Field-Induced Single Molecule Magnets of Phosphine- and Arsine-Oxides
 Matilde Fondo1*
Matilde Fondo1*  Julio Corredoira-Vázquez1
Julio Corredoira-Vázquez1  Ana M. García-Deibe1
Ana M. García-Deibe1  Jesús Sanmartín-Matalobos1
Jesús Sanmartín-Matalobos1  Juan Manuel Herrera2
Juan Manuel Herrera2  Enrique Colacio2
Enrique Colacio2- 1Departamento de Química Inorgánica, Facultade de Química, Universidade de Santiago de Compostela, Santiago de Compostela, Spain
- 2Departamento de Química Inorgánica, Facultad de Ciencias, Universidad de Granada, Granada, Spain
The coordination chemistry of dysprosium and terbium toward phosphine and arsine oxides was further explored. Thus, the new nitrate [M(NO3)3(Ph3PO)3] (M = Tb, 1; Dy, 2), [Dy(NO3)3(EtOH)(Ph3XO)2] (X = P, 3; As, 4), chloride [DyCl2(Ph3AsO)4]Cl (5), triflate [Dy(OTf)2(MePh2PO)4]OTf (6; OTf = triflate) and hexafluoroacetylacetonate [M(hfa)3(Ph3PO)2] (hfa = hexafluoroacetylacetonate; M = Tb, 7; Dy, 8) complexes were isolated and fully characterized. The crystal structures of 1·CH3CN, 2·CH3CN, 4, 5·2.75EtOH·1.25H2O, 6, 7, and 8 show MO9 cores in 1, 2, and 4, with highly distorted geometry, between spherical capped square antiprism and muffin-like, hexacoordinated environments for the dysprosium ions in 5 and 6, with octahedral geometry, and octa-coordination for the lanthanoid metals in 7 and 8, with geometry closer to square antiprism. Comparison of the magnetic behavior of all the complexes allows analyzing which metal ion (Tb or Dy), phosphine or arsine oxide, or anionic ligand favor more the slow relaxation of the magnetization. Alternating current magnetic measurements show that only 2, 4, and 8 present slow relaxation of the magnetization in the presence of an external magnetic field, 8 being the complex with the highest Ueff (44.85 K) of those described herein.
Introduction
The observation for the first time of slow magnetic relaxation in mononuclear lanthanoid complexes (TBA)[Pc2Ln] (TBA = N+; Pc = phthalocyanide; LnIII = Tb or Dy) (Ishikawa et al., 2003) provided a real breakthrough in molecular magnetism, opening the field of single ion magnets (SIMs) in 2003. This field has received growing attention since its origins, given that the non-trivial memory effect and quantum character in SIMs renders them as potential ultra-high density data storage medium and spintronic devices (Woodruff et al., 2013; Shiddiq et al., 2016; Lu et al., 2017). Accordingly, the huge amount of work devoted to this research during the last 15 years has led to many remarkable advances. Thus, Rinehart and Long published in 2011 (Rinehart and Long, 2011) a benchmark study where they provided a clear explanation of how the electronic structure of f-elements can in theory be manipulated to create new single molecule magnets (SMMs). In this study, they give the relationship between the coordination environment of lanthanoid ions and the magnetic anisotropy of the complex and, therefore, they suggest that one can match an appropriate ligand field to maximize magnetic anisotropy on the basis of the shapes (oblate or prolate) of the 4f-shell electron density distributions. For the oblate Tb(III) and Dy(III) ions, theoretical calculations established that the optimum environment to maximize the anisotropy is the axial one (Ungur and Chibotaru, 2011). Thus, the optimum geometry will be lineal but the coordination number two seems too low to be stabilized by the lanthanoid ions. Accordingly, in the absence of this possibility, the coordination number 7, with pentagonal bipyramidal geometry, was the most explored one.
On the basis of these starting theoretical studies, much experimental work was done and continuous attainments relating to anisotropic energy barriers (Ueff) and blocking temperatures (TB) were achieved in the field of SIMs. Hence, the highest Ueff described up to now is 1815 K (Ding et al., 2016), a value significantly higher than 331 K reported for the first SIM (N)[Tb(Pc)2] (Ishikawa et al., 2003). Besides, the blocking temperatures have been continuously increasing, from the initial 1.7 K (Ishikawa et al., 2005) thorough the 20 K achieved in 2016 for an-air stable [Dy(Cy3PO)2(H2O)5]Br3 complex (Chen et al., 2016) to the astonishing 60 K recently reported for the metallocene complex [(Cpttt)2Dy][B(C6F5)4] (Goodwin et al., 2017; Guo et al., 2017).
