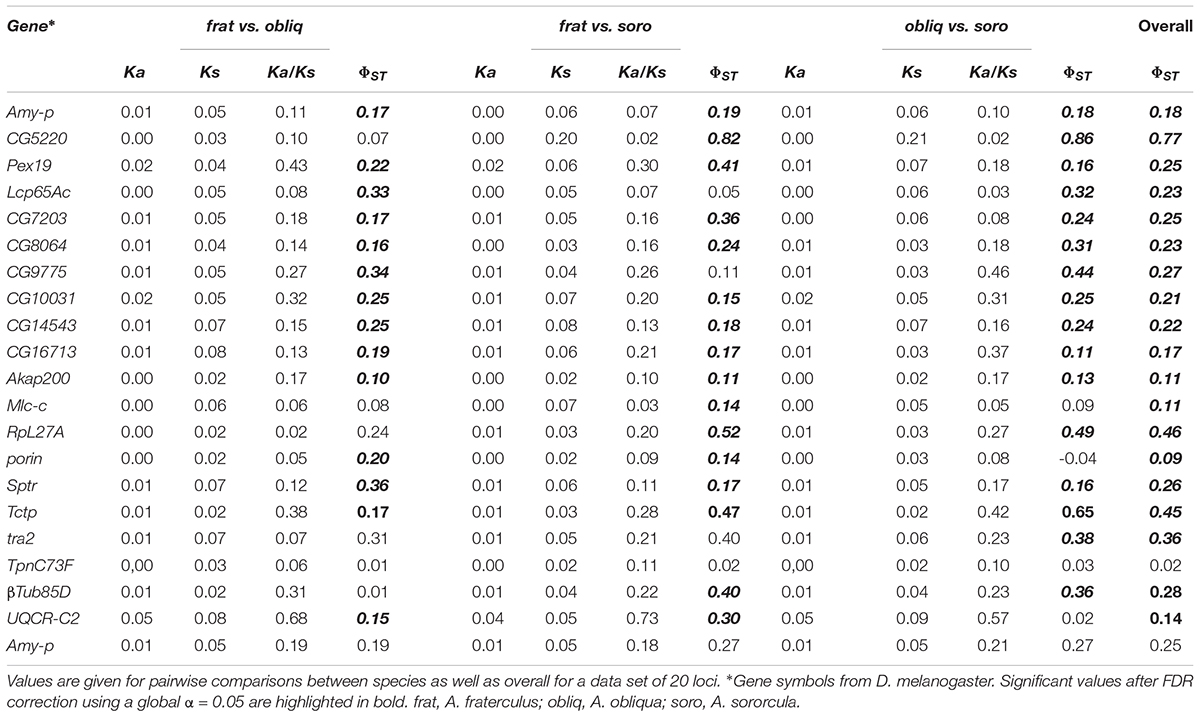
TABLE 2. Divergence among Anastrepha species at synonymous (Ks) and non-synonymous (Ka) sites, and genetic structure among species estimated by ΦST.
All genes here studied failed to show fixed differences that separated all three species when comparing species pairs (Table 3), though there were a few fixed differences that differentiates A. sororcula from each of the other two species. The number of shared polymorphisms showed substantial heterogeneity distribution that ranged between 0 and 22 across loci (Table 3), which was much higher than expected under random parallel mutation with an upper limit of 2.91 shared polymorphisms (Table 3).
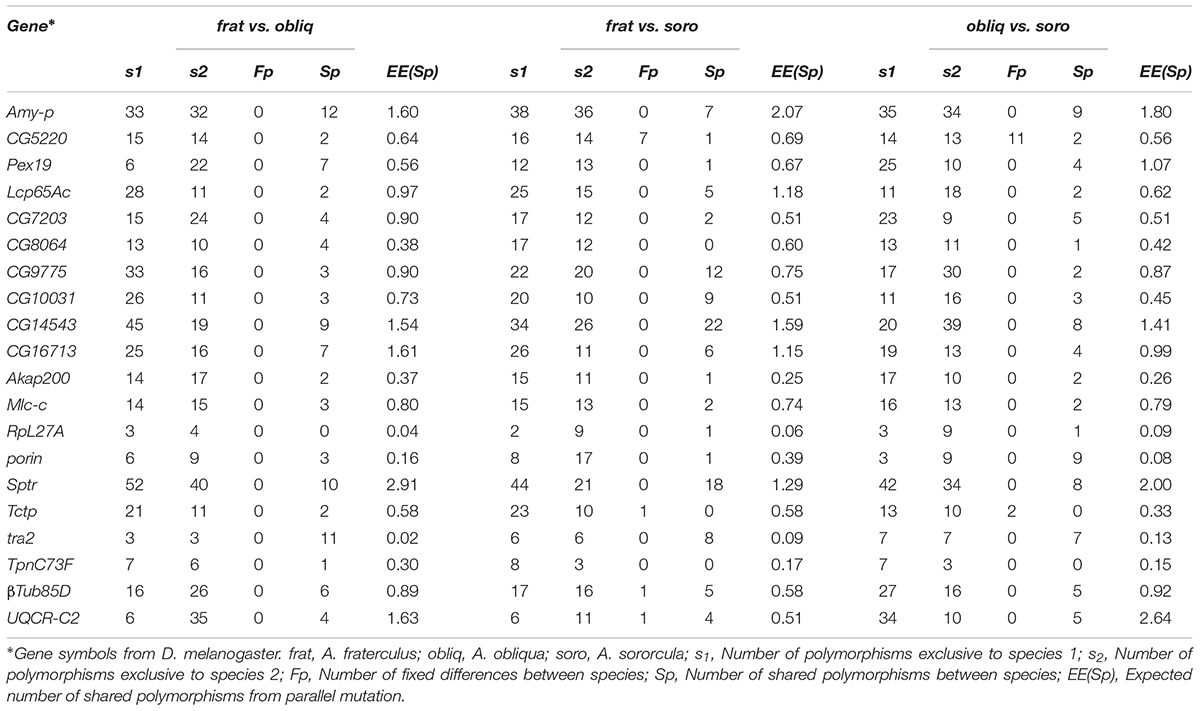
TABLE 3. Shared polymorphism and fixed differences between pairwise comparisons of Anastrepha species.
Phylogenetic Analyses
We performed a comparative analysis with ∗BEAST, which accounts for stochasticity in the coalescent process, and BUCKy, a method that accounts for discordance among gene trees without making any assumption about the cause of discordance, in order to disentangle the leading causes of discordance among gene trees. Several genes here studied showed evidence of substitution saturation following Xia (2013) when considering distant taxa but not for closely related Anastrepha species. Coalescent-based species tree inferences derived from analyses in ∗BEAST had well supported branches and resolved phylogenetic relationships (Figure 2). The topology of this inference is in most part compatible with the best phylogenetic inferences of the species here studied. The sole discordance is the position of D. willistoni as basal to the Drosophila genus, which would make the subgenus Sophophora paraphyletic with regards to true Drosophila (Seetharam and Stuart, 2013). On the other hand, the phylogenetic inferences among Tephritidae agree with what has been inferred elsewhere (Mazzon et al., 2010; Virgilio et al., 2015). More relevant to this study, particularly because there is evidence of saturation for more distantly related taxa, is that ∗BEAST resolved the relationship among the Anastrepha species here studied (Figure 2).

FIGURE 2. Density species tree obtained by ∗BEAST analysis using all 20 genes, showing posterior probabilities for each node. Branch lengths are proportional to genetic distance.
The lack of concordance among different gene topologies suggests great influence of introgressive hybridization or ILS, which was supported by results from BUCKy, since the concordance factors (CF < 0.1) in the PCT indicated many possible alternative topologies for each node (Figure 3). In addition, PT and PCT had different topologies, with the latter showing monophyletic lineages for Anastrepha species whereas the former indicating admixture between A. fraterculus and A. sororcula samples (Figure 3). In this case, the PT, in which Anastrepha species are reciprocally monophyletic, is more likely to represent the species tree (Larget et al., 2010). However, both trees showed reduced support for species branches separating Anastrepha species, suggesting hybridization among the three species and a very recent divergence, a signal that seems to be stronger between A. fraterculus and A. sororcula.
