Synthesis of the 6-Substituted Imidazo[1,2-a]Pyridine-3-yl-2- Phosphonopropionic Acids as Potential Inhibitors of Rab Geranylgeranyl Transferase
 Damian Kusy1†
Damian Kusy1†  Aleksandra Marchwicka2† Joanna Małolepsza1†
Aleksandra Marchwicka2† Joanna Małolepsza1†  Katarzyna Justyna1,2
Katarzyna Justyna1,2  Edyta Gendaszewska-Darmach2
Edyta Gendaszewska-Darmach2  Katarzyna Magdalena Błażewska1*
Katarzyna Magdalena Błażewska1*- 1Faculty of Chemistry, Institute of Organic Chemistry, Lodz University of Technology, Łódź, Poland
- 2Faculty of Biotechnology and Food Sciences, Institute of Molecular and Industrial Biotechnology, Lodz University of Technology, Łódź, Poland
Twelve phosphonopropionates derived from 2-hydroxy-3-imidazo[1,2-a]pyridin-3-yl-2-phosphonopropionic acid (3-IPEHPC) were synthesized and evaluated for their activity as inhibitors of protein geranylgeranylation. The nature of the substituent in the C6 position of imidazo[1,2-a]pyridine ring was responsible for the compound's activity against Rab geranylgeranyl transferase (RGGT). The most active inhibitors disrupted Rab11A prenylation in the human cervical carcinoma HeLa cell line. The esterification of carboxylic acid in the phosphonopropionate moiety turned the inhibitor into an inactive analog.
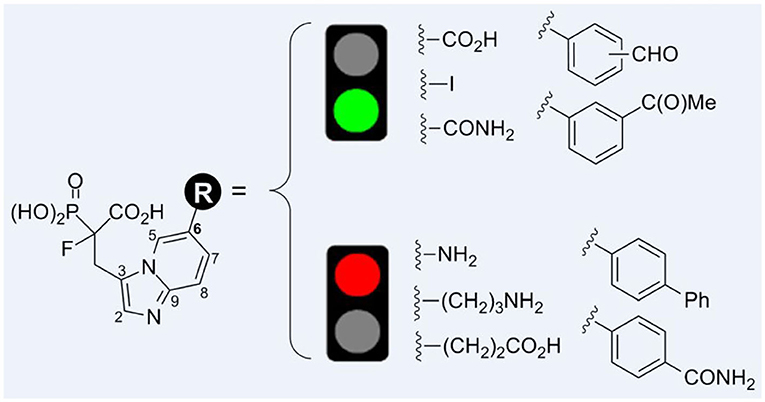
Graphical Abstract.
Introduction
Rab geranylgeranyl transferase (RGGT) constitutes a potential therapeutic target, as it is responsible for posttranslational prenylation of Rab GTPases (Kelly et al., 2012). The perturbed balance of Rabs' modification is observed in several diseases (Mitra et al., 2011; Banworth and Li, 2018), due to their involvement in a wide variety of critical cell processes connected with membrane and vesicle trafficking. The RGGT transfers a geranylgeranyl moiety derived from the mevalonate pathway, which is targeted by two blockbuster classes of drugs: statins and bisphosphonates (Istvan and Deisenhofer, 2001; Russell, 2011). Therefore, the inhibitors of RGGT, as the more downstream enzyme, are gaining on importance as invaluable tools necessary for in-depth studies of molecular bases of these validated drugs' activity (Ali et al., 2015; Jeong et al., 2018).
Currently, a small number of RGGT inhibitors have been identified (Watanabe et al., 2008; Tan et al., 2009; McKenna et al., 2010; Bon et al., 2011; Deraeve et al., 2012; Stigter et al., 2012; Kazmierczak et al., 2017). Among them, only phosphonocarboxylic acids bearing heterocyclic motif were demonstrated to prevent the introduction of the second geranylgeranyl group into Rab GTPases (Baron et al., 2009). Two first representatives of this class, 3-PEHPC and 3-IPEHPC, were identified as a mixed-type inhibitor with respect to GGPP and an uncompetitive inhibitor to Rab substrates (Rab11A was tested in the study) (Baron et al., 2009).
Up to now, the most potent inhibitors of this class are derived from imidazo[1,2-a]pyridines (Kazmierczak et al., 2017) and N1 and/or C5-substituted imidazole analogs (Joachimiak et al., 2018). The analogs bearing C3-substituted pyridine (Coxon et al., 2001; Marma et al., 2007; Baron et al., 2009) and N-substituted benzimidazole (Bhuiyan et al., 2019) are less active against RGGT, although the latter were tested only as the so-called desoxy analogs, which are known to be less active than Cα-fluorinated analogs (Marma et al., 2007; Coxon et al., 2014). As these compounds bear two ionic groups, we have also undertaken the effort to synthesize their prodrug analogs. However, all thus obtained compounds showed unsatisfactory stability, leading only to trace amounts of the particular drug under studied conditions (Joachimiak et al., 2015). Future efforts would require the development of other prodrug design strategies.
We have previously demonstrated that phosphonocarboxylates with imidazo[1,2-a]pyridine ring substituted in the C6 position of the heterocycle show activity toward RGGT (Kazmierczak et al., 2017). Here, we elaborated on the synthesis of analogs equipped with diverse groups in this privileged position. Besides the varied character of substituents in terms of bulkiness, length, and geometry, we studied the influence of the basic and acidic nature of thus introduced groups on the activity of these compounds against RGGT.
Results and Discussion
Synthesis
In these studies, we synthesized a set of 12 new phosphonocarboxylate derivatives of a previously described potent RGGT inhibitor, namely 3-IPEHPC (McKenna et al., 2010; Błazewska et al., 2011). Novel compounds 1a–l, all bearing a substituent in the privileged C6 position of imidazo[1,2-a]pyridine ring (Kazmierczak et al., 2017), were divided into three groups depending on the type of the functional group (Figure 1). The first group was constituted by five compounds (1a–e), in which the modification is directly attached to the imidazo[1,2-a]pyridine ring. One of the representatives of this group, compound 1a, is equipped with carboxyester group in phosphonopropionate moiety. The second group consisted of five compounds (1f–j) in which the functional group is connected with heterocycle via the phenyl group, disubstituted either in para or meta positions. The third group (1k–l) consisted of compounds bearing either an amine or a carboxylic group connected with the imidazo[1,2-a]pyridine via the two- or three-carbon long linker, respectively.
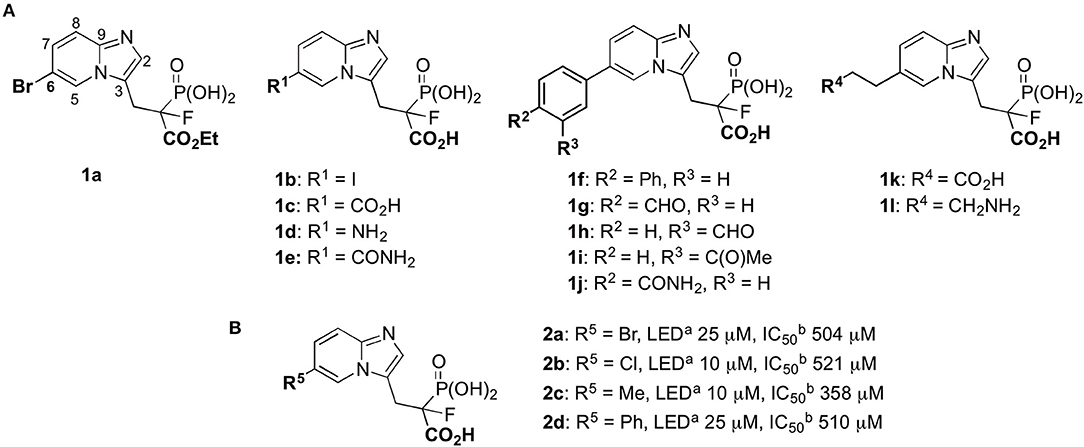
Figure 1. Phosphonocarboxylate inhibitors: (A) structures of the studied compounds; (B) known inhibitors of RGGT (Kazmierczak et al., 2017). aLED = lowest effective dose (inhibition of Rab11A prenylation); bhalf-maximal inhibitory concentration in the HeLa cell line.
The synthesis of compounds representing the first group followed our previously reported strategy (Kazmierczak et al., 2017), which starts from appropriate commercially available 5-substituted-2-aminopyridines 3a–d, which are transferred into imidazo[1,2-a]pyridine aldehydes 4a–d (Kusy et al., 2019), the latter being subject to Knoevenagel condensation with trialkyl phospohonocarboxylate in the presence of TiCl4 and TEA (Scheme 1, details in Supplementary Materials) (Kazmierczak et al., 2017; Joachimiak et al., 2018). Thus, the obtained compounds 5a–c, m were subjected to the reaction with NaBH4 and NiCl2 hydrate in MeOH, giving products 6a–c, m with yields 48–87%. In the case of the synthesis of 6d, it required a three-step procedure, in which reduction of the nitro group (H2, Pd/C), assisted by the partial hydrogenolysis of the double bonds, was followed by protection with Boc2O of the amine group, and then the use of NaBH4 and NiCl2x6H2O. Thus, the obtained compounds 6a–d were subjected to fluorination using N-fluorobenzenesulfonimide (NFSI) and sodium hydride. Only compound 7c was subjected to further transformation of the C6-substituent upon fluorination. In this case, the nitrile group was converted into an amide group using hydrogen peroxide in EtOH (compound 7e, Scheme 2) (Ouvry et al., 2016).