In spite of these advances, it should be noted that most of the SIMs with high Ueff and/or TB are air-unstable, and this is a handicap that must be surpassed. It must be noted that a series of simple, easy to obtain, and air stable phosphine oxide complexes [Dy(R3PO)2(H2O)5]X3 (R = Cy3 or CyPh2, X = Cl, Br or I) show blocking temperatures between 19 and 20 K (Chen et al., 2016, 2017), the largest ones among SMMs if the 60 K reported for the unstable [(Cpttt)2Dy][B(C6F5)4] is excluded. Accordingly, it seems that the coordination chemistry of lanthanoids with phosphine oxides is really interesting from the magnetic point of view. This coordination chemistry has been extensively studied, mainly by Platt (2017), but, in spite of this, the magnetic behavior of these compounds is still poorly investigated. Therefore, taking into account all the above considerations, we have decided to revise and extent the study of the coordination chemistry of Dy and Tb with phosphine and arsine oxides, and to investigate the magnetic behavior of the obtained compounds. The comparison of the magnetic properties of the isolated complexes between them allows establishing some patterns.
Materials and Methods
General
All chemical reagents and solvents were purchased from commercial sources and used as received without further purification. Elemental analyses of C, H and N were recorded on a Carlo Erba EA 1108 analyzer. Infrared spectra were performed in the range 4000–500 cm−1 on a Varian 670 FT/IR spectrophotometer in the ATR mode.
Syntheses of the Complexes
[Tb(NO3)3(Ph3PO)3] (1): To a solution of Tb(NO3)3·5H2O (0.130 g, 0.30 mmol) in acetonitrile (10 mL), triphenylphosphine oxide, (0.167 g, 0.60 mmol) and acetonitrile (10 mL) were added. The mixture was stirred for 4 h at room temperature and the resultant colorless solution was left to slowly evaporate, until single crystals of 1·CH3CN precipitated. The single crystals were filtered, and they lose the acetonitrile solvate on drying to yield 1. Yield (based on Ph3PO): 0.22 g (93%). M.W.: 1179.80. Anal. calcd. for C54H45TbN3O12P3: C 54.97, H 3.84, N 3.56%. Found: C 54.89, H 3.59, N 3.95%. IR (ATR, /cm−1): 1120, 1153 (P = O), 1305 ().
The same product is isolated when Tb(NO3)3·5H2O and Ph3PO are mixed in 1:3 molar ratio in acetonitrile.
[Dy(NO3)3(Ph3PO)3] (2) was obtained in a similar way to 1: amounts of Dy(NO3)3·6H2O (0.23 g, 0.66 mmol) and Ph3PO (0.360 g, 1.32 mmol). Single crystals of 2·CH3CN were isolated in the same way as those of 1·CH3CN, which lose the acetonitrile solvate on drying to yield 2. Yield (based on Ph3PO): 0.403 g (77%). M.W.: 1183.37. Anal. calcd. for C54H45DyN3O12P3: C 54.81, H 3.83, N 3.55%. Found: C 54.84, H 3.40, N 3.53%. IR (ATR, /cm−1): 1120, 1153 (P = O), 1305 ().
The same product is isolated when Dy(NO3)3·6H2O and Ph3PO are mixed in 1:3 molar ratio in acetonitrile.
[Dy(NO3)3(EtOH)(Ph3PO)2] (3): A hot solution of Dy(NO3)3·6H2O (0.137 g, 0.30 mmol) in ethanol (5 mL) was added to a hot solution of triphenylphosphine oxide (0.170 g, 0.60 mmol) in ethanol (5 mL). The mixture was stirred at room temperature for 24 h, when a colorless powder precipitated. The solid was filtered and dried in air. Yield: 0.250 g (88%). M.W.: 951.15. Anal. calcd. for C38H36DyN3O12P2: C 47.98, H 3.81, N 4.42%. Found: C 47.90, H 3.81, N 4.29%. IR (ATR, /cm−1): 1119, 1150 (P = O), 1308 (), 3346 (OH).
