 Assmaa ElSheikh
Assmaa ElSheikh Show-Ling Shyng
Show-Ling Shyng- 1Department of Chemical Physiology and Biochemistry, Oregon Health & Science University, Portland, OR, United States
- 2Department of Medical Biochemistry, Tanta University, Tanta, Egypt
Congenital hyperinsulinism (CHI) is the most common cause of persistent hypoglycemia in infancy/childhood and is a serious condition associated with severe recurrent attacks of hypoglycemia due to dysregulated insulin secretion. Timely diagnosis and effective treatment are crucial to prevent severe hypoglycemia that may lead to life-long neurological complications. In pancreatic β-cells, adenosine triphosphate (ATP)-sensitive K+ (KATP) channels are a central regulator of insulin secretion vital for glucose homeostasis. Genetic defects that lead to loss of expression or function of KATP channels are the most common cause of HI (KATP-HI). Much progress has been made in our understanding of the molecular genetics and pathophysiology of KATP-HI in the past decades; however, treatment remains challenging, in particular for patients with diffuse disease who do not respond to the KATP channel activator diazoxide. In this review, we discuss current approaches and limitations on the diagnosis and treatment of KATP-HI, and offer perspectives on alternative therapeutic strategies.
Introduction
Congenital hyperinsulinism (CHI) is a group of clinically, genetically, and morphologically heterogeneous disorders characterized by recurrent episodes of hyperinsulinemia and hypoglycemia due to dysregulated insulin secretion from pancreatic β-cells (1, 2). This condition can lead to neonatal seizures, developmental delay, and irreversible brain damage if not promptly diagnosed and treated (3). The age of clinical presentation in general correlates with the severity of the disease (4). Severe cases show symptoms of hypoglycemia early in the neonatal life, while milder forms are usually diagnosed later in infancy or childhood with recurrent attacks of hypoglycemia, which manifest following prolonged fasting or other health stress. CHI was first named as “idiopathic hypoglycemia of infancy” (5), which is no longer used after many genetic causes of the disease have been identified (6, 7). It was also once referred to as “nesidioblastosis” based on an early suggestion that the increased insulin secretion is secondary to budding of pancreatic islets observed in histological samples from patients with CHI (8). The use of this name to describe CHI was discontinued after nesidioblastosis was revealed to be a normal fetal and neonatal phenomenon (9, 10). Another discontinued historical term is persistent hyperinsulinemic hypoglycemia of infancy or PHHI, as it is now understood that the disease can be neonatal, infantile or childhood and can persist to adulthood (11). In addition to prototypical CHI, hyperinsulinism can be a pathology of a syndromic disease, including Beckwith-Wiedemann syndrome, Perlman syndrome, Kabuki syndrome, Turner syndrome, Sotos syndrome, and others (7, 12).
The estimated incidence of CHI is 1:28,000–1:50,000 in Western populations but as high as 1:2,500 in populations with higher rates of consanguinity (3, 13, 14). Variants in at least ten genes have now been linked to congenital hyperinsulinism, including genes that encode the KATP channel subunits (ABCC8 and KCNJ11), glucokinase (GCK), glutamate dehydrogenase (GLUD1), the mitochondrial enzyme 3-hydroxyacyl-CoA dehydrogenase (HADH), proton-linked monocarboxylate transporter (SLC16A1), mitochondrial uncoupling protein 2 (UCP2), hepatocyte nuclear factor 1 alpha (HNF1A) and 4 alpha (HNF4A), and hexokinase 1 (HK-1) (15). Defects in these proteins result in dysregulation of insulin secretion and impaired glucose homeostasis. Of the CHI-associated gene mutations, those in ABCC8 or KCNJ11 that lead to loss-of-function of KATP channels are the most common (16). The majority of KATP gene mutations identified to date are in ABCC8, which is much larger than KCNJ11 (16). This review summarizes current approaches and limitations on the diagnosis and treatment of KATP-HI, and offers perspectives on alternative therapeutic strategies. Readers interested in KATP channel structure-function and pharmacology are referred to several recent reviews (17–19).
KATP-HI: Molecular diagnosis
KATP channels have a central role in regulating insulin secretion from pancreatic β-cells in the islets of Langerhans (20, 21). The channel is composed of four pore-forming subunits Kir6.2, encoded by KCNJ11, and four regulatory subunits called sulfonylurea receptor 1 (SUR1), encoded by ABCC8 (22, 23) (Figures 1A, B). SUR1 is so named because it binds sulfonylurea drugs, which inhibit KATP channel activity and are commonly used to treat type 2 diabetes (24). Pancreatic KATP channels are gated physiologically by intracellular ATP and ADP; ATP acts on Kir6.2 to close the channel, while MgADP acts on SUR1 to open the channel (17). This enables KATP channels to serve as metabolic sensors, coupling serum glucose to insulin secretion. At basal glucose levels, the ATP/ADP ratios are relatively low to allow K+ conductance through KATP channels, which sets the plasma membrane in a hyperpolarized state to prevent insulin secretion. When blood glucose levels rise, glucose metabolism increases the ATP/ADP ratio, which favors closure of KATP channels, resulting in cell membrane depolarization, activation of voltage-gated Ca2+ channels, and exocytosis of insulin granules (25) (Figure 1C). The ability of KATP channels to control β-cell membrane excitability in response to blood glucose levels is essential for glucose homeostasis. In CHI, faulty KATP channel genes that reduce or abolish functional channels in the β-cell membranes uncouple blood glucose from insulin secretion, leading to inappropriate insulin secretion despite life-threatening hypoglycemia (21, 26, 27).
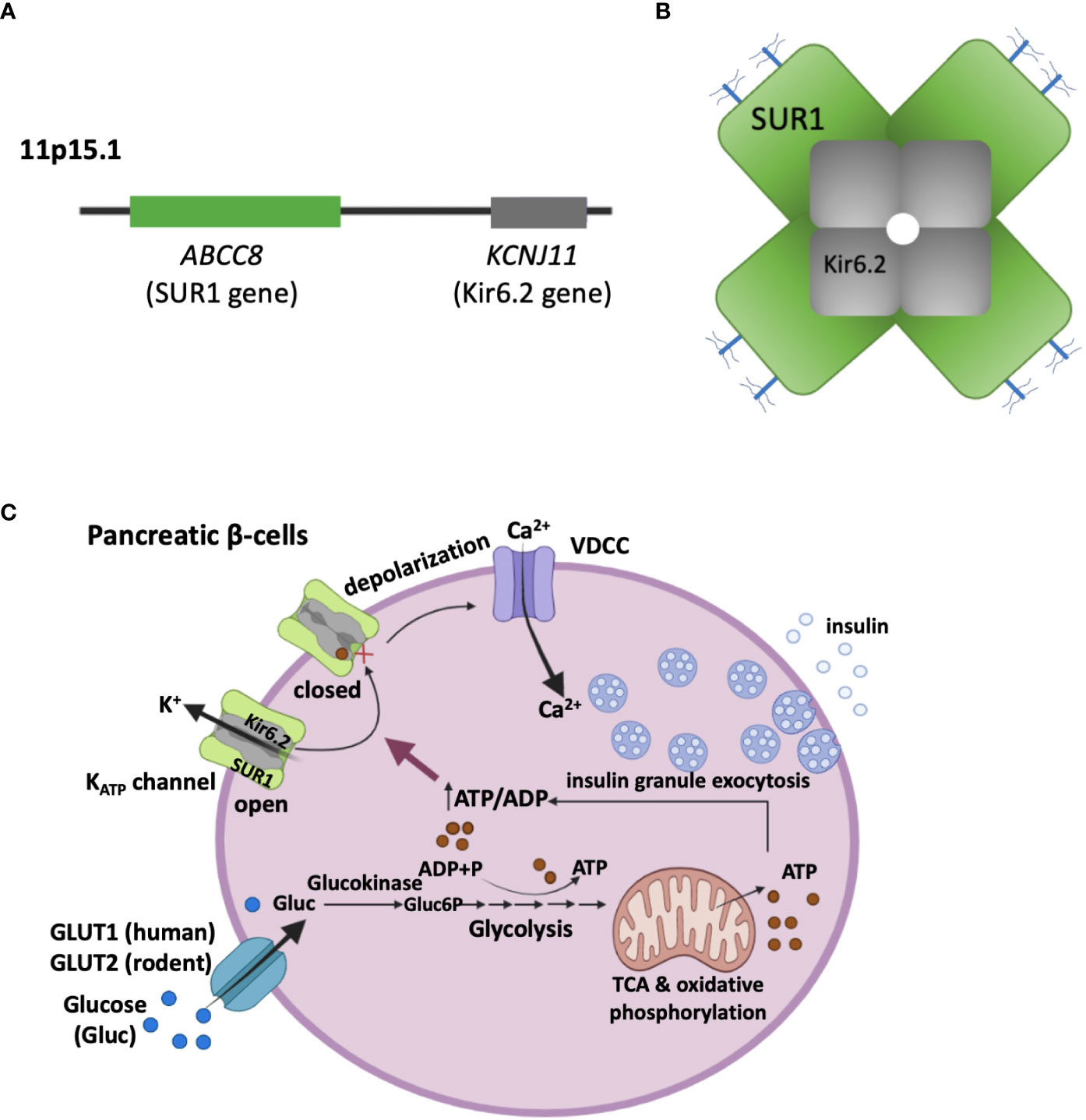
Figure 1 KATP channel composition and its role in coupling glucose metabolism to insulin secretion. (A) ABCC8 and KCNJ11 encoding the two pancreatic KATP channel subunits, SUR1 and Kir6.2 respectively, are located on the short arm of chromosome 11 (11p15.1). (B) Schematic representation of the KATP channel complex viewed in cross section. The K+-conducting pore is formed by four Kir6.2 subunits, which are surrounded by four SUR1 regulatory subunits. The branched blue sticks represent the two N-linked glycosylation sites in each SUR1. (C) β-cell KATP channels couple glucose metabolism to insulin secretion by regulating plasma membrane potential in response to varying blood glucose levels. With high serum blood glucose levels, glucose enters β-cells through glucose transporters (GLUT1 in human, and GLUT2 in rodents). Inside the cell, glucose is catabolized in the cytosol (Glycolysis) and the mitochondria (Tricarboxylic acid cycle, TCA), leading to an elevation of the ATP/ADP ratio. This results in closure of KATP channels, plasma membrane depolarization, opening of voltage-dependent Ca2+ channels (VDCC). The ensuing Ca2+ influx then triggers exocytosis of insulin secretory granules.
