Robustness of Mammalian Gut Microbiota to Humanization in Captivity
 Brian K. Trevelline1,2*
Brian K. Trevelline1,2*  Andrew H. Moeller2*
Andrew H. Moeller2*
- 1Cornell Lab of Ornithology, Cornell University, Ithaca, NY, United States
- 2Department of Ecology and Evolutionary Biology, Cornell University, Ithaca, NY, United States
In mammals, the composition of the gut microbiota is associated with host phylogenetic history, and host-lineage specific microbiota have been shown, in some cases, to contribute to fitness-related traits of their hosts. However, in primates, captivity can disrupt the native microbiota through a process of humanization in which captive hosts acquire gut microbiota constituents found in humans. Despite the potential importance of this process for the health of captive hosts, the degree to which captivity humanizes the gut microbiota of other mammalian taxa has not been explored. Here, we analyzed hundreds of published gut microbiota profiles generated from wild and captive hosts spanning seven mammalian families to investigate the extent of humanization of the gut microbiota in captivity across the mammalian phylogeny. Comparisons of these hosts revealed compositional convergence between captive mammal and human gut microbiota in the majority of mammalian families examined. This convergence was driven by a diversity of microbial lineages, including members of the Archaea, Clostridium, and Bacteroides. However, the gut microbiota of two families—Giraffidae and Bovidae—were remarkably robust to humanization in captivity, showing no evidence of gut microbiota acquisition from humans relative to their wild confamiliars. These results demonstrate that humanization of the gut microbiota is widespread in captive mammals, but that certain mammalian lineages are resistant to colonization by human-associated gut bacteria.
Introduction
The gut microbial communities of wild mammals tend to reflect their hosts’ phylogenetic histories. Across a diversity of mammalian taxa, gut microbial communities are strongly associated with the evolutionary histories of their hosts, in that compositional divergence of the gut microbiota increases with host phylogenetic distance (termed “phylosymbiosis”) (Ley et al., 2008; Ochman et al., 2010; Muegge et al., 2011; Brooks et al., 2016; Moeller et al., 2017; Lim and Bordenstein, 2020). Moreover, there is mounting evidence that certain host-associated microbiota have co-diversified with their hosts (Moeller et al., 2016; Groussin et al., 2017, 2020), indicating the maintenance of specific relationships between gut bacterial and host lineages over evolutionary time. These phylogenetic patterns in the gut microbiota suggest the possibility co-evolution and co-adaptation between gut bacteria and mammals, and experimental evidence in rodents has provided some evidence for this hypothesis. For example, germ-free house mice inoculated with non-native gut microbiota from other species of rodent display stunted immune maturation and growth rates relative to germ-free mice inoculated with a native house-mouse microbiota (Chung et al., 2012; Moeller et al., 2019). These studies suggest that disruption of ancient host-microbe associations can result in adverse fitness consequences for the host.
In contrast to the patterns observed in wild mammals, captive mammals can lose their species-specific gut microbiota and acquire certain gut microbiota constituents found in humans. For example, the gut microbiota of captive primates are more compositionally similar to humans than are those of wild conspecifics (Clayton et al., 2016; Houtz et al., 2021). Humanization of the gut microbiota in captivity may lead to a mismatch between host physiologies and their microbial environments, potentially contributing to the health issues observed in captive mammals such as pathogenic bacterial infections (Wasimuddin et al., 2017) and gastrointestinal dysfunction (Terio et al., 2005). Importantly, attempts to eliminate potentially pathogenic bacteria with antibiotics may further exacerbate adverse health outcomes, as these compounds also reduce the abundance of beneficial microbiota (Dahlhausen et al., 2018). Therefore, humanization of the gut microbiota presents a potential threat to the health of captive mammals and may subvert conservation efforts involving captive breeding and/or reintroductions into the wild (Trevelline et al., 2019). Nevertheless, to date the humanization of gut microbiota has only been investigated in captive primates, and the degree to which captivity humanizes the gut microbiota of other mammalian taxa has not been investigated.
Here, we leverage hundreds of published gut microbiota profiles generated from wild and captive mammals (representing eight families), as well as industrialized and non-industrialized human populations to test the hypotheses: (1) that captivity humanizes the mammalian gut microbiota, and (2) that the degree of humanization varies across mammalian families. Our analyses provided evidence of gut microbiota humanization in the majority of mammalian families examined. However, the gut microbiota of two mammalian families—Giraffidae and Bovidae—were robust to humanization in captivity, showing no evidence of gut microbiota acquisition from humans relative to their wild confamiliars. These results demonstrate that susceptibility of the gut microbiota to humanization appears to be widespread in mammals, but that certain mammalian lineages are resistant to colonization by human-associated gut bacteria.