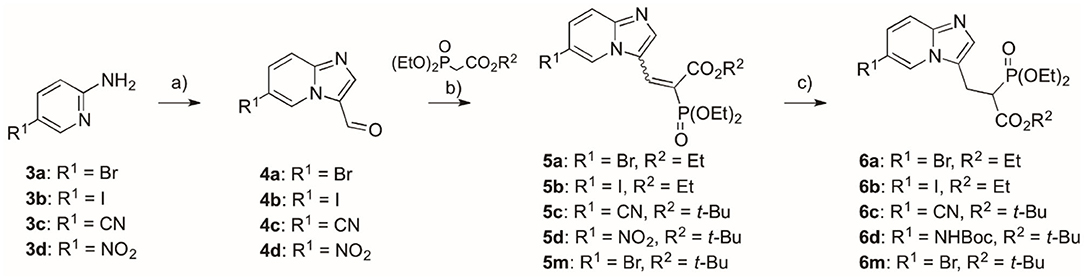
Scheme 1. Reagents and conditions: (a) bromomanoloaldehyde, microwave irradiation, 10 min, 110°C, yield 54–80% (Kusy et al., 2019); (b) tert-butyl diethylphosphonoacetate or triethyl phosphonoacetate, TiCl4, TEA, DCM, overnight, rt, yield 42–66%; (c) for compounds 6a–c, m: NaBH4, NiCl2·6H2O, MeOH, −40°C, yield 74–87%; for compound 6d: (I) 5d, H2, Pd/C, 24 h, rt, (II) Boc2O, DCM, 24 h, rt, (III) NaBH4, NiCl2·6H2O, MeOH, −40°C, yield (three steps) 48%.
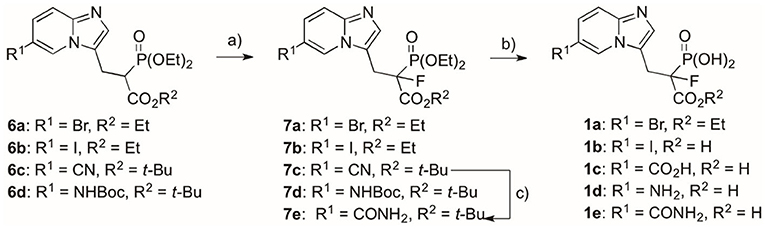
Scheme 2. Reagents and conditions: (a) NaH, NFSI, THF, −70°C/20 min, then rt/overnight, yield 44–90%; (b) for compound 1a: (I) 7a, BTMS, ACN, 2 h, rt, (II) EtOH, 15 min, rt, yield 98%; for compounds 1b–d: 7b–d, 12 M HCl, 5 h, reflux, yield 48–73%; for compound 1e: (I) 7e, BTMS, ACN, 24 h, 35°C, (II) EtOH, 15 min, rt, (III) trifluoroacetic acid, 2 h, rt, yield 68%; (c) 7c, H2O2, DMSO:EtOH 4:1 (v/v), 24 h, rt, yield 72%.
The standard procedure of hydrolysis of the ester groups was applied for most of the compounds 7, and it involved the use of 12 M hydrochloric acid to afford target products 1 with 48–73% yields (Scheme 2). Only compounds 7a and 7e required more cautious conditions in order to retain the carboxyester (7a) or amide groups (7e). Here, we used neutral conditions of McKenna's reaction, subjecting the compound to reaction with BTMS (McKenna and Schmidhuser, 1979; Justyna et al., 2020), followed by TFA-mediated deprotection of tert-butyl carboxyester group, whenever necessary (compound 7e).
The route described above was also used for the synthesis of analogs representing the second and third groups. However, in these cases, the C6-substituents were introduced via Pd-mediated reaction at the later stages of the synthesis, using bromo substituted phosphonoacetate analogs, 5a/5m or 6a/6m.
Compounds 1f–j, bearing the benzene ring in the C6 position, were obtained in the Miyaura–Suzuki reaction between appropriate aryl boronic acid and phosphonoacetates 6a and 6m, in the presence of Na2CO3, Pd(PPh3)4, and toluene:EtOH:H2O (2:1:2), under microwave conditions (Scheme 3). Thus, the obtained compounds 6f–j were subjected to fluorination using the procedure mentioned above, giving appropriate fluoro analogs 7f−7j. The nitrile group in compound 7j was transferred into amide 7j′, using hydrogen peroxide in EtOH. The final acids 1f−1j were obtained as above, either upon hydrolysis in 12 M HCl or using BTMS, followed by TFA (compound 7j′), for dealkylation of phosphonate ester and the cleavage of t-butyl carboxyester, respectively.
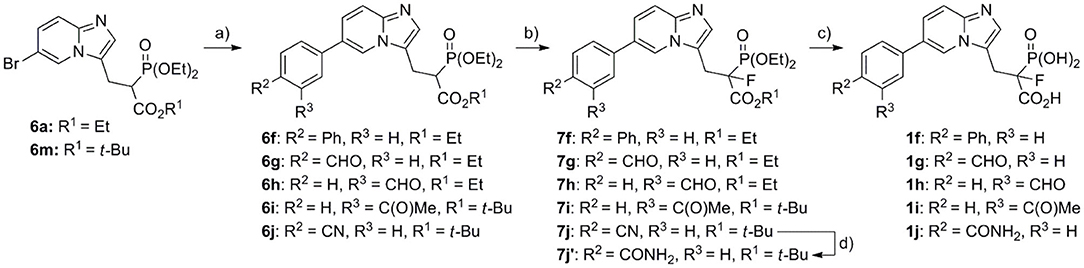
Scheme 3. Reagents and conditions: (a) appropriate boronic acid, Na2CO3, Pd(PPh3)4, toluene:EtOH:H2O (2:1:2), yield 67–83%; (b) NaH, NFSI, THF, −70°C/20 min, then rt/overnight, yield 20–77%; (c) for compounds 1f−1i: 12 M HCl, 5 h, reflux, yield 65–77%; for compound 1j: (I) 7j′, BTMS, ACN, 24 h, 35°C, (II) EtOH, 15 min, rt, (III) trifluoroacetic acid, 2 h, rt, yield 66%; (d) H2O2, DMSO, EtOH, 24 h, rt, yield 57%.
In order to obtain compounds 1k and 1l, equipped with an alkyl chain, the Mizoroki–Heck reaction was applied (Scheme 4), using Pd(OAc)2 and DIPEA and microwave irradiation (Kusy et al., 2020). As we have previously shown, such functionalization with appropriate olefin can be carried out for both types of compounds 5a/5m and 6a/6m. However, depending on the olefin, the type of the functional group present, and its sensitivity to reducing conditions, we chose to use 5m (in the synthesis of amine bearing analog 1l) and 6m (for the synthesis of compound 1k equipped with carboxylic acid). After the Mizoroki–Heck reaction, we subject compound 5l to two-step reduction, obtaining fully saturated analog 6l, which was subject to fluorination with NFSI and NaH (giving compound 7l), followed by hydrolysis in 12 M HCl (see above) (Kazmierczak et al., 2017). In the case of analog with a benzyl carboxylate group, saturated analog 6m was used in the Mizoroki–Heck reaction leading to compound 6k. This product was subject to fluorination (giving compound 7k, Scheme 4), and the newly created double bond was reduced simultaneously with the cleavage of benzyl ester, using hydrogenolytic conditions and Pd/C (giving compound 7k′). The final acids 1k,l were obtained upon hydrolysis (12 M HCl), with the yields 52 and 93%, respectively.
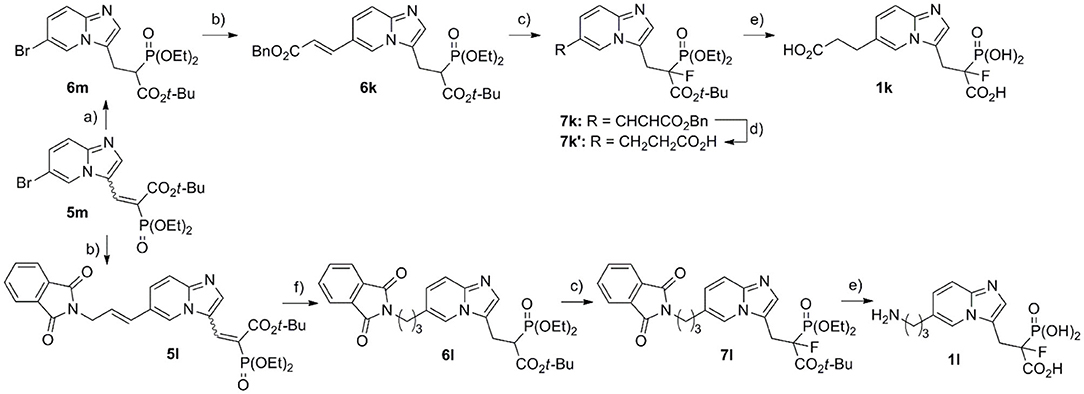
Scheme 4. Reagents and conditions: (a) NaBH4, NiCl2·6H2O, MeOH, −40°C, yield 52–66%; (b) benzyl acrylate or 2-allylisoindoline-1,3-dione, Pd(OAc)2, tri(o-tolyl)phosphine, DIPEA, PCN, microwave irradiation, yield 63–65%, Bn = benzyl; (c) NaH, NFSI, THF, −70°C/20 min, then rt/overnight, yield 52–74%; (d) H2, Pd/C, MeOH, 24 h, rt, yield 85%; (e) 7k′ or 7l, 12 M HCl, 5 h, reflux; (f) (I) H2, Pd/C, 24 h, rt, MeOH (II) NaBH4, NiCl2·6H2O, MeOH, −40°C, yield (two steps) 83%.