Molecular diagnosis of KATP-HI begins with genetic testing. Genomic DNA mutation screening of probands is done using genome isolated from peripheral blood or saliva (7, 28). However, interpreting the effect of novel ABCC8 or KCNJ11 variants can be challenging as they can be dominant or recessive functional mutations, or benign polymorphisms (16, 29). Recent advances combining genetic, clinical, and in vitro biochemical studies to determine which of the genetic variations affect transcription/splicing, translation, and function have significantly improved the diagnosis of KATP-HI (30, 31).
Focal versus diffuse KATP-HI
Histologically, there exist two forms of KATP-HI: focal and diffuse (15, 32). In focal disease, the defect is limited to a focal lesion in the pancreas due to a heterozygous, paternally inherited ABCC8 or KCNJ11 mutation coupled with loss of the normal maternal allele in a subset of pancreatic β-cells during embryonic development (28). In diffuse KATP-HI the entire pancreas is affected. Patients carrying homozygous, heterozygous mutations, or rare compound heterozygous mutations have all been reported (29). The two forms of KATP-HI are not easily distinguishable by clinical presentations (15). Genetic information from the proband and parents can point to possible focal disease. Indeed, in patients whose genetic testing identifies one paternally inherited recessive mutation in ABCC8 or KCNJ11 and no maternal mutation, there is > 95% likelihood of a focal disease (29). This can be directly confirmed by PET imaging using 18F-fluoro-L-DOPA. Localization of focal lesions by 18F-fluoro-L-DOPA PET imaging along with CT-angiography allows for surgical removal of focal lesions in most cases for a complete cure (33). By contrast, the diffuse form, if not responsive to pharmacological treatment or glucose infusion, may require near total pancreatectomy to manage hypoglycemia, leading to future complications (3, 34).
Dominant versus recessive KATP-HI
Determining whether the mutation is dominant or recessive is of great clinical importance, especially for guiding decisions on whether to conduct 18F-fluoro-L-DOPA PET scan to test for focal disease. Moreover, the information is important for genetic counseling concerning recurrence risk as well as for identifying other family members at risk for hypoglycemia (35, 36). In focal CHI, a paternally inherited faulty gene only manifests in a focal region of cells where loss of heterozygosity of the maternal allele occurs, but not in the rest of the pancreas. Thus KATP mutations identified in focal CHI are recessive. Since both inheritance of the paternal mutant allele and loss of the maternal allele are required for disease presentation, the recurrence among the siblings is rare; to our knowledge, it has only been reported once in two siblings (37). However, in consanguineous parents, mothers should be screened for the presence of the paternal mutation responsible for the focal form of CHI to avoid the possibility of diffuse CHI in future pregnancies due to inheritance of the mutation from both parents.
Unlike focal KATP-HI, diffuse KATP-HI may be dominant or recessive, which often correlates with whether the underlying mutation causes KATP channel gating or trafficking defects (see more discussion in “Mechanisms of KATP-HI mutations”). Recessive mutations, whether homozygous or compound heterozygous, are usually found in patients with severe disease and not responsive to diazoxide treatment (38). Dominant mutations have been identified in patients with mild, diazoxide-responsive disease as well as severe, diazoxide-unresponsive disease (39–43). For genetic counseling, homozygous recessive mutations are expected to have a recurrence rate of ~25%, while dominant heterozygous mutations have a recurrence rate up to 50%. Differentiating between dominant and recessive mutations can in some cases be challenging. For example, penetrance of a mutation may not be the same in all carriers (44). Differential expression of a dominant ABCC8 mutation has been observed in lymphocytes from two different carriers and proposed to account for the difference in their clinical presentation of CHI (45). Moreover, the pedigrees of CHI patients are often too small to clarify the inheritance pattern of novel mutations. Combining clinical, genetic, and functional studies using in vitro recombination systems can help resolve ambiguous cases.
Mechanisms of KATP-HI mutations
ABCC8 contains 42 exons encoding 1581 amino acids (or a common alternative isoform with 1582 residues), whereas KCNJ11 contains a single exon encoding 390 amino acids. Genetic variants in ABCC8 and KCNJ11 are found in both non-coding and coding regions. We have divided them into three broad classes: those causing impairment of transcription and translation, those disrupting folding, assembly, and trafficking, and those disrupting gating of KATP channels (Figure 2).
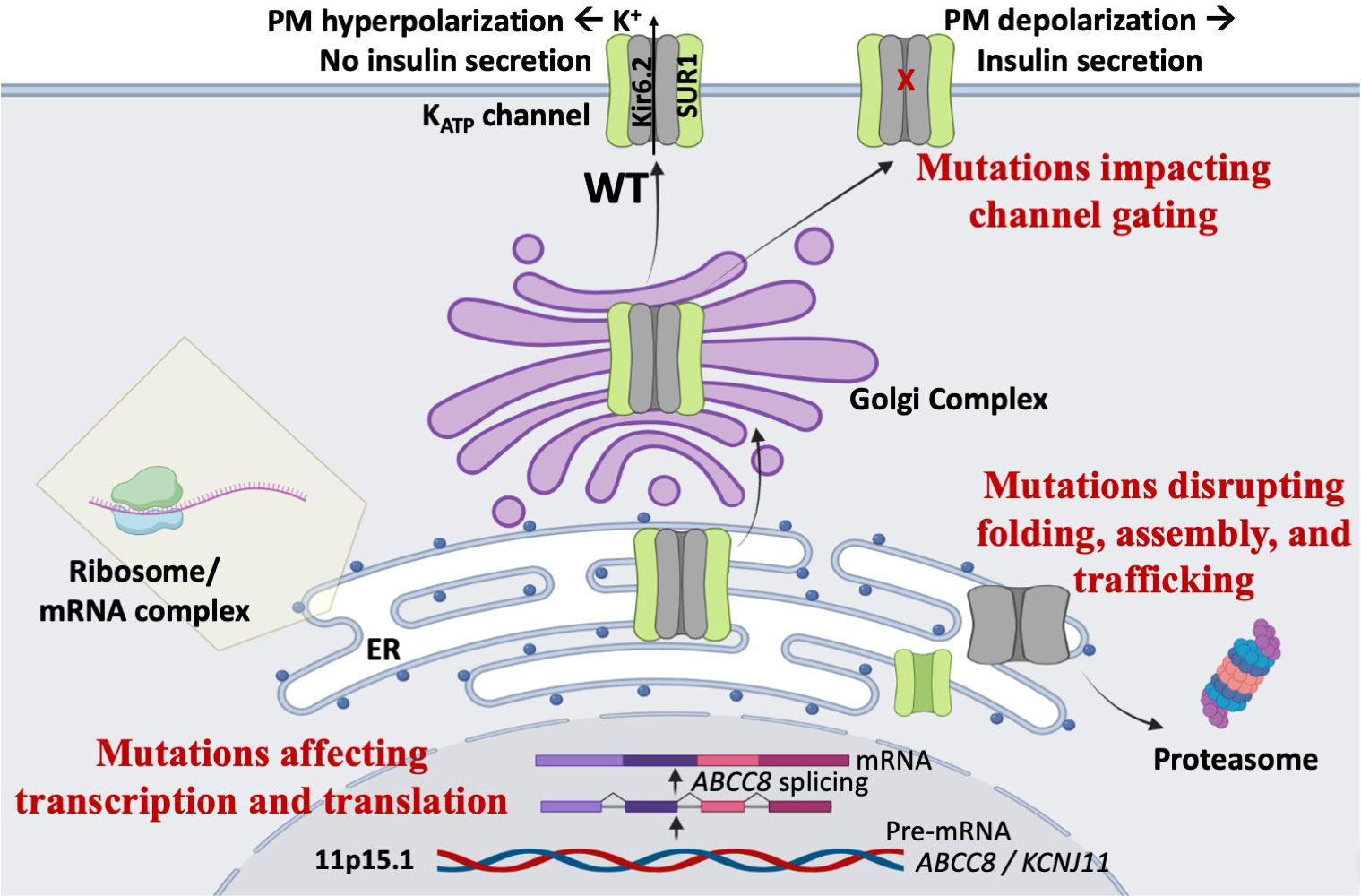
Figure 2 Mechanisms of KATP-HI mutations. Mutations in the KATP channel genes can lead to loss of channel function, persistent plasma membrane (PM) depolarization, and inappropriate insulin secretion via multiple mechanisms. First, mutations may impair gene transcription or protein translation. Second, mutations may disrupt KATP channel protein folding, assembly, and trafficking, thereby compromising surface expression of the channel. Third, mutations may cause gating defects that prevent channel opening when blood glucose levels decline.
In non-coding regions, genetic variations can in principle affect gene expression, or splicing in the case of ABCC8 to reduce transcript copies, and thereby KATP expression and function. Few studies have examined regulation of ABCC8 and KCNJ11 genes in pancreatic β-cells by cis-elements in the non-coding regions (46, 47). Definitive demonstration that a variation in these regions reduce transcript number would be very difficult. Several studies have presented in vitro evidence that genetic variations could disrupt splicing of SUR1 transcripts, especially variants located near the ABCC8 intron-exon boundaries, using a combination of bioinformatics and expression of mini-genes containing the variants or digital droplet PCR of patient lymphocytes (31, 48–51).
Mutations in the coding regions can introduce premature stop codons or frameshift, resulting in truncated, nonfunctional proteins, but can also be silent, i.e. without changing the encoded amino acid. Silent mutations could potentially affect protein translation and folding by altering mRNA structure or codon usage (52); however, this possibility has not been tested. Most commonly, mutations in the coding regions alter primary sequence of SUR1 or Kir6.2. These include missense mutations and indel mutations. Alterations in the primary sequence of channel proteins can reduce or abolish channel function by disrupting channel folding, assembly, trafficking and/or gating response to blood glucose levels (26, 27, 53).
