Prenatal Genetic Diagnosis in Three Fetuses With Left Heart Hypoplasia (LHH) From Three Unrelated Families
 Sukun Luo1†
Sukun Luo1†  Luyi Chen2† Weizhong Wei3† Li Tan1 Meng Zhang3 Zhengrong Duan2 Jiangxia Cao2
Luyi Chen2† Weizhong Wei3† Li Tan1 Meng Zhang3 Zhengrong Duan2 Jiangxia Cao2  Yan Zhou2* Aifen Zhou2*
Yan Zhou2* Aifen Zhou2*  Xuelian He1*
Xuelian He1*- 1Precision Medical Center, Tongji Medical College, Wuhan Children's Hospital (Wuhan Maternal and Child Healthcare Hospital), Huazhong University of Science and Technology, Wuhan, China
- 2Prenatal Diagnosis Center, Tongji Medical College, Wuhan Children's Hospital (Wuhan Maternal and Child Healthcare Hospital), Huazhong University of Science and Technology, Wuhan, China
- 3Ultrasonic Diagnosis Department, Tongji Medical College, Wuhan Children's Hospital (Wuhan Maternal and Child Healthcare Hospital), Huazhong University of Science and Technology, Wuhan, China
Background: Congenital heart defects (CHDs) are the most common birth defects, and left heart hypoplasia (LHH) is a severe form of CHD and responsible for more than 20% cardiac deaths during the first week of life, however, its genetic causes remain largely elusive.
Methods: Three families with fetal LHH were recruited. Genomic DNA from amniotic fluid or peripheral blood, and trio whole exome sequencing (trio-WES) and copy number variation sequencing (CNV-seq) were performed.
Results: All the three couples had no family history, and mid-gestation ultrasound revealed LHH and other variable cardiovascular defects in the fetuses. Trio-WES revealed de novo pathogenic variations in KMT2D (p.Gly3465Aspfs*37) (NM_003482) and WDFY3 (p.Ser117Xfs*) (NM_014991), and CNV-seq identified a deletion of 150 kb encompassing NOTCH1. KMT2D and NOTCH1 previously have been reported to be associated with CHDs, however, WDFY3 is reported for the first time to be possibly related to CHD in human.
Conclusion: Our study suggested that genetic component is an important risk factor for the development of LHH, and next generation sequencing is a powerful tool for genetic diagnosis in fetuses with CHDs and genetic counseling, however, more studies and data are need to establish the correlation of fetal phenotypes and genotypes.
Introduction
Congenital heart defects (CHDs) are a spectrum of diseases involving structural abnormalities of the heart and great vessels that affect about 1% of liveborn infants and occur in 10% of aborted fetuses (1). Left heart hypoplasia (LHH), a severe and complex phenotype of CHDs, refers to the underdeveloped left heart structures including left ventricle, aorta and mitral valve (2). Hypoplastic left heart syndrome (HLHS) is used to describe the most severe condition of LHH, and cause 23% of cardiac deaths in the first week of life without early surgery (3). Although a three-stage surgical palliation has significantly increased the 5-year survival rate (50–70%) (4), the extent of morbidity is considerable and life-expectance remains reduced, and some of them will still develop heart failure and need cardiac transplantation. To achieve substantive progress in the development of therapeutic intervention, the understanding of the formation of the 4-chambered vertebrate heart and the pathogenic mechanism causing LHH is needed.
Several lines of evidence support a genetic contribution to HLHS. A number of reports suggest HLHS is highly associated with chromosomal abnormalities (Turner, trisomy 13, 18, and 21) (5–7), as well as copy number variations (CNV) (DiGeorge, and Jacobsen syndrome) (8, 9). Genetic studies have also identified variants in several candidate genes such as NKX2.5, GJA1, NOTCH1, HAND1, FOXC2, FOXL1, IRX4, MYH6, and RBFOX2, in patients with HLHS (10–15), and these genes are very important for cardiogenesis.
The aim of this study is to identify the pathogenic variants in 3 fetuses with LHH by using next generation sequencing, and we found a de novo loss-of-function variant in KMT2D, and WDFY3 (WD40 repeat and FYVE domain containing 3) gene, respectively, and a de novo heterozygous microdeletion variant in chromosome 9q34.3, encompassing NOTCH1, which aid prenatal genetic counseling and understanding of the pathogenesis of LHH.
