Anthracycline-induced cardiotoxicity: mechanisms, monitoring, and prevention
 Yun Qiu1
Yun Qiu1  Yingmei Huang
Yingmei Huang- 1Department of Cardiology, Jiangxi Provincial People’s Hospital, The First Affiliated Hospital of Nanchang Medical College, Nanchang, China
- 2Department of Oncology, Jiangxi Provincial People’s Hospital, The First Affiliated Hospital of Nanchang Medical College, Nanchang, China
- 3The First Clinical Medical College, Nanchang University, Nanchang, China
Anthracyclines are the most fundamental and important treatment of several cancers especially for lymphoma and breast cancer. However, their use is limited by a dose-dependent cardiotoxicity which may emerge early at the initiation of anthracycline administration or several years after termination of the therapy. A full comprehending of the mechanisms of anthracycline-induced cardiotoxicity, which has not been achieved and is currently under the efforts, is critical to the advance of developing effective methods to protect against the cardiotoxicity, as well as to early detect and treat it. Therefore, we review the recent progress of the mechanism underlying anthracycline-induced cardiotoxicity, as well as approaches to monitor and prevent this issue.
1. Introduction
Anthracyclines are one of the most commonly used chemotherapies. Since found in the 1950s, anthracyclines have become the footing stone of chemotherapies in various cancers, especially in breast cancer and lymphoma. Even though anthracyclines improve long-term survival from cancers, it comes accompanied by increased incidence of cardiovascular complications. Since 1971, a dose-dependent cardiotoxicity induced by anthracyclines has been found (1). Indeed, anthracyclines are the major reason of chemotherapy-mediated cardiotoxicity. Clinically, cardiotoxicity can be asymptomatic or manifest diverse symptoms like arrhythmia, declined systolic function, and myocarditis, which can arise promptly upon anthracycline treatment, or arise after several years. In some cases, cardiotoxicity can even impact the option of cancer therapies (2, 3). When the declined heart functionalities or other evident symptoms arise, patients may be limited to choose ineffective therapies, or the theraputic strategy can even be suspended.
Anthracycline-induced cardiotoxicity refers to a series of cardiovascular complications with no definite definition, as a result, its incidence is different on the basis of its definition. Congestive heart failure (CHF) is found in 2%–4%, subclinical structural alterations in about 10%, arrhythmia in over 12%, and cardiac biomarker elevation in 30%–35% of patients (4). A prospective trial including 2,625 patients with an average follow-up of 5.2 years showed a 9% rate of occurrence of anthracycline-mediated cardiotoxicity (5). In this study, anthracycline-mediated cardiotoxicity was defined as left ventricular ejection fraction (LVEF) <50% and a reduction of >10 percentage. Besides, it's reported that childhood cancer survivors showed a 15-fold augmented risk of CHF as well as a 7-fold augmented risk of premature death (6). It's well validated that anthracycline-induced cardiotoxicity is dose-dependent. Bernstein et al. (7) showed that left ventricular (LV) dysfunction which represented a decrease in ejection fraction of >10% lower than normal, emerged in 10%, 16%, 32%, and 65% at total doxorubicin doses of 250, 300, 400, and 550 mg/m2, separately. Therefore, even at the lowest dose, anthracyclines are capable of causing remarkable LV dysfunction.
Unfortunately, there are no definite guidelines for screening, monitoring, preventing or treating cardiotoxicity in patients receiving anthracycline administration. Also, comprehending the mechanisms of anthracycline-induced cardiotoxicity is critical to the advance of effective methods to protect against the cardiotoxicity, as well as to early detect and treat it. Therefore, we review the recent progress of the mechanism underlying anthracycline-induced cardiotoxicity, as well as approaches to monitor and prevent this issue.
2. Anthracycline-induced cardiotoxicity
It was not until 1967 that the cardiotoxic effects of anthracyclines were identified, following observations by Tan et al. linking them to CHF (8). The cardiotoxicity associated with anthracyclines originally manifest as subclinical cardiomyocyte injuries and gradually lead to early asymptomatic decline of LVEF and eventually symptomatic, and usually intractable, heart failure (HF). Currently, no formal definition of anthracycline-induced cardiotoxicity is achieved. Usually, cardiotoxicity refers to existence of symptoms of HF or asymptomatic reduction of LVEF ≥10% to eventual LVEF <55%.
Generally, anthracycline-induced cardiotoxicity may be categorized into three types: acute cardiotoxicity, early-onset chronic cardiotoxicity, and late-onset chronic cardiotoxicity (9, 10). Acute cardiotoxicity, which occurs in only 1% of patients and typically manifests within the first few days following anthracycline administration, is characterized by supraventricular arrhythmia, transient LV dysfunction, and electrocardiographic alterations (11). Although acute cardiotoxicity manifestations are generally reversible, they are possible to progress into chronic cardiotoxicity. Unfortunately, there are currently no valid approaches for predicting the cardiotoxicity progression.
Early-onset chronic cardiotoxicity, which is the most common type of anthracycline-induced cardiotoxicity and typically manifests within the first year subsequent to termination of anthracycline therapies, is featured by LV dysfunction, reduced LVEF as well as symptomatic HF in about 5% of patients (12).
As for late-onset chronic cardiotoxicity, which is typically irreversible, it may occur a few years or decades after anthracycline therapies (13). This toxicity has been considered as an advanced outcome of early-onset cardiotoxicity with superimposed cardiac injury, primarily manifesting as cardiomyopathy, which features cytoplasmic vacuolization of cardiomyocytes due to sarcoplasmic and mitochondrial swelling, organelle destruction, cardiomyocyte death, as well as muscle fiber disorder (14). These alterations result in an irreversible decrease of LVEF in approximately 26% of patients and symptomatic HF in over 7% of patients (9, 15).
Interestingly, Cardinale et al. questioned the current classification of anthracycline-induced chronic cardiotoxicity which regards early-onset and late-onset chronic cardiotoxicity as two different entities on the basis of retrospective studies in which LVEF decrease was tested either after HF development, or on random assessment in pediatric cancer patients presumed to have no other cardiac complications. Nevertheless, Cardinale et al. demonstrated that HF may be preceded by asymptomatic LVEF reduction featured by early onset with a slow and gradual deterioration that may sustain months or years after the termination of anthracycline treatment (5).
3. Mechanism of anthracyclines-induced cardiotoxicity
Although the mechanism underlying cardiotoxicity caused by anthracyclines is not well comprehended, proposed mechanisms include oxidative stress triggered by various molecular mechanisms, alteration of cell death pathways, and alteration of epigenetics (as shown in Figure 1).
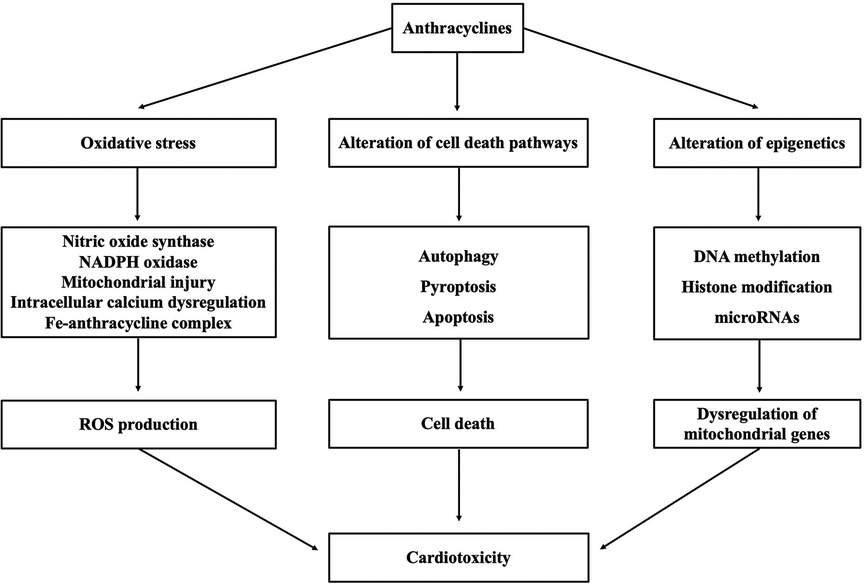
Figure 1. Summary of mechanisms underlying anthracycline-induced cardiotoxicity.
3.1. Oxidative stress
Oxidative stress, caused by the imbalance between reactive oxygen species (ROS) and antioxidants, is the leading concern of anthracycline-induced cardiac muscle cell injuries (16). A certain levels of oxidant species are essential for normal signal transduction process; however, excessive oxidant species have been proven to be related to loads of pathological conditions. As reported, an evident growth of oxidative stress is observed when the total dose of doxorubicin surpasses 500 mg/m2 (17). The molecular mechanisms triggering oxidative stress and its damaging effects on heart tissue are concluded as below.
3.1.1. Effect of nitric oxide synthase on ROS production
Nitric oxide (NO), generated by catalyzation of nitric oxide synthase (NOS), is a primary reason of anthracycline-mediated oxidative stress. And NO levels have been proved elevated in cardiotoxicity associated with anthracycline administration. There are three types of NOS, namely endothelial NOS (eNOS), inducible NOS (iNOS) and neuronal NOS (nNOS), with the first two playing major roles in cardiac NO synthesis. And an upregulated level of iNOS and eNOS has been observed in cardiomyocytes treated with anthracyclines.
Anthracyclines, by binding to the eNOS reductase, boost semiquinone radical generation which reduces the free oxygen into the superoxide (O2–) free radical. An enzymatic single-electron reduction reaction is accomplished with the assistance of flavoenzymes including NADPH-cytochrome P450 reductase and mitochondrial NADH dehydrogenase (18). An imbalance between superoxide free radicals and NO levels is produced after anthracyclines bind to the eNOS reductase. ROS increase the formation of oxidants and afterwards compel the uncoupling of eNOS into monomers, thus promoting the generation of increased superoxide anions as well as declined NO which finally results in cardiotoxicity (19). Neilan et al. verified the important effect of eNOS on doxorubicin-mediated oxidative stress by eNOS knockout mice which exhibited low-level ROS and preserved myocardial function after treated with doxorubicin. Furthermore, an increased deleterious action of anthracyclines on the heart tissue was observed in the eNOS-overexpressed cardiomyocyte (20). Another study suggested that antisense eNOS mRNA decreased the activity of caspase-3, revealing its cardioprotective action on doxorubicin-mediated cardiotoxicity. Similarly, increased anthracycline-mediated ROS generation in the cardiomyocyte was observed in transgenic eNOS mice (21).
In addition to eNOS, anthracycline treatment also promotes the iNOS mRNA and protein levels and contributes to the production of nitrotyrosine (NT) with enhanced mitochondrial superoxide level in heart tissue (22). The generation of superoxide and NT depends on the peroxynitrite level related to the production of powerful oxidants such as nitrogen dioxide and carbonates radical which leads to myocardial cell apoptosis, declined cardiac contractility, and declined glutathione peroxidase activity after anthracycline treatment (23).
3.1.2. Effect of NADPH oxidase on ROS production
NADPH oxidase (Nox), a significant source of cardiac ROS generation, is a multi-component enzyme complex composed of a catalytic core (Nox2/p22phox) and cytosolic regulatory subunits (24). Seven isoforms of NOX have been identified, and NOX2 isoform is the principal one involved in anthracycline-mediated cardiotoxicity and Nox-induced ROS. Research showed that NADPH-dependent superoxide formation was significantly declined in Nox2−/− mouse models compared with that in wild type (WT) mice. Besides, Nox2 mRNA levels were raised after anthracycline treatment in WT hearts but were undetectable in Nox2−/− hearts (25). Noxs, which are usually activated by stimuli like AngII, cyclic stretch and TNF-α which are crucial in cardiac remodeling, can also regulate other ROS sources. For instance, Nox-induced ROS enable the stimulation of oxidative degradation of tetrahydrobiopterin, resulting in NOS uncoupling and superoxide generation (26). Nox is related to anthracycline-induced cardiotoxicity as a significant source of anthracycline-dependent ROS. Like NOS, Nox oxidizes the quinone structure of anthracyclines into a semiquinone radical with NADPH offering electrons. Then semiquinone radicals react with oxygen to produce H2O2 and superoxide, which can impair DNA. On the other hand, anthracyclines stimulate the activation of Nox, leading to ROS production (27).
