Ab initio Study of Anchoring Groups for CuGaO2 Delafossite-Based p-Type Dye Sensitized Solar Cells
 Ana B. Muñoz-García1* Laura Caputo2 Eduardo Schiavo2 Carmen Baiano2 Pasqualino Maddalena1
Ana B. Muñoz-García1* Laura Caputo2 Eduardo Schiavo2 Carmen Baiano2 Pasqualino Maddalena1  Michele Pavone2*
Michele Pavone2*- 1Department of Physics “Ettore Pancini”, University of Naples Federico II, Comp. Univ. Monte Sant'Angelo, Naples, Italy
- 2Department of Chemical Sciences, University of Naples “Federico II”, Comp. Univ. Monte Sant'Angelo, Naples, Italy
Here we report the first theoretical characterization of the interface between the CuGaO2 delafossite oxide and the carboxylic (–COOH) and phosphonic acid (–PO3H2) anchoring groups. The promising use of delafossites as effective alternative to nickel oxide in p-type DSSC is still limited by practical difficulties in sensitizing the delafossite surface. Thus, this work provides atomistic insights on the structure and energetics of all the possible interactions between the anchoring functional groups and the CuGaO2 surface species, including the effects of the Mg doping and of the solvent medium. Our results highlight the presence of a strong selectivity toward the monodentate binding mode on surface Ga atoms for both the carboxylic and phosphonic acid groups. Since the binding modes have a strong influence on the hole injection thermodynamics, these findings have direct implications for further development of delafossite based p-type DSSCs.
Introduction
The increasing world energy demands have boosted research toward the development of technologies that can exploit renewable sources in an efficient way (Hagfeldt et al., 2010). Being inexhaustible and relatively well spread all over the globe, solar energy has the highest potential to satisfy the present and future global energy needs. The photovoltaic (PV) field has rapidly evolved from traditional and commonly used silicon panels based on semiconductor p/n junctions to competitive thin film technologies relying on less abundant elements (Parida et al., 2011). Branching off from these devices, dye-sensitized solar cells (DSSC) arose 20 years ago, initially as cost-effective alternatives to solid-state photovoltaic solar cells. Now that the cost competitiveness with Si is less relevant, DSSCs are still in the spotlight, due to their lightweight, transparency, flexibility and best performance at diffuse and low-intensity light, which make them suitable for portable and indoor applications (Benesperi et al., 2018).
The most studied DSSCs are photoanodes where the electric current arises from the electron injection from the LUMO of a photoexcited dye to the conduction band of the n-type semiconductor (typically TiO2 or ZnO) where such dye is anchored. To date, traditional n-type DSSCs with metallic counterelectrodes have reached ~14% in photo–conversion efficiencies (PCE), while more recently the closely related hybrid organic/inorganic perovskite-based set-ups have exceeded the ~20% PCE (Yang et al., 2017). In order to overcome the intrinsic efficiency drawbacks of first generation DSSCs with only one chromophore, tandem cells have been proposed (He et al., 2000; Green, 2003; Nakasa et al., 2005; Nattestad et al., 2010). In tandem cells, a n-DSSC is coupled to a photocathode made of a p-type semiconductor that is sensitized by a dye with a red-shifted adsorption (with respect to dye at the photoanode), so that a larger portion of the solar spectrum can be harvested. Besides the potential higher photocurrent and PCEs for PV applications, tandem cells can be devised as well as photoelectrochemical devices able to perform photoinduced chemical reactions of great relevance in the energy conversion scenario (e.g., water splitting) (Prévot and Sivula, 2013). The actual application of tandem cells is however hampered for the limited efficiencies of the p-type DSSCs, which reach efficiencies of ~2%, still far below the ~14% of their n-type counterparts (Odobel et al., 2010).
A main culprit behind such poor performances has been ascribed to NiO, which is the most used p-type semiconductor (SC) in p-DSSCs due to its low cost and easy manipulation (Morandeira et al., 2005; Mori et al., 2008; Odobel et al., 2012). Unfortunately, NiO presents substantial intrinsic drawbacks: low electrical conductivity, low hole mobility and high valence band edge potential with respect to the most common I−/ electrolyte, which results in a too low photocathode open circuit potential (VOC) (Odobel and Pellegrin, 2013). The development of new efficient p-DSSC as alternative to NiO is thus of primary relevance.
Copper delafossites with formula CuMO2 (M = Al, Ga, Cr…) (Sullivan et al., 2016) have emerged among the few materials that can indeed outperform NiO as p-SC. Delafossites have wide optical bandgap and low valence band edge (Marquardt et al., 2006; Yu et al., 2012; Kumar et al., 2013). Nattestad et al. reported one of the first consistent comparisons between NiO and CuAlO2 in p-DSSC, obtaining VOC values of 218 and 333 mV, respectively (Nattestad et al., 2011). Later, experimental and theoretical investigations confirmed the lower VBedge position of CuAlO2 compared with NiO (Yu et al., 2014; Das et al., 2015; Schiavo et al., 2018). CuGaO2 and CuCrO2 were also reported as possible alternative to NiO, with lower band edge positions and higher transparency (Yu et al., 2012). With respect to NiO-based p-DSSC, with the new Ga and Cr delafossite oxides the measured increase in VOC was about 160 and 110 mV, respectively (Renaud et al., 2012; Powar et al., 2014). Several works show that the efficiency of these materials in p-DSSCs can be further improved by doping them with a divalent cation (e.g., Mg) at the M site (Scanlon and Watson, 2011; Jiang et al., 2013). This, in fact, enhances the p-type conductivity and has a positive effect on the morphology of the nanoparticles, which can expose a higher surface area resulting in a better light harvesting (Renaud et al., 2014).
However, despite the positive effect on VOC, the replacement of NiO with CuMO2 does not always result in an overall significant increase of photocurrent and/or PCE. While the VOC is determined by the p-SC VBedge vs. the electrolyte redox couple reduction potential, the overall cell efficiency strongly depends also on the interfacial electronic processes that occur between the semiconductor and the sensitizer. First, the lower the position of the valence band, the smaller is the driving force for hole injection to a given dye. For this reason, typical “p-type dyes” used for NiO may not be suitable for delafossites. For example, Renaud et al. tested the C343 prototype coumarin dye on CuGaO2 and did not measure any photocurrent (Renaud et al., 2012). P1, PMI-6T-TPA, or PMI-NDI dyes are used to sensitize CuGaO2, CuAlO2, and CuCrO2 delafossites, delivering small but non-zero current (Yu et al., 2012, 2014).
Another critic issue related to the delafossite-dye interface is the limited light-harvesting arising from poor dye coverage (Renaud et al., 2014). When synthesized via conventional solid-state reaction, CuMO2 tend to form large-size (>1 μn) anisotropic plate-like particles that densely stack along the basal planes, resulting in a limited surface area available for dye sensitization (Yu et al., 2014). With focused but less straightforward synthetic methods (e.g., following the hydrothermal route) it is possible to obtain smaller particles, close to the ideal 20–40 nm (i.e., the typical TiO2 nanoparticle size in n-DSSC) (Xiong et al., 2013; Yu et al., 2014). Nevertheless, these alternative synthetic routes are challenging, also depending on the element at the M site, and advances toward increasing dye coverage are highly desirable, even for particles with non-ideal shape or morphology.
In this context, an open issue is whether the dye-anchoring groups that are used for binding the dye to the rocksalt NiO surfaces are also good for a stable and irreversible binding to the Cu-based delafossite oxide most exposed surfaces. To address this specific issue related to the dye-electrode interface in delafossite-based p-DSSCs, we present here a first-principles study on two anchoring groups on CuGaO2 (001) surface: carboxylic acid (–COOH), which is the most common anchoring group used in both n- and p-type DSSCs, and phosphonic acid (–PO3H2), which is one of the anchoring groups explored in NiO-based p-type DSSCs in search for higher efficiencies (Pellegrin et al., 2011; Klein et al., 2018) and guarantees more stability in aqueous environments where COOH tends to desorb from oxide surfaces (De Angelis et al., 2011; Galliano et al., 2017).
