The Functional Polymorphism of DDAH2 rs9267551 Is an Independent Determinant of Arterial Stiffness
 Carolina Averta1†
Carolina Averta1†  Elettra Mancuso1†
Elettra Mancuso1†  Rosangela Spiga1 Sofia Miceli1
Rosangela Spiga1 Sofia Miceli1  Elena Succurro1,2 Teresa Vanessa Fiorentino1
Elena Succurro1,2 Teresa Vanessa Fiorentino1  Maria Perticone1
Maria Perticone1  Gaia Chiara Mannino1*
Gaia Chiara Mannino1*  Prapaporn Jungtrakoon Thamtarana3
Prapaporn Jungtrakoon Thamtarana3  Angela Sciacqua1,2
Angela Sciacqua1,2  Giorgio Sesti4
Giorgio Sesti4  Francesco Andreozzi1,2
Francesco Andreozzi1,2- 1Department of Medical and Surgical Sciences, University Magna Graecia of Catanzaro, Catanzaro, Italy
- 2Research Center for the Prevention and Treatment of Metabolic Diseases (CR METDIS), University Magna Graecia of Catanzaro, Catanzaro, Italy
- 3Siriraj Center of Research Excellence for Diabetes and Obesity, Division of Molecular Medicine, Department of Research, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand
- 4Department of Clinical and Molecular Medicine, University of Rome-Sapienza, Rome, Italy
Background: The association of circulating asymmetric dimethylarginine (ADMA) levels with cardiovascular risk and arterial stiffness has been reportedly demonstrated, although the causal involvement of ADMA in the pathogenesis of these conditions is still debated. Dimethylaminohydrolase 2 (DDAH2) is the enzyme responsible for ADMA hydrolysis in the vasculature, and carriers of the polymorphism rs9267551 C in the 5′-UTR of DDAH2 have been reported to have higher DDAH2 expression and reduced levels of serum ADMA.
Approach and Results: We genotyped rs9267551 in 633 adults of European ancestry and measured their carotid–femoral pulse wave velocity (cfPWV), the gold-standard method to estimate arterial stiffness. cfPWV resulted significantly lower in rs9267551 C allele carriers (Δ = −1.12 m/s, P < 0.01) after correction for age, sex and BMI, and a univariate regression showed that the presence of rs9267551 C variant was negatively associated with cfPWV (β = −0.110, P < 0.01). In a multivariable regression model, subjects carrying the rs9267551 C allele manifested significantly lower cfPWV than GG carriers (β = −0.098, P = 0.01) independently from several potential confounders. We measured circulating ADMA levels in a subset of 344 subjects. A mediation analysis revealed that the effect of DDAH2 rs9267551 genotype on cfPWV was mediated by the variation in ADMA levels.
Conclusions: These evidences hint that the presence of rs9267551 C allele may explain, at least in part, a reduction in vessel rigidity as measured by cfPWV, and support the attribution of a causative role to ADMA in the pathogenesis of arterial stiffness.
Introduction
The arterial wall is characterized by its high elasticity, which participates to the physiological regulation of this highly pressurized compartment. The stiffness of the arterial wall increases gradually from the central segments to the periphery, determining a gradient of blood pressure, and although brachial blood pressure is commonly measured in clinical practice, aortic blood pressure correlates more strictly with markers of cardiovascular risk (1). An augmented rigidity of the vessels is associated with increased occurrence of cardiovascular disease (2), stroke (3) and kidney disease (4). Several factors contribute to the stiffening of arterial walls as a normal consequence of aging, but arterial elasticity may also be found reduced in younger subjects who carry cardiovascular risk factors such as hypertension, obesity and dyslipidemia. The latter phenomenon suggests that exposure of the vessels to the milieu determined by those pathologic conditions may interfere with the balance of hemodynamic forces (5).
Pulse wave velocity (PWV) is a simple measurement representing the average stiffness between two sites of the arterial system, with higher values indicating the presence of higher resistance (stiffer vessels), and it is considered as a bona fide surrogate early marker of atherosclerosis and cardiovascular morbidity and mortality (6, 7). Indeed, several guidelines propose carotid–femoral PWV (cfPWV), the current gold-standard method used to evaluate arterial stiffness, as a conservative method to determine the presence of aortic function alteration (8–10), and it is an independent predictor of cardiovascular events and all-cause mortality in the general population and in high-risk subjects (2, 11–13).
In the subclinical phases of the atherogenic process the reduced availability of the vasodilator hormone nitric oxide (NO) may foster endothelial dysfunction. The endothelial NO synthase (eNOS) is the enzyme responsible for the synthesis of NO in the vascular district, starting from the substrate L-arginine. Asymmetric dimethylarginine (ADMA) is an endogenous methylated form of arginine able to competing with L-arginine and to inhibit NO production (14, 15). Several research efforts have demonstrated the existence of an association between elevated circulating levels of ADMA and major cardiovascular events or mortality (16–20). In spite of this, the predictive role of ADMA is still subject of debate (21–24). Altogether, there is an evidence gap regarding the causal role of ADMA in the dysregulation of arterial blood pressure and endothelial function. In 2006 a small intervention trial demonstrated that acute exogenous administration of ADMA in healthy male volunteers was able to induce a reduction of cerebral blood flow and arterial compliance due to increased arterial stiffness (25). Very recently, Malle et al. (26) have attempted to resolve this gap and they could not detect the presence in hypertensive patients of a significant association between ADMA and blood pressure or PWV. It is worth noticing, though, that observational studies cannot rule out the masking influence of confounding factors, i.e., due to co-morbidities, or establish whether a marker is the cause or the manifestation of a specific condition, due to their non-interventional nature. Given the contrasting reports, the attribution of a causative role to ADMA in the pathogenesis of arterial hypertension in humans remains difficult.
The enzyme dimethylarginine dimethylaminohydrolase (DDAH) (27, 28) is responsible for the hydrolysis of ADMA to citrulline plus methyl-amine, and is encoded by genes DDAH1 and DDAH2. DDAH1 co-localizes with neuronal NOS, whereas DDAH2 can be found in tissues expressing eNOS, such as the endothelium (29). Mendelian randomization is a method that employs genetic polymorphisms known to be able to modify an exposure of interest to determine the existence of a causal association between this exposure (i.e., a biomarker) and a disease. This approach is arguably less prone to reverse causation than other observational studies, because disease status cannot affect germline DNA sequences (30). Most importantly, since genetic variants persist throughout the lifespan, Mendelian randomization studies provide information on life-long exposition to genetically altered levels of biomarkers (in this case, ADMA). The rs9267551 G/C variant in the 5′-untranslated region (UTR) of DDAH2 has been reportedly demonstrated to affect the quantitative expression of DDAH2, specifically in primary human endothelial cells, and a protective role has been proposed for the C allele due to its association with increased NO production (31). Individuals carrying the rs9267551 C allele were shown to have lower circulating levels of ADMA, better response to insulin, and reduced prevalence of chronic kidney disease (31, 32). In a previous work, we reported a significant association with myocardial infarction in two independent cohorts of subjects with type 2 diabetes mellitus, with the C allele consistently showing a protective effect (33). Therefore, we elected to perform a Mendelian randomization study using the functional rs9267551 polymorphism and to assess its effects on in vivo measurements of arterial stiffness (by cfPWV), in a heterogenic cohort of subjects, assuming that carriers of the C allele have a vascular protection (as a consequence of higher NO availability).