3.1.3. Effect of mitochondrial injury on ROS production
Anthracyclines primarily target the cardiomyocytes' mitochondria, thus resulting in impaired cardiomyocyte. Anthracyclines have a potent affinity with cardiolipin which is a significant composition of the inner mitochondrial membrane, thus generating an irreversible compound through electric force to promote cardiotoxicity (28). After the anthracycline treatment, it binds to cardiolipin, leading to the impaired cardiolipin functioning and the production of superoxide radical (29). Emerging evidence demonstrates that anthracyclines are capable of quickly and directly intercalating into mitochondrial DNA (mtDNA) in vivo, leading to nucleoid aggregation and mtDNA reduction (30). Once anthracyclines intercalate to mtDNA, the mitochondrial oxidative respiratory electron transport chain will be arrested as a result of oxidation of mtDNA proteins like cytochrome C oxidase and NADH dehydrogenase, thus resulting in ROS upregulation, energy decline as well as cell apoptosis. Furthermore, a shift in the metabolism of cardiomyocyte mitochondria after doxorubicin administration have been observed that doxorubicin decreases the oxidation of long-chain-fatty acid but enhances glucose metabolism, which probably cause HF or abnormal cardiac relaxation and contraction (31, 32). In addition, anthracyclines hinder the expression of GATA-4 gene in the mitochondria, thus inhibiting mitochondrial synthesis and metabolism and enhancing apoptosis (33). Recent metabolomic studies highlight the possibility that cardiac metabolic alterations may critically contribute to the development of doxorubicin cardiotoxicity (34) and the metabolic alterations reduced ROS production and improved mitochondrial dysfunction in cardiomyocytes (35).
3.1.4. Intracellular calcium dysregulation
Anthracycline cardiotoxicity is related to impaired calcium homeostasis caused by doxorubicin-induced damage of cell membrane permeability, which results in intracellular calcium upregulation, ROS formation, and resultant apoptosis in cardiomyocytes, whereas Ca2+ chelators significantly attenuate the anthracycline-induced ROS formation and cardiomyocyte injuries (36). Moreover, anthracycline-induced cardiomyopathy is concerned with changes of the calcium exchange regulating gene, which has an effect on ryanodine receptor, calcium storage gene, Sarco/endoplasmic reticulum ATPase (SERCA2a), and phospholamban, eventually leading to damage of cardiac systolic and diastolic abilities. Notably, research shows that the removement of doxorubicin metabolite is rather intractable compared with doxorubicin, which causes the accumulation of doxorubicin metabolite on cardiomyocytes and leads to remarkable calcium dysregulation, damaging heart tissue (37).
In addition, calpains, calcium-dependent proteases, play an important role in intracellular calcium dysregulation. A rise of calcium level in the sarcoplasmic reticulum of cardiomyocytes has been found under oxidative stress and results in leakage of calcium ions which stimulate the calpains. These calpains cleave the caspase-12 and induce cardiomyocyte apoptosis. Anthracycline-induced cardiotoxicity is also associated with the devastation of cardiac muscle proteins, which emerges due to the activation of calpains, which degrade the titin, a major composition of cardiac sarcomere, thus causing cardiotoxicity (38).
3.1.5. Fe-anthracycline complex
Anthracyclines can interact with iron to form compounds which cycle between Fe(II) and Fe(III), leading to remarkable oxidative stress and mitochondrial dysfunction (39). Doxorubicinol, a derivative of doxorubicin, sequesters iron from the iron-sulfur cluster of iron regulatory protein 1 (IRP1), resulting in its inactivation. Inactive IRP1 impairs cellular iron homeostasis via mounting expression of the cellular iron importer protein transferrin receptor but weakening expression of the cellular iron storage protein ferritin, causing enhanced cellular free iron uptake and diminished sequestration, separately (40). And augmented free iron in the myocardial cells are catalysts for intracellular ROS generation. Deficiency of human hereditary hemochromatosis (HFE) gene enhanced mitochondrial ROS generation and evidently exacerbated HF caused by doxorubicin in mice with iron overload in contrast to control group (41). Furthermore, Mfrn-2 is a transporter participated in iron access to the mitochondria. Mfrn-2 level is associated with the absorption in iron mitochondria. And Mfrn-2 deletion destroys the iron homeostasis in the mitochondria (42), indicating the critical effect of free iron on anthracycline-mediated cardiotoxicity and that decreasing iron levels can be a potential way to avoid anthracycline-mediated cardiotoxicity.
3.2. Alteration of cell death pathways
3.2.1. Autophagy
Autophagy is a homeostatic course through which cellular components are degraded and recycled in normal and stress conditions. Indeed, anthracycline can stimulate autophagy, however, it is the deregulation of autophagy that results in extravagant myocardial cell death. It is debatable whether anthracycline enhances or disturbs autophagy in cardiac tissue. In the initiation stage, some evidence demonstrates that anthracycline increases AMPK (43–45), while others demonstrates no alterations (46, 47) or a reduction in AMPK activation (48, 49). Fortunately, recent studies have facilitated to settle this discrepancy via assuming that anthracycline firstly stimulates autophagy but afterwards intercepts it (50, 51) leading to the accumulation of undegraded autophagosomes and autolysosomes that worsen the destruction in myocardial cells. Indeed, a low dose of anthracyclines causes the elevated level of LC3-II, p62, and Beclin1 protein, suggesting stimulation of autophagy. Nevertheless, when evaluating downstream circumstances of autophagy, anthracyclines destroy the autophagic flux and restrict lysosomal acidification in myocardial cells. The deregulation of autophagy causes the accumulation of undegraded autolysosomes, which induces ROS generation and anthracycline-mediated cardiotoxicity (52). Mice with Beclin 1 haplo deficiency were observed declined autophagy initiation ability and thus a decreased amount of undegraded autolysosomes compared with the wildtype mice after anthracycline administration, therefore resulting in a decline of ROS generation and eventual anthracycline-induced cardiotoxicity. Conversely, enhancing the activity of autophagy via upregulating Beclin 1 worsens the cardiotoxicity (52).
3.2.2. Pyroptosis
Pyroptosis has been proven an important effect on the pathogenesis of cardiovascular diseases. Anthracycline-induced pyroptosis proceeds through the increase of Terminal Differentiation-Induced Non-Coding RNA (TINCR), which recruits IGF2BP and enhances the expression of NLRP3 resulting in activation of caspase-1, the cleavage of GMDSD-N, as well as the release of IL-1β, IL-18. The inhibition of NLRP3 with MCC950 prevented the cardiomyocytes from anthracycline-induced cell death (53). Sun et al. showed that Sirtuin 1 activation inhibited NLRP3 and protected cardiomyocytes from anthracycline-induced pyroptosis (54). Protection from anthracycline-induced cardiotoxicity has also been achieved via the inhibition of the NLRP3/caspase 1 signaling with embryonic stem cell-derived exosomes, upregulation of heat shock protein 22, and pharmacologic inhibition of NLRP3 (55). In addition, anthracycline can trigger pyroptosis by the stimulation of Bnip3 in the mitochondria. Anthracycline enhances the expression of Bnip3 which stimulates caspase 3 and results in GSDME-dependent pyroptosis. Destruction of GSDME and silencing of Bnip3 avoids cardiomyocyte death induced by anthracyclines in vitro (56).
3.2.3. Apoptosis
Apoptosis is the most researched programmed cell death pathway in anthracycline-induced cardiotoxicity. Anthracycline administration results in excess oxidative stress (lipid peroxidates, doxorubicin's reduced semiquinone moiety, charged doxorubicin iron complex, respiratory chain failure, peroxynitrites, etc.) and mitochondrial damage which triggers cell death pathways including apoptosis.
Anthracycline can trigger the intrinsic apoptosis pathway through Bax/Bak stimulation and translocation from the cytosol to the outer membrane of mitochondria causing the mitochondrial outer membrane permeabilization, enabling the diffusion of certain proteins to the cytoplasm like pro-apoptosis factor cytochrome c (57). Various mechanisms participate in anthracycline-induced intrinsic apoptosis activation, involving overexpression of p53 that results in Bax overexpression, decline of GATA4 that reduces antiapoptotic Bcl-XL level, activation of JNK and MAPK, and inactivation of PI-3K/Akt pathway (58).
Anthracycline also trigger the extrinsic apoptosis pathway in cardiomyocytes. Death ligands, such as FasL and TNFα, bind to their receptors and stimulate the recruitment of cytosolic proteins Fas-associated via death domain (FADD) and TNFR-associated death domain (TRADD) (59). FADD and TRADD recruit caspase 8, and activated caspase 8 is capable of activating caspase 3 which causes apoptosis. Mechanisms related to anthracycline-induced extrinsic apoptosis activation include activation of nuclear factor-activated T cell-4 (NFAT4) and NF-κB, resulting in the upregulation of Fas/FasL and p53; and, downregulation of FLIP, a FLICE/caspase-8 inhibition protein, which causes Fas-mediated cell death (58).
Recent data indicates that circulating death ligands may serve as serum biomarkers, therefore comprehending the extrinsic apoptosis pathway may be helpful for developing treatment strategies. McSweeney et al. explored doxorubicin-caused transcriptomic alterations by RNA-sequencing (RNAseq) and a cellular model consisting of human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs). Results showed p53 protein was a critical regulator of transcriptomic alterations associated with doxorubicin. Furthermore, overexpressed death receptor and upregulated extrinsic apoptotic pathway were demonstrated obvious correlation with doxorubicin-mediated cardiotoxicity (60). Zhao et al. studied doxorubicin-mediated cardiotoxicity in human induced pluripotent stem cells-derived cardiomyocytes (iPS-CMs). They discovered that doxorubicin remarkably upregulated the expression of death receptors (TNFR1, Fas, DR4 and DR5) in iPS-derived cardiomyocytes at both protein and mRNA levels. The resulting iPS-CMs cells underwent spontaneous apoptosis which was further enhanced by physiologically relevant death ligands including TNF-related apoptosis inducing ligand (TRAIL). Furthermore, TRAIL potentiated doxorubicin-induced decrease in beating rate and amplitude of iPS-derived cardiomyocytes. These data demonstrate that the induction of death receptors in cardiomyocytes is likely a critical mechanism by which doxorubicin causes cardiotoxicity (61).
3.3. Alteration of epigenetics
3.3.1. DNA methylation
Anthracycline downregulates the activity of DNA methyltransferase 1 (DNMT1), thus hindering the DNA methylation procedure. This hypomethylation causes disordered mitochondrial gene expression including peroxisome proliferator-activated receptor-gamma coactivator-1-alpha (PGC-1α), nuclear respiratory factor 1 (NRF-1) and mitochondrial transcription factor A (TFAM) unit in the rat cardiac tissue upon doxorubicin treatment (62), all of which can eventually cause cardiomyocyte dysfunction.
Ferreira et al., used mouse cardiomyocyte H9c2 cells to investigate the role of doxorubicin and found a reduction in glycolysis and basal respiration, as well as disordered mitochondrial DNA transcription after doxorubicin treatment (63). Notably, decrease of DNMT1, along with a global DNA hypomethylation, was found as well. These findings were consistent with earlier research in which global reduction of DNA methylation and tangled mitochondrial gene expression were found in the cardiac tissue of rats with doxorubicin treatment (62). Interestingly, their work showed that pre-exposure of doxorubicin could endow resistance to the following administration of doxorubicin in H9c2 cells, perhaps resulting from mitochondrial adaptation (63).
3.3.2. Histone modifications
Apart from DNA methylation, histone modifications play a role in anthracycline-mediated cardiotoxicity as well. Histone modifications can produce synergistic or antagonistic interactions with chromatin-associated protein, leading to dynamic switching between transcriptionally active and silent status. For instance, histone deacetylase, HDAC6, was proven overexpressed in doxorubicin-treated primary rat cardiomyocytes and mice, leading to deacetylation of α-tubulin. Genetic or pharmacological inhibition of HDAC6 in mice exhibited a cardioprotective effect against doxorubicin by restoring autophagic flux (64). Moreover, Hanf et al., using H9c2 cardiomyocytes, also confirmed that disordered expression of some histone modifiers was related to decrease of global acetylation of histone H3. The expression of histone deacetylases (SIRT1 and HDAC2) was impacted by doxorubicin administration. As for HDAC2, treatment with low-dose doxorubicin reduced its expression, however, no obvious alterations were observed in high dose treatment in contrast to control (65).