First-principles calculations are a valuable tool for obtaining an atomistic insight on the structures, chemical interactions, and electronic features at the dye-electrode interfaces, thus providing a better understanding of the undergoing elementary processes. While electronic, adsorption and excited properties and level alignment of dyes anchored on n-type semiconductors (mainly TiO2) have been studied extensively using density functional theory (DFT) and time-dependent DFT (Martsinovich and Troisi, 2011; Labat et al., 2012; Adamo and Jaquemin, 2013; Bai et al., 2014), p-type semiconductor/dye interfaces have been investigated only more recently, and, in particular, considering NiO as semiconductor. An early work by Preat et al. focuses on computing the electronic excitation and electron injection processes of P1 and derived dyes using as model for NiO a single Ni atom capped by a hydroxyl group to ensure the overall neutrality of the system (Preat et al., 2011). Regarding periodic-slab and spin-polarized DFT calculations, taking into account also the intrinsic magnetism of NiO, only few recent computational studies have addressed the interaction of dye/anchoring groups with the most stable surface of NiO, i.e., the (001) (Muñoz-García and Pavone, 2015; Kontkanen et al., 2016; Wykes et al., 2016; Carella et al., 2018). Wykes et al. (2016) have studied formic and benzoic acid (HCOOH and PhCOOH) as prototypes of the most commonly used carboxyl-based anchoring groups, as well as mono and dimer phenyl-silane [PhSi(OH)3] anchoring candidates, due to the known capacity of alkoxysilanes to form Si-O-metal bonds. Besides different adsorption strengths, they find that such anchoring groups induce different shifts of the NiO VB and CB edges. A previous work by our group on the coumarin dye C343 (which features –COOH as anchoring group) and its analogous with –PO3H2 adsorbed on NiO (001) has shown that not only the nature of the binding group but also the binding modes largely affect the thermodynamic driving force for hole injection, due to the interface dipole generated by the H released to the surface in bi- and tri-dentate cases (Muñoz-García and Pavone, 2015). Piccinin et al. recently reported ab initio molecular dynamics simulations on the coumarin dye with –PO3H2 anchoring group on NiO (001) including explicit water molecules, showing also that the hole injection driving force (NiO-VB/Dye-HOMO level alignment) is very sensitive of the specific anchoring modes and their dynamics in a protic solvent (Piccinin et al., 2017). These works highlight the key role played by the anchoring ligand adsorbed on the surface on the overall performance of the DSSC.
While a few theoretical works have addressed the study of the electronic structure of bulk Cu-based delafossites for DSSC applications (Gillen and Robertson, 2011; Schiavo et al., 2018), the features of the dye-delafossite interface are essentially unexplored. In this work, we perform state-of-the-art DFT-based periodic calculations on anchoring groups –COOH and –PO3H2 on delafossite CuGaO2, which has been proposed as the most convenient for DSSC applications thanks to its wide band gap and easier p-type doping with respect to other delafossites such as CuAlO2 or CuCrO2 (Schiavo et al., 2018). After evaluating the change in the VB edge position upon Mg doping, we report adsorption energies and relevant structural and electronic features of the different anchoring groups/modes on the surface, taking into account the different combination of surface metal sites (Cu or Ga) that can be involved in the dye anchoring. Finally, we present a comparison of our results on CuGaO2 with those on NiO.
Computational details and structural models
We performed periodic spin-polarized density functional theory (DFT) calculations with projector-augmented wave (PAW) potentials (Blöchl, 1994; Kresse and Joubert, 1999) and plane waves (PW) basis set by using the Vienna Ab Initio Simulation Package (VASP, version 5.4.1) (Kresse and Hafner, 1993, 1994; Kresse and Furthmüller, 1996a,b). The generalized-gradient approximation of Perdew Burke and Ernzherof (PBE) has been exploited for the exchange-correlation density functional (Perdew et al., 1996, 1997). To describe the strong-correlated nature of Cu d electrons we applied the rotationally invariant DFT+U approach of Dudarev (Dudarev et al., 1998) as implemented in VASP (Rohrbach et al., 2003). As in previous works on Cu(I)-containing oxides, we applied an average U-J effective value of 6 eV on Cu d electrons (Isseroff and Carter, 2012; D'Arienzo et al., 2017; Schiavo et al., 2018). SCF energy convergence threshold was set to 10−5 eV, and for minimum-energy structural optimizations the total forces on each atom were set to be all below 0.05 eV·Å−1. Regarding numerical parameters, with a kinetic energy cut-off of 750 eV for the PW and a k-point Monkhorst-Pack (Monkhorst and Pack, 1976) sampling grid of 6 × 6 × 2 for bulk CuGaO2 (see below) we obtained convergence of total energies within 5 meV per formula unit. K-point sampling for the slabs used have been scaled accordingly.
Although delafossites can present different polytypes (Marquardt et al., 2006), here we have considered the 3R phase, which is the only one detected with X-ray diffraction in CuGaO2 (Renaud et al., 2014). The hexagonal 3R polytype of CuGaO2 structure has the Ga(III) ions accommodated in a distorted octahedral cavity in the oxygen sublattice, generating layers of Ga(III)O6 units sharing their edges. These layers are separated by Cu(I) ions, linked in a linear geometry with two oxygens in two different layers (Figure 1A). For simplicity, we have built an orthorhombic supercell with aortho = ahexa, bortho = √3 bhexa, and cortho = chexa (Figure 1B). For surface calculations, we considered the (011) surface (012 in the hexagonal system), which has been identified among the preferred facets of delafossite crystals (Das et al., 2015; Alkhayatt et al., 2016) and, in particular, has the lowest surface energy in CuGaO2 (Schiavo et al., 2018). We cleaved such surface from orthorhombic bulk CuGaO2 with the theoretically determined equilibrium lattice constants (a = b = 2.98 Å, c = 17.64 Å), which deviate <3% from the experimental values (a = b = 2.97 Å, c = 17.17Å) (Ishiguro et al., 1981; Köhler and Jansen, 1986; Crottaz et al., 1996) For electronic structure calculations, the (011) surface was represented as slab with a (2 × 1) periodicity in the xy plane, five tri-atom layers of thickness and 10.5 Å of vacuum, in order to avoid images interaction along the z direction (Figure 1C). For Mg-doped CuGaO2, we have placed one Mg substituting one Ga atom on the central atom layer (Figure 1D), as it has been proven that the Mg substitution occurs at the Ga site (Jiang et al., 2013). This delivers a Mg-doped slab with 3.3% content of Mg (all % signs throughout the text are intended as atom %). An additional slab with (1 × 1) lateral periodicity has been used to study Mg-doped CuGaO2 surface with 6.7% of dopant, by substituting with Mg a single Ga atom in the central slab layer. Both in the undoped and Mg-doped cases, atomic positions of all the atoms have been allowed to relax.
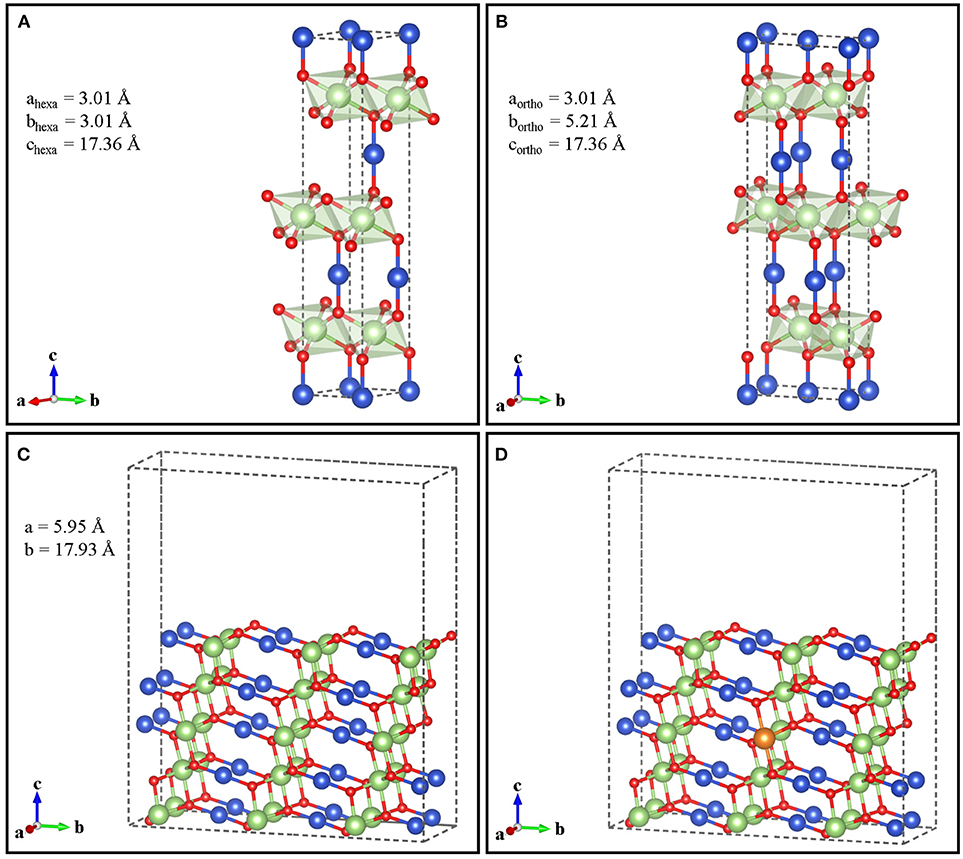
Figure 1. Hexagonal (A) and orthorhombic (B) unit cells of bulk CuGaO2 with calculated cell parameters. Undoped (C) and Mg-doped (D) (011) orthorhombic surface. Color code: Cu (blue), Ga (green), O (red), and Mg (orange).
