 Li-Mei Li
Li-Mei Li Bei-Ge Jiang
Bei-Ge Jiang Liang-Liang Sun
Liang-Liang Sun- 1Research Center for Translational Medicine, Key Laboratory of Arrhythmias of the Ministry of Education of China, Shanghai East Hospital, Tongji University School of Medicine, Shanghai, China
- 2Third Department of Hepatic Surgery, Eastern Hepatobiliary Surgery Hospital, Naval Medical University, Shanghai, China
- 3Department of Endocrinology and Metabolism, Changzheng Hospital, Naval Medical University, Shanghai, China
Diabetes, a disease characterized by hyperglycemia, has a serious impact on the lives and families of patients as well as on society. Diabetes is a group of highly heterogeneous metabolic diseases that can be classified as type 1 diabetes (T1D), type 2 diabetes (T2D), gestational diabetes mellitus (GDM), or other according to the etiology. The clinical manifestations are more or less similar among the different types of diabetes, and each type is highly heterogeneous due to different pathogenic factors. Therefore, distinguishing between various types of diabetes and defining their subtypes are major challenges hindering the precise treatment of the disease. T2D is the main type of diabetes in humans as well as the most heterogeneous. Fortunately, some studies have shown that variants of certain genes involved in monogenic diabetes also increase the risk of T2D. We hope this finding will enable breakthroughs regarding the pathogenesis of T2D and facilitate personalized treatment of the disease by exploring the function of the signal genes involved. Hepatocyte nuclear factor 1 homeobox A (HNF1α) is widely expressed in pancreatic β cells, the liver, the intestines, and other organs. HNF1α is highly polymorphic, but lacks a mutation hot spot. Mutations can be found at any site of the gene. Some single nucleotide polymorphisms (SNPs) cause maturity-onset diabetes of the young type 3 (MODY3) while some others do not cause MODY3 but increase the susceptibility to T2D or GDM. The phenotypes of MODY3 caused by different SNPs also differ. MODY3 is among the most common types of MODY, which is a form of monogenic diabetes mellitus caused by a single gene mutation. Both T2D and GDM are multifactorial diseases caused by both genetic and environmental factors. Different types of diabetes mellitus have different clinical phenotypes and treatments. This review focuses on HNF1α gene polymorphisms, HNF1A-MODY3, HNF1A-associated T2D and GDM, and the related pathogenesis and treatment methods. We hope this review will provide a valuable reference for the precise and individualized treatment of diabetes caused by abnormal HNF1α by summarizing the clinical heterogeneity of blood glucose abnormalities caused by HNF1α mutation.
Introduction
According to the International Diabetes Federation (IDF), there were approximately 463 million adults with diabetes worldwide in 2019. The incidence is increasing, and this value will reach 700 million in 2045 (1). Diabetes has become a considerable public health problem that substantially impacts society and the families of patients (2). The disease is currently divided into four categories: type 1 diabetes (T1D), type 2 diabetes (T2D), gestational diabetes mellitus (GDM) and other special types of diabetes mellitus (3). The clinical manifestations of the different types of diabetes are similar, which greatly hinders the classification of the disease (4). Moreover, there is heterogeneity in the clinical phenotypes of the disease under the same type, which makes the clinical diagnosis of the diabetes type more difficult (4). Correctly diagnosing the type of diabetes is essential for precise treatment. The high heterogeneity of diabetes blurs the classic distinction between diabetes types. Interpreting the heterogeneity has become a major focus in diabetes research. It is necessary to establish a more accurate classification of diabetes. This will enable the precise and personalized treatment of diabetes, including the provision of more appropriate care, the development of hypoglycemic drugs, and the prevention/treatment of complications.
The most common types, i.e., T1D, T2D, and GDM, are multifactorial syndromes related to various gene effects and environmental factors (5). Two rare types, i.e., neonatal diabetes mellitus (NDM) and maturity-onset diabetes of the young (MODY), are caused by a single gene mutation (5, 6). MODY is a type of monogenic diabetes characterized by early-onset (age of diagnosis is usually before 25), autosomal dominant inheritance, no autoimmune process or insulin resistance, retention of endogenous insulin secretion, and no dependence on insulin (7). It is estimated that MODY accounts for approximately 1-2% of all diabetic cases (7). The pathogenesis of T2D and GDM is unclear. T2D is the most important type of diabetes and accounts for more than 90% of the total number of diabetic patients (3). Studies have shown that some monogenic diabetes genes are involved in T2D, with some variants significantly increasing the risk of T2D (8). T2D and GDM have solid genetic factors. Monogenic diabetes provides a good resource for elucidating the pathogenesis and developing personalized care for T2D and GDM. Substantial progress has been made in the research and development of hypoglycemic drugs targeting monogenic diabetes. This has provided the inspiration to further explore the pathogenesis and develop personalized treatments for T2D by studying the monogenic diabetes genes.
Hepatocyte nuclear factor 1α (HNF1α or HNF1A), also known as the MODY3 gene, is the pathogenic gene for MODY3 (7).Common types of HNF1α mutations cause MODY3. Other mutations do not cause MODY3 but significantly increase the risk of T2D, while others increase the risk of GDM. Hyperglycemia in MODY3 is caused by single-gene abnormalities. Unlike the pathogenesis of MODY, a large number of basic and clinical experiments show that T2D and GDM result from genetic and environmental factors. These two factors interact with each other to promote the occurrence of T2D or GDM. Why do different HNF1α mutations cause different types of diabetes? Answering these questions may improve our understanding of diabetes and advance the precise treatment of diabetes. In this review, we first summarize and discuss the relationship between HNF1α gene polymorphisms, MODY3, T2D, and GDM. We then explore the mechanism of hyperglycemia caused by HNF1α mutation by discussing the role of HNF1α in the islets and liver. Finally, we explore the reason why different HNF1α mutations can lead to different types of diabetes by analyzing the protein structure and function. We hope our review will facilitate a more comprehensive understanding of hyperglycemia caused by HNF1α mutation and will be useful for accurate diagnosis and treatment of diabetes, especially the hyperglycemia caused by HNF1α.
Polymorphism of HNF1A Gene
Human HNF1α is located at q24.31 on chromosome 12 (9) and is a widely expressed tissue-specific transcription factor (10). In the liver, HNF1α regulates numerous liver-specific genes and participates in the metabolism of glucose, fat, and other substances (11–13). In the pancreas, HNF1α controls many pancreatic-specific genes involved in β-cell maturation, growth, and insulin secretion (14–16). The HNF1α gene has a large number of polymorphisms with no specific mutation hotspots, and 894 variants are listed in the Exome Aggregation Consortium (ExAC) database. In total, 1231 variants can be queried from Genome Aggregation Database (gnomAD) (17). These variants range from the HNF1α promoter to the 3’ untranslated region (UTR), including missense, translocation, nonsense, splice mutation, in-frame amino acid deletion, insertion, duplication, or partial and whole gene deletion (17).
Although mutations have been observed in all exons, they are most often detected in exons 2 and 4 (Figures 1, 2). Among them, the mutation of exon 4 (p291fsinsc) is the most common (18). In a study of 414 different mutations in 1247 families carrying the HNF1α gene, the most common mutations were missense mutations (55%), frameshifts (22%), splice sites (9%), promoter region mutations (2%) and deletions (1.2%) (19). The mutations caused by these SNPs are as follows: 1) The mutation is located in the exon region and causes the substitution of an amino acid in HNF1α, resulting in a missense mutation (20). 2) The mutation is located in the exon region and causes abnormal shear in the HNF1α transcript (20). For example, although no RNA has been obtained from patients to prove this hypothesis, a synonymous mutation in HNF1α (c.1623 G > A, p.Gln541Gln) involving the last nucleotide of exon 8 was predicted to affect RNA splicing (21). 3) Mutations located in intron regions may generate new splice sites, resulting in pseudoexons (22, 23). 4) The mutation may be located in the promoter region, resulting in reduced gene expression (20).
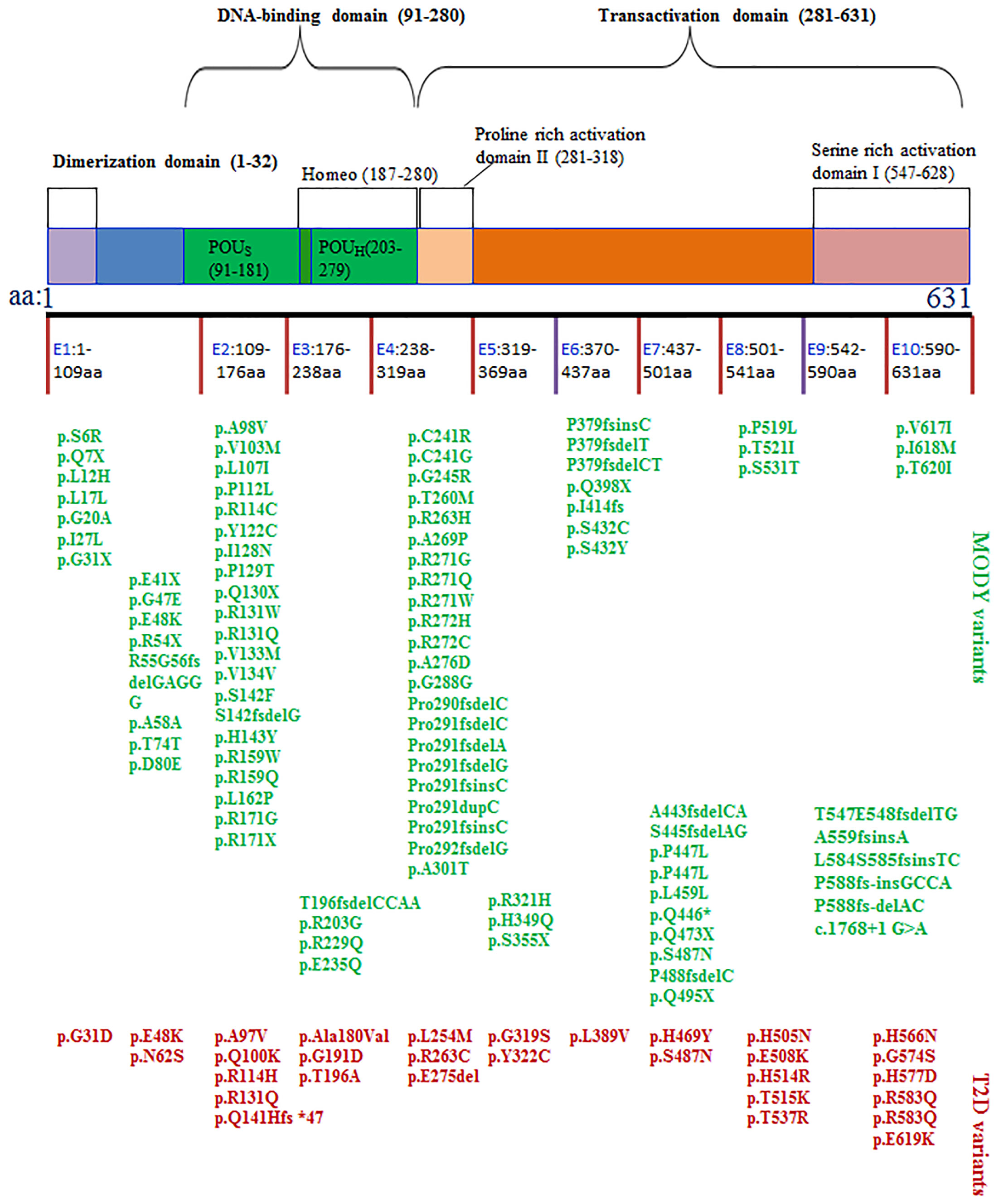
Figure 1 The structure of HNF1α protein and the SNPs associated with MODY3 or T2D. The SNPs in blue often cause MODY3. The SNPs in red often cause T2D.