3.3.3. microRNAs (miRNAs)
Another epigenetic modification involved in anthracycline-induced cardiotoxicity is the regulation of miRNAs. Abnormal levels of some miRNAs have been demonstrated associated with anthracycline-induced cardiotoxicity. For instance, overexpression of miR-15 was found in doxorubicin-mediated apoptotic H9c2 cells (66). This phenomenon was maybe owing to the inhibition of Bmpr1a, a target of miR-15 and BMP receptor, reported to participate in myocardial contractility. As a result, activation of BMP signaling with Bmpr1a agonist is capable of mitigating doxorubicin-induced cardiotoxicity in H9c2 cells. Besides, overexpression of miR-23a, miR-34a, miR-140, miR-146a, and miR-532 were found in vitro and/or in vivo models of anthracycline-caused cardiotoxicity (67). Notably, overexpression of miR-34a, a famous tumor suppressive miRNA, could epigenetically inhibit SIRT1, thereby somewhat elucidating the decrease of the HDAC induced by doxorubicin, in the previously mentioned research (65). Furthermore, adenovirus-mediated upregulation of miR212/132 demonstrated protection against doxorubicin-caused cardiotoxicity in mice (68). Mechanism underlying this may be direct targeting of Fitm2, a transmembrane protein associated with fat storage, via miR-232/132.
4. Monitoring
In 2014, Cancer Therapeutics-Related Cardiac Dysfunction (CTRCD) was formally defined by the American Society of Echocardiography (ASE) and the European Association of Cardiovascular Imaging (EACVI) as a reduction of the LVEF by 10 percentage to below 53% through echocardiography (69, 70). CTRCD can be classified into four groups: “Reversible” when the LVEF recovers to within 5% of the baseline; “Partially reversible” when the LVEF increases by over 10% from the lowest yet still reserves 5% lower than the baseline; “Irreversible” when the LVEF increases by less than 10% from the lowest yet reserves over 5% lower than the baseline; “Indeterminate” when the patient cannot be reevaluated. As a result, the early detection of anthracycline-induced cardiotoxicity in patients prior to the occurrence of clinical manifestations exerts a crucial impact on the prognosis, making cardiac function monitoring before, during, and after anthracycline treatment significantly essential. Cardiotoxicity monitoring modalities generally involve echocardiography, electrocardiogram (ECG), cardiac magnetic resonance (CMR), nuclear cardiac imaging (MUGA), serum biomarkers, etc. Each method has its own superiorities and inferiorities as shown in Table 1.
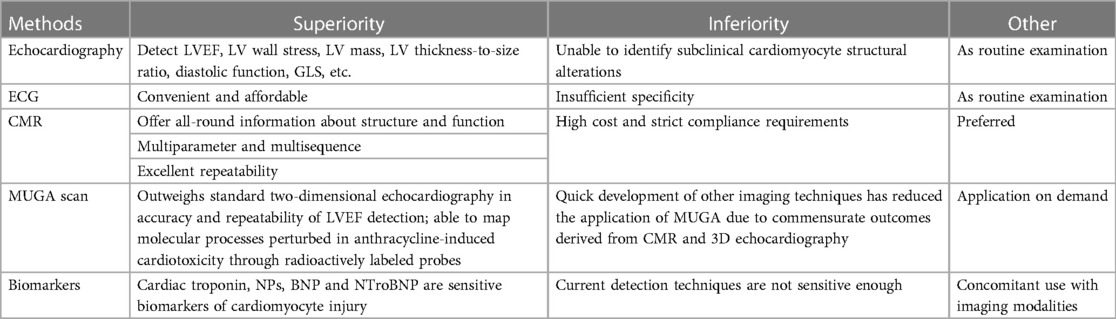
Table 1. Summary of methods for monitoring cardiotoxicity.
4.1. Echocardiography
Echocardiography is the most commonly applied monitoring technique for patients undergoing anthracycline therapy (71). Clinically, the most common parameter for the identification and monitoring of anthracycline-induced cardiotoxicity is LVEF (6). Furthermore, the calculation of two-dimensional echocardiography (2DE) LV volume and LVEF has been proposed by the American Society of Echocardiography (ASE) and the European Association of Echocardiography (EAE) as an upgradation of the Simpson technique. Notably, in comparison to the LVEF alone, LVEF combined with an evaluation of the wall motion score index was proven a more sensitive and effective parameter for monitoring of anthracycline-induced cardiotoxicity (72). When 2 or more out of 16 myocardial segments are not capable of sufficiently exhibiting the endocardial border, echocardiography contrast agents are suggested, which can improve the accuracy and repeatability of echocardiography (73).
Recently, myocardial strain abnormalities have been demonstrated earlier than cardiac dysfunction in anthracycline-induced cardiotoxicity (74). And the ESC guidelines suggest the best parameter for strain is the global longitudinal strain (GLS) which displays prognostic information such as postmyocardial infarction, aortic stenosis, HF and myocarditis (75–77). For instance, 1% improvement in GLS of patients with HF is linked to a 5% declined risk of death (75). Thavendiranathan et al. demonstrated that GLS-guided cardioprotective treatment can significantly defer the impairment of cardiac function compared with LVEF-guided group through comparing the reduction of LVEF between the two groups (78). Furthermore, a 15% decrease in GLS compared to baseline value is regarded an early signal of cardiac toxicity and can forecast subclinical LV systolic dysfunction, but a decrease of <8% excludes the diagnosis (79). Notably, strain packages among distinct manufacturers may be diverse, and there is no definite and standard value for determining the abnormal strain. In view of this condition, it is suggested to carry out follow-up investigations utilizing the same manufacturer to realize reproducibility.
4.2. Electrocardiogram
Electrocardiogram (ECG), a routine evaluation technique, can show the electrophysiological activity of the heart before, during and after anthracycline administration. Cardiovascular toxicity has various manifestations on ECG, such as sinus tachycardia, QT prolongation, ST-T segment alterations, heart block, etc. (80), among which QT prolongation is of great significance. Andreu et al. summarized the incidence, diagnosis, and outcomes of QT prolongation related to antitumor drugs and concluded that the weighted adjusted incidence of QT prolongation was 0%–22% of patients treated with anthracyclines (81). Ventricular premature contractions are the most common class of arrhythmia in patients receiving anthracyclines and ventricular tachycardia emerges in about 73.9% of patients (82). Mulrooney et al. investigated 2,715 childhood cancer survivors, 10.7% had major ECG alterations and 23.3% showed slight alterations, such as premature contraction, non-specific T wave or ST segment alterations, and low QRS (83). As a result, ECG evaluation is of great significance for patients before, during and after anthracycline administration, particularly for high-risk patients considering that early identification of arrhythmias can improve their prognosis.
4.3. Cardiac magnetic resonance (CMR)
Cardiovascular imaging is an important tool in baseline risk assessment and detection of cardiovascular disease in oncology patients receiving cardiotoxic cancer therapies (84). Though echocardiography is a routine examination to detect and monitor the cardiotoxicity caused by anthracyclines, increasing research has confirmed that CMR is an exceedingly reproducible technique in determining LVEF and is the gold standard for the assessment of LV volume and LVEF (85, 86). The advancement of CMR rapid scanning and postprocessing techniques, and the automatic analysis of CMR mapping through machine learning algorithm increases the accuracy and significantly shorten the time for image analysis (87). Jordan et al. investigated whether T1- and T2- weighted measurement of signal intensity was related to declined LVEF in patients receiving anthracyclines and enhanced myocardial contrast echocardiography. Concordant with previous animal studies, the increase in T1-weighted signal intensity was related to myocardial injuries as well as the declined LVEF. The results demonstrated that changes in T1-weighted signal intensity may be an early marker of subclinical myocardial injuries related to the anthracycline treatment (88). Galán-Arriola et al. attempted to detect early cardiotoxicity caused by doxorubicin via serial multiparametric CMR in an animal model. T2 mapping during treatment identified intracardiomyocyte edema generation as the earliest marker of anthracycline-induced cardiotoxicity, in the absence of T1 mapping, ECV, or LV motion defects. The occurrence of these changes at a reversible disease stage indicated the clinical potential of this CMR marker for tailored anthracycline therapy (89).
In addition, CMR exerts a pyramidally conspicuous effect on the identification of cardiotoxicity in multimodal imaging techniques and it may preferably help comprehend the potential mechanisms of anthracycline-induced heart injuries (6). All these results shows that CMR is a very prospective method used to monitor anthracycline-mediated cardiotoxicity. Nevertheless, CMR is limited by high cost, the application of metal devices, serious underlying diseases like advanced renal failure and the like.
Notably, in a lot of research, CMR and echocardiography examinations were generally compared with each other. For, instance, a cohort study of adult survivors from childhood cancers reported approximate average values of LVEF measured by CMR and 3DE respectively, while 2DE values were about 5% higher. Moreover, this research showed that 3DE and 2DE were not the best option for recognizing patients whose LVEFs are lower than a threshold of 50% as determined via CMR (90). In view of this, CMR is the optimal alternative when other methods are useless for determining the LV dysfunction with LVEF lying borderline low.
4.4. MUGA scan
Since 1970s, nuclear imaging (also called MUGA) was employed to determine LVEF reduction prior to the occurrence of CHF in patients treated with anthracyclines. MUGA generally outweighs standard r2DE in terms of the accuracy and repeatability of LVEF detection, and an outstanding merit of MUGA is its help in identifying patients with poor echocardiographic windows (71). Researchers explored MUGA scan in anthracycline-induced cardiotoxicity, from pathobiology to the detection of molecular targets. Actually, MUGA scan can map molecular processes perturbed in anthracycline-induced cardiotoxicity via radioactively labeled probes. Molecular targets in nuclear imaging involve metabolic dysfunction, cell death, sympathetic innervation, cardiac fibrosis detection, myocardial perfusion and blood flow measurement. These researchers offered a clinical framework for prospective use of MUGA scan in forecasting and reclassifying cardiotoxicity in cancer patients receiving anthracyclines, as a supplement to echocardiography (91). Notably, the prompt advance of other imaging techniques has reduced the application of MUGA due to commensurate outcomes derived from CMR and 3D echocardiography.
4.5. Biomarkers
Biomarkers are extremely crucial for risk evaluation and early determination of cardiotoxicity induced by anthracyclines. The highly sensitive cardiac biomarkers, particularly cardiac troponin (cTn) and natriuretic peptide (NPs), are very promising tools for early identification of subclinical cardiomyocyte alterations and forecasting the following LV dysfunction after the anthracycline administration. Besides, cTn and NPs can also be utilized to instruct the starting of cardioprotection treatment during anthracycline administration and monitor its curative effect (92). Actually, troponin increment is a common accompaniment to anthracycline treatment, and patients whose troponin levels increase are at higher risks of LV dysfunction and resultant LVEF reduction (93). Cardinale et al. reported that 65 out of 204 patients receiving high-dose anthracyclines, exhibited a rise in cTnI (>400 ng/L) and a persistent reduction in LVEF (94). Similarly, a study demonstrated that 15% of the investigated patients suffering from hematologic malignancies exhibited an increment in cTnT (≥0.03 ng/ml) with its summit values detected on day 21, and the increment was related to a LVEF reduction (95).
Notably, though troponin is a highly prospective biomarkers for early detection of anthracycline-induced cardiotoxicity, some obstacles impede its extensive use in clinical practice. First, the optimal timing of troponin evaluation is contradictory as it is indefinite whether a singular measurement of elevated cTn owns adequate predictive value or several measurements may be needed (10). Besides, the cutoff point which maximizes the positive and negative predictive value is still disputed (96).
Furthermore, the level of BNP and NTroBNP during anthracycline administration is related to the decline of LVEF and poor prognosis as well (97). Bhuva et al. investigated a cohort of 333 patients receiving anthracyclines undergoing various cancers and suggested that BNP >100 pg/ml was an indication of long-term HF, yet not a risk factor for all-cause mortality. If BNP cutoff point was 30 ng/L, the negative predictive value of occurrence of HF was 98% (87). The continuous rise of NTroBNP in the early stage upon high-dose anthracyclines is also concerned with the progression of myocardial injuries. A study by De Iuliis et al. demonstrated a remarkable correlation between NTroBNP increased values after anthracyclines administration and prediction of mortality at 1 year in breast cancer patients (97). Additionally, some novel serum biomarkers including myeloperoxidase, high-sensitivity C-reactive protein, SFLT1, placental growth factor, arginine nitric oxide metabolites, glycogen phosphorylase BB and topoisomerase 2β all rose upon anthracycline administration (98, 99), indicating that the combination of multiple markers probably enhance the capacity to identify early cardiac structural abnormalities.