The relaxed CuGaO2 surface slab was used to compute the workfunction and vacuum energy level in order to determine the VB edge absolute position (i.e., with respect to the NHE) using the approach proposed by Toroker et al. (2011):
Where BGC is the band-gap center of the slab from our PBE+U calculations and Eg is the eigenvalue gap of CuGaO2 bulk calculated at the HSE level of theory, i.e., 2.1 eV (Schiavo et al., 2018), which is very close to the value of 2.2 eV reported by Iozzi et al. (2015). Evac is the vacuum energy evaluated from the electrostatic potential along the direction normal to the surface plane. For the Mg-doped system, where the BGC is ill-defined due to the typical shoulder of empty valence band states above the Fermi level that appears in the density of states (DOS) in p-type semiconductors, we have calculated the VBedge from the VBedge of un-doped CuGaO2 applying a shift computed from the difference of the workfunctions of the two systems (WFMg:CuGaO2-WFCuGaO2).
To survey the adsorption modes and the corresponding adsorption energies/properties of –COOH and –PO3H2 anchoring groups on CuGaO2 (001), we have chosen as capping substituent the simple methyl group (–CH3). This is a safe approximation for studying anchoring properties since, as we show below, they depend very little on the dye skeleton.
CH3-COOH and CH3-PO3H2 molecules have been placed in one side of CuGaO2 (001) slab with 2 × 1 of lateral periodicity and 3 tri-atom layers of thickness. Adsorption energy vary of <50 meV by increasing the lateral periodicity to 3 × 1 and of <25 meV by increasing the slab thickness to 5 tri-layers. Vacuum was increased to 15 Å to prevent interaction between the slab periodic images. Since molecules are adsorbed onto only one side of the slab, dipole corrections have been applied to avoid long-range polarization from the periodic images along the z direction (Neugebauer and Scheffler, 1992). We have considered all the possible anchoring modes for each group, taking also into account the different surface sites where the anchoring can be bound. Final geometries of these structures have been obtained relaxing all atomic coordinate of the molecule and of the two uppermost CuGaO2 tri-atom layers and by freezing those of the bottommost layers.
After relaxation, we have calculated adsorption energies as follows:
from the energies of the total system (Eslab+CH3−X), the relaxed pristine slab (Eslab) and the isolated molecule (ECH3−X), with X = CO2H or PO3H2. Using the relaxed geometries from gas-phase calculations, we have computed also the adsorption energies in acetonitrile, a common solvent used in p-DSSCs, by performing single-point energy calculations with the implicit solvent scheme implemented in VASP (Mathew et al., 2014) with relative dielectric constant ε = 35.688.
For comparison with NiO, we have studied CH3COOH and CH3PO3H2 molecules anchored on NiO (001) following an analogous procedure. Structural and computational details for NiO (001) surface slab (kinetic energy cut-off, k-point sampling, U-J values, slab thickness, lateral periodicity) are exactly those exploited in a recent previous work on the C343/NiO (001) interface (Muñoz-García and Pavone, 2015).
Results and discussion
In p-type DSSCs, a first key parameter to assess the suitability of a p-semiconductor is the absolute position of its VB edge with respect to the electrolyte redox potential: the difference between these two values determines the open circuit potential VOC of the cell (Qin et al., 2008; Preat et al., 2011). For Mg doped CuGaO2, Renaud et al. (2014) have reported that Mg doping has a positive effect on the photoelectrode by increasing the specific surface area (SSA) of CuGaO2, but only CuGaO2 nanoparticles with low concentration of Mg (1%) deliver higher efficiencies than pristine CuGaO2. Higher concentrations of Mg (up to 5%), which increases further the SSA, do not lead to higher photocurrent. This drawback has been ascribed to the formation of structural defects or electronic features that promote undesired electron-hole recombination processes. Regarding the VOC with respect to the tris(4,4′-bis-tert-butyl-2,2′-bipyridine)cobalt(II/III) redox couple, the same authors (Renaud et al., 2012) measured a decrease in VOC values at increasing Mg contents. To understand this behavior, we computed VB edge positions of Mg-containing CuGaO2 in comparison to that of pristine CuGaO2 so to dissect to what extent Mg doping affects the VOC without any other structural defects. We have evaluated two different Mg concentrations, 3.3 and 6.7%, by substituting one Ga from the central tri-layer of the slab in the 5-tri-layer slabs with (2 × 1) and (1 × 1) periodicity, respectively. Figure 2 shows the resulting band edge potentials vs. NHE of CuGaO2, Mg-doped CuGaO2 and NiO together with common electrolytes employed in dye sensitized solar cells. All the reduction potentials of the redox mediators are measured in an acetonitrile solution, we refer the interested reader to the experimental references (Boschloo and Hagfeldt, 2009; Feldt et al., 2010; Cong et al., 2016) for the exact composition of the electrolyte.
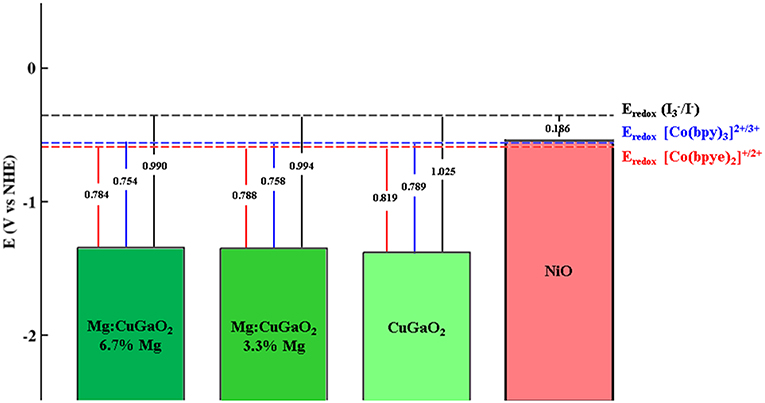
Figure 2. Calculated absolute positions of the valence band edges for, from left to right: Mg:CuGaO2 with two different dopant concentrations (6.7 and 3.3%), undoped CuGaO2 and NiO. Dashed lines represent the redox potentials of the three electrolytes taken from experimental studies: /I− (Boschloo and Hagfeldt, 2009), [Co(bpy)3]2+/3+ (Cong et al., 2016), and [Cu(bpye)2]+/2+ (Feldt et al., 2010). For completeness open circuit voltage calculated with respect to those electrolytes are reported.
The VB edge of CuGaO2 with 3.3% Mg is of 29 meV higher in energy with respect to the undoped case, and it rises up of another 4 meV when increasing Mg concentration from 3.3 to 6.7%. These numbers are of the same order of magnitude of the experimental values reported by Renaud et al. where a decrease of VOC of 30 meV for 1% Mg with respect to the undoped case and a further decrease of 10 meV when doping is increased up to 5% Mg (Renaud et al., 2014). Such little increase of the VB edge by Mg doping is still very low in comparison how low VB edge is in CuGaO2 with respect to NiO, which makes both CuGaO2 and Mg-doped CuGaO2 suitable for p-DSSCs not only with /I− electrolyte (Boschloo and Hagfeldt, 2009) but also with other redox couples that are not compatible with NiO, such as cobalt(II/III) tris(2,2′-bipyridine) [Co(bpy)3]2+/3+ and copper(I/II) bis(1,1-bis(2-pyridyl)ethane) [Cu(bpye)2]+/2+ (Feldt et al., 2010; Cong et al., 2016). It is important to note that these results provide only a qualitative picture of the energy level alignment between the electrode and the electrolyte redox couple, many features are missing in our model (solvent, band bending at the interface, effects of ionic species, etc.). However, the computed trend is consistent with the VOC values observed in experiments and this agreement provides a good assessment on the quality of our surface slab model.
Understanding how the dye anchoring groups interact with the delafossite surface is of primary relevance: high adsorption strengths are needed to provide high dye coverages and, thus, high photoconversion efficiency. Moreover, in the case of H-containing anchoring groups, the individuation of preferred anchoring modes can help to explain and predict the emerging efficiencies, as different anchoring modes of the same anchoring group can deliver different band alignments between the dye HOMO and the p-semiconductor VB edge (Muñoz-García and Pavone, 2015; Zhang and Cole, 2015; Adineh et al., 2016).
We have surveyed all possible anchoring modes for both –COOH and –PO3H2 functional groups on CuGaO2 (011), taking into account the different possible surface adsorption sites and combinations of them. Thus, we have considered monodentate binding on either Ga or Cu surface atoms (M-Ga and M-Cu, respectively), bidentate binding on Ga/Ga, Cu/Cu or Ga/Cu surface atom pairs (B-Ga-Ga, B-Cu-Cu, and B-Ga-Cu) and, for CH3-PO3H2 we have considered also the two possible tridentate binding modes, on two Ga atoms plus one Cu atom or on two Cu atoms plus one Ga atom (T-Ga-Ga-Cu or T-Cu-Cu-Ga). For M cases, we have added the sub index “H” when the OH group forms an H bond to an oxygen surface atom upon relaxation. In B and T cases, the H atoms have been attached to surface oxygen atoms, as far as possible from the anchoring group to avoid artificial overstabilization and distortion of the structures due to H-bond formation between the released H and the O atoms from the anchoring molecules.