Materials and Methods
Processing and Merging 16S rDNA Sequence Data
To assess the evidence for gut microbiota humanization in captive mammals, we merged publicly available V4 16S rRNA gene datasets from wild and captive mammals (McKenzie et al., 2017) and humans (Yatsunenko et al., 2012). These datasets were generated from bead-beating DNA extraction protocol implemented by the same research group, thereby minimizing potential study effects that could confound downstream meta-analyses. Raw sequences were merged into a single analysis using the University of California, San Diego qiita webserver (Gonzalez et al., 2018), and processed for quality using “split libraries” with the following parameters: sequence_max_n = 0, min_per_read_length_fraction = 0.75, max_bad_run_length = 3, and phred_quality_threshold = 3. Filtered sequences were trimmed to a common length of 100 bp to enable comparisons across datasets. Filtered and trimmed sequences were then collapsed into Amplicon Sequence Variants (ASVs) using deblur (Amir et al., 2017) as implemented in qiita. ASVs were determined with Deblur using following parameters: Mean per nucleotide error rate = 0.005, Indel probability = 0.01, Minimum dataset-wide read threshold = 0, Minimum per-sample read threshold = 2, Maximum number of indels = 3. ASVs were classified to taxonomic ranks using the Silva 138 reference (Quast et al., 2012). All samples were rarefied to a common depth of 10,000 reads for downstream analyses.
Beta Diversity Analyses
We calculated the beta diversity dissimilarities between all pairs of samples to test the hypothesis that the gut microbiota of captive mammals was more compositionally similar to humans than was the gut microbiota of wild mammals from the same family. To focus our analyses on differences in community memberships among samples, we calculated Sorensen-Dice distances in QIIME2 (Bolyen et al., 2019)—which in this context measures microbiota dissimilarity between pairs of samples based on the presence or absence of ASVs. Principal coordinates were calculated from the beta diversity dissimilarity matrix using “qiime diversity pcoa,” and samples were plotted against the first two PCs.
Associations Between Beta Diversity and Host Phylogeny in Wild and Captive Mammals
Using the rarefied ASV table described above, we collapsed microbial inventories from wild and captive hosts in the same family to test the hypothesis that captivity altered the association between gut microbiota composition and host phylogenetic history (i.e., phylosymbiosis). This dataset also included average ASV relative abundances of Homo sapiens from Malawi, Venezuela, and the United States. ASVs were averaged across wild or captive hosts in the same family using the command “feature-table group” with “p-mode” = “mean-ceiling” in QIIME2. Average relative abundance of ASVs were used to calculate beta diversity dissimilarity using the binary Sorensen-Dice metric in QIIME2. We used Sorensen-Dice dissimilarities to produce microbial dendrograms using UPGMA hierarchal clustering in QIIME1 (Caporaso et al., 2010). We used the program TreeCmp (Bogdanowicz et al., 2012) and a previously published Python script (Brooks et al., 2016) to investigate the impact of humanization on patterns of phylosymbiosis among captive mammals. We tested for patterns of phylosymbiosis by comparing gut microbiota dendrograms for wild and captive mammals with mammalian host phylogeny (downloaded from TimeTree; Kumar et al., 2017) via the rooted Robinson–Foulds metric with 10,000 random trees. This approach produces a normalized score between 0 (complete congruence) and 1 (complete incongruence). P-values were determined by the probability of 10,000 randomized dendrogram topologies yielding equivalent or more congruent phylosymbiotic patterns than the actual microbiota dendrogram.
Assessing Humanization of Captive Gut Microbiota Within Mammalian Families
To test for significant differences in dissimilarity between wild mammal microbiota vs. human microbiota and captive mammal microbiota vs. human microbiota, we employed non-parametric Monte-Carlo permutation tests using “make_distance_boxplots.py” as implemented in QIIME 1.9. These analyses tested whether the microbiota of captive mammals displayed reduced compositional similarity (based on binary Sorensen-Dice dissimilarities) to microbiota of humans relative to the microbiota of wild-living mammals from the same taxonomic family. Non-parametric p-values from these tests were calculated based on 999 permutations.
Phylogenetic Visualizations
Phylogenetic trees of ASVs were constructed using SEPP insertion against the Greengenes 13–8 reference phylogeny (DeSantis et al., 2006) as implemented in QIIME2. Trees were subsequently pruned in R using the package “phytools” (Revell, 2012) to remove reference sequences. Trees and distributions of ASVs were plotted with EMPress (Cantrell et al., 2021) as implemented in QIIME2.
Results
16S rDNA Datasets
The combined dataset contained gut microbiota profiles from 657 fecal samples collected from humans and other mammals, including 45,394 Amplicon Sequence Variants (ASVs) represented by 569,790,829 reads. In addition to humans (n = 528; Family Hominidae), mammalian families represented in our merged dataset included Bovidae (wild, captive [total] n = 11, 19 [30]), Canidae (n = 4, 5 [9]), Cercopithecidae (n = 33, 8 [41]), Equidae (n = 9, 22 [31]), Giraffidae (n = 2, 4 [6]), Orycteropodidae (n = 5, 18 [23]), and Rhinocerotidae (n = 4, 9 [13]). A complete list of samples and corresponding metadata is presented in Supplementary Table 1. The relative abundances of ASVs across samples are presented in Supplementary Table 2.
Effects of Captivity on Patterns of Phylosymbiosis
We investigated the potential impact of captivity on patterns of phylosymbiosis by comparing microbial community dendrograms and the host phylogenetic tree. Specifically, we tested whether the gut microbiota and host dendrograms exhibited more topological congruence (measure of phylosymbiosis) than expected at random, and whether wild and captive mammals differed in their patterns of phylosymbiosis. Using the UPGMA trees constructed using the Sorenson-Dice dissimilarities, we detected a statistically significant pattern of phylosymbiosis for both captive and wild trees (Figure 1). Two families–Equidae and Rhinocerotidae–clustered more closely with the primates (i.e., Hominidae and Cercopithecidae) in captivity compared to confamiliars in the wild (Figure 1). In contrast, our analysis revealed that the gut microbiota of captive Bovidae and Giraffidae retained the same phylogenetic clustering observed in wild confamiliars (Figure 1).