[Dy(NO3)3(EtOH)(Ph3AsO)2] (4) was obtained in the same way as 3: amounts of Dy(NO3)3·6H2O (0.23 g, 0.66 mmol) and Ph3AsO (0.425 g, 1.32 mmol). The mixture yields a solution that by slow evaporation gives rise to single crystals of 4. Yield: 0.313 g (46%). M.W.: 1039.05. Anal. calcd. for C38H36DyN3O12As2: C 43.92, H 3.49, N 4.04%. Found: C 44.44, H 3.35, N 3.92%. IR (ATR, /cm−1): 902, 924 (As = O), 1313 (), 3334 (OH).
[DyCl2(Ph3AsO)4]Cl·1.25H2O (5·1.25H2O): To an ethanol (10 mL) solution of DyCl3·6H2O (0.460 g, 1.22 mmol), Ph3AsO (0.77 g, 2.395 mmol) was added. The mixture was stirred under reflux for 1 h and the colorless solution was concentrated in a rotaevaporator to ca. 2 mL, yielding an oil. After standing for 24 h, single crystals of 5·2.75EtOH·1.25H2O, suitable for X-ray diffraction studies, were isolated. The crystals were filtered and dried in air, losing the ethanol solvate to give rise to 5·1.25H2O. Yield (based on Ph3AsO): 0.74 g (78%). M.W.: 1579.93. Anal. calcd. for C72H62.5AsCl3DyO5.25: C 54.68, H 3.95%. Found: C 54.27, H 3.33%. IR (ATR, /cm−1): 886 (As = O), 3264 (OH).
The same product is isolated when DyCl3·6H2O and Ph3AsO are mixed in 1:4 molar ratio in ethanol.
[Dy(OTf)2(MePh2PO)4](OTf)·THF (6·THF): Dy(OTf)3 (0.423 g, 0.694 mmol) was dissolved in THF (7 mL), and methyldiphenylphosphine oxide (0.600 g, 1.387 mmol) was added. The mixture was stirred at room temperature for 24 h, and the resultant colorless solution was concentrated in a rotaevaporator up to ca. 2 mL, yielding an oil. The oil was left to stand for 10 days at room temperature until single crystals of 6·THFwere obtained. Yield: 0.250 (47%). M.W.: 1546.63. Anal. calcd. for C59H60DyF9O14P4S3: C 45.78, H 3.88, S 6.21%. Found: C 45.79, H 3.65, S 5.91%. IR (ATR, /cm−1): 633 (δSO3), 1020, 1262 (SO3), 1156, 1220 (CF3), 1132 (P = O).
The same product is isolated when Dy(OTf)3 and MePh2PO are mixed in 1:4 molar ratio in THF.
[Tb(hfa)3(Ph3PO)2] (7): Terbium chloride hexahydrate (0.14 g, 0.36 mmol) was dissolved in distilled water (5 mL). An ethanol solution (20 mL) of hexafluoroacetylacetone (0.22 g, 1.08 mmol) was added to the aqueous solution. An ethanolic tetramethylammonium hydroxide solution (0.1 M) was added dropwise until pH 7 was reached. After stirring the mixture at room temperature for 6 h, the solvent was eliminated in a rotaevaporator. The white solid that precipitated was dissolved in methanol (30 mL), and triphenylphosphine oxide (0.10 g, 0.36 mmol) was added. The mixture was heated under reflux while stirring for 6 h. Slow evaporation of the resultant solution yields single crystals of 7, suitable for X-ray diffraction studies. The crystals were filtered and dried in air. Yield (based on Ph3PO): 0.070 (29%). M.W.: 1336.65. Anal. calcd. for C51H33TbF18O8P2: C 45.83, H 2.49%. Found: C 45.79, H 2.42%. IR (ATR, /cm−1): 1138 (P = O), 1650 (C = O), 1250, 1160 (CF3).
The same product is isolated when TbCl3·6H2O, hfah and Ph3PO are mixed in 1:3:2 molar ratio in water/ethanol.