Methods
Patient Enrollment
This study was approved by the Wuhan Children's Hospital (Wuhan Maternal and Child Healthcare Hospital) ethics committee and informed consent was obtained from the parents of the fetuses. We recruited 3 fetuses with LHH detected by ultrasound during middle gestation. The parents were healthy and non-consanguineous, and denied any adverse history. The maternal age range at diagnosis of LHH was 29–30 years, and the gestation age range at diagnosis was 23–25 weeks. Fetal samples were from amniotic fluid during the invasive prenatal diagnosis. A total of 2 ml of peripheral blood was collected in EDTA-containing tubes from the couples. Multidisciplinary consultation including genetic counseling regarding the risk of LHH and the chance of surgery as well as the benefits and limitations of whole exome sequencing (WES) and CNV sequencing (CNV-seq) were introduced to the couples.
Sample Preparation
Genomic DNA was extracted from 20 ml of amniotic fluid or 2 ml whole blood according to the protocol of the Omega DNA Mini Kit. A NanoDropTM spectrophotometer was used for quality control (QC) of DNA purity and concentration.
Whole Exome Sequencing and Data Analysis
Genomic DNA was sheared by sonication and exome sequences were enriched by IDT xGen® Exome Researcher Panel v1.0, according to the manufacturer's protocol. DNA libraries were sequenced on Illumina Hiseq XTen with a method of pair-end 150 bp reads. Raw image files were processed by the Bcl To Fastq (Illumina) for base calling and raw data production. Low quality variants were filtered out using the quality score ≥ 20 (Q20) standard. The sequencing reads were then mapped to the human reference genome (hg19/GRCh37) with BWA. Single nucleotide variations (SNVs) and Indels were scored by GATK 3.8. All short variants were annotated with databases including 1,000 genomes, dbSNP, Exac, GnomAD, ClinVar, HGMD, and OMIM. Impacts of variants on protein functions were predicted using software packages Provean, SIFT, Polyphen2_HDIV, Polyphen2_HVAR, Mutationtaster, M-CAP, and REVEL. The pathogenicity of candidate variants was evaluated on the base of mutated frequency, conservation of amino acid change and structural or function domain of the protein as well as inheritance pattern. The pathogenicity of identified variants was assessed in accordance with the American College of Medical Genetics (ACMG) guidelines and Sherloc (16, 17).
Generally, the variants detected by Trio-WES were analyzed according to ACMG standards and guidelines, and classified into pathogenic variants, likely pathogenic variants, uncertain significance variants, likely benign variants and benign variants. The former three types were further annotated in OMIM diseases database and filtered through family co-segregation according to genetic rules. Potential pathogenic genes were identified combining disease correlation and clinical observations. In addition, the incidental findings were disposed according to the document of ACMG about prenatal diagnosis (18). Pathogenic and likely pathogenic variants causing neurodevelopmental disorders, intellectual disability or metabolic diseases were reported.
CNV Analysis
A total amount of more than 1.2 μg purified genomic DNA was broken into 200–300 bp fragments by sonication, followed by fragment DNA being sequentially repaired, tailed, ligated with an adaptor and amplified for library construction. The samples were then subjected to Illumina NovaSeq 6000 (Illumina, San Diego, USA). Raw data were processed by the basecall analysis software fastq v0.18.1. The clean data were then mapped to human reference genome (hg19) using BWA. The candidate CNVs were first filtered through normal frequency databases DGV, and, subsequently, were annotated based on scientific literature review and public databases including Decipher, ClinVar, ClinGen, ISCA, and dbVar. According to the ACMG, the candidate CNVs were classified into five categories: pathogenic, likely pathogenic, uncertain clinical significance, likely benign and benign (19).
Sanger Sequencing
The identified variants in the KMT2D and WDFY3 genes were confirmed by Sanger sequencing. Primers were designed to amplify the exon 36 of KMT2D and the exon 6 of WDFY3. PCR products were subjected to ABI 3730XL and analyzed by DNAStar software.
Quantitative PCR Assay
Semi-quantitative qPCR was performed to confirm the presence of the microdeletion identified in case 3 by detecting the copies of DNA fragments within the region on both the fetus and the parents. Primers were designed to quantify the relative copies of PMPCA exon 2 and 13, INPP5E exon 10 as well as NOTCH1 exon 1 and exon 34. The primers for these DNA fragments are listed as following: PMPCA exon 2(Forward: 5′-GTG CCT ATC CCA ACA TCC-3′ and Reverse: 5′-GCG AAG CCC ATT ATC CAA-3′), PMPCA exon 13(Forward: 5′-CCG TTC CCG TGC GTG TTA-3′ and Reverse: 5′-TTC CTG GCT TGC GGT GGT-3′), INPP5E exon 10(Forward: 5′-GCA CCA TCT GCT CCG TTT C-3′ and Reverse: 5′-TTC CTT CCT GGG ACG CTG-3′), NOTCH1 exon 1(Forward: 5′-GAG CGC AGC GAA GGA ACG AG-3′ and Reverse: 5′-CCT CTC TTC CCC GGC TGG CT-3′) and NOTCH1 exon 34 (Forward: 5′-GCA CAG GAG CGC ATG CAT CA-3′ and Reverse: 5′-GCT GGG CTT GCG GAC CTT CT-3′). The 36B4 gene was used as reference gene. q-PCR was performed on a ABI 7500 apparatus using SYBR Green Real-Time PCR Master Mix (Takara) according to the following conditions: 95°C for 2 min, followed by 40 cycles of 95°C for 15 s, 53°C for 15 s and 72°C for 34 s.