Materials and Methods
Study Subjects
For this study we consecutively recruited 633 adults of European ancestry referred to the Department of Medical and Surgical Sciences of the University “Magna Graecia” of Catanzaro (34). The inclusion criteria were: age ≥ 19 years, and presence of one or more cardio-metabolic risk factors including elevated fasting glucose levels, hypertension, dyslipidemia, overweight/obesity, and family history for diabetes. Exclusion criteria were: end-stage renal disease, chronic gastrointestinal diseases or pancreatitis, history of any malignant disease or of alcohol/drug abuse, hepatic failure or positivity for antibodies to hepatitis C virus (HCV) or hepatitis B surface antigen (HBsAg). Venous blood samples were obtained after a 12-h overnight fast. Body mass index (BMI) was calculated as body weight in kilograms divided by the square of height in meters. Readings of blood pressure (BP) were performed in the non-dominant arm with the patient in supine position, after 5 min of rest, with a sphygmomanometer. Type 2 diabetes was defined according to the American Diabetes Association (ADA) criteria (35). Subjects were classified as hypertensive if they had systolic blood pressure ≥ 130 and/or diastolic ≥ 85 mmHg or in presence of antihypertensive treatment and history of hypertension.
As previously reported (36), we adopted a validated system (Sphygmocor™; AtCor Medical, Sydney, Australia), that utilizes high-fidelity applanation tonometry (Millar) and appropriate software for the analysis of pressure waves (Sphygmocor™). Pressure calibration was obtained with patients lying supine, through automatic recording of brachial BP at the dominant arm, after resting for 30 min (Dinamap Compact T; Johnson & Johnson Medical Ltd, Newport, UK). Measurement of BP was repeated five times, and the average of the final three recordings was used for calibration. Pulse wave was measured at the radial artery of the dominant arm with the wrist softly hyperextended, as the average of single pressure waves during eight consecutive seconds. Pulse wave recordings were admitted if peak and bottom values of single waves showed <5% variability. Aortic pulse wave velocity (PWV) was derived from carotid and femoral pressure waveforms. Carotid to femoral transit time (ΔT) was calculated from the foot-to-foot time difference between carotid and femoral waveforms. The distance between the landmark of the sternal notch and femoral artery was used to estimate the path length between the carotid and femoral arteries (L), and PWV was measured as L/ΔT.
The study was approved by the Local Institutional Ethics Committee of the University “Magna Graecia” of Catanzaro (approval code: 2012.63). Written informed consent was obtained from each subject in accordance with the principles of the Declaration of Helsinki.
Analytical Determinations
Glucose, triglyceride, total cholesterol and HDL particles concentration was determined by enzymatic methods (Roche, Basel, Switzerland). Plasma insulin concentration was assessed with a chemiluminescence-based assay (Immulite®, Siemens, Italy). High performance liquid chromatography with a National Glycohemoglobin Standardization Program certified automated analyzer (Adams HA-8160 HbA1C analyzer, Menarini, Italy) was used to measure HbA1c levels. High sensitivity C reactive protein (hsCRP) levels were determined by an automated instrument (CardioPhase® hsCRP, Milan, Italy). Serum ADMA concentration was measured with Human Asymmetric dimethylarginine (ADMA) ELISA Kit (MBS264847, My BioSource, San Diego, CA, USA). The detection range was 5 μmol/L-0.078 μmol/L, with sensitivity up to 0.01 μmol/L, Intra-assay CV ≤ 8% and inter-assay CV ≤ 12%.
Genotyping of DDAH2 Gene Polymorphism
DNA was extracted from whole blood using commercial DNA isolation kits (Promega, Madison, WI and Roche, Mannheim, Germany). rs9267551 DDAH2 genotype calls were assigned by TaqMan allelic discrimination assay (C__27848488_10; Applied Biosystems, Foster City, CA), after amplification on an iCycler Thermal Cycler with iQ5 Multicolor Real-Time PCR Detection System (Bio-Rad Laboratories, Inc., Hercules, CA). 0.05 ng of custom oligo strings (GeneArt® Strings™ DNA Fragments, Invitrogen, Thermo Fisher Scientific) corresponding to ~200 bp around the context sequence of the genotyping assay, differing only for the rs9267551 allele C or G were loaded onto each plate run to represent one heterozygous C/G and two sets of homozygous C/C and G/G controls. Genotyping concordance of the oligo strings was 100%.
Statistical Analysis
The estimation of sample size requirements was performed with the program Quanto (version 1.2) and based upon previously reported data obtained in populations similar to ours. To compute our model we assumed an average cfPWV between 6.7 and 7.5 m/s, with a standard deviation (SD) ranging between 1.8 and 2.5 m/s (37, 38), and a minimum minor allele frequency = 4.5% (31–33). In order to be able to detect a clinically relevant difference in cfPWV between genotypes according to a dominant model [at least 1 m/s (13, 39)], with two-sided α = 0.05 and β = 0.80, the recommended sample size was equal to 607 subjects. Each SNP was coded as 0, 1, or 2 depending on the number of C alleles. Therapies were coded as binary variables, 0 indicated absence of treatment, 1, respectively, meant use of anti-hypertensive, glucose lowering agents, or statins as anti-dyslipidemic therapy. Log transformation was employed when analyzing insulin, triglycerides and hsCRP levels because their distribution did not respect the assumption of normality. Comparison of differences between continuous variables in genotype groups were tested by ANCOVA (general linear model) after adjusting for age, sex, and BMI. Categorical variables were compared by χ2 test. The Hardy–Weinberg equilibrium between genotypes was evaluated by χ2 test. The existing relationships between cfPWV and all collected clinical, biochemical and anthropometrical parameters were explored by univariate regression analysis, with cfPWV as dependent variable. To assess which variables were independently associated with cfPWV we built an exploratory stepwise multivariable linear regression model. The resulting predictive variables were used to compile a predictor factor through principal component analysis. The quality of the reduced-dimension factor was determined through the Bartlett's test for sphercity. The Mediation analysis was conducted through a series of linear regression analyses to calculate the indirect and direct effects and test them for significance (40); we adopted the product of coefficients approach to test the mediating effect of ADMA on the variability of cfPWV, and we tested the significance of the mediation through the Sobel test (41). The significance of indirect and total effects was estimated via bootstrapping (40). A two-sided p-value < 0.05 was considered statistically significant. All calculations were done with SPSS software program Version 22.0 for Windows.
Results
A summary of the clinical and anthropometric characteristics of the population, stratified by rs9267551 polymorphism, is reported in Table 1. The study population consisted of 633 unrelated Caucasian subjects (425 men and 208 women with mean age 52 ± 12 years), who were enrolled in the CATAMERI study (42). The genotype distribution of rs9267551 polymorphism was in Hardy–Weinberg equilibrium (p > 0.10). GC and CC individuals were collectively considered and analyzed as C carriers, according to a dominant genetic model, because we detected only three individuals with rs9267551 CC homozygous genotype, and because previous functional studies performed in endothelial cells (31) had demonstrated a dominant effect of the C allele. When comparing the two groups (GG vs. GC + CC) the rs9267551 polymorphism did not show any significant association with age, sex, BMI, systolic, and diastolic blood pressure, circulating levels of hsCRP, smoking status, and therapy for hypertension (Table 1). After correction for age, sex and BMI, cfPWV was significantly lower in carriers of the C allele as compared with subjects carrying the GG genotype (6.86 vs. 7.98 m/s, respectively; P < 0.01), supporting the hypothesis that constant exposure to higher ADMA levels might lead to reduced vascular elasticity. No differences in lipid profile were observed between groups, despite the higher prevalence of hypolipidemic therapies in the GG group (20.6 vs. 3.8% in the GC + CC group, P = 0.01). Similarly, no differences were reported in fasting plasma glucose and insulin levels, although it is worth noticing that the GC + CC group harbored a significantly lower proportion of diabetic subjects (10.3 vs. 29.2% in the GG group, P = 0.022) and of subjects undergoing a hypoglycemic regimen (3.8 vs. 15.1% in the GG group, P = 0.049).