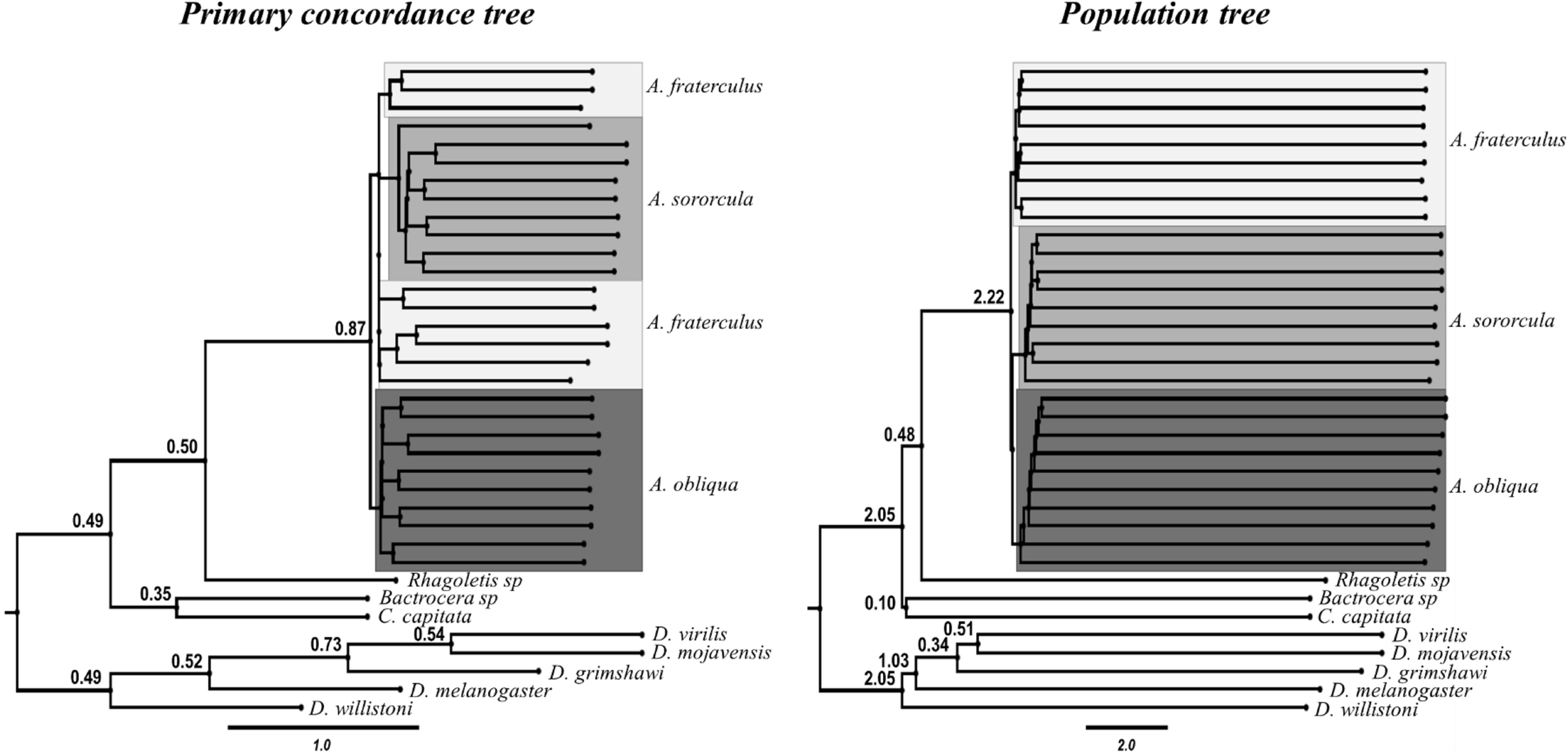
FIGURE 3. Primary concordance tree (PCT) and Population Tree (PT), obtained from the Bayesian concordance analysis in BUCKy. Posterior mean concordance factors (CFs) are displayed above branches. Branches with concordance factors below 0.1 are not shown except in major nodes. Topologies differed among trees, only the PT shows monophyly for the three species. Samples for each Anastrepha species are differentially highlighted. The PCF tree shows poorly supported relationships implying some level of admixture.
Only one gene, Tctp, failed to show a good fit to the multispecies coalescent assumptions in terms of coalescent likelihood or deep coalescent according to the LCWT and NDC statistics, respectively (Supplementary Table S6). ∗BEAST and BUCKy analyses performed after removing this locus showed results similar to what was obtained with its presence, indicating low support for each species monophyly and the PCT tree showing admixture between A. fraterculus and A. sororcula, so we retained the original phylogenetic analysis.
The phylogenetic inferences here performed identified three lineages in ∗BEAST and BUCKy analyses which completely corroborate our species identification. Furthermore, our analyses failed to find sublineages in A. fraterculus or in any of the other species, which suggests that either we failed to sample the other two morphotypes of A. fraterculus, which is not very likely considering our extensive geographical sampling, or, more likely, their close relationship requires a wider molecular set of markers to identify them. Because our analyses only identified three evolutionary lineages that followed the three Anastrepha species sampled, we conservatively treated A. fraterculus as sensu latu for all downstream analyses of speciation models and demographic history of Anastrepha species.
Model Selection and Demographic History
Given the challenges of elucidating the species topology in the presence of introgression, we performed a model selection approach in order to confirm the presence of introgression under alternative topologies in an ABC framework. A comparison of speciation models considering alternative topologies among Anastrepha species (Figure 1) found that the IM model fits our data better than the SI model independent of the topology as was evident from a higher posterior probability in all three cases (Table 4). All three comparisons showed high robustness based on the differentiation of simulation of pseudo-observed data sets under alternative models (Table 4). The Top2 model, which placed A. fraterculus and A. sororcula as sister species, showed the highest posterior density among models and better fitted our data in comparison with the two alternative topologies (Table 4) assuming an IM model. Though comparisons among topology models with IM showed moderate robustness, all comparisons consistently showed higher posterior probabilities for Top2 (P = 1.00) than alternative models, which was also consistent with the phylogenetic analyses and IMa2 results.
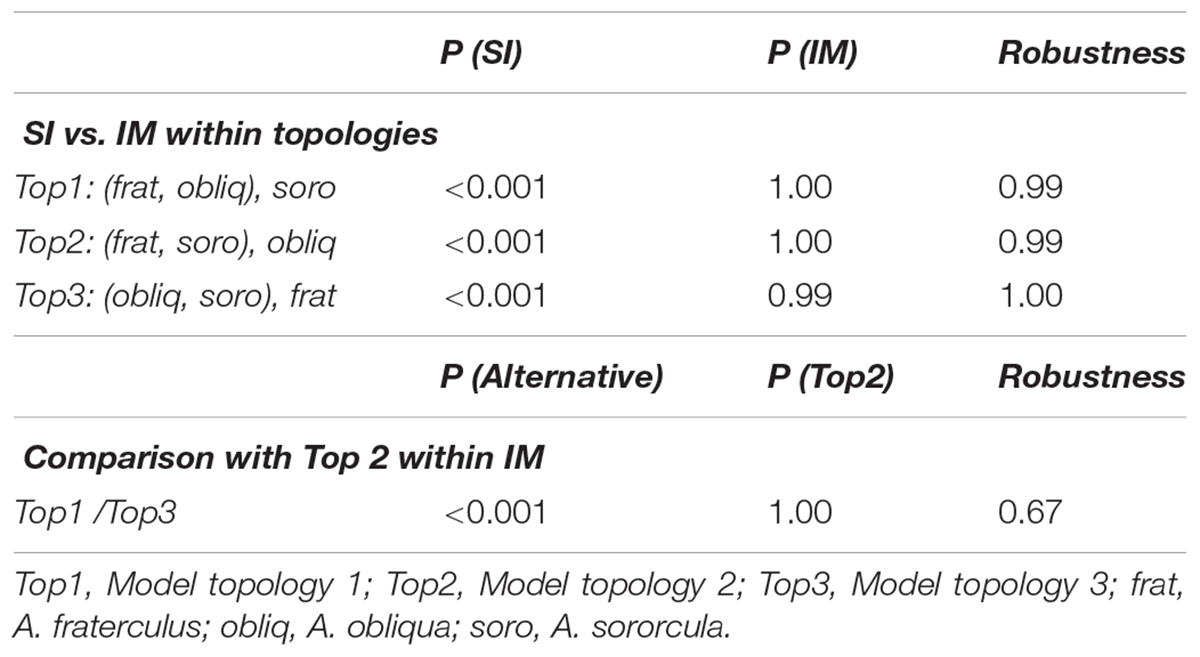
TABLE 4. Posterior probabilities calculated through Bayes factors for speciation models (SI and IM) as well as topology models tested by ABC analysis.
Once the topology and introgression among Anastrepha species was confirmed under an IM framework, specific model details, including direction of migration and demographic parameters for the fraterculus complex were estimated using MIGRATE and IMa2 software. Simulations performed in MIGRATE that tested different migration models between paired species supported the full migration model involving bidirectional migration for all pairwise possibilities (Table 5). An IM model simulated in IMa2 obtained both convergence and posterior probabilities that showed clear peaks for all demographic parameters estimated, suggesting that our data contained sufficient information to estimate that A. fraterculus and A. sororcula diverged ∼1.3 MYA (CI95% = 1.1 – 1.5 MYA), while their common ancestor diverged from A. obliqua ∼2.6 MYA (CI95% = 2.05 - 3.21 MYA) (Figure 4).

TABLE 5. Log marginal likelihoods and posterior probabilities calculated through Bayes factors for migration models tested by MIGRATE using data set of 20 loci.