Results
After performing genetic analysis, variants of two genes, KMT2D (NM_003482) and WDFY3(NM_014991), and a microdeletion involving NOTCH1 were detected in three fetuses, respectively. All the three variants were classified into pathogenic variants because they were LOF and de novo mutations, and absent in control population. The detailed clinical features and genetic information were shown in Table 1. No other pathogenic variants in known pathogenic genes, which were in line with family separation, were identified in the three cases.
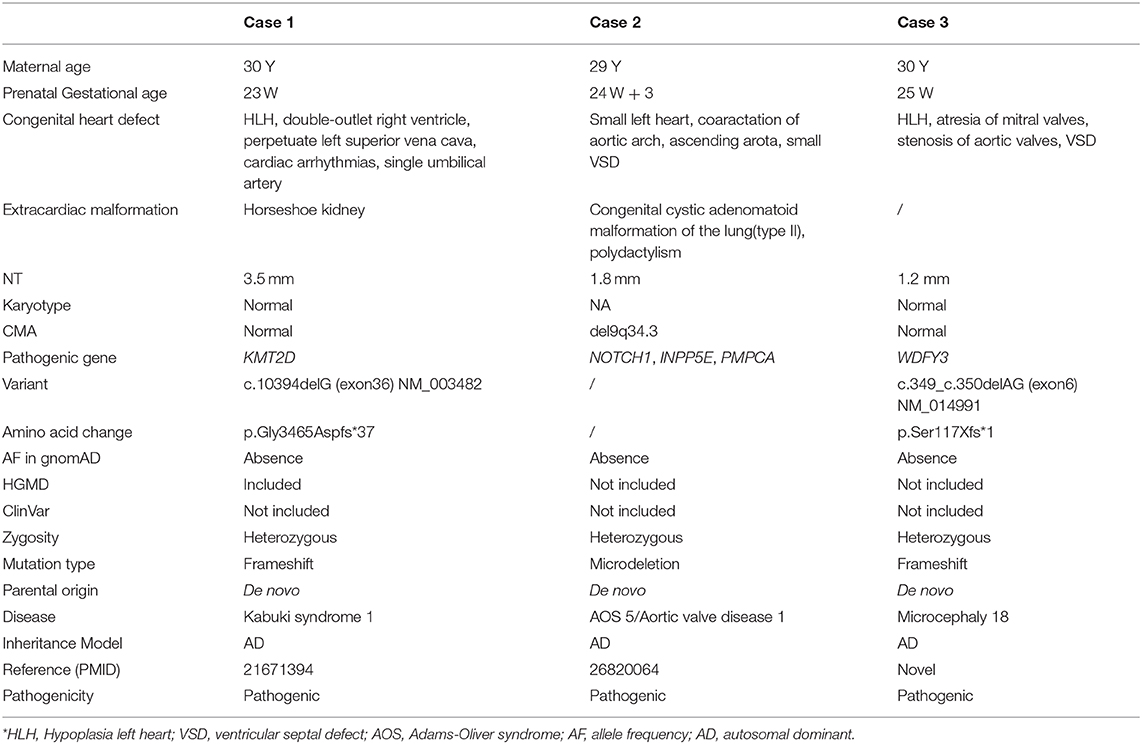
Table 1. Prenatal phenotype and genotype information for the cohort.
Case 1 was identified to be a de novo heterozygous variant (c.10394delG, p.Gly3465Aspfs*37) in the exon 36 of KMT2D gene (Figure 1A), which was reported previously and included in the HGMD (Accession number: CD114940) (20). At 23-week of gestation (wg), the ultrasound, shown in Figures 2A,B, revealed complex congenital heart defects (hypoplastic left heart, ventricular septal defect, double-outlet right ventricle, and perpetuate left superior vena cava), horseshoe kidney, and single umbilical artery, and the fetus presented arrhythmia. Mid-pregnancy ultrasound showed the fetus had increased nuchal translucency of 3.5 mm (normal value <2.5 mm) at 12 wg, and, at 18 wg, the tests of karyotyping and CMA analyses by amniocentesis were negative.