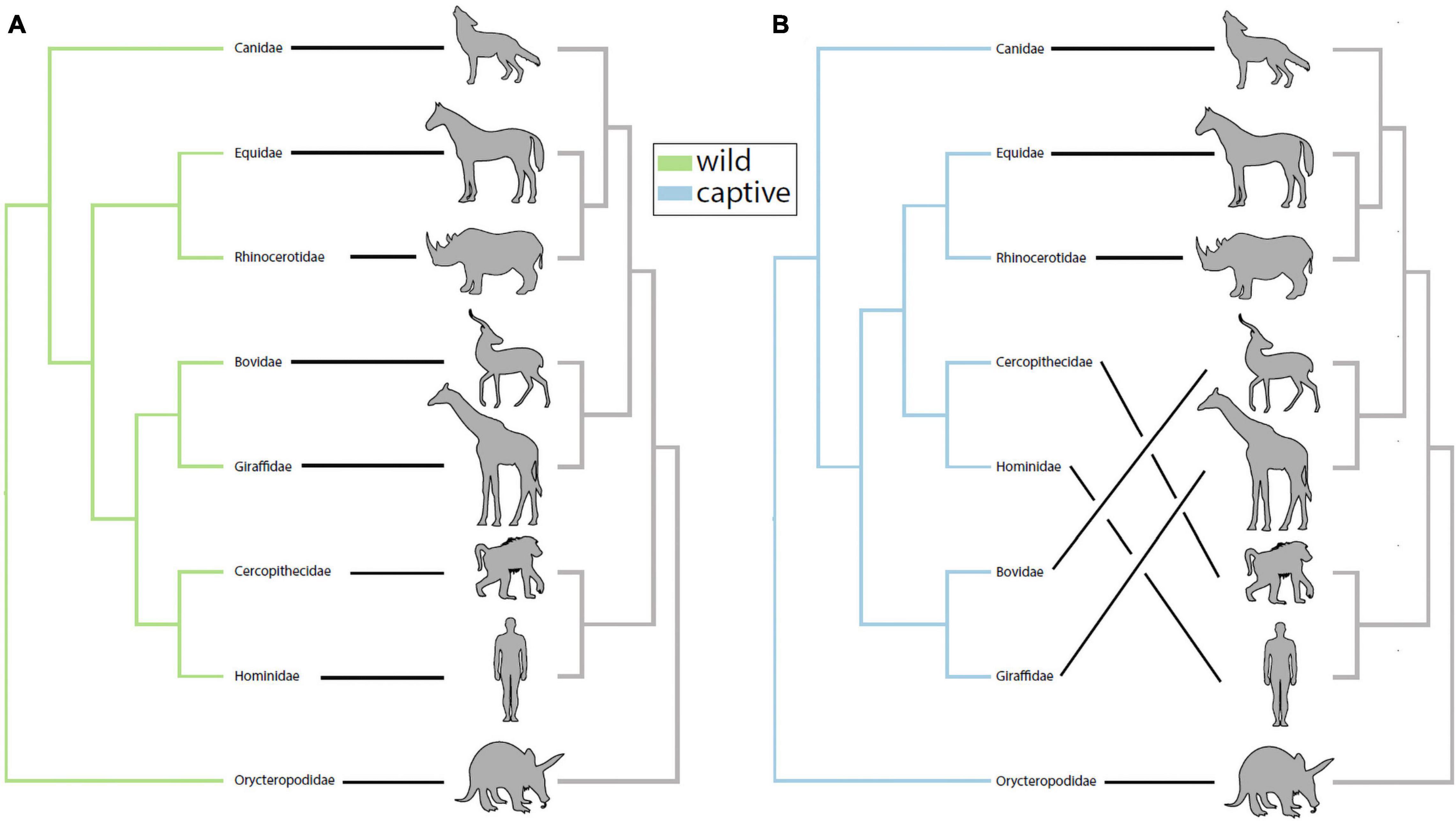
Figure 1. Captivity alters patterns of phylosymbiosis among mammalian families. (A) UPGMA-clustered microbiota dendrograms from wild mammals exhibited significant patterns of phylosymbiosis with host phylogeny (rooted Robinson-Foulds = 0.333, P = 0.0003). (B) UPGMA-clustered microbiota dendrograms from captive mammals also exhibited significant patterns of phylosymbiosis with host phylogeny (rooted Robinson-Foulds = 0.333, P = 0.0002). Although wild and captive trees both exhibited significant patterns of phylosymbiosis, further inspection revealed that captive Equidae and Rhinocerotidae grouped more closely with primates compared to wild confamiliars in (A).