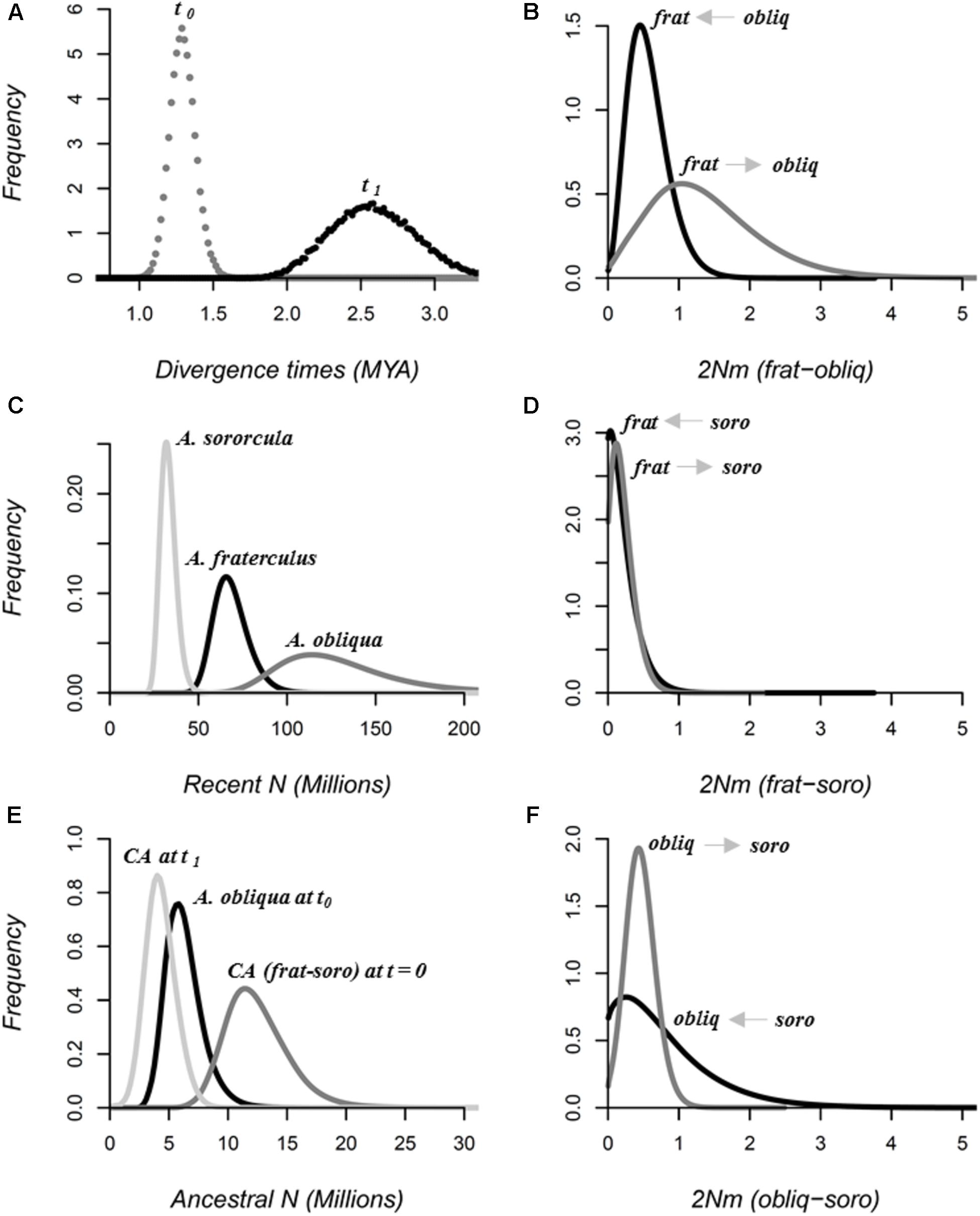
FIGURE 4. Marginal posterior probability distributions for speciation parameters in demographic scales estimated by simulations of the IM model using 20 loci in IMa2 software. (A) Divergence times in MYA, between A. fraterculus and A. sororcula (t0), and between these two species and A. obliqua (t1). Effective population sizes in millions of individuals (N) for (B) each of the species (Recent N) as well as (C) their common ancestors (CA, Ancestral N). Bidirectional migration rates (2Nm) for each species pair: (D) A. fraterculus vs. A. obliqua (frat-obliq), (E) A. fraterculus vs. A. sororcula (frat-soro) and (F) A. obliqua vs. A. sororcula (obliq-soro). Direction of migration is indicated in the graphics. All parameters are scaled using mutation rates per locus per year estimated using the split time reported for Tephritidae (∼ 36 million years).
All species showed large effective population sizes, with values over 30 million individuals. A. obliqua has the highest estimation with 114.0∗106 individuals (CI95% = 79.7 – 200.3∗106 individuals), followed by A. fraterculus with 65.5∗106 (CI95% = 52.0 – 90.5∗106 individuals) and then A. sororcula with 31.9 ∗106 (CI95% = 25.5 – 42.9∗106 individuals). The big difference between recent and ancestral population sizes suggests population expansion for all three species (Figure 4), since Ne of A. fraterculus/A. sororcula common ancestor (t0) was 11.6∗106 individuals (CI95% = 8.1 – 18.4∗106 individuals), about 3–6 times smaller than recent values, while this value was 8–30 times smaller for the common ancestor of all three species (4.0∗106; CI95% = 1.9 – 7.0∗106 individuals) (Figure 4).
Though all species pairs experienced migration, the direction of migration was not symmetrical. A. fraterculus and A. obliqua showed the highest migration rates with around 1.04 migrants per generation (CI95% = 0.20 – 3.15 migrants per generation) from A. obliqua into A. fraterculus and 0.45 (CI95% = 0.13 – 1.23 migrants per generation) in the opposite direction (Figure 4). Gene flow from A. obliqua into A. sororcula was 0.43 migrants per generation (CI95% = 0.09 – 0.91 migrants per generation) while it was 0.25 migrants per generation (CI95% = 0.04 – 2.53 migrants per generation) in the opposite direction (Figure 4). A. fraterculus and A. sororcula on the other hand showed the lowest gene flow with around 0.12 migrants per generation (CI95% = 0.01 – 0.60 migrants per generation) from A. fraterculus into A. sororcula and no significant gene flow detected in the opposite direction (Figure 4).
Since an IM with population expansion better explained the evolution of the Anastrepha species, we then used MIGRATE and IMa2 results as priors for a second ABC analysis to investigate whether the introgression and population expansion have been favored by the recent burst of agriculture activities. Four models of temporal patterns of demographic history (CmigCexp, CmigRexp, RmigCexp and RmigRexp) were tested, considering around 3,000 Anastrepha generations as a temporal point of comparison for recent against continuous patterns of gene flow and population expansion (Figure 1). Although the posterior probability of CmigCexp model was greater than RmigCexp, the two models with recent population expansion CmigRexp and RmigRexp outperformed CmigCexp (Table 6). The CmigRexp model showed the best fit to our data, suggesting that Anastrepha species have experienced gene flow since they split from their common ancestor, and had a population size expansion in the last 300 years (Table 6).
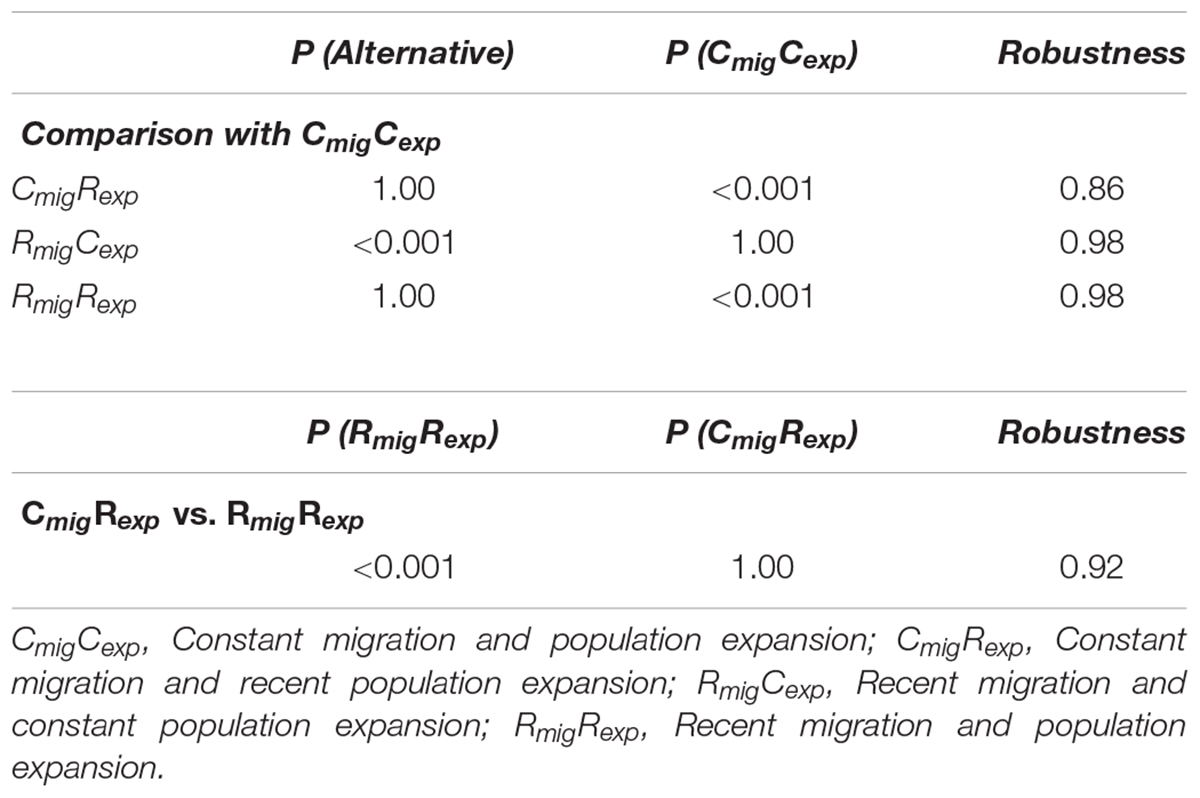
TABLE 6. Posterior probabilities calculated through Bayes factors for speciation models comparing temporal patterns of population expansion and migration (recent vs. continuous) tested by ABC analysis.