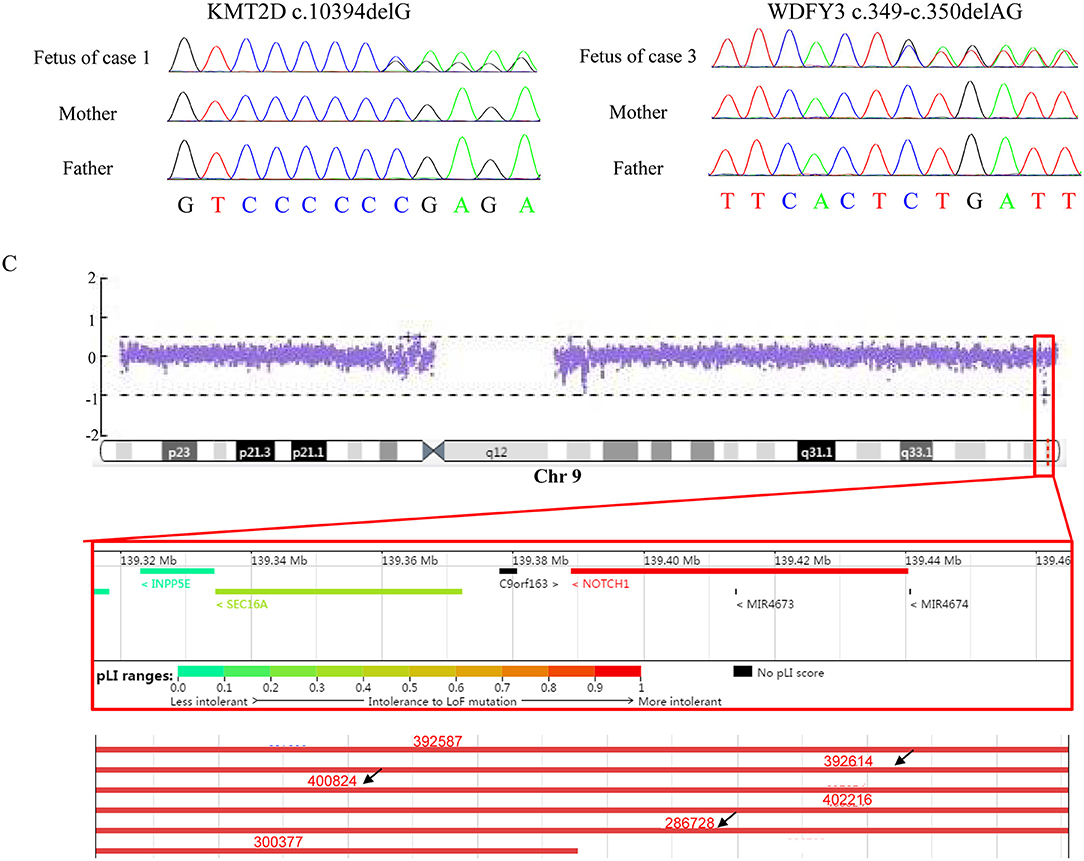
Figure 1. Results of Sanger sequencing and CNV-seq. (A) The mutation, c.10394delG, identified in KMT2D in the fetus (case 1) but not in the parents. (B) The mutation, c.394-c.350delAG, found in WDFY3 in the fetus (case 3) but not in the parents. (C) A 150 kb microdeletion at 9q34.3 resulting in deletion of full-length of NOTCH1 detected in case 2 and part of microdeletion variants encompassing the region in Decipher. Variants pointed by black arrow displayed heart abnormalities.
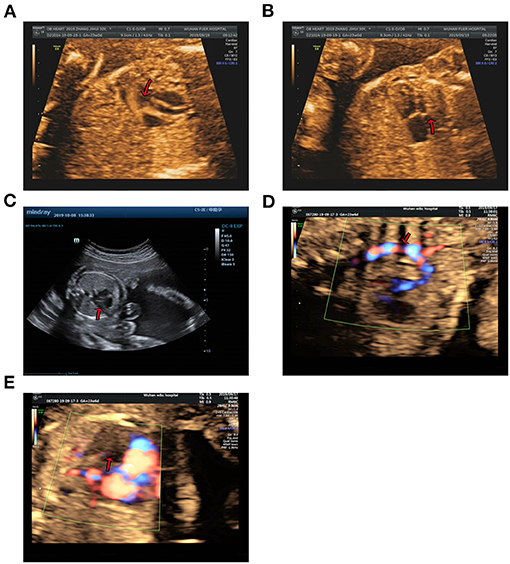
Figure 2. Echocardiographic images of fetal heart structures. (A,B) show fetal ventricular septal defect and hypoplasia of left heart in case 1. (C) displays a smaller left heart in case 2. (D,E) show coarctation of the aorta and mitral atresia in the fetal heart of case 3.
