 Quang-Kim Tran
Quang-Kim Tran- Department of Physiology and Pharmacology, College of Osteopathic Medicine, Des Moines University, Des Moines, IA, United States
17β-Estradiol (E2) is the main estrogenic hormone in the body and exerts many cardiovascular protective effects. Via three receptors known to date, including estrogen receptors α (ERα) and β (ERβ) and the G protein-coupled estrogen receptor 1 (GPER, aka GPR30), E2 regulates numerous calcium-dependent activities in cardiovascular tissues. Nevertheless, effects of E2 and its receptors on components of the calcium signaling machinery (CSM), the underlying mechanisms, and the linked functional impact are only beginning to be elucidated. A picture is emerging of the reciprocality between estrogen biology and Ca2+ signaling. Therein, E2 and GPER, via both E2-dependent and E2-independent actions, moderate Ca2+-dependent activities; in turn, ERα and GPER are regulated by Ca2+ at the receptor level and downstream signaling via a feedforward loop. This article reviews current understanding of the effects of E2 and its receptors on the cardiovascular CSM and vice versa with a focus on mechanisms and combined functional impact. An overview of the main CSM components in cardiovascular tissues will be first provided, followed by a brief review of estrogen receptors and their Ca2+-dependent regulation. The effects of estrogenic agonists to stimulate acute Ca2+ signals will then be reviewed. Subsequently, E2-dependent and E2-independent effects of GPER on components of the Ca2+ signals triggered by other stimuli will be discussed. Finally, a case study will illustrate how the many mechanisms are coordinated to moderate Ca2+-dependent activities in the cardiovascular system.
Main Components of the Calcium Signaling Machinery (CSM) in Cardiovascular Tissues
The CSM herein refers to proteins responsible for the generation or sequestration of intracellular Ca2+ signals and their transduction to target activities. In this section, key CSM components in cardiovascular tissues will be briefly described to facilitate review of the relevant effects and mechanisms of estrogenic agonists and receptors.
Intracellular Ca2+ Stores, Release, and Uptake Mechanisms
Organelles Functioning as Intracellular Ca2+ Stores
The sarcoplasmic/endoplasmic reticulum (SR/ER) is the main Ca2+ store in cardiomyocytes, vascular smooth muscle cells (VSMCs) (1, 2), and endothelial cells (ECs), where the ER stores ~75% Ca2+ and mitochondria house ~25% (3). The Golgi (4, 5) and lysosomes have more recently been recognized as Ca2+ reservoirs (6, 7). Ca2+ reaches 5 × 10−4 M in the ER/SR and lysosomes and 1.3–2.5 × 10−4 M between the trans-Golgi and cis-Golgi (5, 8). The medial Golgi also releases Ca2+ in response to inositol-triphosphate receptor (IP3R) and ryanodine receptor (RyR) stimulation (9). Crosstalk between the ER/SR and other organelles affects their Ca2+ fluxes (10–14). In neonatal cardiomyocytes, beat-to-beat oscillations in mitochondrial and cytosolic Ca2+ occur in parallel (15), and mitochondrial uptake reduces cytosolic Ca2+ (16).
Mechanisms of Ca2+ Uptake Into Ca2+ Stores
SR/ER Ca2+-ATPases (SERCAs) are the key Ca2+ uptake mechanisms. For each ATP hydrolyzed, they pump 2 Ca2+ ions into the ER/SR in exchange for less than four H+ ions (17). SERCA2b is ubiquitously expressed. SERCA2a predominates in cardiomyocytes and is essential for cardiac development (18). SERCA3 is the predominant vascular isoform; its deletion causes smooth muscle relaxation abnormality (19, 20). SERCA3 has lower affinity for Ca2+ and is only active at high Ca2+ levels. Non-phosphorylated phospholamban interacts with SERCA1a, SERCA2a, and SERCA2b and reduces their Ca2+ affinity. Phosphorylation at Ser16 and Thr17 removes phospholamban–SERCA interaction, promoting SERCA activity (21, 22). Sarcolipin also binds SERCAs and reduces their Ca2+ affinity. Its deletion increases SR Ca2+ uptake (23).
The secretory pathway Ca2+ pump (SPCA) mediates Ca2+ uptake into the Golgi with nanomolar affinity for Ca2+. Unlike the SERCA, Ca2+ transport by SPCA is not associated with counter transport of H+. In the medial Golgi, both SERCA and SPCA participate in Ca2+ uptake (9).
Mitochondrial Ca2+ uptake is mediated by the voltage-dependent anion channel (VDAC) and the mitochondrial Ca2+ uniporter (MCU). VDACs are non-selective anion channels in the open state yet in the “closed” state permit influxes of cations such as K+, Na+, and Ca2+ into the mitochondria (24). VDAC isoforms participate equally in transporting Ca2+ triggered by IP3-producing agonists; however, VDAC1 selectively transports apoptotic Ca2+ signals (25). Myocardial VDAC2 regulates rhythmicity by influencing the spatial and temporal properties of cytoplasmic Ca2+ signals (26). The MCU constitutes a low-affinity yet selective Ca2+ channel pore as part of a mitochondrial Ca2+ uptake protein complex (MICU) and the essential MCU regulator (27, 28).
Mechanisms of Ca2+ Release From Ca2+ Stores
In IP3Rs, IP3 binds with IP3R2 > IP3R1 > IP3R3 affinity order (29) and cooperatively switches IP3R tetramers to an open conformation to form clusters and release Ca2+ (30, 31). IP3Rs regulate Ca2+ release from the ER/SR, Golgi apparatus, and nucleus (32). ER/SR Ca2+ release depletes ER Ca2+ and triggers store-operated Ca2+ entry (SOCE). IP3R2 predominates in the cardiomyocytes (33). In failing hearts, IP3R-mediated Ca2+ transients are enhanced, and mitochondrial Ca2+ uptake is reduced, which facilitates contraction and spontaneous action potentials that increase arrhythmogenicity (34). In VSMCs, all IP3Rs are expressed and are important for agonist-induced contraction (35). Endothelial IP3R1 is predominant in the brain (36), whereas IP3R2 and IP3R3 are abundant in the aorta and pulmonary arteries (37, 38).
RyRs (RyR1–RyR3) are the main SR Ca2+ release channels (39). Regulation by cytosolic Ca2+: In cardiomyocytes, RyR2 predominates (40) and is closed, activated, and inhibited, respectively, at Ca2+ <10−7 M, ~10−7-10−5 M, and >10−3 M (41). Entry via voltage-dependent Ca2+ channels (VDCCs) stimulates Ca2+-induced Ca2+ release (CICR) via RyR2, contributing to myocardial contraction. In VSMCs, RyR2 predominates in the aorta and pulmonary and cerebral arteries, while RyR3 is the only isoform in basilar arteries (42–44). CICR also contributes to VSMC contraction, but not as critically as in cardiomyocytes; indeed, skinned smooth muscle fiber bundles can contract at Ca2+ levels that do not activate RyRs (45). In ECs, RyR2 is on the ER and mitochondria (46); however, RyR agonists only cause a slow Ca2+ release that corresponds to a reduction in the IP3-sensitive Ca2+ pool (47, 48). Regulation by SR Ca2+ is important in cardiomyocytes. SR Ca2+ overload triggers spontaneous RyR2-mediated Ca2+ release, a phenomenon called store overload-induced Ca2+ release (SOICR) (49, 50). SOICR can cause delayed afterdepolarizations leading to tachycardias and is abolished by an E4872A mutation in the RyR2 gate (51).
Ca2+ Entry
Store-Operated Ca2+ Entry (SOCE)
SOCE is a ubiquitous mechanism where Ca2+ store depletion triggers Ca2+ influx (52, 53). Proposed in the 1980s, SOCE was confirmed in the mid-2000s with the discoveries of the stromal interaction molecule 1 (STIM1) (54–56) and Orai Ca2+ channels (57–59). STIM1 resides mainly on the ER/SR membrane and has a luminal EF hand that houses a Ca2+-binding loop (60). In Ca2+-full ER/SR, the loop is in a closed conformation. Upon ER/SR Ca2+ depletion, Ca2+ leaving the loop promotes STIM1 oligomerization to interact with Orai1 channels and trigger Ca2+ entry (61–63). STIM1 also interacts with L-type Ca2+ channels (LTCCs) (64), maintains ER/SR structure (65–67), and is upregulated in atherosclerosis and hypertension (68–71). Myocardial SOCE is normally not prominent; however, STIM1 and SOCE are increased in heart failure (67, 72–76). In VSMCs, SOCE contributes significantly to contraction; α1AR-mediated contraction is reduced ~30% in SM-specific STIM1−/− animals (77). In ECs, SOCE is the major Ca2+ entry and is required for many critical functions such as endothelial nitric oxide synthase (eNOS) activity and proliferation (78–82).
Voltage-Dependent Ca2+ Entry (VDCE)
Functional voltage-dependent Ca2+ channels (VDCCs) are the hallmark of tissue excitability and are present in cardiomyocytes and VSMCs, but not ECs. In cardiomyocytes, LTCCs are located mostly in transverse T tubules in apposition to RyR2s (83). Ca2+ entry via LTCCs triggers CICR via RyR2. In VSMCs, LTCCs also play a critical role in Ca2+ entry and contraction (84). The LTCC complex (85) consists of α1, α2, β, δ, and γ subunits. Four LTCC members are named according to their α1 pore-forming subunits: Cav1.1, Cav1.2, Cav1.3, and Cav1.4 (86). Cav1.2 is predominant in cardiac and smooth muscles.
Ca2+ Extrusion via the Plasma Membrane/Sarcolemma
The plasma membrane Ca2+-ATPases (PMCAs) prevail for Ca2+ extrusion in non-excitable tissues while the Na+-Ca2+ exchanger (NCX) is more important in excitable cells. SERCA2a, NCX, and PMCA sequester, respectively, ~70, 28, and 2% of cytosolic Ca2+ in cardiomyocytes (83) and 25, 25, and 50% in ECs (87).
Plasma Membrane Ca2+-ATPase
PMCAs extrude one Ca2+ ion for each ATP used and function as Ca2+-H+ exchangers (88–90). PMCAs are regulated by a Ca2+-dependent interaction with calmodulin (CaM). At low Ca2+, a C-terminal autoinhibitory domain binds to two cytosolic loops and inhibits pump activity. Increased Ca2+ promotes CaM–PMCA interaction, which removes inhibition and activates Ca2+ efflux (91, 92). PSD-95 promotes expression and distribution of PMCA4b via PDZ binding (93). PMCAs are inhibited by C-terminal tyrosine phosphorylation (94). Myocardial PMCAs play a little role under physiological conditions. However, expressions of PMCA1 and PMCA4 are reduced by up to 70 and 50%, respectively, in end-stage heart failure (95), and cardiac-specific overexpression of PMCA4b improved myocardial functions in ischemia–reperfusion injury and heart failure (96). PMCAs concentrate in the caveolae of VSMCs and ECs (97, 98). PMCA1 suppresses VSMC proliferation (99, 100), while PMCA4 mediates cell cycle (101, 102). In ECs, PMCA1b, and PMCA4b are predominant (87, 103, 104).
Na+-Ca2+ Exchanger
The NCX may function in two modes. In the forward mode, myocardial NCX1 balances LTCC-mediated Ca2+ entry and RyR-mediated Ca2+ release during cardiac excitation, extruding ~25% of the Ca2+ needed to activate myofilaments (105). NCX1 also predominates in VSMCs (106, 107). In ECs, NCX accounts for ~25% of Ca2+ removal (87). Endothelial NCX and PMCA dynamically adjust their Ca2+ extrusion rates to maintain sufficient efflux (104). In the reverse mode, upon myocardial depolarization, Na+ entry causes the NCX to transiently operate in this mode, promoting Ca2+ entry. This is much less efficient in triggering SR Ca2+ release compared to LTCC-mediated Ca2+ entry (108, 109). However, it primes the dyad to increase LTCC-mediated CICR (110). In VSMCs, reverse-mode NCX1 facilitates Ca2+ entry and mediates contraction, vascular tone, and blood pressure (111, 112). The reverse mode is not significant in ECs.
Sex Differences in Ca2+ Signaling Proteins
Higher mRNA levels of Cav1.2, RyR, and NCX, but not of phospholamban and SERCA2, have been observed in female than in male rat hearts (113). However, caffeine-induced Ca2+ release is lower in cardiomyocytes from female hearts (114). Cav1.2 mRNA is higher in coronary smooth muscle from male than from female pigs (115). In smooth muscle cells (SMCs), expressions of ERα and ERβ, but not G protein-coupled estrogen receptor 1 (GPER), are higher in female than in male rats (116). These differences and the lower Cav1.2 expression (115) may be responsible for less contraction of VSMCs from females (116). No studies have examined sex differences in Ca2+ handling proteins in ECs.
Transduction of Ca2+ Signals—The Essential Role of Calmodulin (CaM)
While some Ca2+-dependent proteins are activated directly by Ca2+, many are activated by a complex between Ca2+ and CaM. CaM has two lobes linked by a flexible helix and can interact with ~300 target proteins (117, 118). Ca2+-free CaM binds or serves as structural subunits of ~15 proteins (119). However, each CaM lobe has two Ca2+-binding sites, and cooperative Ca2+ binding induces conformations that allow CaM to interact with many proteins, aided by the flexibility of the central helix (120, 121). Thus, CaM is the ubiquitous Ca2+ signal transducer. Activities of Ca2+/CaM-binding proteins depend on the Ca2+ signals, CaM availability, and properties of the interaction between Ca2+-CaM and the target proteins. Many of these factors are subject to estrogenic moderation.
Despite being required for activation of many Ca2+-dependent proteins, up to 50% of cellular CaM is engaged in inseparable interactions, leaving much less available for dynamic target binding (122). This generates an environment of limited CaM (123), as has been demonstrated in ECs (124), VSMCs (125), and cardiomyocytes (126). Consequently, competition for CaM generates a unique crosstalk among CaM-dependent proteins (124, 127), and factors that alter CaM level are predicted to have pervasive functional impact. It is noteworthy that virtually all CSM components interact with CaM and, in the context of reciprocality between estrogenic and Ca2+ signaling pathways, that ERα and GPER are both regulated by direct interactions with Ca2+-CaM.