[Dy(hfa)3(Ph3PO)2] (8) was obtained in a similar way to 7: amounts of DyCl3·6H2O (0.400 g, 1.07 mmol), Ph3PO (0.298, 1.07 mmol) and hfah (0.664 g, 3.21 mmol). Single crystals of 8 are isolated in the same way as those of 7. Yield 0.210 (29%). M.W.: 1340.22. Anal. calcd. for C51H33DyF18O8P2: C 45.70, H 2.48%. Found: C 45.38, H 2.86%. IR (ATR, /cm−1): 1136 (P = O), 1652 (C = O), 1250, 1161(CF3).
The same product is isolated when DyCl3·6H2O, hfah and Ph3PO are mixed in 1:3:2 molar ratio in water/ethanol.
Magnetic Measurements
Magnetic susceptibility direct current (dc) and alternating current (ac) measurements for 1–8 were performed with a Quantum Design SQUID MPMS-XL-5 susceptometer. The magnetic susceptibility dc data were recorded at temperatures ranging from 2 to 300 K, under a magnetic field of 1000 Oe. Magnetization measurements under magnetic fields of 0–50000 Oe at 2.0 K were also recorded. Diamagnetic corrections were estimated from Pascal's Tables. ac susceptibility data were registered with an oscillating ac field of 3.5 Oe and ac frequency of 1400 Hz, under diverse applied static fields (Hdc = 0 or 1,000) for all the reported compounds. In the case of 2 and 8, ac susceptibility measurements were moreover recorded under a dc field of 1000 Oe at ac frequencies in the range 50–1400 Hz.
Crystallographic Refinement and Structure Solution
Crystal data and details of refinement are given in Table S1. Single crystals of 1·CH3CN, 2·CH3CN, 4, 5·2.75EtOH·1.25H2O, 6·THF, 7 and 8 were obtained as detailed above. Data were collected at 100 K on a Bruker Kappa APEXII CCD diffractometer, employing graphite monochromated Mo-kα (λ = 0.71073 Å) radiation. Multi-scan absorption corrections were applied using SADABS (Blessing, 1995; Krause et al., 2015). The structures were solved by standard direct methods, using SHELXT (Sheldrick, 2015a), and then refined by full-matrix least-squares techniques on F2, using the program package SHELXL (Sheldrick, 2015b) from the program package SHELX (Sheldrick, 2008). All non-hydrogen atoms were refined anisotropically, with the exception of some atoms with low occupation sites. Most of the hydrogen atoms were included in the structure factor calculations in geometrically idealized positions. However, non-disordered hydrogen atoms, which could be potentially involved in H-bonding schemes, were mostly located in the corresponding Fourier maps. These H atoms were freely refined, or with thermal parameters derived from their parent atoms.
CCDC 1850933–1850938 and 1851725 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/structures.
Results and Discussion
Synthesis and Spectroscopic Characterization
As previously stated, the chemistry of lanthanoids with phosphine oxides is quite well studied and many complexes with different metal ion:phosphine oxide ratios were isolated (Platt, 2017). These previous studies show that the stoichiometry of the complexes seems to be dependent on the nature of the metal salt employed, and on the molar quantities of salt and phosphine oxide in the reaction mixture, but also on the cone angle of the phosphine (Tolman, 1977). Recently, it has been reported that complexes of formulas [Ln(Cy3PO)2(H2O)5]X3 (Ln = La, Dy, Er, Yb, Lu; X = Cl or Br) (Lees and Platt, 2014, 2015) can be easily obtained. These complexes have pentagonal bipyramidal geometry, and some of them are air stable single ion magnets, with high blocking temperatures (Chen et al., 2016). Nevertheless, similar pentagonal bipyramidal complexes were not reported for Ph3PO, what would lead to evaluate the influence of the aromatic vs. the aliphatic ring in the magnetic behavior of the complexes. With these considerations in mind, different dysprosium and/or terbium salts were initially mixed with Ph3XO (X = P or As) or MePh2PO in 1:2 molar ratio, with the intention of isolating complexes of stoichiometry Ln(R3XO)2Y5 (R3 = Ph3 or MePh2; X = P or As; Y = OH2 and/or monodentate anion).