Case 2 was found to carry a de novo 150 kb microdeletion at 9q34.3 (chr9:139, 315, 643-139, 465, 759), resulting in deletion of full-length of NOTCH1, INPP5E, and SEC16A as well as part of PMPCA (Figure 1C). The ultrasound at 24 wg detected severe congenital heart defects, including small left heart (Figure 2C), coarctation of ascending aorta and aortic ache, and a small ventricular septal defect. Polydactyly and cystic adenomatoid of lung were also observed (Table 1). The nuchal translucency measurement and noninvasive prenatal testing were unremarkable.
Case 3 was identified to have a de novo heterozygous frameshift variant (c.349-c.350delAG, p.Ser117Xfs*1) in the exon 6 of WDFY3 gene (Figure 1B), which was a novel pathogenic variant. Ultrasound of this fetus at 25 wg showed left heart dysplasia with mitral atresia and aortic valve stenosis (Figures 2D,E), and ventricular septum defect. No other abnormality was found during mid-pregnancy examination. Although, WDFY3 pathogenic variants were reported to cause microcephaly or macrocephaly (21, 22), the described fetus in this case had normal head circumference.
Discussion
Although increasing evidence supports a genetic etiology (23–25), the precise pathogenesis of LHH or HLHS is unknown. In this study, we reported 2 de novo frameshift variations in the KMT2D and WDFY3 genes, respectively, and a 9q34.3 microdeletion of 150 kb encompassing NOTCH1, in three fetuses with LHH and other structural malformations.
WES identified a de novo heterozygous frameshift variant (c.10394delG) in the KMT2D gene, resulting in a truncated protein, in Case 1. The KMT2D encodes a histone H3K4 methytransferases that is associated with transcriptionally active genes. Mutations in KMT2D are most frequently, up to 70%, found in patients with Kabuki syndrome (KS, MIM: #147920), which is a rare, autosomal dominant multiple congenital anomaly syndrome characterized by recognizable facial features, global developmental delay, intellectual disability, short stature, and musculoskeletal abnormalities (26). Approximately 55% of patients with KMT2D mutations have been reported to present CHDs, including both septal defects and aortic coarctation (27). Studies in animal models have indicated KMT2D is essential for regulating cardiac gene expression during heart development and is required for the establishment of the primary and secondary heart fields (28, 29). Recently, a study including 80 fetuses with congenital cardiac left-sided lesions showed that KMT2D was the most frequently mutated gene (10.6%) followed by NOTCH1 (6.1%) (30). NOTCH1, encoding one of core receptors in Notch signaling pathway, is a well-known CHD-related gene, and loss-of-function variants in this gene confer a higher risk for and segregates with left-sided-CHD (31, 32). Deletion of the whole NOTCH1 gene was previously reported to cause non-syndromic Tetralogy of Fallot and HLHS (32, 33). Our Case 2 carried a 9q34.3 microdeletion surrounding full-length NOTCH1, and presented underdeveloped left heart and ventricular septal defect. In addition to NOTCH1, the microdeletion contained three other protein-coding genes, PMPCA, INPP5E, and SEC16A according to human hg19/GRCh37. However, our q-PCR assay confirmed that the microdeletion might not destroy the PMPCA gene which contained 13 exons, since the relative copies of PMPCA exon2 and exon 13 in the fetus was comparable to that in its parents (Figure 3). INPP5E were associated with autosomal recessive diseases, while SEC16A had not been reported to be related to a known disease. Deletion of the whole NOTCH1 gene was the most likely cause for LHH of the fetus in case 2.
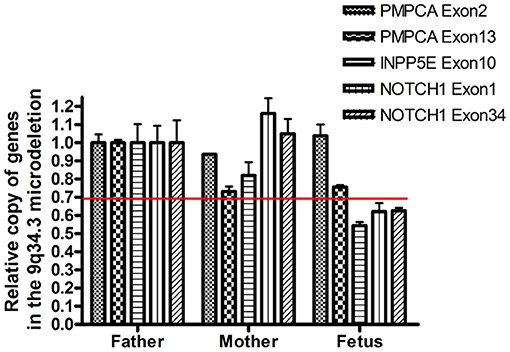
Figure 3. Confirmation of the presence of the deletion by semi-quantitative q-PCR. The copy number of PCPMA, INPP5E, NOTCH1, and 36B4 was evaluated by semi-quantitative PCR. The relative copies of PCPMA (exon 2 and 13), INPP5E (exon 10) and NOTCH1 (exon 1 and 34) were normalized using, a valid single copy gene.