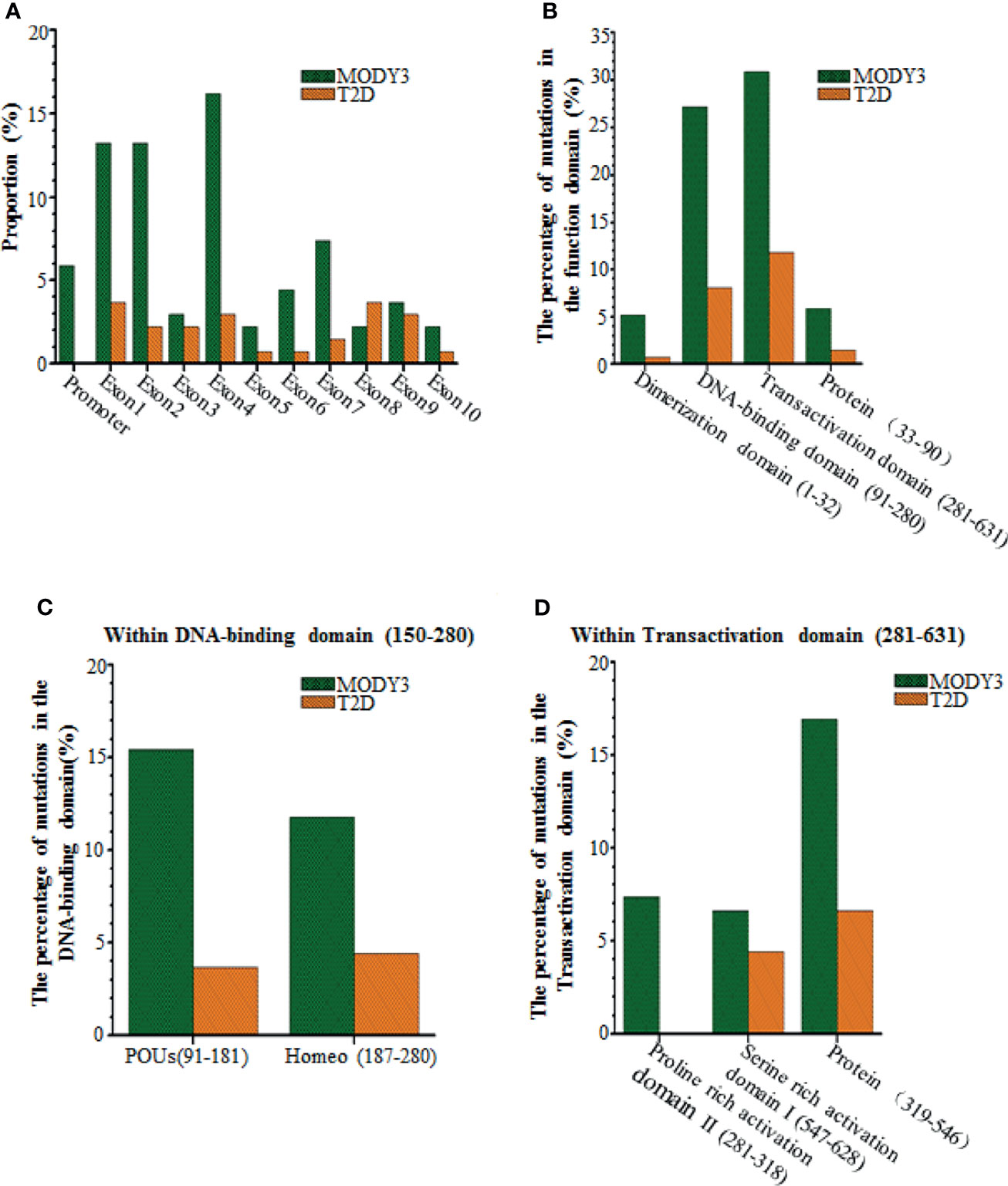
Figure 2 Comparisons of the HNF1a SNPs causing MODY3 with the HNF1a SNPs causing T2D. (A) Location within the promoter and 10 exons, (B) Location within the functional domains of HNF1α protein, (C) Location within DNA-binding domain of HNF1α protein, (D) Location within transactivation domain of HNF1α protein.
Association Between HNF1A Polymorphism and MODY3
HNF1A-MODY3 is characterized as familial diabetes (9, 24). Hyperglycemia usually becomes evident and deteriorates during puberty or early adulthood. Approximately 25% of HNF1A-MODY patients have typical polydipsia, polyuria, polyphagia, and emaciation symptoms in the initial stage, but most patients have none of the above clinical manifestations and only present elevated postprandial blood glucose, usually without ketosis (24, 25). Such patients usually show mild osmotic symptoms (polyuria and polydipsia) or asymptomatic postprandial hyperglycemia, without ketosis or ketoacidosis at age 6-25 (26). However, the clinical symptoms of HNF1A-MODY are very different. The clinical characteristics of HNF1A-MODY differ between and within families, which complicates the diagnostic process. In addition, different MODY3 patients exhibit great heterogeneity in the first-line use of sulfonylureas and the occurrence of complications. Therefore, we hope to provide some valuable information for MODY3 diagnosis, accurate treatment, and early intervention for other carriers of the same mutation in the same family before the occurrence of relevant clinical symptoms by summarizing some significant clinical features of MODY3.
Clinical Heterogeneity in MODY3
Thus far, hundreds of distinct HNF1α SNPs have been found to cause MODY3 (Figure 1 and Supplementary Table 1). The genetic diversity of MODY3 leads to heterogeneity in the MODY3 clinical phenotype. However, environmental factors also contribute to the clinical heterogeneity of MODY3. The main clinical heterogeneities in MODY3 are listed below.
Family History
As a case of autosomal dominant inherited disease, MODY3 patients usually have a family history of diabetes (9, 27, 28). Some studies suggest that the probability of a family history of MODY3 is more than 20 times higher than that of T1D (29). However, some MODY patients may lack a family history of diabetes, possibly due to the following factors (1): the probability of new HNF1A-MODY mutations may be more frequent than expected, and (2) the family members of the patients were not diagnosed because of mild clinical symptoms and signs (29, 30).
Age of Onset
Comparing the age of diabetes induced by HNF1α mutation in the literature revealed the age of onset of diabetes was generally 10-16 years (Table 1). This finding may be attributed to the high genetic penetrance of the HNF1α mutation and is consistent with previously reported conclusions. It has been reported that 63% of carriers are younger than 25 years, 79% are younger than 35 years, and 96% are younger than 55 years (39). The average age of an HNF1A-MODY diagnosis is 14 years, and the disease is rarely diagnosed in children under 10 years old (40). This finding is consistent with our statistical data (Table 1).
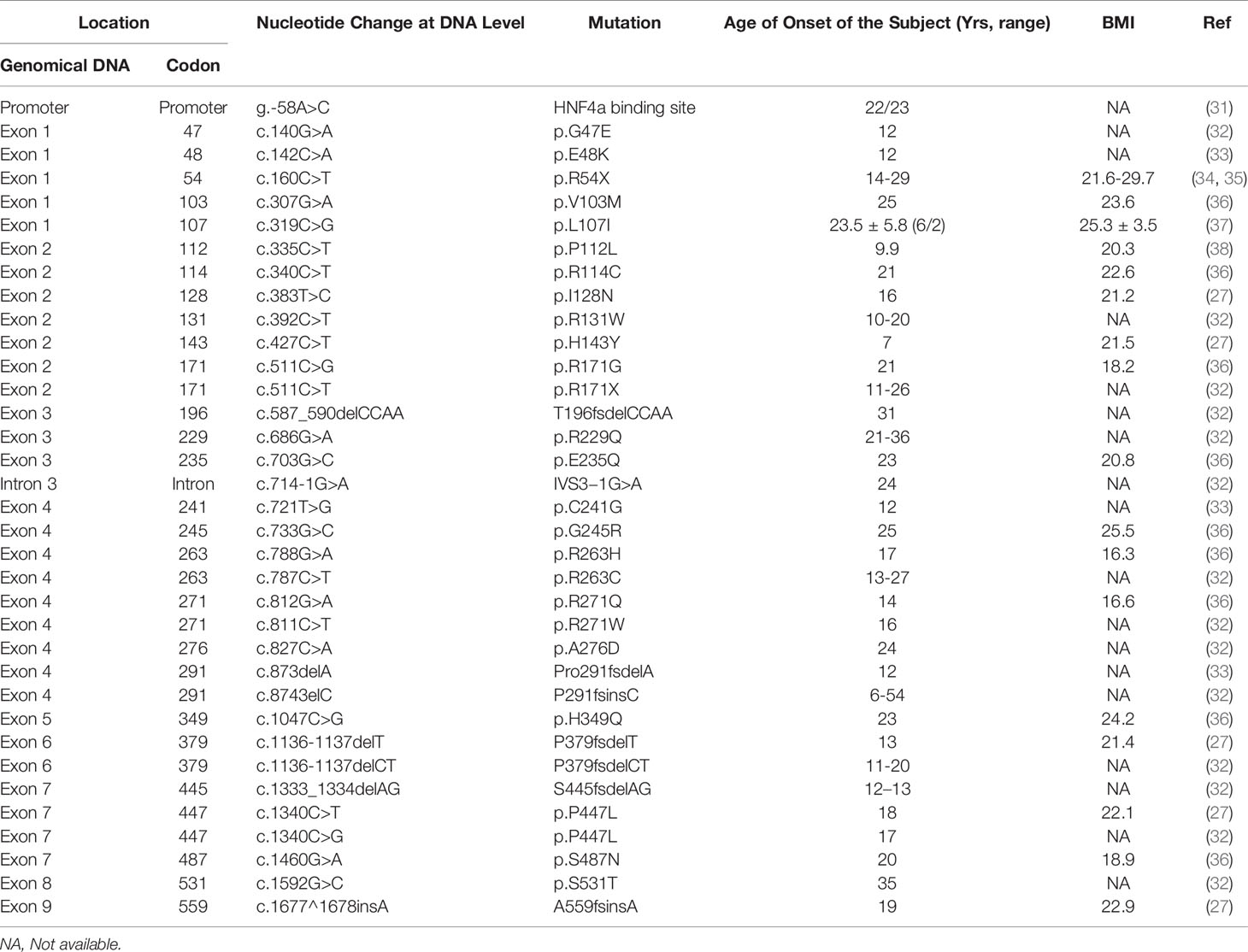
Table 1 Comparison of the onset ages of diabetes caused by different MODY3 associated SNPs.
The location of the mutation determines the age of diagnosis of abnormal blood glucose. As shown in Supplementary Table 1 and Table 1, the SNPs that cause MODY3 are concentrated in exons 1, 2, and 4, with relatively few SNPs in exons 8-10. Bellanne-Chantelot C et al. reported that patients with exon 8-10 mutations were diagnosed with HNF1A-MODY3 8 years later than those with exon 1-6 mutations (41). We found that patients with mutations affecting the dimerization domain or the DNA binding domain had a lower onset age (Table 1 and Figure 1). Patients with truncated mutations had a lower onset age than those with missense mutations. The above findings may indicate that the DNA binding region of HNF1α plays a more important role in regulating blood glucose, and the domain that forms a dimer plays the second most important role.
An individual’s genetic background and environment may also affect the onset age of HNF1A-MODY3. For example, intrauterine exposure (mutation inherited by the mother and hyperglycemia during pregnancy) can lead to an early onset age of less than 12 years (25). There are also differences in the age of onset with the same SNP. For example, a survey of MOD3 in Britain uncovered five diabetes patients in three generations of the same family with the R54X genotype (34, 35). Among them, the proband and his brother, mother, and grandmother were diagnosed with diabetes at puberty, while their uncle carried the same SNP and was only diagnosed with T2D at the age of 29 (24). Another family from China also exhibited a difference in the onset age of diabetes with R54X SNPs (35). The daughter was diagnosed with MODY3 at the age of 19, while the mother was first diagnosed with T2D at the age of 27. Among the above six R54X carriers (Table 2), 50% were first diagnosed with diabetes before the age of 25 years, which conforms to the standard age of diagnosis for MODY3. However, 50% of the diagnoses occurred at an age over 25, but all occurred before the age of 30. Therefore, HNF1α may play decisive roles in the development of abnormal blood glucose, but personal living habits, including eating, daily life, physical exercise, and other genetic factors that can cause abnormal metabolism, also play a role. Therefore, although a single HNF1α gene mutation is the cause of MODY3, environmental factors also play a role in its pathogenesis.