The obtained results indicate that the salt:R3XO molar ratio plays a secondary role in the stoichiometry of the isolated complexes (Figure 1), and that in this case the obtaining of compounds with coordination number 7 and pentagonal bipyramidal geometry was not possible. Thus, mixing of Ln(NO3)2·6H2O (Ln = Dy or Tb) with Ph3XO (X = P or As) in 1:2 molar ratio leads to isolate [Ln(NO3)3(Ph3PO)3] or [Dy(NO3)3(EtOH)(Ph3XO)2] compounds, as a function of the solvent employed in the reaction, as previously described for Ph3PO (Cousins and Hart, 1967). Accordingly, it also seems that the nature of the X atom (P or As) in the Ph3XO donor does not play any role in the stoichiometry of the isolated complexes. Besides, it should be noted that the compounds [Ln(NO3)3(Ph3PO)3] 1 and 2 are also obtained when the salts and oxides are mixed in 1:3 molar ratio, and that these complexes present the same stoichiometry as those reported for Ln = La, Nd, Eu, Er, Tm, and Yb with Cy3PO (Hunter et al., 2007).
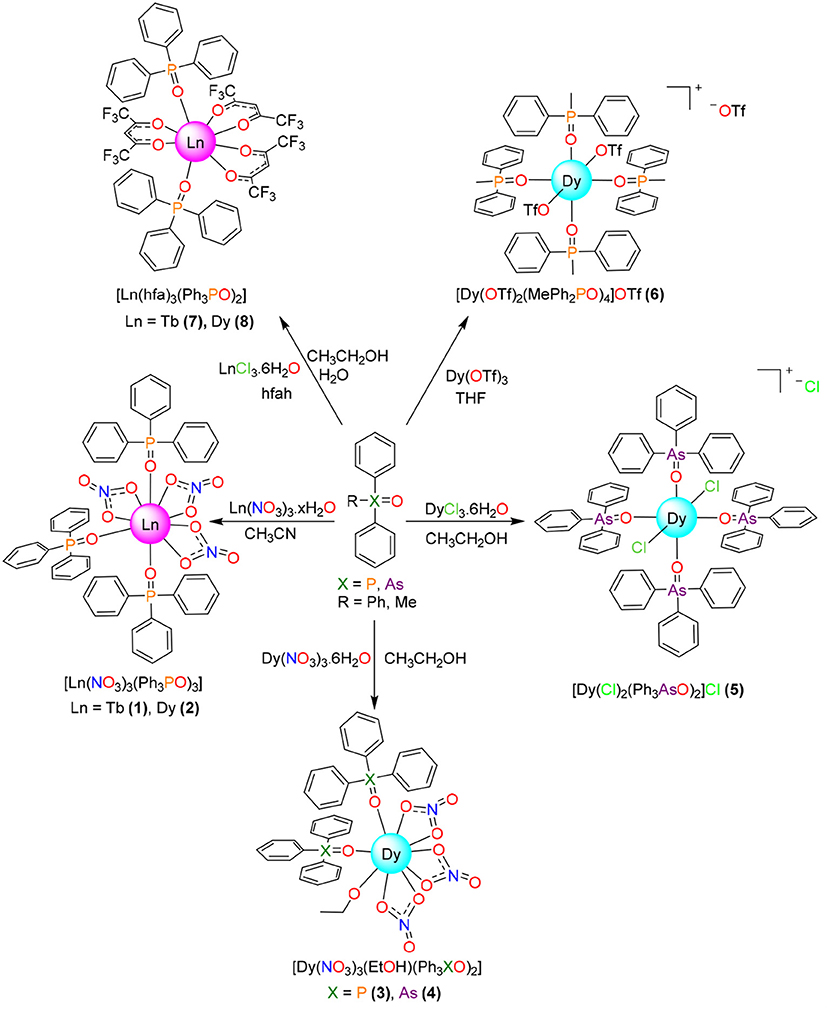
Figure 1. Reaction scheme for isolation of the metal complexes.