5. Prevention
5.1. Anthracycline administration
Strategies used to avoid cardiotoxicity caused by anthracycline whereas keeping antitumor efficacy involving extended infusion delivery times, limited lifetime dose and the application of liposomal-encapsulated anthracyclines. Given the linear correlation between peak anthracycline levels and cardiotoxicity, continuous infusion of anthracyclines decreases the risk of cardiotoxicity in contrast to prompt bolus dosing (100, 101). van Dalen et al. investigated various pharmacokinetic infusion durations among 557 patients, and found anthracycline infusion for 6 h or more decreased the risk of HF in contrast to bolus dosing (102). The presumable reason for these phenomena is that the pharmacokinetic determinates of cardiotoxicity are different from the antitumor efficacy. Though the anticancer efficacy is primarily impacted by overall dose of anthracyclines, cardiotoxicity is related to the peak plasma levels (101). However, continuous doxorubicin infusion more than 48 h for childhood leukemia showed no cardioprotection superiority in contrast to bolus infusion (103). Given its dose dependence of anthracycline-induced cardiotoxicity, the total dose is usually confined to 450 mg/m2 of doxorubicin, 600 mg/m2 of daunorubicin or 900 mg/m2 of epirubicin. Analogs of doxorubicin like epirubicin cause comparatively less cardiac injuries, hence possessing higher dose limits. Smith et al. performed a meta-analysis and summarized that epirubicin had lower risks of both clinical and subclinical cardiotoxicity compared with doxorubicin (104). Though cutting down the overall dose of anthracyclines decreases short-term cardiotoxicity, no influence in long-term cardiotoxicity was observed. In children receiving a maximum dose of doxorubicin of 100 mg/m2, myocardial structure abnormalities were observed in 30% of patients a few years later (105). Another way to decrease anthracycline-mediated cardiotoxicity is to utilize liposomal encapsulation that enables site-specific delivery of anthracyclines. van Dalen et al. carried out a meta-analysis which showed an evidently declined rate of both clinical HF and clinical and subclinical HF combined in patients receiving liposomal-encapsulated doxorubicin (RR = 0.20, 95% CI 0.05–0.75 and RR = 0.38, 95% CI 0.24–0.59 respectively) compared with conventional doxorubicin (106). Despite their safety and no compromise of antitumor activity, the application of liposomal anthracyclines is confined to ovarian cancer, Kaposi's sarcoma, and multiple myeloma due to their high cost.
Interestingly, research has verified that natural bioactive compounds (NBACs) have a wide range of biological activities related to anticancer outcomes. Combining NBACs with anthracyclines like doxorubicin has been considered to be an ideal candidate that might increase the effectiveness of doxorubicin therapy and decreases its unfavorable adverse consequences like anthracycline-induced cardiotoxicity. Elfadadny et al. (107) showed that NBACs possess a high binding affinity to topoisomerase II-alpha ranged from 2.42 to 7.41 pKi that enhance the anticancer effect when combined to doxorubicin. Also, they demonstrated that NBACs-doxorubicin combination alleviates the doxorubicin-induced cardiotoxicity through reduction of ROS production and increases the activity of antioxidant enzymes, indicating that combination of NBACs and doxorubicin may be a very promising antineoplastic and cardioprotective agent.
5.2. Dexrazoxane
Dexrazoxane is the first approved treatment by FDA specific to anthracycline-induced cardiotoxicity. It is a water-soluble analog of the iron chelator ethylenediaminetetraacetic acid (EDTA), and is capable of decreasing anthracycline-induced ROS generation through iron sequestration and iron replacement from anthracycline binding sites. Furthermore, increasing studies have demonstrated that the suppression and reduction of topoisomerase IIβ caused by dexrazoxane and its iron chelating metabolite ADR-925 also plays an important role in its cardioprotective effect (108). Besides, it's reported that dexrazoxane can prevent myocardial cell apoptosis and necrosis through suppressing the MAPK/NF-kB pathways, thus decreasing doxorubicin-mediated inflammation (109).
Recently, Noel et al. reported that mouse models receiving dexrazoxane during doxorubicin therapy showed dramatically reduced myocardial edema, improved GLS, and decreased myocardial deformation compared with no dexrazoxane through continuous monitoring of cardiac function by MRI (110). The result confirmed the cardiac protective effect of dexrazoxane. Clinical trials also validated the effect of dexrazoxane in lowering the incidence of HF (11% vs. 1%) and cardiac events (39% vs. 13%) in advanced or metastatic breast cancer (111).
Notably, early studies have shown that dexrazoxane may elevate the risk of secondary malignant tumors and reduce the efficacy of concomitant anti-tumor drugs (112). Nevertheless, increasing studies demonstrated the cardiac protection effect of dexrazoxane with no interference in the risk for secondary malignancies or the action of anticancer drugs. For instance, Asselin et al. demonstrated that the use of dexrazoxane rescued the LV wall thickness and the LVEF reduction measured by echocardiogram in children and adolescents suffering from acute lymphocytic leukemia or non-Hodgkin lymphoma undergoing anthracycline treatment, with no compromised antitumor efficacy or increased risk of second malignancies (113). Notably, the FDA approved application of dexrazoxane is confined to cardioprotection in advanced or metastatic breast cancer with ongoing anthracycline administration upon an overall dose of over 300 mg/m2 of doxorubicin (114).
Encouragingly, Chow et al. recently provided longer-term data of dexrazoxane on HF and cardiomyopathy in survivors of childhood cancer treated with doxorubicin. Results showed that amid young childhood cancer survivors, dexrazoxane was proven a cardioprotective role approximately 20 years later than first anthracycline treatment (NCT01790152) (115). Macedo et al. performed a meta-analysis including 7 randomized trials and 2 retrospective studies with an overall of 2,177 patients who were treated with anthracyclines. Dexrazoxane reduced the risk of clinical HF and cardiac events in patients with breast cancer undergoing anthracycline chemotherapy with or without trastuzumab and did not significantly impact cancer outcomes such as oncological response, overall survival, and progression-free survival. However, the quality of available evidence is low, and further randomized trials are warranted before the systematic implementation of this therapy in clinical practice (114).
5.3. Statins
Statins are drugs that lower cholesterol and prevent heart attacks and strokes. Given their cardiac protective effect, statins have been studied as a primary prevention of anthracycline-induced cardiotoxicity. Huelsenbeck et al. suggested that, in mouse models, lovastatin reduced doxorubicin-caused acute heart injuries, which was verified by decreased mRNA levels of the pro-fibrotic cytokine connective tissue growth factor (CTGF). And lovastatin prevented doxorubicin-induced subacute cardiac injuries as well, which was verified by declined mRNA levels of CTGF and atrial natriuretic peptide. In addition, increased serum cTnI level and enhanced cardiac fibrosis after doxorubicin administration were also alleviated through lovastatin (116). Kim et al. found that compared with carvedilol, rats pre-treated with rosuvastatin were observed less LV fibrosis, LVEF decline, and cTn increase upon four-week doxorubicin withdrawal (117).
Apart from these pre-clinical studies, some clinical trials have investigated the role of statins in primary prevention of anthracycline-induced cardiotoxicity in various cancers. In a randomized controlled trial of 40 patients with various cancers, prophylactic use of statin for 6 months after anthracycline therapy exerted a protective role on myocardium, with the placebo group suffering from evidently decreased LVEF (from 62.9% to 55.0%, p < 0.0001), but no alteration in LVEF was observed in the atorvastatin arm (from 61.3% to 62.6%, p = 0.144) (118). Likewise, in a prospective study involving 51 patients with different tumors, patients with no statin administration exhibited a declined LVEF (from 57.5% to 52.4%, p = 0.0003), however no remarkable alteration in LVEF was found in patients with statin treatment (from 56.6% to 54.1%, p = 0.15) after 6 months on CMR (119). Recently, Abdel-Qadir et al. studied the relationship between statin treatment and hospitalization or emergency department visits (hospital presentations) for HF after receiving anthracyclines for early breast cancer. Their work indicated that women treated with statin showed a lower rate of HF hospital presentations upon receiving anthracyclines (120).
Although previous studies provided some evidence supporting statins, current research is not adequate to recommend statin as prevention of anthracycline-caused cardiotoxicity. Therefore, further studies are demanded to determine whether there is a clear role of statins in preventing anthracycline-induced cardiotoxicity.
5.4. Angiotensin converting enzyme inhibitors (ACEIs) and angiotensin receptor blockers (ARB)
Clinical trial data for ACEIs and ARB treatment in preventing anthracycline-induced cardiotoxicity has been mixed. Wittayanukorn et al. (121) employed the Surveillance, Epidemiology and End-Results-Medicare-linked (SEER) database to choose 6,542 women with breast cancer undergoing anthracyclines or trastuzumab administration. Patients receiving ACEIs before or within 12 months upon the beginning of anthracyclines/trastuzumab (n = 508) were compared to those who never received ACEIs treatment (n = 6,034). Patients exposed to ACEIs showed 23% lower rate of suffering from cardiotoxicity and 21% lower rate of all-cause death, though these patients appeared to be less healthy with more preexisting cardiovascular risk factors and more comorbidities. Starting ACEIs ≤6 months after the beginning of anthracyclines or trastuzumab and having exposed duration ≥6 months were related to the reduced risk of cardiotoxicity and death as well.
Cardinale et al. performed a large investigation on enalapril in preventing anthracycline-induced cardiotoxicity—the International Cardio Oncology Society-one trial (ICOS-ONE), testifying two strategies: enalapril in all patients starting prior to anthracycline therapies (prevention group) and enalapril started merely in patients with an elevation in cTn during or after anthracycline therapies (cTn-triggered group) (122). Enalapril was administered for 1 year. Results showed that low overall doses of anthracyclines in patients with low cardiovascular risk could elevate cTn, with no difference between the two strategies of enalapril administration. Given the effect of enalapril on protecting against LV dysfunction, a cTn-triggered strategy is probably more convenient. On the other hand, considering that cTn level may rise even after low dose of anthracyclines and is not closely related to the incidence of cardiotoxicity, enalapril exposure independent from cTn level may be more suitable in clinical practice.
A large trial assessing angiotensin antagonists in preventing anthracycline-induced cardiotoxicity called PRADA trial (123) was carried out on 120 breast cancer patients undergoing anthracycline administration. Gulati et al. detected and compared the levels of cardiac cTnI, cTnT, BNP, NT-proBNP, C-reactive protein (CRP) and galectin-3 between control and candesartan arms. The average levels of these biomarkers elevated until accomplishment of anthracyclines, however, merely cTn and CRP levels were proportional to the dose of anthracyclines. Moreover, the trial demonstrated that combined treatment with candesartan avoided LVEF reduction caused by anthracyclines—in the control arm, the reduction was 2.6% and 0.8% in the experimental arm. No difference in right ventricular ejection fraction was observed, as identified through MRI, neither in the LV peak systolic global longitudinal strain via 2D speckle tracking imaging. Notably, the degree of LVEF reduction was lower than what the researchers originally expected; therefore, the potence of the research was diluted to determine the differences between arms.
In conclusions, most of studies (as shown in Table 2) demonstrated the protective role of RAS inhibitors, but requiring to be further confirmed in larger randomized trials with great emphasis on the clinically relevant end points such as mortality and occurrence of HF. Longer follow-up are essential to accurately determine the degree of cardiotoxicity prevention realized by ACEIs/ARB. And it is required to further characterize of high-risk patients and conduct studies in these populations.
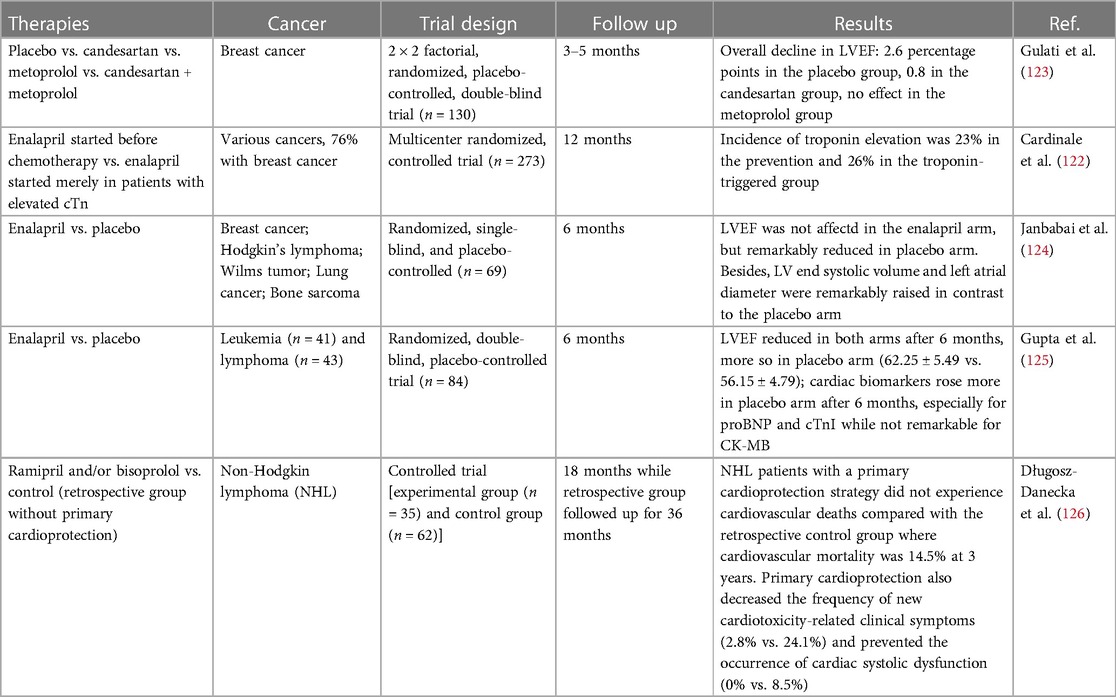
Table 2. Clinical trials evaluating ACEIs/ARB for preventing anthracycline-induced cardiotoxicity.