Understanding how novel KATP channel missense/indel mutations affect channel expression and function greatly facilitates molecular diagnosis and therapeutic management of CHI. Currently, in silico methods are unable to accurately predict the functional impact of a mutation, and native β-cells from patients are mostly unavailable. Endocrinologists around the world collaborate with several academic laboratories including ours to characterize effects of these mutations in recombinant expression systems using biochemical and electrophysiological assays (54). For these studies, recombinant mutant channels are transiently expressed in a mammalian cell line that does not express endogenous KATP channels, such as COSm6 or HEK293 cells. To assess the impact of a mutation on channel properties, mutant channels are first expressed and evaluated as a homogeneous population mimicking homozygous state. For heterozygous mutations, follow-up studies where the mutant is co-expressed with the wild-type at 1:1 ratio to simulate the heterozygous state may be performed to determine whether a mutation has a dominant effect over the wild-type allele on channel expression and function (30).
A rapid and informative method to determine whether a mutation is pathogenic is the Rb+ efflux assay (54, 55). In this assay, Rb+, which passes through KATP channels, is a tracer ion that acts as a surrogate K+ and can be detected using a radioactive form of Rb+, 86Rb+ (54), or by atomic absorption spectroscopy (56), to monitor KATP channel activity. In β-cells, KATP channels open in response to glucose deprivation, which lowers the intracellular ATP/ADP ratio. In COSm6 cells, which are not glucose-responsive, reduction of ATP/ADP ratios can be triggered by incubating cells with metabolic inhibitors including the glycolysis inhibitor 2-deoxyglucose and the oxidative phosphorylation inhibitor oligomycin, which reduce ATP production. In COSm6 cells transiently expressing KATP channels and preloaded with Rb+, opening of KATP channels leads to increased Rb+ efflux. Reduced efflux observed in cells expressing channels harboring a mutation compared to cells expressing wild-type channels would indicate that the mutation causes loss of channel function, therefore has a pathogenic role in CHI. In addition to its utility in evaluating the pathogenic role of a mutation, the Rb+ efflux assay is also useful for assessing whether a mutant form of the channel functionally responds to the KATP channel opener diazoxide, a frontline treatment for CHI (57). Restoration of Rb+ efflux by diazoxide would indicate a clinical response of patients with the mutation to diazoxide treatment. Indeed, there is in general good agreement between response in Rb+ efflux assays and clinical response to diazoxide based on published work (29, 40, 42, 58). However, phenotypical variations caused by the same mutation could result in patient response that deviates from prediction based on Rb+ efflux assays using recombinant mutant channels expressed in cultured cells.
Mutations which reduce KATP channel activity in Rb+ efflux assays can disrupt the ability of the channel to open at low glucose concentrations (gating defects) and/or reduce the number of channels present in the plasma membrane (trafficking defects). The precise mechanisms can be further determined using biochemical and electrophysiological assays, which will aid in the decision on disease treatment plans.
Mutations disrupting channel gating
Accurate response of KATP channels to changes in intracellular ATP and MgADP concentrations is essential for glucose-insulin secretion coupling (25). In addition, channel activity relies on interactions with membrane phospholipids, in particular PI4,5P2 (PIP2) (59, 60). Patch-clamp recordings of KATP channels using the inside-out configuration allows for precise control of the solution on the intracellular face of channels contained in a membrane patch to evaluate channel response to the above physiological ligands. The most common gating defect seen in CHI-associated mutations is impaired response to MgADP/MgATP (61, 62) (Figure 3A), which stimulates KATP channels by binding to the SUR1 nucleotide binding domains (NBDs). Accordingly, these mutations are almost exclusively located in SUR1, and many in the NBDs (62). In general, disease severity correlates with the extent of nucleotide response impairment (30, 40, 42, 58). Moreover, mutations that impair MgADP response also tend to impair channel response to diazoxide (40, 58), which acts by stabilizing SUR1 in MgADP/MgATP stimulated conformation (23). As diazoxide is the only KATP-targeting drug currently available to treat CHI, patients with mutations that cause severe impairment of MgADP response often fail to respond to diazoxide and require alternative interventions (63). Less common are mutations which reduce the open probability of the channel, for example Kir6.2 mutations that reduce channel response to membrane PIP2 (64) or render the current unstable by disrupting Kir6.2 subunit-subunit interactions (65) (Figure 3A). Channels with these mutations generally remain responsive to MgADP and diazoxide (64, 65).
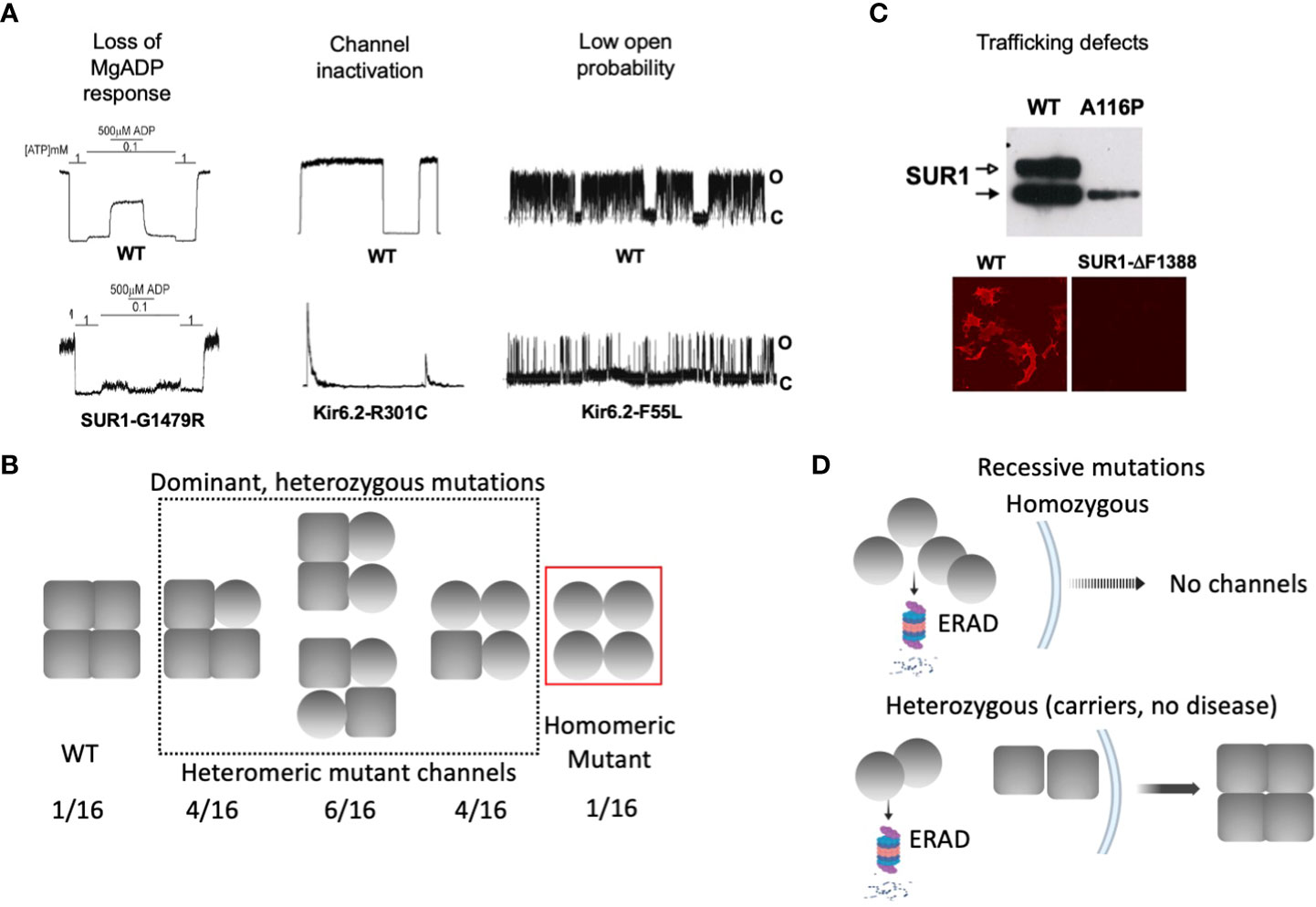
Figure 3 Biochemical and functional characteristics of KATP channel mutations associated with dominant versus recessive diffuse CHI. (A) Gating defects that have been observed in CHI-associated mutations, including loss of MgADP response, spontaneous current decay. i.e. channel inactivation, and reduced channel open probability. For each defect, representative inside-out patch-clamp recordings of WT and homomeric mutant channels are shown. For monitoring MgADP response, channels were exposed to 1 mM ATP, 0.1 mM ATP, or 0.1 mM ATP plus 0.5 mM MgADP as indicated. In all recordings, channel openings (O) are shown as upward deflections, and channel closures (C) as flat baselines. (B) Schematic showing that gating mutations tend to be associated with dominant disease, where heterozygous patients are expected to express a mixed population of channels containing 0-4 mutant subunits. In these cases, the mutation causes gating defects but not defects in channel folding, assembly, and trafficking. (C) Top: A western blot showing both core-glycosylated and complex-glycosylated wild-type SUR1 band when co-expressed with Kir6.2, indicating channel assembly and ability to traffic to the cell surface. In contrast, a trafficking mutation SUR1-A116P, fails to generate the mature complex-glycosylated band. Bottom: Surface immunofluorescence staining showing lack of expression of a trafficking mutant SUR1-ΔF1388, in contrast to WT. (D) Schematic showing trafficking mutations are usually seen in recessive disease. In patients carrying homozygous mutations, mutant protein is targeted for ER-associated proteasomal degradation (ERAD) and is unable to form channels and traffic to the plasma membrane. In heterozygous individuals, mutant protein is degraded and unable to assemble with WT subunit, leaving WT protein to assemble into functional channels that traffic to the plasma membrane, escaping the disease.