Robustness and Susceptibility to Humanization of Gut Microbiota in Captive Mammals
For each mammalian family, we tested whether gut microbiota in captive individuals displayed more similar community memberships to human gut microbiota than did those of wild individuals. These tests allowed us to assess the degree of humanization of the gut microbiota in captivity for each mammalian family. Monte-Carlo non-parametric permutation tests of pairwise beta-diversity (Binary Sorensen-Dice) dissimilarities indicated that the gut microbiota of most mammalian families were more similar to the gut microbiota of humans in captivity than in the wild (Figure 2). Mammalian families that displayed significant evidence of gut microbiota humanization in captivity included the Rhinocerotidae (p = 0.001), Equidae (p = 0.001), Canidae (p = 0.001), and Cercopithecidae (p = 0.006). The sequences and taxonomic assignments of ASVs shared by captive individuals of each family and humans to the exclusion of wild individuals from the family are presented in Supplementary Table 2.
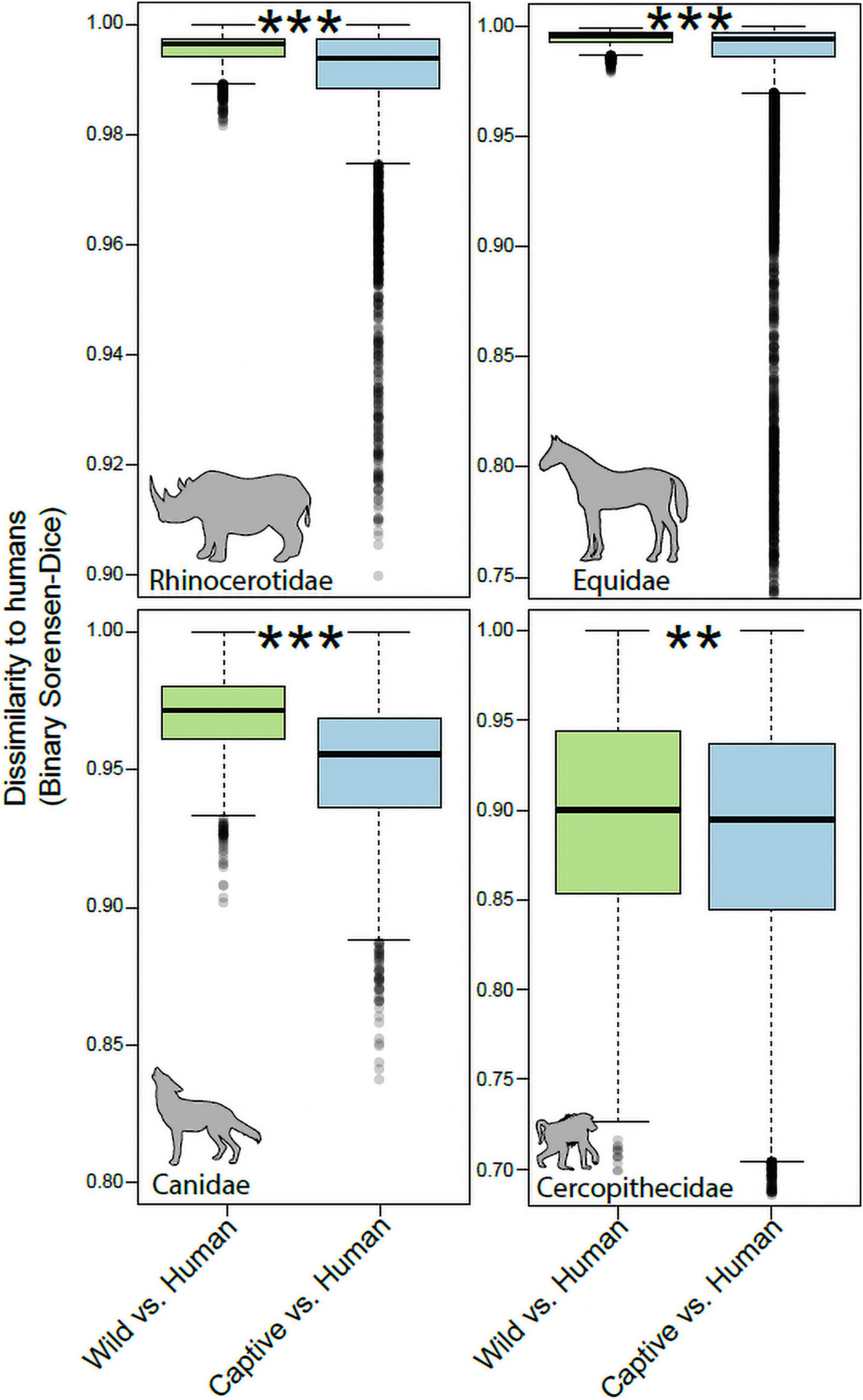
Figure 2. Humanization of the gut microbiota in captive Rhinocerotidae, Equidae, Canidae, and Cercopithecidae. Boxplots show microbiota dissimilarities (binary Sorensen-Dice) between humans and wild mammals (green boxes) and between humans and captive mammals (purple boxes). Each box shows median and interquartile range for a comparison including a single mammalian family. Significant differences between boxplots within each panel are denoted with asterisks; **p < 0.01, ***p = 0.001; non-parametric Monte-Carlo permutation tests.
In contrast to the pattern observed in the Rhinocerotidae, Equidae, Canidae, and Cercopithecidae, the Bovidae and Giraffidae displayed no evidence of humanization of the gut microbiota in captivity (Figure 3). The community memberships of the microbiota in captive individuals of these families were no more similar to that of human microbiota than were those of the microbiota in wild individuals (p = 0.54 for Bovidae, p = 0.81 for Giraffidae).

