 Yu-Peng Han
Yu-Peng Han Li-Juan Liu
Li-Juan Liu Jia-Lin Yan
Jia-Lin Yan Meng-Yuan Chen
Meng-Yuan Chen Xiang-Fei Meng
Xiang-Fei Meng Ling-Bo Qian
Ling-Bo Qian- School of Basic Medical Sciences & Forensic Medicine, Hangzhou Medical College, Hangzhou, China
Diabetic nephropathy (DN), the leading cause of end-stage renal disease, is the most significant microvascular complication of diabetes and poses a severe public health concern due to a lack of effective clinical treatments. Autophagy is a lysosomal process that degrades damaged proteins and organelles to preserve cellular homeostasis. Emerging studies have shown that disorder in autophagy results in the accumulation of damaged proteins and organelles in diabetic renal cells and promotes the development of DN. Autophagy is regulated by nutrient-sensing pathways including AMPK, mTOR, and Sirt1, and several intracellular stress signaling pathways such as oxidative stress and endoplasmic reticulum stress. An abnormal nutritional status and excess cellular stresses caused by diabetes-related metabolic disorders disturb the autophagic flux, leading to cellular dysfunction and DN. Here, we summarized the role of autophagy in DN focusing on signaling pathways to modulate autophagy and therapeutic interferences of autophagy in DN.
1 Introduction
Diabetic nephropathy (DN), a major cause contributing to end-stage renal disease (ESRD), is one of the microvascular complications of diabetes and is commonly rendered by persistent hyperglycemia and the subsequent chronic inflammatory response (1, 2). Almost 35%-40% of diabetic patients finally lead to DN (3), which poses a huge number of diabetic death and a serious threat to the quality of life in diabetes (4). International Diabetes Federation (IDF) Diabetes Atlas (the 10th edition) showed that the number of adult diabetes worldwide will increase from 537 million in 2021 to 643 million by 2030 and over 6.7 million diabetes aged 20-79 years died from diabetes-related diseases in 2021 (http://diabetesatlas.org/atlas/tenth-edition/). Long-term diabetes can damage many organs to cause disabling and life-threatening complications including cardiovascular diseases, neuropathy, and nephropathy. DN, with clinical manifestations including progressive proteinuria as well as decreased glomerular filtration rate (3), and pathological features such as glomerular hypertrophy, glomerular basement membrane (GBM) thickening, mesangial proliferation, and podocyte loss (5), is one of the early complications in diabetes. Though keeping blood pressure, blood glucose, and the renin-angiotensin system (RAS) under control is a primary therapy to relieve proteinuria in diabetes, treatment-resistant proteinuria and ESRD have not been fully avoided (6). Exploring the underlying mechanism of DN and finding novel targets to effectively prevent DN have become urgent for improving the quality of life in diabetes.
The pathogenesis of DN is multifactorial (4), including oxidative stress, inflammatory cascade reaction, and other disorders of metabolic pathways under persistent hyperglycemia (7). Growing evidence reveals that along with diabetes, the accumulation of damaged organelles and proteins owing to impaired autophagy has been reported to disrupt cellular homeostasis and result in the development of DN (3, 7–10). Autophagy normally is activated to degrade impaired organelles or misfolded proteins as a recycling response to nutrition deprivation or starvation (10). The metabolic disorder manifested as persistent high blood glucose and lipids causes a state of overnutrition and suppresses autophagy in diabetic renal cells (11–13), while promoting autophagy lessens renal injury in diabetes (14, 15). All these clues suggest that activating autophagy may be a novel therapeutic target to prevent DN and shed light on treating DN based on the balance of autophagy.
Although the relationship between autophagy and DN has not been fully clarified, numerous studies have confirmed that the development of DN is linked to autophagy. Detailed exploration of autophagy in the pathogenesis of DN can provide new ideas for preventing DN. Thus, this review aims to understand the cellular and molecular bases of autophagy, the role of autophagy in the development of DN, and therapeutic strategies targeting autophagy for the prevention of DN by summarizing current evidence.
2 Profile of autophagy in DN
Autophagy is a highly conserved cellular mechanism by which cytoplasmic constituents including proteins and organelles are transported to lysosomes for degradation and preserving cellular homeostasis (9, 16). Basal cellular autophagy is necessary for keeping physiological functions, whereas autophagy in response to stress serves as an adaptive reaction to ensure cell survival (16). Autophagy is a multistep process that involves the formation of isolation membrane, extension, formation of autophagosome, and final fusion with lysosomes to degrade phagocytic materials and is regulated by multiple protein kinase complexes and autophagy-related proteins, such as autophagy-related gene 5 (Atg5), Atg7, Atg12 and so on (8, 17). Among them, activation of the unc-51-like kinase 1 (ULK1) complex is responsible for the initiation of autophagy (3, 10). The class III phosphatidylinositol 3-kinase (PI3K) complex generates phosphatidylinositol 3-phosphate at the neogenetic autophagosomal membrane to facilitate phagophore nucleation (18). Two ubiquitin-like coupling systems, Atg5-Atg12-Atg16L and Atg8/microtubule-associated protein 1A/1B-light chain 3 (LC3) are involved in autophagosome extension and autolysosome formation (19). Atg4 cleaves LC3 to form cytosolic LC3I, which is then ubiquitinated by Atg7 and Atg3 and binds to phosphatidyl ethanolamine to form autophagosome membrane-bound LC3II (17). Thus, LC3II is evidenced as a marker for autophagosome formation in cells. This conjugated response of LC3II is positively regulated by Atg5-Atg12-Atg16L. Sequestosome 1, known as p62, interacts with LC3II to confine autophagosomes and is repeatedly digested by the autophagy-lysosome system. Significantly, malfunctional autophagy during diabetes causes intracellular accumulation of p62 leading to further inhibition of autophagic flux, thus forming a vicious cycle to promote diabetic complications including diabetic cardiomyopathy, diabetic peripheral neuropathy and DN (20–22).
Autophagy can be triggered by various intracellular stresses, such as reactive oxygen species (ROS), endoplasmic reticulum (ER) stress, and hypoxia (23–25), all of which are involved in the development of DN. Increasing evidence indicates that the abnormal alteration of autophagy appears to be directly linked to the emergence of DN (26, 27). Autophagy is closely associated with nutrient-sensing signal pathways and stress metabolism and is essential to maintain homeostasis in the kidney (3). Although the mechanism of autophagy in DN remains to be elucidated, it has been known that the impaired autophagy is evidenced by the increased collection of p62 and the decreased expression of autophagy-related proteins in diabetic kidney tissues and cells (28–30). The shortage of autophagy results in the accumulation of misfolded or aging proteins and dysfunctional organelles to deteriorate kidney disease in diabetes (19). Activation of autophagy alleviates kidney lesions in diabetes (31, 32) while inhibition of autophagy worsens these diabetic injuries (33, 34), indicating that autophagy might be a promising therapeutic target for DN.
3 Autophagy in renal cells during diabetes
Though different types of renal cells are all damaged by the dysfunctional autophagy in the progression of DN, as shown in Figure 1, these four resident renal cells including podocytes, renal tubular epithelial cells (RTECs), glomerular mesangial cells (GMCs), and glomerular endothelial cells (GEnCs) may be particularly vulnerable to attack from the disorder of autophagy and contribute to DN. Thus, we summarized recent findings of renal cells in diabetic environments to better understand autophagy in DN (Table 1).
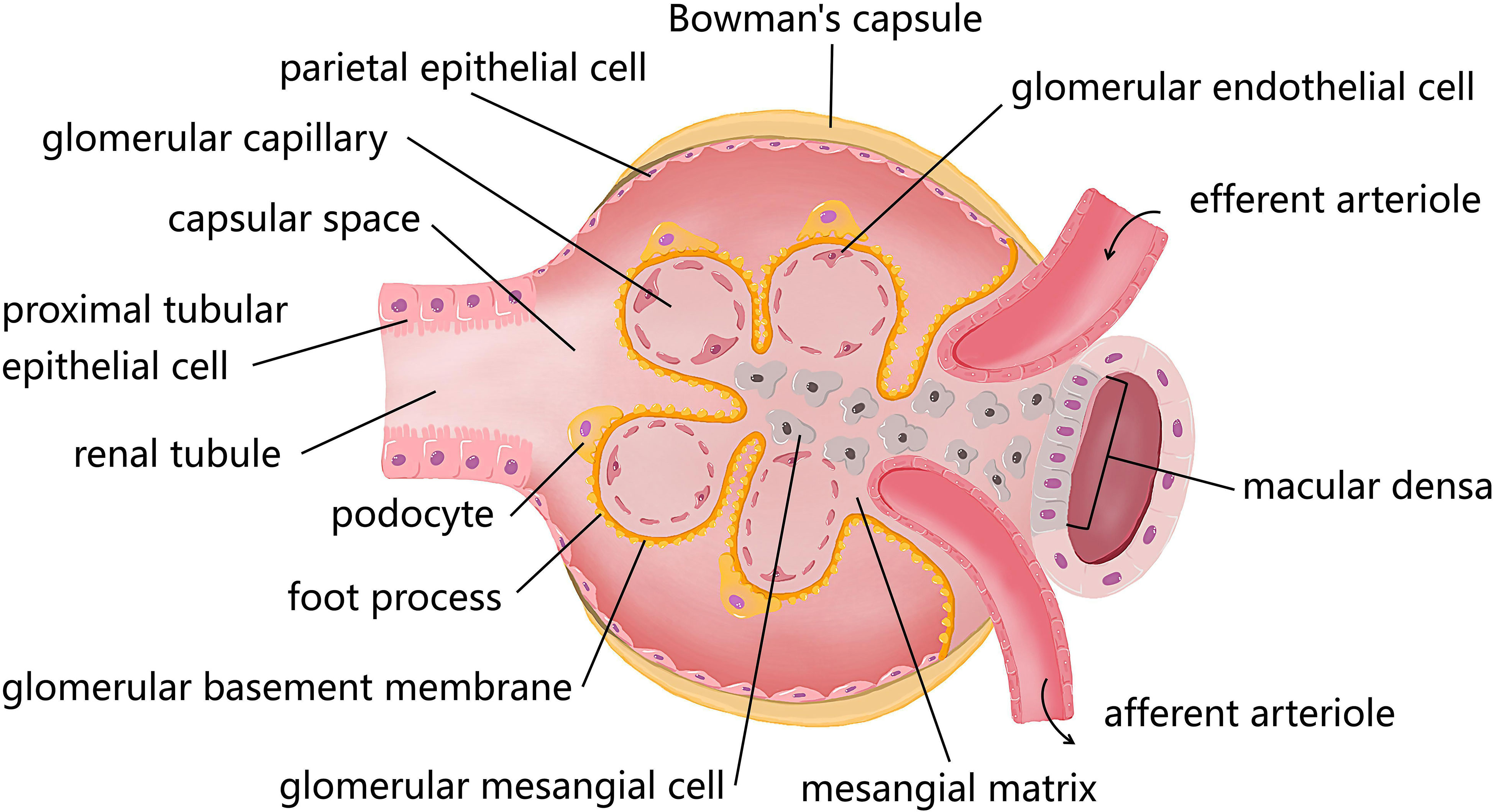
Figure 1 The diagram of resident cells in the glomerulus and proximal tubule. There are four kinds of major resident cells in the glomerulus, glomerular endothelial cells (GEnCs), podocytes, glomerular mesangial cells (GMCs), and parietal epithelial cells. Tubular epithelial cells form the extension of Bowman’s capsule, that is, the renal tubule. Podocytes with their interdigitating foot processes are arranged on the lateral side of the glomerular basement membrane (GBM). GMCs located between glomerular capillary loops, adjacent to endothelial cells or basement membranes are irregularly shaped. GEnCs are flat cells attached to the GBM. GEnCs and podocytes form the glomerular filtration barrier.