Although a clear prenatal genetic diagnosis can be made only when definitely pathogenic genes and variants are identified, prenatal WES is invaluable for the diagnosis of causal genes but also for the discovery of candidate genes. In case 3, we did not detect any pathogenic variants in well-established CHD pathogenic genes, but identified a frameshift (c.349-c.350delAG, p.Ser117Xfs*1) variant in WDFY3, thus, we propose novel candidate gene for early cardiac development supported by functional and phenotyping evidence in animal model (34). WDFY3 is located on chromosome 4q21.23, encompassing 74 exons and encoding a protein with 3,526 amino acids. The encoded protein is an adaptor protein involved in selective degradation of protein aggregates by autophagy, and is critical for brain development (35, 36). It was included in OMIM and associated with microcephaly 18, an autosomal dominant disorder. It is extremely intolerant to loss-of-function (LOF) mutation with a pLI score of 1. Pathogenic WDFY3 variants caused mild to moderate neurodevelopmental delay and intellectual disability, most of which (8/13) were loss-of-function variant and putatively lead to haploinsufficiency (21).
Homozygous knockout of this gene in mice model would lead to variable cardiac anomalies, such as ventricular septum defect, double outlet right ventricle, aortic overriding, thinning of ventricular wall, ventricular dilation and disorganized trabeculation, and abnormal NOTCH1 signaling in the heart of mice (34), suggesting that WDFY3 is critical for cardiac development. However, this report did not mention whether heterozygous WDFY3 knockout mice presented visible heart abnormalities (34). Unfortunately, we did not obtain the heart tissue of the aborted fetus to examine the impacts of WDFY3 variant on signaling pathways, such as NOTCH1 and Wnt pathways related to early cardiac development. This is the first report of a human phenotype caused by a frameshift variant in WDFY3, more cases and further functional studies are needed before we make a conclusion that WDFY3 is pathogenic candidate gene for LHH. In this study, the pregnancy was terminated due to the poor prognosis of LHH and the risk of neurodevelopmental delay. The pregnant woman and her family were informed that the recurrence risk is very low except that the variant is moasic in germline cells.
In conclusion, next generation sequencing is a powerful tool for prenatal genetic diagnosis, in particular, for fetuses with LHH. Apart from two known genes, KMT2D and NOTCH1, we reported a potential pathogenic gene WDFY3, and this is the first case of a fetal CHD with a frameshift variant in WDFY3. Before this genetic variant is used to genetic counseling, more sequencing data and functional studies are needed to determine the contribution of this genetic variant to CHDs.
Bulleted Statements
Genetic variants in KMT2D and NOTCH1 previously have been reported in patients with CHDs, however, WDFY3 is reported for the first time to be possibly related to CHD in human. Genetic factor is an important factor for the pathogenesis of LHH.
Data Availability Statement
The data of KMT2D p.Gly3465Aspfs* and WDFY3 p.Ser117Xfs*1 presented in the study are deposited in the ClinVar database, and the accession numbers were SCV001449147 and SCV001449148, respectively.
Ethics Statement
The studies involving human participants were reviewed and approved by Wuhan Children's Hospital (Wuhan Maternal and Child Healthcare Hospital) ethics committee. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
HX and ZA conceived the study. CL, WW, ZM, DZ, CJ, and ZY clinically analyzed the patients. LS and TL conducted genetic data interpretation. LS and HX performed literature review and prepared the manuscript. ZA, ZY, and HX revised the manuscript. All authors reviewed and approved the manuscript.
Funding
This work was supported by the grants of Wuhan Municipal Health Commission (No. WX19C19, WX14A06, and WX16C17); Youth Program of National Natural Science Foundation of China (No. 81700302); Natural Science Foundation of Hubei Province (2017CFB322); Hubei Health and Family Planning Commission (WJ2015MB247).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We gratefully acknowledge our patients in this study and their families for their consent and participation in this study.