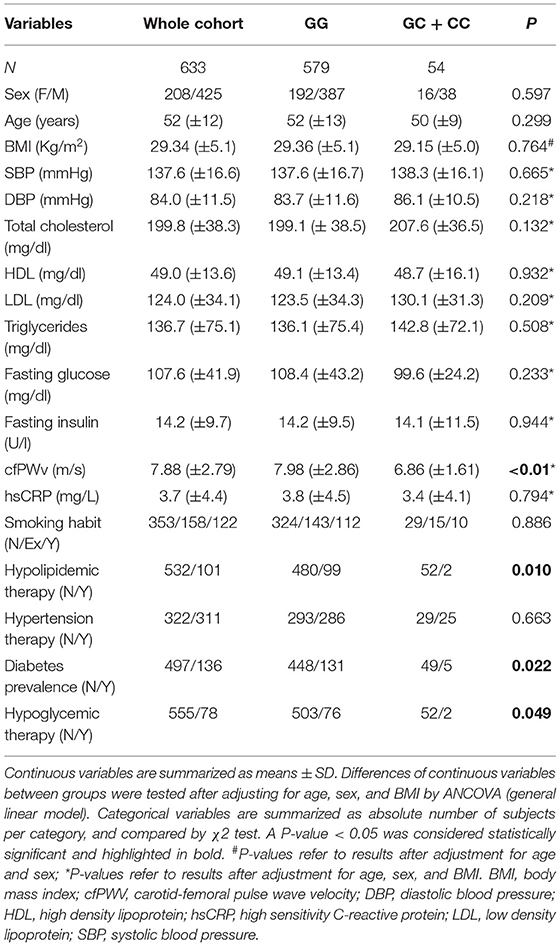
Table 1. Clinical features of 633 study subjects according to the rs9267551 polymorphism of DDAH2.
The association of cfPWV with clinical, biochemical and anthropometrical parameters is reported in Table 2. cfPWV resulted positively and significantly associated with age (β = 0.230, P < 0.001), systolic blood pressure (β = 0.152, P < 0.001), hsCRP (β = 0.116, P < 0.01), fasting plasma glucose (β = 0.133, P < 0.001), fasting plasma insulin (β = 0.142, P = 0.001), prevalence of diabetes (β = 0.157, P < 0.001), and prevalence of treatment with hypoglycemic agents (β = −0.138, P < 0.001). On the other hand, circulating HDL levels (β = −0.091, P = 0.024) and DDAH2 rs9267551 C allele (β = −0.110, P < 0.01) showed a negative effect on cfPWV, consistent with their protective role exerted on the vasculature. Sex is notoriously associated with cardiovascular risk, but in our population we found that male subjects had only a fringe association with higher cfPWV (β = 0.077, P = 0.053). No significant associations were detected between values of cfPWV in our population and BMI, diastolic blood pressure, total cholesterol, LDL particles, circulating triglycerides, prevalence of treatment for hypertension or dyslipidemia, and smoking habit (Table 2).
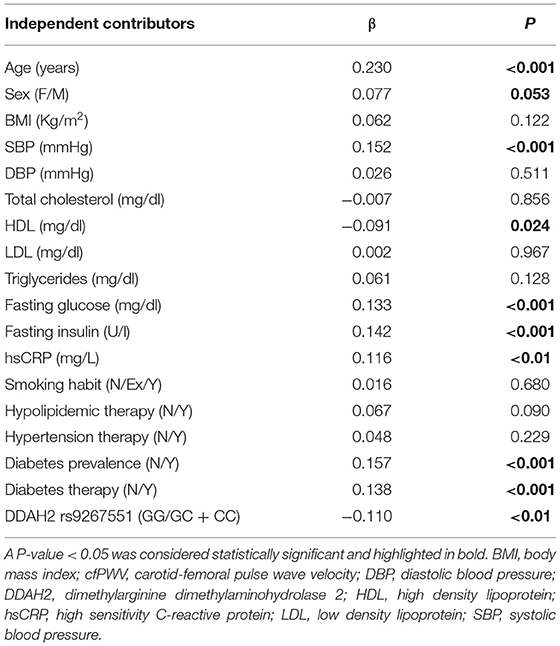
Table 2. Univariate regression analysis with cfPWv as dependent variable.
To estimate the independent contribution of rs9267551 polymorphism to arterial stiffness evaluated through cfPWV, we carried out a multivariable linear regression analysis. The covariates were selected as follows: traditional confounder factors affecting PWV (age, sex, BMI, and smoking habit), variables that resulted significantly associated to cfPWV in our population in the univariate regression analysis mentioned above (SBP, HDL, fasting plasma glucose and insulin, hsCRP, diabetes prevalence and hypoglycemic therapy, Table 2). In order to avoid conflict due to the fact that fasting glucose and insulin levels are strongly correlated to diabetes diagnosis, we built two independent statistical models featuring, respectively, fasting glucose and fasting insulin in Model A and diabetes prevalence in Model B (Table 3). Additionally, when we assessed a third Model, in which treatment with hypoglycemic agents was used as a possible confounding variable in the stead of diabetes prevalence or fasting glucose and insulin levels, we observed no significant influence exerted by the pharmacological treatment on cfPWV (data not shown). The major independent determinants of cfPWV according to Model A were (listed from strongest to weakest): age (β = 0.201, P < 0.001), fasting insulin (β = 0.134, P < 0.001), systolic blood pressure (β = 0.111, P < 0.01) and the rs9267551 polymorphism, with carriers of the C allele having significantly lower vessel rigidity as compared with GG individuals (β = −0.098, P = 0.01) (Table 3). Model B revealed a similar pattern of association, with age (β = 0.199, P < 0.001), systolic blood pressure (β = 0.105, P < 0.01), DDAH2 rs9267551 genotype (β = −0.100, P < 0.01), plus hsCRP (β = 0.078, P = 0.044), and HDL (β = −0.076, P = 0.049) (Table 3). As a dependent variable, cfPWV did not show any significant association with sex, BMI or smoking habit in neither Model A or B (Table 3).