In diffuse KATP-HI, gating mutations often follow a dominant inheritance pattern (Figure 3B). Because each KATP channel contains four Kir6.2 and four SUR1 subunits, heterozygous mutations presumably generate a mixed channel population containing 0, 1, 2, 3, or 4 mutant subunits at a ratio of 1:4:6:4:1, with the gating defect more pronounced as the number of mutant subunit increases. A subunit with a mutation that affects channel gating, but that is able to co-assemble and traffic to the cell surface, would have its gating defect manifested in the total surface channel population. The extent of the MgADP and diazoxide gating defect has been correlated with disease severity and clinical response to diazoxide in CHI children with dominant SUR1 mutations (40, 58). Thus, response of mutant channels to MgADP and diazoxide in electrophysiology experiments may be helpful in predicting disease phenotype and clinical response to diazoxide.
Mutations disrupting channel folding, assembly and trafficking
Many CHI mutations cause improper channel folding, assembly, and trafficking to the plasma membrane (53). The consequent reduction in KATP currents leads to a state of persistent β-cell membrane depolarization and uncontrolled insulin secretion. Translation, folding, and assembly of KATP channel subunits occur in the endoplasmic reticulum (ER) membrane. Upon correct assembly into a hetero-octameric complex, channels exit the ER and traffic to the Golgi. In the Golgi apparatus, SUR1 becomes complex glycosylated at its two N-linked glycosylation sites, giving rise to a mature form that migrates slower on SDS gel compared to the core-glycosylated immature form found in the ER (66, 67). The appearance and intensity of the mature SUR1 band can be used to infer channel proteins that are competent to traffic to the plasma membrane. Conversely, the absence or weakened mature SUR1 band indicates folding/assembly/trafficking defects (Figure 3C). More direct assessment of KATP channel surface expression can be achieved by surface immunofluorescence staining, surface biotinylation followed by pulldown of biotinylated protein and immunoblotting with anti-SUR1 antibody, or by electrophysiological measurement of current density (68–71).
To date, about fifty missense/indel SUR1 and Kir6.2 mutations have been reported to impair KATP channel expression at the cell surface, collectively referred to as trafficking mutations (29, 30, 53, 70, 72, 73). The extent of the impairment varies, with some mutations completely abolishing the mature SUR1 band and others reducing but not eliminating mature SUR1 (71, 74). A prominent example of the former is SUR1ΔF1388 (68), a common recessive mutation found in the Ashkenazi Jewish population (75). The mutant protein is retained in the ER, unable to reach the mature complex-glycosylated state likely because it is misfolded (68), akin to the most prevalent cystic fibrosis-causing mutation CFTRΔF508 (76). Although trafficking mutations are mapped throughout the SUR1 and Kir6.2 proteins, a high percentage are found in the first transmembrane domain of SUR1, called TMD0, and the first transmembrane helix (TM1) of Kir6.2 (53, 72). Recent high resolution 3D structures of the channel complex show that SUR1-TMD0 makes direct contact with Kir6.2-TM1, forming the primary anchor between the two subunits (77–79) (Figure 4). Mutations in these domains likely interfere with channel assembly, and thereby the trafficking of channels to the plasma membrane.
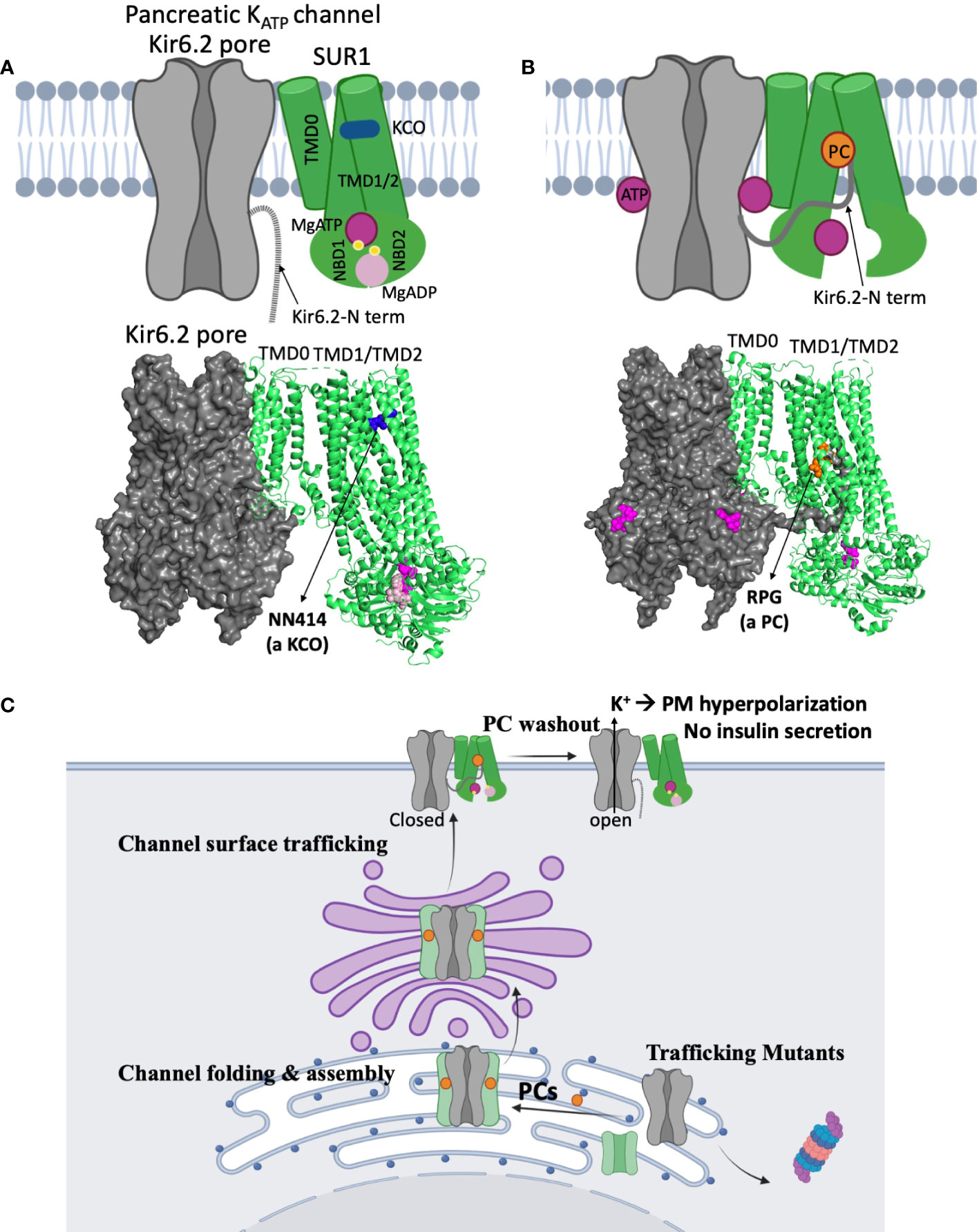
Figure 4 Structural insights on the mechanisms of KATP channel pharmacological modulators. (A) A cartoon model (top) illustrating the mechanism by which potassium channel openers (KCOs) stimulate channel activity, based on a recent structure of a channel bound to MgATP/MgADP and a KCO NN414 shown below (PDB ID: 7W4O; only one SUR1 subunit shown for clarity). In the structure, the SUR1 NBD1 and NBD2 are bound to MgATP and MgADP respectively and dimerized, a conformation that stimulates channel activity. NN414, which binds at a transmembrane domain pocket further stabilizes channels in the SUR1-NBDs dimerized, activated conformation to potentiate channel opening. By inference, diazoxide, a KCO used to treat some CHI patients, promotes channel opening via a similar mechanism. (B) A cartoon (top) showing how KATP channel inhibitors work as pharmacological chaperones (PC) based on cryoEM structures. A channel structure determined in the presence of ATP and a PC repaglinide is shown at the bottom (PDB ID: 7U1S). The structure shows that the PC (orange circle), binds in a transmembrane pocket in SUR1 formed by helices from TMD1 and TMD2. The Kir6.2 N-terminus is in a cavity formed by the two transmembrane helix bundles (TMD1/2) above the two NBDs, and is adjacent to the bound PC. This stabilizes the interaction between the N-terminal domain of Kir6.2 and SUR1 to facilitate the formation of mature hetero-octameric complex of the mutant channel. However, in this conformation the NBDs of SUR1 are separate, unable to respond to MgADP stimulation upon glucose deprivation. This explains why KATP PCs also inhibit channel activity. (C) A cartoon showing a proposed mechanism of how reversible inhibitor PCs can enhance channel surface expression without permanently compromising channel function. Inhibitor PCs bind to the mutant channel subunits and enhance the interaction between SUR1 central cavity and Kir6.2 N-terminus to promote mutant channel assembly and surface trafficking. Reversible inhibitor PCs are released upon washout after mutant channels are rescued to the surface, allowing the channels to open under low blood glucose conditions and inhibit insulin secretion.
In contrast to gating mutations, trafficking mutations reported to date have been associated with recessive CHI and do not respond to diazoxide treatment (15). The recessive nature of these mutations implies that under heterozygous condition the mutant allele may be too defective or is outcompeted by the wild-type allele to form a channel complex. Indeed, some trafficking mutations have been shown to reduce subunit association, and some have been shown to cause rapid degradation of channel proteins (72, 80). This leaves the wild-type subunit to assemble and form normal functional channels, resulting in a haplosufficiency phenotype (Figure 3D). It is possible that certain mutations that cause only mild trafficking defects such that mutant proteins are still able to assemble with the wild-type allele and reach the cell surface, can remain undetected in heterozygous conditions where the mutation does not disrupt gating sufficiently to cause disease. In this regard, it is interesting to note that many heterozygous KATP mutations identified in neonatal diabetes have been shown to cause trafficking defects in addition to gain-of-function gating defects. These mutant proteins are still able to form complex with wild-type subunit and exert their gain-of-function gating effect to cause dominant disease (81–83).