When the nitrate salt was changed by dysprosium chloride, and Ph3AsO was added in 1:2 molar ratio, the hydrated complex [DyCl2(Ph3AsO)4]Cl·1.25H2O was obtained (Figure 1). Its stoichiometry is the same described for [DyCl2(Ph3PO)4]Cl·2H2O (Glazier et al. 2004), obtained by mixing of DyCl3 and Ph3PO in 1:4 molar ratio, but clearly differs from that of [Dy(Cy3PO)2(H2O)5]Cl3 (Lees and Platt, 2014), which also has been obtained by mixing of DyCl3 and Cy3PO in 1:4 molar ratio in ethanol. Accordingly, comparison of this experiment with the reactions with dysprosium nitrate and with previous results (Glazier et al., 2004; Hunter et al., 2007; Lees and Platt, 2014) clearly show that the cone angle of the phosphine plays a fundamental role in the stoichiometry of phosphine or arsine oxide complexes, but that the similarity between the chemistry of Cy3PO and Ph3XO also depends on the nature of the anion of the salt employed.
Since three nitrate donors or two of the chloride anions are coordinated to the lanthanoid metal center in 1-4 and related complexes (Glazier et al., 2004; Hunter et al., 2007; Bowden et al., 2014), a salt of a non-coordinating anion, such as triflate, was chosen, in order to see the influence of the coordinating ability of the anion of the salt in the final stoichiometry, and in the hope of isolating complexes of stoichiometry [Dy(R3XO)2(H2O)5]Y3. In this way, mixing of dysprosium triflate with MePh2PO in 1:2 molar ratio, and in a non-dried non-coordinating solvent such as THF, yields [Dy(OTf)2(MePh2PO)4]OTf, which also has a molar ratio metal:oxide of 1:4. Thus, this experiment shows that, in spite of the quantity of water present in the non-dried THF solvent, the dysprosium atom links to two poorly coordinating triflate ions, preventing the coordination of water to the metal center. Accordingly, this result resembles in part those obtained for Ce, Nd and Lu triflate complexes of Ph3PO (Fawcett et al., 2002; Berthet et al., 2003;), where the stoichiometry is the same, but for the Ce and Nd derivatives the coordination number is 7, with one triflate acting as a bidentate chelate ligand and the other one as a monodentate donor.
Finally, the reactivity of Dy and Tb toward Ph3PO, in 1:1 molar ratio and in the presence of hexafluoroacetylacetone, was studied, given that a [Eu(tmh)3(RPh3PO)] (tmh = 2,2,6,6-tetramethylheptane-3,5-dione, R = H, m-Me or p-Me) complex, with coordination number seven, was previously reported (Yanagisawa et al., 2017). As Eu3+ is significantly bigger than Tb3+ and Dy3+, the isolation of Tb and Dy complexes with coordination number 7 was expected even in the presence of the less sterically hindered hexafluoroacetylacetone donor. Nevertheless, this synthesis leads to complexes [M(hfa)3(Ph3PO)2] (M = Tb, Dy) where the metal:oxide ratio is 1:2, as it occurs in the previously described [Eu(hfa)3(Ph3PO)2] and related compounds (Hasegawa et al., 2013), suggesting that for this kind of complex the size of the lanthanoid ion has a poor influence in the stoichiometry of the isolated compound.
As a summary, comparison of the results described herein with those previously described for Cy3PO (Chen et al., 2016, 2017) seems to indicate that the lower cone angle for triphenylphosphine compared with tricyclohexylphosphine prevents the isolation of pentagonal bipyramidal complexes of triphenylphosphine oxide with dysprosium or terbium. In the same way, hexafluoroacetylacetone allows coordination of two phosphine oxide ligands, leading to coordination number of 8 for the lanthanoid atom while the more sterically hindered 2,2,6,6-tetramethylheptane-3,5-dione only allows linking one phosphine ligand, leading to coordination number 7 (Yanagisawa et al., 2017). Therefore, this study clearly shows that not only the cone angle of the phosphine oxides but also the volume of the auxiliary ligands are fundamental factors in controlling the coordination number in this type of complex, the salt:oxide metal ratio, and even the size of the lanthanoid ion, playing secondary roles.
All the compounds described herein were fully characterized by analytical techniques, IR spectroscopy and by single crystal X-ray diffraction studies, except 3. In addition, direct and alternating current magnetic measurements were recorded for all the compounds.
The IR spectra of all the phosphine oxide complexes show strong bands in the range 1119–1153 cm−1, which can be assigned to the P = O stretching vibration (Bowden et al., 2012). For complexes 1, 2, and 3 there are two bands in this range, indicating the existence of two types of non-equivalent phosphine oxide. Nevertheless, complexes 6-8 present just one band at ca. 1135 cm−1, which suggest that all the phosphine oxide ligands present in the compounds are equivalent.