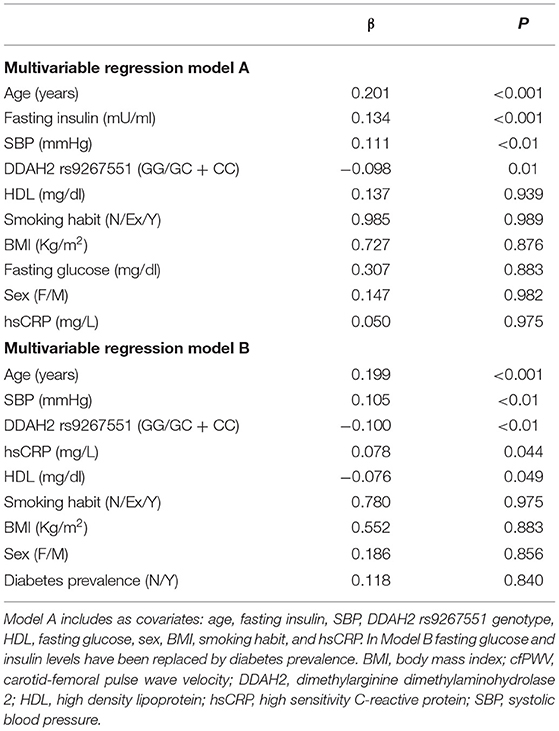
Table 3. Stepwise multiple regression analysis with cfPWv as dependent variable.
In order to exclude any confounding influence deriving from other factors associated with DDAH2 rs9267551 genotype (by chance, or pleiotropy), we constructed a final multivariable regression model (Supplementary Table) encompassing traditional risk factors for arterial stiffness and covariates which resulted associated either with cfPWV, as in Model B, plus the remaining variables associated to rs9267551 in our population (hypolypidemic therapy, Table 1). The independent contribution of each trait to the variability of cfPWV resulting from this supplementary model was almost superimposable to the results obtained from Model B. Serum ADMA concentrations were assessed for a subset of 344 subjects whose biological specimen were available. In this subset there were 321 GG genotypes, and 23 carriers of the C allele (21 subjects with GC genotype and 2 CC homozygous). Consistent with previous data, circulating ADMA was higher in GG individuals than in C allele carriers (0.65 ± 0.31 vs. 0.52 ± 0.21 μmol/l, respectively; P = 0.045 after correction for age, sex, and BMI, Figure 1). At first, we elected to replicate the analysis as in multivariable regression Model B in order to ensure that the subset was a bona fide representative of the full cohort, and we confirmed that the observed genetic effect was preserved (Model C, Table 4) with DDAH2 rs9267551 C allele exerting a comparable protective action (β = −0.102, P = 0.04) over cfPWV. When we added the measurement of serum ADMA levels to the statistical model (Model D, Table 4), it manifested a stronger independent effect on cfPWV (β = 0.139, P = 0.006), and the contribution of DDAH2 rs9267551 genotype became non-significant (β = 0.088, P = 0.969).
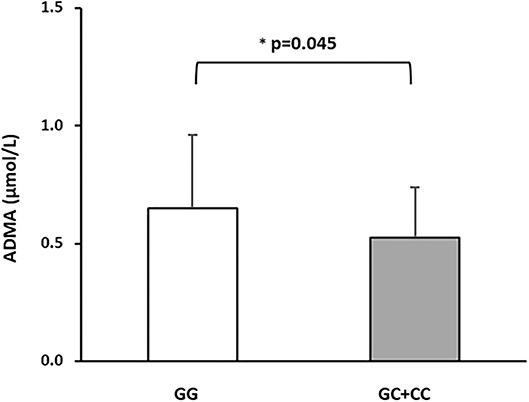
Figure 1. Differences in circulating ADMA concentrations in a subset of 344 subjects stratified based on DDAH2 rs9267551 genotypes. The white bar represents the rs9267551 GG genotype (n = 321) and the grey bar represents the rs9267551 GC + CC genotype (21 subjects with GC genotype and 2 with CC homozygous). *p-value after correction of age, sex, and BMI. Data are means ± Standard Deviation.
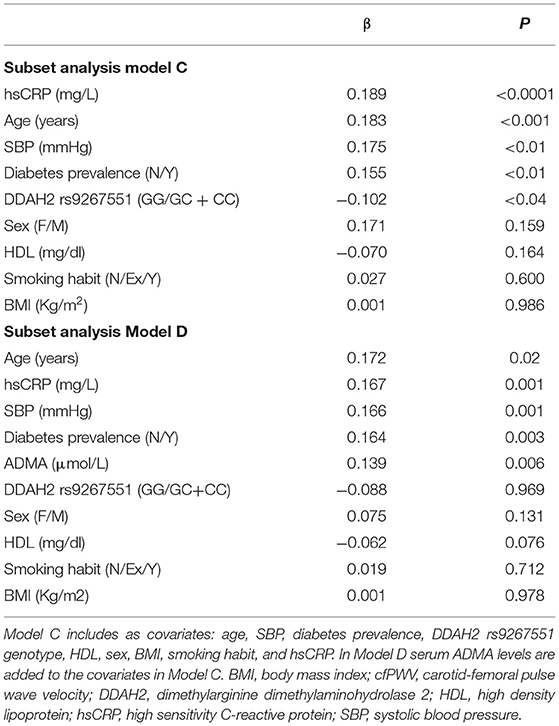
Table 4. Stepwise multiple regression analysis in the subset sample, with cfPWv as dependent variable.
This observation prompted us to the performance of a mediation analysis, graphically summarized in Figure 2. As predictor variable we adopted a factor produced by principal component analysis encompassing age, hsCRP, SBP, diagnosis of diabetes and DDAH2 rs9267551 genotype (χ2 = 90.8, P < 0.0001). This predictor factor had eigenvalue = 1.490 and explained 29.806% of total variance. The total effect of the predictor factor on cfPWV, including the indirect and direct effect, is represented by the coefficient c (β = 0.742, SE = 0.116, P < 0.00001). The coefficient a (β = 0.046, SE = 0.016, P = 0.006) represents the effect of the predictor factor on ADMA, and the coefficient b (β = 1.307, SE = 0.374, P = 0.0005) represents the effect of ADMA on cfPWV. The coefficient c' (β = 0.682, SE = 0.115, P < 0.00001) represents the direct effect of the predictor factor on cfPWV, after correcting for the effect of ADMA. Finally, the mediated (or indirect) effect of the predictor factor on cfPWV through ADMA is represented as a*b (β = 0.059, SE = 0.028), and it was statistically significant via the bootstrapped estimation approach of the Sobel test, (P = 0.034). Thus, as both the direct and the mediated effects between the predictor factor and cfPWV were statistically significant, it may be suggested that a partial mediation exists, and this hypothesis is further supported by the fact that the direct effect c' has a smaller magnitude than the total effect c. Because DDAH2 polymorphism rs9267551 was the only parameter included in the predictor factor to lose its association with cfPWV when circulating ADMA levels were added in the multivariable regression Model D, it is possible to infer that the effect of rs9267551 genotype on cfPWV is mediated by ADMA.
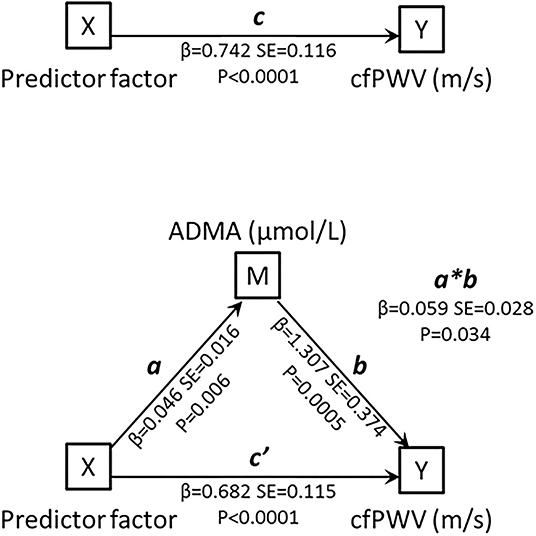
Figure 2. Schematic representation of the mediation model. The mediation analyses were conducted using a series of linear regression models to estimate the total effect of X on Y (c), the indirect effect of X on Y (a*b), and the direct effect of X on Y with removal of the effect of the mediator (c'). M, mediating variable; X, independent variable; Y, dependent variable.