References
1. van der Linde D, Konings EE, Slager MA, Witsenburg M, Helbing WA, Takkenberg JJ, et al. Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J Am Coll Cardiol. (2011) 58:2241–7. doi: 10.1016/j.jacc.2011.08.025
2. Hickey EJ, Caldarone CA, McCrindle BW. Left ventricular hypoplasia: a spectrum of disease involving the left ventricular outflow tract, aortic valve, and aorta. J Am Coll Cardiol. (2012) 59 (1 Suppl.):S43–54. doi: 10.1016/j.jacc.2011.04.046
3. Khairy P, Fernandes SM, Mayer JE Jr, Triedman JK, Walsh EP, Lock JE, et al. Long-term survival, modes of death, and predictors of mortality in patients with Fontan surgery. Circulation. (2008) 117:85–92. doi: 10.1161/CIRCULATIONAHA.107.738559
4. Feinstein JA, Benson DW, Dubin AM, Cohen MS, Maxey DM, Mahle WT, et al. Hypoplastic left heart syndrome: current considerations and expectations. J Am Coll Cardiol. (2012) 59 (1 Suppl.):S1–42. doi: 10.1016/j.jacc.2011.09.022
5. Allen RH, Benson CB, Haug LW. Pregnancy outcome of fetuses with a diagnosis of hypoplastic left ventricle on prenatal sonography. J Ultrasound Med. (2005) 24:1199–203. doi: 10.7863/jum.2005.24.9.1199
6. Madriago E, Nguyen T, McFerson M, Larson EV, Airhart N, Moller JH, et al. Frequency and outcomes of cardiac operations and catheter interventions in Turner syndrome. Am J Cardiol. (2012) 110:580–5. doi: 10.1016/j.amjcard.2012.04.036
7. Zakaria D, Tang X, Bhakta R, ElHassan NO, Prodhan P. Chromosomal abnormalities affect the surgical outcome in infants with hypoplastic left heart syndrome: a large cohort analysis. Pediatr Cardiol. (2018) 39:11–8. doi: 10.1007/s00246-017-1717-3
8. Mattina T, Perrotta CS, Grossfeld P. Jacobsen syndrome. Orphanet J Rare Dis. (2009) 4:9. doi: 10.1186/1750-1172-4-9
9. Warburton D, Ronemus M, Kline J, Jobanputra V, Williams I, Anyane-Yeboa K, et al. The contribution of de novo and rare inherited copy number changes to congenital heart disease in an unselected sample of children with conotruncal defects or hypoplastic left heart disease. Hum Genet. (2014) 133:11–27. doi: 10.1007/s00439-013-1353-9
10. Iascone M, Ciccone R, Galletti L, Marchetti D, Seddio F, Lincesso AR, et al. Identification of de novo mutations and rare variants in hypoplastic left heart syndrome. Clin Genet. (2012) 81:542–54. doi: 10.1111/j.1399-0004.2011.01674.x
11. Elliott DA, Kirk EP, Yeoh T, Chandar S, McKenzie F, Taylor P, et al. Cardiac homeobox gene NKX2-5 mutations and congenital heart disease: associations with atrial septal defect and hypoplastic left heart syndrome. J Am Coll Cardiol. (2003) 41:2072–6. doi: 10.1016/S0735-1097(03)00420-0
12. Dasgupta C, Martinez AM, Zuppan CW, Shah MM, Bailey LL, Fletcher WH. Identification of connexin43 (alpha1) gap junction gene mutations in patients with hypoplastic left heart syndrome by denaturing gradient gel electrophoresis (DGGE). Mutat Res. (2001) 479:173–86. doi: 10.1016/S0027-5107(01)00160-9
13. Reamon-Buettner SM, Ciribilli Y, Inga A, Borlak J. A loss-of-function mutation in the binding domain of HAND1 predicts hypoplasia of the human hearts. Hum Mol Genet. (2008) 17:1397–405. doi: 10.1093/hmg/ddn027
14. Homsy J, Zaidi S, Shen Y, Ware JS, Samocha KE, Karczewski KJ, et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science. (2015) 350:1262–6. doi: 10.1126/science.aac9396
15. Tomita-Mitchell A, Stamm KD, Mahnke DK, Kim MS, Hidestrand PM, Liang HL, et al. Impact of MYH6 variants in hypoplastic left heart syndrome. Physiol Genomics. (2016) 48:912–21. doi: 10.1152/physiolgenomics.00091.2016
16. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
17. Nykamp K, Anderson M, Powers M, Garcia J, Herrera B, Ho YY, et al. Correction: Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria. Genet Med. (2020) 22:240. doi: 10.1038/s41436-019-0624-9
18. Monaghan KG, Leach NT, Pekarek D, Prasad P, Rose NC, on behalf of the APP, Guidelines Committee. The use of fetal exome sequencing in prenatal diagnosis: a points to consider document of the American College of Medical Genetics and Genomics (ACMG). Genet Med. (2020) 22:675–80. doi: 10.1038/s41436-019-0731-7
19. Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med. (2011) 13:680–5. doi: 10.1097/GIM.0b013e3182217a3a