Figure 3. Robustness to humanization of the gut microbiota in captive Giraffidae and Bovidae. Boxplots show microbiota dissimilarities (binary Sorensen-Dice) between humans and wild mammals (green boxes) and between humans and captive mammals (purple boxes). Each box shows median and interquartile range for a comparison including a single mammalian family. Significant differences between boxplots within each panel are denoted with asterisks; NS p > 0.05; non-parametric Monte-Carlo permutation tests.
Taxonomic Distributions of Amplicon Sequence Variants Shared by Captive Mammals and Humans but Not by Wild Mammals
We visualized the distributions of ASVs belonging to Archaea, Clostridium sensu stricto, and Bacteroides (based on Silva 138 assignments) across humans as well as wild and captive mammals belonging to the families Rhinocerotidae, Equidae, Canidae, and Cercopithecidae. These trees and abundance plots (Figure 4) demonstrated that captive mammals exhibit a greater degree of humanization (i.e., ASVs shared by humans and captive mammals but not by wild mammals) than the opposite pattern (i.e., shared by humans and wild mammals but not by captive mammals). For example, seven ASVs belonging to Clostridium sensu stricto were shared by humans and captive mammals to the exclusion of wild mammals, but only a single ASV was shared by humans and wild mammals to the exclusion of captive mammals (Figure 4B).
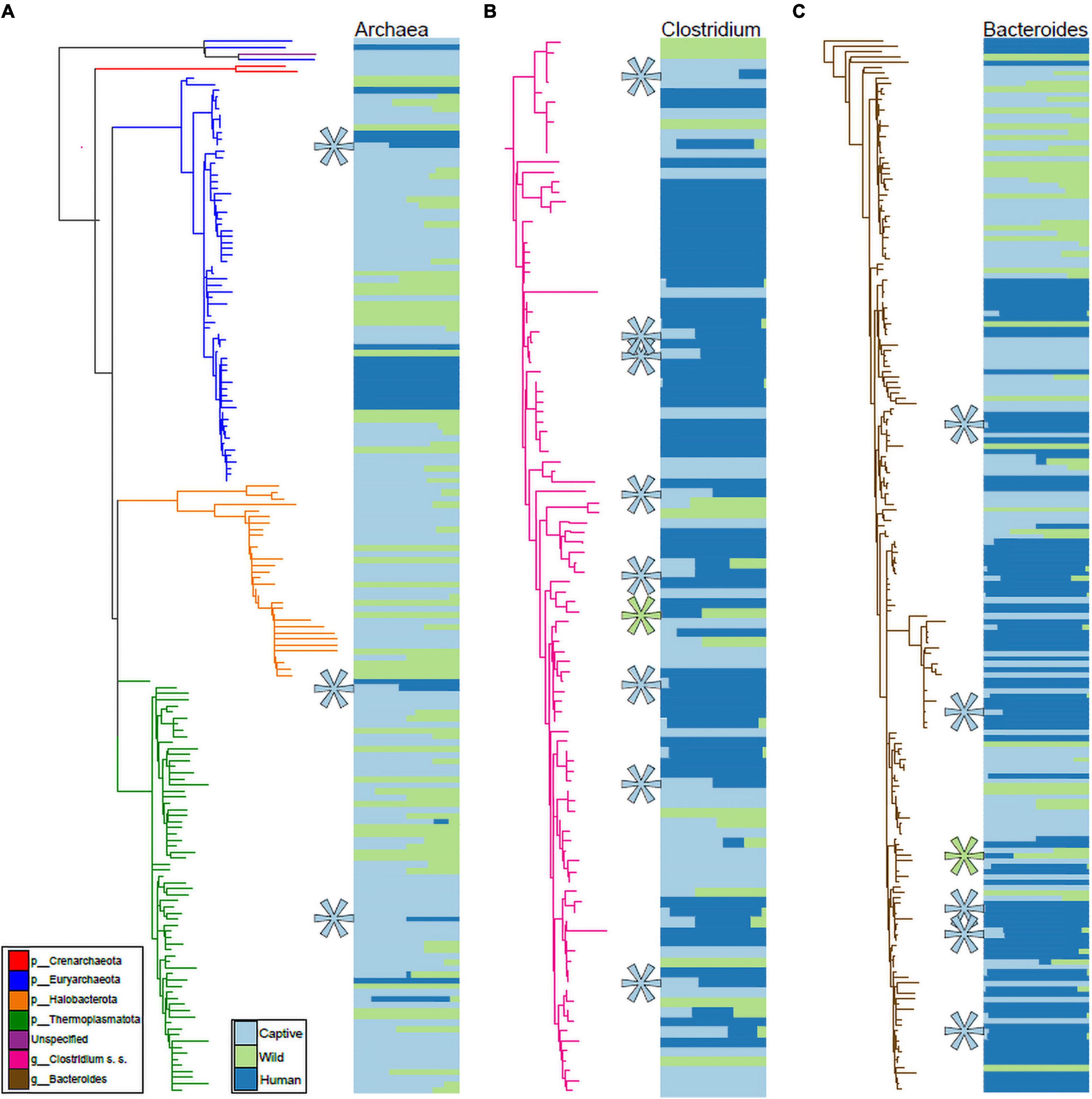
Figure 4. Phylogenetic distributions of Archaeal, Clostridium, and Bacteroides ASVs shared exclusively by humans and captive mammals. Phylogenies in (A–C) show relationships among ASVs in humans, Rhinocerotidae, Equidae, Canidae, and Cercopithecidae belonging to the Archaea (A), Clostridium sensu stricto (B), and Bacteroides (C). Colors of branches correspond to bacterial taxa as indicated by the leftmost inset in (A). Colors of horizontal bars indicate the relative abundance of each ASV in captive mammal, wild mammal, or human hosts, as indicated by the rightmost inset in (A). Relative sizes of horizontal bars are scaled to reflect the mean relative abundance of the ASV in each of the three sample categories. Teal asterisks in (A–C) indicate ASVs shared by captive mammals and humans but not found in wild mammals (i.e., ASVs displaying distributions consistent with transfer from humans to captive mammals), whereas green asterisks indicate ASVs shared by wild mammals and humans but not found in captive mammals.