Complexes 4 and 5·1.25H2O show strong bands about 900 cm−1, which can be assigned to As = O vibrations (Levason et al., 2001). In 4 two bands are observable, also suggesting two non-equivalent arsine oxide ligands, while 5·1.25H2O only shows an As = O band, in agreement with the equivalence of the four arsine oxide donors present in the complex.
In addition, the synthetized complexes also present bands related to the auxiliary nitrate, triflate or hexafluoroacetylacetonate ligands. Thus, complexes 1-4 show a strong band at ca. 1305 cm−1, assigned to the ν1(N-O) vibration of the nitrate ligand, acting as bidentate (Bowden et al., 2012). Complex 6 show many bands (633, 1020, 1262, 1156, and 1220 cm−1) related to S-O or C-F vibrations, which suggest the presence of triflate (Johnston and Shiver, 1993). Finally, the IR spectra of 7 and 8 present strong bands at 1652 cm−1, assigned to the stretching frequency of the C = O group, and at 1250 and 1161, in agreement with vibrations of the CF3 and C-H moieties, all of which clearly suggest the presence of the hexafluoroacetylacetonate ligand (Richardson et al., 1968).
X-Ray Diffraction Studies
[Tb(NO3)3(Ph3PO)3]·CH3CN (1·CH3CN) and [Dy(NO3)3(Ph3PO)3]·CH3CN (2·CH3CN)
The crystal structures of 1·CH3CN and 2·CH3CN are very similar and they will be discussed together. The unit cell of each complex contains neutral [M(NO3)3(Ph3PO)3] (M = Tb or Dy) molecules, in addition to acetonitrile as solvate. Ellipsoid diagrams for 1 and 2 are shown in Figure 2, Figure S1, respectively, and main distances and angles in Table S2.
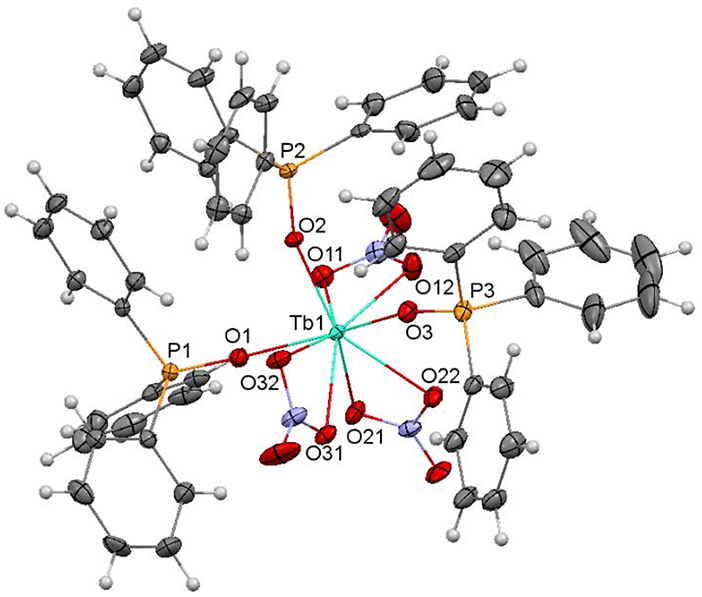
Figure 2. Ellipsoids diagram (50% probability) for 1. Only the Tb, P, and donor O-atoms have been labeled, for clarity. Color code: Tb, light blue; C, gray; H, light gray; N, dark blue; P, orange; O, red.
Both complexes crystallize in the P21/c group, with no symmetry elements relating the different ligands.
In the [M(NO3)3(Ph3PO)3] molecules, three phosphine oxide monodentate ligands and three bidentate chelate nitrate donors complete the coordination sphere of the metal ions. Thus, in both complexes, the lanthanoid ion is in an O9 environment. Calculations of the degree of distortion of the LnO9 core with respect to a perfect nine-vertex polyhedron using the SHAPE software (Llunell et al., 2005, 2010; Ruiz-Martínez et al., 2008) lead to shape measurements between spherical capped square antiprism and muffin-like, but closer to spherical capped square antiprism (Figure 3, Table S3). In these polyhedra, all the distances and angles are within their usual range for lanthanoid complexes with phosphine oxide and nitrate donors (Bowden et al., 2011), showing M-OPh3PO distances notably shorter than the M-Onitrate ones (Table S2).