Discussion
We used a data set of 20 nuclear genes to study the evolutionary relationships among three closely related species of the Anastrepha fraterculus group. Multilocus analyses combined populational, phylogenetic and model selection approaches to reveal that these species retain independent evolutionary lineages despite the occurrence of substantial levels of gene flow. Most neutrality tests failed to show significant departures from neutral expectations, with the exception of significant Fu’s Fs tests that may reflect a general pattern compatible with a recent population expansion, which was supported by our IMa2 and ABC simulations.
Although we recognize that A. fraterculus is a species complex in itself and our sampling might include representatives from some of the three cryptic species identified in Brazil (Selivon and Perondini, 1998; Selivon et al., 1999, 2005; Hernández-Ortiz et al., 2012; Hendrichs et al., 2015; Manni et al., 2015; Roriz et al., 2017), we failed to find genetic evidence for such lineages in our analyses. Thus, we failed to find significant departures from neutrality other than the suggested population size expansions. Our phylogenetic inference performed in BUCKy using the complete gene set identified A. fraterculus, A. obliqua and A. sororcula as different lineages but failed to find sublineages that would be compatible with putative cryptic species. Because this analysis performs a posteriori clustering of samples considering only the phylogenetic signal (based on Concordance Factors) derived from all genes sequenced in a coalescent framework (Larget et al., 2010), the absence of sublineages in A. fraterculus is not impacted by our decision to treat A. fraterculus as sensu latu, since the program does not use a priori species information. Furthermore, all downstream speciation and demographic history simulations failed to show population size heterogeneities that might suggest mixed or heterogeneous sampling. Rather, the analyses performed in Migrate and IMa2 consistently showed clear convergence peaks for the three species. This could either indicate that there is no evidence of a genetic differentiation among the three Brazilian A. fraterculus cryptic species, or that the differentiation among cryptic species is too recent and limited in the genome to be identified by our sampling. Considering what has been described in the literature about several differences across the species’ distribution, it is possible that only by considering an integrative taxonomy approach (Hendrichs et al., 2015; Dias et al., 2016; Schutze et al., 2017) which would require much more than the limited data here used might we be able to correctly identify these species.
The three species here considered showed a large number of very low frequency haplotypes, resulting in several species-specific haplotypes and high levels of shared polymorphic variation. One of the processes that might produce excessive common variants between species is convergence by parallel mutation (Clark, 1997; Kliman et al., 2000; Hedrick, 2013), but we were able to reject this hypothesis, since the number of shared polymorphisms for the majority of genes here investigated was higher than that estimated assuming only parallel mutation. Shared variation could also be caused by differential retention of ancestral polymorphisms due to incomplete lineage sorting (Clark, 1997; Maddison, 1997; Holder et al., 2001) or introgression (Harrison and Larson, 2014). There are examples of closely related species in Drosophila that speciated without subsequent gene flow which show very low levels of interspecific polymorphism (Kliman et al., 2000), while species experiencing introgression show high levels of shared polymorphisms as well as low levels of fixed polymorphisms among species (Bachtrog et al., 2006; Herrig et al., 2014; Beck et al., 2015). We investigated the relative relevance of ILS and introgression to explain the genetic patterns of shared variation across Anastrepha species by combining Bayesian phylogenetic analyses as well as inferring the demographic history of Anastrepha species under a model selection framework.
Results from BUCKy clearly suggest that introgression is relevant to explain disagreement among gene trees, which violates the multispecies coalescent model implemented in ∗BEAST, since this method considers that the discrepancies among different topologies is solely caused by ILS (Heled et al., 2013). However, this model was mainly used to test other coalescent assumptions such as coalescent likelihood and deep coalescent (Reid et al., 2014; Gruenstaeudl et al., 2015), which were rejected, suggesting that ILS (Larget et al., 2010) and introgression are more likely explanations for the discrepancies among gene trees. Since these two scenarios are difficult to differentiate, we implemented a model selection approach in order to investigate different models of speciation, topologies and levels of introgression, by testing each scenario using an ABC framework. These results showed strong evidence of Anastrepha species evolving under an Isolation with Migration model (IM) that favored a more closely relationship between A. fraterculus and A. sororcula when compared to A. obliqua. This topology is in agreement with data based on morphology (Zuchi, 1979; Norrbom et al., 1999), behavioral and host preferences (Aluja, 1994; Aluja et al., 1999; Sivinski et al., 1999; Raga et al., 2011), cytogenetics, allozymes (Selivon, 1996) as well as mtDNA data (McPheron et al., 1999; Smith-Caldas et al., 2001). Despite this potential relationship, this is the first time that the topology in this set of species is confirmed by a multilocus approach in the face of introgression. These results are also consistent with our simulations in MIGRATE and IMa2 which favored models that considered migration involving all species pairs. These three model approximations with no assumptions consistently identified the IM model as the best scenario to explain molecular variation of Anastrepha species, highlighting the important role of introgression in shaping the evolution of these species.
Evidence of introgression among Anastrepha species is not surprising since reciprocal hybrids have been produced in laboratory, even though there is a Haldane’s rule when crossing A. fraterculus or A. sororcula females and A. obliqua males (Selivon et al., 1999; Santos et al., 2001; Rull et al., 2017). This is why introgression between A. fraterculus and A. obliqua has been recently proposed as a potential cause of some discrepancies in the relationships among A. obliqua populations (Scally et al., 2016). However, introgression patterns here observed are more compatible with pre-zygotic rather than post-zygotic barriers since reproductive experiments reported have shown higher reproductive isolation between A. fraterculus/A. sororcula and A. obliqua than between A. fraterculus and A. sororcula, a pattern that apparently is due to phylogenetic distance (Santos et al., 2001). We inferred lower gene flow between the latter species pair, which suggests pre-zygotic isolation driving speciation between A. fraterculus and A. sororcula, though this could also be influenced by higher levels of ILS in these more closely related species.
Our results have indicated presence of gene flow involving all species pairs since they split from their common ancestor ∼2.6 MYA and our estimations of substantial levels of gene flow suggest that their current overlapping distributions may have been a common feature across their history. Considering the wide array of molecular functions associated with the genes here studied, it is hard to envision a common adaptive scenario that would maintain these introgressed loci by selection. Genetic drift on the other hand is a demographic process with a genome-wide impact, which can be differentiated from selection when sets of genes follow particular patterns given a geographic context (Bachtrog et al., 2006; Harrison and Larson, 2014). The lack of a genetic map, or genome information on these species, prevents us from inferring a genome-wide detection of introgression, but the high effective population sizes inferred, together with the evidence for continuous gene flow, favors an interpretation of introgression influenced by drift, which may have been favored by their natural polyphagia and not necessarily natural or sexual selection.
It has been suggested that the fraterculus complex is likely to be in expansion (Vaníèková et al., 2015). That suggestion has been corroborated by our results since we obtained high effective population sizes as well as evidence for population size expansion, which is more likely a consequence of anthropic activities on crops and predators during the recent burst of population expansion in Brazil, as indicated from our simulations in IMa2 and corroborated by an ABC approach.
Although we failed to detect evidence for multiple lineages in A. fraterculus in this study, we consider more likely their differentiation may be still too incipient for the sampling and markers here used to detect them. We should point out though that the potential existence of cryptic species in A. fraterculus does not influence our findings, since most of the demographic history here described probably antedated the divergence of putative cryptic species, e.g., the divergence time estimates between A. fraterculus and the other two species, with the exception of the recent population size expansion. Thus, the data here analyzed enabled the estimation of ancient population sizes and gene flow, which indicates that the introgression patterns here described seem to be a common phenomenon along the demographic history of these species since they split from their common ancestor ∼2.6 MYA and is not a consequence of the recent population size expansion these species experienced. These results should be considered carefully though, particularly for the recent population expansion detected because the past history is the same for the lineages, but the ongoing speciation indicates that their current population sizes and consequently their potential expansion could be species specific.