Relaxed geometries of CH3COOH on CuGaO2 (011) in monodentate (M) and bidentate (B) binding modes are shown in Figure 3 and corresponding selected structural parameters together with binding energies (Eads) are listed in Table 1. xyz files of the intermediates structures are reported in Supplementary Information. These geometries and Eads are compared to those of CH3COOH on NiO (001).
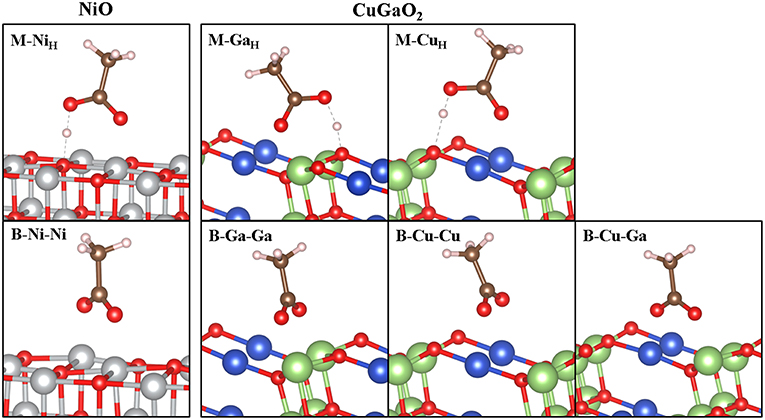
Figure 3. Optimized structures of CH3COOH on CuGaO2 (011) 2 × 1 × 3 L slab (right) and on NiO (001) (left). Labels according to the anchoring mode [monodentate (M) and bidentate (B)] and to the surface atoms involved in the adsorption process. Sub index “H” indicates H bonding (dashed gray line) between the OH group and surface oxygen atom upon relaxation in M cases. Color legend: Ni (gray), Cu (blue), Ga (green), O (red), C (brown), and H (light pink).
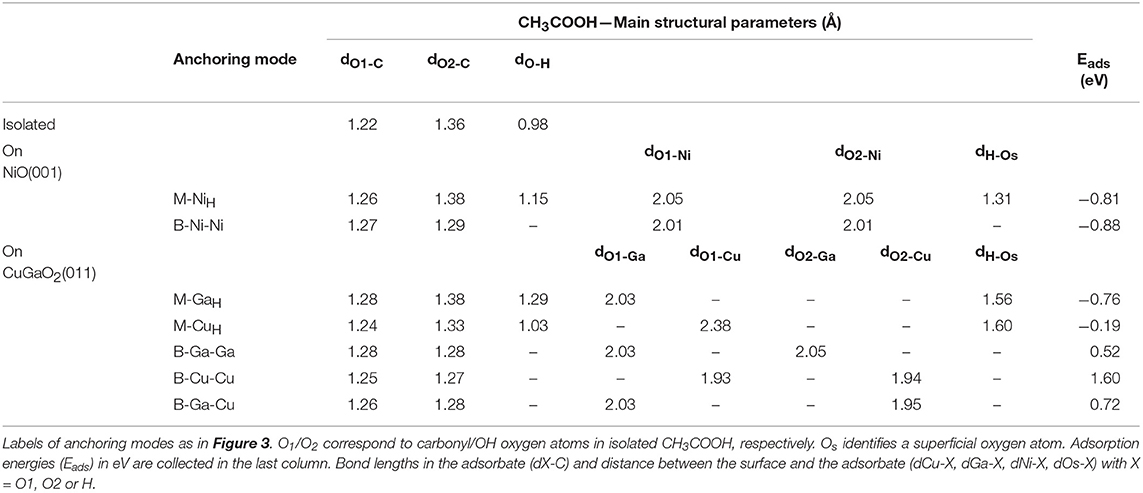
Table 1. Selected structural parameters for relaxed CH3COOH anchored on CuGaO2 (011) and on NiO (001) together with those of the isolated molecule calculated at the DFT-PBE+U level of theory in vacuum.
Adsorbed CH3-COOH in M binding present similar structural features in CuGaO2 (011) and NiO (001), with the molecule not suffering any significant distortion from the isolated minimum. The main difference resides in the different M-carbonyl oxygen distance to the electrode surface (dO1−Xin Table 1), which is much shorter for bonding to a surface Ga (2.03 Å) than to a surface Cu (2.38 Å). The Ga-O1 distance in CuGaO2 is also very similar to the Ni-O1 distance in NiO (2.05 Å) and both values are similar to typical Ga-Olattice and Ni-Olattice in parent solids (2.00 and 2.11 Å in CuGaO2 and NiO, respectively). Contrarily, Cu-O1 distance is significantly longer than Cu-Olattice distances in CuGaO2 (1.87 Å). In CuGaO2, our calculations predict slightly longer H bonds between the OH and surface oxygen atoms (dH−Osin Table 1) in CuGaO2 with respect to NiO. In bidentate modes, the two oxygen atoms of CH3COOH molecule become equivalent and, accordingly, we obtain two equal M-O distances for B-Ga-Ga and B-Cu-Cu cases of CuGaO2 and B-Ni-Ni of NiO. Differently for the M case, here the Cu-O1 distances are much smaller and similar to those of CuGaO2 bulk. In the mixed B-Ga-Cu case, each Ga-O and Cu-O distance is equal to those in B-Ga-Ga and B-Cu-Cu. In spite of the different surface patterns of CuGaO2 (011) and NiO (001), M-M distances are very similar in both solids, i.e., d(Ni-Ni)NiO = 2.99 Å and d(Ga-Ga)CuGaO2 = d(Cu-Cu)CuGaO2 = 2.98 Å, hence the similarities in the binding geometries. Only a slight tilting of the C-C bond with respect to the z axis is predicted for B-Ga-Ga and in B-Cu-Cu in CuGaO2, having the CuGaO2 (011) surface a non-planar pattern.
In spite of these structural analogies between CuGaO2 and NiO, the binding energies (Eads in Table 1) of the different anchoring modes of CH3COOH to the two surfaces present substantial differences. In NiO, Eads of both M and B modes are negative and close, being Eads(B) 0.07 eV more stable than Eads(M). We must note that the binding energies reported here for CH3-COOH (as well as those indicated below for CH3-PO3H2) on NiO are similar to those considering the full C343 dye (Muñoz-García and Pavone, 2015) within 0.1 eV, and deliver the same result of B being more stable than M [Eads(M)-Eads(B) = 0.12 eV for C343]. Thus, we can consider the CH3- capping group as a good approximation for studying adsorption properties of different anchoring groups on the electrode surfaces. Such negative and similar Eads values can be translated into expecting NiO covered by the dye in the two anchoring modes in approximately equal ratio in experimental conditions. In CuGaO2, instead, there is a strong preference for the monodentate binding because all B modes surveyed present a positive (i.e., not favorable) Eads. In particular, M binding is stronger at the Ga site than at the Cu site, with Eads values similar to those for NiO. This is consistent with the trend of transition metal-oxo complex bond dissociation energies (BDE) (Luo, 2007) or Ni-O and Ga-O (−366 and −374 KJ/mol), with a smaller value for Cu-O BDE (−287 kJ/mol). Moreover, while surface Ni/Ga atoms are unsaturated species in NiO (001)/CuGaO2 (011), surface Cu retains its bulk-like linear coordination with two oxygen atoms in CuGaO2 (011).
Analogously, we have explored all possible binding modes of the phosphonic acid group on CuGaO2 (011). Relaxed geometries of CH3PO3H2 on CuGaO2 (011) in monodentate (M), bidentate (B), and tridentate (T) binding modes are shown in Figure 4, while corresponding structural parameters and Eads are listed in Table 2 together with those of CH3PO3H2 on NiO (001).