Table 1 Autophagy in four types of renal cells during diabetes.
3.1 Podocytes
Podocytes, highly differentiated epithelial cells with a limited capacity for proliferation, tightly attach to the GBM (62) and work as an important part of the glomerular filtration barrier (GFB) (63, 64). The damage and apoptosis of podocytes can destroy the integrity of the GFB (31), leading to proteinuria, renal lesions, and finally DN (7, 8, 65).
A high level of autophagy in podocytes is necessary to keep the physiological function (8, 39), which is regulated by the adenosine 5’-monophosphate-activated protein kinase (AMPK) pathway rather than the mammalian target of rapamycin (mTOR) (66). The impairment of autophagy in diabetic podocytes as evidenced by the decreased expression of autophagy-related proteins (beclin1, LC3II/I, Atg12, Atg7, etc.) and the accumulation of the autophagic substrate p62 (40, 67) exacerbates the loss of podocytes with the help of the increased cellular lipid accumulation, oxidative stress, and inflammation (11, 41). Knockout of the Atg5 in podocytes has been reported to cause glomerular lesions accompanied by podocyte loss and albuminuria (68). These findings imply that the shortage of autophagy mediates podocyte damage in diabetes (22). It is interesting to note that the increased autophagosomes in high glucose-treated podocytes was not consistent with the impaired autophagy in the diabetic rat kidney characterized by glomerular hypertrophy, renal tubular expansion, and mesangial cell proliferation (44). To further clarify whether the rise in autophagosomes is caused by autophagy induction or the obstructed fusion of autophagosomes and lysosomes, the fusion inhibitor such as chloroquine can be adopted or the colocalization of LC3 and lysosomes need to be explored. In addition, this contradiction in different diabetic kidney models might be related to the different roles of autophagy in each stage of diabetes (3).
Nutrient signaling pathways are involved in the disorder of autophagy in diabetic podocytes. Increased mTOR activity and decreased expression of AMPK and silent information regulator of transcription 1 (Sirt1) in diabetes can inhibit autophagy to aggravate cellular dysfunction and the progression of DN (69, 70). The silence of AMPK or Sirt1 was reported to inhibit autophagy and promote the loss of podocyte function in a high glucose environment (12, 42, 43). Furthermore, the up-regulation AMPK/mTOR signaling pathway-mediated autophagy prevents the loss of podocyte markers (nephrin, podocin) and ameliorates diabetic kidney injury (46–48). Liver X receptor and high mobility group box 1 also induce podocyte injury by altering autophagy through the nutrient-sensing signal pathway (34, 49).
3.2 Renal tubular epithelial cells
The enhancement of autophagy in proximal tubular epithelial cells (PTECs) in response to multiple stresses such as ischemia and nephrotoxic medications has been reported to protect the kidney (71). Morphological alterations including hypertrophy, hyperplasia and epithelial-mesenchymal transition (EMT) in RTECs, especially in PTECs, primarily owing to the shortage of autophagy in diabetes, are regarded as an early sign of DN, which can easily cause renal dysfunction and even ESRD if not corrected in time (51, 72, 73).
It is noteworthy that the interaction of autophagy with EMT in RTECs is complicated, various factors and signaling pathways are associated with the effect of autophagy-related EMT on the progression of DN (33, 74). The role of rapamycin in reducing profibrotic cytokines, fibroblast proliferation, tubulointerstitial inflammation, and EMT confirms that mTOR-regulated autophagy is necessary for EMT in diabetic RTECs (69, 75). Interestingly, hyperglycemia-induced miR-22 promotes EMT by suppressing autophagy via targeting phosphatase and tensin homolog/protein kinase B (Akt)/mTOR signaling pathway, which suggests that targeting miRNA may be a promising therapeutic approach in preventing DN (32). Recently, mesenchymal stem cell-derived exosomes was reported to activate autophagy to inhibit transforming growth factor-β (TGF-β)-induced EMT progression in RTECs (76). Thus, the role of exosomes on the EMT in diabetic RTECs is worth further investigation.
In the presence of diabetes, carbonyl compounds created by advanced glycation end-products (AGEs) are filtered by the glomerulus and then reabsorbed by the proximal tubule, easily resulting in tubular toxicity (77, 78). Through interaction with the receptor for AGEs (RAGE), accumulation of AGEs triggers various abnormal cellular cascades like oxidative stress, inflammation, and apoptosis and inhibits the protective effect of autophagy in the diabetic kidney (79). The impairment of the autophagy-lysosomal pathway in diabetes promotes the accumulation of AGEs and the excessive AGEs aggravates lysosomal dysfunction, thus forming positive feedback to allow tubulointerstitial inflammation and fibrosis, which might be crucial to the development of DN (17, 80). Inhibiting AGEs/RAGE signaling is reported to restore the disturbed autophagy in glomerular endothelial cells and attenuate DN (59). It is said that AGEs can enhance the expression of profibrotic molecules linked to EMT and ER stress in the human renal tubular epithelial cell line to gradually render renal fibrosis (81), which is prevented by the enhancement of autophagy in RTECs (54). Therefore, the specific role of the AGEs/RAGE axis in DN is worthy of exploring.
3.3 Glomerular mesangial cells
Proliferation and hypertrophy in GMCs and mesangial expansion manifested as excess extracellular matrix (ECM) derived from GMCs are two pathological characteristics of DN, which lead to glomerulosclerosis and tubulointerstitial fibrosis (82, 83). Hyperglycemia, AGEs, and ROS all effectively activate TGF-β to cause ECM accumulation both in Smad-dependent and -independent pathways (84–86), which can be reversed by the up-regulation of autophagy (33, 57).
Sirt1 has been revealed to inhibit ECM accumulation in high glucose-treated GMCs via enhancing autophagy (33) and blocking mTOR-suppressed autophagy has also been documented to effectively reduce inflammation, proliferation, and fibrosis in diabetic GMCs (15, 28, 57). All of the above indicate that autophagy is important for maintaining the structural and functional integrity of GMCs to resist DN.
3.4 Glomerular endothelial cells
GEnCs, the first barrier of glomerular filtration, are vulnerable to hyperglycemia. The abnormal structure manifested as endothelial glycocalyx and endothelial-mesenchymal transition usually occur in the early stage of DN (87). Severe damage to the glomerular endothelium owing to autophagy reduction has been reported in endothelial-specific autophagy-deficient mice and Atg16L-knockdown GEnCs (88, 89). In addition, activation of calcium/calmodulin-dependent protein kinase kinase β (CAMKKβ)/liver kinase B1 (LKB1)/AMPK signaling (60) and inhibition of miR-34a/Atg4b signaling (61) promote autophagy in GEnCs to attenuate DN. It is well established that the interplay of podocytes, GEnCs, and GMCs is key to keep the integrity of the GFB and the pathological alteration in one component evidently affects the other two (87, 90, 91). These results imply that appropriate autophagy in GEnCs can minimize DN by preserving glomerular structural integrity.
4 Autophagic pathways in DN
Autophagy in eukaryotic cells is tightly regulated to adapt or counteract cellular stresses through multiple signaling pathways (17) because both insufficient and excessive autophagy are harmful (92). Nutrient-sensing pathways including AMPK, mTOR, and Sirt1 are well-recognized to regulate autophagy in diabetic complications (10). Moreover, various cellular stresses such as ROS, ER stress, and hypoxia are involved in pathogenic autophagy in DN (Figure 2) (93). Thus, autophagy in the development of DN is precisely regulated.
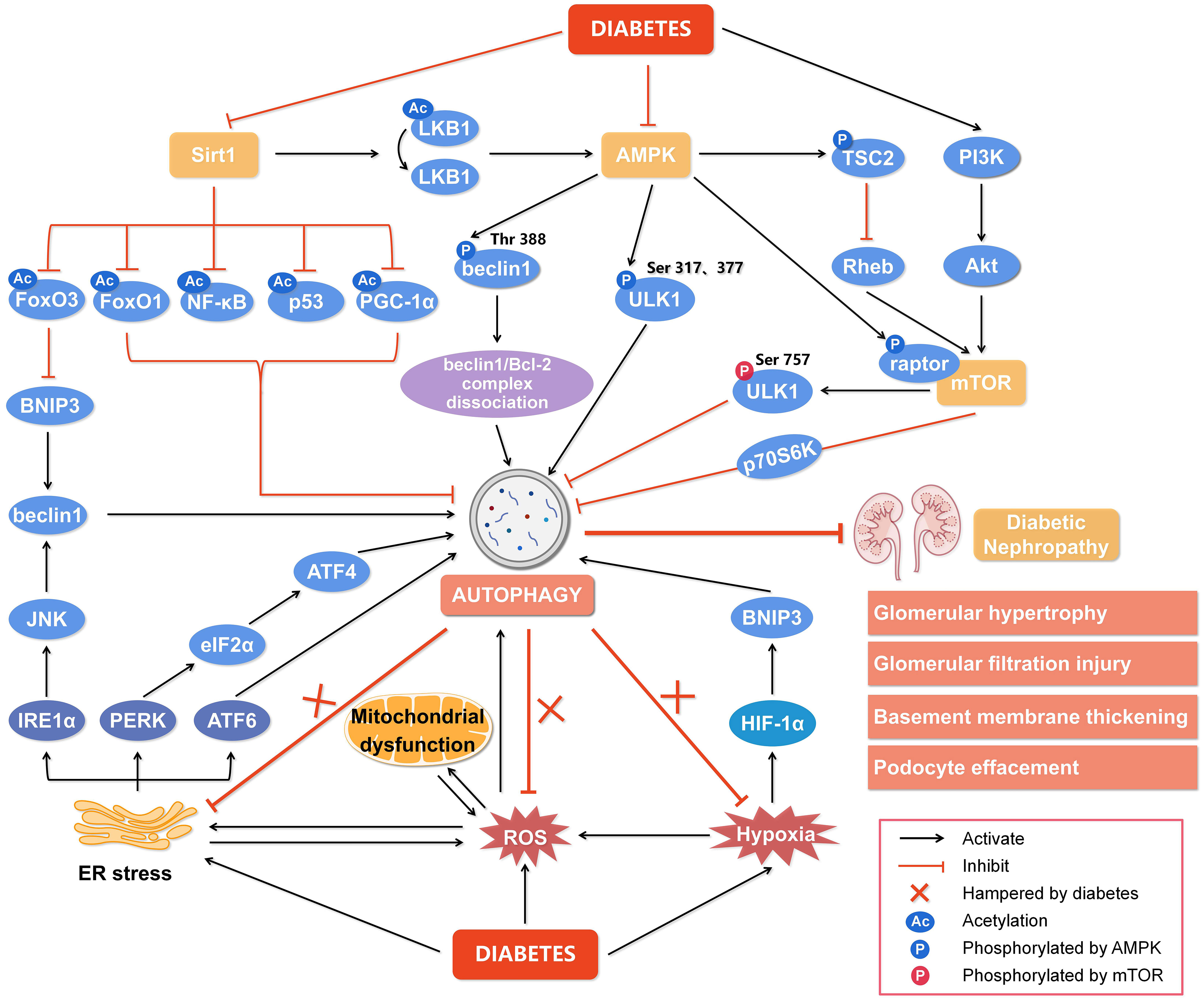
Figure 2 Regulation of autophagy during diabetic nephropathy. Hyperglycemia is considered a state of overnutrition, leading to over-activation of the mammalian target of rapamycin (mTOR) and inhibition of adenosine 5’-monophosphate-activated protein kinase (AMPK) and silent information regulator of transcription 1 (Sirt1). The activated mTOR inhibits autophagy by blocking unc-51-like kinase 1 (ULK1) activation by AMPK and its downstream target phosphoprotein 70 ribosomal protein S6 kinase (p70S6K). The inhibition of AMPK blocks the dissociation of the beclin1/Bcl-2 (B-cell lymphoma-2) complex and the phosphorylation of ULK1, while promotes mTOR activity to reduce autophagy. The inactivated Sirt1 reduces the deacetylation of several target genes like forkhead box O3 (FoxO3), FoxO1, nuclear factor kappa-B (NF-κB), p53, and peroxisome proliferator-activated receptor-gamma coactivator-1α (PGC-1α) to inhibit autophagy. In addition, other cellular events, including reactive oxygen species (ROS), endoplasmic reticulum (ER) stress, and hypoxia, can also regulate autophagy to affect the development of diabetic nephropathy. Hypoxia-inducible factor 1α (HIF-1α) induced by hypoxia promotes the transcription of Bcl-2/adenovirus E1V19-kDa interacting protein 3 (BNIP3) and induces autophagy. ER stress enhances the expression of ER membrane proteins like protein kinase RNA-like ER kinase (PERK), inositol-requiring enzyme 1α (IRE1α), and activating transcription factor 6 (ATF6), leading to autophagy. In addition, autophagy under ER stress may be associated with the signaling pathway of PERK/α-subunit of eukaryotic initiation factor 2 (eIF2α)/ATF4 and IRE1α/c-Jun N-terminal kinase (JNK)/beclin1. Significantly, the endogenous autophagy induced by ER stress, oxidative stress and hypoxia in diabetes is hampered, which aggravates the progression of diabetic nephropathy. Thus, impaired autophagy accelerates the progression of diabetic nephropathy, resulting in a series of renal pathological damages. Rheb, ras homolog enriched in brain; PI3K, class III phosphatidylinositol-3-kinase; Akt, protein kinase B.