5.5. Beta-blockers
Beta blockers have a well-established effect on guideline-directed treatment of HF given their neurohormonal action such as lowering heart rate and alleviating arrhythmias. Besides, certain beta blockers like carvedilol and nebivolol, show prominent antioxidant properties which deplete ROS and protect against mitochondrial disorders, indicating a robust potential in avoiding anthracycline-induced cardiotoxicity (127). A clinical trial reported that the addition of nebivolol for patients with breast cancer undergoing anthracyclines led to improved LVEF, reduced LV end-systolic and end-diastolic diameters, and declined BNP levels after 6 months compared with the control arm (128).
Notably, remarkable heterogeneity exists between studies, most of which are conducted on small samples with different methodology, various definitions of cardiotoxicity and absence of long-term monitoring of clinical effect, so the statistical power to determine the action of beta blockers in protecting against anthracycline-mediated cardiomtoxicity is inadequate. For instance, a study by Nabati and coworkers studied the effect of carvedilol on protecting against doxorubicin-caused cardiotoxicity. Ninety-one women with recently diagnosed breast cancer receiving doxorubicin treatment were randomized into arms giving either carvedilol or placebo. No alteration in LVEF was found in the carvedilol arm, while LVEF was remarkably declined in the placebo arm. Furthermore, echocardiography indicated that both LV end systolic volume and LA diameter were remarkably raised in contrast to the placebo arm (129).
Nevertheless, a large prospective randomized clinical trial called CECCY study evaluated the effect of carvedilol on protecting against anthracycline-induced cardiotoxicity, and testified that in contrast to placebo, carvedilol attenuated the progression of anthracycline-mediated cardiomyocyte injuries, accompanied by decreased troponin levels and alleviated diastolic dysfunction (130). In another study, Huang et al. suggested that the prophylactic application of carvedilol had no influence in the early asymptomatic LVEF reduction while probably could lower the incidence of evident cardiotoxicity and protect against ventricular remodeling (131). Interestingly, in contrast to third generation beta blockers like nebivolol and carvedilol, metoprolol hasn't demonstrated prominent antioxidant effects. In the beta blocker monotherapy arm of the PRADA study, metoprolol succinate exerted no influence in decrease of LVEF or GLS in short or long-term follow up (132). Collectively, current studies (as shown in Table 3) suggest a prospective effect of beta blockers, especially carvedilol and nebivolol, but the small sample and short follow up of the most research confine the extensive use to all patients receiving anthracyclines.
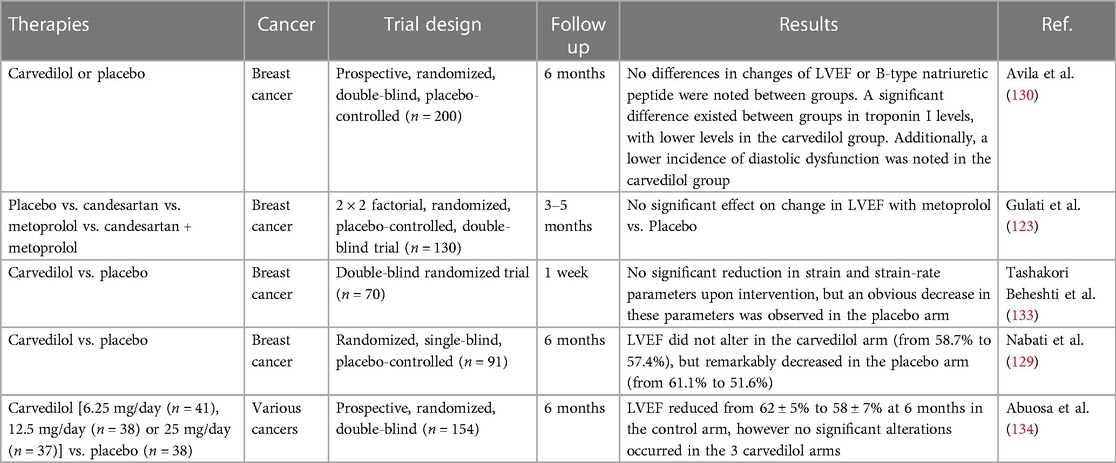
Table 3. Clinical trials evaluating beta-blockers for the preventing anthracycline-induced cardiotoxicity.
Additionally, it has been proposed that combined beta blocker and ACEIs treatment may exhibit an improved synergistic action compared with monotherapy, nevertheless, the outcomes are contradictory among studies. In the PRADA study, no remarkable alterations in LVEF, GLS, or LVESD were observed in breast cancer patients receiving concomitant treatment with candesartan and metoprolol compared with the control arm both in short and long-term follow up (132). However, the OVERCOME study observed a benefit of concomitant treatment with enalapril and carvedilol in acute leukemia patients without remarkable alterations in LVEF after 6 months, as compared to 3% LVEF decline in the control arm (135). Altogether, larger studies are demanded to confirm the clinical relevance of this strategy for preventing anthracycline-induced cardiotoxicity through combined ACEIs and beta blocker treatment.
6. Conclusions and perspectives
The incidence of cardiotoxicity related to anthracycline chemotherapy is anticipated to increase steadily in the near future, considering an increasing number of cancer survivors and the gradual ageing of the population which lead to a growing number of patients aged 65 years or above who have elevated cardiovascular risk and probably suffer from another cardiac damage caused by anthracycline-mediated cardiotoxicity. Therefore, early detection and long-term follow up of anthracycline-induced cardiotoxicity via monitor means, particularly the advanced multimodality imaging modalities reviewed in this article may enable intervention at an earlier stage, which is vital to improve patient's prognosis and reduction in death risk. Prevention measures have not been fully verified. Research has found that ergothioneine may be a viable co-therapy to alleviate the cardiotoxic effects of anthracyclines in the treatment of cancers (136). Dexrazoxane has showed its effect on the attenuation of anthracycline-caused cardiotoxicity, but its safety and influence in cancer-associated outcomes are still debatable. A limited number of clinical trials carried out with statins showed prospective outcomes. Trials studying ACEIs and ARB exhibit supportive outcomes in protecting against LV function reduction, but they didn't mitigate the elevation of cTn, indicating that RAS is not participated in the direct cardiotoxicity induced by anthracyclines but exerts an effect on the myocardial remodeling which occurs after anthracycline treatment. In contrast, betablockers were capable of mitigating the elevation of troponin in some trials, however they showed inconsistent outcomes in preventing LVEF reduction. Therefore, multicenter large clinical trials on the aforementioned drug are vital to achieve better understanding and prevention of anthracycline-induced cardiotoxicity. Further advance of oncological cardiology needs more emphasis on high-risk groups (as show in Table 4), heart function monitoring, and preventive and therapeutic measures. Moreover, the mechanism of anthracycline-induced cardiotoxicity should be explored further to identify methods for better preventing anthracycline-induced cardiotoxicity. We should upgrade existing anthracycline, or develop more efficient and low toxicity anthracycline.
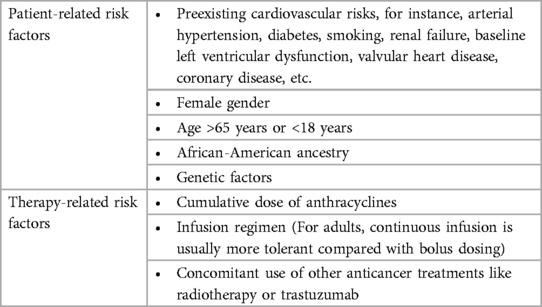
Table 4. Risk factors for developing anthracycline-induced cardiotoxicity.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
The authors declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Middleman E, Luce J, Frei E 3rd. Clinical trials with adriamycin. Cancer. (1971) 28:844–50. doi: 10.1002/1097-0142(1971)28:4%3C844::AID-CNCR2820280407%3E3.0.CO;2-9
2. Herrmann J. Adverse cardiac effects of cancer therapies: cardiotoxicity and arrhythmia. Nat Rev Cardiol. (2020) 17:474–502. doi: 10.1038/s41569-020-0348-1
3. Lenneman CG, Sawyer DB. Cardio-oncology. Circ Res. (2016) 118:1008–20. doi: 10.1161/CIRCRESAHA.115.303633
4. Mcgowan JV, Chung R, Maulik A, Piotrowska I, Walker JM, Yellon DM. Anthracycline chemotherapy and cardiotoxicity. Cardiovasc Drugs Ther. (2017) 31:63–75. doi: 10.1007/s10557-016-6711-0
5. Cardinale D, Colombo A, Bacchiani G, Tedeschi I, Meroni CA, Veglia F, et al. Early detection of anthracycline cardiotoxicity and improvement with heart failure therapy. Circulation. (2015) 131:1981–8. doi: 10.1161/CIRCULATIONAHA.114.013777
6. Armenian SH, Hudson MM, Mulder RL, Chen MH, Constine LS, Dwyer M, et al. Recommendations for cardiomyopathy surveillance for survivors of childhood cancer: a report from the international late effects of childhood cancer guideline harmonization group. Lancet Oncol. (2015) 16:e123–36. doi: 10.1016/S1470-2045(14)70409-7
7. Bernstein D. Anthracycline cardiotoxicity: worrisome enough to have you quaking? Circ Res. (2018) 122:188–90. doi: 10.1161/CIRCRESAHA.117.312395
8. Tan C, Tasaka H, Yu KP, Murphy ML, Karnofsky DA. Daunomycin, an antitumor antibiotic, in the treatment of neoplastic disease. Clinical evaluation with special reference to childhood leukemia. Cancer. (1967) 20:333–53. doi: 10.1002/1097-0142(1967)20:3%3C333::AID-CNCR2820200302%3E3.0.CO;2-K
9. Sawicki KT, Sala V, Prever L, Hirsch E, Ardehali H, Ghigo A. Preventing and treating anthracycline cardiotoxicity: new insights. Annu Rev Pharmacol Toxicol. (2021) 61:309–32. doi: 10.1146/annurev-pharmtox-030620-104842
10. Saleh Y, Abdelkarim O, Herzallah K, Abela GS. Anthracycline-induced cardiotoxicity: mechanisms of action, incidence, risk factors, prevention, and treatment. Heart Fail Rev. (2021) 26:1159–73. doi: 10.1007/s10741-020-09968-2
11. Zamorano JL, Lancellotti P, Rodriguez Muñoz D, Aboyans V, Asteggiano R, Galderisi M, et al. 2016 ESC position paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC committee for practice guidelines: the task force for cancer treatments and cardiovascular toxicity of the European society of cardiology (ESC). Eur J Heart Fail. (2017) 19:9–42. doi: 10.1002/ejhf.654
12. Curigliano G, Cardinale D, Dent S, Criscitiello C, Aseyev O, Lenihan D, et al. Cardiotoxicity of anticancer treatments: epidemiology, detection, and management. CA Cancer J Clin. (2016) 66:309–25. doi: 10.3322/caac.21341
13. Bhagat A, Kleinerman ES. Anthracycline-Induced cardiotoxicity: causes, mechanisms, and prevention. Adv Exp Med Biol. (2020) 1257:181–92. doi: 10.1007/978-3-030-43032-0_15
14. Berry GJ, Jorden M. Pathology of radiation and anthracycline cardiotoxicity. Pediatr Blood Cancer. (2005) 44:630–7. doi: 10.1002/pbc.20346
15. Swain SM, Whaley FS, Ewer MS. Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer. (2003) 97:2869–79. doi: 10.1002/cncr.11407
16. Horenstein MS, Vander Heide RS, L’ecuyer TJ. Molecular basis of anthracycline-induced cardiotoxicity and its prevention. Mol Genet Metab. (2000) 71:436–44. doi: 10.1006/mgme.2000.3043
17. Kaneto H, Katakami N, Matsuhisa M, Matsuoka TA. Role of reactive oxygen species in the progression of type 2 diabetes and atherosclerosis. Mediators Inflamm. (2010) 2010:453892. doi: 10.1155/2010/453892
18. Vásquez-Vivar J, Martasek P, Hogg N, Masters BS, Pritchard KA Jr, Kalyanaraman B. Endothelial nitric oxide synthase-dependent superoxide generation from Adriamycin. Biochemistry. (1997) 36:11293–7. doi: 10.1021/bi971475e