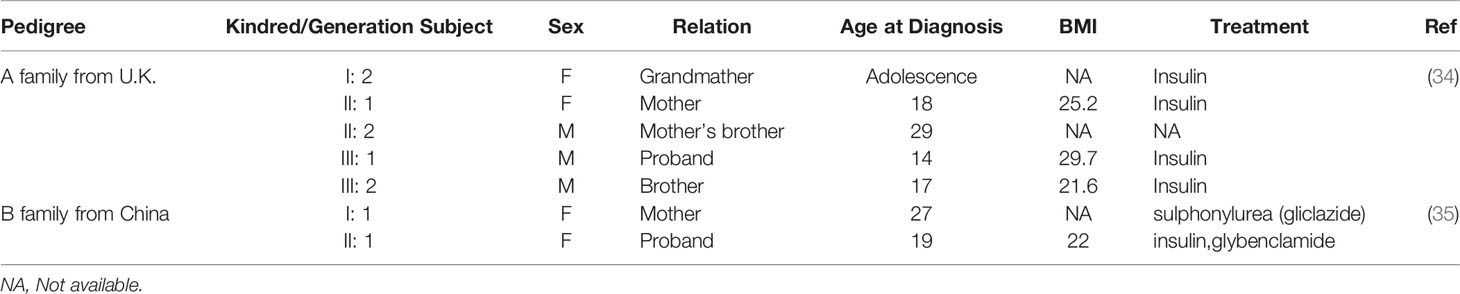
Table 2 The clinical characteristics of two R54X variants from different countries.
Insulin
The basal insulin level and insulin response to corresponding high glucose stimulation differ among patients with HNF1A-MODY3. Byrne MM et al. found that some patients with MODY3 had normal fasting blood glucose, while others had very high fasting blood glucose (42). Compared to those with normal blood glucose, the basal insulin secretion of the MODY3 patients was lower, but their insulin secretion increased in response to high-glucose stimulation; some patients showed a limited change, while others patients showed a decrease (42). This finding may indicate that HNF1α plays a direct role in regulating insulin secretion in islet β cells in response to high glucose or other stimuli. The difference may be a result of different mutations in HNF1α.
Glycosuria
Patients with HNF1α mutations may exhibit glycosuria; however, not all HNF1A-MODY patients do. Some studies have found that diabetes accounts for approximately 30-40% of all HNF1A-MODY patients (40, 43). For example, a study involving 11 HNF1A-MODY patients in Japan found that about 36% of HNF1A-diabetic patients experienced renal dysfunction (43). Another survey by Amanda Stride et al. also showed similar results (40). These authors found that 38% of mutation carriers developed glycosuria (40) 2 hours after oral glucose. Glycosuria in MODY3 may be caused by an impaired renal tubular transport of glucose and reduced glucose reabsorption in the proximal renal tubules (40, 44, 45). The renal glucose threshold of glucose reabsorption is low.
The renal glucose threshold of HNF1A-MODY patients is lower than that of healthy individuals with positive urine glucose, likely because HNF1α regulates the expression of the glucose transporter sodium-glucose cotransporter 2 (SGLT2) in the kidney because HNF1α can bind the promoter region of SGLT2 (46). In general, HNF1α needs to form a dimer to regulate the transcriptional expression of its target gene (11, 47). Both HNF1α and HNF1β are the most common transcription factors in the HNF transcription factor family and play important roles in the pancreas, liver, and kidney (10). These factors often form heterodimers (48). It has been reported by several groups that all diabetic patients with HNF1β mutations exhibit renal dysfunction (43, 49, 50). These research results imply that HNF1β plays a more dominant role in the kidney than HNF1α.
We found that HNF1α SNPs related to glycosuria were mainly concentrated in the dimer domain and DNA binding domain of HNF1α (Table 3), indicating that HNF1α regulates the transcription of genes that control the renal glucose threshold along with other transcription factors, likely by forming a dimer with HNF1β to regulate the genes involved in glucose transport or reabsorption in the kidney. However, this hypothesis is only our speculation and needs to be further confirmed via by experiments.

Table 3 The HNF1A SNPs associated glycosuria.
Cardiovascular System
HNF1A-MODY is a type of nonketotic diabetes characterized by progressive hyperglycemia in childhood, adolescence, and early adulthood and has a high risk of chronic microvascular complications (56, 57). The plasma glucose of patients deteriorates with age, and the risk of microvascular complications increases. In addition to the cardiovascular burden caused by uncontrolled blood glucose, some studies have found that some SNPs can increase the risk of vascular and cardiac diseases, such as I27L, A98V, and S487N (57). Some SNPs are closely related to dyslipidemia, a significant independent risk factor for cardiovascular abnormalities (58). This association may be related to the involvement of HNF1α in the synthesis of liver-related lipoproteins and liver lipid metabolism.
Some researchers reported that the incidence of cardiovascular and microvascular complications is similar to that in patients with T1D and T2D and is associated with poor glycemic control (25, 56, 59). However, Babaya N, et al. reported that the concentration of high-density lipoprotein cholesterol (HDL-C) in HNF1A-MODY (I27L carrier) is higher than that in normal individuals (60). HDL-C can reduce cardiovascular risk; thus, the incidence rate of coronary heart disease in HNF1A-MODY patients with I27L carrier may be lower than that in T1D patients and T2D patients.
Cancer
Recent studies showed that MODY3 is a risk factor for pancreatic cancer (61, 62). HNF1α gene mutation is related to pancreatic, liver, and kidney tumors (63–66). It has been reported that HNF1α mutations (p.E32 * and p.L214Q) are related to hepatocellular tumors (67). Somatic HNF1α mutations are found in approximately 1% to 2% of hepatocellular carcinomas and usually occur in adenomas. HNF1α mutations increase the risk of the malignant transformation of hepatocellular adenomas (67).
Treatment
HNF1A-MODY3 can cause severe diabetic retinopathy, diabetic nephropathy, diabetic peripheral neuropathy, and other complications (59). Therefore, early diagnosis and timely treatment are very important for blood glucose control, delaying the occurrence and development of complications and improving the quality of life.
The phenotype of HNF1A-MODY is characterized by mild nonprogressive hyperglycemia, progressive hyperglycemia, and hyperglycemia with extra-pancreatic characteristics (25, 26). In patients diagnosed with mild hyperglycemia, diet seems to be a reasonable and effective treatment strategy; however, in the case of progressive hyperglycemia, pharmacological methods should be attempted (68, 69). The treatment of HNF1A-MODY patients depends on their age and HbA1c level (64). Patients with HNF1α mutations are very sensitive to the oral hypoglycemic drug sulfonylurea (70, 71). It is speculated that this may be due to the decreased liver clearance of some sulfonylurea derivatives in patients with HNF1α gene mutations, resulting in an increase in serum levels (72). The increased circulating levels of these drugs could explain the enhanced efficacy. The response of HNF1A-MODY patients to sulfonylureas is five times higher than that to standard metformin (73). However, in T2D, the efficacy of the two drugs has been demonstrated to be almost the same (73). Sulfonylureas usually control blood glucose better than insulin therapy in patients with HNF1A-MODY, and the fasting hypoglycemic effect is also good (70). HNF1A-MODY patients show obvious sensitivity to oral sulfonylurea drugs. The failure of sulfonylurea treatment is rare and occurs in only a few patients with the c.618G>A mutation (74). Therefore, low-dose sulfonylurea drugs (such as 20-40 mg/day gliclazide) are preferred for long-term treatment and should be regarded as the first-line treatment for HNF1A-MODY (73).
However, studies have shown that patients with some variants no longer respond to the above treatments. Patients with p.His126Asp do not respond to sulfonylureas (low and high doses), dipeptidyl peptidase-4 inhibitors (DPP-4 inhibitors), or glucagon-like peptide-1 receptor agonists (GLP-1Raa), also known as incretin analogs (75). Low-dose sulfonylurea therapy is the first-line therapy for MODY3 but does not show special sensitivity to HNF1α-related T2D (76). For example, Mexican carriers of the HNF1α p.E508K variant have no increased sensitivity to sulfonylureas (76).
In a word, HNF1A-MODY is a disease with genetic and clinical heterogeneity. However, MODY3 still has strong common characteristics. Patients with HNF1A-MODY usually have low high-sensitivity C-reactive protein (hsCRP) levels (77, 78). In addition, although MODY3 is a type of monogenic diabetes, the severity can differ with different genetic backgrounds and environmental factors. Previously, MODY3 patients were generally not overweight or obese and lacked other risk factors for T2D, such as hypertension or dyslipidemia (79). However, as an increasing number of people have become overweight or obese, individuals with MODY3 may also be overweight or obese. According to statistics, nearly 30% of HNF1A-MODY patients in the United States are overweight or obese, rendering the differential diagnosis between HNF1A-MODY3 and T2D more difficult (80). Therefore, although some clinical guidelines for MODY3 diagnosis exist, HNF1A-MODY3 patients often fail to meet all diagnostic criteria or are misdiagnosed with T1D or T2D.
Association Between HNF1A Polymorphism and T2D
Studies have found that some HNF1α SNPs do not cause MODY3 but increase the susceptibility to T2D (Supplementary Table 1). A large-scale association analysis of a group of people mainly of European ancestry showed that the T2D susceptibility loci existed near HNF1α (81). Moreover, SNPs related to T2D are closely related to race. The p.E508K variant was found only in T2D patients in Mexico (82). The rare p.A98V allele may be associated with T2D in the Caucasian population (83). However, the p.A98V allele does not appear to be associated with T2D in Asian populations (84). Among them, the most famous locus is G319S. The G319S polymorphism of HNF1α is positively correlated with the high prevalence of T2D in Canadian Aborigines (85, 86).
Gene sequencing revealed that the G319S mutation in HNF1α was associated with an increased incidence rate of T2D in the Oji Cree ethnic group in Canada. The specific and positive predictive values of G319S carriers suffering from T2D before the age of 50 were 97% and 95%, respectively (87). G319S is associated with a distinct form of T2D characterized by onset at an earlier age, higher postprandial plasma glucose, and lower body mass index (BMI) (85). In patients with T2D, compared to those with G319/G319, the BMIs of individuals with S319/S319 and S319/G319 were significantly lower, and postprandial blood glucose was significantly higher (85). In nondiabetic individuals, the plasma insulin of S319/G319 heterozygotes was significantly lower than that of G319/G319 homozygotes (85). A lower BMI coupled with the decrease of insulin secretion before the onset of diabetes is the pre-diabetic physiologic state of individuals with HNF1A-T2D. This group is different from those with other types of T2D caused by other genes. The latter group is often generally obese and has a high BMI with insulin resistance before obvious diabetes. Moreover, smoking appears to increase the risk of diabetes in HNF1α G319S carriers (88). G319s is located in HNF1α transactivation sites, which are rich in proline II domains (Figure 1), with changes in conserved glycine residues. The function of the protein carrying the G319S mutation was found to be impaired in vitro (87), and the transcription ability was reduced by approximately 50% (88). However, this mutation did not affect DNA binding or protein stability. There is no evidence that the mutant protein has a dominant-negative effect. The G319S mutation affected the transcriptional shear of HNF1α. Two abnormal transcripts and an alteration in the relative balance of normal splicing products were produced by the G319S variant (89). Two abnormal transcripts present only in the G319S cells included premature termination codons resulting from the inclusion of seven nucleotides from intron 4 or the deletion of exon 8. A novel isoform lacking the terminal 12 bases of exon 4 was increased compared with that in control cell lines and human pancreatic tissue. The combination of the reduced activity of the G319S protein and abnormal splicing transcripts may increase the susceptibility to diabetes.
HNF1A-SNPs associated with T2D only increase the risk of T2D. Obvious T2D requires other factors, such as genetic and/or environmental factors. An interesting example can be found in stories of a new HNF1α variant c.539C >T (p.Ala180Val) in two families in Norway (90). There was an obvious difference in the probability of T2D between these two families (90). p.Ala180Val is a mutation that affects highly conserved amino acid residues in proteins. The HNF1α mutant p. Ala180Val does not cause MODY3 but may increase the risk of T2D. This variation was found to be completely separate from diabetes in one family (family A), but the data did not support its role as a pathogenic factor of MODY3. In the other family (family B), there was no clear genotype/phenotype correlation. Two diabetic patients and one individual with normal plasma glucose levels in this family were homozygous mutation carriers. In family A, the mutation carriers had similar metabolic syndromes, including obesity, diabetes, hypertension, and dyslipidemia. Moreover, the nonmutation carriers in the family were overweight or obese, although they had normal blood glucose levels, but they were as overweight or obese as the family members carrying the mutation. Genetic factors may be related to the metabolic abnormalities in family A. Therefore, it can be speculated that HNF1α p.Ala180Val could lead to a certain degree of β cell dysfunction; however, it does not cause significant glycemic abnormalities. The genetic background of family A associated with metabolic abnormalities increased insulin resistance in the HNF1α p.Ala180Val carriers, leading to marked diabetes. In contrast, family B members did not present with obesity or metabolic syndrome, but some female mutation carriers had a history of GDM. Therefore, we can speculate that p.Ala180Val can increase the susceptibility to hyperglycemia but cannot lead to obvious diabetes. Under the influence of additional stress, such as other genetic factors responsible for obesity or pregnancy, p.Ala180Val carriers develop peripheral insulin resistance or permanent impairment in β cellular function, resulting in marked diabetes. The difference between the two families may be due to the accumulation of the higher-risk variant for T2D in family A. Overall, lifestyle and environmental factors may also play a role in the phenotypic differences.