Despite the lack of information on the multilocus structure in our data set, our pooling approach increased our capacity to sequence a larger multilocus set. This allowed us to perform extensive and unbiased Bayesian genealogy sampler methods (Cruickshank and Hahn, 2014; Hey et al., 2015), in order to elucidate the demographic history of the Anastrepha fraterculus species complex. Our method has proved to be highly effective as was evidenced from the analyses of haplotype networks, which followed coalescent expectations rather than a potential pooling bias, as well as from the consistency of several methods pointing to the same final conclusions. Furthermore, these results are in agreement with what has been suggested elsewhere from multiple disciplines (McPheron et al., 1999; Selivon et al., 1999; Santos et al., 2001; Smith-Caldas et al., 2001; Vaníèková et al., 2015; Scally et al., 2016; Rull et al., 2017).
Our results showed that the species of Anastrepha here investigated have been evolving as distinct lineages despite incongruences across individual gene tree topologies which are more likely a consequence of introgression rather than ILS. These species may have had overlapping distributions since their differentiation, which may have been favored by their natural polyphagia. We also corroborated that the species in the fraterculus group have experienced a recent population expansion driven by anthropic activities of the past 300 years in Brazil. The combination of recent divergence and substantial ongoing gene flow seems to produce a situation similar to what has been described for other Tephritids (Feder et al., 1999; Xie et al., 2007; Michel et al., 2010; Arcella et al., 2015). A speciation with migration, in which few genes diverge whereas the remainder genome homogenizes by gene flow, the so called islands of speciation (Feder and Nosil, 2010; Michel et al., 2010). Our findings then suggest that portions of the genome across species in the fraterculus group may be evolving as a common entity, which highlights the importance of considering the whole complex not only to understand their evolution, but also for pest management of these fruit flies, rather that treat them as individual species. In this scenario, species identification would demand efforts on integrative taxonomy applying multiple lines of evidence on the same specimens, integrating molecular with phenotypic approaches across the entire geographic range, as proposed by Schutze et al. (2017).
Data Availability Statement
Data for this study are available at GenBank genetic sequence database (https://www.ncbi.nlm.nih.gov/genbank/) (Supplementary Table S4).
Author Contributions
RdB conceived and designed the project. AL, AN, FF, and IS performed the majority of the laboratory work. AL performed statistical analyses for individual genes. FD performed multilocus and speciation modeling analyses. All the authors participated in the interpretation of results. FD and RdB wrote the manuscript and all authors read and approved the final manuscript.
Funding
This project was supported by FAPESP (Fundaçao do Amparo à Pesquisa do Estado de São Paulo) Grant #2010/20455-4 and the Science without Borders program at CAPES (Processo PVE 056/2013). CAPES SWB supported a post-doctorate fellowship to FD.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank FAPESP (Fundaçao do Amparo à Pesquisa do Estado de São Paulo) Grant #2010/20455-4 and the Science without Borders program at CAPES (Processo PVE 056/2013) for financial support. We would also like to thank Drs. Roberto Zucchi and Keiko Uramoto, from University of São Paulo, for help in identifying several of specimens here used.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2018.00359/full#supplementary-material
Footnotes
References
Aluja, M. (1994). Bionomics and management of Anastrepha. Annu. Rev. Entomol. 39, 155–178. doi: 10.1146/annurev.en.39.010194.001103
Aluja, M., Piñero, J., Jácome, I., Díaz-Fleischer, F., and Sivinski, J. (1999). “Behavior of flies in the genus Anastrepha (Trypetinae: Toxotrypanini),” in Fruit Flies (Tephritidae): Phylogeny and Evolution of Behavior, eds M. Aluja and A. L. Norrbom (Boca Ratón, FL: CRC Press), 375–406. doi: 10.1201/9781420074468
Ané, C., Larget, B., Baum, D. A., Smith, S. D., and Rokas, A. (2007). Bayesian estimation of concordance among gene trees. Mol. Biol. Evol. 24, 412–426. doi: 10.1093/molbev/msl170
Arcella, T., Hood, G. R., Powell, T. H. Q., Sim, S. B., Yee, W. L., Schwarz, D., et al. (2015). Hybridization and the spread of the apple maggot fly, Rhagoletis pomonella (Diptera: Tephritidae), in the northwestern United States. Evol. Appl. 8, 834–846. doi: 10.1111/eva.12298
Bachtrog, D., Thornton, K., Clark, A., and Andolfatto, P. (2006). Extensive introgression of mitochondrial DNA relative to nuclear genes in the Drosophila yakuba species group. Evolution 60, 292–302. doi: 10.1111/j.0014-3820.2006.tb01107.x
Barr, N., Cui, L., and McPheron, B. (2005). Molecular systematics of nuclear gene period in genus Anastrepha (Tephritidae). Ann. Entomol. Soc. Am. 98, 173–180. doi: 10.1603/0013-87462005098[0173:MSONGP]2.0.CO;2
Beaumont, M. A., Zhang, W., and Balding, D. J. (2002). Approximate bayesian computation in population genetics. Genetics 162, 2025–2035.
Beck, E. A., Thompson, A. C., Sharbrough, J., Brud, E., and Llopart, A. (2015). Gene flow between Drosophila yakuba and Drosophila santomea in subunit V of cytochrome c oxidase: a potential case of cytonuclear cointrogression. Evolution 69, 1973–1986. doi: 10.1111/evo.12718
Beerli, P., and Palczewski, M. (2010). Unified framework to evaluate panmixia and migration direction among multiple sampling locations. Genetics 185, 313–326. doi: 10.1534/genetics.109.112532
Beltran, M., Jiggins, C., Bull, V., McMillan, W., Mallet, J., and Bermingham, E. (2002). Phylogenetic discordance at the species boundary: gene genealogies in Heliconius butterflies. Mol. Biol. Evol. 19, 2176–2190. doi: 10.1093/oxfordjournals.molbev.a004042
Beverley, S. M., and Wilson, A. C. (1984). Molecular evolution in Drosophila and the higher Diptera. II. A time scale for fly evolution. J. Mol. Evol. 21, 1–13. doi: 10.1007/BF02100622
Buckley, T. R., Cordeiro, M., Marshall, D. C., and Simon, C. (2006). Differentiating between hypotheses of lineage sorting and introgression in New Zealand alpine cicadas (Maoricicada Dugdale). Syst. Biol. 55, 411–425. doi: 10.1080/10635150600697283
Carstens, B. C., and Knowles, L. L. (2007). Estimating species phylogeny from gene-tree probabilities despite incomplete lineage sorting: an example from Melanoplus grasshoppers. Syst. Biol. 56, 400–411. doi: 10.1080/10635150701405560
Castelloe, J., and Templeton, A. R. (1994). Root probabilities for intraspecific gene trees under neutral coalescent theory. Mol. Phylogenet. Evol. 3, 102–113. doi: 10.1006/mpev.1994.1013
Celedonio-Hurtado, H., Liedo, P., Aluja, M., Guillen, J., Berrigan, D., and Carey, J. (1988). Demography of Anastrepha ludens, A. obliqua and A. serpentina (Diptera: Tephritidae) in Mexico. Florida Entomol. 71, 111–120. doi: 10.2307/3495357
Chomczynski, P., and Sacchi, N. (1987). Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol chloroform extraction. Anal. Biochem. 162, 156–159. doi: 10.1006/abio.1987.9999
Clark, A. G. (1997). Neutral behavior of shared polymorphism. Proc. Natl. Acad. Sci. U.S.A. 94, 7730–7734. doi: 10.1073/pnas.94.15.7730
Clarkson, C. S., Weetman, D., Essandoh, J., Yawson, A. E., Maslen, G., Manske, M., et al. (2014). Adaptive introgression between Anopheles sibling species eliminates a major genomic island but not reproductive isolation. Nat. Commun. 5:4248. doi: 10.1038/ncomms5248
Cline, J., Braman, J., and Hogrefe, H. (1996). PCR fidelity of pfu DNA polymerase and other thermostable DNA polymerases. Nucl. Acids Res. 24, 3546–3551. doi: 10.1093/nar/24.18.3546
Cruickshank, T. E., and Hahn, M. W. (2014). Reanalysis suggests that genomic islands of speciation are due to reduced diversity, not reduced gene flow. Mol. Ecol. 23, 3133–3157. doi: 10.1111/mec.12796
Dias, V. S., Silva, J. G., Lima, K. M., Petitinga, C. S. C. D., Hernández-Ortiz, V., Laumann, R. A., et al. (2016). An integrative multidisciplinary approach to understanding cryptic divergence in Brazilian species of the Anastrepha fraterculus complex (Diptera: Tephritidae). Biol. J. Linn. Soc. 117, 725–746. doi: 10.1111/bij.12712
Drummond, A., and Rambaut, A. (2007). BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7:214. doi: 10.1186/1471-2148-7-214
Excoffier, L., and Foll, M. (2011). Fastsimcoal: a continuous-time coalescent simulator of genomic diversity under arbitrarily complex evolutionary scenarios. Bioinformatics 27, 1332–1334. doi: 10.1093/bioinformatics/btr124
Excoffier, L., and Lischer, H. E. L. (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567. doi: 10.1111/j.1755-0998.2010.02847.x
Excoffier, L., Smouse, P. E., and Quattro, J. M. (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131, 479–491.