19. Griffith OW, Stuehr DJ. Nitric oxide synthases: properties and catalytic mechanism. Annu Rev Physiol. (1995) 57:707–36. doi: 10.1146/annurev.ph.57.030195.003423
20. Neilan TG, Blake SL, Ichinose F, Raher MJ, Buys ES, Jassal DS, et al. Disruption of nitric oxide synthase 3 protects against the cardiac injury, dysfunction, and mortality induced by doxorubicin. Circulation. (2007) 116:506–14. doi: 10.1161/CIRCULATIONAHA.106.652339
21. Kalivendi SV, Kotamraju S, Zhao H, Joseph J, Kalyanaraman B. Doxorubicin-induced apoptosis is associated with increased transcription of endothelial nitric-oxide synthase. Effect of antiapoptotic antioxidants and calcium. J Biol Chem. (2001) 276:47266–76. doi: 10.1074/jbc.M106829200
22. Akolkar G, Bagchi AK, Ayyappan P, Jassal DS, Singal PK. Doxorubicin-induced nitrosative stress is mitigated by vitamin C via the modulation of nitric oxide synthases. Am J Physiol Cell Physiol. (2017) 312:C418–c427. doi: 10.1152/ajpcell.00356.2016
23. Liu B, Li H, Qu H, Sun B. Nitric oxide synthase expressions in ADR-induced cardiomyopathy in rats. J Biochem Mol Biol. (2006) 39:759–65. doi: 10.5483/bmbrep.2006.39.6.759
24. Elnakish MT, Hassanain HH, Janssen PM, Angelos MG, Khan M. Emerging role of oxidative stress in metabolic syndrome and cardiovascular diseases: important role of Rac/NADPH oxidase. J Pathol. (2013) 231:290–300. doi: 10.1002/path.4255
25. Zhao Y, Mclaughlin D, Robinson E, Harvey AP, Hookham MB, Shah AM, et al. Nox2 NADPH oxidase promotes pathologic cardiac remodeling associated with doxorubicin chemotherapy. Cancer Res. (2010) 70:9287–97. doi: 10.1158/0008-5472.CAN-10-2664
26. Antoniades C, Shirodaria C, Warrick N, Cai S, De Bono J, Lee J, et al. 5-methyltetrahydrofolate rapidly improves endothelial function and decreases superoxide production in human vessels: effects on vascular tetrahydrobiopterin availability and endothelial nitric oxide synthase coupling. Circulation. (2006) 114:1193–201. doi: 10.1161/CIRCULATIONAHA.106.612325
27. Yu J, Wang C, Kong Q, Wu X, Lu JJ, Chen X. Recent progress in doxorubicin-induced cardiotoxicity and protective potential of natural products. Phytomedicine. (2018) 40:125–39. doi: 10.1016/j.phymed.2018.01.009
28. Nicolay K, Timmers RJ, Spoelstra E, Van Der Neut R, Fok JJ, Huigen YM, et al. The interaction of adriamycin with cardiolipin in model and rat liver mitochondrial membranes. Biochim Biophys Acta. (1984) 778:359–71. doi: 10.1016/0005-2736(84)90380-8
29. Schlame M, Rua D, Greenberg ML. The biosynthesis and functional role of cardiolipin. Prog Lipid Res. (2000) 39:257–88. doi: 10.1016/S0163-7827(00)00005-9
30. Ashley N, Poulton J. Mitochondrial DNA is a direct target of anti-cancer anthracycline drugs. Biochem Biophys Res Commun. (2009) 378:450–5. doi: 10.1016/j.bbrc.2008.11.059
31. Carvalho RA, Sousa RP, Cadete VJ, Lopaschuk GD, Palmeira CM, Bjork JA, et al. Metabolic remodeling associated with subchronic doxorubicin cardiomyopathy. Toxicology. (2010) 270:92–8. doi: 10.1016/j.tox.2010.01.019
32. Raj S, Franco VI, Lipshultz SE. Anthracycline-induced cardiotoxicity: a review of pathophysiology, diagnosis, and treatment. Curr Treat Options Cardiovasc Med. (2014) 16:315. doi: 10.1007/s11936-014-0315-4
33. Suliman HB, Carraway MS, Ali AS, Reynolds CM, Welty-Wolf KE, Piantadosi CA. The CO/HO system reverses inhibition of mitochondrial biogenesis and prevents murine doxorubicin cardiomyopathy. J Clin Invest. (2007) 117:3730–41. doi: 10.1172/JCI32967
34. Russo M, Della Sala A, Tocchetti CG, Porporato PE, Ghigo A. Metabolic aspects of anthracycline cardiotoxicity. Curr Treat Options Oncol. (2021) 22:18. doi: 10.1007/s11864-020-00812-1
35. Mercurio V, Cuomo A, Cadeddu Dessalvi C, Deidda M, Di Lisi D, Novo G, et al. Redox imbalances in ageing and metabolic alterations: implications in cancer and cardiac diseases. An overview from the working group of cardiotoxicity and cardioprotection of the Italian society of cardiology (SIC). Antioxidants (Basel). (2020) 9(7):641. doi: 10.3390/antiox9070641
36. Kalivendi SV, Konorev EA, Cunningham S, Vanamala SK, Kaji EH, Joseph J, et al. Doxorubicin activates nuclear factor of activated T-lymphocytes and Fas ligand transcription: role of mitochondrial reactive oxygen species and calcium. Biochem J. (2005) 389:527–39. doi: 10.1042/BJ20050285
37. Menna P, Salvatorelli E, Gianni L, Minotti G. Anthracycline cardiotoxicity. Top Curr Chem. (2008) 283:21–44. doi: 10.1007/128_2007_11
38. Rawat PS, Jaiswal A, Khurana A, Bhatti JS, Navik U. Doxorubicin-induced cardiotoxicity: an update on the molecular mechanism and novel therapeutic strategies for effective management. Biomed Pharmacother. (2021) 139:111708. doi: 10.1016/j.biopha.2021.111708
39. Keizer HG, Pinedo HM, Schuurhuis GJ, Joenje H. Doxorubicin (Adriamycin): a critical review of free radical-dependent mechanisms of cytotoxicity. Pharmacol Ther. (1990) 47:219–31. doi: 10.1016/0163-7258(90)90088-J
40. Minotti G, Recalcati S, Mordente A, Liberi G, Calafiore AM, Mancuso C, et al. The secondary alcohol metabolite of doxorubicin irreversibly inactivates aconitase/iron regulatory protein-1 in cytosolic fractions from human myocardium. FASEB J. (1998) 12:541–52. doi: 10.1096/fasebj.12.7.541
41. Miranda CJ, Makui H, Soares RJ, Bilodeau M, Mui J, Vali H, et al. Hfe deficiency increases susceptibility to cardiotoxicity and exacerbates changes in iron metabolism induced by doxorubicin. Blood. (2003) 102:2574–80. doi: 10.1182/blood-2003-03-0869
42. Ichikawa Y, Ghanefar M, Bayeva M, Wu R, Khechaduri A, Naga Prasad SV, et al. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J Clin Invest. (2014) 124:617–30. doi: 10.1172/JCI72931
43. Wang X, Wang XL, Chen HL, Wu D, Chen JX, Wang XX, et al. Ghrelin inhibits doxorubicin cardiotoxicity by inhibiting excessive autophagy through AMPK and p38-MAPK. Biochem Pharmacol. (2014) 88:334–50. doi: 10.1016/j.bcp.2014.01.040
44. Lv X, Yu X, Wang Y, Wang F, Li H, Wang Y, et al. Berberine inhibits doxorubicin-triggered cardiomyocyte apoptosis via attenuating mitochondrial dysfunction and increasing Bcl-2 expression. PLoS One. (2012) 7:e47351. doi: 10.1371/journal.pone.0047351
45. Chen MB, Wu XY, Gu JH, Guo QT, Shen WX, Lu PH. Activation of AMP-activated protein kinase contributes to doxorubicin-induced cell death and apoptosis in cultured myocardial H9c2 cells. Cell Biochem Biophys. (2011) 60:311–22. doi: 10.1007/s12013-011-9153-0
46. Sun A, Cheng Y, Zhang Y, Zhang Q, Wang S, Tian S, et al. Aldehyde dehydrogenase 2 ameliorates doxorubicin-induced myocardial dysfunction through detoxification of 4-HNE and suppression of autophagy. J Mol Cell Cardiol. (2014) 71:92–104. doi: 10.1016/j.yjmcc.2014.01.002
47. Wu R, Wang HL, Yu HL, Cui XH, Xu MT, Xu X, et al. Doxorubicin toxicity changes myocardial energy metabolism in rats. Chem Biol Interact. (2016) 244:149–58. doi: 10.1016/j.cbi.2015.12.010
48. Gu J, Hu W, Song ZP, Chen YG, Zhang DD, Wang CQ. Resveratrol-induced autophagy promotes survival and attenuates doxorubicin-induced cardiotoxicity. Int Immunopharmacol. (2016) 32:1–7. doi: 10.1016/j.intimp.2016.01.002
49. Andreadou I, Mikros E, Ioannidis K, Sigala F, Naka K, Kostidis S, et al. Oleuropein prevents doxorubicin-induced cardiomyopathy interfering with signaling molecules and cardiomyocyte metabolism. J Mol Cell Cardiol. (2014) 69:4–16. doi: 10.1016/j.yjmcc.2014.01.007
50. Abdullah CS, Alam S, Aishwarya R, Miriyala S, Bhuiyan MAN, Panchatcharam M, et al. Doxorubicin-induced cardiomyopathy associated with inhibition of autophagic degradation process and defects in mitochondrial respiration. Sci Rep. (2019) 9:2002. doi: 10.1038/s41598-018-37862-3
51. Bartlett JJ, Trivedi PC, Yeung P, Kienesberger PC, Pulinilkunnil T. Doxorubicin impairs cardiomyocyte viability by suppressing transcription factor EB expression and disrupting autophagy. Biochem J. (2016) 473:3769–89. doi: 10.1042/BCJ20160385
52. Li DL, Wang ZV, Ding G, Tan W, Luo X, Criollo A, et al. Doxorubicin blocks cardiomyocyte autophagic flux by inhibiting lysosome acidification. Circulation. (2016) 133:1668–87. doi: 10.1161/CIRCULATIONAHA.115.017443
53. Meng L, Lin H, Zhang J, Lin N, Sun Z, Gao F, et al. Doxorubicin induces cardiomyocyte pyroptosis via the TINCR-mediated posttranscriptional stabilization of NLR family pyrin domain containing 3. J Mol Cell Cardiol. (2019) 136:15–26. doi: 10.1016/j.yjmcc.2019.08.009
54. Sun Z, Lu W, Lin N, Lin H, Zhang J, Ni T, et al. Dihydromyricetin alleviates doxorubicin-induced cardiotoxicity by inhibiting NLRP3 inflammasome through activation of SIRT1. Biochem Pharmacol. (2020) 175:113888. doi: 10.1016/j.bcp.2020.113888
55. Tavakoli Dargani Z, Singla DK. Embryonic stem cell-derived exosomes inhibit doxorubicin-induced TLR4-NLRP3-mediated cell death-pyroptosis. Am J Physiol Heart Circ Physiol. (2019) 317:H460–71. doi: 10.1152/ajpheart.00056.2019
56. Zheng X, Zhong T, Ma Y, Wan X, Qin A, Yao B, et al. Bnip3 mediates doxorubicin-induced cardiomyocyte pyroptosis via caspase-3/GSDME. Life Sci. (2020) 242:117186. doi: 10.1016/j.lfs.2019.117186
57. An J, Li P, Li J, Dietz R, Donath S. ARC is a critical cardiomyocyte survival switch in doxorubicin cardiotoxicity. J Mol Med (Berl). (2009) 87:401–10. doi: 10.1007/s00109-008-0434-z
58. Christidi E, Brunham LR. Regulated cell death pathways in doxorubicin-induced cardiotoxicity. Cell Death Dis. (2021) 12:339. doi: 10.1038/s41419-021-03614-x
59. Shi J, Abdelwahid E, Wei L. Apoptosis in anthracycline cardiomyopathy. Curr Pediatr Rev. (2011) 7:329–36. doi: 10.2174/157339611796892265
60. Mcsweeney KM, Bozza WP, Alterovitz WL, Zhang B. Transcriptomic profiling reveals p53 as a key regulator of doxorubicin-induced cardiotoxicity. Cell Death Discov. (2019) 5:102. doi: 10.1038/s41420-019-0182-6
61. Zhao L, Zhang B. Doxorubicin induces cardiotoxicity through upregulation of death receptors mediated apoptosis in cardiomyocytes. Sci Rep. (2017) 7:44735. doi: 10.1038/srep44735
62. Ferreira A, Cunha-Oliveira T, Simões RF, Carvalho FS, Burgeiro A, Nordgren K, et al. Altered mitochondrial epigenetics associated with subchronic doxorubicin cardiotoxicity. Toxicology. (2017) 390:63–73. doi: 10.1016/j.tox.2017.08.011