Biological Studies
Cytotoxicity Assay
Inhibition of geranylgeranylation is known to reduce cell viability (Wiemer et al., 2009; Kazmierczak et al., 2017). Therefore, 6-substituted imidazo[1,2-a]pyridine analogs of α-phosphonocarboxylates generated in this work were initially screened using the human cervical carcinoma HeLa cell line by PrestoBlue® fluorescent viability assay. To find the most active compounds, HeLa cells were exposed to the incremental concentrations (25–2,000 μM) of phosphonocarboxylates to determine the half-maximal inhibitory concentration (IC50) (Supplementary Figure 1). Half of the 12 studied compounds turned out to be highly cytotoxic (IC50 < 150 μM or below), while three of them (1a, 1d, and 1l) displayed negligible inhibitory effects. The remaining analogs demonstrated a cytotoxic effect with IC50 values from 386 to 735 μM (Table 1).
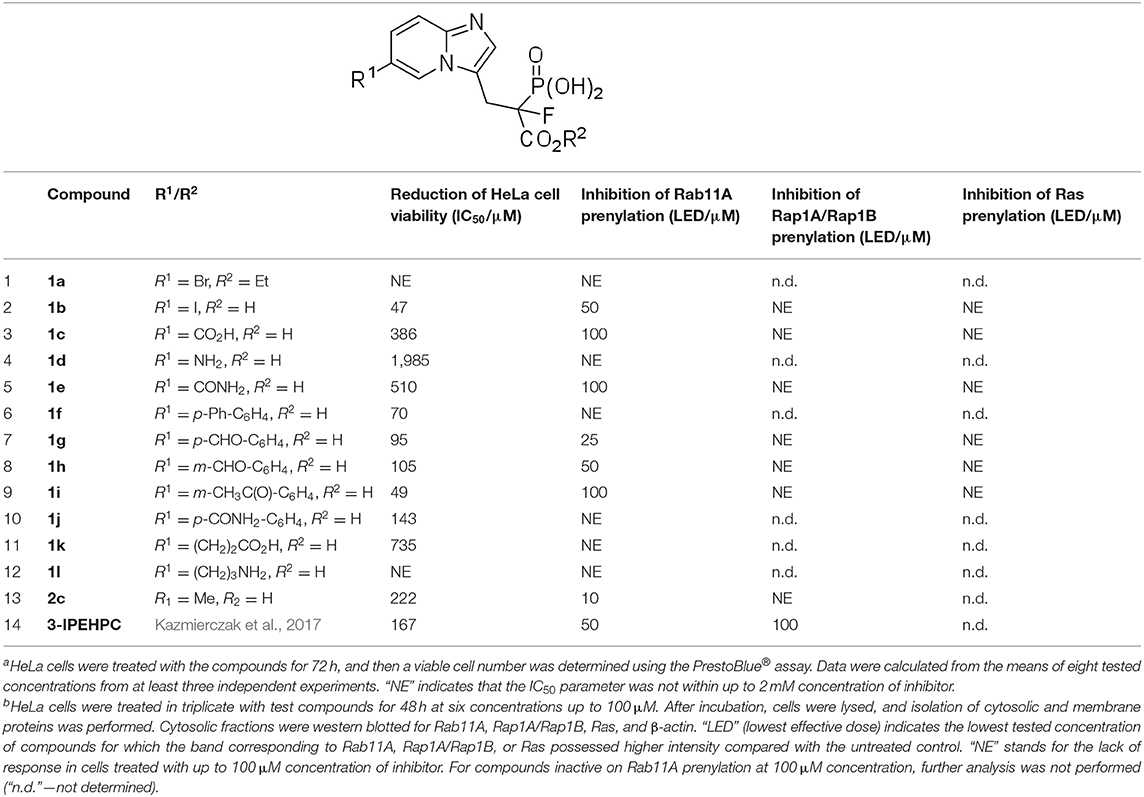
Table 1. Cytotoxic activitya of novel 6-substituted imidazo[1,2-a]pyridine analogs of α-phosphonocarboxylates and their influence on Rab11A, Rap1A/Rap1B, and Ras prenylationb in the HeLa cell line.
Structure–Activity Relationship (SAR)
Previously, we have identified the C6 position in the imidazo[1,2-a]pyridine ring of phosphonocarboxylates 2a–d as the privileged one (Figure 1). Modifications only in this specific position are allowed to retain the activity against RGGT (Kazmierczak et al., 2017). Therefore, here we have extended the library of phosphonocarboxylate analogs, studying the effect of different bulkiness, length, and geometry as well as the acidic/basic character of the introduced substituents. Indeed, the nature of the substituents evaluated in this study influenced their activity.
Cytotoxic effect was correlated with inhibition of Rab11A prenylation, except for compounds 1f, 1j, and 1k, bearing biphenyl, 3-carbamoyl phenyl moiety, or a carboxylic group attached via a two-carbon linker, respectively (Table 1, Figure 2). The latter three compounds showed a cytotoxic effect resulted from different than RGGT inhibition mode of action. Among the non-active and non-cytotoxic compounds, we can differentiate compound 1a, the only example in which carboxylic acid from phosphonoacetate residue was in the form of an ester, and analogs 1d and 1l, both equipped with a free amine group, either directly or via a three-carbon chain connected with imidazo[1,2-a]pyridine ring. Compounds that showed no inhibition of Rab11A prenylation at 100 μM screen (1a, 1d, 1f, 1j, 1k, 1l) were excluded from further analysis.
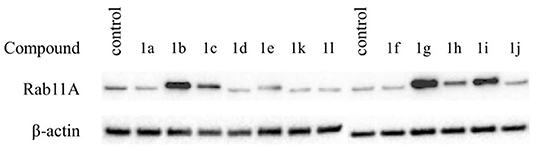
Figure 2. Effect of the studied compounds (1a–l) on Rab11A prenylation in HeLa cells. Cells were treated with phosphonocarboxylates at 100 μM for 48 h, lysed, and fractionated into cytosolic and membrane-rich fractions. Cytosolic fractions containing unprenylated proteins were separated by electrophoresis and western blotted for Rab11A and β-actin.
The active analogs were selected for full panel six-dose assay to determine the lowest effective dose of inhibition of Rab11A, Rap1A/Rap1B, and Ras prenylation (Table 1), proteins modified by three prenyl transferases, RGGT, GGT-I (geranylgeranyl transferase type I), and FT (farnesyl transferase), respectively. This assessment allowed us to define the potency and selectivity of inhibitors against RGGT. Lovastatin, a hydroxymethylglutaryl (HMG)-coenzyme A (CoA) reductase inhibitor, was included as a positive control. Competitive displacement of the natural substrate by lovastatin prevents the downstream biosynthesis of cholesterol and prenylation with the geranylgeranyl or farnesyl moieties (Greenwood et al., 2006).
Compound 1g, which possessed a formyl group at the para position of the phenyl ring, appeared to be the most potent inhibitor of RGGT in the presented set of compounds, as measured by the lowest effective dose (LED). It inhibited Rab11A prenylation at a concentration of 25 μM (Figure 3), which makes this compound more potent than 3-IPEHPC, but slightly less potent than the previously reported most active compound of this class, 2c (Kazmierczak et al., 2017). Shifting a formyl group to the meta position resulted in a two-fold weaker inhibition (1h LED = 50 μM; Figure 3), while the potency of methyl ketone analog 1i was even lower, decreasing to 100 μM (Figure 3). The analog with an amide group in the para position of the phenyl ring, compound 1j, was not active up to 100 μM concentration. This result might imply that either the amide substituent in this position is too bulky (compared with the formyl group) or the electron-dispersing character of the amide group does not promote interaction with the active site.