Feder, J. L., and Nosil, P. (2010). The efficacy of divergence hitchhiking in generating genomic islands during ecological speciation. Evolution 64, 1729–1747. doi: 10.1111/j.1558-5646.20
Feder, J. L., Williams, S. M., Berlocher, S. H., McPheron, B. A., and Bush, G. L. (1999). The population genetics of the apple maggot fly, Rhagoletis pomonella and the snowberry maggot, R. zephyria: implications for models of sympatric speciation. Entomol. Exp. Appl. 90, 9–24. doi: 10.1046/j.1570-7458.1999.00419.x
Fu, Y.-X. (1997). Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147, 915–925.
Fu, Y.-X., and Li, W.-H. (1993). Statistical tests of neutrality of mutations. Genetics 133, 693–709.
Furtado, C. (1959). Formação Econômica do Brasil, 32nd Edn. Rio de Janeiro: Companhia Editora Naciona. doi: 10.1590/S0103-40142001000300025
Garrigan, D., Kingan, S. B., Geneva, A. J., Andolfatto, P., Clark, A. G., Thornton, K. R., et al. (2012). Genome sequencing reveals complex speciation in the Drosophila simulans clade. Genome Res. 22, 1499–1511. doi: 10.1101/gr.130922.111.22
Giraldo, N., Salazar, C., Jiggins, C. D., Bermingham, E., and Linares, M. (2008). Two sisters in the same dress: heliconius cryptic species. BMC Evol. Biol. 8:324. doi: 10.1186/1471-2148-8-324
Gonçalves, V. R., Sobrinho, I. S., Malagó-Jr, W., Henrique-Silva, F., and de Brito, R. A. (2013). Transcriptome analysis of female reproductive tissues of Anastrepha obliqua and molecular evolution of eggshell proteins in the fraterculus group. Insect Mol. Biol. 22, 551–561. doi: 10.1111/imb.12045
Gruenstaeudl, M., Reid, N. M., Wheeler, G. L., and Carstens, B. C. (2015). Posterior predictive checks of coalescent models: P2C2M, an R package. Mol. Ecol. Resour. 16, 193–205. doi: 10.1111/1755-0998.12435
Hall, T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41, 95–98.
Han, H. Y., and Ro, K. E. (2016). Molecular phylogeny of the superfamily Tephritoidea (Insecta: Diptera) reanalysed based on expanded taxon sampling and sequence data. J. Zool. Syst. Evol. Res. 54, 276–288. doi: 10.1111/jzs.12139
Harrison, R. G., and Larson, E. L. (2014). Hybridization, introgression, and the nature of species boundaries. J. Hered. 105, 795–809. doi: 10.1093/jhered/esu033
Hasegawa, M., Kishino, H., and Yano, T-a. (1985). Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 22, 160–174. doi: 10.1007/BF02101694
Hedrick, P. W. (2013). Adaptive introgression in animals: examples and comparison to new mutation and standing variation as sources of adaptive variation. Mol. Ecol. 22, 4606–4618. doi: 10.1111/mec.12415
Heled, J., Bryant, D., and Drummond, A. J. (2013). Simulating gene trees under the multispecies coalescent and time-dependent migration. BMC Evol. Biol. 13:44. doi: 10.1186/1471-2148-13-44
Hendrichs, J., Teresa Vera, M., De Meye, M., and Clarke, A. R. (2015). Resolving cryptic species complexes of major tephritid pests. Zookeys 540, 5–39. doi: 10.3897/zookeys.540.9656
Hernández-Ortiz, V., Bartolucci, A. F., Morales-Valles, P., Frías, D., and Selivon, D. (2012). Cryptic species of the Anastrepha fraterculus complex (Diptera: Tephritidae): a multivariate approach for the recognition of South American morphotypes. Ann. Entomol. Soc. Am. 105, 305–318. doi: 10.1603/AN11123
Hernández-Ortiz, V., Canal, N. A., Tigrero Salas, J. O., Ruíz-Hurtado, F. M., and Dzul-Cauich, J. F. (2015). Taxonomy and phenotypic relationships of the Anastrepha fraterculus complex in the mesoamerican and pacific neotropical dominions (Diptera, tephritidae). Zookeys 540, 95–124. doi: 10.3897/zookeys.540.6027
Hernández-Ortiz, V., Gómez-Anaya, J. A., Sánchez, A., McPheron, B. A., and Aluja, M. (2004). Morphometric analysis of Mexican and South American populations of the Anastrepha fraterculus complex (Diptera: Tephritidae) and recognition of a distinct Mexican morphotype. Bull. Entomol. Res. 94, 487–499. doi: 10.1079/BER2004325
Herrig, D. K., Modrick, A. J., Brud, E., and Llopart, A. (2014). Introgression in the Drosophila subobscura-D. madeirensis sister species: fvidence of gene flow in nuclear genes despite mitochondrial differentiation. Evolution 68, 705–719. doi: 10.1111/evo.12295
Hey, J. (2006). Recent advances in assessing gene flow between diverging populations and species. Curr. Opin. Genet. Dev. 16, 592–596. doi: 10.1016/j.gde.2006.10.005
Hey, J. (2010a). Isolation with migration models for more than two populations. Mol. Biol. Evol. 27, 905–920. doi: 10.1093/molbev/msp296
Hey, J. (2010b). The divergence of Chimpanzee species and subspecies as revealed in multipopulation isolation-with-migration analyses. Mol. Biol. Evol. 27, 921–933. doi: 10.1093/molbev/msp298
Hey, J., Chung, Y., and Sethuraman, A. (2015). On the occurance of false positives in tests of migration under an isolation with migration model. Mol. Ecol. 24, 5078–5083. doi: 10.1007/128
Hey, J., and Nielsen, R. (2004). Multilocus methods for estimating population sizes, migration rates and divergence time, with applications to the divergence of Drosophila pseudoobscura and D. persimilis. Genetics 167, 747–760. doi: 10.1534/genetics.103.024182
Hey, J., and Nielsen, R. (2007). Integration within the Felsenstein equation for improved Markov chain Monte Carlo methods in population genetics. Proc. Natl. Acad. Sci. U.S.A. 104, 2785–2790. doi: 10.1073/pnas.0611164104
Holder, M. T., Anderson, J. A., and Holloway, A. K. (2001). Difficulties in detecting hybridization. Syst. Biol. 50, 978–982. doi: 10.1080/106351501753462911
Huang, H., Sukumaran, J., Smith, S. A., and Knowles, L. L. (2017). Cause of gene tree discord? Distinguishing incomplete lineage sorting and lateral gene transfer in phylogenetics. PeerJ Prepr 5:e3489v1. doi: 10.7287/PEERJ.PREPRINTS.3489V1
Huelsenbeck, J. P., and Ronquist, F. (2001). MRBAYES: bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755. doi: 10.1093/bioinformatics/17.8.754
Joachim-Bravo, I. S., Magalhaes, T. C., Silva Neto, A. M., Guimaraes, A. N., and Nascimento, A. S. (2003). Longevity and fecundity of four species of Anastrepha (Diptera: Tephritidae). Neotrop. Entomol. 32, 543–549. doi: 10.1590/S1519-566X2003000400003
Juárez, M. L., Devescovi, F., Bøízová, R., Bachmann, G., Segura, D. F., Kalinová, B., et al. (2015). Evaluating mating compatibility within fruit fly cryptic species complexes and the potential role of sex pheromones in pre-mating isolation. Zookeys 540, 125–155. doi: 10.3897/zookeys.540.6133
Kamneva, O. K., and Rosenberg, N. A. (2017). Simulation-based evaluation of hybridization network reconstruction methods in the presence of incomplete lineage sorting. Evol. Bioinforma 13:1176934317691935. doi: 10.1177/1176934317691935
Kliman, R. M., Andolfatto, P., Coyne, J. A., Depaulis, F., Kreitman, M., Berry, A. J., et al. (2000). The population genetics of the origin and divergence of the Drosophila simulans complex species. Genetics 156, 1913–1931.