Estrogen Receptors and Their Calcium-Dependent Regulation
Estrogen Receptor α (ERα)
ERα (128–130) is a nuclear receptor that, upon E2 binding (Kd ~ 10−10 M), assumes an active conformation to bind estrogen-responsive elements (EREs) in the promoters of target genes, modulating their transcription (131). Its N-terminus has a transcriptional activation function (AF-1) domain, a DNA-binding domain, and a hinge region; the C-terminus houses the ligand-binding domain and a second AF-2 domain. ERα is robustly expressed in the heart (132), VSMCs, and ECs (133–136).
ERα activities are strongly regulated by the Ca2+-dependent interaction with CaM. ERα binds CaM in a Ca2+-dependent fashion with a Kd of 1.6 ×10−10 M and an EC50(Ca2+) value of ~3 × 10−7 M (137). When ERα from Wistar rats' uteri is used, CaM decreases ERα-E2 binding but increases liganded ERα-ERE interaction (138, 139). A comparison of the CaM-bound/CaM-unbound ERα ratio in the cytosolic (unliganded) and nuclear (liganded) ERα pools isolated from MCF-7 cells suggests that E2 binding induces a conformation that favors ERα-CaM interaction (138). The CaM-binding domain was initially predicted to be a.a. 298–310 (137) but was later determined to be a.a. 298–317, with a.a. 248–317 required for maximal interaction (140). Further studies revealed that a.a. 287–311 is required to interact with both CaM lobes (141). CaM binding promotes ERα homodimerization that is critical for transcription activity (140, 142). With two lobes, each CaM binds two ERα molecules and thus stabilizes ERα dimerization (143). Notably, analogs of ERα17p (a.a. 295–311) that are unable to bind CaM downregulates ERα, stimulates ERα-dependent transcription, and enhances proliferation of MCF-7 cells, as does the wild-type ERα17p, indicating that this domain may also be involved in CaM-independent posttranslational regulation of ERα (144).
Estrogen Receptor β (ERβ)
ERβ has ~96% and 55–58% sequence homology with ERα in the DNA- and ligand-binding domains, respectively (145, 146). ERβ binds E2 with a Kd of ~4–6 × 10−10 M. ERβ forms homodimers but more preferentially forms heterodimers with ERα, which bind E2 with a Kd of ~2 × 10−9 M and are transcriptionally active (147). ERβ is abundantly expressed in the vasculature (133–136). However, its expression and direct actions in the heart are controversial; cardiac manifestations in ERβ−/− animals have been attributed to indirect effects from vascular changes (148). ERβ is not regulated by Ca2+ or CaM (149).
GPER
GPER (150), aka GPR30, was cloned from various tissues in the 1990s (151–156). GPR30 is required for estrogenic activation of extracellular signal-related kinase (ERK)1/2 via transactivation of the epidermal growth factor receptor (EGFR) and release of the heparan-bound epidermal growth factor (EGF) (157, 158). It was shown to bind E2 in 2005 (159, 160), and the designation GPER was adopted by the International Union of Basic and Clinical Pharmacology in 2007 (161). A host of steroidal and non-steroidal agents and specific GPER agonists can activate GPER (150). GPER couples with Gαs or Gαi/o. Supporting Gαs coupling are data that (1) most membrane-bound [35S]GTPγ-S from cells overexpressing GPER and treated with E2 coimmunoprecipitate with Gαs (159), (2) GPER is present in Gαs-pull-down fraction from GPER-expressing cells, and (3) E2 promotes GPER-dependent cyclic adenosine monophosphate (cAMP) production (162). Supporting GPER-Gαi/o association are results that pertussis toxin prevents (1) E2-induced, GPER-mediated ERK1/2 phosphorylation in cells transfected with GPER (134, 157); (2) upregulation of c-fos in ERα/ERβ-negative, GPER-positive SKBr3 cells (163); and (3) E2-induced Ca2+ signals in ECs (164).
GPER is robustly expressed in cardiovascular tissues (133–136). In ECs, GPER mRNA is increased 8-fold by shear stress (154). GPER is localized on the ER/SR membrane (160) and responds to cell-permeable ligands (165). However, it also resides on the plasma membrane (166) and requires its C-terminal PDZ-binding motif to do so (167). The plasmalemmal GPER pool seems to constitutively undergo clathrin-dependent endocytosis and accumulate in the trans-Golgi network for ubiquitination in the proteasome without recycling to the plasma membrane, a process unaffected by agonist stimulation (168). Despite its predominant expression in the ER/SR, the sequence that drives GPER localization here has not been identified.
GPER is directly regulated by Ca2+-CaM complexes. In VSMCs and ECs, GPER coimmunoprecipitates with CaM in a constitutive association that is promoted by treatment with E2, G-1, or receptor-independent stimulation of Ca2+ entry (169, 170). GPER is the first G protein-coupled receptor (GPCR) shown to possess four CaM-binding sites on its respective four submembrane domains (SMDs) (169). Fluorescence resonance energy transfer (FRET) biosensors based on SMDs of GPER bind CaM with Kd from 0.4 to 136 × 10−6 M and affinity ranking SMD2 > SMD4 > SMD3 > SMD1. These interactions are Ca2+ dependent, with an EC50 (Ca2+) of 1.3 × 10−7-5 × 10−6 M, values within the physiological Ca2+ range (169). Due to technical challenges with purifying full-length GPCRs, the KCaM for GPER as a holoreceptor is not available. The presence of four CaM-binding sites makes this task even more challenging and, in some way, not useful functionally. Functionally, mutations that reduce CaM binding but that do not perturb GPER-Gβγ preassociation drastically prevent GPER-mediated ERK1/2 phosphorylation (170).
Stimulation of Calcium Signals by Estrogen and GPER Agonists
Observations
In rat hearts, E2 (10−12-10−8 M) triggers 45Ca2+ uptake that is inhibited by LTCC antagonists (171). In VSMCs, GPER agonist G-1 triggers a slow-rising Ca2+ signal that is <2 × 10−7 M (172). In MCF-7 cells, E2 (10−7 M) induces Ca2+ store release and entry, yet only the former is required to activate mitogen-activated protein kinase (MAPK) (173). Interestingly, the ERα/ERβ antagonist ICI182,780 (10−6 M) also triggered Ca2+ signals in these cells. In ECs, E2 (10−10-10−9 M) triggers Ca2+ store release and entry, effects not affected by ERα/ERβ inhibitor tamoxifen (164, 174). The data with ICI182,780 and tamoxifen implicate a receptor other than ERα or ERβ in mediating the Ca2+ signal. Both reagents were later shown to be GPER agonists, triggering ERK1/2 phosphorylation only in cells expressing GPER (157, 159). Later studies confirmed Ca2+ signals stimulated by E2, GPER agonist G-1, and ICI182,780 in cells expressing GPER endogenously and absence of this effect in GPER−/− cells (160, 175, 176).
Mechanisms (Figure 1)
Direct E2-Cav1.2 Interaction
E2 (10−11-10−9 M) potentiates ICa,L in neurons and HEK293 cells overexpressing the α1C subunit; nifedipine displaces membrane E2 binding; and E2's effect is reduced by a dihydropyridine-insensitive LTCC mutant, indicating that E2 binds to the dihydropyridine-binding site (177). Intriguingly, E2 and the dihydropyridines exert opposite effects on ICa,L.
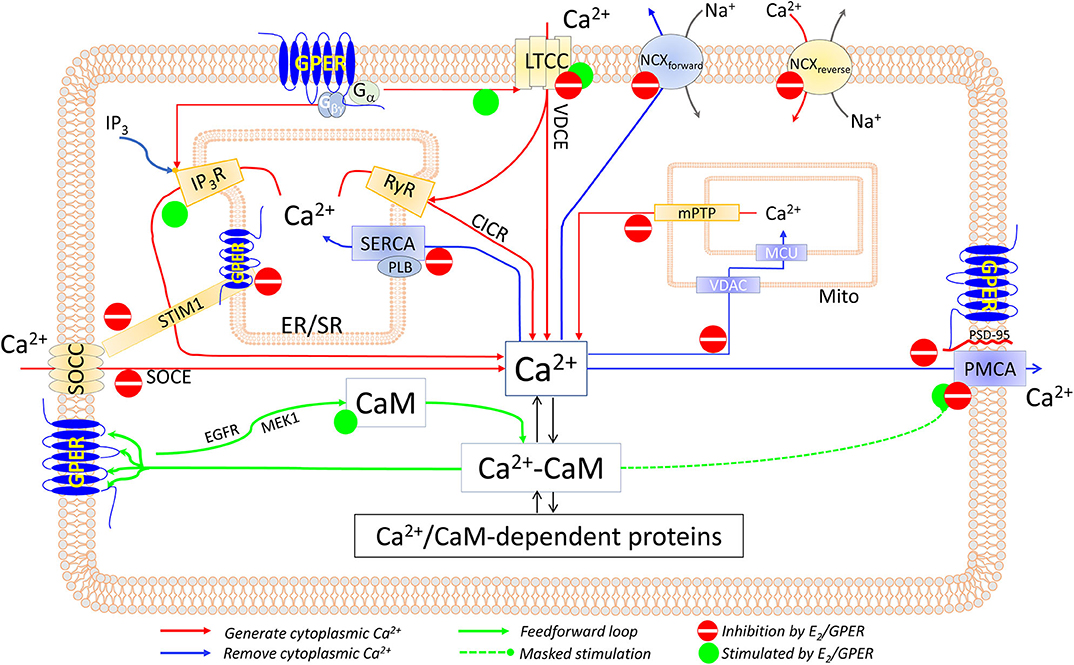
Figure 1. Components of the CSM that are affected by E2 and/or GPER in excitable and non-excitable cardiovascular tissues. See text for details.
Direct, Membrane-Delimited Activation of Ca2+ Channels by Gα Subunits
GPCR stimulation can trigger Ca2+ signals independently of the second messenger (178–180). GPER couples with Gαs and Gαi/o, which can interact with LTCC (178, 181, 182) and trigger Ca2+ entry.
Release of Gβγ Subunit Upon GPER-Associated Gαi Stimulation
Gβγ stimulates PLCβ (183–185) and activates IP3R1 (186), both of which trigger Ca2+ store depletion and SOCE. Consistently, E2-induced Ca2+ store release and entry in ECs are completely inhibited by pertussis toxin and PLCβ inhibitor U73122 (164). Also, HEK293 cells only produce a Ca2+ response to E2 when expressing HA-tagged GPER (162). Since (1) Ca2+ entry channels are located on the membrane and (2) Gβγ activates IP3Rs by interacting with the IP3-binding sites (186) on IP3Rs' cytosolic domains, both the membrane-delimited/Gα-mediated and Gβγ-mediated mechanisms should only be operable by the plasmalemmal GPER pool. A distinguishing feature is that the former mechanism would not trigger SR/ER Ca2+ release in the absence of extracellular Ca2+, whereas the latter would. Based on this feature, data fitting the former are available from renal tubular cells (176); and data fitting the latter, from vascular ECs (164).
Functional Impact
Do Ca2+ signals stimulated by estrogenic agonists activate Ca2+-dependent activities? When reported, the concentration of a Ca2+ signal allows for prediction of proteins that may or may not be affected by it. For example, E2 induces ER Ca2+ release signals of ~2 × 10−7 M and activates MAPK (173), because this Ca2+ level is sufficient for MAPK activity (187); indeed, Ca2+ chelation abolishes E2's effect (173). Considering that GPER mediates the effect of E2 to trigger Ca2+ signals that activate MAPK, GPER activity can promote many downstream effects (163, 170, 188). In ECs, E2 (10−9-10−6 M) stimulates very small Ca2+ signals (<10−7 M) (174). One can predict that only proteins with very high Ca2+ sensitivity, for example, phosphorylated eNOS (170, 189, 190), would be activated by these signals. Whether a Ca2+ signal can produce a predicted effect also depends on other factors. For example, the Ca2+ signal of ~2 × 10−7 M triggered by G-1 in VSMCs (172) would be sufficient to activate myosin light-chain kinase (MLCK) and cause vasoconstriction, based on MLCK's properties (191). However, G-1 causes vasodilation (172, 192–194), likely by activating eNOS (170, 193, 195–198), inhibiting VSMC Ca2+ (199), and stimulating SMC K+ efflux (200).
Calcium Entry Inhibition by Estrogenic Agonists and Estrogen Receptors
To a large extent, estrogenic regulation of Ca2+ signaling involves effects of estrogenic agonists and receptors on the Ca2+ signals triggered by other stimuli, via both E2-dependent and E2-independent mechanisms.
Store-Operated Ca2+ Entry (Figure 1)
In VSMCs, E2 (10−8-10−5 M) inhibits norepinephrine- and phenylephrine-induced arterial constriction in the presence of extracellular Ca2+ but not that induced in Ca2+-free medium (201, 202). These effects may be attributed to inhibition of both VDCE and SOCE, as α1 adrenoceptor agonists can activate both (77). GPER-mediated inhibition of SOCE has been shown in ECs, where G-1 (10−8-10−6 M) suppresses SOCE induced by thapsigargin or bradykinin (203). Interestingly, the observations that in the absence of any treatment with agonists, thapsigargin-induced SOCE is increased by 80% in GPER-knockdown ECs and is reduced by 40% in GPER-overexpressing HEK293 cells implicate E2-independent mechanisms (203).
How E2/GPER suppresses SOCE seems to involve STIM1. G-1 treatment prevents thapsigargin-induced STIM1 puncta, indicating inhibition of STIM1's association with the Ca2+ channel; and Ser575/608/621Ala mutations of STIM1 reduce the inhibitory effect of G-1 (203). Consistently, E2 inhibits Ser575 STIM1 phosphorylation in bronchial epithelial cells, thus suppressing STIM1 mobility and SOCE (204). Our initial data also indicate that dynamic physical interaction between them contributes importantly to GPER's inhibition of SOCE (205).