Treatment of KATP-HI: Challenges and opportunities
Early diagnosis and effective treatment are critical to prevent serious neurocognitive impairments that dramatically impact CHI patients and their families due to severe associated morbidity with lifelong disability (3, 57). For focal KATP-HI, complete surgical removal of the lesion often leads to a cure (33). However, treatment for diffuse KATP-HI remains challenging. Current mainstay treatments include diazoxide, somatostatin analogues, continuous enteral feedings/dextrose, and surgery (3, 57). Diazoxide treatment would be preferred if patients have β-cell KATP channels that have sufficient response to the drug. However, many patients do not respond to diazoxide. Even with patients who do respond to diazoxide, there is often significant side effects (3, 11, 84). Currently, diazoxide is the only KATP channel opener approved in the US and Europe. Diazoxide not only activates pancreatic KATP channels but also vascular KATP channels, causing excess hair growth and other cardiovascular complications resembling Cantú syndrome patients carrying gain of function mutations in vascular KATP channels (85). Continuous feeding/dextrose infusion can restore normal glycemia, but is a heavy burden on caretakers and patients and is associated with many complications including rapid weight gain and food aversion (3). For patients with diffuse disease and unresponsive to diazoxide and somatostatin analogues, pancreatectomy is often performed to correct the life-threatening hypoglycemia. This leads to insulin dependency later in life and digestive complications from removal of exocrine tissues (86).
With better understanding of the molecular mechanism underlying insulin secretion regulation and CHI, off label use of other drugs, such as somatostatin analogues including longer-acting octreotide (sandostatin LP, monthly injections rather than daily), glucagon, GLP-1 receptor antagonists (exendin 9-39), mTOR inhibitors (sirolimus), the calcium channel blocker nifedipine, and anti-insulin receptor antibody have been considered for CHI patients who are unresponsive to the maximum dose of diazoxide (87). However, these alternative treatments have limited success or are still in clinical trials, and do not target the root causes of KATP dysfunction in diazoxide-unresponsive diffuse CHI, namely, severe MgADP/diazoxide response defects or impaired channel trafficking to the plasma membrane. Overcoming these limitations requires better understanding of the structural mechanisms underlying channel function, dysfunction, and pharmacology.
KATP channel structures and implications for KATP-HI
A major advance in KATP channel research in recent years is our ability to visualize 3D channel structures at near atomic resolution. Using single-particle cryogenic electron microscopy (cryoEM), structures of pancreatic KATP bound to various inhibitors and activators have been determined (53, 56, 77–79, 88–91). These structures not only reveal the binding sites of key physiological and pharmacological ligands of KATP channels but also provide insights to the mechanisms of how ligands regulate channel assembly and function (17). The knowledge is of great value in future efforts to expand KATP channel pharmacology and overcome current therapeutic challenges.
Of particular interest are recent studies showing the pancreatic, cardiac, and vascular KATP channel SUR subunits, SUR1, SUR2A, and SUR2B, respectively, bound to their selective activators, NN414 (for SUR1) and P1075 or levcromakalim (for SUR2A and SUR2B) (56, 92). In the structure of SUR1 bound to NN414, NN414 sits in a transmembrane pocket that is stabilized by dimerization of the MgADP/MgATP bound NBDs of SUR1 (Figure 4A). A similar binding location for P1075 or levcromakalim is observed in SUR2A/2B. Although no diazoxide bound structure is yet available, diazoxide most likely binds to the same pocket. The structures help to explain why the effect of diazoxide requires MgADP/MgATP and why mutations that disrupt MgADP response also impair diazoxide response. Of note, NN414 stimulates KATP channel activity like diazoxide, but is more potent and selective for SUR1 containing pancreatic KATP channels (93, 94). NN414 was previously tested in clinical trials for type 2 diabetes based on the idea that opening of pancreatic KATP will allow β-cell rest and restore insulin secretion; however, the trial was stopped due to concerns over elevated liver enzymes (94). Whether NN414 can be used to treat CHI at concentrations that do not elicit hepatic or Cantú-like side effects remains untested. Regardless, the structures of different SUR isoforms bound to different openers (17, 19) provide a framework for rational drug design with improved specificity for pancreatic KATP channels without undesirable side effects to expand the medical options for CHI.
A significant number of KATP gating mutations result in channels with little or no MgADP and diazoxide response (30, 40, 58). For these mutations, drugs that open channels independent of the stimulatory effect of MgADP on SUR1 may be explored. For example, recent channel structures showing an open Kir6.2 pore conformation (56, 89) could be used to search for compounds, via virtual screening, that may stabilize the channel in an open state.
Lastly, mutations which impair channel expression at the cell surface also require novel approaches. Here, pharmacological chaperones, small molecules that bind to KATP channel proteins to facilitate biogenesis, correct misfolding, and restore full or partial function of the affected channels represents a promising approach (53, 95). This approach has been highly successful in cystic fibrosis, a disease caused by mutations in the CFTR gene. CFTR is a chloride channel that shares structural similarity with SUR1 (96), the regulatory subunit of KATP channels. The most common cystic fibrosis-causing mutation is ΔF508, which impairs folding, and thereby surface expression of the protein (76). Several small molecules that correct the folding and trafficking defect of mutant CFTR (lumacaftor, tezacaftor, and elexacaftor) have been recently approved for clinical use (76, 97, 98).
Sulfonylureas were the first reported KATP pharmacological chaperones (99). These antidiabetics, which inhibit KATP channels and stimulate insulin secretion, include the high affinity sulfonylurea glibenclamide and the low affinity sulfonylurea tolbutamide. Subsequently, glinides, a second class of KATP inhibiting antidiabetics including repaglinide, was found to have similar pharmacological chaperone effects (100). More recently, carbamazepine, an anticonvulsant known to block voltage-gated sodium channels was also discovered as a KATP pharmacological chaperone (69). Interestingly, these compounds are all KATP channel inhibitors (69, 101). Moreover, each of these KATP channel inhibitors are effective for only a subset of trafficking mutations. In particular, they promote trafficking of channels harboring mutations in the TMD0 domain of SUR1, which is the primary domain that interacts with Kir6.2 (69, 100), suggesting drug binding corrects trafficking defects by facilitating subunit assembly (102). By examining cryoEM structures of channels bound to glibenclamide, repaglinide, or carbamazepine, it was discovered that the distal N-terminus of Kir6.2 cooperates with SUR1 for drug binding, and drug binding in turn stabilizes Kir6.2-N terminus interaction with SUR1 (103) (Figure 4B). Thus, these drugs likely exert their channel chaperoning and inhibition effects via the same mechanism (Figure 4B), i.e. by stabilizing the Kir6.2 N-terminus in the SUR1 ABC core cavity. By contrast, channel openers such as diazoxide and NN414, which stabilize SUR1-NBD dimerization and exclude Kir6.2 N-terminus from the SUR1 ABC core cavity (Figure 4A), diminish Kir6.2-SUR1 interactions and have been shown to lack chaperoning activity (74).
While promising, translation of the above basic science finding to CHI treatment has a number of challenges. First, all KATP pharmacological chaperones reported to date are inhibitors. The most potent chaperones like glibenclamide and repaglinide are also the most potent inhibitors. The nearly irreversible inhibition of channels by these high affinity inhibitors precludes functional recovery of mutant channels rescued to the cell surface (99). To date, tolbutamide is by far the most reversible inhibitor that is also effective in rescuing mutant channels to the cell surface, albeit at significantly higher concentrations (99). Tolbutamide, a first-generation sulfonylurea which has been around since the 1950s, has largely been replaced with other oral hypoglycemics and is no longer available in the US. While early randomized trials associated its use with increased cardiovascular and all-cause mortality, the increased cardiovascular and all-cause mortality was not statistically significant (104). Off label use of tolbutamide as a potential pharmacologic therapy for CHI patients with diffuse disease and KATP trafficking mutations is an intriguing possibility (Figure 4C). Secondly, some trafficking mutations also disrupt channel gating. For example, two CHI-causing SUR1 trafficking mutations, R74W and E128K, also render channels less sensitive to ATP inhibition such that upon pharmacological rescue to the plasma membrane mutant channels cause membrane hyperpolarization and decreased glucose-stimulated insulin secretion in cultured insulinoma cells that resemble neonatal diabetes mutation phenotypes (105). Thirdly, not all trafficking mutations respond to the KATP pharmacological chaperones identified to date (71, 99). These mutations, largely located outside TMD0 of SUR1, likely cause severe misfolding such that mutant proteins are triaged for degradation (99, 106). Additional drug screening studies such as those done for CFTRΔF508 will be required to overcome defects caused by such mutations. Finally, there is currently no animal models to test the in vivo feasibility of pharmacological chaperone therapy. Recent development of induced pluripotent stem cells (iPSCs) derived from a CHI patient carrying a homozygous trafficking mutation SUR1-V187D (107) could serve as an intermediate experimental model to test the effect of reversible KATP pharmacological chaperones such as tolbutamide.
Concluding remarks
Much progress has been made in the diagnosis and management of KATP-HI since the first report linking KATP channel gene mutations to CHI (108). There are now hundreds of mutations that have been identified, and there has been steady progress in our understanding of genotype-phenotype correlation, mutation mechanisms, and drug response. The growing knowledge base facilitates rapid diagnosis and treatment. Despite the progress, timely and accurate molecular diagnosis of patients carrying variants of unknown significance, and treatment of diazoxide-unresponsive diffuse disease caused by severe gating and trafficking mutations remain challenging. With recent rapid technical advances in gene sequencing, bioinformatics, channel structure determination, machine-learning based drug design, and gene therapy, there is great optimism that new and personalized therapies for KATP-HI will become a reality in the not-too-distant future.
Author contributions
All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Institutes of Health grants DK066485 and GM145784 (to Show-Ling Shyng) and a predoctoral scholarship from the Egyptian Government to Assmaa ElSheikh.