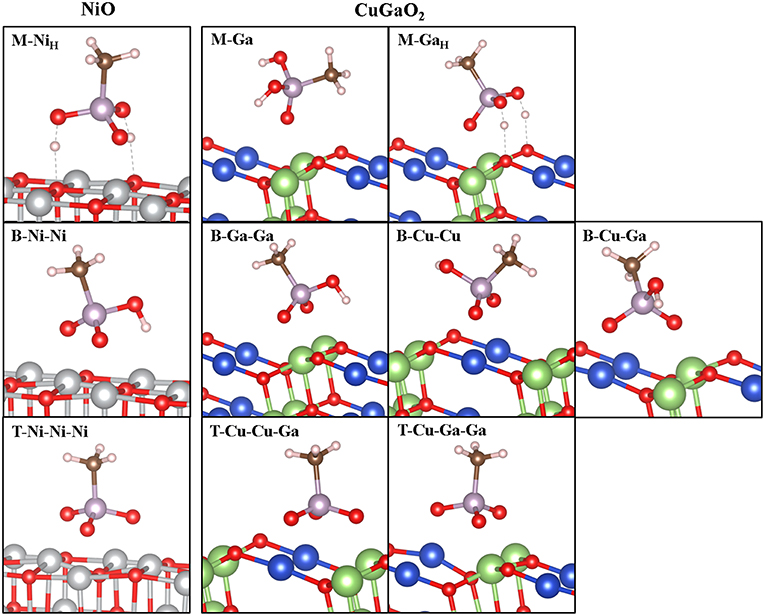
Figure 4. Optimized structures of CH3PO3H2 on CuGaO2 (011) 2 × 1 × 3 L slab (right) and comparison to NiO (001) (left). Labels according to the anchoring mode [monodentate (M), bidentate (B), and tridentate (T)] and to the surface atoms involved in the adsorption process. Sub index “H” indicates H bonding (dashed gray line) between the OH group and surface oxygen atom upon relaxation in M cases. Color legend: Ni (gray), Cu (blue), Ga (green), O (red), C (brown), H (light pink), and P (lavender).
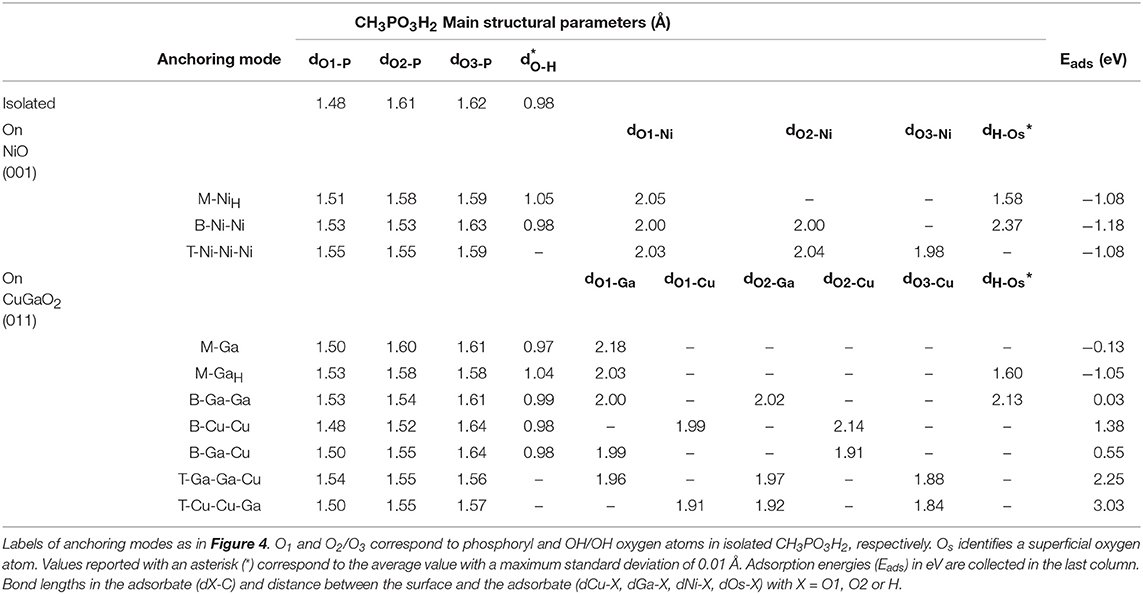
Table 2. Selected structural parameters for relaxed CH3PO3H2 anchored on CuGaO2 (011) in comparison to those on NiO (001) and of the isolated molecule calculated at the DFT(PBE)+U level of theory in vacuum.
In this case, only the monodentate adsorption modes on surface Ga atoms are favored. In particular, we find two stable M geometries, one without any H-bonding to the surface (M-Ga) and one where both the two OH groups form H-bonds with the delafossite surface O atoms (M-GaH). Such H bonds stabilize the latter by 0.92 eV with respect to M-Ga. We explored different starting geometries with phosphoryl oxygen O1 linked to surface Cu but all evolve into either M-Ga or M-GaH upon relaxation. As in NiO, the –PO3H2 group in M binding (with H bonding) is more strongly bound than –COOH. As for –COOH, we find that only M binding of the –PO3H2 group is stable on the delafossite surface, differently from NiO, where M, B, and T modes are all strongly anchored to the surface with Eads lying in a narrow window of energy. In CuGaO2, B modes present positive Eads that go from the few eV of the B-Ga-Ga case to the 1.38 eV in the B-Cu-Cu case. In all the tridentate modes, adsorption energies are very high. So, we can conclude that –PO3H2 anchoring group will bind to CuGaO2 (001) preferentially through a M-GaH-like anchoring mode, with small amounts of M-Ga-like and negligible presence of B-Ga-Ga.
In order to evaluate to what extent the selectivity toward monodentate binding on CuGaO2 for both anchoring groups described above is affected by Mg-doping and the presence of the solvent, we have also calculated Eads of each anchoring group/mode in the pristine delafossite slab considering acetonitrile as solvent and on the slab containing 3.3% of Mg, both in vacuum and in acetonitrile. We chose acetonitrile as solvent since it is one of the most widely employed solvents in DSSCs and it was also employed as a solvent in the experimental electrolyte compositions we refer to for open circuit voltage determination.
These three additional sets of Eads, together with those calculated in vacuum on the pristine slabs are gathered in Table 3.
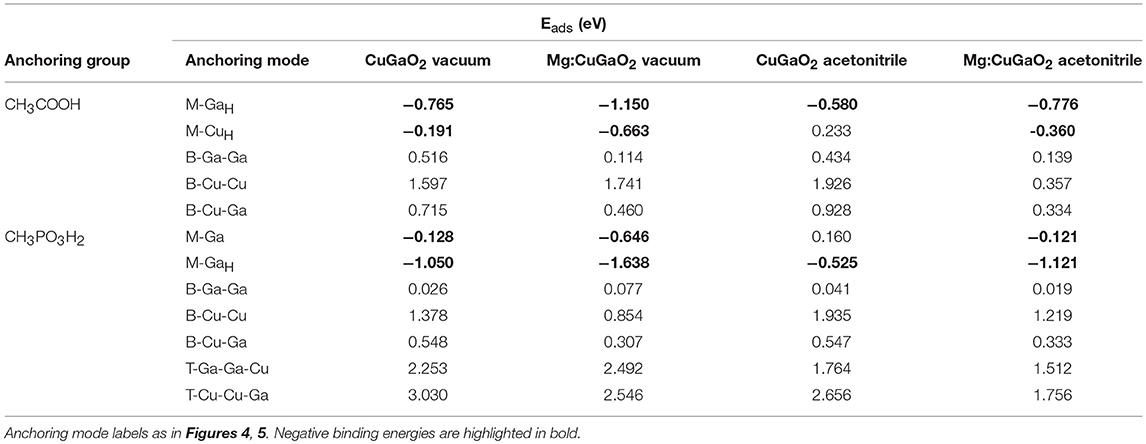
Table 3. Calculated adsorption energies (Eads) for CH3COOH and CH3PO3H2 on CuGaO2 (001) at the DFT(PBE)+U level of theory in vacuum and acetonitrile (implicit solvent), without and with Mg doping (3.3%).
From our calculations, both anchoring groups bind significantly more strongly to the Mg-doped surface than on the undoped surface with Eads that decrease almost uniformly around ~0.5 eV. There are very few exceptions where Eads suffers a negligible increase (max. ~0.05 eV) upon Mg-doping as CH3COOH-B-Cu-Cu and CH3PO3H2-B-Ga-Ga/T-Ga-Ga-Cu. This stabilizing effect can be explained to the higher Lewis acidity of oxide surfaces upon p-type doping, where a hole has been introduced, increasing the ability of such oxide to accept electron-donating species (Metiu et al., 2012). We must note here that, as in bulk CuGaO2 (Schiavo et al., 2018), the presence of Mg does not lead to a significant localized oxidation of any particular cation but to the hole being distributed among all Cu atoms of the system (each Cu losing max. 0.08 e−), with even smaller involvement of Ga. This explains the uniform decrease in Eads in all surface sites and not only on Cu sites. Regarding the solvent, as common trend to all the M and B anchoring modes, the dielectric continuum increases Eads but not to the same extent. Only in the cases of T-modes in CH3PO3H2, it decreases by ~0.5 eV but this decrease is not enough to stabilize the very unfavorable T anchoring modes. From a general perspective, the acetonitrile solvent medium with a mild dielectric constant is expected to weaken the ionic contribution to the bonding between anchoring group O atoms and Ga surface sites. At the same time, for the T modes, the solvent is able to stabilize the dipole moments of the –OH surface species that are formed during the anchoring. In any case, the overall adsorption energy landscape is not qualitatively changed when both the electrolyte solvent and the Mg-dopant are taken into account. Eads values are analogous to those calculated for the pristine slab in vacuum, as the two effects balance each other. Thus, we can state that M binding is likely the preferred anchoring mode for CH3COOH and CH3PO3H2 in operating conditions. This result is of utmost importance for the cell performance since it has been shown that the driving force for hole injection between the dye HOMO and the VB of the semiconductor is maximized in M binding modes where no H has been released to the surface and the interfacial dipole at the electrode surface is minimum (Muñoz-García and Pavone, 2015). Moreover, we can state that Mg doping, besides having an effect on improving p-type conductivity and nanoparticle morphology (Renaud et al., 2014), improves dye coverage by increasing the affinity between the delafossite and dye anchoring groups.