Low-dose sulfonylurea therapy is the first-line therapy for MODY3 but does not show special sensitivity to T2D. For carriers of HNF1α variants that may cause T2D, dietary treatment is the first recommendation.
Association Between HNF1A Polymorphism and GDM
GDM is a common complication of pregnancy that has adverse effects on the short-term and long-term health of women and their children (91). Approximately 2-5% of pregnant women develop GDM during pregnancy, and the incidence rate has significantly increased over the past 10 years (92). GDM is diagnosed when any degree of glucose intolerance occurs during pregnancy for the first time. GDM is also heterogeneous diabetes, with varying degrees of diabetes caused by β cell dysfunction (92). When pancreatic β cells can no longer compensate for the increased insulin resistance during pregnancy, they show varying degrees of glucose intolerance. Although the pathogenesis of the disease is still largely unknown, GDM is considered the result of an interaction between genetic and environmental risk factors. Age, obesity, and a high-fat diet are some important nongenetic factors (93).
Many studies have found that HNF1α SNP increases the risk of GDM. HNF1α is one of the pathogenic genes of GDM in Danish women. The sequencing of 354 Danish GDM patients revealed five diabetes-susceptible variants of HNF1α in seven HNF1α mutation carriers (94). Only those with the Gly288fs* variant were diagnosed with diabetes before the follow-up period and received insulin treatment. The remaining HNF1α mutation carriers were not diagnosed with diabetes before the follow-up period. p.A98V was associated with significant impairment of serum insulin and C-peptide secretion during an oral glucose tolerance test in previously GDM-free glucose-tolerant women (95). The p.I27L polymorphism of HNF1α seems to increase the risk of GDM in Scandinavian women (96). The p.I27L gene was found to increase GDM by increasing insulin resistance in Turkish women (97). In Scandinavian women, the p.I27L polymorphism of HNF1α also increased the risk of GDM (96). The p.I27L TT genotype was associated with an increased risk of preeclampsia in patients with GDM by increasing blood pressure and urinary protein (97). No difference in weight was observed compared to non-diabetic pregnant women with HNF1α mutation during the entire pregnancy.
Pregnant women with HNF1α mutation may develop GDM due to islet dysfunction. Pregnancy is a significant source of stress for women, which leads to the increase of insulin demand. If the need for insulin cannot be met, GDM will gradually develop. Dyslipidemia during pregnancy increases the risk of pregnancy complications. The lipid profile has a strong genetic determinant. In 2017, Xiaojing Wang et al. found that the total cholesterol levels of pregnant women carrying the T alleles of rs1169309 in the HNF1α gene were elevated, which could significantly increase the risk of GDM (98). Insulin resistance may also be involved in the occurrence of GDM.
Compared with other gene mutation carriers, GDM HNF1α mutation carriers exhibit a significant reduction in hsCRP expression (94). hsCRP is encoded by the CRP gene, which has an HNF1α transcription factor-specific binding site (65, 99). SNPs in HNF1α have been associated with CRP levels in different populations (100, 101). The expression of hsCRP in GDM patients is higher than that in HNF1A-MODY patients (84). This finding indicates that GDM caused by HNF1α results in the same susceptibility to diabetes as the T2D variant, but insufficient penetrance leads to clinical MODY.
Dyslipidemia in pregnancy increases the risk of pregnancy complications. Therefore, pregnant women who are HNF1α gene mutation carriers should pay special attention to their health management and consume a reasonable diet during pregnancy. If HNF1α mutation carriers have hyperglycemia during pregnancy, they should not be treated with sulfonylureas and need to be treated with insulin.
Mechanism of Abnormal Blood Glucose Associated With HNF1A Gene Polymorphism
HNF1α is expressed in embryonic development, infancy, and adulthood. Moreover, the expression distribution has strong tissue specificity, mainly concentrated in the tissues responsible for metabolism, such as the pancreas and liver. Through transcriptomics and antibody-based proteomics, the analysis of human tissue-specific expression showed that the expression level of HNF1α varies in human tissue and controls the transcriptional expression of many genes in the tissue (102). According to incomplete statistics, HNF1α can bind at least 106 target genes in the pancreas (103), which may explain why the mutation location of the HNF1α gene determines the age of diabetes onset. The above results indicated that HNF1α may play varied and important roles in the tissues. Below, we focus on the possible mechanism of HNF1α for glucose homeostasis in the pancreas and liver.
Functions in Pancreas
Maintain the Mature β Cell Function
The dysfunction of mature β cells is the main reason for the hyperglycemia caused by HNF1α. Basal insulin secretion and insulin production corresponding to high glucose stimuli are the basic functions of mature β cells. MODY3 patients have insulin secretion disorder, and the islet secretion function gradually declines as the disease worsens (104). Fasting insulin and glucose-stimulated insulin secretion were found to be abnormal in 40 HNF1α mutation carries from Britain and France (105). The insulin sensitivity was elevated in these individuals as well as the proinsulin to insulin ratio. The results indicated that HNF1α mutation directly altered basal insulin secretion, rather than glucose sensing insulin secretion. However, a recent islet study of a 33-year-old patient with MODY3, who was misdiagnosed with T1D for 17 years, found that HNF1α causes insulin deficiency diabetes by affecting glucose-stimulated insulin secretion (106). The two results were different, possibly due to different HNF1α variants. In addition, G319S carriers reportedly showed a decrease in insulin secretion before diabetes appeared (85). This indicated islet dysfunction also exists in HNF1A-T2D patients. Therefore, we think HNF1α plays very important and different roles in basal insulin secretion and insulin production corresponding to high glucose stimuli. In order to better study the role of HNF1α in β cell dysfunction, it is necessary to compare the islet function of HNF1α mutants with different degrees of clinical severity to identify the domain of HNF1α involved in the secretion of basic insulin and the domain involved in the secretion of insulin stimulated by high glucose.
HNF1α can regulate many genes involved in insulin secretion (16, 42) and directly bind to the promoter region of the insulin gene, positively regulating the transactivation of the latter. In addition to directly regulating insulin transcription, HNF1α was found to directly regulate the GLUT2 and HNF4 pyruvate kinase genes, which are the key genes involved in cellular insulin secretion (103). In addition, heterozygous HNF1α variations change the expression of key enzymes involved in mitochondrial glucose metabolism (107). In conclusion, HNF1α is an important transcription factor for the maintenance of β cell function, but the specific mechanism needs to be further studied.
Development and Maturation of Islets
HNF1α is involved in the development and maturation of islets. In normal embryonic mouse pancreas, the expression of HNF1α was detected on embryonic day 10.5 (E10.5) (108). When the dominant negative p291fsinsc HNF1α mutation is specifically expressed in β cells (driven by the rat Ins2 promoter), the islets are gradually disordered with reduced cells numbers, and the cells are dispersed in the islet (109, 110). Signs of serious cell damage can be observed, including vacuolization, immature secretory granules, swollen mitochondria, and expanded endoplasmic reticulum (109, 110). Nkx6.1 is a homologous domain transcription factor that plays a role in pancreatic development and the maintenance of mature β cellular function (111). Studies have shown that Nkx6 can directly activate the expression of HNF1α (112). HNF1α in turn directly activates the expression of MafA, which encodes transcription factors produced later in the developing pancreatic transcription program and is expressed only in differentiated insulin hormone positive cells (113).
Some studies indicated that HNF1α is not limited to β cell development and may also affect α cell development. HNF1α whole-body knockout mice died at 6 weeks after birth with small islets and a high α/β ratio (114). This phenomenon is consistent with the islet results for diabetic patients with HNF1α mutation (106). The increased α/β cell ratio may be caused by the slightly higher quality of α cells in the pancreas of these patients. The manifestations of cell hyperfunction, excessive glucagon secretion, weakened negative feedback to glucose, and decreased intestinal glucagon effect are observed in MODY3 patients (115). In 2020, Kazuya Yamagata et al. found HNF1α can inhibit glucagon secretion by regulating SGLT1 expression in α cells (116). HNF1α was found to inhibit α cell characteristics in modeling monogenic diabetes using human embryonic stem cells through mutations in HNF1α (117).
Although further research is needed, it is clear that HNF1α plays a role in pancreatic organogenesis, endocrine and exocrine cell differentiation, and growth by influencing islet specific transcription factors.
Function in Liver
In the liver, HNF1α may affect the balance of blood glucose through regulated glucose and lipid metabolism. HNF1α directly binds to the promoter region of the glucose 6-phosphate transporter (G6PT) gene, the key enzyme of the glucose-6-phosphatase (G6Pase) system, to promote the transcription of the latter (118). Compared with HNF1α (+/+) and HNF1α (+/-) littermates, hepatic G6PT mRNA levels and microsomal G6P transport activity are markedly reduced in HNF1α (-/-) mice (118). The G6Pase system is essential for the maintenance of glucose homeostasis. Thus the HNF1α variant may cause abnormal glucose homeostasis through the G6Pase system. After HNF1α deletion in the liver, the expression of genes encoding fatty acid synthetic enzymes (fatty acid synthase and acyl-CoA carboxylase) and peroxisomal β-oxidation enzymes (CYP4A3, bifunctional enzyme, and thiolase) increased (119). However, the expression of the hepatic fatty acid binding protein (L-FABP) gene decreased significantly (119). Two HNF1α binding sites were found in the 5’ promoter region of L-FABP by sequence analysis. Cell experiments confirmed that HNF1α was necessary for the transactivation of L-FABP (119). We speculated that HNF1α mutation may disrupt the balance of the blood glucose through regulating glucose metabolism and adipogenesis.
HNF1α also participates in the transcription of apolipoprotein genes (120, 121). The HNF1α G319S genotype was significantly correlated with the total plasma cholesterol, low-density lipoprotein cholesterol (LDL-C), and apolipoprotein (Apo) B concentration in Oji Cree individuals with T2D (122). In Oji Cree people who did not have T2D, we found that the HNF1α G319S genotype was significantly associated with the plasma concentrations of HDL-C and apolipoprotein AI (123). The phenotype was not related to plasma triglyceride or lipoprotein (a). I27L had a protective effect on hypertriglyceridemia in these individual samples (57). The above studies all indicated that HNF1α plays a role in the lipid profile of diabetic individuals. These data enhance our understanding of the complex interactions among genes, hyperglycemia, and cardiovascular risk factors in T2D. Some studies have shown that a decreased expression of HNF1α increases the risk of fatty liver, which is closely related to insulin resistance (122, 124).
In summary, HNF1α SNPs responsible for the abnormality of blood glucose may be caused by changes in the development, promotion, and death of β cells, the maintenance of mature pancreatic function, as well as glucose and lipid metabolism in the liver.
Relationship Between HNF1A and Different Types of Diabetes
HNF1A-MODY3 is characterized by not only gene heterogeneity but also phenotype heterogeneity. For example, the two human variants HNF1α (P408H) and HNF1α (P409H) can be considered different variants (125, 126). A possible reason is that these two adjacent sites regulate the promoters of two different HNF1A-targeted genes. This is not surprising and can be explained by the interactions between HNF1α and different coactivators, which are necessary for the complete activation of different HNF1α downstream target promoters (127). The above phenomena show that HNF1α plays many roles and is widely involved in the strict and fine regulation of many genes responsible for metabolism.