Voltage-Dependent Ca2+ Entry (Figure 1)
Electrically induced Ca2+ signals are increased in cardiomyocytes from ovariectomized (OVX) animals (206–208). Many lines of evidence indicate that GPER mediates the inhibitory effect of E2 on ICa,L. These include inhibitory effects of E2 (1–3 × 10−5 M) and combined ERα/ERβ antagonists/GPER agonists (ICI182,780, tamoxifen, or raloxifene) on ICa,L in cardiomyocytes from both WT and ERα−/−/ERβ−/− animals, as reviewed in (132). Similarly, in VSMCs, E2 inhibits electrically induced ICa,L (209, 210), and ERα/ERβ antagonists/GPER agonists tamoxifen and ICI164,384 inhibit high-K+-induced contraction (202). GPER agonist G-1 (10−6 M) inhibits nifedipine-sensitive Ca2+ spikes in LTCC-expressing A7R5 SMCs, an effect prevented by GPER antagonist G-15 (10−6 M) (199); these concentrations are specific for GPER (175, 211). Consistently, ERα knockout does not affect E2's inhibition of KCl-induced 45Ca2+ uptake in VSMCs and vasorelaxation (212).
How E2 inhibits electrically induced VDCE is still unknown. Hypothetically, at high levels, E2 binding to the dihydropyridine-binding site on LTCC (177) may instead inhibit ICa,L. As for prevention of β adrenoceptor (βAR)-mediated potentiation of VDCE, recent evidence suggests that GPER may be an intrinsic component of β1AR activation. Thus, G-1 inhibits isoproterenol-induced increases in left ventricle (LV) pressure, heart rate, ectopic contractions, ICa,L, LTCC phosphorylation, and total myocardial Ca2+ signal, while the GPER inhibitor G-36 promotes ISO-induced Ca2+ signal and LTCC phosphorylation (213). Speculatively, GPER may do so in part by interacting with β1AR or with A kinase-anchoring protein 5, thus inhibiting cAMP production (167). These may represent some E2-independent effects of GPER. Studies in GPER-knockout tissues are needed to further clarify the mechanisms.
Estrogenic Regulation of Cytoplasmic Calcium Removal Mechanisms
SERCA Activity
Few studies, mostly in cardiac tissues, have examined the effects of E2 on SERCA activity, with somewhat conflicting results. E2 (1–30 × 10−6 M) does not affect the Vmax of SR vesicle Ca2+ uptake in canine LV tissue (214). However, ovariectomy reduces the Vmax but increases the Ca2+ sensitivity for SR Ca2+ uptake of rat LV homogenates or SR-enriched membrane fractions; mechanistically, these effects appear to be associated with reduced Thr17 phosphorylation of phospholamban and are restored by treatment with either E2 or progesterone (215) (Figure 1). How E2 and progesterone promote Thr17 phosphorylation of phospholamban is unknown, perhaps by inhibiting CaM kinase II (216), the enzyme that phosphorylates phospholamban (21). The effect of E2 on SERCA activity in VSMCs has not been examined.
NCX Activity
As with SERCA activity, few studies have measured the effects of E2 on NCX activity. Na+-dependent 45Ca2+ uptake in rat LV myocytes is increased by ~3-fold after 60 days of ovariectomy, which is restored by replenishment with E2 (1.5 mg/60 days) (208). During myocardial ischemia, intracellular Na+ concentration is higher in male than in female cardiomyocytes and is associated with increased Ca2+ concentration as a result of increased NCX activity (217). These studies are consistent with an inhibitory effect of E2 on NCX activity in both the forward and reverse modes (Figure 1). However, the mechanisms of this inhibition are unclear.
Mitochondrial Ca2+ Uptake
In the heart, diethylstilbestrol (0.9–1.8 × 10−3 M) inhibits mitochondrial 45Ca2+ uptake (218). Mitochondrial Ca2+ retention capacity (mCRC), a combination of mitochondrial Ca2+ uptake, total mitochondrial Ca2+-binding sites, and mitochondrial Ca2+ release mechanisms, is a determinant of the protective role of the mitochondria during cytoplasmic Ca2+ overload. E2 (4 × 10−8 M) increases myocardial mCRC following ischemia–reperfusion, an effect abolished by genetic deletion of GPER but not of ERα or ERβ; mechanistically, this effect seems to involve PKC-dependent, MAPK-dependent phosphorylation of glycogen synthase kinase (GSK)-3β, leading to inhibition of the mitochondrial permeability transition pore (219). Consistently, E2 (10−8 M) inhibits high Ca2+-induced cytochrome c release from myocardial mitochondria (220). In ECs, 48-h E2 (10−8 M) treatment inhibits mitochondrial Ca2+ uptake, an effect abolished by the ERα/ERβ antagonist ICI182,780 (10−8 M) (221). The mechanisms whereby E2 inhibits mitochondrial Ca2+ uptake are still unknown (Figure 1).
PMCA Activity
Recent data show that GPER inhibits PMCA activity via both E2-dependent and E2-independent mechanisms (Figure 1). E2-dependent mechanisms are evidenced by the effects of G-1 (10−8-10−6 M) and E2 (1–5 × 10−9 M) to inhibit PMCA-mediated efflux in primary ECs without affecting PMCA expression levels and to promote PMCA phosphorylation at Tyr1176 (135, 170), which is known to inhibit pump activity (94). Notably, this phosphorylation masks the stimulatory effect of enhancing the PMCA–CaM interaction produced by 48-h E2 treatment (170). E2-independent mechanisms are indicated by the findings that (1) GPER constitutively interacts with PMCA4b via the anchoring action of PSD-95 at their C-terminal PDZ-binding motifs; (2) overexpression of GPER decreases PMCA activity; (3) GPER knockdown promotes PMCA activity; and (4) PSD-95 knockdown or truncation of the PDZ-binding motif on GPER releases GPER–PMCA association and promotes PMCA activity (135). Functionally, these mechanisms collectively prolong agonist-induced Ca2+ signal and enhance eNOS activity in ECs (135, 170, 203). Consistent with suppressed Ca2+ efflux, the Ca2+ signals stimulated by E2 and the GPER agonist G-1 in cells overexpressing GPER reported by various laboratories display much more prolonged plateau phases compared to Ca2+ signals in cells not overexpressing GPER or those stimulated by other agonists such as ATP or bradykinin (160, 162, 164, 175). GPER–PMCA4b interaction seems to be mutually influential, such that knockdown of PMCA decreases GPER-mediated ERK1/2 phosphorylation, while GPER knockdown does the opposite on PMCA activity (135).
Estrogenic Regulation of Calcium Signal Transduction—The Calmodulin Network
Since CaM is the universal Ca2+ signal transducer for numerous proteins (117, 118), is insufficiently expressed for its targets (122, 125, 126), and is a source of competition among target proteins (124, 127), factors that regulate its expression and target interactions are predicted to have a pervasive impact. The effects of E2 on the CaM network have been examined in some detail in vascular ECs in recent studies (135, 169, 170). E2 treatment (1–5 × 10−9 M, 48 h) upregulates total CaM by around 7-fold and free Ca2+-CaM by ~15-fold in primary ECs. Data obtained using specific estrogen receptor agonists, gene silencing, and receptor overexpression indicate that GPER, but not ERα or ERβ, mediates this effect. Thus, the GPER agonist G-1 (10−9-10−7 M), but not the ERα agonist propyl pyrazole triol (PPT) (3 × 10−10-2 × 10−7 M) or the ERβ agonist diarylpropionitrile (DPN) (10−10-5 × 10−8 M), increases CaM expression; GPER knockdown reduces the effect of E2 to upregulate CaM; and E2 upregulates CaM in SKBR3 cells that express only GPER and not ERα or ERβ (170). Consistently, the ERα/ERβ antagonist/GPER agonist ICI182,780 dose-dependently upregulates CaM. Mechanistically, GPER exerts this action via the activities of EGFR and MAPK/ERK kinase 1 (MEK1). Functionally, E2 upregulates CaM and promotes the PMCA–CaM interaction; however, the predicted stimulatory effect on Ca2+ extrusion is masked by E2-induced inhibitory phosphorylation at Tyr1176 of PMCA (170); additionally, GPER exerts E2-dependent and E2-independent effects to inhibit PMCA (135). These collective actions prolong Ca2+ signals, promote Ca2+-CaM complex formation, and increase Ca2+-CaM associations with low- to high-affinity CaM network members, represented by GPER itself, ERα, and eNOS (170). Considering that CaM binding stabilizes ERα homodimers, these effects are expected to promote other genomic actions of E2 as well. Thus, a feedforward mechanism exists in which GPER mediates E2's effects to increase CaM and inhibits Ca2+ efflux, prolonging cytoplasmic Ca2+ signals, and the resultant increases in Ca2+-CaM complexes in turn promote the activities of GPER itself and other CaM network members (170) (Figure 1).
Estrogenic Moderation of Calcium-Dependent Activities
How do the various mechanisms discussed so far come together in regulating cardiovascular functions? An immediate challenge is how to reconcile the effects of estrogenic agonists to both trigger acute Ca2+ signals by themselves and inhibit otherwise stimulated Ca2+ signals. The Ca2+ signals triggered by estrogenic agonists in primary cardiovascular cells are generally of very low amplitude. Furthermore, as in experiments testing their effects on Ca2+ signals otherwise triggered, estrogenic agonists are present in situ with other stimuli whose Ca2+ signals they inhibit. Thus, for mechanisms that generate cytoplasmic Ca2+ signals, E2 and GPER exert ultimate inhibitory effects. For cytoplasmic Ca2+ removal mechanisms, estrogenic agonists and GPER also are inhibitory. For Ca2+ signal transduction, E2, via a feedforward at GPER, increases CaM expression and enhances linkage in the CaM-binding proteome.
All things considered, E2 and GPER, via both E2-dependent and E2-independent mechanisms, act to moderate Ca2+-dependent activities in the cardiovascular system. They “clamp” cytoplasmic Ca2+ signals by lowering peaks (inhibition of signal generation) and raising troughs (inhibition of signal removal), collectively confining tissues in a narrower yet more sustained operating range of Ca2+. Also, GPER-mediated increases in CaM expression and CaM network linkage improve Ca2+ signal transduction efficiency. Considering the Ca2+ sensitivity of Ca2+-dependent proteins in this context, one can predict that those with low Ca2+ sensitivity (requiring high Ca2+ for activation) are more likely to be affected by the inhibition of Ca2+ signal generation. On the other hand, proteins with high Ca2+ sensitivity (requiring low Ca2+ for activation) are more likely to be promoted by the inhibition of Ca2+ removal and less affected by the suppression of Ca2+ signal generation (Figure 2).
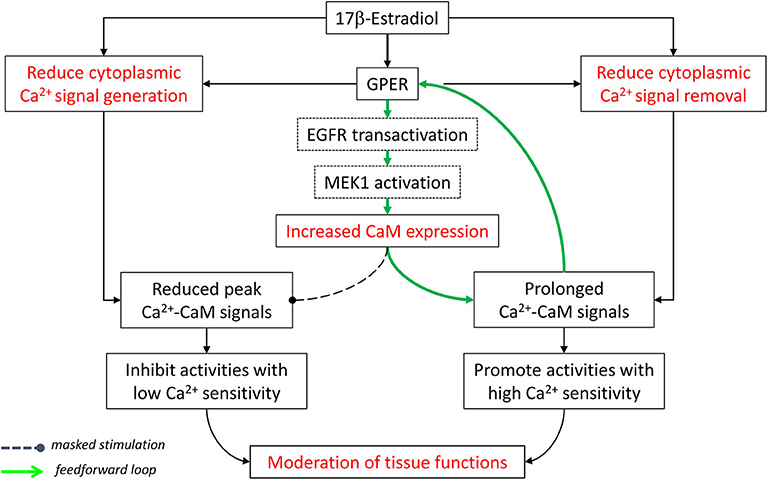
Figure 2. Moderation of cardiovascular functions by E2 and GPER via effects on Ca2+ signal generation, Ca2+ signal removal, and Ca2+ signal transduction. See text for details. Modified with permission from the author's previous publication (170).
This notion has been demonstrated experimentally via the case of eNOS, a Ca2+-dependent CaM-binding protein (222) with sub-nanomolar affinity for CaM (127). CaM interaction and subsequent activation of wild-type eNOS have high Ca2+ sensitivities, with respective EC50(Ca2+) values ~1.8 × 10−7 and 4 × 10−7 M (190). eNOS is also regulated by multisite phosphorylation (223). Notably, its bi-phosphorylation at Ser617 and Ser1179 promotes NO production by increasing the Ca2+ sensitivity for both CaM binding and enzyme activation, reducing their respective EC50 (Ca2+) values to ~0.7 × 10−7 and 1.3 × 10−7 M, thus rendering the synthase active at resting cytoplasmic Ca2+ (189). E2 and GPER (1) prolong endothelial cytoplasmic Ca2+ signal by inhibiting Ca2+ efflux (135, 170), (2) promote eNOS phosphorylation at Ser617 and Ser1179 (170, 198), (3) increase CaM expression and eNOS–CaM interaction (170), and (4) suppress endothelial SOCE (203). When we incorporate these effects into a verified sequential “CaM binding eNOS activation” model (189, 190), eNOS activity and NO accumulation are shown to substantially increase across the time course of bradykinin-induced Ca2+ signal in ECs by treatment with G-1 (203). Importantly, major contributions to this outcome include the increases in CaM binding, phosphorylation, Ca2+ sensitivity, and duration of Ca2+ signals due to Ca2+ efflux inhibition, but little or no effect of the inhibition of SOCE (203), due obviously to the synthase's high Ca2+ sensitivity (Figure 3). Thus, via multifaceted actions on components of the CSM, E2 and GPER moderate Ca2+-dependent activities by differentially affecting the continuum of Ca2+-dependent proteins based on their Ca2+ sensitivities for Ca2+ or Ca2+-CaM complexes.