Discussion
In the current study, we report, for the first time, evidences hinting that the presence of rs9267551 C allele may protect, at least in part, from an increase in arterial stiffness as measured by cfPWV, independently from the presence of traditional cardiovascular risk factors including age, sex, BMI, smoking habit and type 2 diabetes. Furthermore, our observations support the attribution of a causative role to ADMA in the pathogenesis of arterial rigidity, in line with evidences that long-term ADMA infusion is able to induce arteriosclerotic damage in an animal model of eNOS deficient mice (43), and that the acute infusion of subpressor doses of ADMA (0.10 mg/kg/min) increases vascular stiffness and decreases cerebral perfusion in healthy humans, independently from changes in blood pressure (25).
The measurement of PWV is a simple, non-invasive, and trustworthy method to evaluate the rigidity of a segment in the arterial system, and its use is advised by several guidelines (8, 10). More specifically, cfPWV is considered the gold-standard method for the measurement of aortic stiffness in a simple and non-invasive way (44). Several studies have shown that high cfPWV predict the occurrence of cardiovascular diseases even more than other traditional major risk factors (2, 6, 11, 12, 45, 46). Our population showed a difference in cfPWV of more than 1 m/s when the groups based on rs9267551 polymorphism were compared (Table 1). This difference is clinically relevant, as discussed in 2010 by the meta-analysis authored by Vlachopoulos et al. (13) that evaluated 15,877 ethnically etherogenous subjects with variable high baseline risk (populations with hypertension, diabetes, end-stage renal disease, coronary artery disease, and subjects from the general population). The results of the meta-analysis revealed that the relative risk associated with an increase of 1 m/s in aortic PWV was 1.14 (95% CI: 1.09-1.20) for total CV events, 1.15 (95% CI: 1.09-1.21) for cardiovascular mortality, and 1.15 (95% CI: 1.09-1.21) for all-cause mortality.
ADMA is an endogenous inhibitor of eNOS activity, it competes for the binding of eNOS active site with L-Arginine, which is the substrate for the synthesis of NO. There is substantial evidence that NO bioavailability is associated with the levels of various components of the L-arginine/NO pathway (47), and several studies have documented a significant association between ADMA and hypertension (48–50). Furthermore, increased levels of ADMA are considered predictors of cardiovascular morbidity and mortality (18) and have been proposed as an explanation for the reduced production of NO typically observed in insulin resistance (50–52). Hence, serum ADMA levels have the potential to serve as diagnostic markers and therapeutic targets, however, animal and human studies have supplied conflicting results, and could not ultimately determine whether ADMA has a causal role in the pathogenesis of arterial stiffness and hypertension (26, 53, 54).
The hydrolysis of ADMA catalyzed by the DDAH enzyme is a major determinant of plasma ADMA concentration (27). Notably, animal models in which the modulation of ADMA had been constricted through overexpression or knockout of DDAH have provided support to the hypothesis that ADMA might play a pathogenic role (53–55), whereas other models have failed to report consistent evidences (56–58). This discrepancy suggests that pharmacological lowering of ADMA by targeting DDAH could be the key to understand whether ADMA directly plays a role in human cardiovascular pathology.
Demonstrating the existence of a causal role for ADMA in vessel dysfunction has the potential to determine meaningful changes in the clinical practice, and few therapeutic approaches specifically targeting ADMA pathways have already been proposed (59, 60). Among them, there is one experimental molecule with DDAH properties (M-DDAH) that resulted effective at lowering ADMA in preclinical models (61). Therefore, it is important to clarify the pathological role of ADMA in humans, in order to set the foundation in support of future studies. To achieve this objective we applied a Mendelian randomization study design, with the final goal of understanding if ADMA levels are associated with higher vascular stiffness, measured through cfPWV. This approach requires that the selected instrument (in this case SNP rs9267551) associates with the exposure. In our study, ADMA is the exposure and, although previous researches evaluating the impact of genetic variation of the DDAH2 locus with cardiovascular risk have not supplied consistent results (62–64), the rs9267551 variant in the 5′-UTR of DDAH2 has been proven to have a robust functional impact: primary human endothelial cells obtained from donors carrying the C allele showed increased transcription of DDAH2; and circulating ADMA levels were reduced in adult human subjects carrying the C allele when compared to the rs9267551 GG homozygous group (31). These evidences suggest that the rs9267551 C polymorphism in the locus of DDAH2 may protect eNOS activity and NO production through its effects on ADMA levels. Indeed, in a previous published paper, we reported a role for our instrument, the functional polymorphism rs9267551, in modulating the risk of myocardial infarction even among diabetic patients (33). To date, no data are available in the literature about the impact of polymorphism rs9267551 on arterial stiffness.
Our results support the hypothesis that rs9267551 genotype may affect cfPWV, and suggest that this effect is mediated by circulating ADMA levels. In addition to this, it is worth noticing that the association between rs9267551 and cfPWV was independent from blood pressure, an observation that, if confirmed, might help elucidate, at least partially, the discrepancies in the literature about the causal role of ADMA in the pathogenesis of arterial stiffness and hypertension. Indeed, our data hint that long-term exposure to higher ADMA levels determines a specific vascular risk via the alteration of vessel elasticity (as measured by cfPWV) without affecting arterial hypertension.
The present study has some strengths including the exclusion of possible biases deriving from the presence of infectious or malignant conditions by study design, the accurate characterization of all enrolled subjects (no self-reported data were used) that permitted to encompass multiple recognized cardio-metabolic risk factors in the analyses, and the compliance with the assumptions of Mendelian randomization. Moreover, the homogeneity of the ethnicity of the population (all Italians of European descent), the accuracy of data collection via standardized protocol, and the centralized laboratory determinations, represent other points of strength of our analysis.
Notwithstanding this, several limitations are also present. First, the cross-sectional nature of the design does not permit to evaluate incident vascular dysfunction; therefore the present results require replication in prospective studies for validation. Furthermore, unfortunately, due to the low minor allele frequency, we were not able to establish the effect relative to allelic dosage (“per allele” effect), and biological specimen for laboratory determination of circulating ADMA levels were only available for a subset of our population. However, we have been able to confirm our previous report that ADMA levels were significantly higher in individuals with rs9267551 GG genotype than in C allele carriers (31). Although the robustness of our main claim (the association between rs9267551 genotype and cfPWV) is significantly reinforced by the positive independent linear relationship between circulating ADMA levels and cfPWV, we cannot completely exclude the influence of residual confounding. Additionally, caution should be exerted in extending the present findings to the general population, because they were based upon observational data collected from Caucasian subjects of European ancestry with mild to high cardiometabolic risk.