Some HNF1α SNPs cause obvious MODY3, while others do not cause MODY3 but increase the risk of T2D or GDM. These are very interesting phenomena. By determining the underlying mechanism of these phenomena, we may improve our ability to individualize diabetes treatment. First, what is the difference between MODY, T2D, and GDM? The biggest difference is the age/time of onset. The onset age of MODY is below 25 years old, while T2D occurs in adulthood, and the onset of GDM is obvious diabetes in the middle and later stages of pregnancy. The earlier the onset, the more dominant is the role of genetic factors in the occurrence of diseases. The impact of environmental and other physical factors is greater with later onset. In essence, pregnancy is a stressor on the female body. The second major difference is whether the family history is obvious. The more obvious the family history is, the more important is the role of genetic factors. HNF1α heterozygous mutations lead to MODY3. Since an HNF1α allele is normal in MODY patients, it can be deduced that the expression level of HNF1α is important in this group. MODY plays a vital role in cell function, especially in β cells. A lack of sufficient protein levels leads to a significant loss of function of mutant alleles (104), and dominant negative effects caused by interference between mutant products and wild-type forms lead to the formation of inactive heterodimers (128). HNF1A-MODY-related variants function through one of the above mechanisms, i.e., a simple loss of function or a dominant-negative mechanism. T2D and GDM are diabetes types caused by multiple factors. The HNF1α SNP, which triggers T2D and GDM, has a certain impact on the process of abnormal blood glucose. Therefore, we speculate that HNF1α SNPs related to T2D or GDM partially impair the function of the HNF1α protein.
The HNF1α protein contains the following three functional domains: an N-terminal dimer domain, a DNA binding region containing a nuclear localization signal, and a C-terminal transactivation domain (Figure 1). The N-terminal dimeric domain (residues 1-32) forms a four-helix bundle, two of which separate the α-helix in a circle to form a dimer (129, 130). Usually, two HNF1αs form a homodimer or one HNF1α associates with the HNF1β transcription factor, which has a similar structure, to form a heterodimer (47). The DNA binding domain (DBD) of HNF1α binds reverse palindrome 5’-gttaatnataac-3’ and forms a helix-to-helix structure (131). The DBD includes two POU subdomains, i.e., POU-specific domain (POUs, amino acids 91-181) and POU-homologous domain (POUH, amino acids 203-279) (104). The amino acid positions of DBDs differ slightly. POUs are an integral part of HNF1α and play a vital role in maintaining protein stability (132). Comparing the common HNF1α SNPs that cause MODY3 with those that cause T2D, most of the former are concentrated in exon 1, 2, and 4, which encode the DNA binding region of the C-terminal transactivation domain of HNF1α (Figure 1). The HNF1α SNP leading to T2D is concentrated in exons 8 and 9. The above sites of the HNF1α SNP that lead to T2D are almost outside the proline rich activation domain II of HNF1α, which is located in the transactivation domain.
SNP mutations may affect the function of the HNF1α protein through the following mechanisms: 1) Affecting the ability of DNA to bind the transcriptional regulatory region of the target gene; 2) Affecting the transcriptional activity of HNF1α; 3) Affecting the nuclear entry ability of the HNF1α protein; 4) Affecting the stability of the HNF1α protein; and 5) Affecting the expression of the HNF1α protein, especially SNPs located in the promoter region (107, 125, 133). In 2017, Najmi et al. found that the transcriptional activity or DNA binding ability of HNF1α variants that cause T2D was between those of normal wild-type protein and the HNF1A-MODY variant (134). The above interesting study may indicate that HNF1α participates in multiple signaling pathways involved in abnormal blood glucose and that the HNF1α variants identified among T2D patients may lack sufficient penetrance to drive diabetes but still increase the susceptibility to diabetes.
In summary, the HNF1α gene is highly polymorphic, and the clinical phenotypes caused by different SNPs may vary greatly. Therefore, to better manage the abnormal blood glucose levels caused by HNF1α mutations, genotype identification should be performed to obtain detailed information concerning HNF1α.
Conclusion and Future Perspectives
To date, many HNF1α SNPs have been identified and are widely distributed in the HNF1α gene. HNF1α-associated diabetes mellitus has larger clinical heterogeneity. Significant differences have been found in abnormal plasma glucose caused by SNPs at different sites. Patients with some variants do not have diabetes throughout their lives, some with other variants show serious hyperglycemia in childhood, while others show hyperglycemia in their older years. HNF1α is a tissue-specific transcription factor mainly expressed in the pancreas and liver. Hundreds of target genes have been found in these tissues. In the pancreas, HNF1α not only maintains the function of mature pancreatic β cells, but also affects the development and maturation of β cells. In the liver, HNF1α abnormality inhibits hepatic glycogen decomposition and promotes lipolysis. Uncovering the mechanism underlying why different HNF1α mutations cause different types of diabetes will provide a theoretical basis for personalized prevention and treatment of HNF1α-associated hyperglycemia and will also benefit research on T2D, which is the main type of diabetes. HNF1α may cause different forms of diabetes due to different levels of penetrance and genetic backgrounds. These differences have a certain correlation with the location of the SNPs. The SNP for HNF1A-MODY is often located in the DNA binding region, while those for T2D and GDM are located in a different region. Functional studies have shown that the transcriptional activity or DNA binding ability of the HNF1α variant of T2D is between those of the normal wild-type protein and the HNF1A-MODY variant. This finding may indicate that the HNF1α variants identified among T2D patients may lack sufficient penetrance to drive diabetes but still increase the susceptibility to diabetes. Accordingly, there must be differences in the treatment of diabetes caused by different SNPs. Low-dose sulfonylurea therapy is the first-line therapy for MODY3 but does not show special sensitivity to T2D. For carriers of the HNF1α variants that may cause T2D or GDM, dietary treatment is the first recommendation. We hope research on the pathogenesis and drug treatments for HNF1A-T2D and GDM will progress in the near future with studies on the function of HNF1α.
Author Contributions
L-ML and B-GJ collected information. L-LS and L-ML wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by grants from the National Natural Science Foundation of China (81672721), National Key Research and Development Projection (No. 2018YFC1314100), Shanghai Science Foundation (17DZ1910604 and 19ZR1456900), Diabetes Talent Research Project of China International Medical Foundation (2018-N-01-26), Key Laboratory Development Project of Minimally Invasive Techniques & Rapid Rehabilitation of Digestive System Tumor (21SZDSYS05).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2022.829565/full#supplementary-material
References
1. Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N, et al. Global and Regional Diabetes Prevalence Estimates for 2019 and Projections for 2030 and 2045: Results From the International Diabetes Federation Diabetes Atlas, 9(Th) Edition. Diabetes Res Clin Pract (2019) 157:107843. doi: 10.1016/j.diabres.2019.107843
2. Bommer C, Heesemann E, Sagalova V, Manne-Goehler J, Atun R, Barnighausen T, et al. The Global Economic Burden of Diabetes in Adults Aged 20-79 Years: A Cost-of-Illness Study. Lancet Diabetes Endocrinol (2017) 5:423–30. doi: 10.1016/S2213-8587(17)30097-9
3. American Diabetes Association. Diagnosis and Classification of Diabetes Mellitus. Diabetes Care (2014) 37 Suppl 1:S81–90. doi: 10.2337/dc14-S081
4. Redondo MJ, Hagopian WA, Oram R, Steck AK, Vehik K, Weedon M, et al. The Clinical Consequences of Heterogeneity Within and Between Different Diabetes Types. Diabetologia (2020) 63:2040–8. doi: 10.1007/s00125-020-05211-7
5. Udler MS, McCarthy MI, Florez JC, Mahajan A. Genetic Risk Scores for Diabetes Diagnosis and Precision Medicine. Endocr Rev (2019) 40:1500–20. doi: 10.1210/er.2019-00088
6. Hattersley AT, Patel KA. Precision Diabetes: Learning From Monogenic Diabetes. Diabetologia (2017) 60:769–77. doi: 10.1007/s00125-017-4226-2
7. Stanik JJ, Dusatkova P, Cinek O, Valentinova L, Huckova M, Skopkova M, et al. De Novo Mutations of GCK, HNF1A and HNF4A may be More Frequent in MODY Than Previously Assumed. Diabetologia (2014) 57:480–4. doi: 10.1007/s00125-013-3119-2
8. Redondo MJ, Balasubramanyam A. Toward an Improved Classification of Type 2 Diabetes: Lessons From Research Into the Heterogeneity of a Complex Disease. J Clin Endocrinol Metab (2021) 106:e4822–33. doi: 10.1210/clinem/dgab545
9. Yamagata K, Oda N, Kaisaki PJ, Menzel S, Furuta H, Vaxillaire M, et al. Mutations in the Hepatocyte Nuclear Factor-1alpha Gene in Maturity-Onset Diabetes of the Young (MODY3). Nature (1996) 384:455–8. doi: 10.1038/384455a0
10. Harries LW, Brown JE, Gloyn AL. Species-Specific Differences in the Expression of the HNF1A, HNF1B and HNF4A Genes. PloS One (2009) 4:e7855. doi: 10.1371/journal.pone.0007855
11. Ktistaki E, Talianidis I. Modulation of Hepatic Gene Expression by Hepatocyte Nuclear Factor 1. Science (1997) 277:109–12. doi: 10.1126/science.277.5322.109
12. Shih DQ, Bussen M, Sehayek E, Ananthanarayanan M, Shneider BL, Suchy FJ, et al. Hepatocyte Nuclear Factor-1alpha is an Essential Regulator of Bile Acid and Plasma Cholesterol Metabolism. Nat Genet (2001) 27:375–82. doi: 10.1038/86871