63. Ferreira LL, Cunha-Oliveira T, Veloso CD, Costa CF, Wallace KB, Oliveira PJ. Single nanomolar doxorubicin exposure triggers compensatory mitochondrial responses in H9c2 cardiomyoblasts. Food Chem Toxicol. (2019) 124:450–61. doi: 10.1016/j.fct.2018.12.017
64. Song R, Yang Y, Lei H, Wang G, Huang Y, Xue W, et al. HDAC6 inhibition protects cardiomyocytes against doxorubicin-induced acute damage by improving α-tubulin acetylation. J Mol Cell Cardiol. (2018) 124:58–69. doi: 10.1016/j.yjmcc.2018.10.007
65. Hanf A, Oelze M, Manea A, Li H, Münzel T, Daiber A. The anti-cancer drug doxorubicin induces substantial epigenetic changes in cultured cardiomyocytes. Chem Biol Interact. (2019) 313:108834. doi: 10.1016/j.cbi.2019.108834
66. Wan GX, Cheng L, Qin HL, Zhang YZ, Wang LY, Zhang YG. MiR-15b-5p is involved in doxorubicin-induced cardiotoxicity via inhibiting Bmpr1a signal in H9c2 cardiomyocyte. Cardiovasc Toxicol. (2019) 19:264–75. doi: 10.1007/s12012-018-9495-6
67. Kumari H, Huang WH, Chan MWY. Review on the role of epigenetic modifications in doxorubicin-induced cardiotoxicity. Front Cardiovasc Med. (2020) 7:56. doi: 10.3389/fcvm.2020.00056
68. Gupta SK, Garg A, Avramopoulos P, Engelhardt S, Streckfuss-Bömeke K, Batkai S, et al. miR-212/132 cluster modulation prevents doxorubicin-mediated atrophy and cardiotoxicity. Mol Ther. (2019) 27:17–28. doi: 10.1016/j.ymthe.2018.11.004
69. Lang RM, Badano LP, Mor-Avi V, Afilalo J, Armstrong A, Ernande L, et al. Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American society of echocardiography and the European association of cardiovascular imaging. Eur Heart J Cardiovasc Imaging. (2015) 16:233–70. doi: 10.1093/ehjci/jev014
70. Plana JC, Galderisi M, Barac A, Ewer MS, Ky B, Scherrer-Crosbie M, et al. Expert consensus for multimodality imaging evaluation of adult patients during and after cancer therapy: a report from the American society of echocardiography and the European association of cardiovascular imaging. J Am Soc Echocardiogr. (2014) 27:911–39. doi: 10.1016/j.echo.2014.07.012
71. Su N, Sun J, Zhang G, Meng Y. New advances in medical imaging technology for the evaluation of anthracycline-induced cardiotoxicity. Chin Med J (Engl). (2022) 135:1883–5. doi: 10.1097/CM9.0000000000002123
72. Lang RM, Bierig M, Devereux RB, Flachskampf FA, Foster E, Pellikka PA, et al. Recommendations for chamber quantification: a report from the American society of echocardiography’s guidelines and standards committee and the chamber quantification writing group, developed in conjunction with the European association of echocardiography, a branch of the European society of cardiology. J Am Soc Echocardiogr. (2005) 18:1440–63. doi: 10.1016/j.echo.2005.10.005
73. Bountioukos M, Doorduijn JK, Roelandt JR, Vourvouri EC, Bax JJ, Schinkel AF, et al. Repetitive dobutamine stress echocardiography for the prediction of anthracycline cardiotoxicity. Eur J Echocardiogr. (2003) 4:300–5. doi: 10.1016/S1525-2167(03)00017-9
74. Thavendiranathan P, Poulin F, Lim KD, Plana JC, Woo A, Marwick TH. Use of myocardial strain imaging by echocardiography for the early detection of cardiotoxicity in patients during and after cancer chemotherapy: a systematic review. J Am Coll Cardiol. (2014) 63:2751–68. doi: 10.1016/j.jacc.2014.01.073
75. Park JJ, Park JB, Park JH, Cho GY. Global longitudinal strain to predict mortality in patients with acute heart failure. J Am Coll Cardiol. (2018) 71:1947–57. doi: 10.1016/j.jacc.2018.02.064
76. Kasner M, Aleksandrov A, Escher F, Al-Saadi N, Makowski M, Spillmann F, et al. Multimodality imaging approach in the diagnosis of chronic myocarditis with preserved left ventricular ejection fraction (MCpEF): the role of 2D speckle-tracking echocardiography. Int J Cardiol. (2017) 243:374–8. doi: 10.1016/j.ijcard.2017.05.038
77. Caspar T, Fichot M, Ohana M, El Ghannudi S, Morel O, Ohlmann P. Late detection of left ventricular dysfunction using two-dimensional and three-dimensional speckle-tracking echocardiography in patients with history of nonsevere acute myocarditis. J Am Soc Echocardiogr. (2017) 30:756–62. doi: 10.1016/j.echo.2017.04.002
78. Thavendiranathan P, Negishi T, Somerset E, Negishi K, Penicka M, Lemieux J, et al. Strain-guided management of potentially cardiotoxic cancer therapy. J Am Coll Cardiol. (2021) 77:392–401. doi: 10.1016/j.jacc.2020.11.020
79. Ye L, Yang ZG, Selvanayagam JB, Luo H, Yang TZ, Perry R, et al. Myocardial strain imaging by echocardiography for the prediction of cardiotoxicity in chemotherapy-treated patients: a meta-analysis. JACC Cardiovasc Imaging. (2020) 13:881–2. doi: 10.1016/j.jcmg.2019.09.013
80. Schwartz PJ, Wolf S. QT interval prolongation as predictor of sudden death in patients with myocardial infarction. Circulation. (1978) 57:1074–7. doi: 10.1161/01.CIR.57.6.1074
81. Porta-Sánchez A, Gilbert C, Spears D, Amir E, Chan J, Nanthakumar K, et al. Incidence, diagnosis, and management of QT prolongation induced by cancer therapies: a systematic review. J Am Heart Assoc. (2017) 6(12):e007724. doi: 10.1161/JAHA.117.007724
82. Mazur M, Wang F, Hodge DO, Siontis BL, Beinborn DS, Villarraga HR, et al. Burden of cardiac arrhythmias in patients with anthracycline-related cardiomyopathy. JACC Clin Electrophysiol. (2017) 3:139–50. doi: 10.1016/j.jacep.2016.08.009
83. Mulrooney DA, Soliman EZ, Ehrhardt MJ, Lu L, Duprez DA, Luepker RV, et al. Electrocardiographic abnormalities and mortality in aging survivors of childhood cancer: a report from the St Jude lifetime cohort study. Am Heart J. (2017) 189:19–27. doi: 10.1016/j.ahj.2017.03.023
84. Celutkiene J, Pudil R, Lopez-Fernandez T, Grapsa J, Nihoyannopoulos P, Bergler-Klein J, et al. Role of cardiovascular imaging in cancer patients receiving cardiotoxic therapies: a position statement on behalf of the heart failure association (HFA), the European association of cardiovascular imaging (EACVI) and the cardio-oncology council of the European society of cardiology (ESC). Eur J Heart Fail. (2020) 22:1504–24. doi: 10.1002/ejhf.1957
85. Houard L, Militaru S, Tanaka K, Pasquet A, Vancraeynest D, Vanoverschelde JL, et al. Test-retest reliability of left and right ventricular systolic function by new and conventional echocardiographic and cardiac magnetic resonance parameters. Eur Heart J Cardiovasc Imaging. (2021) 22:1157–67. doi: 10.1093/ehjci/jeaa206
86. Lambert J, Lamacie M, Thampinathan B, Altaha MA, Esmaeilzadeh M, Nolan M, et al. Variability in echocardiography and MRI for detection of cancer therapy cardiotoxicity. Heart. (2020) 106:817–23. doi: 10.1136/heartjnl-2019-316297
87. Bhuva AN, Bai W, Lau C, Davies RH, Ye Y, Bulluck H, et al. A multicenter, scan-rescan, human and machine learning CMR study to test generalizability and precision in imaging biomarker analysis. Circ Cardiovasc Imaging. (2019) 12:e009214. doi: 10.1161/CIRCIMAGING.119.009214
88. Jordan JH, D’agostino RB Jr, Hamilton CA, Vasu S, Hall ME, Kitzman DW, et al . Longitudinal assessment of concurrent changes in left ventricular ejection fraction and left ventricular myocardial tissue characteristics after administration of cardiotoxic chemotherapies using T1-weighted and T2-weighted cardiovascular magnetic resonance. Circ Cardiovasc Imaging. (2014) 7:872–9. doi: 10.1161/CIRCIMAGING.114.002217
89. Galán-Arriola C, Lobo M, Vílchez-Tschischke JP, López GJ, De Molina-Iracheta A, Pérez-Martínez C, et al. Serial magnetic resonance imaging to identify early stages of anthracycline-induced cardiotoxicity. J Am Coll Cardiol. (2019) 73:779–91. doi: 10.1016/j.jacc.2018.11.046
90. Armstrong GT, Plana JC, Zhang N, Srivastava D, Green DM, Ness KK, et al. Screening adult survivors of childhood cancer for cardiomyopathy: comparison of echocardiography and cardiac magnetic resonance imaging. J Clin Oncol. (2012) 30:2876–84. doi: 10.1200/JCO.2011.40.3584
91. Jong J, Pinney JR, Packard RRS. Anthracycline-induced cardiotoxicity: from pathobiology to identification of molecular targets for nuclear imaging. Front Cardiovasc Med. (2022) 9:919719. doi: 10.3389/fcvm.2022.919719
92. Pudil R, Mueller C, Čelutkienė J, Henriksen PA, Lenihan D, Dent S, et al. Role of serum biomarkers in cancer patients receiving cardiotoxic cancer therapies: a position statement from the cardio-oncology study group of the heart failure association and the cardio-oncology council of the European society of cardiology. Eur J Heart Fail. (2020) 22:1966–83. doi: 10.1002/ejhf.2017
93. Michel L, Mincu RI, Mahabadi AA, Settelmeier S, Al-Rashid F, Rassaf T, et al. Troponins and brain natriuretic peptides for the prediction of cardiotoxicity in cancer patients: a meta-analysis. Eur J Heart Fail. (2020) 22:350–61. doi: 10.1002/ejhf.1631
94. Cardinale D, Sandri MT, Martinoni A, Tricca A, Civelli M, Lamantia G, et al. Left ventricular dysfunction predicted by early troponin I release after high-dose chemotherapy. J Am Coll Cardiol. (2000) 36:517–22. doi: 10.1016/S0735-1097(00)00748-8
95. Auner HW, Tinchon C, Linkesch W, Tiran A, Quehenberger F, Link H, et al. Prolonged monitoring of troponin T for the detection of anthracycline cardiotoxicity in adults with hematological malignancies. Ann Hematol. (2003) 82:218–22. doi: 10.1007/s00277-003-0615-3
96. Plana JC, Galderisi M, Barac A, Ewer MS, Ky B, Scherrer-Crosbie M, et al. Expert consensus for multimodality imaging evaluation of adult patients during and after cancer therapy: a report from the American society of echocardiography and the European association of cardiovascular Imaging. Eur Heart J Cardiovasc Imaging. (2014) 15:1063–93. doi: 10.1093/ehjci/jeu192
97. De Iuliis F, Salerno G, Taglieri L, De Biase L, LanzA R, Cardelli P, et al. Serum biomarkers evaluation to predict chemotherapy-induced cardiotoxicity in breast cancer patients. Tumour Biol. (2016) 37:3379–87. doi: 10.1007/s13277-015-4183-7
98. Finkelman BS, Putt M, Wang T, Wang L, Narayan H, Domchek S, et al. Arginine-nitric oxide metabolites and cardiac dysfunction in patients with breast cancer. J Am Coll Cardiol. (2017) 70:152–62. doi: 10.1016/j.jacc.2017.05.019
99. Beer LA, Kossenkov AV, Liu Q, Luning Prak E, Domchek S, Speicher DW, et al. Baseline immunoglobulin E levels as a marker of doxorubicin- and trastuzumab-associated cardiac dysfunction. Circ Res. (2016) 119:1135–44. doi: 10.1161/CIRCRESAHA.116.309004