Acknowledgments
We thank Dr. Bruce L. Patton for editing the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Giri D, Hawton K, Senniappan S. Congenital hyperinsulinism: recent updates on molecular mechanisms, diagnosis and management. J Pediatr Endocrinol Metab (2022) 35(3):279–96. doi: 10.1515/jpem-2021-0369
2. Thornton PS, Stanley CA, De Leon DD. Congenital hyperinsulinism: An historical perspective. Horm Res Paediatr (2022) 95(6):631–7. doi: 10.1159/000526442
3. Banerjee I, Raskin J, Arnoux J.B, Leon De D.D, Weinzimer S.A, Hammer M, et al. Congenital hyperinsulinism in infancy and childhood: challenges, unmet needs and the perspective of patients and families. Orphanet J Rare Dis (2022) 17(1):61. doi: 10.1186/s13023-022-02214-y
4. de Lonlay P, Fournet JC, Touati G, Groos MS, Martin D, Sevin C, et al. Heterogeneity of persistent hyperinsulinaemic hypoglycaemia. a series of 175 cases. Eur J Pediatr (2002) 161(1):37–48. doi: 10.1007/s004310100847
5. Mc QI. Idiopathic spontaneously occurring hypoglycemia in infants; clinical significance of problem and treatment. AMA Am J Dis Child (1954) 87(4):399–428.
6. Galcheva S, Demirbilek H, Al-Khawaga S, Hussain K. The genetic and molecular mechanisms of congenital hyperinsulinism. Front Endocrinol (Lausanne) (2019) 10:111. doi: 10.3389/fendo.2019.00111
7. Hewat TI, Johnson MB, Flanagan SE. Congenital hyperinsulinism: Current laboratory-based approaches to the genetic diagnosis of a heterogeneous disease. Front Endocrinol (Lausanne) (2022) 13:873254. doi: 10.3389/fendo.2022.873254
8. Yakovac WC, Baker L, Hummeler K. Beta cell nesidioblastosis in idiopathic hypoglycemia of infancy. J Pediatr (1971) 79(2):226–31. doi: 10.1016/S0022-3476(71)80105-1
9. Jaffe R, Hashida Y, Yunis EJ. Pancreatic pathology in hyperinsulinemic hypoglycemia of infancy. Lab Invest (1980) 42(3):356–65.
10. Rahier J, Wallon J, Henquin JC. Cell populations in the endocrine pancreas of human neonates and infants. Diabetologia (1981) 20(5):540–6. doi: 10.1007/BF00252762
11. Arnoux J-B, Verkarre V, Saint-Martin C, Montravers F, Brassier A, Valayannopoulos V. F, et al. Congenital hyperinsulinism: current trends in diagnosis and therapy. Orphanet J Rare Dis (2011) 6(1):63. doi: 10.1186/1750-1172-6-63
12. Kostopoulou E, Dastamani A, Guemes M, Clement E, Caiulo S, Shanmugananda P, et al. Syndromic forms of hyperinsulinaemic hypoglycaemia-a 15-year follow-up study. Clin Endocrinol (Oxf) (2021) 94(3):399–412. doi: 10.1111/cen.14393
13. Pasquini TLS, Mesfin M, Schmitt J, Raskin J. Global registries in congenital hyperinsulinism. Front Endocrinol (Lausanne) (2022) 13:876903. doi: 10.3389/fendo.2022.876903
14. Amaratunga SA, Tayeb TH, Dusatkova P, Pruhova S, Lebl J. Invaluable role of consanguinity in providing insight into paediatric endocrine conditions: Lessons learnt from congenital hyperinsulinism, monogenic diabetes, and short stature. Horm Res Paediatr (2022) 95(1):1–11. doi: 10.1159/000521210
15. Rosenfeld E, Ganguly A, De Leon DD. Congenital hyperinsulinism disorders: Genetic and clinical characteristics. Am J Med Genet C Semin Med Genet (2019) 181(4):682–92. doi: 10.1002/ajmg.c.31737
16. De Franco E, Saint-Martin C, Brusgaard K, Johnson Knight AE, Aguilar-Bryan L, Bowman P, et al. Update of variants identified in the pancreatic beta-cell K(ATP) channel genes KCNJ11 and ABCC8 in individuals with congenital hyperinsulinism and diabetes. Hum Mutat (2020) 41(5):884–905. doi: 10.1002/humu.23995
17. Driggers CM, Shyng SL. Mechanistic insights on KATP channel regulation from cryo-EM structures. J Gen Physiol (2023) 155(1):e202113046. doi: 10.1085/jgp.202113046
18. Wu JX, Ding D, Chen L. The emerging structural pharmacology of ATP-sensitive potassium channels. Mol Pharmacol (2022) 102(5):234–9. doi: 10.1124/molpharm.122.000570
19. Martin GM, Patton BL, Shyng SL. K(ATP) channels in focus: Progress toward a structural understanding of ligand regulation. Curr Opin Struct Biol (2023) 79:102541. doi: 10.1016/j.sbi.2023.102541
20. Aguilar-Bryan L, Bryan J. Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocr Rev (1999) 20(2):101–35. doi: 10.1210/edrv.20.2.0361
21. Ashcroft FM. K(ATP) channels and insulin secretion: a key role in health and disease. Biochem Soc Trans (2006) 34(Pt 2):243–6. doi: 10.1042/BST0340243
22. Inagaki N, Gonoi T, Clement JPT, Namba N, Inazawa J, Gonzalez G, et al. Reconstitution of IKATP: An inward rectifier subunit plus the sulfonylurea receptor. Science (1995) 270(5239):1166–70. doi: 10.1126/science.270.5239.1166
23. Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature (2006) 440(7083):470–6. doi: 10.1038/nature04711
24. Aguilar-Bryan L, Nichols C.G, Wechsler S.W, Clement J. P. t, Boyd A.E 3rd, Gonzalez G, et al. Cloning of the beta cell high-affinity sulfonylurea receptor: a regulator of insulin secretion. Science (1995) 268(5209):423–6. doi: 10.1126/science.7716547
25. Ashcroft FM. The walter b. cannon physiology in perspective lecture, 2007. ATP-sensitive k+ channels and disease: from molecule to malady. Am J Physiol Endocrinol Metab (2007) 293(4):E880–9. doi: 10.1152/ajpendo.00348.2007
26. Huopio H, Shyng SL, Otonkoski T, Nichols CG. K(ATP) channels and insulin secretion disorders. Am J Physiol Endocrinol Metab (2002) 283(2):E207–16. doi: 10.1152/ajpendo.00047.2002
27. Sharma N, Crane A, Gonzalez G, Bryan J, Aguilar-Bryan L. Familial hyperinsulinism and pancreatic beta-cell ATP-sensitive potassium channels. Kidney Int (2000) 57(3):803–8. doi: 10.1046/j.1523-1755.2000.00918.x
28. De Leon DD, Stanley CA. Congenital hypoglycemia disorders: New aspects of etiology, diagnosis, treatment and outcomes: Highlights of the proceedings of the congenital hypoglycemia disorders symposium, Philadelphia April 2016. Pediatr Diabetes (2017) 18(1):3–9. doi: 10.1111/pedi.12453
29. Snider KE, Becker S, Boyajian L, Shyng SL, MacMullen C, Hughes N, et al. Genotype and phenotype correlations in 417 children with congenital hyperinsulinism. J Clin Endocrinol Metab (2013) 98(2):E355–63. doi: 10.1210/jc.2012-2169
30. Boodhansingh KE, Kandasamy B, Mitteer L, Givler S, Leon De DD, Shyng SL, et al. Novel dominant K(ATP) channel mutations in infants with congenital hyperinsulinism: Validation by in vitro expression studies and in vivo carrier phenotyping. Am J Med Genet A (2019) 179(11):2214–27. doi: 10.1002/ajmg.a.61335
31. Saint-Martin C, Miere Cauchois-Le M, Rex E, Soukarieh O, Arnoux JB, Buratti J, et al. Functional characterization of ABCC8 variants of unknown significance based on bioinformatics predictions, splicing assays, and protein analyses: Benefits for the accurate diagnosis of congenital hyperinsulinism. Hum Mutat (2021) 42(4):408–20. doi: 10.1002/humu.24164
32. Bellanne-Chantelot C, Saint-Martin C, Ribeiro MJ, Vaury C, Verkarre V, Arnoux JB, et al. ABCC8 and KCNJ11 molecular spectrum of 109 patients with diazoxide-unresponsive congenital hyperinsulinism. J Med Genet (2010) 47(11):752–9. doi: 10.1136/jmg.2009.075416
33. Pizzoferro M, Masselli G, Maiorana A, Casciani E, Sollaku S, Dionisi-Vici C, et al. PET/CT in congenital hyperinsulinism: transforming patient’s lives by molecular hybrid imaging. Am J Nucl Med Mol Imaging (2022) 12(2):44–53.