13. Rufibach LE, Duncan SA, Battle M, Deeb SS. Transcriptional Regulation of the Human Hepatic Lipase (LIPC) Gene Promoter. J Lipid Res (2006) 47:1463–77. doi: 10.1194/jlr.M600082-JLR200
14. Boj SF, Parrizas M, Maestro MA, Ferrer J. A Transcription Factor Regulatory Circuit in Differentiated Pancreatic Cells. Proc Natl Acad Sci USA (2001) 98:14481–6. doi: 10.1073/pnas.241349398
15. Wang H, Antinozzi PA, Hagenfeldt KA, Maechler P, Wollheim CB. Molecular Targets of a Human HNF1 Alpha Mutation Responsible for Pancreatic Beta-Cell Dysfunction. EMBO J (2000) 19:4257–64. doi: 10.1093/emboj/19.16.4257
16. Malakauskas SM, Kourany WM, Zhang XY, Lu D, Stevens RD, Koves TR, et al. Increased Insulin Sensitivity in Mice Lacking Collectrin, a Downstream Target of HNF-1alpha. Mol Endocrinol (2009) 23:881–92. doi: 10.1210/me.2008-0274
17. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of Protein-Coding Genetic Variation in 60,706 Humans. Nature (2016) 536:285–91. doi: 10.1038/nature19057
18. Ellard S, Colclough K. Mutations in the Genes Encoding the Transcription Factors Hepatocyte Nuclear Factor 1 Alpha (HNF1A) and 4 Alpha (HNF4A) in Maturity-Onset Diabetes of the Young. Hum Mutat (2006) 27:854–69. doi: 10.1002/humu.20357
19. Colclough K, Bellanne-Chantelot C, Saint-Martin C, Flanagan SE, Ellard S. Mutations in the Genes Encoding the Transcription Factors Hepatocyte Nuclear Factor 1 Alpha and 4 Alpha in Maturity-Onset Diabetes of the Young and Hyperinsulinemic Hypoglycemia. Hum Mutat (2013) 34:669–85. doi: 10.1002/humu.22279
20. Zhao Z, Fu YX, Hewett-Emmett D, Boerwinkle E. Investigating Single Nucleotide Polymorphism (SNP) Density in the Human Genome and its Implications for Molecular Evolution. Gene (2003) 312:207–13. doi: 10.1016/s0378-1119(03)00670-x
21. Alvelos MI, Goncalves CI, Coutinho E, Almeida JT, Bastos M, Sampaio ML, et al. Maturity-Onset Diabetes of the Young (MODY) in Portugal: Novel GCK, HNFA1 and HNFA4 Mutations. J Clin Med (2020) 9:288. doi: 10.3390/jcm9010288
22. Tilgner H, Nikolaou C, Althammer S, Sammeth M, Beato M, Valcarcel J, et al. Nucleosome Positioning as a Determinant of Exon Recognition. Nat Struct Mol Biol (2009) 16:996–1001. doi: 10.1038/nsmb.1658
23. Dhir A, Buratti E. Alternative Splicing: Role of Pseudoexons in Human Disease and Potential Therapeutic Strategies. FEBS J (2010) 277:841–55. doi: 10.1111/j.1742-4658.2009.07520.x
24. Peixoto-Barbosa R, Reis AF, Giuffrida FMA. Update on Clinical Screening of Maturity-Onset Diabetes of the Young (MODY). Diabetol Metab Syndr (2020) 12:50. doi: 10.1186/s13098-020-00557-9
25. Hattersley AT, Greeley SAW, Polak M, Rubio-Cabezas O, Njolstad PR, Mlynarski W, et al. ISPAD Clinical Practice Consensus Guidelines 2018: The Diagnosis and Management of Monogenic Diabetes in Children and Adolescents. Pediatr Diabetes (2018) 19(Suppl 27):47–63. doi: 10.1111/pedi.12772
26. Frayling TM, Evans JC, Bulman MP, Pearson E, Allen L, Owen K, et al. Beta-Cell Genes and Diabetes: Molecular and Clinical Characterization of Mutations in Transcription Factors. Diabetes (2001) 50(Suppl 1):S94–100. doi: 10.2337/diabetes.50.2007.s94
27. Hansen T, Eiberg H, Rouard M, Vaxillaire M, Moller AM, Rasmussen SK, et al. Novel MODY3 Mutations in the Hepatocyte Nuclear Factor-1alpha Gene: Evidence for a Hyperexcitability of Pancreatic Beta-Cells to Intravenous Secretagogues in a Glucose-Tolerant Carrier of a P447L Mutation. Diabetes (1997) 46:726–30. doi: 10.2337/diab.46.4.726
28. Vaxillaire M, Rouard M, Yamagata K, Oda N, Kaisaki PJ, Boriraj VV, et al. Identification of Nine Novel Mutations in the Hepatocyte Nuclear Factor 1 Alpha Gene Associated With Maturity-Onset Diabetes of the Young (MODY3). Hum Mol Genet (1997) 6:583–6. doi: 10.1093/hmg/6.4.583
29. Salzano G, Passanisi S, Mammi C, Priolo M, Pintomalli L, Caminiti L, et al. Maturity Onset Diabetes of the Young is Not Necessarily Associated With Autosomal Inheritance: Case Description of a De Novo HFN1A Mutation. Diabetes Ther (2019) 10:1543–8. doi: 10.1007/s13300-019-0633-3
30. Li M, Riviere JB, Polychronakos C. Why All MODY Variants are Dominantly Inherited: A Hypothesis. Trends Genet (2021) S0168-9525(21):00285–7. doi: 10.1016/j.tig.2021.10.001
31. Gragnoli C, Lindner T, Cockburn BN, Kaisaki PJ, Gragnoli F, Marozzi G, et al. Maturity-Onset Diabetes of the Young Due to a Mutation in the Hepatocyte Nuclear Factor-4 Alpha Binding Site in the Promoter of the Hepatocyte Nuclear Factor-1 Alpha Gene. Diabetes (1997) 46:1648–51. doi: 10.2337/diacare.46.10.1648
32. Bjørkhaug L, Sagen JV, Thorsby P, Søvik O, Molven A, Njølstad PR. Hepatocyte Nuclear Factor-1 Alpha Gene Mutations and Diabetes in Norway. J Clin Endocrinol Metab (2003) 88:920–31. doi: 10.1210/jc.2002-020945
33. Møller AM, Dalgaard LT, Pociot F, Nerup J, Hansen T, Pedersen O. Mutations in the Hepatocyte Nuclear Factor-1alpha Gene in Caucasian Families Originally Classified as Having Type I Diabetes. Diabetologia (1998) 41:1528–31. doi: 10.1007/s001250051101
34. Lambert AP, Ellard S, Allen LI, Gallen IW, Gillespie KM, Bingley PJ, et al. Identifying Hepatic Nuclear Factor 1alpha Mutations in Children and Young Adults With a Clinical Diagnosis of Type 1 Diabetes. Diabetes Care (2003) 26:333–7. doi: 10.2337/diacare.26.2.333
35. Fang C, Huang J, Huang Y, Chen L, Chen X, Hu J. A Novel Nonsense Mutation of the HNF1alpha in Maturity-Onset Diabetes of the Young Type 3 in Asian Population. Diabetes Res Clin Pract (2015) 109:e5–7. doi: 10.1016/j.diabres.2015.05.026
36. Balamurugan K, Bjørkhaug L, Mahajan S, Kanthimathi S, Njølstad PR, Srinivasan N, et al. Structure-Function Studies of HNF1A (MODY3) Gene Mutations in South Indian Patients With Monogenic Diabetes. Clin Genet (2016) 90:486–95. doi: 10.1111/cge.12757
37. Cervin C, Orho-Melander M, Ridderstråle M, Lehto M, Barg S, Groop L, et al. Characterization of a Naturally Occurring Mutation (L107I) in the HNF1 Alpha (MODY3) Gene. Diabetologia (2002) 45:1703–8. doi: 10.1007/s00125-002-0977-4
38. Hameed S, Ellard S, Woodhead HJ, Neville KA, Walker JL, Craig ME, et al. Persistently Autoantibody Negative (PAN) Type 1 Diabetes Mellitus in Children. Pediatr Diabetes (2011) 12:142–9. doi: 10.1111/j.1399-5448.2010.00681.x
39. Harries LW, Ellard S, Stride A, Morgan NG, Hattersley AT. Isomers of the TCF1 Gene Encoding Hepatocyte Nuclear Factor-1 Alpha Show Differential Expression in the Pancreas and Define the Relationship Between Mutation Position and Clinical Phenotype in Monogenic Diabetes. Hum Mol Genet (2006) 15:2216–24. doi: 10.1093/hmg/ddl147
40. Stride A, Ellard S, Clark P, Shakespeare L, Salzmann M, Shepherd M, et al. Beta-Cell Dysfunction, Insulin Sensitivity, and Glycosuria Precede Diabetes in Hepatocyte Nuclear Factor-1alpha Mutation Carriers. Diabetes Care (2005) 28:1751–6. doi: 10.2337/diacare.28.7.1751
41. Bellanne-Chantelot C, Carette C, Riveline JP, Valero R, Gautier JF, Larger E, et al. The Type and the Position of HNF1A Mutation Modulate Age at Diagnosis of Diabetes in Patients With Maturity-Onset Diabetes of the Young (MODY)-3. Diabetes (2008) 57:503–8. doi: 10.2337/db07-0859
42. Byrne MM, Sturis J, Menzel S, Yamagata K, Fajans SS, Dronsfield MJ, et al. Altered Insulin Secretory Responses to Glucose in Diabetic and Nondiabetic Subjects With Mutations in the Diabetes Susceptibility Gene MODY3 on Chromosome 12. Diabetes (1996) 45:1503–10. doi: 10.2337/diab.45.11.1503
43. Iwasaki N, Ogata M, Tomonaga O, Kuroki H, Kasahara T, Yano N, et al. Liver and Kidney Function in Japanese Patients With Maturity-Onset Diabetes of the Young. Diabetes Care (1998) 21:2144–8. doi: 10.2337/diacare.21.12.2144
44. Menzel R, Kaisaki PJ, Rjasanowski I, Heinke P, Kerner W, Menzel S. A Low Renal Threshold for Glucose in Diabetic Patients With a Mutation in the Hepatocyte Nuclear Factor-1alpha (HNF-1alpha) Gene. Diabetes Med (1998) 15:816–20. doi: 10.1002/(SICI)1096-9136(199810)15:10<816::AID-DIA714>3.0.CO;2-P
45. Bingham C, Ellard S, Nicholls AJ, Pennock CA, Allen J, James AJ, et al. The Generalized Aminoaciduria Seen in Patients With Hepatocyte Nuclear Factor-1alpha Mutations is a Feature of All Patients With Diabetes and is Associated With Glucosuria. Diabetes (2001) 50:2047–52. doi: 10.2337/diabetes.50.9.2047
46. Pontoglio M, Prie D, Cheret C, Doyen A, Leroy C, Froguel P, et al. HNF1alpha Controls Renal Glucose Reabsorption in Mouse and Man. EMBO Rep (2000) 1:359–65. doi: 10.1093/embo-reports/kvd071
47. Soutoglou E, Papafotiou G, Katrakili N, Talianidis I. Transcriptional Activation by Hepatocyte Nuclear Factor-1 Requires Synergism Between Multiple Coactivator Proteins. J Biol Chem (2000) 275:12515–20. doi: 10.1074/jbc.275.17.12515
48. Tronche F, Yaniv M. HNF1, a Homeoprotein Member of the Hepatic Transcription Regulatory Network. Bioessays (1992) 14:579–87. doi: 10.1002/bies.950140902
49. Horikawa Y, Iwasaki N, Hara M, Furuta H, Hinokio Y, Cockburn BN, et al. Mutation in Hepatocyte Nuclear Factor-1 Beta Gene (TCF2) Associated With MODY. Nat Genet (1997) 17:384–5. doi: 10.1038/ng1297-384
50. Nishigori H, Yamada S, Kohama T, Tomura H, Sho K, Horikawa Y, et al. Frameshift Mutation, A263fsinsGG, in the Hepatocyte Nuclear Factor-1beta Gene Associated With Diabetes and Renal Dysfunction. Diabetes (1998) 47:1354–5. doi: 10.2337/diab.47.8.1354
51. Maltoni G, Zucchini S, Scipione M, Mantovani V, Salardi S, Cicognani A. Onset of Type 1 Diabetes Mellitus in Two Patients With Maturity Onset Diabetes of the Young. Pediatr Diabetes (2012) 13:208–12. doi: 10.1111/j.1399-5448.2011.00788.x
52. Karaoglan M, Nacarkahya G. Clinical and Laboratory Clues of Maturity-Onset Diabetes of the Young and Determination of Association With Molecular Diagnosis. J Diabetes (2021) 13:154–63. doi: 10.1111/1753-0407.13097
53. Oliveira RV, Bernardo T, Martins S, Sequeira A. Monogenic Diabetes: A New Pathogenic Variant of HNF1A Gene. BMJ Case Rep (2021) 14:e231837. doi: 10.1136/bcr-2019-231837
54. Park HS, Kim IJ, Kim EG, Ryu CS, Lee JY, Ko EJ, et al. A Study of Associations Between CUBN, HNF1A, and LIPC Gene Polymorphisms and Coronary Artery Disease. Sci Rep (2020) 10:16294. doi: 10.1038/s41598-020-73048-6