20. Hannibal MC, Buckingham KJ, Ng SB, Ming JE, Beck AE, McMillin MJ, et al. Spectrum of MLL2 (ALR) mutations in 110 cases of Kabuki syndrome. Am J Med Genet A. (2011) 155a:1511–6. doi: 10.1002/ajmg.a.34074
21. Le Duc D, Giulivi C, Hiatt SM, Napoli E, Panoutsopoulos A, Harlan De Crescenzo A, et al. Pathogenic WDFY3 variants cause neurodevelopmental disorders and opposing effects on brain size. Brain. (2019) 142:2617–30. doi: 10.1093/brain/awz198
22. Kadir R, Harel T, Markus B, Perez Y, Bakhrat A, Cohen I, et al. ALFY-controlled DVL3 autophagy regulates Wnt signaling, determining human brain size. PLoS Genet. (2016) 12:e1005919. doi: 10.1371/journal.pgen.1005919
23. Grossfeld PD. Hypoplastic left heart syndrome: it is all in the genes. J Am Coll Cardiol. (2007) 50:1596–7. doi: 10.1016/j.jacc.2007.06.045
24. Hinton RB Jr, Martin LJ, Tabangin ME, Mazwi ML, Cripe LH, Benson DW. Hypoplastic left heart syndrome is heritable. J Am Coll Cardiol. (2007) 50:1590–5. doi: 10.1016/j.jacc.2007.07.021
25. McBride KL, Zender GA, Fitzgerald-Butt SM, Koehler D, Menesses-Diaz A, Fernbach S, et al. Linkage analysis of left ventricular outflow tract malformations (aortic valve stenosis, coarctation of the aorta, and hypoplastic left heart syndrome). Eur J Hum Genet. (2009) 17:811–9. doi: 10.1038/ejhg.2008.255
26. Bögershausen N, Wollnik B. Unmasking Kabuki syndrome. Clin Genet. (2013) 83:201–11. doi: 10.1111/cge.12051
27. Armstrong L, Abd El Moneim A, Aleck K, Aughton DJ, Baumann C, Braddock SR, et al. Further delineation of Kabuki syndrome in 48 well-defined new individuals. Am J Med Genet A. (2005) 132A:265–72. doi: 10.1002/ajmg.a.30340
28. Schwenty-Lara J, Nürnberger A, Borchers A. Loss of function of Kmt2d, a gene mutated in Kabuki syndrome, affects heart development in Xenopus laevis. Dev Dyn. (2019) 248:465–76. doi: 10.1002/dvdy.39
29. Ang SY, Uebersohn A, Spencer CI, Huang Y, Lee JE, Ge K, et al. KMT2D regulates specific programs in heart development via histone H3 lysine 4 di-methylation. Development. (2016) 143:810–21. doi: 10.1242/dev.132688
30. Sun H, Yi T, Hao X, Yan H, Wang J, Li Q, et al. Contribution of single-gene defects to congenital cardiac left-sided lesions in the prenatal setting. Ultrasound Obstet Gynecol. (2020) 56:225–32. doi: 10.1002/uog.21883
31. Helle E, Córdova-Palomera A, Ojala T, Saha P, Potiny P, Gustafsson S, et al. Loss of function, missense, and intronic variants in NOTCH1 confer different risks for left ventricular outflow tract obstructive heart defects in two European cohorts. Genet Epidemiol. (2019) 43:215–26. doi: 10.1002/gepi.22176
32. Kerstjens-Frederikse WS, van de Laar IM, Vos YJ, Verhagen JM, Berger RM, Lichtenbelt KD, et al. Cardiovascular malformations caused by NOTCH1 mutations do not keep left: data on 428 probands with left-sided CHD and their families. Genet Med. (2016) 18:914–23. doi: 10.1038/gim.2015.193
33. Greenway SC, Pereira AC, Lin JC, DePalma SR, Israel SJ, Mesquita SM, et al. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat Genet. (2009) 41:931–5. doi: 10.1038/ng.415
34. Zhang S, Song Z, An L, Liu X, Hu XW, Naz A, et al. WD40 repeat and FYVE domain containing 3 is essential for cardiac development. Cardiovasc Res. (2019) 115:1320–31. doi: 10.1093/cvr/cvy285
35. Isakson P, Holland P, Simonsen A. The role of ALFY in selective autophagy. Cell Death Differ. (2013) 20:12–20. doi: 10.1038/cdd.2012.66
Keywords: prenatal diagnosis, congenital heart defects, KMT2D, NOTCH1, WDFY3
Citation: Luo S, Chen L, Wei W, Tan L, Zhang M, Duan Z, Cao J, Zhou Y, Zhou A and He X (2021) Prenatal Genetic Diagnosis in Three Fetuses With Left Heart Hypoplasia (LHH) From Three Unrelated Families. Front. Cardiovasc. Med. 8:631374. doi: 10.3389/fcvm.2021.631374
Received: 19 November 2020; Accepted: 04 March 2021;
Published: 09 April 2021.
Edited by:
Seitaro Nomura, The University of Tokyo, JapanReviewed by:
Olivier M. Vanakker, Ghent University, BelgiumDouglas A. Marchuk, Duke University, United States
Copyright © 2021 Luo, Chen, Wei, Tan, Zhang, Duan, Cao, Zhou, Zhou and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xuelian He, hexuelian2013@hotmail.com; Aifen Zhou, 937577332@qq.com; Yan Zhou, zhouyan030118@126.com
†These authors have contributed equally to this work