34. Rasmussen AG, Melikian M, Globa E, Detlefsen S, Rasmussen L, Petersen H, et al. The difficult management of persistent, non-focal congenital hyperinsulinism: A retrospective review from a single, tertiary center. Pediatr Diabetes (2020) 21(3):441–55. doi: 10.1111/pedi.12989
35. Sait H, Sharma L, Dabadghao P, Phadke SR. Congenital hyperinsulinemia of infancy: Role of molecular testing in management and genetic counseling. Indian J Pediatr (2022) 89(4):395–8. doi: 10.1007/s12098-021-04014-x
36. Stanley CA. Perspective on the genetics and diagnosis of congenital hyperinsulinism disorders. J Clin Endocrinol Metab (2016) 101(3):815–26. doi: 10.1210/jc.2015-3651
37. Ismail D, Smith VV, Lonlay P, Ribeiro MJ, Rahier J, Blankenstein O, et al. Familial focal congenital hyperinsulinism. J Clin Endocrinol Metab (2011) 96(1):24–8. doi: 10.1210/jc.2010-1524
38. Faletra F, Snider K, Shyng SL, Bruno I, Athanasakis E, Gasparini P, et al. Co-Inheritance of two ABCC8 mutations causing an unresponsive congenital hyperinsulinism: clinical and functional characterization of two novel ABCC8 mutations. Gene (2013) 516(1):122–5. doi: 10.1016/j.gene.2012.12.055
39. Flanagan SE, Kapoor RR, Banerjee I, Hall C, Smith VV, Hussain K, et al. Dominantly acting ABCC8 mutations in patients with medically unresponsive hyperinsulinaemic hypoglycaemia. Clin Genet (2011) 79(6):582–7. doi: 10.1111/j.1399-0004.2010.01476.x
40. Macmullen CM, Zhou Q, Snider KE, Tewson PH, Becker SA, Aziz AR, et al. Diazoxide-unresponsive congenital hyperinsulinism in children with dominant mutations of the beta-cell sulfonylurea receptor SUR1. Diabetes (2011) 60(6):1797–804. doi: 10.2337/db10-1631
41. Nessa A, Aziz QH, Thomas AM, Harmer SC, Tinker A, Hussain K. Molecular mechanisms of congenital hyperinsulinism due to autosomal dominant mutations in ABCC8. Hum Mol Genet (2015) 24(18):5142–53. doi: 10.1093/hmg/ddv233
42. Pinney SE, MacMullen C, Becker S, Lin YW, Hanna C, Thornton P, et al. Clinical characteristics and biochemical mechanisms of congenital hyperinsulinism associated with dominant KATP channel mutations. J Clin Invest (2008) 118(8):2877–86. doi: 10.1172/JCI35414
43. Huopio H, Reimann F, Ashfield R, Komulainen J, Lenko HL, Rahier J, et al. Dominantly inherited hyperinsulinism caused by a mutation in the sulfonylurea receptor type 1. J Clin Invest (2000) 106(7):897–906. doi: 10.1172/JCI9804
44. Perge K, Nicolino M. Variable phenotypes of individual and family monogenic cases with hyperinsulinism and diabetes: a systematic review. Rev Endocr Metab Disord (2022) 23(5):1063–78. doi: 10.1007/s11154-022-09749-2
45. Shemer R, Avnon Ziv C, Laiba E, Zhou Q, Gay J, Tunovsky-Babaey S, et al. Relative expression of a dominant mutated ABCC8 allele determines the clinical manifestation of congenital hyperinsulinism. Diabetes (2012) 61(1):258–63. doi: 10.2337/db11-0984
46. Kim JW, Seghers V, Cho JH, Kang Y, Kim S, Ryu Y, et al. Transactivation of the mouse sulfonylurea receptor I gene by BETA2/NeuroD. Mol Endocrinol (2002) 16(5):1097–107. doi: 10.1210/mend.16.5.0934
47. Moritz W, Leech CA, Ferrer J, Habener JF. Regulated expression of adenosine triphosphate-sensitive potassium channel subunits in pancreatic beta-cells. Endocrinology (2001) 142(1):129–38. doi: 10.1210/endo.142.1.7885
48. Flanagan SE, Xie W, Caswell R, Damhuis A, Vianey-Saban C, Akcay T, et al. Next-generation sequencing reveals deep intronic cryptic ABCC8 and HADH splicing founder mutations causing hyperinsulinism by pseudoexon activation. Am J Hum Genet (2013) 92(1):131–6. doi: 10.1016/j.ajhg.2012.11.017
49. Kiff S, Babb C, Guemes M, Dastamani A, Gilbert C, Flanagan SE, et al. Partial diazoxide responsiveness in a neonate with hyperinsulinism due to homozygous ABCC8 mutation. Endocrinol Diabetes Metab Case Rep (2019) 2019:18–0120. doi: 10.1530/EDM-18-0120
50. Reyes Diaz JV, Jin Y, Garber K, Cossen KM, Li Y, Jin P, et al. A homozygous exonic variant leading to exon skipping in ABCC8 as the cause of severe congenital hyperinsulinism. Am J Med Genet A (2022) 188(8):2429–33. doi: 10.1002/ajmg.a.62843
51. Takasawa K, Miyakawa Y, Saito Y, Adachi E, Shidei T, Sutani A, et al. Marked clinical heterogeneity in congenital hyperinsulinism due to a novel homozygous ABCC8 mutation. Clin Endocrinol (Oxf) (2021) 94(6):940–8. doi: 10.1111/cen.14443
52. Liu Y, Yang Q, Zhao F. Synonymous but not silent: The codon usage code for gene expression and protein folding. Annu Rev Biochem (2021) 90:375–401. doi: 10.1146/annurev-biochem-071320-112701
53. Martin GM, Sung MW, Shyng SL. Pharmacological chaperones of ATP-sensitive potassium channels: Mechanistic insight from cryoEM structures. Mol Cell Endocrinol (2020) 502:110667. doi: 10.1016/j.mce.2019.110667
54. Kandasamy B, Shyng SL. Methods for characterizing disease-associated ATP-sensitive potassium channel mutations. Methods Mol Biol (2018) 1684:85–104. doi: 10.1007/978-1-4939-7362-0_8
55. Thornton PS, MacMullen C, Ganguly A, Ruchelli E, Steinkrauss L, Crane A, et al. Clinical and molecular characterization of a dominant form of congenital hyperinsulinism caused by a mutation in the high-affinity sulfonylurea receptor. Diabetes (2003) 52(9):2403–10. doi: 10.2337/diabetes.52.9.2403
56. Wang M, Wu JX, Ding D, Chen L. Structural insights into the mechanism of pancreatic K(ATP) channel regulation by nucleotides. Nat Commun (2022) 13(1):2770. doi: 10.1038/s41467-022-30430-4
57. Thornton PS. Recent updates in the management of infants and children with hyperinsulinism. Curr Opin Pediatr (2021) 33(4):424–9. doi: 10.1097/MOP.0000000000001022
58. Saint-Martin C, Zhou Q, Martin GM, Vaury C, Leroy G, Arnoux JB, et al. Monoallelic ABCC8 mutations are a common cause of diazoxide-unresponsive diffuse form of congenital hyperinsulinism. Clin Genet (2015) 87(5):448–54. doi: 10.1111/cge.12428
59. Baukrowitz T, Schulte U, Oliver D, Herlitze S, Krauter T, Tucker SJ, et al. PIP2 and PIP as determinants for ATP inhibition of KATP channels. Science (1998) 282(5391):1141–4. doi: 10.1126/science.282.5391.1141
60. Shyng SL, Nichols CG. Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science (1998) 282(5391):1138–41. doi: 10.1126/science.282.5391.1138
61. Nichols CG, Shyng SL, Nestorowicz A, Glaser B, Clement JPT, Gonzalez G, et al. Adenosine diphosphate as an intracellular regulator of insulin secretion. Science (1996) 272(5269):1785–7. doi: 10.1126/science.272.5269.1785
62. Shyng SL, Ferrigni T, Shepard JB, Nestorowicz A, Glaser B, Permutt MA, et al. Functional analyses of novel mutations in the sulfonylurea receptor 1 associated with persistent hyperinsulinemic hypoglycemia of infancy. Diabetes (1998) 47(7):1145–51. doi: 10.2337/diabetes.47.7.1145
63. Worth C, Yau D, Estebanez Salomon ME, Cosgrove K, Dunne M, et al. Complexities in the medical management of hypoglycaemia due to congenital hyperinsulinism. Clin Endocrinol (Oxf) (2020) 92(5):387–95. doi: 10.1111/cen.14152
64. Lin YW, MacMullen C, Ganguly A, Stanley CA, Shyng SL. A novel KCNJ11 mutation associated with congenital hyperinsulinism reduces the intrinsic open probability of beta-cell ATP-sensitive potassium channels. J Biol Chem (2006) 281(5):3006–12. doi: 10.1074/jbc.M511875200
65. Lin YW, Bushman JD, Yan FF, Haidar S, MacMullen C, Ganguly A, et al. Destabilization of ATP-sensitive potassium channel activity by novel KCNJ11 mutations identified in congenital hyperinsulinism. J Biol Chem (2008) 283(14):9146–56. doi: 10.1074/jbc.M708798200
66. Raab-Graham KF, Cirilo LJ, Boettcher AA, Radeke CM, Vandenberg CA. Membrane topology of the amino-terminal region of the sulfonylurea receptor. J Biol Chem (1999) 274(41):29122–9. doi: 10.1074/jbc.274.41.29122
67. Zerangue N, Schwappach B, Jan YN, Jan LY. A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K(ATP) channels. Neuron (1999) 22(3):537–48. doi: 10.1016/S0896-6273(00)80708-4
68. Cartier EA, Conti LR, Vandenberg CA, Shyng SL. Defective trafficking and function of KATP channels caused by a sulfonylurea receptor 1 mutation associated with persistent hyperinsulinemic hypoglycemia of infancy. Proc Natl Acad Sci U.S.A. (2001) 98(5):2882–7. doi: 10.1073/pnas.051499698
69. Chen PC, Olson EM, Zhou Q, Kryukova Y, Sampson HM, Thomas DY, et al. Carbamazepine as a novel small molecule corrector of trafficking-impaired ATP-sensitive potassium channels identified in congenital hyperinsulinism. J Biol Chem (2013) 288(29):20942–54. doi: 10.1074/jbc.M113.470948
70. Reimann F, Huopio H, Dabrowski M, Proks P, Gribble FM, Laakso M, et al. Characterisation of new KATP-channel mutations associated with congenital hyperinsulinism in the Finnish population. Diabetologia (2003) 46(2):241–9. doi: 10.1007/s00125-002-1014-3