Discussion
We found surprising variation across mammalian families in the robustness of the gut microbiota to humanization in captivity. Most of the mammalian families examined here harbored gut microbiota that were more similar to human gut microbiota in captivity than in the wild. These results are consistent with previous results from non-human primates, which display evidence of gut microbiota humanization in captivity (Clayton et al., 2016), and extend these observations to a broader diversity of host lineages spanning the mammalian phylogeny. However, in contrast to previous results from primates and the majority of mammalian families studied here, the gut microbiota of two mammalian families—Bovidae and Giraffidae—displayed robustness to humanization in captivity. This result indicates that certain mammalian taxa, in this case herbivorous Artiodactyla, did not exhibit evidence of acquisition of human-associated gut microbiota when living in captivity compared to wild confamiliars, raising questions about the mechanisms underlying this robustness and the potential implications for captive hosts.
In the Rhinocerotidae, Equidae, Canidae, and Cercopithecidae, both bacteria and archaea displayed evidence of humanization in captivity, and these patterns were driven by several bacterial taxa within these domains (Figure 4). Clostridium sensu stricto displayed a particularly pronounced signal of ASV sharing between humans and captive mammals to the exclusion of wild mammals (Figure 4B). This taxon contains a diversity of opportunistic pathogens, including the leading cause of mortality from bacterial gastrointestinal infections in humans C. difficile. Previous work has found that C. difficile infections can also cause mortality in captive mammals (Rolland et al., 1997). Our results suggest the possibility that these infections in captivity may stem from human sources. However, it is also possible that lifestyle factors in captivity promote the proliferation of these C. difficile lineages. Further metagenomic sequencing and strain tracking will be required to differentiate among these hypotheses. Moreover, this observation motivates further research into fecal transplants from wild conspecifics into captive mammals as a strategy for mitigating the effects of captivity on the gut microbiota, as fecal microbiota transplant experiments have been shown to be effective treatments for Clostridium infections in humans (Hvas et al., 2019).
Humanization of the gut microbiota in captivity may have consequences for animal health that subvert the aims of wildlife rehabilitation and conservation programs. For example, the routine administration of antibiotics in captivity can disrupt native microbiota (Dahlhausen et al., 2018) and therefore could allow for human-associated microbiota to opportunistically colonize the host. Intraspecific co-habitation can homogenize microbiomes between species (e.g., Song et al., 2013; Lemieux-Labonté et al., 2016), which could potentially exacerbate the mismatch between hosts and their native microbiota. Conversely, previous work has shown that facilitating conspecific interactions increases microbial transmission between individuals and improve microbial community diversity (Nelson et al., 2013), and thus could help to maintain natural microbiota and ameliorate the negative effects of captivity. Further, recent work has demonstrated that captive housing that incorporates native microbial reservoirs and diets can mitigate the loss of species-specific microbiota (Kohl and Dearing, 2014; Loudon et al., 2014). However, it is important to note that there are many host-specific factors that influence the microbiome, and thus characterization and screening of microbial reservoirs must be conducted to ensure that introduction of these materials will not result in off-target effects on native microbial communities or adverse health outcomes.
In addition to the potential adverse consequences for animal health, the observation that certain microbial lineages appear to colonize both humans and animals raises the possibility for gut microbiota transmission from captive animals into humans, a processes that could lead to zoonotic infections in humans. However, this possibility of gut microbiota transmission from captive mammals into humans has not been tested. Future work that samples humans and captive mammals at multiple facilities will have the opportunity to test whether human microbiota converge with captive mammals within these facilities.
The observation that populations of Bovidae and Giraffidae studied here do not display evidence of gut microbiota humanization indicates that certain host taxa are more robust to this effect of captivity than others. One possibility underlying this result is that the herbivorous diets of Bovidae and Giraffidae may differ substantially from those of humans, whereas omnivorous mammalian families may be less susceptible to colonization by human-derived bacteria. However, previous work in primates has shown that, at least for the genera Alouatta, Colobus, Cercopithecus, Gorilla, and Pan, folivore gut microbiota experience larger compositional shifts in captivity than do non-folivore gut microbiota (Frankel et al., 2019). This previous result appears to contradict the result reported here that folivorous Bovidae and Giraffidae displayed relative robustness to gut microbiota humanization in captivity, possibly due to major differences in digestive physiology between primates and artiodactyls. Resolving the association between host diet and the propensity for gut microbiota humanization in captivity will require further studies that sample a broader range of host taxa and assess this relationship in a comparative phylogenetic framework.