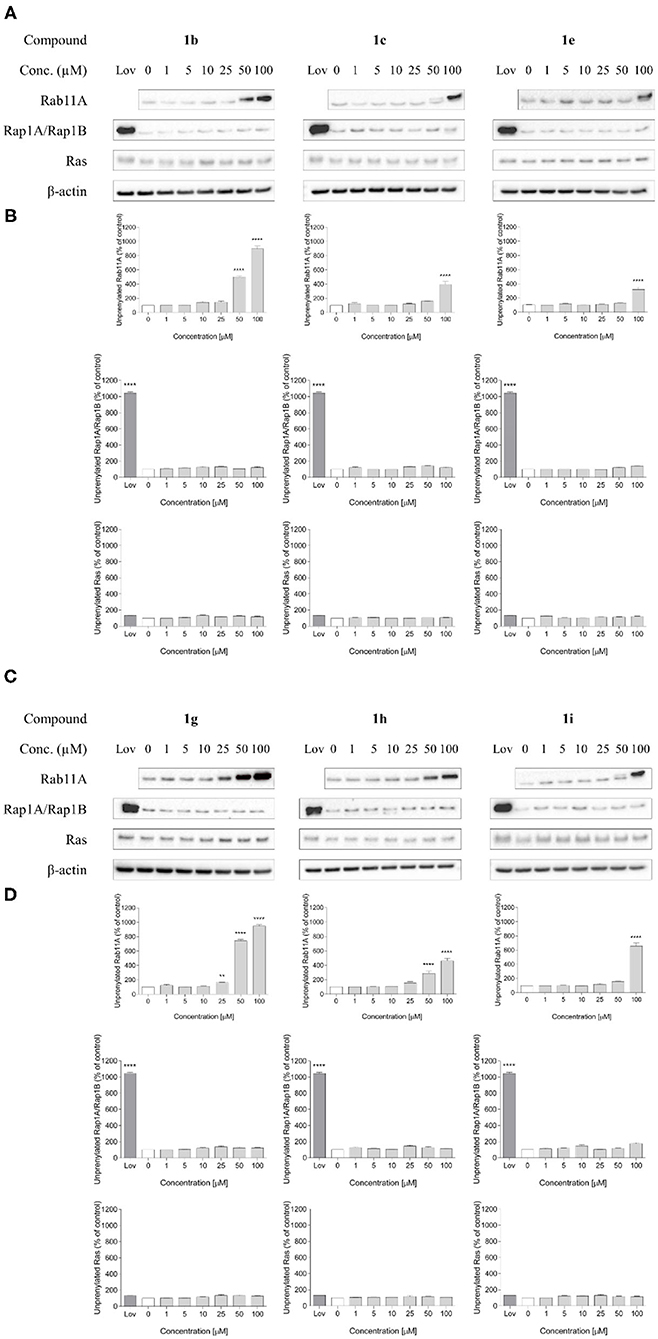
Figure 3. Effect of 6-substituted 3-IPEHPC analogs with iodo (1b), carboxyl (1c), amide (1e), 3- and 4-formylphenyl (1g–h), or 3-acetylphenyl (1i) group on Rab11A, Rap1A/Rap1B, and Ras prenylation in HeLa cells. Cells were exposed to indicated concentrations of compounds or 10 μM lovastatin (Lov) for 48 h. Subsequently, cells were lysed and isolation of cytosolic and membrane proteins was performed. Cytosolic fractions containing unprenylated proteins were western blotted for Rab11A, Rap1A/Rap1B, Ras, and β-actin (A,C), and the immunoblot bands were quantified by densitometry analysis, normalized to β-actin, and presented as a percentage of controls (B,D). Lovastatin was used as control. Even though it is the inhibitor of more upstream enzyme of mevalonate pathway, HMG-CoA reductase, and it is expected that treatment of cells with lovastatin should prevent the Ras accumulation at the membrane, we observed the failure of lovastatin to cause mislocalization of Ras. However, our observations of the lack of Ras inhibition by lovastatin in HeLa cells is in accordance with the results of Tennakoon et al. (2019). They compared how different statins (fluvastatin, atorvastatin, lovastatin) attenuate prenylation of Ras superfamily members in HeLa cells, showing that HeLa cells exposed to lovastatin exhibited negligible inhibition of both Ras members. Data represented mean ± SEM from at least three independent experiments; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001.
Three other active analogs possessed functional groups attached directly to the C6 of imidazo[1,2-a]pyridine ring. Compounds 1c and 1e with carboxylic acid and amide substituent, respectively, influenced RGGT activity only at the highest concentration tested: LED = 100 μM (Figure 3). The third inhibitor, compound 1b, with iodine atom in the C6 position, exhibited a weaker activity toward inhibition of Rab11A prenylation compared to chlorine (2b) and bromine (2a) analogs (Figures 3A,B) (Kazmierczak et al., 2017), setting a trend of decreasing activity with increasing size and polarizability of the electronegative substituent. Its lowest effective dose was 50 μM (Figures 3A,B).
All tested compounds did not influence GGT-I or FT activity, because they affected neither Rap1A/Rap1B nor Ras prenylation (LED > 100 μM, Table 1, Figure 3), which makes them selective inhibitors of RGGT.
Conclusions
A series of 12 novel 6-substituted imidazo[1,2-a]pyridine analogs of α-phosphonocarboxylates bearing structurally different moieties were designed as new RGGT inhibitors. In order to obtain such structurally different compounds, we designed three distinct synthetic routes, including such that allowed late-stage functionalization of the scaffold already bearing phosphonoacetate moiety.
Among the synthesized compounds, nine derivatives, 1b–c, 1e−1k, exhibited cytotoxic activity against cancer HeLa cells. Six compounds, 1b–c, 1e, and 1g–i, displayed inhibitory properties toward Rab11A prenylation with LED ranging from 25 to 100 μM.
The most active compound bears para-substituted formylphenyl substituent. Since its meta-substituted analogs, bearing either formyl or methylketone groups, were also active, it may imply that there is an additional stabilization of the interaction between the inhibitor and an enzyme, thanks to a carbonyl group, possibly via arginine moiety in the active side (Kazmierczak et al., 2017). Other para-substituted phenyl analogs did not show the activity against RGGT in the studied concentration range. The studies clearly show that both aryl and alkyl amine residues have a detrimental effect on the activity against RGGT. On the other hand, the substituent's acidic character in this position may promote the activity (when directly connected with imidazo[1,2-a]pyridine ring) or suppress it (when connected with heterocyclic ring via alkyl chain). Interestingly, the trend observed previously for an electronegative spherical halogen substituent in the C6 position was confirmed, showing retainment of activity, which decreases with the increasing size of the atom. Irrespective of the activity of the synthesized analogs, most of them constitute an interesting point of departure for their further functionalization, thanks to the presence of easily derivatized amine, carboxylic, or carbonyl groups.
Experimental Section
General Synthesis
All reagents were purchased from commercial sources and used as obtained unless specified otherwise. Thin-layer chromatography was performed using silica gel 60 with F254 indicator on alumina plates. Compounds were purified using flash chromatography and 40–63 μm silica gel. Solvent ratios for the purification of compounds by flash chromatography are reported as volume ratios (v/v). Preparative HPLC for purification of compounds 1 was performed using Gilson Prep equipped with a UV–vis-156 detector and semipreparative column Kromasil 100–5-C18 (5 μm, 10 × 250 mm). NMR spectra were measured at 250.13 or 700 MHz for 1H NMR, 62.90 or 170 MHz for 13C NMR, and 283 or 101.30 MHz for 31P NMR on Bruker Avance DPX 250 and Bruker Avance II Plus 700 spectrometers, respectively. Chemical shifts (δ) are reported in parts per million (ppm) relative to internal residual CHCl3 in CDCl3 (δ 7.26 1H NMR) or CDCl3 signal in 13C NMR (δ 77.16) or external 85% H3PO4 (δ 0 ppm 31P NMR). 31P NMR and 13C NMR spectra were proton-decoupled. Coupling constants (J) are quoted in hertz. The assignment of the signals in 1H NMR and 13C NMR was supported by two-dimensional experiments (COZY, HMQC, HMBC, DEPT-135).
General Procedure for the Deprotection of Phosphonate Ester Groups via McKenna Reaction
Synthesis of Compounds 1a, 1e, and 1j
Compounds (7a, 7e, or 7j′) were dissolved in acetonitrile. Then, bromotrimethylsilane (8–10 equiv.) was added. The mixture was stirred for 24 h at room temperature. Then, the solvent was evaporated under reduced pressure and EtOH (1 ml) was added. After 15 min of stirring, the solvent was evaporated. For compounds 1e and 1j, an additional step was required for deprotection of tert-butyl ester. Trifluoroacetic acid (2 ml) was added and the mixture was stirred for 1 h at 30°C. The product was concentrated under reduced pressure using co-evaporation with toluene. Products 1a, 1e, and 1j were isolated by crystallization.
General Procedure for the Hydrolysis With HCl
Synthesis of Compounds 1b–d, 1f–i, and 1k–l
Compounds (7b–d, 7f–i, or 7k–l) (35 mg) were dissolved in 12 M HCl (2 ml). After 4–5 h of reflux, the mixture was evaporated to dryness. The products were obtained as white solids with 48–93% yields upon precipitation using water (2–4 ml). If needed, HPLC was performed, as detailed below.