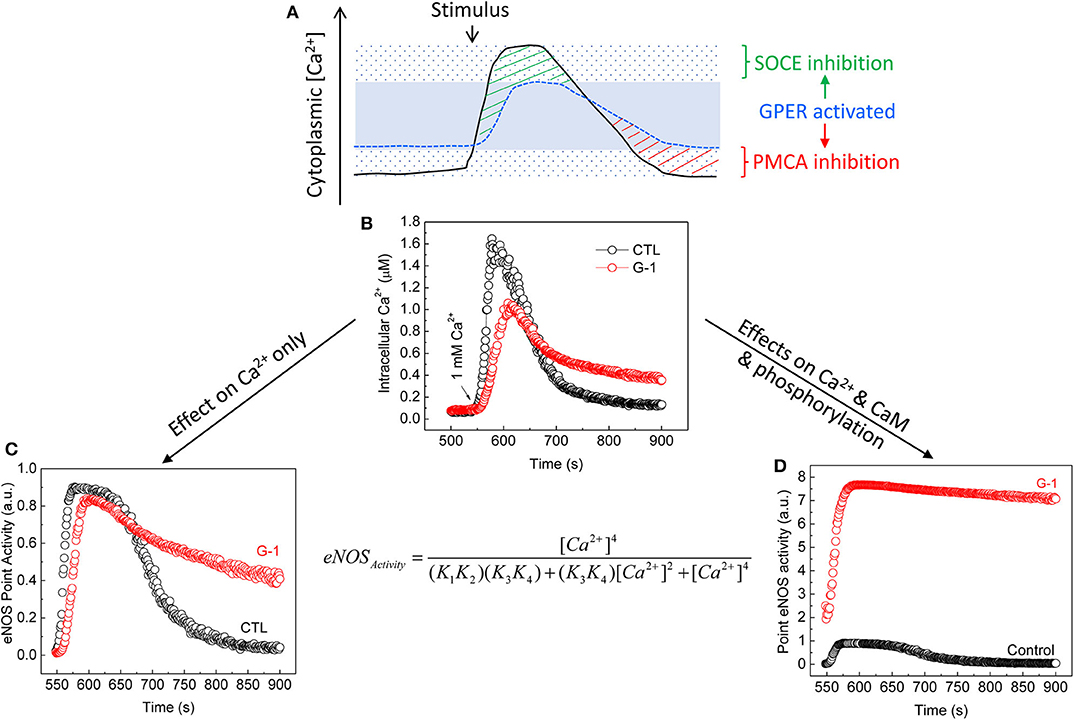
Figure 3. Moderation of Ca2+-dependent eNOS activity by GPER activation. (A) Cytoplasmic Ca2+ clamping by GPER activation in ECs (203). The solid line represents Ca2+ signals produced in response to agonist stimulation in the absence of GPER activation. The sparsely dotted area represents the range of cytoplasmic Ca2+ signals, in which peak and trough are seen due to maximal effects of Ca2+ entry and Ca2+ efflux. The stippled blue line represents Ca2+ signals produced in the presence of GPER and its activation. These signals are clamped in a narrower range (the blue area) due to inhibitory effects on both SOCE [green stripes (203)] and PMCA4b-mediated Ca2+ efflux [red stripes (135, 170)]. (B) Average time courses of cytoplasmic Ca2+ signals measured in primary ECs treated with bradykinin in the absence of extracellular Ca2+ followed by treatment with vehicle or G-1; total Ca2+ signals were triggered by re-addition of extracellular Ca2+ [arrow (203)]. (C) Calculated eNOS point activity corresponding to each Ca2+ value in (B) considering only changes in Ca2+ due to GPER activation using a verified sequential eNOS–CaM binding eNOS activation model [equation, where (K1, K2) and (K3, K4) are derived products of the binding constants of Ca2+ at the Ca2+-binding sites on the N and C lobes of CaM in binding to CaM and interaction of Ca2+-CaM and eNOS (189, 190). (D) Calculated eNOS point activity corresponding to each Ca2+ value measured in (B), factoring in changes in Ca2+, CaM binding, and eNOS phosphorylation (170, 203). See details in text and (170, 203). Reproduced with permission from the author's previous publication (203).
Considering the two Ca2+-dependent estrogen receptors—ERα and GPER—how does the presence of one influence the effects of the other on Ca2+ signaling? A complex relationship is predicted to exist in which ERα transcriptional activities affect the expression of certain Ca2+ signaling proteins but are themselves influenced by the amplitudes and dynamics of Ca2+ signals limited by GPER activation and the availability of CaM that is promoted by GPER action (170). In turn, as CaM is limited in cells (122, 124, 126, 127), the high affinity binding of CaM by ERα and GPER further limits CaM availability and will influence CaM-dependent regulation of each other at the receptor level, a predictable outcome of the functional crosstalk via competition for limited CaM (124, 127). These relationships may represent but a small aspect of the reciprocality between estrogen and Ca2+ signaling.
Conclusion and Future Perspectives
Reciprocality between estrogen signaling and Ca2+-dependent activities is becoming evident. Considering the impact of estrogen and its receptors on Ca2+ signaling, E2, and in many cases, GPER exert inhibitory effects on many components of the CSM in cardiovascular tissues, from Ca2+ store release and uptake (214, 215, 221) and Ca2+ entry (199, 201–210, 212, 213) to cytosolic Ca2+ removal mechanisms (135, 170, 208, 217–221). Considering the impact of Ca2+ signaling on estrogen biology, both ERα and GPER are strongly regulated by direct Ca2+-dependent interactions with CaM. These interactions serve to stabilize receptor dimerization and enhance subsequent transcriptional activities [the case of ERα (137, 138, 142, 143)] or promote receptor-mediated downstream signaling [the case of GPER (169, 170)]. Also, E2-induced MAPK activation has long been known to be dependent on the Ca2+ signal produced (173). Reciprocality between estrogen biology and Ca2+ signaling is further evidenced by the demonstration of a feedforward mechanism, in which E2, via GPER activation, upregulates total cellular CaM expression and free intracellular Ca2+-CaM concentration, which promotes functions of GPER and ERα and other classes of Ca2+-CaM-dependent proteins (170). The combination of these various actions is predicted to affect Ca2+-dependent functions depending on the affinity and Ca2+ sensitivities of the proteins involved, as exemplified by the case of eNOS (Figures 2, 3) (170, 203).
The moderating effects that estrogenic agonists and receptors exert on the CSM can explain many of their cardiovascular effects, such as preventing excessive cardiac contraction during sympathetic stress, limiting adverse outcomes related to Ca2+ overload, and reducing vascular tone. Nevertheless, the effects of E2 and estrogen receptors on many CSM components have not been examined. Additionally, many questions remain regarding mechanisms of the observed effects that estrogenic agonist and receptors produce on the CSM. For example, how do E2 and GPER inhibit ICa,L? What are the mechanisms that position GPER as an intrinsic component of β1AR signaling in the myocardium? What are the mechanisms whereby E2 inhibits the activities of SERCA and NCX? What are the mechanisms whereby E2 inhibits mitochondrial Ca2+ uptake? Further studies are needed to answer these questions. Through many examples, however, it is clear that GPER produces both E2-dependent and E2-independent effects on the CSM. While the search is ongoing for approaches to apply specific estrogen receptor agonists to the prevention of cardiovascular disease, the therapeutic potential of E2-independent effects of GPER and other estrogen receptors is as yet an unexplored territory.
Author Contributions
Q-KT conceived the ideas, generated the figures, and wrote the manuscript.
Funding
The publication cost of this review article is covered by a fund from Des Moines University to the author.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AF domain, transcriptional activation function domain; CaM, calmodulin; Ca2+-CaM, Ca2+-bound calmodulin; cAMP, cyclic adenosine monophosphate; CICR, Ca2+-induced Ca2+ release; CRAC, Ca2+ release-activated channels; CSM, Ca2+ signaling machinery; E2, 17β-estradiol; ECs, endothelial cells; EGFR, epidermal growth factor receptor; eNOS, endothelial nitric oxide synthase; ERβ, estrogen receptor β; ERα, estrogen receptor α; ERK1/2, extracellular signal-related kinases 1 and 2; FRET, fluorescence resonance energy transfer; GPER, G protein-coupled estrogen receptor 1; GPR30, G protein-coupled estrogen receptor 1; HEK293 cells, human embryonic kidney 293 cells; ICa, L, L-type Ca2+ channel current; IP3Rs, inositol-trisphosphate receptors; LTCC, L-type Ca2+ channels; LV, left ventricle; MAPK, mitogen-activated protein kinases; mCRC, mitochondrial Ca2+ retention capacity; MCU, mitochondrial Ca2+ uniporter; MEK1, MAP (mitogen-activated protein) kinase/ERK (extracellular signal-regulated kinase) kinase 1; mPTP, mitochondrial permeability transition pore; NCX, Na+-Ca2+ exchanger; OVX, ovariectomy/ovariectomized; PDZ, PSD-95/Dlg/ZO; PKC, protein kinase C; PLCβ, phospholipase C-β; PMCA, plasma membrane Ca2+-ATPase; PSD-95, post-synaptic density protein 95; RMP, resting membrane potential; RyRs, ryanodine receptors; SCPA, secretory pathway Ca2+ pump; SERCA, sarcoplasmic/endoplasmic reticulum-ATPase; SMD, submembrane domains of G protein-coupled receptors; SOCE, store-operated Ca2+ entry; SOICR, store overload-induced Ca2+ release; SR/ER, sarco-endoplasmic reticulum; STIM1, stromal interaction molecule 1; VDAC, voltage-dependent anion channel; VDCC, voltage-dependent Ca2+ channels; VDCE, voltage-dependent Ca2+ entry; VSMCs, vascular smooth muscle cells.
References
1. Somlyo AP, Somlyo AV. Electron probe analysis of calcium content and movements in sarcoplasmic reticulum, endoplasmic reticulum, mitochondria, and cytoplasm. J Cardiovasc Pharmacol. (1986) 8(Suppl. 8):S42–47. doi: 10.1097/00005344-198600088-00009
2. Wray S, Burdyga T. Sarcoplasmic reticulum function in smooth muscle. Physiol Rev. (2010) 90:113–78. doi: 10.1152/physrev.00018.2008
3. Wood PG, Gillespie JI. Evidence for mitochondrial Ca2+ induced Ca2+ release in permeabilised endothelial cells. Biochem Biophys Res Commun. (1998) 246:543–8. doi: 10.1006/bbrc.1998.8661
4. Pinton P, Pozzan T, Rizzuto R. The Golgi apparatus is an inositol 1,4,5-trisphosphate-sensitive Ca2+ store, with functional properties distinct from those of the endoplasmic reticulum. EMBO J. (1998) 17:5298–308. doi: 10.1093/emboj/17.18.5298
5. Pizzo P, Lissandron V, Capitanio P, Pozzan T. Ca2+ signalling in the golgi apparatus. Cell Calcium. (2011) 50:184–92. doi: 10.1016/j.ceca.2011.01.006
6. Lopez-Sanjurjo CI, Tovey SC, Prole DL, Taylor CW. Lysosomes shape Ins(1,4,5)P3-evoked Ca2+ signals by selectively sequestering Ca2+ released from the endoplasmic reticulum. J Cell Sci. (2013) 126(Pt 1):289–300. doi: 10.1242/jcs.116103
7. Morgan AJ, Platt FM, Lloyd-Evans E, Galione A. Molecular mechanisms of endolysosomal Ca2+ signalling in health and disease. Biochem J. (2011) 439:349–74. doi: 10.1042/BJ20110949
8. Pizzo P, Lissandron V, Pozzan T. The trans-golgi compartment: a new distinct intracellular Ca store. Communicat Integr Biol. (2010) 3:462–4. doi: 10.4161/cib.3.5.12473
9. Wong AK, Capitanio P, Lissandron V, Bortolozzi M, Pozzan T, Pizzo P. Heterogeneity of Ca2+ handling among and within Golgi compartments. J Mol Cell Biol. (2013) 5:266–76. doi: 10.1093/jmcb/mjt024
10. Kilpatrick BS, Eden ER, Schapira AH, Futter CE, Patel S. Direct mobilisation of lysosomal Ca2+ triggers complex Ca2+ signals. J Cell Sci. (2013) 126(Pt 1):60–66. doi: 10.1242/jcs.118836
11. Kilpatrick BS, Yates E, Grimm C, Schapira AH, Patel S. Endo-lysosomal TRP mucolipin-1 channels trigger global ER Ca2+ release and Ca2+ influx. J Cell Sci. (2016) 129:3859–67. doi: 10.1242/jcs.190322
12. Morgan AJ, Davis LC, Wagner SK, Lewis AM, Parrington J, Churchill GC, et al. Bidirectional Ca2+ signaling occurs between the endoplasmic reticulum and acidic organelles. J Cell Biol. (2013) 200:789–805. doi: 10.1083/jcb.201204078
13. Csordas G, Varnai P, Golenar T, Roy S, Purkins G, Schneider TG, et al. Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol Cell. (2010) 39:121–32. doi: 10.1016/j.molcel.2010.06.029
14. Rizzuto R, Marchi S, Bonora M, Aguiari P, Bononi A, De Stefani D, et al. Ca2+ transfer from the ER to mitochondria: when, how and why. Biochim Biophys Acta. (2009) 1787:1342–51. doi: 10.1016/j.bbabio.2009.03.015
15. Robert V, Gurlini P, Tosello V, Nagai T, Miyawaki A, Di Lisa F, et al. Beat-to-beat oscillations of mitochondrial [Ca2+] in cardiac cells. EMBO J. (2001) 20:4998–5007. doi: 10.1093/emboj/20.17.4998
16. Drago I, De Stefani D, Rizzuto R, Pozzan T. Mitochondrial Ca2+ uptake contributes to buffering cytoplasmic Ca2+ peaks in cardiomyocytes. Proc Natl Acad Sci USA. (2012) 109:12986–91. doi: 10.1073/pnas.1210718109
17. Yu X, Carroll S, Rigaud JL, Inesi G. H+ countertransport and electrogenicity of the sarcoplasmic reticulum Ca2+ pump in reconstituted proteoliposomes. Biophys J. (1993) 64:1232–42. doi: 10.1016/S0006-3495(93)81489-9
18. Ver Heyen M, Heymans S, Antoons G, Reed T, Periasamy M, Awede B, et al. Replacement of the muscle-specific sarcoplasmic reticulum Ca2+-ATPase isoform SERCA2a by the nonmuscle SERCA2b homologue causes mild concentric hypertrophy and impairs contraction-relaxation of the heart. Circ Res. (2001) 89:838–46. doi: 10.1161/hh2101.098466
19. Anger M, Samuel JL, Marotte F, Wuytack F, Rappaport L, Lompre AM. The sarco(endo)plasmic reticulum Ca2+-ATPase mRNA isoform, SERCA 3, is expressed in endothelial and epithelial cells in various organs. FEBS Lett. (1993) 334:45–48. doi: 10.1016/0014-5793(93)81677-R
20. Wuytack F, Dode L, Baba-Aissa F, Raeymaekers L. The SERCA3-type of organellar Ca2+ pumps. Biosci Rep. (1995) 15:299–306. doi: 10.1007/BF01788362
21. Wegener AD, Simmerman HK, Lindemann JP, Jones LR. Phospholamban phosphorylation in intact ventricles. Phosphorylation of serine 16 and threonine 17 in response to beta-adrenergic stimulation. J Biol Chem. (1989) 264:11468–74.