Conclusion
Here, we leveraged hundreds of publicly available gut microbial inventories to demonstrate that captivity humanizes the mammalian gut microbiome and that the degree of humanization differs across the mammalian phylogeny. Specifically, we showed that herbivorous artiodactyls are remarkably robust to humanization compared to other mammalian families. These findings indicate that the mechanisms influencing susceptibility to humanization in captivity vary across the mammalian phylogeny. While more work is needed to elucidate the mechanistic basis of this phenomenon, our study advances understanding of microbiome humanization with implications for future experimental work and wildlife management programs.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
AHM and BKT designed the study, conducted the analyses, and wrote the manuscript. Both authors contributed to the article and approved the submitted version.
Funding
This work was funded by a grant from the National Institutes of Health to AHM (R35 GM138284) and a Rose Fellowship from the Cornell Lab of Ornithology to BKT.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank the editors for the invitation to contribute to this special issue and to reviewers for their constructive comments on the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2021.785089/full#supplementary-material
References
Amir, A., McDonald, D., Navas-Molina, J. A., Kopylova, E., Morton, J. T., Zech Xu, Z., et al. (2017). Deblur rapidly resolves single-nucleotide community sequence patterns. mSystems 2, e00191–16. doi: 10.1128/mSystems.00191-16
Bogdanowicz, D., Giaro, K., and Wróbel, B. (2012). TreeCmp: comparison of Trees in Polynomial Time. Evol. Bioinform. Online 8, 475–487. doi: 10.4137/EBO.S9657
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Brooks, A. W., Kohl, K. D., Brucker, R. M., van Opstal, E. J., and Bordenstein, S. R. (2016). Phylosymbiosis: relationships and functional effects of microbial communities across host evolutionary history. PLoS Biol. 14:e2000225. doi: 10.1371/journal.pbio.2000225
Cantrell, K., Fedarko, M. W., Rahman, G., McDonald, D., Yang, Y., Zaw, T., et al. (2021). EMPress enables tree-guided, interactive, and exploratory analyses of multi-omic data sets. mSystems 6, e01216–20. doi: 10.1128/mSystems.01216-20
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Chung, H., Pamp, S. J., Hill, J. A., Surana, N. K., Edelman, S. M., Troy, E. B., et al. (2012). Gut immune maturation depends on colonization with a host-specific microbiota. Cell 149, 1578–1593. doi: 10.1016/j.cell.2012.04.037
Clayton, J. B., Vangay, P., Huang, H., Ward, T., Hillmann, B. M., Al-Ghalith, G. A., et al. (2016). Captivity humanizes the primate microbiome. Proc. Natl. Acad. Sci. U. S. A. 113, 10376–10381. doi: 10.1073/pnas.1521835113
Dahlhausen, K. E., Doroud, L., Firl, A. J., Polkinghorne, A., and Eisen, J. A. (2018). Characterization of shifts of koala (Phascolarctos cinereus) intestinal microbial communities associated with antibiotic treatment. PeerJ 6:e4452. doi: 10.7717/peerj.4452
DeSantis, T. Z., Hugenholtz, P., Larsen, N., Rojas, M., Brodie, E. L., Keller, K., et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072. doi: 10.1128/AEM.03006-05
Frankel, J. S., Mallott, E. K., Hopper, L. M., Ross, S. R., and Amato, K. R. (2019). The effect of captivity on the primate gut microbiome varies with host dietary niche. Am. J. Primatol. 81:e23061. doi: 10.1002/ajp.23061
Gonzalez, A., Navas-Molina, J. A., Kosciolek, T., McDonald, D., Vázquez-Baeza, Y., Ackermann, G., et al. (2018). Qiita: rapid, web-enabled microbiome meta-analysis. Nat. Methods 15, 796–798. doi: 10.1038/s41592-018-0141-9
Groussin, M., Mazel, F., and Alm, E. J. (2020). Co-evolution and co-speciation of host-gut bacteria systems. Cell Host Microbe 28, 12–22. doi: 10.1016/j.chom.2020.06.013
Groussin, M., Mazel, F., Sanders, J. G., Smillie, C. S., Lavergne, S., Thuiller, W., et al. (2017). Unraveling the processes shaping mammalian gut microbiomes over evolutionary time. Nat. Commun. 8:14319. doi: 10.1038/ncomms14319
Houtz, J., Sanders, J. G., Denice, A., and Moeller, A. (2021). Predictable and host-species specific humanization of the gut microbiota in captive primates. Mol. Ecol. 30, 3677–3687. doi: 10.1111/mec.15994
Hvas, C. L., Dahl Jørgensen, S. M., Jørgensen, S. P., Storgaard, M., Lemming, L., Hansen, M. M., et al. (2019). Fecal microbiota transplantation is superior to fidaxomicin for treatment of recurrent clostridium difficile infection. Gastroenterology 156, 1324–1332.e3. doi: 10.1053/j.gastro.2018.12.019
Kohl, K. D., and Dearing, M. D. (2014). Wild-caught rodents retain a majority of their natural gut microbiota upon entrance into captivity: effect of captivity on rodent gut microbiota. Environ. Microbiol. Reports 6, 191–195. doi: 10.1111/1758-2229.12118
Kumar, S., Stecher, G., Suleski, M., and Hedges, S. B. (2017). TimeTree: a resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 34, 1812–1819. doi: 10.1093/molbev/msx116
Lemieux-Labonté, V., Tromas, N., Shapiro, B. J., and Lapointe, F.-J. (2016). Environment and host species shape the skin microbiome of captive neotropical bats. PeerJ 4:e2430. doi: 10.7717/peerj.2430
Ley, R. E., Hamady, M., Lozupone, C., Turnbaugh, P. J., Ramey, R. R., Bircher, J. S., et al. (2008). Evolution of mammals and their gut microbes. Science 320, 1647–1651.