In conclusion, to the best of our knowledge, we are supplying the first evidences in support of the hypothesis that the variant rs9267551 in the DDAH2 gene can modulate cfPWV in Italian adult subjects, and we have been able to demonstrate the existence of an interaction between rs9267551 genotype and circulating ADMA levels, able to affect arterial stiffness. Hopefully, replication of these results in future studies will allow their extension to other populations of different ethnic background, with the final goal of definitively unraveling the role of ADMA in the pathogenesis of vascular risk and if intervening on DDAH2 may turn out to be a fruitful therapeutic/preventive strategy.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The study was approved by the Institutional Ethics Committee of the University Magna Graecia of Catanzaro (Approval Code: 2012.63). Written informed consent was obtained from each subject in accordance with the principles of the Declaration of Helsinki.
Author Contributions
GM, CA, and EM researched data, reviewed, and edited the article. EM, RS, SM, TF, GM, ES, and AS researched data. GM, PJ, and GS contributed to the discussion and reviewed the article. GM and GS analyzed the data and reviewed the article. FA designed the study, analyzed the data, and wrote the article. All authors contributed to the article and approved the submitted version.
Funding
GM was supported by funds from the EU project AIM1829805-3.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2021.811431/full#supplementary-material
References
1. Avolio AP, Van Bortel LM, Boutouyrie P, Cockcroft JR, McEniery CM, Protogerou AD, et al. Role of pulse pressure amplification in arterial hypertension: experts' opinion and review of the data. Hypertension. (2009) 54:375–83. doi: 10.1161/HYPERTENSIONAHA.109.134379
2. Willum-Hansen T, Staessen JA, Torp-Pedersen C, Rasmussen S, Thijs L, Ibsen H, et al. Prognostic value of aortic pulse wave velocity as index of arterial stiffness in the general population. Circulation. (2006) 113:664–70. doi: 10.1161/CIRCULATIONAHA.105.579342
3. Mattace-Raso FUS, van der Cammen TJM, Hofman A, van Popele NM, Bos ML, Schalekamp MADH, et al. Arterial stiffness and risk of coronary heart disease and stroke: the Rotterdam Study. Circulation. (2006) 113:657–63. doi: 10.1161/CIRCULATIONAHA.105.555235
4. Safar ME, London GM, Plante GE. Arterial stiffness and kidney function. Hypertension. (2004) 43:163–8. doi: 10.1161/01.HYP.0000114571.75762.b0
5. Mitchell GF, Parise H, Benjamin EJ, Larson MG, Keyes MJ, Vita JA, et al. Changes in arterial stiffness and wave reflection with advancing age in healthy men and women: the Framingham Heart Study. Hypertension. (2004) 43:1239–45. doi: 10.1161/01.HYP.0000128420.01881.aa
6. Boutouyrie P, Tropeano AI, Asmar R, Gautier I, Benetos A, Lacolley P, et al. Aortic stiffness is an independent predictor of primary coronary events in hypertensive patients: a longitudinal study. Hypertension. (2002) 39:10–5. doi: 10.1161/hy0102.099031
7. Vlachopoulos C, Aznaouridis K, Terentes-Printzios D, Ioakeimidis N, Stefanadis C. Prediction of cardiovascular events and all-cause mortality with brachial-ankle elasticity index: a systematic review and meta-analysis. Hypertension. (2012) 60:556–62. doi: 10.1161/HYPERTENSIONAHA.112.194779
8. Ogihara T, Kikuchi K, Matsuoka H, Fujita T, Higaki J, Horiuchi M, et al. The Japanese Society of Hypertension Guidelines for the Management of Hypertension (JSH 2009). Hypertens Res. (2009) 32:3–107. doi: 10.1038/hr.2009.34
9. Covic A, Siriopol D. Pulse wave velocity ratio: the new “gold standard” for measuring arterial stiffness. Hypertension. (2015) 65:289–90. doi: 10.1161/HYPERTENSIONAHA.114.04678
10. Williams B, Mancia G, Spiering W, Agabiti Rosei E, Azizi M, Burnier M, et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension: the task force for the management of arterial hypertension of the European Society of Cardiology and the European Society of Hypertension: the task force for the management of arterial hypertension of the European Society of Cardiology and the European Society of Hypertension. J Hypertens. (2018) 36:1953–2041. doi: 10.1097/HJH.0000000000001940
11. Laurent S, Boutouyrie P, Asmar R, Gautier I, Laloux B, Guize L, et al. Aortic stiffness is an independent predictor of all-cause and cardiovascular mortality in hypertensive patients. Hypertension. (2001) 37:1236–41. doi: 10.1161/01.HYP.37.5.1236
12. Laurent S, Katsahian S, Fassot C, Tropeano A-I, Gautier I, Laloux B, et al. Aortic stiffness is an independent predictor of fatal stroke in essential hypertension. Stroke. (2003) 34:1203–6. doi: 10.1161/01.STR.0000065428.03209.64
13. Vlachopoulos C, Aznaouridis K, Stefanadis C. Prediction of cardiovascular events and all-cause mortality with arterial stiffness: a systematic review and meta-analysis. J Am Coll Cardiol. (2010) 55:1318–27. doi: 10.1016/j.jacc.2009.10.061
14. Cooke JP. Does ADMA cause endothelial dysfunction? Arterioscler Thromb Vasc Biol. (2000) 20:2032–7. doi: 10.1161/01.ATV.20.9.2032
15. Perticone F, Sciacqua A, Maio R, Perticone M, Maas R, Boger RH, et al. Asymmetric dimethylarginine, L-arginine, and endothelial dysfunction in essential hypertension. J Am Coll Cardiol. (2005) 46:518–23. doi: 10.1016/j.jacc.2005.04.040
16. Böger RH, Maas R, Schulze F, Schwedhelm E. Asymmetric dimethylarginine (ADMA) as a prospective marker of cardiovascular disease and mortality–an update on patient populations with a wide range of cardiovascular risk. Pharmacol Res. (2009) 60:481–7. doi: 10.1016/j.phrs.2009.07.001
17. Willeit P, Freitag DF, Laukkanen JA, Chowdhury S, Gobin R, Mayr M, et al. Asymmetric dimethylarginine and cardiovascular risk: systematic review and meta-analysis of 22 prospective studies. J Am Heart Assoc. (2015) 4:e001833. doi: 10.1161/JAHA.115.001833
18. Schlesinger S, Sonntag SR, Lieb W, Maas R. Asymmetric and symmetric dimethylarginine as risk markers for total mortality and cardiovascular outcomes: a systematic review and meta-analysis of prospective studies. PLoS ONE. (2016) 11:e0165811. doi: 10.1371/journal.pone.0165811
19. Zhou S, Zhu Q, Li X, Chen C, Liu J, Ye Y, et al. Asymmetric dimethylarginine and all-cause mortality: a systematic review and meta-analysis. Sci Rep. (2017) 7:44692. doi: 10.1038/srep44692