4.1 Nutrient-sensing pathways
4.1.1 mTOR pathway
Rapamycin-sensitive type of mTOR (mTORC1), a master inhibitor of autophagy, is inhibited by starvation to reduce the phosphorylation of ULK1 at Ser757, which frees ULK1 to be activated by AMPK and then initiates autophagy to provide nutrients for the cell’s use by degrading the captured cytoplasmic components (94, 95). mTOR is over-mobilized in the diabetic kidney to promote the inflammatory response and exacerbate renal impairment (96, 97), which is reversed by rapamycin (98). In addition, the mTOR signaling pathway can be activated by vascular endothelial growth factor via PI3K/Akt cascade, which suppresses autophagy via phosphorylating its downstream phosphoprotein 70 ribosomal protein S6 kinase (p70S6K) and exacerbates DN (99, 100). All of these suggest that the overactivation of the mTOR pathway is extremely detrimental to the development of DN (101, 102). Numerous studies have demonstrated the critical role that long noncoding RNAs (LncRNAs) play in the pathophysiology of DN (103). LncRNAs potently affect the pathological alteration in the diabetic kidney by inhibiting the autophagy-related Akt/mTOR pathway, which has been supported by growing evidence that LncRNA silencing sperm-associated antigen 5 antisense RNA1 promotes hyperglycemia-induced injury in podocytes targeting Akt/mTOR signaling (37), and LncRNA nuclear enriched abundant transcript 1 accelerates (58), whereas LncRNA SOX2 overlapping transcript inhibits (15), proliferation and fibrosis in diabetic GMCs via modulating Akt/mTOR signaling-related autophagy. Thus, the effect of LncRNAs is diversified depending on the type of LncRNAs in the development of DN though the same target of Akt/mTOR signaling-related autophagy may be involved.
4.1.2 AMPK pathway
AMPK belongs to the serine/threonine protein kinase family and is composed of the catalytic subunit α and the regulatory subunits β and γ (104). The phosphorylation of the threonine 172 (Thr172) site on the subunit α is necessary for the activation of AMPK (105). AMPK is regulated by the AMP/ATP ratio as an energy sensor (3). Under harmful conditions like hunger and hypoxia, the ratio of AMP/ATP ratio rises and renders AMP binding to the subunit γ of AMPK, which promotes Thr172 phosphorylation by LKB1 (106). In addition, AMPK is even activated by CAMKKβ and TGF-β-activated kinase by the action of hormones, drugs, or proinflammatory cytokines (106, 107) to trigger autophagy for keeping cellular energy homeostasis under starvation.
It has been shown that AMPK and autophagy are deactivated in the diabetic kidney accompanied by proteinuria and renal pathological alterations (11, 45, 56, 108). As shown in Figure 2, AMPK can phosphorylate ULK1 at Ser317 and Ser377 to directly initiate autophagy (109, 110) or indirectly promote autophagy by blocking mTORC1 to release ULK1 through phosphorylating tuberous sclerosis complex 2 (TSC2) and raptor, the critical mTORC1-binding subunit (111), which benefits to hinder the progression of DN (112). In addition, AMPK activates Sirt1 by increasing cellular NAD+ levels (56) or phosphorylating and redistributing glyceraldehyde 3-phosphate dehydrogenase into the nucleus to free Sirt1 (111), which promotes autophagy and alleviates DN (28, 56). AMPK can promote the dissociation of the beclin1/B-cell lymphoma-2 (Bcl-2) complex via phosphorylating beclin1 at Thr388 to initiate autophagy (113). Thus, AMPK-regulated autophagy is key to the development of DN and AMPK may be a promising target for preventing DN.
4.1.3 Sirt1 pathway
Sirt1, the most widely studied NAD-dependent deacetylase in the Sirtuin family (114, 115), is highly expressed in renal tubular cells and podocytes (115) and has been reported to attenuate diabetic kidney disease by reducing the phosphorylation and acetylation levels of NF-κB and signal transducer and activator of transcription 3 (33, 44, 116). In addition, Sirt1 reduces acetylation or phosphorylation of several target genes such as AMPK, forkhead box O1 (FoxO1), p53, and peroxisome proliferator-activated receptor-gamma coactivator-1α (PGC-1α) to enhance autophagy (Figure 2) (28, 50, 117). As a positive regulator of autophagy, Sirt1 has been revealed to up-regulate Bcl-2/adenovirus E1V19-kDa interacting protein 3 (BNIP3) by deacetylating the transcription factor FoxO3 to enhance autophagy and inhibit DN (118, 119). LKB1 deacetylated by Sirt1 activates AMPK to enhance autophagy (120, 121). In addition, deacetylation of p53 by Sirt1 potently activates AMPK-dependent autophagy to ameliorate DN (12) and this protective effect of Sirt1 against DN is inhibited by several miRNAs including miR-135a-5p (122), miR-138 (65), miR-150-5p (12), miR-155-5p (123), and miR-217 (124) targeting the 3’ untranslated region of Sirt1. The relationship between miRNAs and Sirt1 is complicated in the progression of DN and more efforts are needed to clarify the underlying mechanism by which Sirt1-regulated autophagy prevents DN.
4.2 Cellular stress signaling
4.2.1 Oxidative stress
Excessive production of ROS and/or reactive nitrogen species beyond the endogenous scavenging capacity leads to oxidative stress. Oxidative damage of cellular lipids, proteins, nucleic acids, and carbohydrates breaks the structural integrity and results in physiological dysfunction (125). Oxidative stress induced by hyperglycemia through de novo ROS generation and suppression of the antioxidant defense system promotes mitochondria swelling, cristae breakage, and mitochondrial disintegration in the diabetic kidney, which can be reversed by the enhancing autophagy to eliminate damaged mitochondria (126).
It should be noted that autophagy and oxidative stress are interactive. ROS are reported as an early inducer for autophagy initiation and execution, which may be a crucial adaptive response to reduce oxidative stress and obtain the nutrient for reuse through autophagy-dependent degrading oxidative damaged cellular components (127). On the contrary, oxidative modification of key upstream autophagy regulators and autophagy core proteins including AMPK, Sirt1, Atg4, and Parkin impair autophagy (128). Thus, oxidative stress affects autophagy in the development of DN as a two-edged sword and antioxidant therapy may protect the kidney against diabetes through activating autophagy. This notion has been supported by some evidence that antioxidant compounds derived from plants such as betulinic acid, ursolic acid, genistein, and luteolin effectively attenuate the kidney injury induced by diabetes or poisons by promoting autophagy (38, 129–132).
4.2.2 Endoplasmic reticulum stress
The accumulation of unfolded or misfolded proteins in the ER lumen leads to ER stress which is evident in DN (24, 133). Overproduction of ROS due to chronic hyperglycemia disrupts intracellular Ca2+ homeostasis and oxidation of ER-resident proteins to trigger ER stress, in turn, hyperactivates the oxidative folding machinery to correct improper disulfide bonds, further producing ROS (134, 135). This vicious cycle leads to the disruption of cellular homeostasis (Figure 2). Emerging evidence suggests that autophagy is linked to the unfolded protein response (UPR) to relieve ER stress by clearing misfolded proteins (24, 136, 137). Under ER stress, the UPR is triggered by three protein sensors, protein kinase RNA-like ER kinase (PERK), inositol-requiring enzyme 1α (IRE1α), and activating transcription factor 6 (ATF6) after accumulation of misfolded proteins (24). As shown in Figure 2, all these three sensors of the UPR under ER stress can induce autophagy via activating signaling pathways of PERK/α-subunit of eukaryotic initiation factor 2/activating transcription factor 4 (PERK/eIF2α/ATF4) (138), IRE1α/c-Jun N-terminal kinase (JNK)/beclin1 and ATF6 (24, 139). The negative regulator of autophagy mTOR in diabetic PTECs is activated accompanying the increase of ER stress (140) and activating autophagy by Jujuboside A potently attenuates ER stress and cell death in the diabetic kidney (141). The autophagy in the kidney is usually inhibited under diabetic status (142, 143), which is reversed by the ER stress inhibitors salubrinal and tauroursodeoxycholic acid (143). Since ER stress inhibitors such as tauroursodeoxycholic acid, ursodeoxycholic acid, and 4-phenylbutyrate potently rescue diabetic renal tubules and podocytes (144, 145), investigating in detail the interaction between ER stress and autophagy in the progression of DN is promising.
4.2.3 Hypoxia stress
Kidney hypoxia, preceding the onset of albuminuria (146) and correlating with reduced glomerular filtration rate, runs through the whole stage of DN owing to the limited capacity of enhancing renal plasma flow and oxygen delivery (147). Hypoxia-inducible factor (HIF) is key to adaptively maintain cellular homeostasis by transcriptionally activating the expression of several target genes in response to hypoxia (148, 149).