55. Malecki MT, Skupien J, Gorczynska-Kosiorz S, Klupa T, Nazim J, Moczulski DK, et al. Renal Malformations may be Linked to Mutations in the Hepatocyte Nuclear Factor-1α (MODY3) Gene. Diabetes Care (2005) 28:2774–6. doi: 10.2337/diacare.28.11.2774
56. Steele AM, Shields BM, Shepherd M, Ellard S, Hattersley AT, Pearson ER. Increased All-Cause and Cardiovascular Mortality in Monogenic Diabetes as a Result of Mutations in the HNF1A Gene. Diabetes Med (2010) 27:157–61. doi: 10.1111/j.1464-5491.2009.02913.x
57. Giuffrida FM, Furuzawa GK, Kasamatsu TS, Oliveira MM, Reis AF, Dib SA. HNF1A Gene Polymorphisms and Cardiovascular Risk Factors in Individuals With Late-Onset Autosomal Dominant Diabetes: A Cross-Sectional Study. Cardiovasc Diabetol (2009) 8:28. doi: 10.1186/1475-2840-8-28
58. Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, et al. Biological, Clinical and Population Relevance of 95 Loci for Blood Lipids. Nature (2010) 466:707–13. doi: 10.1038/nature09270
59. Isomaa B, Henricsson M, Lehto M, Forsblom C, Karanko S, Sarelin L, et al. Chronic Diabetic Complications in Patients With MODY3 Diabetes. Diabetologia (1998) 41:467–73. doi: 10.1007/s001250050931
60. Babaya N, Ikegami H, Fujisawa T, Nojima K, Itoi-Babaya M, Inoue K, et al. Association of I27L Polymorphism of Hepatocyte Nuclear Factor-1 Alpha Gene With High-Density Lipoprotein Cholesterol Level. J Clin Endocrinol Metab (2003) 88:2548–51. doi: 10.1210/jc.2002-021891
61. Hoskins JW, Jia J, Flandez M, Parikh H, Xiao W, Collins I, et al. Transcriptome Analysis of Pancreatic Cancer Reveals a Tumor Suppressor Function for HNF1A. Carcinogenesis (2014) 35:2670–8. doi: 10.1093/carcin/bgu193
62. Lu Y, Xu D, Peng J, Luo Z, Chen C, Chen Y, et al. HNF1A Inhibition Induces the Resistance of Pancreatic Cancer Cells to Gemcitabine by Targeting ABCB1. EBioMedicine (2019) 44:403–18. doi: 10.1016/j.ebiom.2019.05.013
63. Willson JS, Godwin TD, Wiggins GA, Guilford PJ, McCall JL. Primary Hepatocellular Neoplasms in a MODY3 Family With a Novel HNF1A Germline Mutation. J Hepatol (2013) 59:904–7. doi: 10.1016/j.jhep.2013.05.024
64. Fu J, Wang T, Zhai X, Xiao X. Primary Hepatocellular Adenoma Due to Biallelic HNF1A Mutations and its Co-Occurrence With MODY 3: Case-Report and Review of the Literature. Endocrine (2020) 67:544–51. doi: 10.1007/s12020-019-02138-x
65. Rebouissou S, Vasiliu V, Thomas C, Bellanne-Chantelot C, Bui H, Chretien Y, et al. Germline Hepatocyte Nuclear Factor 1alpha and 1beta Mutations in Renal Cell Carcinomas. Hum Mol Genet (2005) 14:603–14. doi: 10.1093/hmg/ddi057
66. Zhang J, Liu Y, Xu B, Li F, Wang Y, Li M, et al. Circulating Tumor DNA Analysis of Metastatic Renal Cell Carcinoma. Mol Clin Oncol (2021) 14:16. doi: 10.3892/mco.2020.2178
67. Hechtman JF, Abou-Alfa GK, Stadler ZK, Mandelker DL, Roehrl MHA, Zehir A, et al. Somatic HNF1A Mutations in the Malignant Transformation of Hepatocellular Adenomas: A Retrospective Analysis of Data From MSK-IMPACT and TCGA. Hum Pathol (2019) 83:1–6. doi: 10.1016/j.humpath.2018.08.004
68. Tang J, Tang CY, Wang F, Guo Y, Tang HN, Zhou CL, et al. Genetic Diagnosis and Treatment of a Chinese Ketosis-Prone MODY 3 Family With Depression. Diabetol Metab Syndr (2017) 9:5. doi: 10.1186/s13098-016-0198-5
69. Delvecchio M, Pastore C, Giordano P. Treatment Options for MODY Patients: A Systematic Review of Literature. Diabetes Ther (2020) 11:1667–85. doi: 10.1007/s13300-020-00864-4
70. Pearson ER, Liddell WG, Shepherd M, Corrall RJ, Hattersley AT. Sensitivity to Sulphonylureas in Patients With Hepatocyte Nuclear Factor-1alpha Gene Mutations: Evidence for Pharmacogenetics in Diabetes. Diabetes Med (2000) 17:543–5. doi: 10.1046/j.1464-5491.2000.00305.x
71. Pearson ER, Starkey BJ, Powell RJ, Gribble FM, Clark PM, Hattersley AT. Genetic Cause of Hyperglycaemia and Response to Treatment in Diabetes. Lancet (2003) 362:1275–81. doi: 10.1016/S0140-6736(03)14571-0
72. Boileau P, Wolfrum C, Shih DQ, Yang TA, Wolkoff AW, Stoffel M. Decreased Glibenclamide Uptake in Hepatocytes of Hepatocyte Nuclear Factor-1alpha-Deficient Mice: A Mechanism for Hypersensitivity to Sulfonylurea Therapy in Patients With Maturity-Onset Diabetes of the Young, Type 3 (MODY3). Diabetes (2002) 51(Suppl 3):S343–8. doi: 10.2337/diabetes.51.2007.s343
73. McDonald TJ, Ellard S. Ellard. Maturity Onset Diabetes of the Young: Identification and Diagnosis. Ann Clin Biochem (2013) 50:403–15. doi: 10.1177/0004563213483458
74. Demol S, Lebenthal Y, Bar-Meisels M, Phillip M, Gat-Yablonski G, Gozlan Y. A Family With a Novel Termination Mutation in Hepatic Nuclear Factor 1alpha in Maturity-Onset Diabetes of the Young Type 3 Which is Unresponsive to Sulphonylurea Therapy. Horm Res Paediatr (2014) 81:280–4. doi: 10.1159/000356925
75. Tan C, Ang SF, Lim SC. Response to Multiple Glucose-Lowering Agents in a Sib-Pair With a Novel HNF1alpha (MODY3) Variant. Eur J Hum Genet (2020) 28:518–20. doi: 10.1038/s41431-019-0561-8
76. Martagon AJ, Bello-Chavolla OY, Arellano-Campos O, Almeda-Valdes P, Walford GA, Cruz-Bautista I, et al. Mexican Carriers of the HNF1A P.E508K Variant Do Not Experience an Enhanced Response to Sulfonylureas. Diabetes Care (2018) 41:1726–31. doi: 10.2337/dc18-0384
77. Tin A, Marten J, Halperin Kuhns VL, Li Y, Wuttke M, Kirsten H, et al. Polymorphisms of the HNF1A Gene Encoding Hepatocyte Nuclear Factor-1 Alpha are Associated With C-Reactive Protein. Am J Hum Genet (2008) 82:1193–201. doi: 10.1038/s41588-019-0504-x
78. Curocichin G, Wu Y, McDade TW, Kuzawa CW, Borja JB, Qin L, et al. Single-Nucleotide Polymorphisms at Five Loci are Associated With C-Reactive Protein Levels in a Cohort of Filipino Young Adults. J Hum Genet (2011) 56:823–7. doi: 10.1038/jhg.2011.106
79. Yahaya TO, Ufuoma SB. Genetics and Pathophysiology of Maturity-Onset Diabetes of the Young (MODY): A Review of Current Trends. Oman Med J (2020) 35:e126. doi: 10.5001/omj.2020.44
80. Bellanne-Chantelot C, Levy DJ, Carette C, Saint-Martin C, Riveline JP, Larger E, et al. French Monogenic Diabetes Study, Clinical Characteristics and Diagnostic Criteria of Maturity-Onset Diabetes of the Young (MODY) Due to Molecular Anomalies of the HNF1A Gene. J Clin Endocrinol Metab (2011) 96:E1346–1351. doi: 10.1210/jc.2011-0268
81. BellanneWiltshire S, Frayling TM, Groves CJ, Levy JC, Hitman GA, Sampson M, et al. Evidence From a Large U.K. Family Collection That Genes Influencing Age of Onset of Type 2 Diabetes Map to Chromosome 12p and to the MODY3/NIDDM2 Locus on 12q24. Diabetes (2004) 53:855–60. doi: 10.2337/diabetes.53.3.855
82. SIGMA Type 2 Diabetes Consortium, Estrada K, Aukrust I, Bjørkhaug L, Burtt NP, Mercader JM, et al. Association of a Low-Frequency Variant in HNF1A With Type 2 Diabetes in a Latino Population. JAMA (2014) 311:2305–14. doi: 10.1001/jama.2014.6511
83. Weedon MN, Owen KR, Shields B, Hitman G, Walker M, McCarthy MI, et al. A Large-Scale Association Analysis of Common Variation of the HNF1alpha Gene With Type 2 Diabetes in the U.K. Caucasian Population. Diabetes (2005) 54:2487–91. doi: 10.2337/diabetes.54.8.2487
84. Lee HJ, Ahn CW, Kim SJ, Song YD, Lim SK, Kim KR, et al. Mutation in Hepatocyte Nuclear Factor-1alpha is Not a Common Cause of MODY and Early-Onset Type 2 Diabetes in Korea. Acta Diabetol (2001) 38:123–7. doi: 10.1007/s005920170008
85. Hegele RA, Cao H, Harris SB, Hanley AJ, Zinman B. The Hepatic Nuclear Factor-1alpha G319S Variant is Associated With Early-Onset Type 2 Diabetes in Canadian Oji-Cree. J Clin Endocrinol Metab (1999) 84:1077–82. doi: 10.1210/jcem.84.3.5528
86. Hegele RA, Hanley AJ, Zinman B, Harris SB, Anderson CM. Disparity Between Association and Linkage Analysis for HNF1A G319S in Type 2 Diabetes in Canadian Oji-Cree. J Hum Genet (2000) 45:184–7. doi: 10.1007/s100380050208
87. Hegele RA, Zinman B, Hanley AJ, Harris SB, Barrett PH, Cao H. Genes, Environment and Oji-Cree Type 2 Diabetes. Clin Biochem (2003) 36:163–70. doi: 10.1016/s0009-9120(03)00004-3
88. Triggs-Raine BL, Kirkpatrick RD, Kelly SL, Norquay LD, Cattini PA, Yamagata K, et al. HNF-1alpha G319S, a Transactivation-Deficient Mutant, is Associated With Altered Dynamics of Diabetes Onset in an Oji-Cree Community. Proc Natl Acad Sci USA (2002) 99:4614–9. doi: 10.1073/pnas.062059799
89. Harries LW, Sloman MJ, Sellers EA, Hattersley AT, Ellard S. Diabetes Susceptibility in the Canadian Oji-Cree Population is Moderated by Abnormal mRNA Processing of HNF1A G319S Transcripts. Diabetes (2008) 57:1978–82. doi: 10.2337/db07-1663
90. Sagen JV, Bjørkhaug L, Haukanes BI, Grevle L, Molnes J, Nedrebo BG, et al. The HNF1A Mutant Ala180Val: Clinical Challenges in Determining Causality of a Rare HNF1A Variant in Familial Diabetes. Diabetes Res Clin Pract (2017) 133:142–9. doi: 10.1016/j.diabres.2017.08.001
91. Słupecka-Ziemilska M, Wychowański P, Puzianowska-Kuznicka M. Gestational Diabetes Mellitus Affects Offspring’s Epigenome. Is There a Way to Reduce the Negative Consequences? Nutrients (2020) 12:2792. doi: 10.3390/nu12092792
92. Rosik J, Szostak B, Machaj F, Pawlik A. The Role of Genetics and Epigenetics in the Pathogenesis of Gestational Diabetes Mellitus. Ann Hum Genet (2020) 84:114–24. doi: 10.1111/ahg.12356
93. Dalfrà MG, Burlina S, Del Vescovo GG, Lapolla A. Genetics and Epigenetics: New Insight on Gestational Diabetes Mellitus. Front Endocrinol (Lausanne) (2020) 11:602477. doi: 10.3389/fendo.2020.602477
94. Gjesing AP, Rui G, Lauenborg J, Have CT, Hollensted M, Andersson E, et al. High Prevalence of Diabetes-Predisposing Variants in MODY Genes Among Danish Women With Gestational Diabetes Mellitus. J Endocr Soc (2017) 1:681–90. doi: 10.1210/js.2017-00040
95. Bergmann A, Li J, Selisko T, Reimann M, Fischer S, Grässler J, et al. The A98V Single Nucleotide Polymorphism (SNP) in Hepatic Nuclear Factor 1 Alpha (HNF-1alpha) is Associated With Insulin Sensitivity and Beta-Cell Function. Exp Clin Endocrinol Diabetes (2008) 116(Suppl 1):S50–55. doi: 10.1055/s-2008-1081492