20. Xuan C, Tian Q-W, Li H, Zhang B-B, He G-W, Lun L-M. Levels of asymmetric dimethylarginine (ADMA), an endogenous nitric oxide synthase inhibitor, and risk of coronary artery disease: a meta-analysis based on 4713 participants. Eur J Prev Cardiol. (2016) 23:502–10. doi: 10.1177/2047487315586094
21. Krzyzanowska K, Mittermayer F, Wolzt M, Schernthaner G. Asymmetric dimethylarginine predicts cardiovascular events in patients with type 2 diabetes. Diabetes Care. (2007) 30:1834–9. doi: 10.2337/dc07-0019
22. Lu T-M, Chung M-Y, Lin M-W, Hsu C-P, Lin S-J. Plasma asymmetric dimethylarginine predicts death and major adverse cardiovascular events in individuals referred for coronary angiography. Int J Cardiol. (2011) 153:135–40. doi: 10.1016/j.ijcard.2011.06.120
23. Tarnow L, Hovind P, Teerlink T, Stehouwer CDA, Parving H-H. Elevated plasma asymmetric dimethylarginine as a marker of cardiovascular morbidity in early diabetic nephropathy in type 1 diabetes. Diabetes Care. (2004) 27:765–9. doi: 10.2337/diacare.27.3.765
24. Zobel EH, von Scholten BJ, Reinhard H, Persson F, Teerlink T, Hansen TW, et al. Symmetric and asymmetric dimethylarginine as risk markers of cardiovascular disease, all-cause mortality and deterioration in kidney function in persons with type 2 diabetes and microalbuminuria. Cardiovasc Diabetol. (2017) 16:88. doi: 10.1186/s12933-017-0569-8
25. Kielstein JT, Donnerstag F, Gasper S, Menne J, Kielstein A, Martens-Lobenhoffer J, et al. ADMA increases arterial stiffness and decreases cerebral blood flow in humans. Stroke. (2006) 37:2024–9. doi: 10.1161/01.STR.0000231640.32543.11
26. Malle O, Trummer C, Theiler-Schwetz V, Meinitzer A, Keppel MH, Grübler MR, et al. NO synthesis markers are not significantly associated with blood pressure and endothelial dysfunction in patients with arterial hypertension: a cross-sectional study. J Clin Med. (2020) 9:3895. doi: 10.3390/jcm9123895
27. Ogawa T, Kimoto M, Watanabe H, Sasaoka K. Metabolism of NG,NG-and NG,N'G-dimethylarginine in rats. Arch Biochem Biophys. (1987) 252:526–37. doi: 10.1016/0003-9861(87)90060-9
28. Ogawa T, Kimoto M, Sasaoka K. Purification and properties of a new enzyme, NG,NG-dimethylarginine dimethylaminohydrolase, from rat kidney. J Biol Chem. (1989) 264:10205–9. doi: 10.1016/S0021-9258(18)81786-0
29. Leiper JM, Santa Maria J, Chubb A, MacAllister RJ, Charles IG, Whitley GS, et al. Identification of two human dimethylarginine dimethylaminohydrolases with distinct tissue distributions and homology with microbial arginine deiminases. Biochem J. (1999) 343:209–14. doi: 10.1042/bj3430209
30. Thanassoulis G. Mendelian randomization: how genetics is pushing the boundaries of epidemiology to identify new causes of heart disease. Can J Cardiol. (2013) 29:30–6. doi: 10.1016/j.cjca.2012.09.014
31. Andreozzi F, Presta I, Mannino GC, Scarpelli D, Di Silvestre S, Di Pietro N, et al. A functional variant of the dimethylarginine dimethylaminohydrolase-2 gene is associated with insulin sensitivity. PLoS ONE. (2012) 7:e36224. doi: 10.1371/journal.pone.0036224
32. Sesti G, Mannino GC, De Lorenzo C, Greco A, Sciacqua A, Marini MA, et al. A functional variant of the dimethylarginine dimethylaminohydrolase-2 gene is associated with chronic kidney disease. Atherosclerosis. (2013) 231:141–4. doi: 10.1016/j.atherosclerosis.2013.08.041
33. Mannino GC, Pezzilli S, Averta C, Fuoco A, Spiga R, Mancuso E, et al. A functional variant of the dimethylarginine dimethylaminohydrolase-2 gene is associated with myocardial infarction in type 2 diabetic patients. Cardiovasc Diabetol. (2019) 18:102. doi: 10.1186/s12933-019-0906-1
34. Scarpelli D, Cardellini M, Andreozzi F, Laratta E, Hribal ML, Marini MA, et al. Variants of the interleukin-10 promoter gene are associated with obesity and insulin resistance but not type 2 diabetes in caucasian italian subjects. Diabetes. (2006) 55:1529–33. doi: 10.2337/db06-0047
35. American Diabetes Association. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2019. Diabetes Care. (2019) 42(Suppl 1):S13–28. doi: 10.2337/dc19-S002
36. Perticone M, Maio R, Tassone EJ, Tripepi G, Di Cello S, Miceli S, et al. Insulin-resistance HCV infection-related affects vascular stiffness in normotensives. Atherosclerosis. (2015) 238:108–12. doi: 10.1016/j.atherosclerosis.2014.11.025
37. Sciacqua A, Maio R, Miceli S, Pascale A, Carullo G, Grillo N, et al. Association between one-hour post-load plasma glucose levels and vascular stiffness in essential hypertension. PLoS ONE. (2012) 7:e44470. doi: 10.1371/journal.pone.0044470
38. Cassano V, Crescibene D, Hribal ML, Pelaia C, Armentaro G, Magurno M, et al. Uric acid and vascular damage in essential hypertension: role of insulin resistance. Nutrients. (2020) 12:E2509. doi: 10.3390/nu12092509
39. Kousios A, Kouis P, Hadjivasilis A, Panayiotou A. Cardiovascular risk assessment using ultrasonographic surrogate markers of atherosclerosis and arterial stiffness in patients with chronic renal impairment: a narrative review of the evidence and a critical view of their utility in clinical practice. Can J Kidney Health Dis. (2020) 7:2054358120954939. doi: 10.1177/2054358120954939
40. Preacher KJ, Hayes AF. SPSS and SAS procedures for estimating indirect effects in simple mediation models. Behav Res Methods Instrum Comput. (2004) 36:717–31. doi: 10.3758/BF03206553
41. MacKinnon DP, Lockwood CM, Hoffman JM, West SG, Sheets V. A comparison of methods to test mediation and other intervening variable effects. Psychol Methods. (2002) 7:83–104. doi: 10.1037/1082-989X.7.1.83
42. Andreozzi F, Succurro E, Mancuso MR, Perticone M, Sciacqua A, Perticone F, et al. Metabolic and cardiovascular risk factors in subjects with impaired fasting glucose: the 100 versus 110 mg/dL threshold. Diabetes Metab Res Rev. (2007) 23:547–50. doi: 10.1002/dmrr.724
43. Suda O, Tsutsui M, Morishita T, Tasaki H, Ueno S, Nakata S, et al. Asymmetric dimethylarginine produces vascular lesions in endothelial nitric oxide synthase-deficient mice: involvement of renin-angiotensin system and oxidative stress. Arterioscler Thromb Vasc Biol. (2004) 24:1682–8. doi: 10.1161/01.ATV.0000136656.26019.6e
44. Laurent S, Cockcroft J, Van Bortel L, Boutouyrie P, Giannattasio C, Hayoz D, et al. Expert consensus document on arterial stiffness: methodological issues and clinical applications. Eur Heart J. (2006) 27:2588–605. doi: 10.1093/eurheartj/ehl254