Accumulating evidence shows that hypoxia is an important pathogenic factor for DN. Deficiency of HIF-1α has been reported to aggravate renal dysfunction (150), while up-regulation of HIF-1α effectively enhances autophagy to mitigate DN, which may associate with the increased expression of Sirt1, FoxO3, and BNIP3 (119, 151, 152). Recent studies demonstrate that up-regulation of sestrin2 by HIF-1α is involved in hypoxia-related diseases (153), which may modulate AMPK and mTORC1-dependent autophagy to reduce the production of ROS and attenuate DN (154, 155). Thus, the deteriorating effect of hypoxia on the diabetic kidney is not ignored and HIF-1α-related autophagy may be a potential target for treating DN.
5 Therapeutic strategies targeting autophagy for DN
The symptomatic treatment for DN usually includes glycemic control, reducing albuminuria, and blocking RAS with the usage of angiotensin-converting enzyme inhibitors (ACEI) and angiotensin receptor antagonists (ARB) (156, 157). New hypoglycemic agents such as sodium-glucose cotransporter 2 (SGLT2) inhibitors, glucagon-like peptide 1 receptor (GLP-1R) agonists, and dipeptidyl peptidase-4 (DPP-4) inhibitors have been shown to protect the diabetic kidney via modulating autophagy (Table 2).
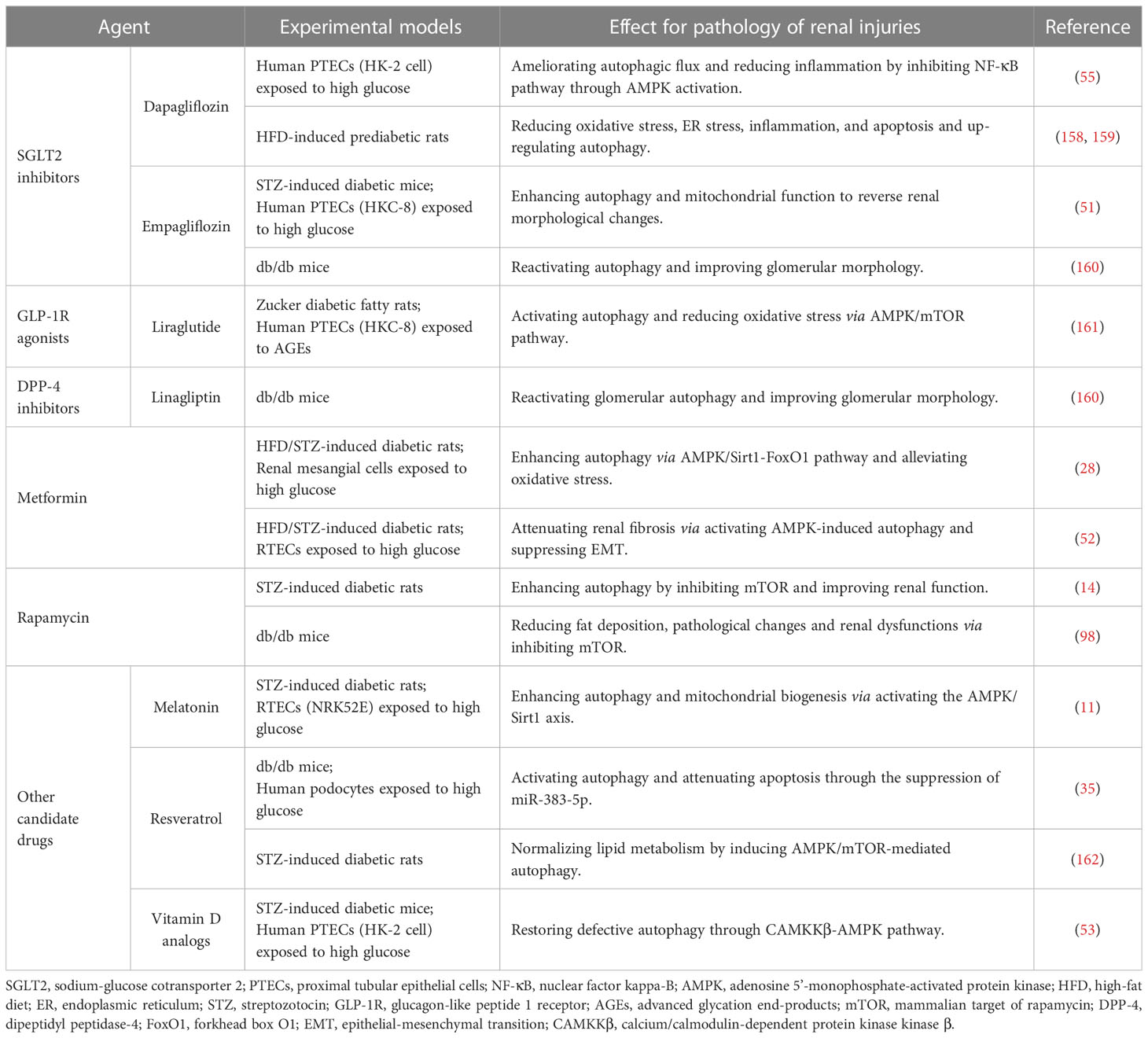
Table 2 Agents targeting autophagy for diabetic nephropathy.
Inhibiting SGLT2, located on the lumen surface of PTECs, potently lowers blood glucose by reducing the reabsorption of glucose (163). SGLT2 inhibitors empagliflozin and dapagliflozin have been shown to enhance autophagy depending on AMPK/mTOR pathway to attenuate diabetic kidney injury (51, 55, 158). Additionally, the progression of renal complications in pre-diabetes is slowed by dapagliflozin through the suppression of renal inflammation, ER stress, and apoptosis (159). Although the commercially available SGLT2 inhibitors including empagliflozin, dapagliflozin, and canagliflozin have been used in clinics (147), the protective effect against DN has not been fully elucidated (164).
Liraglutide, a GLP-1R analogue to lower blood glucose, has been shown to significantly improve the prognosis for DN (165), which may be related to reducing apoptosis and oxidative stress through promoting AMPK-regulated autophagy (161, 166). DPP-4 inhibitor linagliptin not only hinders the degradation of endogenous GLP-1 to lower blood glucose, but also alleviates mesangial expansion, podocyte foot process effacement, and albuminuria excretion in the diabetic kidney by reactivating autophagy (160). Additionally, the hypoglycemic agent metformin was reported to mitigate tubulointerstitial fibrosis and oxidative stress in diabetes by enhancing autophagy through AMPK/Sirt1/FoxO1 pathway (28, 52). Rapamycin has been shown to improve the short-term pathological alterations in DN by enhancing autophagy by blocking the mTORC1/ULK1 pathway (9). However, the serious side effect of rapamycin limits its use in long-term clinical treatment (75). Animal studies showed that melatonin, resveratrol, and vitamin D analogs prevent DN by modulating AMPK-regulated autophagy (11, 35, 53, 162), which may be the candidate drug for treating DN in the clinic.
Recently, exosome is becoming a promising therapeutic target for DN treatment (167). Exosome, as a kind of extracellular vesicles, is involved in intercellular communication by carrying various biomolecules and may be a novel biomarker for evaluating the progression of DN (168, 169). MiRNAs contained in the exosome derived from different cells attenuate high glucose-induced renal cell injury by promoting autophagy (36, 170, 171). Additionally, mesenchymal stem cell-derived exosomes induce autophagy via inhibiting mTOR to attenuate diabetic renal fibrosis (172). It is evident that the more we understand DN, the more we can do about DN. Exosome therapy combined with autophagy regulation may be promising for treating DN.
6 Conclusion
The significant increase in the incidence of diabetes has become a serious worldwide health issue. The high mortality of diabetes is strongly correlated with DN and the subsequent ESRD. Due to the complexity and diversity of the pathogenesis of DN, both rigorous control of blood glucose and cholesterol and blocking RAS with the usage of ACEI and ARB do not improve the endpoint of DN. The role of autophagy in the progression of DN sheds light on treating DN and how to keep the balance of autophagy in the diabetic kidney may be a new direction for prevention and management of DN though more efforts should be paid to exploring the precise regulation of autophagy in DN.