Lim, S. J., and Bordenstein, S. R. (2020). An introduction to phylosymbiosis. Proc. Biol. Soc. 287:20192900. doi: 10.1098/rspb.2019.2900
Loudon, A. H., Woodhams, D. C., Parfrey, L. W., Archer, H., Knight, R., McKenzie, V., et al. (2014). Microbial community dynamics and effect of environmental microbial reservoirs on red-backed salamanders (Plethodon cinereus). ISME J. 8, 830–840. doi: 10.1038/ismej.2013.200
McKenzie, V. J., Song, S. J., Delsuc, F., Prest, T. L., Oliverio, A. M., Korpita, T. M., et al. (2017). The effects of captivity on the mammalian gut microbiome. Integr. Comp. Biol. 57, 690–704. doi: 10.1093/icb/icx090
Moeller, A. H., Caro-Quintero, A., Mjungu, D., Georgiev, A. V., Lonsdorf, E. V., Muller, M. N., et al. (2016). Cospeciation of gut microbiota with hominids. Science 353, 380–382. doi: 10.1126/science.aaf3951
Moeller, A. H., Gomes-Neto, J. C., Mantz, S., Kittana, H., Segura Munoz, R. R., Schmaltz, R. J., et al. (2019). Experimental evidence for adaptation to species-specific gut microbiota in house mice. mSphere 4, e00387–19. doi: 10.1128/mSphere.00387-19
Moeller, A. H., Suzuki, T. A., Lin, D., Lacey, E. A., Wasser, S. K., and Nachman, M. W. (2017). Dispersal limitation promotes the diversification of the mammalian gut microbiota. Proc. Natl. Acad. Sci. U. S. A. 114, 13768–13773.
Muegge, B. D., Kuczynski, J., Knights, D., Clemente, J. C., González, A., Fontana, L., et al. (2011). Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 332, 970–974.
Nelson, T., Rogers, T., Carlini, A., and Brown, M. (2013). Diet and phylogeny shape the gut microbiota of Antarctic seals: a comparison of wild and captive animals. Environ. Microbiol. 15, 1132–1145.
Ochman, H., Worobey, M., Kuo, C. H., Ndjango, J. B., Peeters, M., Hahn, B. H., et al. (2010). Evolutionary relationships of wild hominids recapitulated by gut microbial communities. PLoS Biol. 8:e1000546. doi: 10.1371/journal.pbio.1000546
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2012). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Revell, L. J. (2012). phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 3, 217–223. doi: 10.1111/j.2041-210X.2011.00169.x
Rolland, R., Chalifoux, L., Snook, S., Ausman, L., and Johnson, L. (1997). Five spontaneous deaths associated with Clostridium difficile in a colony of cotton-top tamarins (Saguinus oedipus). Lab. Anim. Sci. 47, 472–476.
Song, S. J., Lauber, C., Costello, E. K., Lozupone, C. A., Humphrey, G., Berg-Lyons, D., et al. (2013). Cohabiting family members share microbiota with one another and with their dogs. Elife 2:e00458. doi: 10.7554/eLife.00458
Terio, K. A., Munson, L., Marker, L., Aldridge, B. M., and Solnick, J. V. (2005). Comparison of Helicobacter spp. in Cheetahs (Acinonyx jubatus) with and without Gastritis. J. Clin. Microbiol. 43, 229–234. doi: 10.1128/JCM.43.1.229-234.2005
Trevelline, B. K., Fontaine, S. S., Hartup, B. K., and Kohl, K. D. (2019). Conservation biology needs a microbial renaissance: a call for the consideration of host-associated microbiota in wildlife management practices. Proc. Biol. Soc. 286:20182448. doi: 10.1098/rspb.2018.2448
Wasimuddin, Menke, S., Melzheimer, J., Thalwitzer, S., Heinrich, S., Wachter, B., et al. (2017). Gut microbiomes of free-ranging and captive Namibian cheetahs: diversity, putative functions and occurrence of potential pathogens. Mol. Ecol. 26, 5515–5527. doi: 10.1111/mec.14278
Keywords: mammals, conservation, metagenomics, microbiome, bacteria, transmission
Citation: Trevelline BK and Moeller AH (2022) Robustness of Mammalian Gut Microbiota to Humanization in Captivity. Front. Ecol. Evol. 9:785089. doi: 10.3389/fevo.2021.785089
Received: 28 September 2021; Accepted: 16 November 2021;
Published: 03 January 2022.
Edited by:
Katherine Amato, Northwestern University, United StatesReviewed by:
Huan Li, Lanzhou University, ChinaJorge Doña, University of Illinois at Urbana–Champaign, United States
Copyright © 2022 Trevelline and Moeller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brian K. Trevelline, brian.trevelline@gmail.com; Andrew H. Moeller, ahm226@cornell.edu