45. Ben-Shlomo Y, Spears M, Boustred C, May M, Anderson SG, Benjamin EJ, et al. Aortic pulse wave velocity improves cardiovascular event prediction: an individual participant meta-analysis of prospective observational data from 17,635 subjects. J Am Coll Cardiol. (2014) 63:636–46. doi: 10.1016/j.jacc.2013.09.063
46. Mitchell GF, Hwang S-J, Vasan RS, Larson MG, Pencina MJ, Hamburg NM, et al. Arterial stiffness and cardiovascular events: the Framingham Heart Study. Circulation. (2010) 121:505–11. doi: 10.1161/CIRCULATIONAHA.109.886655
47. Mels CMC, Huisman HW, Smith W, Schutte R, Schwedhelm E, Atzler D, et al. The relationship of nitric oxide synthesis capacity, oxidative stress, and albumin-to-creatinine ratio in black and white men: the SABPA study. Age. (2016) 38:9. doi: 10.1007/s11357-016-9873-6
48. Surdacki A, Nowicki M, Sandmann J, Tsikas D, Boeger RH, Bode-Boeger SM, et al. Reduced urinary excretion of nitric oxide metabolites and increased plasma levels of asymmetric dimethylarginine in men with essential hypertension. J Cardiovasc Pharmacol. (1999) 33:652–8. doi: 10.1097/00005344-199904000-00020
49. Achan V, Broadhead M, Malaki M, Whitley G, Leiper J, MacAllister R, et al. Asymmetric dimethylarginine causes hypertension and cardiac dysfunction in humans and is actively metabolized by dimethylarginine dimethylaminohydrolase. Arterioscler Thromb Vasc Biol. (2003) 23:1455–9. doi: 10.1161/01.ATV.0000081742.92006.59
50. Perticone F, Sciacqua A, Maio R, Perticone M, Galiano Leone G, Bruni R, et al. Endothelial dysfunction, ADMA and insulin resistance in essential hypertension. Int J Cardiol. (2010) 142:236–41. doi: 10.1016/j.ijcard.2008.12.131
51. Stühlinger MC, Abbasi F, Chu JW, Lamendola C, McLaughlin TL, Cooke JP, et al. Relationship between insulin resistance and an endogenous nitric oxide synthase inhibitor. JAMA. (2002) 287:1420–6. doi: 10.1001/jama.287.11.1420
52. McLaughlin T, Stühlinger M, Lamendola C, Abbasi F, Bialek J, Reaven GM, et al. Plasma asymmetric dimethylarginine concentrations are elevated in obese insulin-resistant women and fall with weight loss. J Clin Endocrinol Metab. (2006) 91:1896–900. doi: 10.1210/jc.2005-1441
53. Leiper J, Nandi M, Torondel B, Murray-Rust J, Malaki M, O'Hara B, et al. Disruption of methylarginine metabolism impairs vascular homeostasis. Nat Med. (2007) 13:198–203. doi: 10.1038/nm1543
54. Hasegawa K, Wakino S, Tatematsu S, Yoshioka K, Homma K, Sugano N, et al. Role of asymmetric dimethylarginine in vascular injury in transgenic mice overexpressing dimethylarginie dimethylaminohydrolase 2. Circ Res. (2007) 101:e2-10. doi: 10.1161/CIRCRESAHA.107.156901
55. Xu X, Zhang P, Kwak D, Fassett J, Yue W, Atzler D, et al. Cardiomyocyte dimethylarginine dimethylaminohydrolase-1 (DDAH1) plays an important role in attenuating ventricular hypertrophy and dysfunction. Basic Res Cardiol. (2017) 112:55. doi: 10.1007/s00395-017-0644-z
56. Nakayama Y, Ueda S, Yamagishi S, Obara N, Taguchi K, Ando R, et al. Asymmetric dimethylarginine accumulates in the kidney during ischemia/reperfusion injury. Kidney Int. (2014) 85:570–8. doi: 10.1038/ki.2013.398
57. Rodionov RN, Martens-Lobenhoffer J, Brilloff S, Hohenstein B, Jarzebska N, Jabs N, et al. Role of alanine:glyoxylate aminotransferase 2 in metabolism of asymmetric dimethylarginine in the settings of asymmetric dimethylarginine overload and bilateral nephrectomy. Nephrol Dial Transplant. (2014) 29:2035–42. doi: 10.1093/ndt/gfu236
58. Jacobi J, Maas R, Cordasic N, Koch K, Schmieder RE, Böger RH, et al. Role of asymmetric dimethylarginine for angiotensin II-induced target organ damage in mice. Am J Physiol Heart Circ Physiol. (2008) 294:H1058-66. doi: 10.1152/ajpheart.01103.2007
59. Xuan C, Lun L-M, Zhao J-X, Wang H-W, Wang J, Ning C-P, et al. L-citrulline for protection of endothelial function from ADMA-induced injury in porcine coronary artery. Sci Rep. (2015) 5:10987. doi: 10.1038/srep10987
60. Reule CA, Goyvaerts B, Schoen C. Effects of an L-arginine-based multi ingredient product on endothelial function in subjects with mild to moderate hypertension and hyperhomocysteinemia - a randomized, double-blind, placebo-controlled, cross-over trial. BMC Complement Altern Med. (2017) 17:92. doi: 10.1186/s12906-017-1603-9
61. Lee Y, Mehrotra P, Basile D, Ullah M, Singh A, Skill N, et al. Specific lowering of asymmetric dimethylarginine by pharmacological dimethylarginine dimethylaminohydrolase improves endothelial function, reduces blood pressure and ischemia-reperfusion injury. J Pharmacol Exp Ther. (2021) 376:181–9. doi: 10.1124/jpet.120.000212
62. Anderssohn M, McLachlan S, Lüneburg N, Robertson C, Schwedhelm E, Williamson RM, et al. Genetic and environmental determinants of dimethylarginines and association with cardiovascular disease in patients with type 2 diabetes. Diabetes Care. (2014) 37:846–54. doi: 10.2337/dc13-0546
63. Xuan C, Xu L-Q, Tian Q-W, Li H, Wang Q, He G-W, et al. Dimethylarginine dimethylaminohydrolase 2 (DDAH 2) gene polymorphism, asymmetric dimethylarginine (ADMA) concentrations, and risk of coronary artery disease: a case-control study. Sci Rep. (2016) 6:33934. doi: 10.1038/srep33934
Keywords: pulse wave velocity, arterial stiffness, dimethylarginine dimethylaminohydrolase, rs9267551, ADMA
Citation: Averta C, Mancuso E, Spiga R, Miceli S, Succurro E, Fiorentino TV, Perticone M, Mannino GC, Thamtarana PJ, Sciacqua A, Sesti G and Andreozzi F (2022) The Functional Polymorphism of DDAH2 rs9267551 Is an Independent Determinant of Arterial Stiffness. Front. Cardiovasc. Med. 8:811431. doi: 10.3389/fcvm.2021.811431
Received: 08 November 2021; Accepted: 29 November 2021;
Published: 03 January 2022.
Edited by:
Manfredi Tesauro, University of Rome Tor Vergata, ItalyReviewed by:
Oren Rom, Louisiana State University Health Shreveport, United StatesAlessandro De Stefano, Catholic University of the Sacred Heart, Italy
Copyright © 2022 Averta, Mancuso, Spiga, Miceli, Succurro, Fiorentino, Perticone, Mannino, Thamtarana, Sciacqua, Sesti and Andreozzi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gaia Chiara Mannino, gaiamannino@unicz.it
†These authors have contributed equally to this work and share first authorship