Author contributions
Y-PH wrote the manuscript. Y-PH, L-JL, J-LY, M-YC, X-FM, X-RZ, and L-BQ designed the figures and edited the manuscript. X-RZ and L-BQ supervised the writing. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Natural Science Foundation of China (81772035), Scientific Research Project of Education Department of Zhejiang Province (Y202045357), Basic Research Fee for Basic Research Business of Hangzhou Medical College (KYQN202005) and Program of Cultivating Zhejiang Provincial High-level Personnel in Health (Innovative Talent in 2021).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
References
1. Reidy K, Kang HM, Hostetter T, Susztak K. Molecular mechanisms of diabetic kidney disease. J Clin Invest (2014) 124:2333–40. doi: 10.1172/JCI72271
2. Lassén E, Daehn IS. Molecular mechanisms in early diabetic kidney disease: glomerular endothelial cell dysfunction. Int J Mol Sci (2020) 21:E9456. doi: 10.3390/ijms21249456
3. Yang D, Livingston MJ, Liu Z, Dong G, Zhang M, Chen J-K, et al. Autophagy in diabetic kidney disease: regulation, pathological role and therapeutic potential. Cell Mol Life Sci (2018) 75:669–88. doi: 10.1007/s00018-017-2639-1
4. Cao Z, Cooper ME. Pathogenesis of diabetic nephropathy. J Diabetes Investig (2011) 2:243–7. doi: 10.1111/j.2040-1124.2011.00131.x
5. Fu H, Liu S, Bastacky SI, Wang X, Tian X-J, Zhou D. Diabetic kidney diseases revisited: A new perspective for a new era. Mol Metab (2019) 30:250–63. doi: 10.1016/j.molmet.2019.10.005
6. Yamahara K, Yasuda M, Kume S, Koya D, Maegawa H, Uzu T. The role of autophagy in the pathogenesis of diabetic nephropathy. J Diabetes Res (2013) 2013:193757. doi: 10.1155/2013/193757
7. Wang W, Sun W, Cheng Y, Xu Z, Cai L. Role of sirtuin-1 in diabetic nephropathy. J Mol Med Berl Ger (2019) 97:291–309. doi: 10.1007/s00109-019-01743-7
8. Lin T-A, Wu VC-C, Wang C-Y. Autophagy in chronic kidney diseases. Cells (2019) 8:E61. doi: 10.3390/cells8010061
9. Gonzalez CD, Carro Negueruela MP, Nicora Santamarina C, Resnik R, Vaccaro MI. Autophagy dysregulation in diabetic kidney disease: From pathophysiology to pharmacological interventions. Cells (2021) 10:2497. doi: 10.3390/cells10092497
10. Parmar UM, Jalgaonkar MP, Kulkarni YA, Oza MJ. Autophagy-nutrient sensing pathways in diabetic complications. Pharmacol Res (2022) 184:106408. doi: 10.1016/j.phrs.2022.106408
11. Siddhi J, Sherkhane B, Kalavala AK, Arruri V, Velayutham R, Kumar A. Melatonin prevents diabetes-induced nephropathy by modulating the AMPK/Sirt1 axis: Focus on autophagy and mitochondrial dysfunction. Cell Biol Int (2022) 46:2142–57. doi: 10.1002/cbin.11899
12. Dong W, Zhang H, Zhao C, Luo Y, Chen Y. Silencing of miR-150-5p ameliorates diabetic nephropathy by targeting Sirt1/p53/AMPK pathway. Front Physiol (2021) 12:624989. doi: 10.3389/fphys.2021.624989
13. Ayinde KS, Olaoba OT, Ibrahim B, Lei D, Lu Q, Yin X, et al. AMPK allostery: A therapeutic target for the management/treatment of diabetic nephropathy. Life Sci (2020) 261:118455. doi: 10.1016/j.lfs.2020.118455
14. Liu L, Yang L, Chang B, Zhang J, Guo Y, Yang X. The protective effects of rapamycin on cell autophagy in the renal tissues of rats with diabetic nephropathy via mTOR-S6K1-LC3II signaling pathway. Ren Fail (2018) 40:492–7. doi: 10.1080/0886022X.2018.1489287
15. Chen K, Yu B, Liao J. LncRNA SOX2OT alleviates mesangial cell proliferation and fibrosis in diabetic nephropathy via Akt/mTOR-mediated autophagy. Mol Med Camb Mass (2021) 27:71. doi: 10.1186/s10020-021-00310-6
16. Shamekhi Amiri F. Intracellular organelles in health and kidney disease. Nephrol Ther (2019) 15:9–21. doi: 10.1016/j.nephro.2018.04.002
17. Tang C, Livingston MJ, Liu Z, Dong Z. Autophagy in kidney homeostasis and disease. Nat Rev Nephrol (2020) 16:489–508. doi: 10.1038/s41581-020-0309-2
18. Bhattacharya D, Mukhopadhyay M, Bhattacharyya M, Karmakar P. Is autophagy associated with diabetes mellitus and its complications? a review. EXCLI J (2018) 17:709–20. doi: 10.17179/excli2018-1353
19. Koch EAT, Nakhoul R, Nakhoul F, Nakhoul N. Autophagy in diabetic nephropathy: a review. Int Urol Nephrol (2020) 52:1705–12. doi: 10.1007/s11255-020-02545-4
20. Xiao C, Chen M-Y, Han Y-P, Liu L-J, Yan J-L, Qian L-B. The protection of luteolin against diabetic cardiomyopathy in rats is related to reversing JNK-suppressed autophagy. Food Funct (2023). doi: 10.1039/d2fo03871d
21. Abdelkader NF, Elbaset MA, Moustafa PE, Ibrahim SM. Empagliflozin mitigates type 2 diabetes-associated peripheral neuropathy: a glucose-independent effect through AMPK signaling. Arch Pharm Res (2022) 45:475–93. doi: 10.1007/s12272-022-01391-5
22. Su P-P, Liu D-W, Zhou S-J, Chen H, Wu X-M, Liu Z-S. Down-regulation of risa improves podocyte injury by enhancing autophagy in diabetic nephropathy. Mil Med Res (2022) 9:23. doi: 10.1186/s40779-022-00385-0
23. Koya D, Hayashi K, Kitada M, Kashiwagi A, Kikkawa R, Haneda M. Effects of antioxidants in diabetes-induced oxidative stress in the glomeruli of diabetic rats. J Am Soc Nephrol (2003) 14:S250–3. doi: 10.1097/01.asn.0000077412.07578.44
24. Cybulsky AV. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nat Rev Nephrol (2017) 13:681–96. doi: 10.1038/nrneph.2017.129
25. Liu H, Li Y, Xiong J. The role of hypoxia-inducible factor-1 alpha in renal disease. Mol Basel Switz (2022) 27:7318. doi: 10.3390/molecules27217318
26. Takabatake Y, Kimura T, Takahashi A, Isaka Y. Autophagy and the kidney: health and disease. Nephrol Dial Transplant (2014) 29:1639–47. doi: 10.1093/ndt/gft535
27. Gonzalez CD, Lee M-S, Marchetti P, Pietropaolo M, Towns R, Vaccaro MI, et al. The emerging role of autophagy in the pathophysiology of diabetes mellitus. Autophagy (2011) 7:2–11. doi: 10.4161/auto.7.1.13044
28. Ren H, Shao Y, Wu C, Ma X, Lv C, Wang Q. Metformin alleviates oxidative stress and enhances autophagy in diabetic kidney disease via AMPK/Sirt1-FoxO1 pathway. Mol Cell Endocrinol (2020) 500:110628. doi: 10.1016/j.mce.2019.110628
29. Han Y, Xiong S, Zhao H, Yang S, Yang M, Zhu X, et al. Lipophagy deficiency exacerbates ectopic lipid accumulation and tubular cells injury in diabetic nephropathy. Cell Death Dis (2021) 12:1031. doi: 10.1038/s41419-021-04326-y
30. Yang C, Chen X-C, Li Z-H, Wu H-L, Jing K-P, Huang X-R, et al. SMAD3 promotes autophagy dysregulation by triggering lysosome depletion in tubular epithelial cells in diabetic nephropathy. Autophagy (2021) 17:2325–44. doi: 10.1080/15548627.2020.1824694
31. Ding Y, Choi ME. Autophagy in diabetic nephropathy. J Endocrinol (2015) 224:R15–30. doi: 10.1530/JOE-14-0437
32. Zhang Y, Zhao S, Wu D, Liu X, Shi M, Wang Y, et al. MicroRNA-22 promotes renal tubulointerstitial fibrosis by targeting PTEN and suppressing autophagy in diabetic nephropathy. J Diabetes Res (2018) 2018:4728645. doi: 10.1155/2018/4728645
33. Wang X, Gao Y, Tian N, Zhu Z, Wang T, Xu J, et al. Astragaloside IV represses high glucose-induced mesangial cells activation by enhancing autophagy via Sirt1 deacetylation of NF-κB p65 subunit. Drug Des Devel Ther (2018) 12:2971–80. doi: 10.2147/DDDT.S174058
34. Jin J, Gong J, Zhao L, Zhang H, He Q, Jiang X. Inhibition of high mobility group box 1 (HMGB1) attenuates podocyte apoptosis and epithelial-mesenchymal transition by regulating autophagy flux. J Diabetes (2019) 11:826–36. doi: 10.1111/1753-0407.12914
35. Huang S-S, Ding D-F, Chen S, Dong C-L, Ye X-L, Yuan Y-G, et al. Resveratrol protects podocytes against apoptosis via stimulation of autophagy in a mouse model of diabetic nephropathy. Sci Rep (2017) 7:45692. doi: 10.1038/srep45692
36. Huang H, Liu H, Tang J, Xu W, Gan H, Fan Q, et al. M2 macrophage-derived exosomal miR-25-3p improves high glucose-induced podocytes injury through activation autophagy via inhibiting DUSP1 expression. IUBMB Life (2020) 72:2651–62. doi: 10.1002/iub.2393
37. Xu J, Deng Y, Wang Y, Sun X, Chen S, Fu G. SPAG5-AS1 inhibited autophagy and aggravated apoptosis of podocytes via SPAG5/Akt/mTOR pathway. Cell Prolif (2020) 53:e12738. doi: 10.1111/cpr.12738
38. Xu L, Fan Q, Wang X, Li L, Lu X, Yue Y, et al. Ursolic acid improves podocyte injury caused by high glucose. Nephrol Dial Transplant (2017) 32:1285–93. doi: 10.1093/ndt/gfv382
39. Li Y, Pan Y, Cao S, Sasaki K, Wang Y, Niu A, et al. Podocyte EGFR inhibits autophagy through upregulation of Rubicon in type 2 diabetic nephropathy. Diabetes (2021) 70:562–76. doi: 10.2337/db20-0660
40. Liu X-Q, Jiang L, Li Y-Y, Huang Y-B, Hu X-R, Zhu W, et al. Wogonin protects glomerular podocytes by targeting bcl-2-mediated autophagy and apoptosis in diabetic kidney disease. Acta Pharmacol Sin (2022) 43:96–110. doi: 10.1038/s41401-021-00721-5
41. Zhang Y, Yao H, Li C, Sun W, Chen X, Cao Y, et al. Gandi capsule improved podocyte lipid metabolism of diabetic nephropathy mice through Sirt1/AMPK/HNF4A pathway. Oxid Med Cell Longev (2022) 2022:6275505. doi: 10.1155/2022/6275505
42. Li F, Song L, Chen J, Chen Y, Li Y, Huang M, et al. Effect of genipin-1-β-d-gentiobioside on diabetic nephropathy in mice by activating AMP-activated protein kinase/silencing information regulator-related enzyme 1/ nuclear factor-κB pathway. J Pharm Pharmacol (2021) 73:1201–11. doi: 10.1093/jpp/rgab041
43. Wang S, Huang Y, Luo G, Yang X, Huang W. Cyanidin-3-O-glucoside attenuates high glucose-induced podocyte dysfunction by inhibiting apoptosis and promoting autophagy via activation of Sirt1/AMPK pathway. Can J Physiol Pharmacol (2021) 99:589–98. doi: 10.1139/cjpp-2020-0341