22. Gustavsson M, Verardi R, Mullen DG, Mote KR, Traaseth NJ, Gopinath T, et al. Allosteric regulation of SERCA by phosphorylation-mediated conformational shift of phospholamban. Proc Natl Acad Sci USA. (2013) 110:17338–43. doi: 10.1073/pnas.1303006110
23. Bhupathy P, Babu GJ, Periasamy M. Sarcolipin and phospholamban as regulators of cardiac sarcoplasmic reticulum Ca2+ ATPase. J Mol Cell Cardiol. (2007) 42:903–11. doi: 10.1016/j.yjmcc.2007.03.738
24. Tan W, Colombini M. VDAC closure increases calcium ion flux. Biochim Biophys Acta. (2007) 1768:2510–15. doi: 10.1016/j.bbamem.2007.06.002
25. De Stefani D, Bononi A, Romagnoli A, Messina A, De Pinto V, Pinton P, et al. VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ. (2012) 19:267–73. doi: 10.1038/cdd.2011.92
26. Shimizu H, Schredelseker J, Huang J, Lu K, Naghdi S, Lu F, et al. Mitochondrial Ca2+ uptake by the voltage-dependent anion channel 2 regulates cardiac rhythmicity. Elife. (2015) 4:e04801. doi: 10.7554/eLife.04801
27. Mammucari C, Raffaello A, Vecellio Reane D, Gherardi G, De Mario A, Rizzuto R. Mitochondrial calcium uptake in organ physiology: from molecular mechanism to animal models. Pflugers Arch. (2018) 470:1165–79. doi: 10.1007/s00424-018-2123-2
28. Wacquier B, Combettes L, Dupont G. Cytoplasmic and mitochondrial calcium signaling: a two-way relationship. Cold Spring Harb Perspect Biol. (2019) 11:ea035139. doi: 10.1101/cshperspect.a035139
29. Iwai M, Tateishi Y, Hattori M, Mizutani A, Nakamura T, Futatsugi A, et al. Molecular cloning of mouse type 2 and type 3 inositol 1,4,5-trisphosphate receptors and identification of a novel type 2 receptor splice variant. J Biol Chem. (2005) 280:10305–17. doi: 10.1074/jbc.M413824200
30. Tateishi Y, Hattori M, Nakayama T, Iwai M, Bannai H, Nakamura T, et al. Cluster formation of inositol 1,4,5-trisphosphate receptor requires its transition to open state. J Biol Chem. (2005) 280:6816–22. doi: 10.1074/jbc.M405469200
31. Meyer T, Holowka D, Stryer L. Highly cooperative opening of calcium channels by inositol 1,4,5-trisphosphate. Science. (1988) 240:653–56. doi: 10.1126/science.2452482
32. Vermassen E, Parys JB, Mauger JP. Subcellular distribution of the inositol 1,4,5-trisphosphate receptors: functional relevance and molecular determinants. Biol Cell. (2004) 96:3–17. doi: 10.1016/j.biolcel.2003.11.004
33. Perez PJ, Ramos-Franco J, Fill M, Mignery GA. Identification and functional reconstitution of the type 2 inositol 1,4,5-trisphosphate receptor from ventricular cardiac myocytes. J Biol Chem. (1997) 272:23961–9. doi: 10.1074/jbc.272.38.23961
34. Hohendanner F, Walther S, Maxwell JT, Kettlewell S, Awad S, Smith GL, et al. Inositol-1,4,5-trisphosphate induced Ca2+ release and excitation-contraction coupling in atrial myocytes from normal and failing hearts. J Physiol. (2015) 593:1459–77. doi: 10.1113/jphysiol.2014.283226
35. Lin Q, Zhao G, Fang X, Peng X, Tang H, Wang H, et al. IP3 receptors regulate vascular smooth muscle contractility and hypertension. JCI Insight. (2016) 1:e89402. doi: 10.1172/jci.insight.89402
36. Haorah J, Knipe B, Gorantla S, Zheng J, Persidsky Y. Alcohol-induced blood-brain barrier dysfunction is mediated via inositol 1,4,5-triphosphate receptor (IP3R)-gated intracellular calcium release. J Neurochem. (2007) 100:324–36. doi: 10.1111/j.1471-4159.2006.04245.x
37. Geyer M, Huang F, Sun Y, Vogel SM, Malik AB, Taylor CW, et al. Microtubule-associated protein EB3 regulates IP3 receptor clustering and Ca2+ signaling in endothelial cells. Cell Rep. (2015) 12:79–89. doi: 10.1016/j.celrep.2015.06.001
38. Sundivakkam PC, Kwiatek AM, Sharma TT, Minshall RD, Malik AB, Tiruppathi C. Caveolin-1 scaffold domain interacts with TRPC1 and IP3R3 to regulate Ca2+ store release-induced Ca2+ entry in endothelial cells. Am J Physiol Cell Physiol. (2009) 296:C403–13. doi: 10.1152/ajpcell.00470.2008
39. Zalk R, Clarke OB, des Georges A, Grassucci RA, Reiken S, Mancia F, et al. Structure of a mammalian ryanodine receptor. Nature. (2015) 517:44–49. doi: 10.1038/nature13950
40. Brillantes AM, Allen P, Takahashi T, Izumo S, Marks AR. Differences in cardiac calcium release channel (ryanodine receptor) expression in myocardium from patients with end-stage heart failure caused by ischemic versus dilated cardiomyopathy. Circ Res. (1992) 71:18–26. doi: 10.1161/01.RES.71.1.18
41. Copello JA, Barg S, Onoue H, Fleischer S. Heterogeneity of Ca2+ gating of skeletal muscle and cardiac ryanodine receptors. Biophys J. (1997) 73:141–56. doi: 10.1016/S0006-3495(97)78055-X
42. Yang XR, Lin MJ, Yip KP, Jeyakumar LH, Fleischer S, Leung GP, et al. Multiple ryanodine receptor subtypes and heterogeneous ryanodine receptor-gated Ca2+ stores in pulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. (2005) 289:L338–48. doi: 10.1152/ajplung.00328.2004
43. Westcott EB, Jackson WF. Heterogeneous function of ryanodine receptors, but not IP3 receptors, in hamster cremaster muscle feed arteries and arterioles. Am J Physiol Heart Circ Physiol. (2011) 300:H1616–30. doi: 10.1152/ajpheart.00728.2010
44. Salomone S, Soydan G, Moskowitz MA, Sims JR. Inhibition of cerebral vasoconstriction by dantrolene and nimodipine. Neurocrit Care. (2009) 10:93–102. doi: 10.1007/s12028-008-9153-0
45. Iino M. Calcium release mechanisms in smooth muscle. Jpn J Pharmacol. (1990) 54:345–54. doi: 10.1254/jjp.54.345
46. Lesh RE, Marks AR, Somlyo AV, Fleischer S, Somlyo AP. Anti-ryanodine receptor antibody binding sites in vascular and endocardial endothelium. Circ Res. (1993) 72:481–8. doi: 10.1161/01.RES.72.2.481
47. Wang X, Lau F, Li L, Yoshikawa A, van Breemen C. Acetylcholine-sensitive intracellular Ca2+ store in fresh endothelial cells and evidence for ryanodine receptors. Circ Res. (1995) 77:37–42. doi: 10.1161/01.RES.77.1.37
48. Sasajima H, Wang X, van Breemen C. Fractional Ca2+ release from the endoplasmic reticulum activates Ca2+ entry in freshly isolated rabbit aortic endothelial cells. Biochem Biophys Res Commun. (1997) 241:471–5. doi: 10.1006/bbrc.1997.7844
49. Kass RS, Tsien RW. Fluctuations in membrane current driven by intracellular calcium in cardiac purkinje fibers. Biophys J. (1982) 38:259–69. doi: 10.1016/S0006-3495(82)84557-8
50. Orchard CH, Eisner DA, Allen DG. Oscillations of intracellular Ca2+ in mammalian cardiac muscle. Nature. (1983) 304:735–8. doi: 10.1038/304735a0
51. Chen W, Wang R, Chen B, Zhong X, Kong H, Bai Y, et al. The ryanodine receptor store-sensing gate controls Ca2+ waves and Ca2+-triggered arrhythmias. Nat Med. (2014) 20:184–92. doi: 10.1038/nm.3440
52. Putney JW Jr. A model for receptor-regulated calcium entry. Cell Calcium. (1986) 7:1–12. doi: 10.1016/0143-4160(86)90026-6
53. Prakriya M, Lewis RS. Store-operated calcium channels. Physiol Rev. (2015) 95:1383–436. doi: 10.1152/physrev.00020.2014
54. Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE, et al. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. (2005) 15:1235–41. doi: 10.1016/j.cub.2005.05.055
55. Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. (2005) 169:435–45. doi: 10.1083/jcb.200502019
56. Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, et al. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. (2005) 437:902–5. doi: 10.1038/nature04147
57. Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, et al. A mutation in orai1 causes immune deficiency by abrogating CRAC channel function. Nature. (2006) 441:179–85. doi: 10.1038/nature04702
58. Peinelt C, Vig M, Koomoa DL, Beck A, Nadler MJ, Koblan-Huberson M, et al. Amplification of CRAC current by STIM1 and CRACM1 (Orai1). Nat Cell Biol. (2006) 8:771–3. doi: 10.1038/ncb1435
59. Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG. Orai1 is an essential pore subunit of the CRAC channel. Nature. (2006) 443:230–33. doi: 10.1038/nature05122
60. Soboloff J, Rothberg BS, Madesh M, Gill DL. STIM proteins: dynamic calcium signal transducers. Nat Rev Mol Cell Biol. (2012) 13:549–65. doi: 10.1038/nrm3414
61. Liou J, Fivaz M, Inoue T, Meyer T. Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc Natl Acad Sci USA. (2007) 104:9301–6. doi: 10.1073/pnas.0702866104
62. Yuan JP, Zeng W, Dorwart MR, Choi YJ, Worley PF, Muallem S. SOAR and the polybasic STIM1 domains gate and regulate Orail channels. Nat Cell Biol. (2009) 11:337–43. doi: 10.1038/ncb1842
63. Park CY, Hoover PJ, Mullins FM, Bachhawat P, Covington ED, Raunser S, et al. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell. (2009) 136:876–90. doi: 10.1016/j.cell.2009.02.014
64. Park CY, Shcheglovitov A, Dolmetsch R. The CRAC channel activator STIM1 binds and inhibits L-type voltage-gated calcium channels. Science. (2010) 330:101–5. doi: 10.1126/science.1191027
65. Grigoriev I, Gouveia SM, van der Vaart B, Demmers J, Smyth JT, Honnappa S, et al. STIM1 is a MT-plus-end-tracking protein involved in remodeling of the ER. Curr Biol. (2008) 18:177–82. doi: 10.1016/j.cub.2007.12.050
66. Hooper R, Samakai E, Kedra J, Soboloff J. Multifaceted roles of STIM proteins. Pflugers Arch. (2013) 465:1383–96. doi: 10.1007/s00424-013-1270-8
67. Collins HE, He L, Zou L, Qu J, Zhou L, Litovsky SH, et al. Stromal interaction molecule 1 is essential for normal cardiac homeostasis through modulation of ER and mitochondrial function. Am J Physiol Heart Circ Physiol. (2014) 306:H1231–9. doi: 10.1152/ajpheart.00075.2014
68. Edwards JM, Neeb ZP, Alloosh MA, Long X, Bratz IN, Peller CR, et al. Exercise training decreases store-operated Ca2+entry associated with metabolic syndrome and coronary atherosclerosis. Cardiovasc Res. (2010) 85:631–40. doi: 10.1093/cvr/cvp308
69. Varga-Szabo D, Braun A, Kleinschnitz C, Bender M, Pleines I, Pham M, et al. The calcium sensor STIM1 is an essential mediator of arterial thrombosis and ischemic brain infarction. J Exp Med. (2008) 205:1583–91. doi: 10.1084/jem.20080302
70. Giachini FR, Chiao CW, Carneiro FS, Lima VV, Carneiro ZN, Dorrance AM, et al. Increased activation of stromal interaction molecule-1/Orai-1 in aorta from hypertensive rats: a novel insight into vascular dysfunction. Hypertension. (2009) 53:409–16. doi: 10.1161/HYPERTENSIONAHA.108.124404
71. Kassan M, Ait-Aissa K, Radwan E, Mali V, Haddox S, Gabani M, et al. Essential role of smooth muscle STIM1 in hypertension and cardiovascular dysfunction. Arterioscler Thromb Vasc Biol. (2016) 36:1900–9. doi: 10.1161/ATVBAHA.116.307869
72. Hulot JS, Fauconnier J, Ramanujam D, Chaanine A, Aubart F, Sassi Y, et al. Critical role for stromal interaction molecule 1 in cardiac hypertrophy. Circulation. (2011) 124:796–805. doi: 10.1161/CIRCULATIONAHA.111.031229