(3-(6-Bromoimidazo[1,2-a]pyridin-3-yl)-1-ethoxy-2-fluoro-1-oxopropan-2-yl)phosphonic acid (1a)
Scale of the reaction: compound 7a 80 mg. Product 1a was purified by crystallization from H2O. Yield 98%. HRMS m/z: calculated 394.9802 (C12H13BrFN2O5P+H)+, found 394.9800. 1H NMR (700 MHz, CD3OD) δ 1.26 (t, 3JHH = 7.1, CO2CH2CH3, 3H), 3.87–4.20 (m, CH2CPF, 2H), 4.26 (q, 3JHH = 7.1, CO2CH2, 2H), 7.93 (dd, 3JHH = 9.5, 5JHH = 0.9, CHAr(8), 1H), 8.00 (s, CHAr(2), 1H), 8.13 (dd, 3JHH = 9.5, 4JHH = 1.7, CHAr(7), 1H), 9.10 [bs, CHAr(5), 1H]. 31P NMR (284 MHz, CD3OD) δ 6.01 (d, 2JPF = 70.5). 13C NMR (176 MHz, CD3OD) δ 13.2 (s, CO2CH2CH3, 1C), 27.5 (bd, 2JFC = 20.6, CH2CPF, 1C), 63.8 (s, CO2CH2, 1C), 96.8 (dd, 1JFC = 193.9, 1JPC = 143.2, CPF, 1C), 111.7 [s, CAr(6), 1C], 112.9 [s, CHAr(8), 1C], 121.5 [d, 3JPC = 13.5, CAr(3), 1C], 122.1 [s, CHAr(2), 1C], 127.1 [bs, CHAr(5), 1C], 136.7 [s, CHAr(7), 1C], 138.9 [, CAr(9), 1C], 169.7 (d, 2JPC = 24.1, CO2, 1C).
(3-(6-Iodoimidazo[1,2-a]pyridin-3-yl)-1-ethoxy-2-fluoro-1-oxopropan-2-yl)phosphonic acid (1b)
Scale of the reaction: compound 7b 60 mg. Yield 48%. H NMR (700 MHz, D2O, pH 7) δ 3.59 (dt, 2JHH = 16.0, 3JFH = 3JPH = 6.4, CH2CFP, 1H), 3.89 (dd, 3JFH = 41.9, 2JHH = 16.0, CH2CFP, 1H), 7.38 [d, 3JHH = 9.4, CHAr(7), 1H], 7.41 [s, CHAr(2), 1H], 7.56 [m, 3JHH = 9.4, CHAr(8), 1H], 8.84 [s, CHAr(5), 1H]. 31P NMR (283 MHz, D2O, pH 7) δ 10.08 (d, 2JPF = 70.2). 13C NMR (176 MHz, D2O, pH 7) δ 29.1 (dd, 2JFC = 21.3, 2JPC = 3.4, CH2CFP, 1C), 75.0 [s, CHAr(6), 1C], 100.8 (dd, 1JFC = 190.3, 1JPC = 138.1, CH2CFP, 1C), 116.7 [s, CHAr(7), 1C], 122.0 [d, 3JPC = 13.9, CAr(3), 1C], 130.5 [d, 5JFC = 4.4, CHAr(5), 1C], 131.2 [s, CHAr(8), 1C], 133.2 [s, CAr(2), 1C], 144.2 [s, CAr(9), 1C], 176.5 (dd, 2JFC = 21.0, 2JPC = 3.4, CO2H, 1C).
3-(2-Carboxy-2-fluoro-2-phosphonoethyl)imidazo[1,2-a]pyridine-6-carboxylic acid (1c)
Scale of the reaction: compound 7c: 100 mg. Yield 73%. 1H NMR (700 MHz, D2O, pH 8) δ 3.71–3.78 (m, CH2CPF, 1H), 3.99 (ddd, 3JFH = 39.9, 2JHH = 16.3, 3JPH = 2.7, CH2CPF, 1H), 7.65 [s, CHAr(2), 1H], 7.70 [d, 3JHH = 9.3, 5JHH = 0.8, CHAr(8), 1H], 8.00 [dd, 3JHH = 9.3, 4JHH = 1.5 CHAr(7), 1H], 9.00 [bs, CHAr(5), 1H]. 31P NMR (284 MHz, D2O, pH 8) δ 10.18 (d, 2JPF = 75.2). 13C NMR (176 MHz, D2O) δ 28.3 (dd, 2JFC = 21.3, 2JPC = 3.7, CH2CPF, 1C), 99.1 (dd, 1JFC = 191.8, 1JPC = 144.0, CPF, 1C), 113.2 [s, CHAr(8), 1C], 122.7 [d, 3JPC = 15.1, CAr(3), 1C], 124.2 [s, CAr(6), 1C], 127.4 [bs, CHAr(2), 1C], 128.1 [d, 5JFC = 4.6 CHAr(5), 1C], 129.2 [s, CHAr(7), 1C], 143.5 [s, CAr(9), 1C], 171.9 [s, CAr(6)CO2H, 1C], 174.9 (d, 2JPC = 20.7, FCCO2H, 1C). 19F NMR (659 MHz, D2O, pH 8) δ −165.29 (ddd, 2JPF = 75.2, 3JHH = 39.9, 3JHH = 7.5, 1F).
3-(2-Carboxy-2-fluoro-2-phosphonoethyl)imidazo[1,2-a]pyridin-6-aminium chloride (1d)
Scale of the reaction: compound 7d 60 mg. Yield 63%. Product 1d was purified by preparative HPLC (eluent A: H2O:ACN:TFA 95:5:0.1 (v/v/v), isocratic, retention time 2.8 min) as eluent followed by lyophilization from 0.1 M HCl (repeated three times). 1H NMR (700 MHz, D2O, pH 2) δ 3.89 (ddd, 2JHH = 17.1, 3JFH = 11.1, 3JPH = 6.6, CH2C(F)P, 1H), 4.04 (ddd, 3JFH = 37.4, 2JHH = 16.6, 3JPH = 4.8, CH2C(F)P, 1H), 7.85 [d, 3JHH = 9.6, CHAr(8), 1H], 7.92 [s, CHAr(2), 1H], 7.96 [d, 3JHH = 9.6, CHAr(7), 1H], 8.64 [s, CHAr(5), 1H]. 13C NMR (176 MHz, D2O, pH 2) δ 27.6 (dd, 2JFC = 20.3, 2JPC = 2.9, CH2C(F)P, 1C), 96.9 (dd, 1JFC = 193.2, 1JPC = 144.1, C(F)P, 1C), 113.2 [s, CHAr(7), 1C], 117.9 [s, CHAr(5), 1C], 121.9 [d, 3JPC = 12.8, CAr(3), 1C], 122.4 [s, CHAr(2), 1C], 128.4 [s, CAr(6), 1C], 128.4 [s, CHAr(8), 1C], 138.0 [s, CAr(9), 1C], 171.6 (d, 2JFC = 23.9, CO2, 1C). 31P NMR (284 MHz, D2O, pH 2) δ 6.79 (d, 2JPF = 67.1). HRMS m/z: (C10H12FN3O5P)+ calculated 304.0493, found 304.0485.
3-(6-Carbamoylimidazo[1,2-a]pyridin-3-yl)-2-fluoro-2-phosphonopropanoic acid (1e)
Scale of the reaction: compound 7e 100 mg. Product 1e was purified by crystallization from H2O. Yield 68%. 1H NMR (700 MHz, D2O, pH 7) δ 3.74 (ddd, 2JHH = 15.7, 3JFH = 7.7, 3JPH = 7.7, CH2CFP, 1H), 4.01 (ddd, 3JFH = 39.4, 2JHH = 16.3, 3JPH = 3.0, CH2CFP, 1H), 7.71 [s, CHAr(2), 1H], 7.77 (d, 3JHH = 9.4, CHAr, 1H), 7.94 (d, 3JHH = 9.1, CHAr, 1H), 9.10 [bs, CHAr(5), 1H]. 31P NMR (283 MHz, D2O, pH 7) δ 10.19 (d, 2JPF = 76.7). 13C NMR (176 MHz, D2O, pH 7) δ 28.4 (d, 2JFC = 21.3, 2JPC = 4.0, CH2CFP, 1C), 98.58 (dd, 1JFC = 192.3, 1JPC = 146.9, CH2CFP, 1C), 114.05 (s, CHAr, 1C), 121.11 [s, CHAr(6), 1C], 122.85 [d, 3JPC = 15.3, CAr(3), 1C], 127.3 [d, 5JFC = 4.4, CHAr(5), 1C], 127.5 (s, CHAr, 1C), 127.6 [s, CHAr(2), 1C], 143.2 [s, CAr(9), 1C], 166.2 (s, CONH2, 1C), 174.2 (dd, 2JFC = 20.3, 2JPC = 3.3, CO2, 1C). 19F NMR (659 MHz, D2O, pH 7) δ −165.91 (bdd, 2JPF = 75, 3JFH = 41, CH2CFP, 1F).