44. Liu Y, Liu W, Zhang Z, Hu Y, Zhang X, Sun Y, et al. Yishen capsule promotes podocyte autophagy through regulating Sirt1/NF-κB signaling pathway to improve diabetic nephropathy. Ren Fail (2021) 43:128–40. doi: 10.1080/0886022X.2020.1869043
45. Chen J, Yang Y, Lv Z, Shu A, Du Q, Wang W, et al. Study on the inhibitive effect of catalpol on diabetic nephropathy. Life Sci (2020) 257:118120. doi: 10.1016/j.lfs.2020.118120
46. Zhang X, Zhang L, Chen Z, Li S, Che B, Wang N, et al. Exogenous spermine attenuates diabetic kidney injury in rats by inhibiting AMPK/mTOR signaling pathway. Int J Mol Med (2021) 47:27. doi: 10.3892/ijmm.2021.4860
47. Liu H, Wang Q, Shi G, Yang W, Zhang Y, Chen W, et al. Emodin ameliorates renal damage and podocyte injury in a rat model of diabetic nephropathy via regulating AMPK/mTOR-mediated autophagy signaling pathway. Diabetes Metab Syndr Obes Targets Ther (2021) 14:1253–66. doi: 10.2147/DMSO.S299375
48. Yang L, Liang B, Li J, Zhang X, Chen H, Sun J, et al. Dapagliflozin alleviates advanced glycation end product induced podocyte injury through AMPK/mTOR mediated autophagy pathway. Cell Signal (2022) 90:110206. doi: 10.1016/j.cellsig.2021.110206
49. Zhang Z, Tang S, Gui W, Lin X, Zheng F, Wu F, et al. Liver X receptor activation induces podocyte injury via inhibiting autophagic activity. J Physiol Biochem (2020) 76:317–28. doi: 10.1007/s13105-020-00737-1
50. Zhou D, Zhou M, Wang Z, Fu Y, Jia M, Wang X, et al. PGRN acts as a novel regulator of mitochondrial homeostasis by facilitating mitophagy and mitochondrial biogenesis to prevent podocyte injury in diabetic nephropathy. Cell Death Dis (2019) 10:524. doi: 10.1038/s41419-019-1754-3
51. Lee YH, Kim SH, Kang JM, Heo JH, Kim D-J, Park SH, et al. Empagliflozin attenuates diabetic tubulopathy by improving mitochondrial fragmentation and autophagy. Am J Physiol Renal Physiol (2019) 317:F767–80. doi: 10.1152/ajprenal.00565.2018
52. Wang F, Sun H, Zuo B, Shi K, Zhang X, Zhang C, et al. Metformin attenuates renal tubulointerstitial fibrosis via upgrading autophagy in the early stage of diabetic nephropathy. Sci Rep (2021) 11:16362. doi: 10.1038/s41598-021-95827-5
53. Li A, Yi B, Han H, Yang S, Hu Z, Zheng L, et al. Vitamin d-VDR (vitamin d receptor) regulates defective autophagy in renal tubular epithelial cell in streptozotocin-induced diabetic mice via the AMPK pathway. Autophagy (2022) 18:877–90. doi: 10.1080/15548627.2021.1962681
54. Takahashi A, Takabatake Y, Kimura T, Maejima I, Namba T, Yamamoto T, et al. Autophagy inhibits the accumulation of advanced glycation end products by promoting lysosomal biogenesis and function in the kidney proximal tubules. Diabetes (2017) 66:1359–72. doi: 10.2337/db16-0397
55. Xu J, Kitada M, Ogura Y, Liu H, Koya D. Dapagliflozin restores impaired autophagy and suppresses inflammation in high glucose-treated HK-2 cells. Cells (2021) 10:1457. doi: 10.3390/cells10061457
56. Shati AA. Salidroside ameliorates diabetic nephropathy in rats by activating renal AMPK/Sirt1 signaling pathway. J Food Biochem (2020) 44:e13158. doi: 10.1111/jfbc.13158
57. Gao C, Fan F, Chen J, Long Y, Tang S, Jiang C, et al. FBW7 regulates the autophagy signal in mesangial cells induced by high glucose. BioMed Res Int (2019) 2019:6061594. doi: 10.1155/2019/6061594
58. Huang S, Xu Y, Ge X, Xu B, Peng W, Jiang X, et al. Long noncoding RNA NEAT1 accelerates the proliferation and fibrosis in diabetic nephropathy through activating Akt/mTOR signaling pathway. J Cell Physiol (2019) 234:11200–7. doi: 10.1002/jcp.27770
59. Hou B, Qiang G, Zhao Y, Yang X, Chen X, Yan Y, et al. Salvianolic acid a protects against diabetic nephropathy through ameliorating glomerular endothelial dysfunction via inhibiting AGE-RAGE signaling. Cell Physiol Biochem (2017) 44:2378–94. doi: 10.1159/000486154
60. Lim Jih, Kim Hw, Kim My, Kim Tw, Kim En, Kim Y, et al. Cinacalcet-mediated activation of the CaMKKβ-LKB1-AMPK pathway attenuates diabetic nephropathy in db/db mice by modulation of apoptosis and autophagy. Cell Death Dis (2018) 9:270. doi: 10.1038/s41419-018-0324-4
61. Jianbing H, Xiaotian L, Jie T, Xueying C, Honge J, Bo Z, et al. The effect of allograft inflammatory factor-1 on inflammation, oxidative stress, and autophagy via miR-34a/Atg4b pathway in diabetic kidney disease. Oxid Med Cell Longev (2022) 2022:1668000. doi: 10.1155/2022/1668000
62. Nagata M. Podocyte injury and its consequences. Kidney Int (2016) 89:1221–30. doi: 10.1016/j.kint.2016.01.012
63. Hartleben B, Gödel M, Meyer-Schwesinger C, Liu S, Ulrich T, Köbler S, et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest (2010) 120:1084–96. doi: 10.1172/JCI39492
64. Hong Q, Zhang L, Das B, Li Z, Liu B, Cai G, et al. Increased podocyte sirtuin-1 function attenuates diabetic kidney injury. Kidney Int (2018) 93:1330–43. doi: 10.1016/j.kint.2017.12.008
65. Liu F, Guo J, Qiao Y, Pan S, Duan J, Liu D, et al. MiR-138 plays an important role in diabetic nephropathy through Sirt1-p38-TTP regulatory axis. J Cell Physiol (2021) 236:6607–18. doi: 10.1002/jcp.30238
66. Bork T, Liang W, Yamahara K, Lee P, Tian Z, Liu S, et al. Podocytes maintain high basal levels of autophagy independent of mtor signaling. Autophagy (2020) 16:1932–48. doi: 10.1080/15548627.2019.1705007
67. Wang Z, Choi ME. Autophagy in kidney health and disease. Antioxid Redox Signal (2014) 20:519–37. doi: 10.1089/ars.2013.5363
68. Yin L, Yu L, He JC, Chen A. Controversies in podocyte loss: death or detachment? Front Cell Dev Biol (2021) 9:771931. doi: 10.3389/fcell.2021.771931
69. Kume S. Pathophysiological roles of nutrient-sensing mechanisms in diabetes and its complications. Diabetol Int (2019) 10:245–9. doi: 10.1007/s13340-019-00406-9
70. Kume S, Thomas MC, Koya D. Nutrient sensing, autophagy, and diabetic nephropathy. Diabetes (2012) 61:23–9. doi: 10.2337/db11-0555
71. Sugawara H, Moniwa N, Kuno A, Ohwada W, Osanami A, Shibata S, et al. Activation of the angiotensin II receptor promotes autophagy in renal proximal tubular cells and affords protection from ischemia/reperfusion injury. J Pharmacol Sci (2021) 145:187–97. doi: 10.1016/j.jphs.2020.12.001
72. Haraguchi R, Kohara Y, Matsubayashi K, Kitazawa R, Kitazawa S. New insights into the pathogenesis of diabetic nephropathy: Proximal renal tubules are primary target of oxidative stress in diabetic kidney. Acta Histochem Cytochem (2020) 53:21–31. doi: 10.1267/ahc.20008
73. Wu M, Zhang M, Zhang Y, Li Z, Li X, Liu Z, et al. Relationship between lysosomal dyshomeostasis and progression of diabetic kidney disease. Cell Death Dis (2021) 12:958. doi: 10.1038/s41419-021-04271-w
74. Shin JH, Kim KM, Jeong JU, Shin JM, Kang JH, Bang K, et al. Nrf2-heme oxygenase-1 attenuates high-glucose-induced epithelial-to-mesenchymal transition of renal tubule cells by inhibiting ROS-mediated PI3K/Akt/GSK-3β signaling. J Diabetes Res (2019) 2019:2510105. doi: 10.1155/2019/2510105
75. Lieberthal W, Levine JS. The role of the mammalian target of rapamycin (mTOR) in renal disease. J Am Soc Nephrol (2009) 20:2493–502. doi: 10.1681/ASN.2008111186
76. Yin S, Zhou S, Ren D, Zhang J, Xin H, He X, et al. Mesenchymal stem cell-derived exosomes attenuate epithelial-mesenchymal transition of HK-2 cells. Tissue Eng Part A (2022) 28:651–9. doi: 10.1089/ten.TEA.2021.0190
77. Kuwahara S, Hosojima M, Kaneko R, Aoki H, Nakano D, Sasagawa T, et al. Megalin-mediated tubuloglomerular alterations in high-fat diet-induced kidney disease. J Am Soc Nephrol (2016) 27:1996–2008. doi: 10.1681/ASN.2015020190
78. Saito A, Takeda T, Sato K, Hama H, Tanuma A, Kaseda R, et al. Significance of proximal tubular metabolism of advanced glycation end products in kidney diseases. Ann N Y Acad Sci (2005) 1043:637–43. doi: 10.1196/annals.1333.072
79. Wu X-Q, Zhang D-D, Wang Y-N, Tan Y-Q, Yu X-Y, Zhao Y-Y. AGE/RAGE in diabetic kidney disease and ageing kidney. Free Radic Biol Med (2021) 171:260–71. doi: 10.1016/j.freeradbiomed.2021.05.025
80. Wendt T, Tanji N, Guo J, Hudson BI, Bierhaus A, Ramasamy R, et al. Glucose, glycation, and RAGE: implications for amplification of cellular dysfunction in diabetic nephropathy. J Am Soc Nephrol (2003) 14:1383–95. doi: 10.1097/01.asn.0000065100.17349.ca
81. Jeon GY, Nam M-H, Lee K-W. Inhibitory effect of caffeic acid on advanced glycation end product-induced renal fibrosis in vitro: A potential therapeutic target. J Food Sci (2021) 86:579–86. doi: 10.1111/1750-3841.15588
82. Liu H-F, Liu H, Lv L-L, Ma K-L, Wen Y, Chen L, et al. CCN3 suppresses TGF-β1-induced extracellular matrix accumulation in human mesangial cells. vitro. Acta Pharmacol Sin (2018) 39:222–9. doi: 10.1038/aps.2017.87
83. Thomas HY, Ford Versypt AN. Pathophysiology of mesangial expansion in diabetic nephropathy: mesangial structure, glomerular biomechanics, and biochemical signaling and regulation. J Biol Eng (2022) 16:19. doi: 10.1186/s13036-022-00299-4
84. Baccora MHA, Cortes P, Hassett C, Taube DW, Yee J. Effects of long-term elevated glucose on collagen formation by mesangial cells. Kidney Int (2007) 72:1216–25. doi: 10.1038/sj.ki.5002517
85. Donate-Correa J, Luis-Rodríguez D, Martín-Núñez E, Tagua VG, Hernández-Carballo C, Ferri C, et al. Inflammatory targets in diabetic nephropathy. J Clin Med (2020) 9:458. doi: 10.3390/jcm9020458
86. Zhang Y, Jin D, Kang X, Zhou R, Sun Y, Lian F, et al. Signaling pathways involved in diabetic renal fibrosis. Front Cell Dev Biol (2021) 9:696542. doi: 10.3389/fcell.2021.696542
87. Sol M, Kamps JAAM, van den Born J, van den Heuvel MC, van der Vlag J, Krenning G, et al. Glomerular endothelial cells as instigators of glomerular sclerotic diseases. Front Pharmacol (2020) 11:573557. doi: 10.3389/fphar.2020.573557