Figures 5, 6 show the projected density of states of CuGaO2(011) with anchored CH3COOH and CH3PO3H2, respectively, in the most stable binding mode (M-GaH). We have considered all the four systems for which the adsorption energies were computed: pristine/vacuum, Mg-doped/vacuum, pristine/acetonitrile, and Mg-doped/acetonitrile. In all these cases, the molecular states of the anchoring groups are far from the semiconductor VB edge and, hence, they would not contribute to electron/hole transport process. These results are consistent with those recently computed for Ph-COOH and silanes on NiO (Wykes et al., 2016), in line with what is expected for the proper functioning of the solar cell.
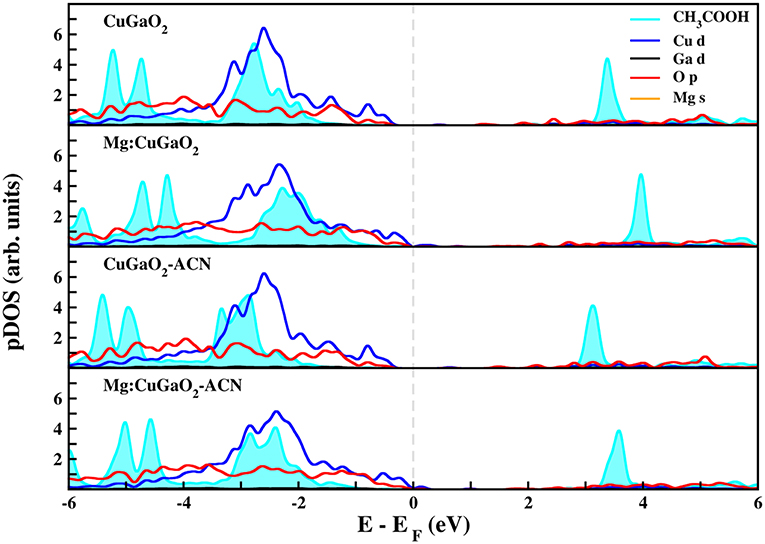
Figure 5. Projected density of states (pDOS) calculated at the DFT(PBE)+U level of theory of CH3COOH adsorbed on CuGaO2 (011). Fermi level (dashed gray line) has been set to zero.
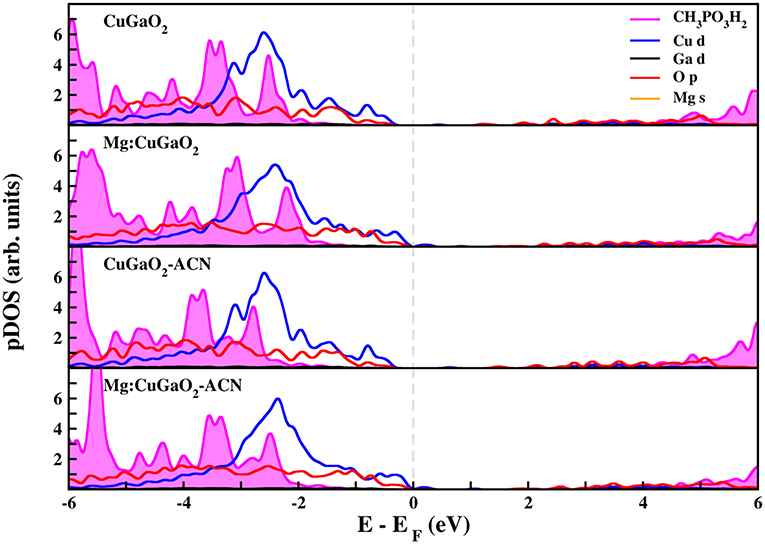
Figure 6. Projected density of states (pDOS) calculated at the DFT(PBE)+U level of theory of CH3PO3H2 adsorbed on CuGaO2 (011). Fermi level (dashed gray line) has been set to zero.
Conclusions
This work reports an ab initio study of CuGaO2 delafossite as alternative to NiO in p-type DSSCs. The semiconductive copper delafossite with chemical formula CuMO2 presents a lower absolute position of the valence band edge than NiO. Thus, besides providing a higher open circuit potential (VOC) when used with traditional I−/ than NiO, it enables the use of new and high performing Co- and Cu-based electrolytes. Gallium delafossite CuGaO2 is of particular interest since it can be easily doped with Mg, which enhances the p-type conductivity and the shape and morphology of delafossite nanoparticles. First, we have focused here on studying the change in the valence band absolute position after Mg doping: with ~3 and ~6% dopant contents the VOC of the copper gallium delafossite is mostly unchanged, in agreement with experiments.
Due to the practical difficulties of sensitizing delafossite oxides, we have addressed the adsorption properties of two anchoring groups widely used for grafting dye molecules on semiconductor surfaces: –COOH and –PO3H2 (carboxylic acid and phosphonic acid, respectively) on the most stable CuGaO2 surface, i.e., the (011). We characterized the interaction between these anchoring groups and the surface in terms of anchoring mode minimum-energy structures and corresponding adsorption energies. We have dissected the effects of Mg doping and of the presence of the solvent on these features, as well as on the electronic structure, which is of key importance to DSSC operation. Contrary to what happens in the case of NiO, on delafossite surface there is a strong selectivity toward monodentate binding modes for both the carboxylic and phosphonic anchoring groups, with a particular affinity toward the Ga surface sites. Since it has been shown that driving force for hole injection from the dye to the semiconductor is jeopardized when protic groups release H to oxide surfaces in bidentate and tridentate modes, our results point out that the combination of –COOH or –PO3H2 anchoring groups with CuGaO2 might deliver much better performance than with NiO. Besides, our calculations show that Mg doping increases the affinity of CuGaO2 surface for both anchoring groups and, thus, the better performances of Mg-containing samples can be also ascribed to a higher sensitization driven by more favorable adsorption energies.
In conclusion, this work provides a first theoretical characterization of the interface between delafossite oxide and the –COOH or –PO3H2 anchoring groups, thus paving the route to further studies on full dye-sensitized delafossite-based photocathodes in order to help the development of p-type DSSC technologies with first-principles derived rational design guidelines.
Data Availability
The datasets generated for this study are available on request to the corresponding author.