3-(6-([1,1′-Biphenyl]-4-yl)imidazo[1,2-a]pyridin-3-yl)-2-fluoro-2-phosphonopropanoic acid (1f)
Scale of the reaction: compound 7f 47 mg. Yield 77%. HRMS m/z: calculated 441.1011 (C22H18FN2O5P+H)+, found 441.1005. 1H NMR (700 MHz, D2O, pH 8) δ 3.71–3.80 (m, CH2CPF, 1H), 4.01 (ddd, 3JFH = 40.4, 2JHH = 16.1, 3JPH = 1.7, CH2CPF, 1H), 7.42 (t, 3JHH = 7.4, CHAr, 1H), 7.50 (t, 3JHH = 7.4, CHAr, 2H), 7.56 [s, CHAr(2), 1H], 7.64–7.73 (m, CHAr, 3H), 7.73–7.79 (m, CHAr, 4H), 7.86 (bd, 3JHH = 9.3, CHAr, 1H), 8.74 [bs, CHAr(5), 1H]. 31P NMR (284 MHz, D2O, pH 8) δ 10.06 (d, 2JPF = 72.9).
2-Fluoro-3-(6-(4-formylphenyl)imidazo[1,2-a]pyridin-3-yl)-2-phosphonopropanoic acid (1g)
Scale of the reaction: compound 7g 35 mg. Yield 65%. HRMS m/z: calculated 393.0647 (C17H14FN2O6P+H)+, found 393.0632. 1H NMR (700 MHz, D2O, pH 8) δ 3.67–3.77 (m, CH2CPF, 1H), 4.00 (bdd, 3JFH = 41.9, 2JHH = 16.2, CH2CPF, 1H), 7.51 [s, CHAr(2), 1H], 7.65 [bd, 3JHH = 9.1, CHAr(7), 1H], 7.73 [bd, 3JHH = 9.1, CHAr(8), 1H], 7.96–8.02 (m, C6H4, 2H), 8.05–8.09 (m, C6H4, 2H), 8.84 [bs, CHAr(5), 1H], 10.00 (s, CHO), 1H). 31P NMR (284 MHz, D2O, pH 8) δ 10.16 (d, 2JPF = 70.3).
2-Fluoro-3-(6-(3-formylphenyl)imidazo[1,2-a]pyridin-3-yl)-2-phosphonopropanoic acid (1h)
Scale of the reaction: compound 7h 40 mg. Yield 61%. HRMS m/z: calculated 393.0647 (C17H14FN2O6P+H)+, found 393.0630. 1H NMR (700 MHz, D2O, pH 8) δ 3.64–3.72 (m, CH2CFP, 1H), 3.91–4.04 (m, CH2CFP, 1H), 7.55 [s, CHAr(2), 1H], 7.57–7.65 [m, CHAr(14, 8), 1H], 7.71–7.77 [m, CHAr(7), 1H], 7.82 [d, 3JHH = 7.5, CHAr(15), 1H], 7.94 [d, 3JHH = 7.5, CHAr(13), 1H], 7.99–8.02 [m, CHAr(11), 1H], 8.65 [bs, CHAr(5), 1H], 9.89 (s, CHO, 1H). 31P NMR (283 MHz, D2O, pH 8) δ 10.11 (d, 2JPF = 73.5). 13C NMR (176 MHz, D2O, pH 8) δ 28.8 (dd, 2JFC = 21.3, 2JPC = 3.4, CH2CPF, 1C), 99.5 (dd, 1JFC = 191.7, 1JPC = 142.0, CPF, 1C), 114.2 [s, CHAr(8), 1C], 122.5 [d, 3JPC = 14.8, CAr(3), 1C], 123.0 [d, 5JFC = 3.9, CHAr(5), 1C], 126.5 [s, CAr(6), 1C], 127.2 [s, CHAr(2), 1C], 128.2 [s, CHAr(7, 11), 2C], 129.3 [s, CHAr(15), 1C], 129.9 [s, CHAr(14), 1C], 133.4 [s, CHAr(13), 1C], 136.1 [s, CAr(12), 1C], 136.5 [s, CAr(10), 1C], 141.9 [s, CAr(9), 1C], 175.2 (dd, 2JFC = 20.3, 2JPC = 2.7, CO2H, 1C), 195.9 (s, CHO, 1C).
3-(6-(3-Acetylphenyl)imidazo[1,2-a]pyridin-3-yl)-2-fluoro-2-phosphonopropanoic acid (1i)
Scale of the reaction: compound 7i 60 mg. Yield 70%. 1H NMR (700 MHz, D2O, pH 7) δ 3.70 (dt, 2JHH = 16.0, 3JFH = 3JPH = 6.7, CH2CFP, 1H), 3.99 (dd, 3JFH = 42.1, 2JHH = 16.0, CH2CFP, 1H), 7.51 [s, CHAr(2), 1H], 7.63–7.71 [m, CHAr(7, 8, 15), 3H], 8.03 [d, 3JHH = 7.8 Hz, CHAr(13), 1H], 8.05 [d, 3JHH = 7.7 Hz, CHAr(14), 1H], 8.28 [t, 4JHH = 1.8 Hz, CHAr(11), 1H], 8.75 [s, CHAr(5), 1H]. 31P NMR (283 MHz, D2O, pH 7) δ 10.22 (d, 2JPF = 70.0). 13C NMR (176 MHz, D2O, pH 7) δ 29.2 (m, CH2CFP, 1C), 99.8–101.8 (m, CH2CFP, 1C), 115.9 [s, CHAr(8), 1C], 122.6 [d, 3JPC = 14.5, CAr(3), 1C], 123.1 [d, 5JFC = 3.5, CHAr(5), 1C], 125.4 [s, CHAr(12), 1C], 125.6 (s, CHAr, 1C), 127.0 [s, CHAr(11), 1C], 127.8 [s, CHAr(13), 1C], 129.5 (s, CHAr, 1C), 131.6 [s, CHAr(2), 1C], 132.6 [s, CHAr(14), 1C], 137.1 [s, CAr(6), 1C], 137.8 [s, CAr(10), 1C], 144.8 [s, CAr(9), 1C], 176.7 (dd, 2JFC = 20.8, 2JPC = 3.2, CO2H, 1C), 203.9 (s, C(O)CH3, 1C).
3-(6-(4-Carbamoylphenyl)imidazo[1,2-a]pyridin-3-yl)-2-fluoro-2-phosphonopropanoic acid (1j)
Scale of the reaction: compound 7j′ 40 mg. Product 1j was purified by crystallization from a mixture of MeOH:H2O 2:1 (v/v). Yield 66%. 1H NMR (700 MHz, D2O, pH 7) δ 3.67 (ddd, 2JHH = 16.1, 3JFH = 6.7, 3JPH = 6.7, CH2CFP, 1H), 3.96 (ddd, 3JFH = 42.0, 2JHH = 16.4, 3JPH = 1.8, CH2CFP, 1H), 7.46 [s, CHAr(2), 1H], 7.53 (d, 3JHH = 9.3, CHAr, 1H), 7.59 (dd, 3JHH = 9.3, 5JHH = 1.7, CHAr, 1H), 7.75 [d, 3JHH = 8.2, CHAr(11), 2H], 7.80 [d, 3JHH = 8.4, CHAr(12), 2H], 8.65 [bs, CHAr(5), 1H]. 31P NMR (284 MHz, D2O, pH 7) δ 10.28 (d, 2JPF = 70.2). 13C NMR (176 MHz, D2O, pH 7) δ 29.2 (dd, 2JFC = 21.2, 2JPC = 3.1, CH2CPF, 1C), 100.8 (dd, 1JFC = 190.6, 1JPC = 138.1, CPF, 1C), 115.7 (s, CHAr, 1C), 122.5 [d, 3JPC = 14.0, CAr(3), 1C], 123.0 [d, 5JFC = 3.8, CHAr(5), 1C], 124.9 (s, CHAr, 1C), 125.3 [s, CAr(6), 1C], 126.8 [s, CHAr(11), 2C], 127.9 [s, CHAr(12), 2C], 131.2 [s, CAr(10), 1C], 131.4 [s, CHAr(2), 1C], 140.7 [s, CAr(13), 1C], 144.6 [s, CAr(9), 1C], 172.2 (s, CONH2, 1C), 176.6 (dd, 2JFC = 20.8, 2JPC = 3.3, CO2, 1C). 19F NMR (659 MHz, D2O, pH 7) δ −164.52 (ddd, 2JPF = 70.2, 3JFH = 42.0, 3JFH = 6.4, CH2CFP, 1F).
3-(6-(2-Carboxyethyl)imidazo[1,2-a]pyridin-3-yl)-2-fluoro-2-phosphonopropanoic acid (1k)
Scale of the reaction: compound 7k′ 40 mg. Yield 52%. 1H NMR (700 MHz, D2O, pH 8) δ 2.78 (t, 3JHH = 7.2, CH2CO2, 2H), 3.07 (t, 3JHH = 7.2, CH2CH2CO2, 2H), 3.71–4.22 (m, CH2CPF, 2H), 7.69–7.89 [m, CHAr(2, 7, 8), 3H], 8.54 [bs, CHAr(5), 1H]. 31P NMR (284 MHz, D2O, pH 8) δ 7.05 (d, 2JPF = 73.7). 13C NMR (176 MHz, D2O, pH 8) δ 27.0 (s, CH2CH2CO2, 1C), 27.6 (bd, 2JFC = 21.3, CH2CPF, 1C), 34.4 (s, CH2CO2, 1C), 97.5 (m, CPF, 1C), 111.7 [s, CHAr(8), 1C], 121.2 [d, 3JPC = 14.7, CAr(3), 1C], 121.3 [s, CHAr(2), 1C], 124.8 [d, 5JFC = 4.2, CHAr(5), 1C], 130.2 [s, CHAr(6), 1C], 135.2 [s, CHAr(7), 1C], 139.1 [s, CAr(9), 1C], 171.6 (bs, FCCO2H, 1C), 176.9 (s, CH2CO2H, 1C).