100. El-Kareh AW, Secomb TW. A mathematical model for comparison of bolus injection, continuous infusion, and liposomal delivery of doxorubicin to tumor cells. Neoplasia. (2000) 2:325–38. doi: 10.1038/sj.neo.7900096
101. Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. (2004) 56:185–229. doi: 10.1124/pr.56.2.6
102. Van Dalen EC, Van Der Pal HJ, Kremer LC. Different dosage schedules for reducing cardiotoxicity in people with cancer receiving anthracycline chemotherapy. Cochrane Database Syst Rev. (2016) 3:Cd005008. doi: 10.1002/14651858.CD005008.pub4
103. Lipshultz SE, Giantris AL, Lipsitz SR, Kimball Dalton V, Asselin BL, Barr RD, et al. Doxorubicin administration by continuous infusion is not cardioprotective: the Dana-Farber 91-01 acute lymphoblastic leukemia protocol. J Clin Oncol. (2002) 20:1677–82. doi: 10.1200/JCO.2002.20.6.1677
104. Smith LA, Cornelius VR, Plummer CJ, Levitt G, Verrill M, Canney P, et al. Cardiotoxicity of anthracycline agents for the treatment of cancer: systematic review and meta-analysis of randomised controlled trials. BMC Cancer. (2010) 10:337. doi: 10.1186/1471-2407-10-337
105. Leger K, Slone T, Lemler M, Leonard D, Cochran C, Bowman WP, et al. Subclinical cardiotoxicity in childhood cancer survivors exposed to very low dose anthracycline therapy. Pediatr Blood Cancer. (2015) 62:123–7. doi: 10.1002/pbc.25206
106. Van Dalen EC, Michiels EM, Caron HN, Kremer LC. Different anthracycline derivates for reducing cardiotoxicity in cancer patients. Cochrane Database Syst Rev. (2010) 2010(5):Cd005006. doi: 10.1002/14651858.CD005006.pub3
107. Elfadadny A, Ragab RF, Hamada R, Al Jaouni SK, Fu J, Mousa SA, et al. Natural bioactive compounds-doxorubicin combinations targeting topoisomerase II-alpha: anticancer efficacy and safety. Toxicol Appl Pharmacol. (2023) 461:116405. doi: 10.1016/j.taap.2023.116405
108. Jirkovský E, Jirkovská A, Bavlovič-Piskáčková H, Skalická V, Pokorná Z, Karabanovich G, et al. Clinically translatable prevention of anthracycline cardiotoxicity by dexrazoxane is mediated by topoisomerase II Beta and not metal chelation. Circ Heart Fail. (2021) 14:e008209. doi: 10.1161/CIRCHEARTFAILURE.120.008209
109. Wallace KB, Sardão VA, Oliveira PJ. Mitochondrial determinants of doxorubicin-induced cardiomyopathy. Circ Res. (2020) 126:926–41. doi: 10.1161/CIRCRESAHA.119.314681
110. Noel CV, Rainusso N, Robertson M, Romero J, Masand P, Coarfa C, et al. Early detection of myocardial changes with and without dexrazoxane using serial magnetic resonance imaging in a pre-clinical mouse model. Cardiooncology. (2021) 7:23. doi: 10.1186/s40959-021-00109-8
111. Marty M, Espié M, Llombart A, Monnier A, Rapoport BL, Stahalova V. Multicenter randomized phase III study of the cardioprotective effect of dexrazoxane (cardioxane) in advanced/metastatic breast cancer patients treated with anthracycline-based chemotherapy. Ann Oncol. (2006) 17:614–22. doi: 10.1093/annonc/mdj134
112. Tebbi CK, London WB, Friedman D, Villaluna D, De Alarcon PA, Constine LS, et al. Dexrazoxane-associated risk for acute myeloid leukemia/myelodysplastic syndrome and other secondary malignancies in pediatric Hodgkin’s disease. J Clin Oncol. (2007) 25:493–500. doi: 10.1200/JCO.2005.02.3879
113. Asselin BL, Devidas M, Chen L, Franco VI, Pullen J, Borowitz MJ, et al. Cardioprotection and safety of dexrazoxane in patients treated for newly diagnosed T-cell acute lymphoblastic leukemia or advanced-stage lymphoblastic non-Hodgkin lymphoma: a report of the children’s oncology group randomized trial pediatric oncology group 9404. J Clin Oncol. (2016) 34:854–62. doi: 10.1200/JCO.2015.60.8851
114. Macedo AVS, Hajjar LA, Lyon AR, Nascimento BR, Putzu A, Rossi L, et al. Efficacy of dexrazoxane in preventing anthracycline cardiotoxicity in breast cancer. JACC CardioOncol. (2019) 1:68–79. doi: 10.1016/j.jaccao.2019.08.003
115. Chow EJ, Aggarwal S, Doody DR, Aplenc R, Armenian SH, Baker KS, et al. Dexrazoxane and long-term heart function in survivors of childhood cancer. J Clin Oncol. (2023) 41:2248–57. doi: 10.1200/JCO.22.02423
116. Huelsenbeck J, Henninger C, Schad A, Lackner KJ, Kaina B, Fritz G. Inhibition of Rac1 signaling by lovastatin protects against anthracycline-induced cardiac toxicity. Cell Death Dis. (2011) 2:e190. doi: 10.1038/cddis.2011.65
117. Kim YH, Park SM, Kim M, Kim SH, Lim SY, Ahn JC, et al. Cardioprotective effects of rosuvastatin and carvedilol on delayed cardiotoxicity of doxorubicin in rats. Toxicol Mech Methods. (2012) 22:488–98. doi: 10.3109/15376516.2012.678406
118. Acar Z, Kale A, Turgut M, Demircan S, Durna K, Demir S, et al. Efficiency of atorvastatin in the protection of anthracycline-induced cardiomyopathy. J Am Coll Cardiol. (2011) 58:988–9. doi: 10.1016/j.jacc.2011.05.025
119. Chotenimitkhun R, D’agostino R Jr, Lawrence JA, Hamilton CA, Jordan JH, Vasu S, et al . Chronic statin administration may attenuate early anthracycline-associated declines in left ventricular ejection function. Can J Cardiol. (2015) 31:302–7. doi: 10.1016/j.cjca.2014.11.020
120. Abdel-Qadir H, Bobrowski D, Zhou L, Austin PC, Calvillo-Argüelles O, Amir E, et al. Statin exposure and risk of heart failure after anthracycline- or trastuzumab-based chemotherapy for early breast cancer: a propensity score‒matched cohort study. J Am Heart Assoc. (2021) 10:e018393. doi: 10.1161/JAHA.119.018393
121. Wittayanukorn S, Qian J, Westrick SC, Billor N, Johnson B, Hansen RA. Prevention of trastuzumab and anthracycline-induced cardiotoxicity using angiotensin-converting enzyme inhibitors or β-blockers in older adults with breast cancer. Am J Clin Oncol. (2018) 41:909–18. doi: 10.1097/COC.0000000000000389
122. Cardinale D, Ciceri F, Latini R, Franzosi MG, Sandri MT, Civelli M, et al. Anthracycline-induced cardiotoxicity: a multicenter randomised trial comparing two strategies for guiding prevention with enalapril: the international CardioOncology society-one trial. Eur J Cancer. (2018) 94:126–37. doi: 10.1016/j.ejca.2018.02.005
123. Gulati G, Heck SL, Ree AH, Hoffmann P, Schulz-Menger J, Fagerland MW, et al. Prevention of cardiac dysfunction during adjuvant breast cancer therapy (PRADA): a 2×2 factorial, randomized, placebo-controlled, double-blind clinical trial of candesartan and metoprolol. Eur Heart J. (2016) 37:1671–80. doi: 10.1093/eurheartj/ehw022
124. Janbabai G, Nabati M, Faghihinia M, Azizi S, Borhani S, Yazdani J. Effect of enalapril on preventing anthracycline-induced cardiomyopathy. Cardiovasc Toxicol. (2017) 17:130–9. doi: 10.1007/s12012-016-9365-z
125. Gupta V, Kumar Singh S, Agrawal V, Bali Singh T. Role of ACE inhibitors in anthracycline-induced cardiotoxicity: a randomized, double-blind, placebo-controlled trial. Pediatr Blood Cancer. (2018) 65:e27308. doi: 10.1002/pbc.27308
126. Długosz-Danecka M, Gruszka AM, Szmit S, Olszanecka A, Ogórka T, Sobociński M, et al. Primary cardioprotection reduces mortality in lymphoma patients with increased risk of anthracycline cardiotoxicity, treated by R-CHOP regimen. Chemotherapy. (2018) 63:238–45. doi: 10.1159/000492942
127. Graffagnino J, Kondapalli L, Arora G, Hawi R, Lenneman CG. Strategies to prevent cardiotoxicity. Curr Treat Options Oncol. (2020) 21:32. doi: 10.1007/s11864-020-0722-6
128. Kaya MG, Ozkan M, Gunebakmaz O, Akkaya H, Kaya EG, Akpek M, et al. Rotective effects of nebivolol against anthracycline-induced cardiomyopathy: a randomized control study. Int J Cardiol. (2013) 167:2306–10. doi: 10.1016/j.ijcard.2012.06.023
129. Nabati M, Janbabai G, Baghyari S, Esmaili K, Yazdani J. Cardioprotective effects of carvedilol in inhibiting doxorubicin-induced cardiotoxicity. J Cardiovasc Pharmacol. (2017) 69:279–85. doi: 10.1097/FJC.0000000000000470
130. Avila MS, Ayub-Ferreira SM, De Barros Wanderley MR Jr, Das Dores Cruz F, Gonçalves Brandão SM, Rigaud VOC, et al. Carvedilol for prevention of chemotherapy-related cardiotoxicity: the CECCY trial. J Am Coll Cardiol. (2018) 71:2281–90. doi: 10.1016/j.jacc.2018.02.049
131. Huang S, Zhao Q, Yang ZG, Diao KY, He Y, Shi K, et al. Protective role of beta-blockers in chemotherapy-induced cardiotoxicity-a systematic review and meta-analysis of carvedilol. Heart Fail Rev. (2019) 24:325–33. doi: 10.1007/s10741-018-9755-3
132. Bosch X, Sitges M, Rovira M, Ortiz-Pérez JT, Esteve J. Reply: enalapril and carvedilol for preventing chemotherapy-induced left ventricular systolic dysfunction in patients with malignant hemopathies. J Am Coll Cardiol. (2013) 62:2452. doi: 10.1016/j.jacc.2013.08.712
133. Tashakori Beheshti A, Mostafavi Toroghi H, Hosseini G, Zarifian A, Homaei Shandiz F, Fazlinezhad A. Carvedilol administration can prevent doxorubicin-induced cardiotoxicity: a double-blind randomized trial. Cardiology. (2016) 134:47–53. doi: 10.1159/000442722
134. Abuosa AM, Elshiekh AH, Qureshi K, Abrar MB, Kholeif MA, Kinsara AJ, et al. Prophylactic use of carvedilol to prevent ventricular dysfunction in patients with cancer treated with doxorubicin. Indian Heart J. (2018) 70(Suppl 3):S96–100. doi: 10.1016/j.ihj.2018.06.011
135. Bosch X, Rovira M, Sitges M, Domènech A, Ortiz-Pérez JT, De Caralt TM, et al. Enalapril and carvedilol for preventing chemotherapy-induced left ventricular systolic dysfunction in patients with malignant hemopathies: the OVERCOME trial (preventiOn of left ventricular dysfunction with enalapril and caRvedilol in patients submitted to intensive ChemOtherapy for the treatment of malignant hEmopathies). J Am Coll Cardiol. (2013) 61:2355–62. doi: 10.1016/j.jacc.2013.02.072
Keywords: anthracycline, cardiotoxicity, mechanisms, monitoring, prevention
Citation: Qiu Y, Jiang P and Huang Y (2023) Anthracycline-induced cardiotoxicity: mechanisms, monitoring, and prevention. Front. Cardiovasc. Med. 10:1242596. doi: 10.3389/fcvm.2023.1242596
Received: 22 June 2023; Accepted: 4 December 2023;
Published: 19 December 2023.
Edited by:
Jiong-Wei Wang, National University of Singapore, SingaporeReviewed by:
Ahmed Elfadadny, Tokyo University of Agriculture and Technology, JapanClaudia Penna, University of Turin, Italy
Yanqiu Lai, National University of Singapore, Singapore
© 2023 Qiu, Jiang and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yingmei Huang hym57872021@163.com