88. Matsuda J, Namba T, Takabatake Y, Kimura T, Takahashi A, Yamamoto T, et al. Antioxidant role of autophagy in maintaining the integrity of glomerular capillaries. Autophagy (2018) 14:53–65. doi: 10.1080/15548627.2017.1391428
89. Gui Z, Suo C, Wang Z, Zheng M, Fei S, Chen H, et al. Impaired Atg16L-dependent autophagy promotes renal interstitial fibrosis in chronic renal graft dysfunction through inducing EndMT by NF-κB signal pathway. Front Immunol (2021) 12:650424. doi: 10.3389/fimmu.2021.650424
90. Casalena GA, Yu L, Gil R, Rodriguez S, Sosa S, Janssen W, et al. The diabetic microenvironment causes mitochondrial oxidative stress in glomerular endothelial cells and pathological crosstalk with podocytes. Cell Commun Signal (2020) 18:105. doi: 10.1186/s12964-020-00605-x
91. Lenoir O, Jasiek M, Hénique C, Guyonnet L, Hartleben B, Bork T, et al. Endothelial cell and podocyte autophagy synergistically protect from diabetes-induced glomerulosclerosis. Autophagy (2015) 11:1130–45. doi: 10.1080/15548627.2015.1049799
92. Wang L, Wang J, Cretoiu D, Li G, Xiao J. Exercise-mediated regulation of autophagy in the cardiovascular system. J Sport Health Sci (2020) 9:203–10. doi: 10.1016/j.jshs.2019.10.001
93. Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell (2010) 40:280–93. doi: 10.1016/j.molcel.2010.09.023
94. Russell RC, Yuan H-X, Guan K-L. Autophagy regulation by nutrient signaling. Cell Res (2014) 24:42–57. doi: 10.1038/cr.2013.166
95. Chan EYW, Kir S, Tooze SA. siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J Biol Chem (2007) 282:25464–74. doi: 10.1074/jbc.M703663200
96. Packer M. Interplay of adenosine monophosphate-activated protein kinase/sirtuin-1 activation and sodium influx inhibition mediates the renal benefits of sodium-glucose co-transporter-2 inhibitors in type 2 diabetes: A novel conceptual framework. Diabetes Obes Metab (2020) 22:734–42. doi: 10.1111/dom.13961
97. Packer M. Role of impaired nutrient and oxygen deprivation signaling and deficient autophagic flux in diabetic CKD development: Implications for understanding the effects of sodium-glucose cotransporter 2-inhibitors. J Am Soc Nephrol (2020) 31:907–19. doi: 10.1681/ASN.2020010010
98. Mori H, Inoki K, Masutani K, Wakabayashi Y, Komai K, Nakagawa R, et al. The mTOR pathway is highly activated in diabetic nephropathy and rapamycin has a strong therapeutic potential. Biochem Biophys Res Commun (2009) 384:471–5. doi: 10.1016/j.bbrc.2009.04.136
99. Zhang M-Z, Wang Y, Paueksakon P, Harris RC. Epidermal growth factor receptor inhibition slows progression of diabetic nephropathy in association with a decrease in endoplasmic reticulum stress and an increase in autophagy. Diabetes (2014) 63:2063–72. doi: 10.2337/db13-1279
100. Wang Y, Lu Y-H, Tang C, Xue M, Li X-Y, Chang Y-P, et al. Calcium dobesilate restores autophagy by inhibiting the VEGF/PI3K/Akt/mTOR signaling pathway. Front Pharmacol (2019) 10:886. doi: 10.3389/fphar.2019.00886
101. Inoki K, Mori H, Wang J, Suzuki T, Hong S, Yoshida S, et al. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J Clin Invest (2011) 121:2181–96. doi: 10.1172/JCI44771
102. Grahammer F, Huber TB, Artunc F. Role of mTOR signaling for tubular function and disease. Physiology (2021) 36:350–8. doi: 10.1152/physiol.00021.2021
103. Guo J, Liu Z, Gong R. Long noncoding RNA: an emerging player in diabetes and diabetic kidney disease. Clin Sci Lond Engl 1979 (2019) 133:1321–39. doi: 10.1042/CS20190372
104. Steinberg GR, Kemp BE. AMPK in health and disease. Physiol Rev (2009) 89:1025–78. doi: 10.1152/physrev.00011.2008
105. Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol (2018) 19:121–35. doi: 10.1038/nrm.2017.95
106. Hardie DG. AMPK: positive and negative regulation, and its role in whole-body energy homeostasis. Curr Opin Cell Biol (2015) 33:1–7. doi: 10.1016/j.ceb.2014.09.004
107. Momcilovic M, Hong S-P, Carlson M. Mammalian TAK1 activates Snf1 protein kinase in yeast and phosphorylates AMP-activated protein kinase. vitro. J Biol Chem (2006) 281:25336–43. doi: 10.1074/jbc.M604399200
108. Wei X, Lu Z, Li L, Zhang H, Sun F, Ma H, et al. Reducing NADPH synthesis counteracts diabetic nephropathy through restoration of AMPK activity in type 1 diabetic rats. Cell Rep (2020) 32:108207. doi: 10.1016/j.celrep.2020.108207
109. Mack HID, Zheng B, Asara JM, Thomas SM. AMPK-dependent phosphorylation of ULK1 regulates Atg9 localization. Autophagy (2012) 8:1197–214. doi: 10.4161/auto.20586
110. Kim S-J, Tang T, Abbott M, Viscarra JA, Wang Y, Sul HS. AMPK phosphorylates Desnutrin/AtgL and hormone-sensitive lipase to regulate lipolysis and fatty acid oxidation within adipose tissue. Mol Cell Biol (2016) 36:1961–76. doi: 10.1128/MCB.00244-16
111. Chang C, Su H, Zhang D, Wang Y, Shen Q, Liu B, et al. AMPK-dependent phosphorylation of GAPDH triggers Sirt1 activation and is necessary for autophagy upon glucose starvation. Mol Cell (2015) 60:930–40. doi: 10.1016/j.molcel.2015.10.037
112. Dusabimana T, Park EJ, Je J, Jeong K, Yun SP, Kim HJ, et al. Geniposide improves diabetic nephropathy by enhancing ULK1-mediated autophagy and reducing oxidative stress through AMPK activation. Int J Mol Sci (2021) 22:1651. doi: 10.3390/ijms22041651
113. Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, et al. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell (2013) 152:290–303. doi: 10.1016/j.cell.2012.12.016
114. Juszczak F, Caron N, Mathew AV, Declèves A-E. Critical role for AMPK in metabolic disease-induced chronic kidney disease. Int J Mol Sci (2020) 21:E7994. doi: 10.3390/ijms21217994
115. Morigi M, Perico L, Benigni A. Sirtuins in renal health and disease. J Am Soc Nephrol (2018) 29:1799–809. doi: 10.1681/ASN.2017111218
116. Liu R, Zhong Y, Li X, Chen H, Jim B, Zhou M-M, et al. Role of transcription factor acetylation in diabetic kidney disease. Diabetes (2014) 63:2440–53. doi: 10.2337/db13-1810
117. Zhang H, Yang X, Pang X, Zhao Z, Yu H, Zhou H. Genistein protects against ox-LDL-induced senescence through enhancing Sirt1/LKB1/AMPK-mediated autophagy flux in HUVECs. Mol Cell Biochem (2019) 455:127–34. doi: 10.1007/s11010-018-3476-8
118. Zhang N, Li L, Wang J, Cao M, Liu G, Xie G, et al. Study of autophagy-related protein light chain 3 (LC3)-II expression levels in thyroid diseases. BioMed Pharmacother (2015) 69:306–10. doi: 10.1016/j.biopha.2014.12.021
119. Ma L, Fu R, Duan Z, Lu J, Gao J, Tian L, et al. Sirt1 is essential for resveratrol enhancement of hypoxia-induced autophagy in the type 2 diabetic nephropathy rat. Pathol Res Pract (2016) 212:310–8. doi: 10.1016/j.prp.2016.02.001
120. Lan F, Cacicedo JM, Ruderman N, Ido Y. Sirt1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. possible role in AMP-activated protein kinase activation. J Biol Chem (2008) 283:27628–35. doi: 10.1074/jbc.M805711200
121. Ghosh HS, McBurney M, Robbins PD. Sirt1 negatively regulates the mammalian target of rapamycin. PloS One (2010) 5:e9199. doi: 10.1371/journal.pone.0009199
122. Zhang J, Zhang L, Zha D, Wu X. Inhibition of miRNA−135a−5p ameliorates TGF−β1−induced human renal fibrosis by targeting Sirt1 in diabetic nephropathy. Int J Mol Med (2020) 46:1063–73. doi: 10.3892/ijmm.2020.4647
123. Wang Y, Zheng Z-J, Jia Y-J, Yang Y-L, Xue Y-M. Role of p53/miR-155-5p/Sirt1 loop in renal tubular injury of diabetic kidney disease. J Transl Med (2018) 16:146. doi: 10.1186/s12967-018-1486-7
124. Shao Y, Lv C, Wu C, Zhou Y, Wang Q. Mir-217 promotes inflammation and fibrosis in high glucose cultured rat glomerular mesangial cells via Sirt1/HIF-1α signaling pathway. Diabetes Metab Res Rev (2016) 32:534–43. doi: 10.1002/dmrr.2788
125. Sies H. Oxidative stress: a concept in redox biology and medicine. Redox Biol (2015) 4:180–3. doi: 10.1016/j.redox.2015.01.002
126. Kaushal GP, Chandrashekar K, Juncos LA. Molecular interactions between reactive oxygen species and autophagy in kidney disease. Int J Mol Sci (2019) 20:3791. doi: 10.3390/ijms20153791
127. Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ (2015) 22:377–88. doi: 10.1038/cdd.2014.150
128. Pajares M, Cuadrado A, Engedal N, Jirsova Z, Cahova M. The role of free radicals in autophagy regulation: Implications for ageing. Oxid Med Cell Longev (2018) 2018:2450748. doi: 10.1155/2018/2450748
129. Ogura Y, Kitada M, Xu J, Monno I, Koya D. CD38 inhibition by apigenin ameliorates mitochondrial oxidative stress through restoration of the intracellular NAD+/NADH ratio and Sirt3 activity in renal tubular cells in diabetic rats. Aging (2020) 12:11325–36. doi: 10.18632/aging.103410
130. Hu Q, Qu C, Xiao X, Zhang W, Jiang Y, Wu Z, et al. Flavonoids on diabetic nephropathy: advances and therapeutic opportunities. Chin Med (2021) 16:74. doi: 10.1186/s13020-021-00485-4
131. Xu X, Yu Z, Han B, Li S, Sun Y, Du Y, et al. Luteolin alleviates inorganic mercury-induced kidney injury via activation of the AMPK/mTOR autophagy pathway. J Inorg Biochem (2021) 224:111583. doi: 10.1016/j.jinorgbio.2021.111583
132. Lu X, Fan Q, Xu L, Li L, Yue Y, Xu Y, et al. Ursolic acid attenuates diabetic mesangial cell injury through the up-regulation of autophagy via miRNA-21/PTEN/Akt/mTOR suppression. PloS One (2015) 10:e0117400. doi: 10.1371/journal.pone.0117400
133. Volpe CMO, Villar-Delfino PH, Dos Anjos PMF, Nogueira-Machado JA. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis (2018) 9:119. doi: 10.1038/s41419-017-0135-z
134. Görlach A, Klappa P, Kietzmann T. The endoplasmic reticulum: folding, calcium homeostasis, signaling, and redox control. Antioxid Redox Signal (2006) 8:1391–418. doi: 10.1089/ars.2006.8.1391