3-(3-(2-Carboxy-2-fluoro-2-phosphonoethyl)imidazo[1,2-a]pyridin-6-yl)propan-1-aminium chloride (1l)
Scale of the reaction: compound 7l 75 mg. Product 1l was purified by preparative HPLC (eluent A: H2O:ACN:TFA 95:5:0.1 (v/v/v), isocratic, retention time 3.3 min) as eluent followed by lyophilization from 0.1 M HCl (repeated three times). Yield 93%. 1H NMR (700 MHz, D2O, pH 2) δ 2.10 [tt, 3JHH = 7.4 CAr(6)CH2CH2, 2H], 2.93 [dd, 3JHH = 7.5, 6.5 CAr(6)CH2, 1H], 3.08 (t, 3JHH = 7.8, CH2NH2, 2H), 3.89 (ddd, 2JHH = 16.6, 3JFH = 10.1, 3JPH = 6.7, CH2C(F)P, 1H), 4.05 (ddd, 3JFH = 38.0, 2JHH = 16.5, 3JPH = 4.1, CH2C(F)P, 1H), 7.84 [d, 3JHH = 9.1, CHAr(8), 1H], 7.84 [s, CHAr(2), 1H], 7.88 [d, 3JHH = 9.2, CHAr(7), 1H], 8.60 [s, CHAr(5), 1H]. 13C NMR (176 MHz, D2O, pH 2) δ 27.5 (s, CH2CH2NH2, 1C), 27.5 (d, 2JFC = 19.5, CH2C(F)P, 1C), 28.6 [s, CAr(6)CH2, 1C], 38.6 (s, CH2NH2, 1C), 97.0 (dd, 1JFC = 192.8, 1JPC = 144.2, C(F)P, 1C), 111.8 [s, CHAr(8), 1C], 121.1 [d, 3JPC = 13.7, CAr(3), 1C], 121.4 [s, CHAr(2), 1C], 124.6 [d, JFC = 3.4, CHAr(5), 1C], 130.2 [s, CAr(6), 1C], 135.1 [s, CHAr(7), 1C], 139.1 [s, CAr(9), 1C], 171.7 (d, 2JFC = 23.7, CO2, 1C). 31P NMR (284 MHz, D2O, pH 2) δ 6.79 (d, 2JPF = 72.0). HRMS m/z: (C13H18FN3O5P)+ calculated 346.0963, found 346.3316.
Biological Studies—General
PrestoBlue® Cell Viability Reagent, Mem-PER™ Plus Membrane Protein Extraction Kit, and all reagents for cell culture were purchased from Life Technologies (Carlsbad, CA, USA). Bradford Protein Assay and Clarity™ Western ECL Substrate were obtained from Bio-Rad (Hercules, CA, USA). Protease inhibitor cocktail and lovastatin were purchased from Sigma (Saint Louis, MO, USA). Primary antibodies against Rab11A, Rap1A/Rap1B, and Ras were obtained from Abcam (Cambridge, UK), whereas primary antibodies against β-actin and secondary HRP-linked antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA).
HeLa Cell Culture
The cervical epithelial carcinoma HeLa cell line was purchased from the American Type Cell Collection (ATCC). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS), 100 IU/ml penicillin, 0.25 μg/ml amphotericin B, and 50 μg/ml neomycin. Cells were incubated in a humidified incubator at 37°C and 5% CO2. Stock solutions of all compounds have been prepared freshly in phosphate-buffered saline (PBS) (10 mM) and directly diluted in culture medium.
In vitro Assessment of Cytotoxicity
HeLa cells were seeded into 96-well plates at a density of 4 × 103 cells/well in 100 μl of culture medium. After 24 h, cells were washed with PBS and 100 μl of serum-free medium was added. Subsequently, HeLa cells were treated with compounds at eight concentrations (25, 50, 100, 250, 500, 750, 1,000, and 2,000 μM) for 72 h. After incubation, PrestoBlue® Cell Viability Reagent was applied to each well and fluorescence at Ex/Em = 530/590 nm was measured in a Synergy 2 Microplate Reader (BioTek, Vermont, USA) following 50 min incubation. The obtained fluorescence magnitudes were used to calculate cell viability expressed as a percentage of the untreated control cells' viability. The data expressed as the mean of at least three independent experiments were used to calculate the IC50 parameter.
Determination of RGGT, GGT-I, and FT Activity Inhibition
HeLa cells were seeded into six-well cell culture plates at a density of 4 × 105 cells/well in 3 ml of culture medium. On the following day, 1.5 ml of fresh serum-free medium was added after washing cells with 1 ml of PBS. Subsequently, cells were supplemented with studied compounds as well as lovastatin, which served as a positive control. After 48 h of incubation, cell monolayers were rinsed with PBS and detached using a trypsin–EDTA solution. To obtain cell fractions enriched with cytosolic or membrane proteins, containing protease inhibitor cocktail, Mem-PER™ Plus Membrane Protein Extraction Kit was applied according to the manufacturer's instructions. The concentration of proteins in both fractions was measured using the Bradford protein assay. Equal amounts of cytosolic proteins (30 μg) were resolved by 12% SDS-PAGE gels and transferred on 0.2 μm nitrocellulose membrane. Membranes were incubated with β-actin, Rab11A, Rap1A/Rap1B, or Ras primary antibodies overnight, and then an appropriate HRP-conjugated secondary antibody was applied. Anti-Ras antibodies are predicted to react with H-, N-, and K-Ras. Visualization of protein bands was obtained using enhanced chemiluminescence (ECL) method and was performed using the ChemiDoc™ MP Imaging System (Bio-Rad). Densitometry analysis was performed with ImageLab™ Software (Bio-Rad), and relative unprenylated protein band intensity was normalized to β-actin and quantified relative to controls (untreated cells).
Statistical Analysis
Unless stated otherwise, biological results are presented as means ± standard error of the mean (SEM) from at least three independent experiments. Statistical analysis was performed using GraphPad Prism software (version 6.01 for Windows, GraphPad Software, La Jolla, CA, USA, www.graphpad.com). Statistical differences between mean values of inhibitor-treated and untreated control samples were evaluated using one-way analysis of variance (ANOVA) followed by Dunnett's multiple comparisons test. Confidence p levels are indicated by asterisks, with * denoting p ≤ 0.05, ** denoting p ≤ 0.01, *** denoting p ≤ 0.001, and **** denoting p ≤ 0.0001.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
KB conceived the study and was in charge of overall direction and planning. KB and EG-D supervised the project. DK, AM, JM, and KJ carried out the experiments. DK, JM, and KJ performed the synthesis and analyzed the NMR spectra. AM performed the biological studies. JM drafted the manuscript. JM and AM designed the figures and schemes. KB and JM took the lead in writing the manuscript, while EG-D and AM provided the biological part. All authors provided critical feedback and helped shape the research, analysis, and manuscript.