96. Shaat N, Karlsson E, Lernmark A, Ivarsson S, Lynch K, Parikh H, et al. Common Variants in MODY Genes Increase the Risk of Gestational Diabetes Mellitus. Diabetologia (2006) 49:1545–51. doi: 10.1007/s00125-006-0258-8
97. Beysel S, Pinarli FA, Eyerci N, Kizilgul M, Hepsen S, Alhan A, et al. HNF1A Gene P.I27L is Associated With Co-Existing Preeclampsia in Gestational Diabetes Mellitus. Gynecol Endocrinol (2020) 36:530–4. doi: 10.1080/09513590.2019.1698023
98. Wang X, Li W, Ma L, Ping F, Liu J, Wu X, et al. Variants in MODY Genes Associated With Maternal Lipids Profiles in Second Trimester of Pregnancy. J Gene Med (2017) 19:6–7. doi: 10.1002/jgm.2962
99. Wu Y, McDade TW, Kuzawa CW, Borja J, Li Y, Adair LS, et al. Genome-Wide Association With C-Reactive Protein Levels in CLHNS: Evidence for the CRP and HNF1A Loci and Their Interaction With Exposure to a Pathogenic Environment. Inflammation (2012) 35:574–83. doi: 10.1007/s10753-011-9348-y
100. McDonald TJ, Shields BM, Lawry J, Owen KR, Gloyn AL, Ellard S, et al. High-Sensitivity CRP Discriminates HNF1A-MODY From Other Subtypes of Diabetes. Diabetes Care (2011) 34:1860–2. doi: 10.2337/dc11-0323
101. Shah N, Thanabalasingham G, Owen KR, James TJ. Comparability of High-Sensitivity CRP Methods to Detect Maturity-Onset Diabetes of the Young Due to HNF1A Mutations. Br J BioMed Sci (2014) 71:84–5. doi: 10.1080/09674845.2014.11978288
102. Fagerberg L, Hallström BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, et al. Analysis of the Human Tissue-Specific Expression by Genome-Wide Integration of Transcriptomics and Antibody-Based Proteomics. Mol Cell Proteomics (2014) 13:397–406. doi: 10.1074/mcp.M113.035600
103. Odom DT, Zizlsperger N, Gordon DB, Bell GW, Rinaldi NJ, Murray HL, et al. Control of Pancreas and Liver Gene Expression by HNF Transcription Factors. Science (2004) 303:1378–81. doi: 10.1126/science.1089769
104. Vaxillaire M, Abderrahmani A, Boutin P, Bailleul B, Froguel P, Yaniv M, et al. Anatomy of a Homeoprotein Revealed by the Analysis of Human MODY3 Mutations. J Biol Chem (1999) 274:35639–46. doi: 10.1074/jbc.274.50.35639
105. Pearson ER, Velho G, Clark P, Stride A, Shepherd M, Frayling TM, et al. Beta-Cell Genes and Diabetes: Quantitative and Qualitative Differences in the Pathophysiology of Hepatic Nuclear Factor-1alpha and Glucokinase Mutations. Diabetes (2001) 50(Suppl 1):S101–7. doi: 10.2337/diabetes.50.2007.s101
106. Haliyur R, Tong X, Sanyoura M, Shrestha S, Lindner J, Saunders DC, et al. Human Islets Expressing HNF1A Variant Have Defective β Cell Transcriptional Regulatory Networks. J Clin Invest (2019) 129:246–51. doi: 10.1172/JCI121994
107. Magaña-Cerino JM, Luna-Arias JP, Labra-Barrios ML, Avendaño-Borromeo B, Boldo-León XM, Martínez-López MC. Identification and Functional Analysis of C.422_423inst, a Novel Mutation of the HNF1A Gene in a Patient With Diabetes. Mol Genet Genomic Med (2017) 5:50–65. doi: 10.1002/mgg3.261
108. Nammo T, Yamagata K, Hamaoka R, Zhu Q, Akiyama TE, Gonzalez FJ, et al. Expression Profile of MODY3/HNF-1alpha Protein in the Developing Mouse Pancreas. Diabetologia (2002) 45:1142–53. doi: 10.1007/s00125-002-0892-8
109. Hagenfeldt-Johansson KA, Herrera PL, Wang H, Gjinovci A, Ishihara H, Wollheim CB. Beta-Cell-Targeted Expression of a Dominantnegative Hepatocyte Nuclear Factor-1 Alpha Induces a Maturity-Onset Diabetes of the Young (MODY)3-Like Phenotype in Transgenic Mice. Endocrinology (2001) 142:5311–20. doi: 10.1210/endo.142.12.8592
110. Yamagata K, Nammo T, Moriwaki M, Ihara A, Iizuka K, Yang Q, et al. Overexpression of Dominant-Negative Mutant Hepatocyte Nuclear Fctor-1alpha in Pancreatic Beta-Cells Causes Abnormal Islet Architecture With Decreased Expression of E-Cadherin, Reduced Beta-Cell Proliferation, and Diabetes. Diabetes (2002) 51:114–23. doi: 10.2337/diabetes.51.1.114
111. Lyttle BM, Li J, Krishnamurthy M, Fellows F, Wheeler MB, Goodyer CG, et al. Transcription Factor Expression in the Developing Human Fetal Endocrine Pancreas. Diabetologia (2008) 51:1169–80. doi: 10.1007/s00125-008-1006-z
112. Donelan W, Koya V, Li SW, Yang LJ. Distinct Regulation of Hepatic Nuclear Factor 1alpha by NKX6.1 in Pancreatic Beta Cells. J Biol Chem (2010) 285:12181–9. doi: 10.1074/jbc.M109.064238
113. Hunter CS, Maestro MA, Raum JC, Guo M, Thompson 3FH, Ferrer J, et al. Hnf1α (MODY3) Regulates β-Cell-Enriched MafA Transcription Factor Expression. Mol Endocrinol (2011) 25:339–47. doi: 10.1210/me.2010-0362
114. Pontoglio M, Sreenan S, Roe M, Pugh W, Ostrega D, Doyen A, et al. Defective Insulin Secretion in Hepatocyte Nuclear Factor 1alpha-Deficient Mice. J Clin Invest (1998) 101:2215–22. doi: 10.1172/JCI2548
115. Østoft SH, Bagger JI, Hansen T, Pedersen O, Holst JJ, Knop FK, et al. Incretin Effect and Glucagon Responses to Oral and Intravenous Glucose in Patients With Maturity-Onset Diabetes of the Young–Type 2 and Type 3. Diabetes (2014) 63:2838–44. doi: 10.2337/db13-1878
116. Sato Y, Rahman MM, Haneda M, Tsuyama T, Mizumoto T, Yoshizawa T, et al. Hnf1α Controls Glucagon Secretion in Pancreatic α-Cells Through Modulation of SGLT1. Biochim Biophys Acta Mol Basis Dis (2020) 1866:165898. doi: 10.1016/j.bbadis.2020.165898
117. Cardenas-Diaz FL, Osorio-Quintero C, Diaz-Miranda MA, Kishore S, Leavens K, Jobaliya C, et al. Modeling Monogenic Diabetes using Human ESCs Reveals Developmental and Metabolic Deficiencies Caused by Mutations in HNF1A. Cell Stem Cell (2019) 25:273–89.e5. doi: 10.1016/j.stem.2019.07.007
118. Hiraiwa H, Pan CJ, Lin B, Akiyama TE, Gonzalez FJ, Chou JY. A Molecular Link Between the Common Phenotypes of Type 1 Glycogen Storage Disease and HNF1alpha-Null Mice. J Biol Chem (2001) 276:7963–7. doi: 10.1074/jbc.M010523200
119. Akiyama TE, Ward JM, Gonzalez FJ. Regulation of the Liver Fatty Acid-Binding Protein Gene by Hepatocyte Nuclear Factor 1alpha (HNF1alpha). Alterations in Fatty Acid Homeostasis in HNF1alpha-Deficient Mice. J Biol Chem (2000) 275:27117–2722. doi: 10.1074/jbc.M004388200
120. Richter S, Shih DQ, Pearson ER, Wolfrum C, Fajans SS, Hattersley AT, et al. Regulation of Apolipoprotein M Gene Expression by MODY3 Gene Hepatocyte Nuclear Factor-1alpha: Haploinsufficiency is Associated With Reduced Serum Apolipoprotein M Levels. Diabetes (2003) 52:2989–95. doi: 10.2337/diabetes.52.12.2989
121. Ma X, Hu YW, Zhao ZL, Zheng L, Qiu YR, Huang JL, et al. Anti-Inflammatory Effects of Propofol are Mediated by Apolipoprotein M in a Hepatocyte Nuclear Factor-1alpha-Dependent Manner. Arch Biochem Biophys (2013) 533:1–10. doi: 10.1016/j.abb.2013.03.002
122. Ni Q, Ding K, Wang KQ, He J, Yin C, Shi J, et al. Deletion of HNF1alpha in Hepatocytes Results in Fatty Liver-Related Hepatocellular Carcinoma in Mice. FEBS Lett (2017) 591:1947–57. doi: 10.1002/1873-3468.12689
123. Hegele RA, Cao H, Harris SB, Hanley AJ, Zinman B, Connelly PW. The Private Hepatocyte Nuclear Factor-1alpha G319S Variant is Associated With Plasma Lipoprotein Variation in Canadian Oji-Cree. Arterioscler Thromb Vasc Biol (2000) 20:217–22. doi: 10.1161/01.atv.20.1.217
124. Sakellariou S, Morgan Y, Heaton N, Portmann B, Quaglia A, Tobal K. New Monoallelic (Partial Tandem Duplication) Mutation of HNF1a Gene in Steatotic Hepatocellular Adenoma. Eur J Gastroenterol Hepatol (2011) 23:623–7. doi: 10.1097/MEG.0b013e328347964d
125. Nocera D, Menniti M, Belviso S, Bond HM, Lanzillotta D, Spoleti CB, et al. Functional Characterization of P.Pro409His Variant in HNF1A, a Hypomorphic Mutation Involved in Pancreatic Beta-Cell Dysfunction. Acta Diabetol (2019) 56:883–8. doi: 10.1007/s00592-019-01298-6
126. Kim KA, Kang K, Chi YI, Chang I, Lee MK, Kim KW, et al. Identification and Functional Characterization of a Novel Mutation of Hepatocyte Nuclear Factor-1alpha Gene in a Korean Family With MODY3. Diabetologia (2003) 46:721–7. doi: 10.1007/s00125-003-1079-7
127. Galán M, García-Herrero CM, Azriel S, Gargallo M, Duran M, Gorgojo JJ, et al. Differential Effects of HNF-1alpha Mutations Associated With Familial Young-Onset Diabetes on Target Gene Regulation. Mol Med (2011) 17:256–65. doi: 10.2119/molmed.2010.00097
128. Chi YI, Frantz JD, Oh BC, Hansen L, Dhe-Paganon S, Shoelson SE. Diabetes Mutations Delineate an Atypical POU Domain in HNF-1alpha. Mol Cell (2002) 10:1129–37. doi: 10.1016/s1097-2765(02)00704-9
129. Narayana N, Hua Q, Weiss MA. The Dimerization Domain of HNF-1alpha: Structure and Plasticity of an Intertwined Four-Helix Bundle With Application to Diabetes Mellitus. J Mol Biol (2001) 310:635–58. doi: 10.1006/jmbi.2001.4780
130. Rose RB, Endrizzi JA, Cronk JD, Holton J, Alber T. High-Resolution Structure of the HNF-1alpha Dimerization Domain. Biochemistry (2000) 39:15062–70. doi: 10.1021/bi001996t
131. Rose RB, Bayle JH, Endrizzi JA, Cronk JD, Crabtree GR, Alber T. Structural Basis of Dimerization, Coactivator Recognition and MODY3 Mutations in HNF-1alpha. Nat Struct Biol (2000) 7:744–8. doi: 10.1038/78966
132. Sneha P, Thirumal Kumar D, George Priya Doss C, Siva R, Zayed H. Determining the Role of Missense Mutations in the POU Domain of HNF1A That Reduce the DNA-Binding Affinity: A Computational Approach. PloS One (2017) 12:e0174953. doi: 10.1371/journal.pone.0174953
133. Malikova J, Kaci A, Dusatkova P, Aukrust I, Torsvik J, Vesela K, et al. Functional Analyses of HNF1A-MODY Variants Refine the Interpretation of Identified Sequence Variants. J Clin Endocrinol Metab (2020) 105:dgaa051. doi: 10.1210/clinem/dgaa051
Keywords: HNF1α, polymorphism, heterogeneity, MODY3, type 2 diabetes, gestational diabetes mellitus
Citation: Li LM, Jiang BG and Sun LL (2022) HNF1A:From Monogenic Diabetes to Type 2 Diabetes and Gestational Diabetes Mellitus. Front. Endocrinol. 13:829565. doi: 10.3389/fendo.2022.829565
Received: 06 December 2021; Accepted: 03 February 2022;
Published: 01 March 2022.
Edited by:
Ming Liu, Tianjin Medical University General Hospital, ChinaReviewed by:
Michael A. Weiss, Indiana University, United StatesJuan Zheng, Sun Yat-sen University, China
Copyright © 2022 Li, Jiang and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bei-Ge Jiang, jiang_beige@aliyun.com; Liang-Liang Sun, sun_lliang@smmu.edu.cn