Kosakovsky-Pond, S. L., Posada, D., Gravenor, M. B., Woelk, C. H., and Frost, S. D. W. (2006a). Automated phylogenetic detection of recombination using a genetic algorithm. Mol. Biol. Evol. 23, 1891–1901. doi: 10.1093/molbev/msl051
Kosakovsky-Pond, S. L., Posada, D., Gravenor, M. B., Woelk, C. H., and Frost, S. D. W. (2006b). GARD: a genetic algorithm for recombination detection. Bioinformatics 22, 3096–3098. doi: 10.1093/bioinformatics/btl474
Kulathinal, R. J., Stevison, L. S., and Noor, M. A F. (2009). The genomics of speciation in Drosophila: diversity, divergence, and introgression estimated using low-coverage genome sequencing. PLoS Genet. 5:e1000550. doi: 10.1371/journal.pgen.1000550
Larget, B. R., Kotha, S. K., Dewey, C. N., and Ané, C. (2010). BUCKy: gene tree/species tree reconciliation with Concordance analysis. Bioinformatics 26, 2910–2911. doi: 10.1093/bioinformatics/btq539
Leuenberger, C., and Wegmann, D. (2010). Bayesian computation and model selection without likelihoods. Genetics 184, 243–252. doi: 10.1534/genetics.109.109058
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Lis, J. T., and Schleif, R. (1975). Size fractionation of double-stranded DNA by precipitation with polyethylene glycol. Nucleic Acids Res. 2, 383–389. doi: 10.1093/nar/2.3.383
Llopart, A., Lachaise, D., and Coyne, J. A. (2005). Multilocus analysis of introgression between two sympatric sister species of Drosophila: Drosophila yakuba and D. santomea. Genetics 171, 197–210. doi: 10.1534/genetics.104.033597
Machado, C. A., Kliman, R. M., Markert, J. A., and Hey, J. (2002). Inferring the history of speciation from multilocus DNA sequence Data: the case of Drosophila pseudoobscura and close relatives. Mol. Biol. Evol. 19, 472–488. doi: 10.1093/oxfordjournals.molbev.a004103
Maddison, W. P., and Knowles, L. L. (2006). Inferring phylogeny despite incomplete lineage sorting. Syst. Biol. 55, 21–30. doi: 10.1080/10635150500354928
Mallet, J., Beltrán, M., Neukirchen, W., and Linares, M. (2007). Natural hybridization in heliconiine butterflies: the species boundary as a continuum. BMC Evol. Biol. 7:28. doi: 10.1186/1471-2148-7-28
Manni, M., Lima, K. M., Guglielmino, C. R., Lanzavecchia, S. B., Juri, M., Vera, T., et al. (2015). Relevant genetic differentiation among brazilian populations of Anastrepha fraterculus (Diptera, tephritidae). Zookeys 540, 157–173. doi: 10.3897/zookeys.540.6713
Marsden, C. D., Lee, Y., Nieman, C. C., Sanford, M. R., Dinis, J., Martins, C., et al. (2011). Asymmetric introgression between the M and S forms of the malaria vector, Anopheles gambiae, maintains divergence despite extensive hybridization. Mol. Ecol. 20, 4983–4994. doi: 10.1111/j.1365-294X.2011.05339.x
Martin, D., and Rybicki, E. (2000). RDP: detection of recombination amongst aligned sequences. Bioinformatics 16, 562–563. doi: 10.1093/bioinformatics/16.6.562
Martin, D. P., Murrell, B., Golden, M., Khoosal, A., and Muhire, B. (2015). RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol. 1:vev003. doi: 10.1093/ve/vev003
Martin, S. H., Dasmahapatra, K. K., Nadeau, N. J., Slazar, C., Walters, J. R., Simpson, F., et al. (2013). Heliconius and sympatric speciation. Genome Res. 23, 1817–1828. doi: 10.1101/gr.159426.113
Mazzon, L., Martinez-Sañudo, I., Simonato, M., Squartini, A., Savio, C., and Girolami, V. (2010). Phylogenetic relationships between flies of the Tephritinae subfamily (Diptera, Tephritidae) and their symbiotic bacteria. Mol. Phylogenet. Evol. 56, 312–326. doi: 10.1016/j.ympev.2010.02.016
McPheron, B. A., Han,H-Y., Silva, J. G., and Norrbom, A. L. (1999). “Phylogeny of the genera Anastrepha and Toxotrypana (Trypetinae: Toxotrypanini) based upon 16S rRNA mitochondrial DNA sequences,” in Fruitlfies (Tephritidae): Phylogeny and Evolution of Behavior, eds M. Aluja and A. L. Norrbom (Boca Raton, FL: CRC), 343–362.
Mengual, X., Kerr, P., Norrbom, A. L., Barr, N. B., Lewis, M. L., Stapelfeldt, A. M., et al. (2017). Phylogenetic relationships of the tribe Toxotrypanini (Diptera: Tephritidae) based on molecular characters. Mol. Phylogenet. Evol. 113, 84–112. doi: 10.1016/j.ympev.2017.05.011
Meyer, B. S., Matschiner, M., and Salzburger, W. (2016). Disentangling incomplete lineage sorting and introgression to refine species-tree estimates for Lake Tanganyika cichlid fishes. Syst Biol. 66, 531–550. doi: 10.1101/039396
Michel, A. P., Sim, S., Powell, T. H. Q., Taylor, M. S., Nosil, P., and Feder, J. L. (2010). Widespread genomic divergence during sympatric speciation. Proc. Natl. Acad. Sci. U.S.A. 107, 9724–9729. doi: 10.1073/pnas.1000939107
Nadeau, N. J., Ruiz, M., Salazar, P., Counterman, B., Medina, J. A., Ortiz-Zuazaga, H., et al. (2014). Population genomics of parallel hybrid zones in the mimetic butterflies. H. melpomene and H. erato. Genome Res. 24, 1316–1333. doi: 10.1101/gr.169292.113
Nascimento, A. S., Morgante, J. S., Malavasi, A., and Uramoto, K. (1993). “Occurrence and distribution in Anastrepha in melon production areas in Brazil,” in Fruit Flies, Biology, and Management, eds M. Aluja and P. Liedo (New York, NY: Springer-Verlag Inc.). doi: 10.1007/978-1-4757-2278-9
Nielsen, R., and Wakeley, J. (2001). Distinguishing migration from isolation: a markov chain monte carlo approach. Genetics 158, 885–896.
Norrbom, A. L. (1994). New genera of Tephritidae (Diptera) from Brazil and Dominican amber, with phylogenetic analysis of the tribe Ortalotrypetini. Insecta Mundi 8:289.
Norrbom, A. L., Korytkowski, C. A., Zucchi, R. A., Uramoto, K., Venable, G. L., McCormick, J., et al. (2012). Anastrepha and Toxotrypana: Descriptions, Illustrations, and Interactive Keys. 28th Version. Available at: http://delta-intkey.com
Norrbom, A. L., Zucchi, R. A., and Hernández-Ortiz, V. (1999). “Phylogeny of the genera Anastrepha and Toxotrypana (Trypetinae: Toxotrypanini) based on morphology,” in Fruit Flies (Tephritidae): Phylogeny and Evolution of Behaviour, eds M. Aluja and A. L. Norrbom (Boca Ratón, FL: CRC Press), 299–342.
Norris, L. C., Main, B. J., Lee, Y., Collier, T. C., Fofana, A., Cornel, A. J., et al. (2015). Adaptive introgression in an African malaria mosquito coincident with the increased usage of insecticide-treated bed nets. Proc. Natl. Acad. Sci. U.S.A. 112:201418892. doi: 10.1073/pnas.1418892112
Nunes, M. D. S., Wengel, P. O. T., Kreissl, M., and Schlotterer, C. (2010). Multiple hybridization events between Drosophila simulans and Drosophila mauritiana are supported by mtDNA introgression. Mol. Ecol. 19, 4695–4707. doi: 10.5061/dryad.1731
Pamilo, P., and Nei, M. (1988). Relationships between gene trees and species trees. Mol. Biol. Evol. 5, 568–583.
Pardo-Diaz, C., Salazar, C., Baxter, S. W., Merot, C., Figueiredo-Ready, W., Joron, M., et al. (2012). Adaptive introgression across species boundaries in Heliconius butterflies. PLoS Genet. 8:e1002752. doi: 10.1371/journal.pgen.1002752.
Perre, P., Faria, F. A., Jorge, L. R., Rocha, A., Torres, R. S., Souza-Filho, M. F., et al. (2016). Toward an automated identification of Anastrepha fruit flies in the fraterculus group (Diptera, Tephritidae). Neotrop. Entomol. 45, 554–558. doi: 10.1007/s13744-016-0403-0
Perre, P., Jorge, L. R., Lewinsohn, T. M., and Zucchi, R. A. (2014). Morphometric differentiation of fruit fly pest species of the Anastrepha fraterculus group (Diptera: Tephritidae). Ann. Entomol. Soc. Am. 107, 490–495. doi: 10.1603/AN13122
Posada, D. (2008). jModelTest: phylogenetic model averaging. Mol. Biol. Evol. 25, 1253–1256. doi: 10.1093/molbev/msn083
Raga, A., Souza-Filho, M. F., De, Machado, R. A., Sato, M. E., and Siloto, R. C. (2011). Host ranges and infestation indices of fruit flies (Tephritidae) and lance flies (Lonchaeidae) in São Paulo State, Brazil. Florida Entomol. 94, 787–794. doi: 10.1653/024.094.0409
Reid, N. M., Hird, S. M., Brown, J. M., Pelletier, T. A., McVay, J. D., Satler, J. D., et al. (2014). Poor fit to the multispecies coalescent is widely detectable in empirical data. Syst. Biol. 63, 322–333. doi: 10.1093/sysbio/syt057
Roriz, A. K. P., Japyassú, H. F., and Joachim-Bravo, I. S. (2017). Incipient speciation in the Anastrepha fraterculus cryptic species complex: reproductive compatibility between A. sp.1 aff. fraterculus and A. sp.3 aff. fraterculus. Entomol. Exp. Appl. 162, 346–357. doi: 10.1111/eea.12526
Ruiz, M. F., Milano, A., Salvemini, M., Eirín-López, J. M., Perondini, A. L. P., Selivon, D., et al. (2007). The gene transformer of Anastrepha fruit flies (Diptera, Tephritidae) and its evolution in insects. PLoS One 2:e1239. doi: 10.1371/journal.pone.0001239.