135. Burgos-Morón E, Abad-Jiménez Z, de Marañón AM, Iannantuoni F, Escribano-López I, López-Domènech S, et al. Relationship between oxidative stress, ER stress, and inflammation in type 2 diabetes: The battle continues. J Clin Med (2019) 8:1385. doi: 10.3390/jcm8091385
136. Cybulsky AV. The intersecting roles of endoplasmic reticulum stress, ubiquitin- proteasome system, and autophagy in the pathogenesis of proteinuric kidney disease. Kidney Int (2013) 84:25–33. doi: 10.1038/ki.2012.390
137. Cunard R. Endoplasmic reticulum stress in the diabetic kidney, the good, the bad and the ugly. J Clin Med (2015) 4:715–40. doi: 10.3390/jcm4040715
138. Xu X, Chen B, Huang Q, Wu Y, Liang T. The effects of puerarin on autophagy through regulating of the PERK/eIF2α/ATF4 signaling pathway influences renal function in diabetic nephropathy. Diabetes Metab Syndr Obes Targets Ther (2020) 13:2583–92. doi: 10.2147/DMSO.S256457
139. Wei Y, Sinha S, Levine B. Dual role of JNK1-mediated phosphorylation of bcl-2 in autophagy and apoptosis regulation. Autophagy (2008) 4:949–51. doi: 10.4161/auto.6788
140. Dorotea D, Jiang S, Pak ES, Son JB, Choi HG, Ahn S-M, et al. Pan-src kinase inhibitor treatment attenuates diabetic kidney injury via inhibition of fyn kinase-mediated endoplasmic reticulum stress. Exp Mol Med (2022) 54:1086–97. doi: 10.1038/s12276-022-00810-3
141. Zhong Y, Luo R, Liu Q, Zhu J, Lei M, Liang X, et al. Jujuboside a ameliorates high fat diet and streptozotocin induced diabetic nephropathy via suppressing oxidative stress, apoptosis, and enhancing autophagy. Food Chem Toxicol (2022) 159:112697. doi: 10.1016/j.fct.2021.112697
142. Ni L, Yuan C, Wu X. Endoplasmic reticulum stress in diabetic nephrology: regulation, pathological role, and therapeutic potential. Oxid Med Cell Longev (2021) 2021:7277966. doi: 10.1155/2021/7277966
143. Fang L, Zhou Y, Cao H, Wen P, Jiang L, He W, et al. Autophagy attenuates diabetic glomerular damage through protection of hyperglycemia-induced podocyte injury. PloS One (2013) 8:e60546. doi: 10.1371/journal.pone.0060546
144. Zhang J, Fan Y, Zeng C, He L, Wang N. Tauroursodeoxycholic acid attenuates renal tubular injury in a mouse model of type 2 diabetes. Nutrients (2016) 8:589. doi: 10.3390/nu8100589
145. Cao A-L, Wang L, Chen X, Wang Y-M, Guo H-J, Chu S, et al. Ursodeoxycholic acid and 4-phenylbutyrate prevent endoplasmic reticulum stress-induced podocyte apoptosis in diabetic nephropathy. Lab Investig J Tech Methods Pathol (2016) 96:610–22. doi: 10.1038/labinvest.2016.44
146. Franzén S, Pihl L, Khan N, Gustafsson H, Palm F. Pronounced kidney hypoxia precedes albuminuria in type 1 diabetic mice. Am J Physiol Renal Physiol (2016) 310:F807–809. doi: 10.1152/ajprenal.00049.2016
147. DeFronzo RA, Reeves WB, Awad AS. Pathophysiology of diabetic kidney disease: impact of SGLT2 inhibitors. Nat Rev Nephrol (2021) 17:319–34. doi: 10.1038/s41581-021-00393-8
148. Semenza GL. Pharmacologic targeting of hypoxia-inducible factors. Annu Rev Pharmacol Toxicol (2019) 59:379–403. doi: 10.1146/annurev-pharmtox-010818-021637
149. Catrina S-B, Zheng X. Hypoxia and hypoxia-inducible factors in diabetes and its complications. Diabetologia (2021) 64:709–16. doi: 10.1007/s00125-021-05380-z
150. Jiang N, Zhao H, Han Y, Li L, Xiong S, Zeng L, et al. HIF-1α ameliorates tubular injury in diabetic nephropathy via HO-1-mediated control of mitochondrial dynamics. Cell Prolif (2020) 53:e12909. doi: 10.1111/cpr.12909
151. Li L, Kang H, Zhang Q, D’Agati VD, Al-Awqati Q, Lin F. FoxO3 activation in hypoxic tubules prevents chronic kidney disease. J Clin Invest (2019) 129:2374–89. doi: 10.1172/JCI122256
152. Lin Q, Li S, Jiang N, Jin H, Shao X, Zhu X, et al. Inhibiting NLRP3 inflammasome attenuates apoptosis in contrast-induced acute kidney injury through the upregulation of HIF1α and BNIP3-mediated mitophagy. Autophagy (2021) 17:2975–90. doi: 10.1080/15548627.2020.1848971
153. Pan C, Chen Z, Li C, Han T, Liu H, Wang X. Sestrin2 as a gatekeeper of cellular homeostasis: Physiological effects for the regulation of hypoxia-related diseases. J Cell Mol Med (2021) 25:5341–50. doi: 10.1111/jcmm.16540
154. Eid AA, Lee D-Y, Roman LJ, Khazim K, Gorin Y. Sestrin 2 and AMPK connect hyperglycemia to Nox4-dependent endothelial nitric oxide synthase uncoupling and matrix protein expression. Mol Cell Biol (2013) 33:3439–60. doi: 10.1128/MCB.00217-13
155. Ala M, Eftekhar SP. Target sestrin2 to rescue the damaged organ: Mechanistic insight into its function. Oxid Med Cell Longev (2021) 2021:8790369. doi: 10.1155/2021/8790369
156. Greco EV, Russo G, Giandalia A, Viazzi F, Pontremoli R, De Cosmo S. GLP-1 receptor agonists and kidney protection. Med Kaunas Lith (2019) 55:233. doi: 10.3390/medicina55060233
157. Abdel-Rahman EM, Saadulla L, Reeves WB, Awad AS. Therapeutic modalities in diabetic nephropathy: standard and emerging approaches. J Gen Intern Med (2012) 27:458–68. doi: 10.1007/s11606-011-1912-5
158. Jaikumkao K, Promsan S, Thongnak L, Swe MT, Tapanya M, Htun KT, et al. Dapagliflozin ameliorates pancreatic injury and activates kidney autophagy by modulating the AMPK/mTOR signaling pathway in obese rats. J Cell Physiol (2021) 236:6424–40. doi: 10.1002/jcp.30316
159. Jaikumkao K, Pongchaidecha A, Chueakula N, Thongnak L-O, Wanchai K, Chatsudthipong V, et al. Dapagliflozin, a sodium-glucose co-transporter-2 inhibitor, slows the progression of renal complications through the suppression of renal inflammation, endoplasmic reticulum stress and apoptosis in prediabetic rats. Diabetes Obes Metab (2018) 20:2617–26. doi: 10.1111/dom.13441
160. Korbut AI, Taskaeva IS, Bgatova NP, Muraleva NA, Orlov NB, Dashkin MV, et al. SGLT2 inhibitor empagliflozin and DPP4 inhibitor linagliptin reactivate glomerular autophagy in db/db mice, a model of type 2 diabetes. Int J Mol Sci (2020) 21:2987. doi: 10.3390/ijms21082987
161. Yang S, Lin C, Zhuo X, Wang J, Rao S, Xu W, et al. Glucagon-like peptide-1 alleviates diabetic kidney disease through activation of autophagy by regulating AMP-activated protein kinase-mammalian target of rapamycin pathway. Am J Physiol Endocrinol Metab (2020) 319:E1019–30. doi: 10.1152/ajpendo.00195.2019
162. Zhu H, Zhong S, Yan H, Wang K, Chen L, Zhou M, et al. Resveratrol reverts streptozotocin-induced diabetic nephropathy. Front Biosci Landmark Ed (2020) 25:699–709. doi: 10.2741/4829
163. Sarafidis P, Ferro CJ, Morales E, Ortiz A, Malyszko J, Hojs R, et al. SGLT-2 inhibitors and GLP-1 receptor agonists for nephroprotection and cardioprotection in patients with diabetes mellitus and chronic kidney disease. a consensus statement by the EURECA-m and the DIABESITY working groups of the ERA-EDTA. Nephrol Dial Transplant (2019) 34:208–30. doi: 10.1093/ndt/gfy407
164. Barutta F, Bernardi S, Gargiulo G, Durazzo M, Gruden G. SGLT2 inhibition to address the unmet needs in diabetic nephropathy. Diabetes Metab Res Rev (2019) 35:e3171. doi: 10.1002/dmrr.3171
165. Su K, Yi B, Yao B-Q, Xia T, Yang Y-F, Zhang Z-H, et al. Liraglutide attenuates renal tubular ectopic lipid deposition in rats with diabetic nephropathy by inhibiting lipid synthesis and promoting lipolysis. Pharmacol Res (2020) 156:104778. doi: 10.1016/j.phrs.2020.104778
166. Miao X, Gu Z, Liu Y, Jin M, Lu Y, Gong Y, et al. The glucagon-like peptide-1 analogue liraglutide promotes autophagy through the modulation of 5’-AMP-activated protein kinase in INS-1 β-cells under high glucose conditions. Peptides (2018) 100:127–39. doi: 10.1016/j.peptides.2017.07.006
167. Thongboonkerd V, Kanlaya R. The divergent roles of exosomes in kidney diseases: Pathogenesis, diagnostics, prognostics and therapeutics. Int J Biochem Cell Biol (2022) 149:106262. doi: 10.1016/j.biocel.2022.106262
168. Mohan A, Singh RS, Kumari M, Garg D, Upadhyay A, Ecelbarger CM, et al. Urinary exosomal microRNA-451-5p is a potential early biomarker of diabetic nephropathy in rats. PloS One (2016) 11:e0154055. doi: 10.1371/journal.pone.0154055
169. Yamamoto CM, Murakami T, Oakes ML, Mitsuhashi M, Kelly C, Henry RR, et al. Uromodulin mRNA from urinary extracellular vesicles correlate to kidney function decline in type 2 diabetes mellitus. Am J Nephrol (2018) 47:283–91. doi: 10.1159/000489129
170. Jin J, Shi Y, Gong J, Zhao L, Li Y, He Q, et al. Exosome secreted from adipose-derived stem cells attenuates diabetic nephropathy by promoting autophagy flux and inhibiting apoptosis in podocyte. Stem Cell Res Ther (2019) 10:95. doi: 10.1186/s13287-019-1177-1
171. Zhao J, Chen J, Zhu W, Qi X-M, Wu Y-G. Exosomal miR-7002-5p derived from highglucose-induced macrophages suppresses autophagy in tubular epithelial cells by targeting Atg9b. FASEB J (2022) 36:e22501. doi: 10.1096/fj.202200550RR
Keywords: diabetic nephropathy, autophagy, nutrient-sensing pathway, cellular stress, renal cell
Citation: Han Y-P, Liu L-J, Yan J-L, Chen M-Y, Meng X-F, Zhou X-R and Qian L-B (2023) Autophagy and its therapeutic potential in diabetic nephropathy. Front. Endocrinol. 14:1139444. doi: 10.3389/fendo.2023.1139444
Received: 07 January 2023; Accepted: 07 March 2023;
Published: 20 March 2023.
Edited by:
Guiting Lin, University of California, San Francisco, United StatesReviewed by:
Zhengbing Zhuge, Karolinska Institutet (KI), SwedenYuanqing Feng, University of Pennsylvania, United States
Kai Wang, First Affiliated Hospital of Wenzhou Medical University, China
Copyright © 2023 Han, Liu, Yan, Chen, Meng, Zhou and Qian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ling-Bo Qian, bioqian@163.com; Xin-Ru Zhou, yorisep9098@163.com