Author Contributions
AM-G and MP designed the research. LC, ES, and CB performed the calculations. AM-G, PM, and MP rationalized the results. All authors contributed in writing and revising the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge funding from the Italian Ministry of University and Research (MIUR) under grant PRIN 2015XBZ5YA.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00158/full#supplementary-material
References
Adamo, C., and Jaquemin, D. (2013). The calculations of excited-state properties with Time-Dependent Density Functional Theory. Chem. Soc. Rev. 42, 845–856. doi: 10.1039/C2CS35394F
Adineh, M., Tahay, P., Ameri, M., Safari, N., and Mohajeranic, E. (2016). Fabrication and analysis of dye-sensitized solar cells (DSSCs) using porphyrin dyes with catechol anchoring groups. RSC Adv. 6, 14512–14521. doi: 10.1039/C5RA23584G
Alkhayatt, A. H. O., Thahab, S. M., and Zgair, I. A. (2016). Structure, surface morphology and optical properties of post-annealed delafossite CuFeO2 thin films. Optik 127, 3745–3749. doi: 10.1016/j.ijleo.2015.12.144
Bai, Y. I., Mora-Sero De Angelis, F., Bisquert, J., and Wang, P. (2014). Titanium dioxide nanomaterials for photovoltaic applications. Chem. Rev. 114, 10095–10130. doi: 10.1021/cr400606n
Benesperi, I., Michaels, H., and Freitag, M. (2018). The researcher's guide to solid-state dye-sensitized solar cells. J. Mater. Chem. C. 6, 11903–11942. doi: 10.1039/C8TC03542C
Blöchl, P. E. (1994). Projector augmented-wave method. Phys. Rev. B 50:17953. doi: 10.1103/PhysRevB.50.17953
Boschloo, G., and Hagfeldt, A. (2009). Characteristics of the iodide/triiodide redox mediator in dye-sensitized solar cells. Acc. Chem. Res. 42, 1819–1826 doi: 10.1021/ar900138m
Carella, A., Centore, R., Borbone, F., Toscanesi, M., Trifuoggi, M., Bella, F., et al. (2018). Tuning optical and electronic properties in novel carbazole photosensitizers for p-type dye-sensitized solar cells. Electrochim. Acta 292, 805–816. doi: 10.1016/j.electacta.2018.09.204
Cong, J., Kinschel, D., Daniel, Q., Safdari, M., Gabrielsson, E., Chen, H., et al. (2016). Bis(1,1-bis(2-pyridyl)ethane)copper(I/II) as an efficient redox couple for liquid dye-sensitized solar cells. J. Mater. Chem. A 4, 14550–14554. doi: 10.1039/C6TA06782D
Crottaz, O., Kubel, F., and Schmid, H. (1996). Preparation of trigonal and hexagonal cuprous chromite and phase transition study based on single crystal structure data. J. Solid State Chem. 122, 247–250. doi: 10.1006/jssc.1996.0109
D'Arienzo, M., Gamba, L., Morazzoni, F., Cosentino, U., Greco, C., Lasagni, M., et al. (2017). Experimental and theoretical investigation on the catalytic generation of environmentally persistent free radicals from benzene. J. Phys. Chem. C 121, 9381–9393. doi: 10.1021/acs.jpcc.7b01449
Das, B., Renaud, A., Volosin, A. M., Yu, L., Newman, N., and Seo, D.-K. (2015). Nanoporous delafossite CuAlO2 from inorganic/polymer double gels: a desirable high-surface-area p-type transparent electrode material. Inorg. Chem. 54, 1100–1108. doi: 10.1021/ic5023906
De Angelis, F., Fantacci, S., and Gebauer, R. (2011). Simulating dye-sensitized TiO2 heterointerfaces in explicit solvent: absorption spectra, energy levels, and dye desorption. J. Phys. Chem. Lett. 2, 813–817. doi: 10.1021/jz200191u
Dudarev, S. L., Botton, G. A., Savrasov, S. Y., Humphreys, C. J., and Sutton, A. P. (1998). Electron-energy-loss spectra and the structural stability of nickel oxide: an LSDA+U study. Phys. Rev. B 57:1505. doi: 10.1103/PhysRevB.57.1505
Feldt, S. M., Gibson, E. A., Gabrielsson, E., Sun, L., Boschloo, G., and Hagfeldt, A. (2010). Design of organic dyes and cobalt polypyridine redox mediators for high-efficiency dye-sensitized solar cells. J. Am. Chem. Soc. 132, 16714–16724. doi: 10.1021/ja1088869
Galliano, S., Bella, F., Gerbaldi, C., Falco, M., Viscardi, G., Grätzel, M., et al. (2017). Photoanode/electrolyte interface stability in aqueous dye-sensitized solar cells. Energy Technol. 5, 300–311. doi: 10.1002/ente.201600285
Gillen, R., and Robertson, J. (2011). Band structure calculations of CuAlO2, CuGaO2, CuInO2, and CuCrO2 by screened exchange. Phys. Rev. B 84:035125. doi: 10.1103/PhysRevB.84.035125
Green, M. A. (2003). Third Generation PhotoVoltaics: Advanced Solar Energy Conversion. Berlin; Heidelberg; Springer-Verlag.
Hagfeldt, A., Boschloo, G., Sun, L., Kloo, L., and Petterson, H. (2010). Dye-sensitized solar cells. Chem. Rev. 110:6595 doi: 10.1021/cr900356p
He, J., Lindström, H., Hagfeldt, A., and Lindquist, S. E. (2000). Iodide management in formamidinium-lead-halide-based perovskite layers for efficient solar cells. Sol. Energy Mater. Sol. Cells 62:265 doi: 10.1016/S0927-0248(99)00168-3
Iozzi, M. F., Vajeeston, P., Vidya, R., Ravindran, P., and Fjellvag, H. (2015). Structural and electronic properties of transparent conducting delafossite: a comparison between the AgBO2 and CuBO2 families (B = Al, Ga, In and Sc, Y). RSC Adv. 5, 1366–1377. doi: 10.1039/C3RA47531J
Ishiguro, T., Kitazawa, A., Mizutani, N., and Kato, M. (1981). Single-crystal growth and crystal structure refinement of CuAlO2. J. Solid State Chem. 40, 170–174. doi: 10.1016/0022-4596(81)90377-7
Isseroff, L. Y., and Carter, E. A. (2012). Importance of reference Hamiltonians containing exact exchange for accurate one-shot GW calculations of Cu2O. Phys. Rev. B: Condens. Matter Mater. Phys. 85:235142 doi: 10.1103/PhysRevB.85.235142
Jiang, H., Wang, X., Zang, X., Wu, W., Sun, S., Xiong, C., et al. (2013). Electronic properties of bivalent cations (Be, Mg and Ca) substitution for Al in delafossite CuAlO2 semiconductor by first-principles calculations. J. Alloys Compd. 553 245–252. doi: 10.1016/j.jallcom.2012.11.101
Klein, Y. M., Marinakis, N., Constable, E. C., and Housecroft, C. E. (2018). A phosphonic acid anchoring analogue of the sensitizer P1 for p-type dye-sensitized solar cells. Crystals 8:389. doi: 10.3390/cryst8100389
Köhler, B. U., and Jansen, M. (1986). Darstellung und Strukturdaten von Delafossiten CuMO2 (M = Al, Ga, Sc, Y). Z. Anorg. Allg. Chem. 543, 73–80.
Kontkanen, O. V., Niskanen, M., Hukka, T. I., and Rantala, T. T. (2016). Electronic structure of p-type perylene monoimide-based donor–acceptor dyes on the nickel oxide (100) surface: a DFT approach. Phys. Chem. Chem. Phys. 18, 14382–14389. doi: 10.1039/C6CP02510B
Kresse, G., and Furthmüller, J. (1996a). Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mat. Sci. 6, 15–50. doi: 10.1016/0927-0256(96)00008-0
Kresse, G., and Furthmüller, J. (1996b). Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set Phys. Rev. B. 54, 11169–11186. doi: 10.1103/PhysRevB.54.11169
Kresse, G., and Hafner, J. (1993). Ab initio molecular dynamics for liquid. Phys. Rev. B 47, 558–561. doi: 10.1103/PhysRevB.47.558
Kresse, G., and Hafner, J. (1994). Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 49, 14251–14269. doi: 10.1103/PhysRevB.49.14251
Kresse, G., and Joubert, J. (1999). From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59:1758. doi: 10.1103/PhysRevB.59.1758
Kumar, M., Zhao, H., and Persson, C. (2013). Study of band-structure, optical properties and native defects in AIBIIIO2 (AI = Cu or Ag, BIII = Al, Ga or In) delafossites. Semicond. Sci. Technol. 28:065003. doi: 10.1088/0268-1242/28/6/065003
Labat, F., Le Bahers, T., Ciofini, I., and Adamo, C. (2012). The calculations of excited-state properties with Time-Dependent Density Functional Theory. Acc. Chem. Res. 45, 1268–1277. doi: 10.1021/ar200327w
Luo, Y. R. (2007). Comprehensive Handbook of Chemical Bond Energies. Boca Raton, FL: CRC Press. doi: 10.1201/9781420007282
Marquardt, M. A., Ashmore, N. A., and Cann, D. P. (2006). Crystal chemistry and electrical properties of the delafossite structure. Thin Solid Films 496, 146–156. doi: 10.1016/j.tsf.2005.08.316
Martsinovich, N., and Troisi, A. (2011). Theoretical studies of dye-sensitised solar cells: from electronic structure to elementary processes. Energy Environ. Sci. 4, 4473–4495. doi: 10.1039/C1EE01906F
Mathew, K., Sundararaman, R., Letchworth-Weaver, K., Arias, T. A., and Hennig, R. G. (2014). Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. J. Chem. Phys. 140:084106. doi: 10.1063/1.4865107
Metiu, H., Chrétien, S., Hu, Z., Bo, L.i, and Ying Sun, X. (2012). Anchoring groups for dye-sensitized solar cells. J. Phys. Chem. C 116, 10439–10450. doi: 10.1021/jp301341t
Monkhorst, H. J., and Pack, J. D. (1976). Special points for Brillouin-zone integrations. Phys. Rev. B 13:5188. doi: 10.1103/PhysRevB.13.5188
Morandeira, A., Boschloo, G., Hagfeld, A., and Hammarström, L. (2005). Photoinduced ultrafast dynamics of coumarin 343 sensitized p-type-nanostructured NiO films. J. Phys. Chem. B 109:19403. doi: 10.1021/jp053230e
Mori, S., Fukuda, S., Sumikura, S., Takeda, Y., Tamaki, Y., Suzuki, E., et al. (2008). Charge-transfer processes in dye-sensitized NiO solar cells. J. Phys. Chem. C 112, 16134–16139. doi: 10.1021/jp803919b
Muñoz-García, A. B., and Pavone, M. (2015). Structure and energy level alignment at the dye–electrode interface in p-type DSSCs: new hints on the role of anchoring modes from ab initio calculations. Phys. Chem. Chem. Phys. 17, 12238–12246. doi: 10.1039/C5CP01020A
Nakasa, A., Usami, H., Sumikura, S., Hasegawa, S., Koyama, T., and Suzuki, E. (2005). A high voltage dye-sensitized solar cell using a nanoporous NiO photocathode. Chem. Lett. 34:500. doi: 10.1246/cl.2005.500
Nattestad, A., Mozer, A. J., Fischer, M. K. R., Cheng, Y. B., Mishra, A., Bäuerle, P., et al. (2010). A high voltage dye-sensitized solar cell using a nanoporous NiO photocathode. Nat. Mater. 9, 31–35. doi: 10.1038/nmat2588
Nattestad, A., Zhang, X., Bach, U. Y., and Cheng, B. (2011). Dye-sensitized CuAlO2 photocathodes for tandem solar cell applications. J. Photon. Energy 1, 11103–11112. doi: 10.1117/1.3528236
Neugebauer, J., and Scheffler, M. (1992). Adsorbate-substrate and adsorbate-adsorbate interactions of Na and K adlayers on Al(111). Phys. Rev. B Condens. Matter Mater. Phys. 46, 16067–16080.