Funding
This work was financially supported by the National Science Centre, Poland (Sonata Bis 2014/14/E/ST5/00491).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Marek Domin (Boston College, USA) for the HRMS of the products. We thank Dr. Barbara Pacholczyk-Sienicka and Mr. Grzegorz Ciepielowski for carrying out the NMR experiments and Ms. Alicja Górczyńska for the synthesis of selected precursor compounds.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2020.596162/full#supplementary-material
References
Ali, N., Jurczyluk, J., Shay, G., Tnimov, Z., Alexandrov, K., Munoz, M. A., et al. (2015). A highly sensitive prenylation assay reveals in vivo effects of bisphosphonate drug on the Rab prenylome of macrophages outside the skeleton. Small GTPases 6, 202–211. doi: 10.1080/21541248.2015.1085485
Banworth, M. J., and Li, G. (2018). Consequences of Rab GTPase dysfunction in genetic or acquired human diseases. Small GTPases 9, 158–181. doi: 10.1080/21541248.2017.1397833
Baron, R. A., Tavaré, R., Figueiredo, A. C., Błazewska, K. M., Kashemirov, B. A., McKenna, C. E., et al. (2009). Phosphonocarboxylates inhibit the second geranylgeranyl addition by Rab geranylgeranyl transferase. J. Biol. Chem. 284, 6861–6868. doi: 10.1074/jbc.M806952200
Bhuiyan, N. H., Varney, M. L., Wiemer, D. F., and Holstein, S. A. (2019). Novel benzimidazole phosphonates as potential inhibitors of protein prenylation. Bioorg. Med. Chem. Lett. 29:126757. doi: 10.1016/j.bmcl.2019.126757
Błazewska, K. M., Ni, F., Haiges, R., Kashemirov, B. A., Coxon, F. P., Stewart, C. A., et al. (2011). Synthesis, stereochemistry, and SAR of a series of minodronate analogues as RGGT inhibitors. Eur. J. Med. Chem. 46, 4820–4826. doi: 10.1016/j.ejmech.2011.04.063
Bon, R. S., Guo, Z., Stigter, E. A., Wetzel, S., Menninger, S., Wolf, A., et al. (2011). Structure-guided development of selective RabGGTase inhibitors. Angew. Chem. 123, 5059–5063. doi: 10.1002/ange.201101210
Coxon, F., Joachimiak, Ł., Najumudeen, A. K., Breen, G., Gmach, J., Oetken-Lindholm, C., et al. (2014). Synthesis and characterization of novel phosphonocarboxylate inhibitors of RGGT. Eur. J. Med. Chem. 84, 77–89. doi: 10.1016/j.ejmech.2014.06.062
Coxon, F. P., Helfrich, M. H., Larijani, B., Muzylak, M., Dunford, J. E., Marshall, D., et al. (2001). Identification of a novel phosphonocarboxylate inhibitor of rab geranylgeranyl transferase that specifically prevents rab prenylation in osteoclasts and macrophages. J. Biol. Chem. 276, 48213–48222. doi: 10.1074/jbc.M106473200
Deraeve, C., Guo, Z., Bon, R. S., Blankenfeldt, W., DiLucrezia, R., Wolf, A., et al. (2012). Psoromic acid is a selective and covalent rab-prenylation inhibitor targeting autoinhibited rabggtase. J. Am. Chem. Soc. 134, 7384–7391. doi: 10.1021/ja211305j
Greenwood, J., Steinman, L., and Zamvil, S. S. (2006). Statin therapy and autoimmune disease: from protein prenylation to immunomodulation. Nat. Rev. Immunol. 6, 358–370. doi: 10.1038/nri1839
Istvan, E. S., and Deisenhofer, J. (2001). Structural mechanism for statin inhibition of HMG-CoA reductase. Science 292, 1160–1164. doi: 10.1126/science.1059344
Jeong, A., Suazo, K. F., Wood, W. G., Distefano, M. D., and Li, L. (2018). Isoprenoids and protein prenylation: implications in the pathogenesis and therapeutic intervention of Alzheimer's disease. Crit. Rev. Biochem. Mol. Biol. 53, 279–310. doi: 10.1080/10409238.2018.1458070
Joachimiak, Ł., Janczewski, Ł., Ciekot, J., Boratyński, J., and Błazewska, K. (2015). Applying the prodrug strategy to α-phosphonocarboxylate inhibitors of Rab GGTase-synthesis and stability studies. Org. Biomol. Chem. 13, 6844–6856. doi: 10.1039/C5OB00281H
Joachimiak, Ł., Marchwicka, A., Gendaszewska-Darmach, E., and Błazewska, K. M. (2018). Synthesis and biological evaluation of imidazole-bearing α-phosphonocarboxylates as inhibitors of Rab Geranylgeranyl Transferase (RGGT). ChemMedChem 13, 842–851. doi: 10.1002/cmdc.201700791
Justyna, K., Małolepsza, J., Kusy, D., Maniukiewicz, W., and Błazewska, K. M. (2020). The McKenna reaction—avoiding side reactions in phosphonate deprotection. Beilstein J. Org. Chem. 16, 1436–1446. doi: 10.3762/bjoc.16.119
Kazmierczak, A., Kusy, D., Niinivehmas, S. P., Gmach, J., Joachimiak, Ł., Pentikäinen, O. T., et al. (2017). Identification of the privileged position in the imidazo[1,2-a]pyridine ring of phosphonocarboxylates for development of Rab Geranylgeranyl Transferase (RGGT) inhibitors. J. Med. Chem. 60, 8781–8800. doi: 10.1021/acs.jmedchem.7b00811
Kelly, E. E., Horgan, C. P., Goud, B., and McCaffrey, M. W. (2012). The Rab family of proteins: 25 years on. Biochem. Soc. Trans. 40, 1337–1347. doi: 10.1042/BST20120203
Kusy, D., Maniukiewicz, W., and Błazewska, K. M. (2019). Microwave-assisted synthesis of 3-formyl substituted imidazo[1,2-a]pyridines. Tetrahedron Lett. 60:151244. doi: 10.1016/j.tetlet.2019.151244
Kusy, D., Wojciechowska, A., Małolepsza, J., and Błazewska, K. M. (2020). Functionalization of the imidazo[1,2- a]pyridine ring in α-phosphonoacrylates and α-phosphonopropionates via microwave-assisted Mizoroki–Heck reaction. Beilstein J. Org. Chem. 16, 15–21. doi: 10.3762/bjoc.16.3
Marma, M. S., Xia, Z., Stewart, C., Coxon, F., Dunford, J. E., Baron, R., et al. (2007). Synthesis and biological evaluation of α-halogenated bisphosphonate and phosphonocarboxylate analogues of risedronate. J. Med. Chem. 50, 5967–5975. doi: 10.1021/jm0702884
McKenna, C. E., Kashemirov, B. A., Błazewska, K. M., Mallard-Favier, I., Stewart, C. A., Rojas, J., et al. (2010). Synthesis, chiral high performance liquid chromatographic resolution, and enantiospecific activity of a potent new geranylgeranyl transferase inhibitor, 2-hydroxy-3-imidazo[1,2-a]pyridin-3-yl-2-phosphonopropionic acid. J. Med. Chem. 53, 3454–3464. doi: 10.1021/jm900232u
McKenna, C. E., and Schmidhuser, J. (1979). Functional selectivity in phosphonate ester dealkylation with bromotrimethylsilane. J. Chem. Soc. Chem. Commun. 870:739. doi: 10.1039/c39790000739
Mitra, S., Cheng, K. W., and Mills, G. B. (2011). Rab GTPases implicated in inherited and acquired disorders. Semin. Cell Dev. Biol. 22, 57–68. doi: 10.1016/j.semcdb.2010.12.005
Ouvry, G., Bouix-Peter, C., Ciesielski, F., Chantalat, L., Christin, O., Comino, C., et al. (2016). Discovery of phenoxyindazoles and phenylthioindazoles as RORγ inverse agonists. Bioorg. Med. Chem. Lett. 26, 5802–5808. doi: 10.1016/j.bmcl.2016.10.023
Russell, R. G. G. (2011). Bisphosphonates: the first 40 years. Bone 49, 2–19. doi: 10.1016/j.bone.2011.04.022
Stigter, E. A., Guo, Z., Bon, R. S., Wu, Y. W., Choidas, A., Wolf, A., et al. (2012). Development of selective, potent RabGGTase inhibitors. J. Med. Chem. 55, 8330–8340. doi: 10.1021/jm300624s
Tan, K. T., Guiu-Rozas, E., Bon, R. S., Guo, Z., Delon, C., Wetzel, S., et al. (2009). Design, synthesis, and characterization of peptide-based Rab geranylgeranyl transferase inhibitors. J. Med. Chem. 52, 8025–8037. doi: 10.1021/jm901117d
Tennakoon, M., Kankanamge, D., Senarath, K., Fasih, Z., and Karunarathne, A. (2019). Statins perturb GBG signaling and cell behavior in a GG subtype dependent manners. Mol. Pharmacol. 95, 361–375. doi: 10.1124/mol.118.114710
Watanabe, M., Fiji, H. D. G., Guo, L., Chan, L., Kinderman, S. S., Slamon, D. J., et al. (2008). Inhibitors of protein geranylgeranyltransferase I and Rab geranylgeranyltransferase identified from a library of allenoate-derived compounds. J. Biol. Chem. 283, 9571–9579. doi: 10.1074/jbc.M706229200
Keywords: phosphonocarboxylate, phosphonocarboxylic acid, phosphonopropionate, phosphonopropionic acid, RGGT, Rab geranylgeranyl transferase, prenylation, imidazo[1, 2-a]pyridine
Citation: Kusy D, Marchwicka A, Małolepsza J, Justyna K, Gendaszewska-Darmach E and Błażewska KM (2021) Synthesis of the 6-Substituted Imidazo[1,2-a]Pyridine-3-yl-2- Phosphonopropionic Acids as Potential Inhibitors of Rab Geranylgeranyl Transferase. Front. Chem. 8:596162. doi: 10.3389/fchem.2020.596162
Received: 18 August 2020; Accepted: 26 November 2020;
Published: 06 January 2021.
Edited by:
Pawel Kafarski, Wrocław University of Technology, PolandReviewed by:
Letizia Crocetti, University of Florence, ItalyStamatia Vassiliou, National and Kapodistrian University of Athens, Greece
Copyright © 2021 Kusy, Marchwicka, Małolepsza, Justyna, Gendaszewska-Darmach and Błażewska. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katarzyna Magdalena Błażewska, katarzyna.blazewska@p.lodz.pl
†These authors have contributed equally to this work