73. Luo X, Hojayev B, Jiang N, Wang ZV, Tandan S, Rakalin A, et al. STIM1-dependent store-operated Ca2+ entry is required for pathological cardiac hypertrophy. J Mol Cell Cardiol. (2012) 52:136–47. doi: 10.1016/j.yjmcc.2011.11.003
74. Correll RN, Goonasekera SA, van Berlo JH, Burr AR, Accornero F, Zhang H, et al. STIM1 elevation in the heart results in aberrant Ca2+ handling and cardiomyopathy. J Mol Cell Cardiol. (2015) 87:38–47. doi: 10.1016/j.yjmcc.2015.07.032
75. Benard L, Oh JG, Cacheux M, Lee A, Nonnenmacher M, Matasic DS, et al. Cardiac stim1 silencing impairs adaptive hypertrophy and promotes heart failure through inactivation of mTORC2/Akt signaling. Circulation. (2016) 133:1458–71. doi: 10.1161/CIRCULATIONAHA.115.020678
76. Collins HE, Pat BM, Zou L, Litovsky SH, Wende AR, Young ME, et al. Novel role of the ER/SR Ca2+ sensor, STIM1, in regulation of cardiac metabolism. Am J Physiol Heart Circ Physiol. (2018) 316:H1014–26. doi: 10.1152/ajpheart.00544.2018
77. Mancarella S, Potireddy S, Wang Y, Gao H, Gandhirajan RK, Autieri M, et al. Targeted STIM deletion impairs calcium homeostasis, NFAT activation, and growth of smooth muscle. FASEB J. (2013) 27:893–906. doi: 10.1096/fj.12-215293
78. Lin S, Fagan KA, Li KX, Shaul PW, Cooper DM, Rodman DM. Sustained endothelial nitric-oxide synthase activation requires capacitative Ca2+ entry. J Biol Chem. (2000) 275:17979–85. doi: 10.1074/jbc.275.24.17979
79. Tran QK, Watanabe H. Calcium signalling in the endothelium. Handb Exp Pharmacol. (2006) 2006(176 Pt 1):145–87. doi: 10.1007/3-540-32967-6_5
80. Abdullaev IF, Bisaillon JM, Potier M, Gonzalez JC, Motiani RK, Trebak M. Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circ Res. (2008) 103:1289–99. doi: 10.1161/01.RES.0000338496.95579.56
81. Moore TM, Norwood NR, Creighton JR, Babal P, Brough GH, Shasby DM, et al. Receptor-dependent activation of store-operated calcium entry increases endothelial cell permeability. Am J Physiol Lung Cell Mol Physiol. (2000) 279:L691–8. doi: 10.1152/ajplung.2000.279.4.L691
82. Chetham PM, Babal P, Bridges JP, Moore TM, Stevens T. Segmental regulation of pulmonary vascular permeability by store-operated Ca2+ entry. Am J Physiol. (1999) 276:L41–50. doi: 10.1152/ajplung.1999.276.1.L41
83. Bers DM. Cardiac excitation-contraction coupling. Nature. (2002) 415:198–205. doi: 10.1038/415198a
84. Tykocki NR, Boerman EM, Jackson WF. Smooth muscle ion channels and regulation of vascular tone in resistance arteries and arterioles. Compr Physiol. (2017) 7:485–581. doi: 10.1002/cphy.c160011
85. Hofmann F, Flockerzi V, Kahl S, Wegener JW. L-type CaV1.2 calcium channels: from in vitro findings to in vivo function. Physiol Rev. (2014) 94:303–26. doi: 10.1152/physrev.00016.2013
86. Ertel EA, Campbell KP, Harpold MM, Hofmann F, Mori Y, Perez-Reyes E, et al. Nomenclature of voltage-gated calcium channels. Neuron. (2000) 25:533–5. doi: 10.1016/S0896-6273(00)81057-0
87. Wang X, Reznick S, Li P, Liang W, van Breemen C. Ca2+ removal mechanisms in freshly isolated rabbit aortic endothelial cells. Cell Calcium. (2002) 31:265–77. doi: 10.1016/S0143-4160(02)00075-1
88. Brini M, Carafoli E. Calcium pumps in health and disease. Physiol Rev. (2009) 89:1341–78. doi: 10.1152/physrev.00032.2008
89. Daugirdas JT, Arrieta J, Ye M, Flores G, Battle DC. Intracellular acidification associated with changes in free cytosolic calcium. Evidence for Ca2+/H+ exchange via a plasma membrane Ca2+-ATPase in vascular smooth muscle cells. J Clin Invest. (1995) 95:1480–89. doi: 10.1172/JCI117819
90. Boczek T, Lisek M, Ferenc B, Kowalski A, Stepinski D, Wiktorska M, et al. Plasma membrane Ca2+-ATPase isoforms composition regulates cellular pH homeostasis in differentiating PC12 cells in a manner dependent on cytosolic Ca2+ elevations. PLoS ONE. (2014) 9:e102352. doi: 10.1371/journal.pone.0102352
91. Strehler EE. The ATP2B plasma membrane Ca2+-ATPase family: regulation in response to changing demands of cellular calcium transport. In: Sajal Chakraborti NSD, editor. Regulation of Ca2+-ATPases,V-ATPases and F-ATPases (Springer) (2015). p. 63–80.
92. Di Leva F, Domi T, Fedrizzi L, Lim D, Carafoli E. The plasma membrane Ca2+ ATPase of animal cells: structure, function and regulation. Arch Biochem Biophys. (2008) 476:65–74. doi: 10.1016/j.abb.2008.02.026
93. Padanyi R, Paszty K, Strehler EE, Enyedi A. PSD-95 mediates membrane clustering of the human plasma membrane Ca2+ pump isoform 4b. Biochim Biophys Acta. (2009) 1793:1023–32. doi: 10.1016/j.bbamcr.2008.11.007
94. Dean WL, Chen D, Brandt PC, Vanaman TC. Regulation of platelet plasma membrane Ca2+-ATPase by cAMP-dependent and tyrosine phosphorylation. J Biol Chem. (1997) 272:15113–9. doi: 10.1074/jbc.272.24.15113
95. Borlak J, Thum T. Hallmarks of ion channel gene expression in end-stage heart failure. FASEB J. (2003) 17:1592–608. doi: 10.1096/fj.02-0889com
96. Sadi AM, Afroze T, Siraj MA, Momen A, White-Dzuro C, Zarrin-Khat D, et al. Cardiac-specific inducible overexpression of human plasma membrane Ca2+ ATPase 4b is cardioprotective and improves survival in mice following ischemic injury. Clin Sci. (2018) 132:641–54. doi: 10.1042/CS20171337
97. Schnitzer JE, Oh P, Jacobson BS, Dvorak AM. Caveolae from luminal plasmalemma of rat lung endothelium: microdomains enriched in caveolin, Ca2+-ATPase, and inositol trisphosphate receptor. Proc Natl Acad Sci USA. (1995) 92:1759–63. doi: 10.1073/pnas.92.5.1759
98. Fujimoto T. Calcium pump of the plasma membrane is localized in caveolae. J Cell Biol. (1993) 120:1147–57. doi: 10.1083/jcb.120.5.1147
99. Afroze T, Husain M. Cell cycle dependent regulation of intracellular calcium concentration in vascular smooth muscle cells: a potential target for drug therapy. Curr Drug Targets Cardiovasc Haematol Disord. (2001) 1:23–40. doi: 10.2174/1568006013338060
100. Husain M, Jiang L, See V, Bein K, Simons M, Alper SL, et al. Regulation of vascular smooth muscle cell proliferation by plasma membrane Ca2+-ATPase. Am J Physiol. (1997) 272(6 Pt 1):C1947–59. doi: 10.1152/ajpcell.1997.272.6.C1947
101. Abramowitz J, Aydemir-Koksoy A, Helgason T, Jemelka S, Odebunmi T, Seidel CL, et al. Expression of plasma membrane calcium ATPases in phenotypically distinct canine vascular smooth muscle cells. J Mol Cell Cardiol. (2000) 32:777–89. doi: 10.1006/jmcc.2000.1120
102. Afroze T, Yang G, Khoshbin A, Tanwir M, Tabish T, Momen A, et al. Calcium efflux activity of plasma membrane Ca2+ ATPase-4 (PMCA4) mediates cell cycle progression in vascular smooth muscle cells. J Biol Chem. (2014) 289:7221–31. doi: 10.1074/jbc.M113.533638
103. Strehler EE, Zacharias DA. Role of alternative splicing in generating isoform diversity among plasma membrane calcium pumps. Physiol Rev. (2001) 81:21–50. doi: 10.1152/physrev.2001.81.1.21
104. Sedova M, Blatter LA. Dynamic regulation of [Ca2+]i by plasma membrane Ca2+-ATPase and Na+/Ca2+ exchange during capacitative Ca2+ entry in bovine vascular endothelial cells. Cell Calcium. (1999) 25:333–43. doi: 10.1054/ceca.1999.0036
105. Shigekawa M, Iwamoto T. Cardiac Na+-Ca2+ exchange: molecular and pharmacological aspects. Circ Res. (2001) 88:864–76. doi: 10.1161/hh0901.090298
106. Nakasaki Y, Iwamoto T, Hanada H, Imagawa T, Shigekawa M. Cloning of the rat aortic smooth muscle Na+/Ca2+ exchanger and tissue-specific expression of isoforms. J Biochem. (1993) 114:528–34. doi: 10.1093/oxfordjournals.jbchem.a124211
107. Quednau BD, Nicoll DA, Philipson KD. Tissue specificity and alternative splicing of the Na+/Ca2+ exchanger isoforms NCX1, NCX2, and NCX3 in rat. Am J Physiol. (1997) 272(4 Pt 1):C1250–61. doi: 10.1152/ajpcell.1997.272.4.C1250
108. Sipido KR, Maes M, Van de Werf F. Low efficiency of Ca2+ entry through the Na+-Ca2+ exchanger as trigger for Ca2+ release from the sarcoplasmic reticulum. A comparison between L-type Ca2+ current and reverse-mode Na+-Ca2+ exchange. Circ Res. (1997) 81:1034–44. doi: 10.1161/01.RES.81.6.1034
109. Sham JS, Cleemann L, Morad M. Functional coupling of Ca2+ channels and ryanodine receptors in cardiac myocytes. Proc Natl Acad Sci USA. (1995) 92:121–5. doi: 10.1073/pnas.92.1.121
110. Litwin SE, Li J, Bridge JH. Na-Ca exchange and the trigger for sarcoplasmic reticulum Ca release: studies in adult rabbit ventricular myocytes. Biophys J. (1998) 75:359–71. doi: 10.1016/S0006-3495(98)77520-4
111. Lee CH, Poburko D, Sahota P, Sandhu J, Ruehlmann DO, van Breemen C. The mechanism of phenylephrine-mediated [Ca(2+)](i) oscillations underlying tonic contraction in the rabbit inferior vena cava. J Physiol. (2001) 534(Pt 3):641–50. doi: 10.1111/j.1469-7793.2001.t01-1-00641.x
112. Zhang J. New insights into the contribution of arterial NCX to the regulation of myogenic tone and blood pressure. Adv Exp Med Biol. (2013) 961:329–43. doi: 10.1007/978-1-4614-4756-6_28
113. Chu SH, Sutherland K, Beck J, Kowalski J, Goldspink P, Schwertz D. Sex differences in expression of calcium-handling proteins and beta-adrenergic receptors in rat heart ventricle. Life Sci. (2005) 76:2735–49. doi: 10.1016/j.lfs.2004.12.013
114. Chen J, Petranka J, Yamamura K, London RE, Steenbergen C, Murphy E. Gender differences in sarcoplasmic reticulum calcium loading after isoproterenol. Am J Physiol Heart Circ Physiol. (2003) 285:H2657–62. doi: 10.1152/ajpheart.00557.2003
115. Bowles DK, Maddali KK, Ganjam VK, Rubin LJ, Tharp DL, Turk JR, et al. Endogenous testosterone increases L-type Ca2+ channel expression in porcine coronary smooth muscle. Am J Physiol Heart Circ Physiol. (2004) 287:H2091–8. doi: 10.1152/ajpheart.00258.2004
116. Ma Y, Qiao X, Falone AE, Reslan OM, Sheppard SJ, Khalil RA. Gender-specific reduction in contraction is associated with increased estrogen receptor expression in single vascular smooth muscle cells of female rat. Cell Physiol Biochem. (2010) 26:457–70. doi: 10.1159/000320569
117. Levitan IB. It is calmodulin after all! mediator of the calcium modulation of multiple ion channels. Neuron. (1999) 22:645–8. doi: 10.1016/S0896-6273(00)80722-9
118. Shen X, Valencia CA, Szostak JW, Dong B, Liu R. Scanning the human proteome for calmodulin-binding proteins. Proc Natl Acad Sci USA. (2005) 102:5969–74. doi: 10.1073/pnas.0407928102
119. Jurado LA, Chockalingam PS, Jarrett HW. Apocalmodulin. Physiol Rev. (1999) 79:661–82. doi: 10.1152/physrev.1999.79.3.661
120. Persechini A, Kretsinger RH. The central helix of calmodulin functions as a flexible tether. J Biol Chem. (1988) 263:12175–8.
121. Persechini A, Blumenthal DK, Jarrett HW, Klee CB, Hardy DO, Kretsinger RH. The effects of deletions in the central helix of calmodulin on enzyme activation and peptide binding. J Biol Chem. (1989) 264:8052–8.