Odobel, F., Le Pleux, L., Pellegrin, Y., and Blart, E. (2010). New photovoltaic devices based on the sensitization of p-type semiconductors: challenges and opportunities. Acc. Chem. Res. 43, 1063–1071. doi: 10.1021/ar900275b
Odobel, F., and Pellegrin, Y. (2013). Recent advances in the sensitization of wide-band-gap nanostructured p-type semiconductors. Photovoltaic and photocatalytic applications. J. Phys. Chem. Lett. 4, 2551–2564. doi: 10.1021/jz400861v
Odobel, F., Pellegrin, Y., Gibson, E. A., Hagfeld, A., Smeigh, A. L., and Hammarström, L. (2012). Recent advances and future directions to optimize the performances of p-type dye-sensitized solar cells. Coord. Chem. Rev. 256, 2414–2423. doi: 10.1016/j.ccr.2012.04.017
Parida, B., Iniyan, S., and Goic, R. (2011). A review of solar photovoltaic technologies. Renew. Sust. Energ. Rev. 15, 1625–1636. doi: 10.1016/j.rser.2010.11.032
Pellegrin, Y., Le Pleux, L., Blart, E., Renaud, A., Chavillon, B., Chavillon Szuwarski, N., Odobel, F., et al. (2011). Ruthenium polypyridine complexes as sensitizers in NiO based p-type dye-sensitized solar cells: effects of the anchoring groups J. Photochem. Photobiol. A Chem. 219, 235–242. doi: 10.1016/j.jphotochem.2011.02.025
Perdew, J. P., Burke, K., and Ernzerhof, M. (1996). Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868. doi: 10.1103/PhysRevLett.77.3865
Perdew, J. P., Burke, K., and Ernzerhof, M. (1997). ERRATA generalized gradient approximation made simple [Phys. Rev. Lett. 77, 3865 (1996)]. Phys. Rev. Lett. 78, 1396–1396. doi: 10.1103/PhysRevLett.78.1396
Piccinin, S., Rocca, D., and Pastore, M. (2017). Role of solvent in the energy level alignment of dye-sensitized NiO interfaces. J. Phys Chem. C 121, 22286–22294. doi: 10.1021/acs.jpcc.7b08463
Powar, S., Xiong, D., Daeneke, T., Ma, M. T., Gupta, A., Lee, G., et al. (2014). Improved photovoltages for p-type dye-sensitized solar cells using CuCrO2 nanoparticles. J. Phys. Chem. C 118 16375–16379. doi: 10.1021/jp409363u
Preat, J., Hagfeldt, A., and Perpète, E. A. (2011). Investigation of the photoinduced electron injection processes for p-type triphenylamine-sensitized solar cells. Energy Environ. Sci. 4:4537. doi: 10.1039/c1ee01638e
Prévot, M. S., and Sivula, K. (2013). Photoelectrochemical tandem cells for solar water splitting. J. Phys. Chem. C 117, 17879–17893. doi: 10.1021/jp405291g
Qin, P., Zhu, H., Edvinsson, T., Boschloo, G., Hagfeldt, A., and Sun, L. (2008). Design of an organic chromophore for P-type dye-sensitized solar cells. J. Am. Chem. Soc. 130, 8570–8571. doi: 10.1021/ja8001474
Renaud, A., Cario, L., Deniard, P., Gautron, E., Rocquefelte, X., Pelegrin, Y., et al. (2014). Impact of Mg doping on performances of CuGaO2 based p-type dye-sensitized solar cells. J. Phys. Chem. C 118, 54–59. doi: 10.1021/jp407233k
Renaud, A., Chavillon, B., Le Pleuc, L., Pellegrin, Y., Blart, E. M., et al. (2012). CuGaO 2: a promising alternative for NiO in p-type dye solar cells. J. Mater. Chem. 22, 14353–14356. doi: 10.1039/c2jm31908j
Rohrbach, A., Hafner, J., and Kresse, G. (2003). Electronic correlation effects in transition-metal sulfides. J. Phys. Condens. Matter 15, 979–996. doi: 10.1088/0953-8984/15/6/325
Scanlon, D. O., and Watson, G. W. (2011). Understanding the p-type defect chemistry of CuCrO2. J. Mater. Chem. 21, 3655–3663. doi: 10.1039/c0jm03852k
Schiavo, E., Latouche, C., Barone, V., Crescenzi, O., Muñoz-García, A. B., and Pavone, M. (2018). An ab initio study of Cu-based delafossites as an alternative to nickel oxide in photocathodes: effects of Mg-doping and surface electronic features. Phys. Chem. Chem. Phys. 20, 14082–14089. doi: 10.1039/C8CP00848E
Sullivan, I., Zoellner, B., and Maggard, P. A. (2016). Copper(I)-based p-type oxides for photoelectrochemical and photovoltaic solar energy conversion. Chem. Mater. 28, 5999–6016. doi: 10.1021/acs.chemmater.6b00926
Toroker, M. C., Kanan, D. K., Alidoust, N., Isseroff, L. Y., Liao, P., and Carter, E. (2011). First principles scheme to evaluate band edge positions in potential transition metal oxide photocatalysts and photoelectrodes. Phys. Chem. Chem. Phys. 13, 16644–16654. doi: 10.1039/c1cp22128k
Wykes, M., Odobel, F., Adamo, C., Ciofini, I., and Labat, F. (2016). Anchoring groups for dyes in p-DSSC application: insights from DFT. J. Mol. Model. 22:289. doi: 10.1007/s00894-016-3155-1
Xiong, D., Zhang, W., Zeng, X., Xu, Z., Chen, W., Cui, J., et al. (2013). Enhanced performance of p-type dye-sensitized solar cells based on ultrasmall Mg-doped CuCrO2 nanocrystals. ChemSusChem 6, 1432–1437. doi: 10.1002/cssc.201300265
Yang, W. S., Park, B. W., Jung, E. H., Jeon, N. J., Kim, Y. C., Lee, D. U., et al. (2017). Iodide management in formamidinium-lead-halide-based perovskite layers for efficient solar cells. Science 356, 1376–1379. doi: 10.1126/science.aan2301
Yu, M., Draskovic, T. I., and Wu, Y. (2014). Cu(I)-based delafossite compounds as photocathodes in p-type dye-sensitized solar cells. Phys. Chem. Chem. Phys. 16, 5026–5033. doi: 10.1039/c3cp55457k
Yu, M., Natu, G., Ji, Z., and Wu, Y. (2012). p-type dye-sensitized solar cells based on delafossite CuGaO2 nanoplates with saturation photovoltages exceeding 460 mV. J. Phys. Chem. Lett. 3, 1074–1078. doi: 10.1021/jz3003603
Keywords: density functional theory, p-type DSSCs, anchoring groups, Cu delafossites, delafossite surface
Citation: Muñoz-García AB, Caputo L, Schiavo E, Baiano C, Maddalena P and Pavone M (2019) Ab initio Study of Anchoring Groups for CuGaO2 Delafossite-Based p-Type Dye Sensitized Solar Cells. Front. Chem. 7:158. doi: 10.3389/fchem.2019.00158
Received: 29 November 2018; Accepted: 04 March 2019;
Published: 29 March 2019.
Edited by:
Marina Freitag, Uppsala University, SwedenCopyright © 2019 Muñoz-García, Caputo, Schiavo, Baiano, Maddalena and Pavone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ana B. Muñoz-García, anabelen.munozgarcia@unina.it
Michele Pavone, michele.pavone@unina.it