71. Yan FF, Lin YW, MacMullen C, Ganguly A, Stanley CA, Shyng SL, et al. Congenital hyperinsulinism associated ABCC8 mutations that cause defective trafficking of ATP-sensitive k+ channels: identification and rescue. Diabetes (2007) 56(9):2339–48. doi: 10.2337/db07-0150
72. Crane A, Aguilar-Bryan L. Assembly, maturation, and turnover of K(ATP) channel subunits. J Biol Chem (2004) 279(10):9080–90. doi: 10.1074/jbc.M311079200
73. Fukuda Y, Aguilar-Bryan L, Vaxillaire M, Dechaume A, Wang Y, Dean M, et al. Conserved intramolecular disulfide bond is critical to trafficking and fate of ATP-binding cassette (ABC) transporters ABCB6 and sulfonylurea receptor 1 (SUR1)/ABCC8. J Biol Chem (2011) 286(10):8481–92. doi: 10.1074/jbc.M110.174516
74. Martin GM, Rex EA, Devaraneni P, Denton JS, Boodhansingh KE, DeLeon DD, et al. Pharmacological correction of trafficking defects in ATP-sensitive potassium channels caused by sulfonylurea receptor 1 mutations. J Biol Chem (2016) 291(42):21971–83. doi: 10.1074/jbc.M116.749366
75. Nestorowicz A, Glaser B, Wilson BA, Shyng SL, Nichols CG, Stanley CA, et al. Genetic heterogeneity in familial hyperinsulinism. Hum Mol Genet (1998) 7(7):1119–28. doi: 10.1093/hmg/7.7.1119
76. Brusa I, Sondo E, Falchi F, Pedemonte N, Roberti M, Cavalli A, et al. Proteostasis regulators in cystic fibrosis: Current development and future perspectives. J Med Chem (2022) 65(7):5212–43. doi: 10.1021/acs.jmedchem.1c01897
77. Lee KPK, Chen J, MacKinnon R. Molecular structure of human KATP in complex with ATP and ADP. Elife (2017) 6:e32481. doi: 10.7554/eLife.32481
78. Li N, Wu JX, Ding D, Cheng J, Gao N, Chen L, et al. Structure of a pancreatic ATP-sensitive potassium channel. Cell (2017) 168(1-2):101–110 e10. doi: 10.1016/j.cell.2016.12.028
79. Martin GM, Yoshioka C, Rex EA, Fay JF, Xie Q, Whorton MR, et al. Cryo-EM structure of the ATP-sensitive potassium channel illuminates mechanisms of assembly and gating. Elife (2017) 6:e24149. doi: 10.7554/eLife.24149
80. Chan KW, Zhang H, Logothetis DE. N-terminal transmembrane domain of the SUR controls trafficking and gating of Kir6 channel subunits. EMBO J (2003) 22(15):3833–43. doi: 10.1093/emboj/cdg376
81. Lin CW, Lin YW, Yan FF, Casey J, Kochhar M, Pratt EB, et al. Kir6.2 mutations associated with neonatal diabetes reduce expression of ATP-sensitive k+ channels: implications in disease mechanism and sulfonylurea therapy. Diabetes (2006) 55(6):1738–46. doi: 10.2337/db05-1571
82. Lin YW, Li A, Grasso V, Battaglia D, Crino A, Colombo C, et al. Functional characterization of a novel KCNJ11 in frame mutation-deletion associated with infancy-onset diabetes and a mild form of intermediate DEND: a battle between K(ATP) gain of channel activity and loss of channel expression. PloS One (2013) 8(5):e63758. doi: 10.1371/journal.pone.0063758
83. Zhou Q, Garin I, Castano L, Argente J, Munoz-Calvo MT, Nanclares Perez G, et al. Neonatal diabetes caused by mutations in sulfonylurea receptor 1: interplay between expression and mg-nucleotide gating defects of ATP-sensitive potassium channels. J Clin Endocrinol Metab (2010) 95(12):E473–8. doi: 10.1210/jc.2010-1231
84. Yildizdas D, Erdem S, Kucukosmanoglu O, Yilmaz M, Yuksel B. Pulmonary hypertension, heart failure and neutropenia due to diazoxide therapy. Adv Ther (2008) 25(5):515–9. doi: 10.1007/s12325-008-0049-3
85. Nichols CG. Personalized therapeutics for K(ATP)-dependent pathologies. Annu Rev Pharmacol Toxicol (2023) 63:541–63. doi: 10.1146/annurev-pharmtox-051921-123023
86. Banerjee I, Salomon-Estebanez M, Shah P, Nicholson J, Cosgrove KE, Dunne MJ, et al. Therapies and outcomes of congenital hyperinsulinism-induced hypoglycaemia. Diabetes Med (2019) 36(1):9–21. doi: 10.1111/dme.13823
87. De Cosio AP, Thornton P. Current and emerging agents for the treatment of hypoglycemia in patients with congenital hyperinsulinism. Paediatr Drugs (2019) 21(3):123–36. doi: 10.1007/s40272-019-00334-w
88. Martin GM, Kandasamy B, DiMaio F, Yoshioka C, Shyng SL. Anti-diabetic drug binding site in a mammalian K(ATP) channel revealed by cryo-EM. Elife (2017) 6:e31054. doi: 10.7554/eLife.31054
89. Zhao C, MacKinnon R. Molecular structure of an open human K(ATP) channel. Proc Natl Acad Sci U.S.A. (2021) 118(48):e2112267118. doi: 10.1073/pnas.2112267118
90. Ding D, Wang M, Wu JX, Kang Y, Chen L. The structural basis for the binding of repaglinide to the pancreatic K(ATP) channel. Cell Rep (2019) 27(6):1848–1857 e4. doi: 10.1016/j.celrep.2019.04.050
91. Wu JX, Ding D, Wang M, Kang Y, Zeng X, Chen L, et al. Ligand binding and conformational changes of SUR1 subunit in pancreatic ATP-sensitive potassium channels. Protein Cell (2018) 9(6):553–67. doi: 10.1007/s13238-018-0530-y
92. Ding D, Wu JX, Duan X, Ma S, Lai L, Chen L, et al. Structural identification of vasodilator binding sites on the SUR2 subunit. Nat Commun (2022) 13(1):2675. doi: 10.1038/s41467-022-30428-y
93. Sikimic J, Hoffmeister T, Gresch A, Kaiser J, Barthlen W, Wolke C, et al. Possible new strategies for the treatment of congenital hyperinsulinism. Front Endocrinol (Lausanne) (2020) 11:545638. doi: 10.3389/fendo.2020.545638
94. Zdravkovic M, Kruse M, Rost KL, Moss J, Kecskes A. The effects of NN414, a SUR1/Kir6.2 selective potassium channel opener in subjects with type 2 diabetes. Exp Clin Endocrinol Diabetes (2007) 115(6):405–6. doi: 10.1055/s-2007-973062
95. Leidenheimer NJ, Ryder KG. Pharmacological chaperoning: a primer on mechanism and pharmacology. Pharmacol Res (2014) 83:10–9. doi: 10.1016/j.phrs.2014.01.005
96. Thomas C, Tampe R. Structural and mechanistic principles of ABC transporters. Annu Rev Biochem (2020) 89:605–36. doi: 10.1146/annurev-biochem-011520-105201
97. Fiedorczuk K, Chen J. Mechanism of CFTR correction by type I folding correctors. Cell (2022) 185(1):158–168 e11. doi: 10.1016/j.cell.2021.12.009
98. Lopes-Pacheco M. CFTR modulators: The changing face of cystic fibrosis in the era of precision medicine. Front Pharmacol (2019) 10:1662. doi: 10.3389/fphar.2019.01662
99. Yan F, Lin CW, Weisiger E, Cartier EA, Taschenberger G, Shyng SL, et al. Sulfonylureas correct trafficking defects of ATP-sensitive potassium channels caused by mutations in the sulfonylurea receptor. J Biol Chem (2004) 279(12):11096–105. doi: 10.1074/jbc.M312810200
100. Yan FF, Casey J, Shyng SL. Sulfonylureas correct trafficking defects of disease-causing ATP-sensitive potassium channels by binding to the channel complex. J Biol Chem (2006) 281(44):33403–13. doi: 10.1074/jbc.M605195200
101. Zhou Q, Chen PC, Devaraneni PK, Martin GM, Olson EM, Shyng SL, et al. Carbamazepine inhibits ATP-sensitive potassium channel activity by disrupting channel response to MgADP. Channels (Austin) (2014) 8(4):376–82. doi: 10.4161/chan.29117
102. Devaraneni PK, Martin GM, Olson EM, Zhou Q, Shyng SL. Structurally distinct ligands rescue biogenesis defects of the KATP channel complex via a converging mechanism. J Biol Chem (2015) 290(12):7980–91. doi: 10.1074/jbc.M114.634576
103. Martin GM, Sung MW, Yang Z, Innes LM, Kandasamy B, David LL, et al. Mechanism of pharmacochaperoning in a mammalian K(ATP) channel revealed by cryo-EM. Elife (2019) 8:e46417. doi: 10.7554/eLife.46417
104. Schwartz TB, Meinert CL. The UGDP controversy: thirty-four years of contentious ambiguity laid to rest. Perspect Biol Med (2004) 47(4):564–74. doi: 10.1353/pbm.2004.0071
105. Pratt EB, Yan FF, Gay JW, Stanley CA, Shyng SL. Sulfonylurea receptor 1 mutations that cause opposite insulin secretion defects with chemical chaperone exposure. J Biol Chem (2009) 284(12):7951–9. doi: 10.1074/jbc.M807012200
106. Yan FF, Lin CW, Cartier EA, Shyng SL. Role of ubiquitin-proteasome degradation pathway in biogenesis efficiency of beta-cell ATP-sensitive potassium channels. Am J Physiol Cell Physiol (2005) 289(5):C1351–9. doi: 10.1152/ajpcell.00240.2005
107. Lithovius V, Otonkoski T. Stem cell based models in congenital hyperinsulinism - perspective on practicalities and possibilities. Front Endocrinol (Lausanne) (2022) 13:837450. doi: 10.3389/fendo.2022.837450
Keywords: ATP-sensitive potassium channel, insulin secretion, sulphonylurea receptor 1 (SUR1), kir6.2, hypoglycemia
Citation: ElSheikh A and Shyng S-L (2023) KATP channel mutations in congenital hyperinsulinism: Progress and challenges towards mechanism-based therapies. Front. Endocrinol. 14:1161117. doi: 10.3389/fendo.2023.1161117
Received: 07 February 2023; Accepted: 13 March 2023;
Published: 28 March 2023.
Edited by:
Klaus Mohnike, University Hospital Magdeburg, GermanyReviewed by:
Stephen F. Betz, Crinetics Pharmaceuticals, United StatesMaria Salomon Estebanez, Royal Manchester Children’s Hospital, United Kingdom
Copyright © 2023 ElSheikh and Shyng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Show-Ling Shyng, shyngs@ohsu.edu