122. Kakiuchi S, Yasuda S, Yamazaki R, Teshima Y, Kanda K, Kakiuchi R, et al. Quantitative determinations of calmodulin in the supernatant and particulate fractions of mammalian tissues. J Biochem. (1982) 92:1041–8. doi: 10.1093/oxfordjournals.jbchem.a134019
123. Persechini A, Stemmer PM. Calmodulin is a limiting factor in the cell. Trends Cardiovasc Med. (2002) 12:32–37. doi: 10.1016/S1050-1738(01)00144-X
124. Tran QK, Black DJ, Persechini A. Intracellular coupling via limiting calmodulin. J Biol Chem. (2003) 278:24247–50. doi: 10.1074/jbc.C300165200
125. Luby-Phelps K, Hori M, Phelps JM, Won D. Ca2+-regulated dynamic compartmentalization of calmodulin in living smooth muscle cells. J Biol Chem. (1995) 270:21532–8. doi: 10.1074/jbc.270.37.21532
126. Wu X, Bers DM. Free and bound intracellular calmodulin measurements in cardiac myocytes. Cell Calcium. (2007) 41:353–64. doi: 10.1016/j.ceca.2006.07.011
127. Tran QK, Black DJ, Persechini A. Dominant affectors in the calmodulin network shape the time courses of target responses in the cell. Cell Calcium. (2005) 37:541–53. doi: 10.1016/j.ceca.2005.02.001
128. Walter P, Green S, Greene G, Krust A, Bornert JM, Jeltsch JM, et al. Cloning of the human estrogen receptor cDNA. Proc Natl Acad Sci USA. (1985) 82:7889–93. doi: 10.1073/pnas.82.23.7889
129. Greene GL, Gilna P, Waterfield M, Baker A, Hort Y, Shine J. Sequence and expression of human estrogen receptor complementary DNA. Science. (1986) 231:1150–4. doi: 10.1126/science.3753802
130. Green S, Walter P, Kumar V, Krust A, Bornert JM, Argos P, et al. Human oestrogen receptor cDNA: sequence, expression and homology to v-erb-A. Nature. (1986) 320:134–9. doi: 10.1038/320134a0
131. Evans RM. The steroid and thyroid hormone receptor superfamily. Science. (1988) 240:889–95. doi: 10.1126/science.3283939
132. Groban L, Tran QK, Ferrario CM, Sun X, Cheng CP, Kitzman DW, et al. Female heart health: is GPER the missing link? Front Endocrinol. (2020) 10:919. doi: 10.3389/fendo.2019.00919
133. Haas E, Meyer MR, Schurr U, Bhattacharya I, Minotti R, Nguyen HH, et al. Differential effects of 17beta-estradiol on function and expression of estrogen receptor alpha, estrogen receptor beta, and GPR30 in arteries and veins of patients with atherosclerosis. Hypertension. (2007) 49:1358–63. doi: 10.1161/HYPERTENSIONAHA.107.089995
134. Ding Q, Gros R, Limbird LE, Chorazyczewski J, Feldman RD. Estradiol-mediated ERK phosphorylation and apoptosis in vascular smooth muscle cells requires GPR 30. Am J Physiol Cell Physiol. (2009) 297:C1178–87. doi: 10.1152/ajpcell.00185.2009
135. Tran QK, VerMeer M, Burgard MA, Hassan AB, Giles J. Hetero-oligomeric complex between the G protein-coupled estrogen receptor 1 and the plasma membrane Ca2+-ATPase 4b. J Biol Chem. (2015) 290:13293–307. doi: 10.1074/jbc.M114.628743
136. Ding Q, Hussain Y, Chorazyczewski J, Gros R, Feldman RD. GPER-independent effects of estrogen in rat aortic vascular endothelial cells. Mol Cell Endocrinol. (2014) 399:60–8. doi: 10.1016/j.mce.2014.07.023
137. Castoria G, Migliaccio A, Nola E, Auricchio F. In vitro interaction of estradiol receptor with Ca2+-calmodulin. Mol Endocrinol. (1988) 2:167–74. doi: 10.1210/mend-2-2-167
138. Bouhoute A, Leclercq G. Modulation of estradiol and DNA binding to estrogen receptor upon association with calmodulin. Biochem Biophys Res Commun. (1995) 208:748–55. doi: 10.1006/bbrc.1995.1401
139. Bouhoute A, Leclercq G. Calmodulin decreases the estrogen binding capacity of the estrogen receptor. Biochem Biophys Res Commun. (1996) 227:651–7. doi: 10.1006/bbrc.1996.1564
140. Li L, Li Z, Sacks DB. The transcriptional activity of estrogen receptor-alpha is dependent on Ca2+/calmodulin. J Biol Chem. (2005) 280:13097–104. doi: 10.1074/jbc.M410642200
141. Carlier L, Byrne C, Miclet E, Bourgoin-Voillard S, Nicaise M, Tabet JC, et al. Biophysical studies of the interaction between calmodulin and the R287-T311 region of human estrogen receptor alpha reveals an atypical binding process. Biochem Biophys Res Commun. (2012) 419:356–61. doi: 10.1016/j.bbrc.2012.02.028
142. Li L, Li Z, Sacks DB. Calmodulin regulates the transcriptional activity of estrogen receptors. Selective inhibition of calmodulin function in subcellular compartments. J Biol Chem. (2003) 278:1195–200. doi: 10.1074/jbc.M210708200
143. Zhang Y, Li Z, Sacks DB, Ames JB. Structural basis for Ca2+-induced activation and dimerization of estrogen receptor alpha by calmodulin. J Biol Chem. (2012) 287:9336–44. doi: 10.1074/jbc.M111.334797
144. Gallo D, Jacquemotte F, Cleeren A, Laios I, Hadiy S, Rowlands MG, et al. Calmodulin-independent, agonistic properties of a peptide containing the calmodulin binding site of estrogen receptor alpha. Mol Cell Endocrinol. (2007) 268:37–49. doi: 10.1016/j.mce.2007.01.012
145. Mosselman S, Polman J, Dijkema R. ER beta: identification and characterization of a novel human estrogen receptor. FEBS Lett. (1996) 392:49–53. doi: 10.1016/0014-5793(96)00782-X
146. Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA. Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci USA. (1996) 93:5925–30. doi: 10.1073/pnas.93.12.5925
147. Cowley SM, Hoare S, Mosselman S, Parker MG. Estrogen receptors alpha and beta form heterodimers on DNA. J Biol Chem. (1997) 272:19858–62. doi: 10.1074/jbc.272.32.19858
148. Forster C, Kietz S, Hultenby K, Warner M, Gustafsson JA. Characterization of the ERbeta-/-mouse heart. Proc Natl Acad Sci USA. (2004) 101:14234–9. doi: 10.1073/pnas.0405571101
149. Garcia Pedrero JM, Del Rio B, Martinez-Campa C, Muramatsu M, Lazo PS, Ramos S. Calmodulin is a selective modulator of estrogen receptors. Mol Endocrinol. (2002) 16:947–60. doi: 10.1210/mend.16.5.0830
150. Prossnitz ER, Arterburn JB. International union of basic and clinical pharmacology. XCVII. G protein-coupled estrogen receptor and its pharmacologic modulators. Pharmacol Rev. (2015) 67:505–40. doi: 10.1124/pr.114.009712
151. Owman C, Blay P, Nilsson C, Lolait SJ. Cloning of human cDNA encoding a novel heptahelix receptor expressed in burkitt's lymphoma and widely distributed in brain and peripheral tissues. Biochem Biophys Res Commun. (1996) 228:285–92. doi: 10.1006/bbrc.1996.1654
152. Feng Y, Gregor P. Cloning of a novel member of the G protein-coupled receptor family related to peptide receptors. Biochem Biophys Res Commun. (1997) 231:651–4 doi: 10.1006/bbrc.1997.6161
153. Kvingedal AM, Smeland EB. A novel putative G-protein-coupled receptor expressed in lung, heart and lymphoid tissue. FEBS Lett. (1997) 407:59–62. doi: 10.1016/S0014-5793(97)00278-0
154. Takada Y, Kato C, Kondo S, Korenaga R, Ando J. Cloning of cDNAs encoding G protein-coupled receptor expressed in human endothelial cells exposed to fluid shear stress. Biochem Biophys Res Commun. (1997) 240:737–41. doi: 10.1006/bbrc.1997.7734
155. Carmeci C, Thompson DA, Ring HZ, Francke U, Weigel RJ. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics. (1997) 45:607–17. doi: 10.1006/geno.1997.4972
156. O'Dowd BF, Nguyen T, Marchese A, Cheng R, Lynch KR, Heng HH, et al. Discovery of three novel G-protein-coupled receptor genes. Genomics. (1998) 47:310–13. doi: 10.1006/geno.1998.5095
157. Filardo EJ, Quinn JA, Bland KI, Frackelton AR Jr. Estrogen-induced activation of Erk-1 Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol. (2000) 14:1649–60. doi: 10.1210/mend.14.10.0532
158. Filardo EJ, Quinn JA, Frackelton AR Jr, Bland KI. Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol Endocrinol. (2002) 16:70–84. doi: 10.1210/mend.16.1.0758
159. Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology. (2005) 146:624–32. doi: 10.1210/en.2004-1064
160. Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. (2005) 307:1625–30. doi: 10.1126/science.1106943
161. Barton M, Filardo EJ, Lolait SJ, Thomas P, Maggiolini M, Prossnitz ER. Twenty years of the G protein-coupled estrogen receptor GPER: historical and personal perspectives. J Steroid Biochem Mol Biol. (2018) 176:4–15. doi: 10.1016/j.jsbmb.2017.03.021
162. Filardo E, Quinn J, Pang Y, Graeber C, Shaw S, Dong J, et al. Activation of the novel estrogen receptor G protein-coupled receptor 30 (GPR30) at the plasma membrane. Endocrinology. (2007) 148:3236–45. doi: 10.1210/en.2006-1605
163. Maggiolini M, Vivacqua A, Fasanella G, Recchia AG, Sisci D, Pezzi V, et al. The G protein-coupled receptor GPR30 mediates c-fos up-regulation by 17beta-estradiol and phytoestrogens in breast cancer cells. J Biol Chem. (2004) 279:27008–16. doi: 10.1074/jbc.M403588200
164. Rubio-Gayosso I, Sierra-Ramirez A, Garcia-Vazquez A, Martinez-Martinez A, Munoz-Garcia O, Morato T, et al. 17Beta-estradiol increases intracellular calcium concentration through a short-term and nongenomic mechanism in rat vascular endothelium in culture. J Cardiovasc Pharmacol. (2000). 36:196–202. doi: 10.1097/00005344-200008000-00009
165. Revankar CM, Mitchell HD, Field AS, Burai R, Corona C, Ramesh C, et al. Synthetic estrogen derivatives demonstrate the functionality of intracellular GPR30. ACS Chem Biol. (2007) 2:536–44. doi: 10.1021/cb700072n
166. Akama KT, Thompson LI, Milner TA, McEwen BS. Post-synaptic density-95 (PSD-95) binding capacity of G-protein-coupled receptor 30 (GPR30), an estrogen receptor that can be identified in hippocampal dendritic spines. J Biol Chem. (2013) 288:6438–50. doi: 10.1074/jbc.M112.412478
167. Broselid S, Berg KA, Chavera TA, Kahn R, Clarke WP, Olde B, et al. G Protein-coupled receptor 30 (GPR30) forms a plasma membrane complex with membrane-associated guanylate kinases (MAGUKs) and AKAP5 that constitutively inhibits cAMP production. J Biol Chem. (2014) 289:22117–27. doi: 10.1074/jbc.M114.566893
168. Cheng SB, Quinn JA, Graeber CT, Filardo EJ. Downmodulation of the G-protein-coupled estrogen receptor, GPER, from the cell surface occurs via a transgolgi-proteasome pathway. J Biol Chem. (2011) 286:22441–55. doi: 10.1074/jbc.M111.224071
169. Tran QK, VerMeer M. Biosensor-based approach identifies four distinct calmodulin-binding domains in the G protein-coupled estrogen receptor 1. PLoS ONE. (2014) 9:e89669 doi: 10.1371/journal.pone.0089669
170. Tran QK, Firkins R, Giles J, Francis S, Matnishian V, Tran P, et al. Estrogen enhances linkage in the vascular endothelial calmodulin network via a feedforward mechanism at the G protein-coupled estrogen receptor 1. J Biol Chem. (2016) 291:10805–23. doi: 10.1074/jbc.M115.697334
171. Buitrago C, Massheimer V, de Boland AR. Acute modulation of Ca2+ influx on rat heart by 17beta-estradiol. Cell Signal. (2000) 12:47–52. doi: 10.1016/S0898-6568(99)00066-2
172. Haas E, Bhattacharya I, Brailoiu E, Damjanovic M, Brailoiu GC, Gao X, et al. Regulatory role of G protein-coupled estrogen receptor for vascular function and obesity. Circ Res. (2009) 104:288–91. doi: 10.1161/CIRCRESAHA.108.190892
173. Improta-Brears T, Whorton AR, Codazzi F, York JD, Meyer T, McDonnell DP. Estrogen-induced activation of mitogen-activated protein kinase requires mobilization of intracellular calcium. Proc Natl Acad Sci USA. (1999) 96:4686–91. doi: 10.1073/pnas.96.8.4686
174. Moini H, Bilsel S, Bekdemir T, Emerk K. 17 beta-Estradiol increases intracellular free calcium concentrations of human vascular endothelial cells and modulates its responses to acetylcholine. Endothelium. (1997) 5:11–19. doi: 10.3109/10623329709044155
175. Bologa CG, Revankar CM, Young SM, Edwards BS, Arterburn JB, Kiselyov AS, et al. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat Chem Biol. (2006) 2:207–12. doi: 10.1038/nchembio775
176. Hofmeister MV, Damkier HH, Christensen BM, Olde B, Fredrik Leeb-Lundberg LM, Fenton RA, et al. 17beta-Estradiol induces nongenomic effects in renal intercalated cells through G protein-coupled estrogen receptor 1. Am J Physiol Renal Physiol. (2012) 302:F358–68. doi: 10.1152/ajprenal.00343.2011
177. Sarkar SN, Huang RQ, Logan SM, Yi KD, Dillon GH, Simpkins JW. Estrogens directly potentiate neuronal L-type Ca2+ channels. Proc Natl Acad Sci USA. (2008) 105:15148–53. doi: 10.1073/pnas.0802379105
178. Yatani A, Codina J, Imoto Y, Reeves JP, Birnbaumer L, Brown AM. A G protein directly regulates mammalian cardiac calcium channels. Science. (1987) 238:1288–92. doi: 10.1126/science.2446390
179. Rosenthal W, Hescheler J, Trautwein W, Schultz G. Receptor- and G-protein-mediated modulations of voltage-dependent calcium channels. Cold Spring Harb Symposia Quanti Biol. (1988) 53(Pt 1):247–54. doi: 10.1101/SQB.1988.053.01.031
180. Rosenthal W, Hescheler J, Trautwein W, Schultz G. Control of voltage-dependent Ca2+ channels by G protein-coupled receptors. FASEB J. (1988) 2:2784–90. doi: 10.1096/fasebj.2.12.2457531
181. Hescheler J, Rosenthal W, Hinsch KD, Wulfern M, Trautwein W, Schultz G. Angiotensin II-induced stimulation of voltage-dependent Ca2+ currents in an adrenal cortical cell line. EMBO J. (1988) 7:619–24. doi: 10.1002/j.1460-2075.1988.tb02855.x
182. Rosenthal W, Hescheler J, Hinsch KD, Spicher K, Trautwein W, Schultz G. Cyclic AMP-independent, dual regulation of voltage-dependent Ca2+ currents by LHRH and somatostatin in a pituitary cell line. EMBO J. (1988) 7:1627–33. doi: 10.1002/j.1460-2075.1988.tb02989.x
183. Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. (2002) 3:639–50. doi: 10.1038/nrm908
184. Camps M, Carozzi A, Schnabel P, Scheer A, Parker PJ, Gierschik P. Isozyme-selective stimulation of phospholipase C-beta 2 by G protein beta gamma-subunits. Nature. (1992) 360:684–6. doi: 10.1038/360684a0
185. Boyer JL, Waldo GL, Harden TK. Beta gamma-subunit activation of G-protein-regulated phospholipase C. J Biol Chem. (1992) 267:25451–6.