Rull, J., Tadeo, E., Lasa, R., Rodríguez, C. L., Altuzar-Molina, A., and Aluja, M. (2017). Experimental hybridization and reproductive isolation between two sympatric species of tephritid fruit flies in the Anastrepha fraterculus species group. Insect Sci. doi: 10.1111/1744-7917.12489 [Epub ahead of print].
Santos, P., Uramoto, K., and Matioli, S. R. (2001). Experimental hybridization among Anastrepha species (Diptera: Tephritidae): production and morphological characterization of F1 Hybrids. Ann. Entomol. Soc. Am. 94, 717–725. doi: 10.1603/0013-87462001094[0717:EHAASD]2.0.CO;2.
Sarno, F., Ruiz, M. F., Eirín-López, J. M., Perondini, A. L. P., Selivon, D., and Sánchez, L. (2010). The gene transformer-2 of Anastrepha fruit flies (Diptera. Tephritidae) and its evolution in insects. BMC Evol. Biol. 10:140. doi: 10.1186/1471-2148-10-140
Scally, M., Into, F., Thomas, D. B., Ruiz-Arce, R., Barr, N. B., and Schuenzel, E. L. (2016). Resolution of inter and intra-species relationships of the West Indian fruit fly Anastrepha obliqua. Mol. Phylogenet. Evol. 101, 286–293. doi: 10.1016/j.ympev.2016.04.020
Schutze, M. K., Virgilio, M., Norrbom, A., and Clarke, A. R. (2017). Tephritid integrative taxonomy: where we are now, with a focus on the resolution of three tropical fruit fly species complexes. Annu. Rev. Entomol. 62, 147–164. doi: 10.1146/annurev-ento-031616-035518
Seetharam, A. S., and Stuart, G. W. (2013). Whole genome phylogeny for 21 Drosophila species using predicted 2b-RAD fragments. PeerJ 1:e226. doi: 10.7717/peerj.226
Selivon, D. (1996). Estudo Sobre a Diferenciação Populacional em Anastrepha fraterculus (Wiedemann) (Diptera: Tephritidae). Ph.D. Thesis, Instituto de Biociências, São Paulo.
Selivon, D., and Perondini, A. L. P. (1998). Eggshell morphology in two cryptic species of the Anastrefa fraterculus complex (Diptera: Tephritidae). Ann. Entomol. Soc. Am. 91, 473–478. doi: 10.1093/aesa/91.4.473
Selivon, D., and Perondini, A. L. P. (2007). “Especies cripticas del complejo Anastrepha fraterculus en Brasil,” in Moscas de la Fruta en Latinoamérica (Díptera: Tephritidae): Diversidad, Biología y Manejo, ed. V. Hernández-Ortiz (México: S y G Editores), 101–118.
Selivon, D., Perondini, A. L. P., and Morgante, J. S. (1999). Haldane’s rule and other aspects of reproductive isolation observed in the Anastrepha fraterculus complex (Diptera: Tephritidae). Genet. Mol. Biol. 22, 507–510. doi: 10.1590/S1415-47571999000400007
Selivon, D., Perondini, A. L. P., and Morgante, J. S. (2005). A Genetic–Morphological Characterization of Two Cryptic Species of the Anastrepha fraterculus Complex (Diptera: Tephritidae). Ann. Entomol. Soc. Am. 98, 367–381. doi: 10.1603/0013-87462005098[0367:AGCOTC]2.0.CO;2.
Sivinski, J., Aluja, M., Dodson, G., Freidberg, A., Headrick, D., Kaneshiro, K., et al. (1999). “Topics in the Evolution of Sexual Behavior in the Tephritidae,” in Fruit Flies (Tephritidae): Phylogeny and Evolution of Behavior, eds M. Aluja and A. L. Norrbom (Boca Ratón, FL: CRC Press), 751–792.
Smith-Caldas, M. R. B., Mcpheron, B. A., Silva, J. G., and Zucchi, R. A. (2001). Phylogenetic relationships among species of the fraterculus group (Anastrepha: Diptera: Tephritidae) inferred from DNA sequences of mitochondrial Cytochrome Oxidase I. Neotrop. Entomol. 30, 565–573. doi: 10.1590/S1519-566X2001000400009
Tajima, F. (1989). Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595.
Tavare, S., Balding, D. J., Griffiths, R. C., and Donnelly, P. (1997). Inferring coalescence times from DNA sequence Data. Genetics 145, 505–518.
Thompson, J. D., Higgins, D. G., and Gibson, T. J. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680. doi: 10.1093/nar/22.22.4673
Vaníčková, L., Hernández-Ortiz, V., Joachim Bravo, I. S., Dias, V., Passos Roriz, A. K., Laumann, R. A., et al. (2015). Current knowledge of the species complex Anastrepha fraterculus (Diptera, Tephritidae) in Brazil. Zookeys 540, 211–237. doi: 10.3897/zookeys.540.9791
Virgilio, M., Jordaens, K., Verwimp, C., White, I. M., and De Meyer, M. (2015). Higher phylogeny of frugivorous flies (Diptera, Tephritidae, Dacini): localised partition conflicts and a novel generic classification. Mol. Phylogenet. Evol. 85, 171–179. doi: 10.1016/j.ympev.2015.01.007
Weetman, D., Wilding, C. S., Steen, K., Pinto, J., and Donnelly, M. J. (2012). Gene flow-dependent genomic divergence between anopheles gambiae M and S forms. Mol. Biol. Evol. 29, 279–291. doi: 10.1093/molbev/msr199
Wegmann, D., Leuenberger, C., and Excoffier, L. (2009). Efficient approximate Bayesian computation coupled with markov chain monte carlo without likelihood. Genetics 182, 1207–1218. doi: 10.1534/genetics.109.102509
Wegmann, D., Leuenberger, C., Neuenschwander, S., and Excoffier, L. (2010). ABCtoolbox: a versatile toolkit for approximate Bayesian computations. BMC Bioinformatics 11:116. doi: 10.1186/1471-2105-11-116
Weir, B. S., and Cockerham, C. C. (1984). Estimating F-statistics for the analysis of population structure. Evolution 38, 1358–1370.
Wright, S. (1951). The genetic structure of populations. Ann. Eugen. 15, 323–354. doi: 10.1111/j.1469-1809.1949.tb02451.x
Xia, X. (2013). DAMBE5: a comprehensive software package for data analysis in molecular biology and evolution. Mol. Biol. Evol. 30, 1720–1728. doi: 10.1093/molbev/mst064
Xie, X., Rull, J., Michel, A. P., Velez, S., Forbes, A. A., Lobo, N. F., et al. (2007). Hawthorn-infesting populations of Rhagoletis pomonella in Mexico and speciation mode plurality. Evolution 61, 1091–1105. doi: 10.1111/j.1558-5646.2007.00091.x
Zhang, W., Dasmahapatra, K. K., Mallet, J., Moreira, G. R. P., and Kronforst, M. R. (2016). Genome-wide introgression among distantly related Heliconius butterfly species. Genome Biol. 17:25. doi: 10.1186/s13059-016-0889-0
Keywords: Anastrepha fraterculus group, speciation, introgression, incomplete lineage sorting, isolation with migration, approximate Bayesian computation, population expansion
Citation: Díaz F, Lima ALA, Nakamura AM, Fernandes F, Sobrinho I Jr and de Brito RA (2018) Evidence for Introgression Among Three Species of the Anastrepha fraterculus Group, a Radiating Species Complex of Fruit Flies. Front. Genet. 9:359. doi: 10.3389/fgene.2018.00359
Received: 12 May 2018; Accepted: 21 August 2018;
Published: 10 September 2018.
Edited by:
Norman A. Johnson, University of Massachusetts Amherst, United StatesReviewed by:
Christen Kerry Mirth, Monash University, AustraliaPeter J. Prentis, Queensland University of Technology, Australia
Copyright © 2018 Díaz, Lima, Nakamura, Fernandes, Sobrinho and de Brito. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fernando Díaz, ferdiazfer@gmail.com