186. Zeng W, Mak DO, Li Q, Shin DM, Foskett JK, Muallem S. A new mode of Ca2+ signaling by G protein-coupled receptors: gating of IP3 receptor Ca2+ release channels by Gbetagamma. Curr Biol. (2003) 13:872–6. doi: 10.1016/S0960-9822(03)00330-0
187. Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature. (1997) 386:855–8. doi: 10.1038/386855a0
188. Santolla MF, Lappano R, De Marco P, Pupo M, Vivacqua A, Sisci D, et al. G protein-coupled estrogen receptor mediates the up-regulation of fatty acid synthase induced by 17beta-estradiol in cancer cells and cancer-associated fibroblasts. J Biol Chem. (2012) 287:43234–45. doi: 10.1074/jbc.M112.417303
189. Tran QK, Leonard J, Black DJ, Nadeau OW, Boulatnikov IG, Persechini A. Effects of combined phosphorylation at Ser-617 and Ser-1179 in endothelial nitric-oxide synthase on EC50(Ca2+) values for calmodulin binding and enzyme activation. J Biol Chem. (2009) 284:11892–9. doi: 10.1074/jbc.M806205200
190. Tran QK, Leonard J, Black DJ, Persechini A. Phosphorylation within an autoinhibitory domain in endothelial nitric oxide synthase reduces the Ca2+ concentrations required for calmodulin to bind and activate the enzyme. Biochemistry. (2008) 47:7557–66. doi: 10.1021/bi8003186
191. Word RA, Tang DC, Kamm KE. Activation properties of myosin light chain kinase during contraction/relaxation cycles of tonic and phasic smooth muscles. J Biol Chem. (1994) 269:21596–602.
192. Meyer MR, Baretella O, Prossnitz ER, Barton M. Dilation of epicardial coronary arteries by the G protein-coupled estrogen receptor agonists G-1 and ICI 182,780. Pharmacology. (2010) 86:58–64. doi: 10.1159/000315497
193. Lindsey SH, Liu L, Chappell MC. Vasodilation by GPER in mesenteric arteries involves both endothelial nitric oxide and smooth muscle cAMP signaling. Steroids. (2014) 81:99–102. doi: 10.1016/j.steroids.2013.10.017
194. Jang EJ, Seok YM, Arterburn JB, Olatunji LA, Kim IK. GPER-1 agonist G1 induces vasorelaxation through activation of epidermal growth factor receptor-dependent signalling pathway. J Pharm Pharmacol. (2013) 65:1488–99. doi: 10.1111/jphp.12113
195. Filice E, Recchia AG, Pellegrino D, Angelone T, Maggiolini M, Cerra MC. A new membrane G protein-coupled receptor (GPR30) is involved in the cardiac effects of 17beta-estradiol in the male rat. J Physiol Pharmacol. (2009) 60:3–10.
196. Arefin S, Simoncini T, Wieland R, Hammarqvist F, Spina S, Goglia L, et al. Vasodilatory effects of the selective GPER agonist G-1 is maximal in arteries of postmenopausal women. Maturitas. (2014) 78:123–30. doi: 10.1016/j.maturitas.2014.04.002
197. Meyer MR, Fredette NC, Howard TA, Hu C, Ramesh C, Daniel C, et al. G protein-coupled estrogen receptor protects from atherosclerosis. Sci Rep. (2014) 4:7564. doi: 10.1038/srep07564
198. Fredette NC, Meyer MR, Prossnitz ER. Role of GPER in estrogen-dependent nitric oxide formation and vasodilation. J Steroid Biochem Mol Biol. (2017) 176:65–72. doi: 10.1016/j.jsbmb.2017.05.006
199. Holm A, Hellstrand P, Olde B, Svensson D, Leeb-Lundberg LM, Nilsson BO. The G protein-coupled estrogen receptor 1 (GPER1/GPR30) agonist G-1 regulates vascular smooth muscle cell Ca2+ handling. J Vasc Res. (2013) 50:421–9. doi: 10.1159/000354252
200. Yu X, Ma H, Barman SA, Liu AT, Sellers M, Stallone JN, et al. Activation of G protein-coupled estrogen receptor induces endothelium-independent relaxation of coronary artery smooth muscle. Am J Physiol. (2011) 301:E882–8. doi: 10.1152/ajpendo.00037.2011
201. Gonzales RJ, Kanagy NL. Endothelium-independent relaxation of vascular smooth muscle by 17beta-estradiol. J Cardiovasc Pharmacol Ther. (1999) 4:227–34. doi: 10.1177/107424849900400404
202. Kitazawa T, Hamada E, Kitazawa K, Gaznabi AK. Non-genomic mechanism of 17 beta-oestradiol-induced inhibition of contraction in mammalian vascular smooth muscle. J Physiol. (1997) 499(Pt 2):497–511. doi: 10.1113/jphysiol.1997.sp021944
203. Terry LE, VerMeer M, Giles J, Tran QK. Suppression of store-operated Ca2+ entry by activation of GPER: contribution to a clamping effect on endothelial Ca2+ signaling. Biochem J. (2017) 474:3627–42. doi: 10.1042/BCJ20170630
204. Sheridan JT, Gilmore RC, Watson MJ, Archer CB, Tarran R. 17beta-estradiol inhibits phosphorylation of stromal interaction molecule 1 (STIM1) protein: implication for store-operated calcium entry and chronic lung diseases. J Biol Chem. (2013) 288:33509–18. doi: 10.1074/jbc.M113.486662
205. Pekas D, Gebert-Oberle B, Giles J, Underwood T, VerMeer M, Tran K, et al. Regulation of the Ca2+-sensing capacity of STIM1 by the G protein-coupled estrogen receptor. Faseb J. (2019) 33:810.4. doi: 10.1096/fasebj.2019.33.1_supplement.810.4
206. Curl CL, Wendt IR, Canny BJ, Kotsanas G. Effects of ovariectomy and 17 beta-oestradiol replacement on [Ca2+]i in female rat cardiac myocytes. Clin Exp Pharmacol Physiol. (2003) 30:489–94. doi: 10.1046/j.1440-1681.2003.03864.x
207. Kam KW, Kravtsov GM, Liu J, Wong TM. Increased PKA activity and its influence on isoprenaline-stimulated L-type Ca2+ channels in the heart from ovariectomized rats. Br J Pharmacol. (2005) 144:972–81. doi: 10.1038/sj.bjp.0706123
208. Kravtsov GM, Kam KW, Liu J, Wu S, Wong TM. Altered Ca2+ handling by ryanodine receptor and Na+-Ca2+ exchange in the heart from ovariectomized rats: role of protein kinase A. Am J Physiol Cell Physiol. (2007) 292:C1625–35. doi: 10.1152/ajpcell.00368.2006
209. Nakajima T, Kitazawa T, Hamada E, Hazama H, Omata M, Kurachi Y. 17βa-estradiol inhibits the voltage-dependent L-type Ca2+ currents in aortic smooth muscle cells. Eur J Pharmacol. (1995) 294:625–35. doi: 10.1016/0014-2999(95)00602-8
210. Zhang F, Ram JL, Standley PR, Sowers JR. 17β-estradiol attenuates voltage-dependent Ca2+ currents in A7r5 vascular smooth muscle cell line. Am J Physiol. (1994) 266(4 Pt 1):C975–80. doi: 10.1152/ajpcell.1994.266.4.C975
211. Dennis MK, Burai R, Ramesh C, Petrie WK, Alcon SN, Nayak TK, et al. In vivo effects of a GPR30 antagonist. Nat Chem Biol. (2009) 5:421–7. doi: 10.1038/nchembio.168
212. Freay AD, Curtis SW, Korach KS, Rubanyi GM. Mechanism of vascular smooth muscle relaxation by estrogen in depolarized rat and mouse aorta. Role of nuclear estrogen receptor and Ca2+ uptake. Circ Res. (1997) 81:242–8. doi: 10.1161/01.RES.81.2.242
213. Whitcomb V, Wauson E, Christian D, Clayton S, Giles J, Tran QK. Regulation of beta adrenoceptor-mediated myocardial contraction and calcium dynamics by the G protein-coupled estrogen receptor 1. Biochem Pharmacol. (2020) 171:113727. doi: 10.1016/j.bcp.2019.113727
214. Dodds ML, Kargacin ME, Kargacin GJ. Effects of anti-oestrogens and beta-estradiol on calcium uptake by cardiac sarcoplasmic reticulum. Br J Pharmacol. (2001) 132:1374–82. doi: 10.1038/sj.bjp.0703924
215. Bupha-Intr T, Wattanapermpool J. Regulatory role of ovarian sex hormones in calcium uptake activity of cardiac sarcoplasmic reticulum. Am J Physiol Heart Circ Physiol. (2006) 291:H1101–8. doi: 10.1152/ajpheart.00660.2005
216. Ma Y, Cheng WT, Wu S, Wong TM. Oestrogen confers cardioprotection by suppressing Ca2+/calmodulin-dependent protein kinase II. Br J Pharmacol. (2009) 157:705–15. doi: 10.1111/j.1476-5381.2009.00212.x
217. Sugishita K, Su Z, Li F, Philipson KD, Barry WH. Gender influences [Ca(2+)](i) during metabolic inhibition in myocytes overexpressing the Na+-Ca2+ exchanger. Circulation. (2001) 104:2101–6. doi: 10.1161/hc4001.097038
218. Batra S, Bengtsson LP. Inhibition by diethyl stilbestrol of calcium uptake by human myometrial mitochondria. Eur J Pharmacol. (1972) 18:281–3. doi: 10.1016/0014-2999(72)90254-3
219. Kabir ME, Singh H, Lu R, Olde B, Leeb-Lundberg LM, Bopassa JC. G protein-coupled estrogen receptor 1 mediates acute estrogen-induced cardioprotection via MEK/ERK/GSK-3beta pathway after ischemia/reperfusion. PLoS ONE. (2015) 10:e0135988. doi: 10.1371/journal.pone.0135988
220. Morkuniene R, Jekabsone A, Borutaite V. Estrogens prevent calcium-induced release of cytochrome c from heart mitochondria. FEBS Lett. (2002) 521:53–6. doi: 10.1016/S0014-5793(02)02820-X
221. Suman M, Giacomello M, Corain L, Ballarin C, Montelli S, Cozzi B, et al. Estradiol effects on intracellular Ca2+ homeostasis in bovine brain-derived endothelial cells. Cell Tissue Res. (2012) 350:109–18. doi: 10.1007/s00441-012-1460-2
222. Bredt DS, Snyder SH. Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc Natl Acad Sci USA. (1990) 87:682–5. doi: 10.1073/pnas.87.2.682
Keywords: estrogen, G protein—coupled estrogen receptor, calcium, calmodulin, calmodulin-binding proteins, cardiomyocytes, vascular smooth muscle, endothelium
Citation: Tran Q-K (2020) Reciprocality Between Estrogen Biology and Calcium Signaling in the Cardiovascular System. Front. Endocrinol. 11:568203. doi: 10.3389/fendo.2020.568203
Received: 31 May 2020; Accepted: 19 August 2020;
Published: 29 September 2020.
Edited by:
Yves Jacquot, Faculté de pharmacie de Paris, Université Paris Descartes, FranceReviewed by:
Nandini Vasudevan, University of Reading, United KingdomCatarina Segreti Porto, Federal University of São Paulo, Brazil
Coralie Fontaine, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Copyright © 2020 Tran. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Quang-Kim Tran, kim.tran@dmu.edu