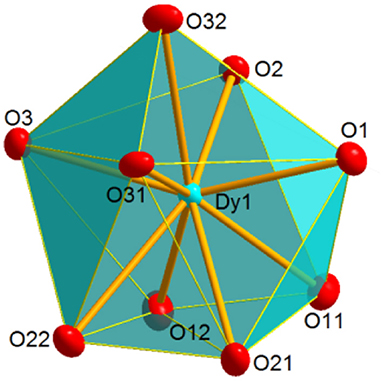
Figure 3. Coordination polyhedron for Dy in 2, illustrating the distorted spherical capped square antiprism geometry shown by the metal ion in 1 and 2.
[Dy(NO3)3(EtOH)(Ph3AsO)2] (4)
An ellipsoid diagram for 4 is shown in Figure 4 and main bond distances and angles in Table S4.
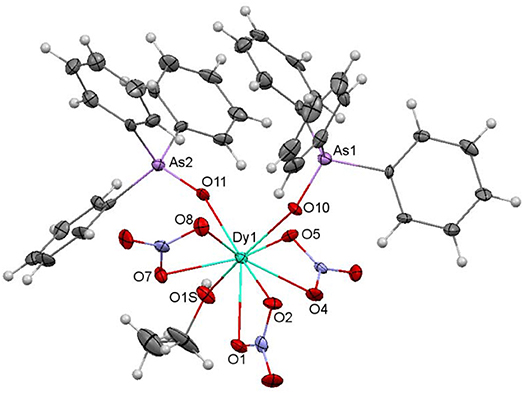
Figure 4. Ellipsoids diagram (50% probability) for 4. Only the Dy, As, and donor O-atoms have been labeled, for clarity. Color code: Dy, light blue; As, violet; C, gray; H, light gray; N, dark blue; O, red.
The crystal structure of 4 resembles that of 2. Besides, it should be noted that 4 is isomorphous with the previously described and crystallographically characterized [La(NO3)3(EtOH)(Ph3AsO)2] complex (Levason et al., 2001). Thus, three bidentate chelate nitrate ligands, two monodentate Ph3AsO donors and an ethanol molecule fill the O9 coordination sphere of the dysprosium ion. SHAPE calculations indicate that the geometry about the metal ion is also between spherical capped square antiprism and muffin-like, as in 1 and 2, but in this case the muffin-like disposition seems to be a bit more stable (Table S2). All the distances and angles in this complex are within the usual range, but it should be noted that the Dy-Oarsine oxide distances are a bit shorter than the corresponding Dy-Ophospine oxide ones in 2, and this can be a consequence of the smaller electronegativity of arsenic with respect to phosphorous, and, therefore, to the greater donor strength of the arsine oxide, given that the geometry of both complexes is very similar.
Finally, it is worth of mention that this complex shows a hydrogen bond between one oxygen atom (O6) of one nitrate donor and the ethanol ligand of a neighboring unit. This hydrogen bond is reciprocal and gives rise to a pseudodimer, with a Dy···Dy distance of 7.1870(9) Å (Figure 5).
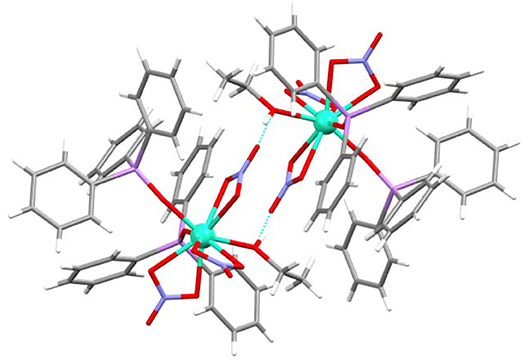
Figure 5. Hydrogen bond for 4, showing the pseudodimer.
[DyCl2(Ph3AsO)4]Cl·2.75CH3CH2OH·1.25H2O (5·2.75 CH3CH2OH·1.25H2O)
An ellipsoid diagram for [Dy(Ph3AsO)4(Cl)2]Cl is shown in Figure 6 and main distances and angles in Table S5.
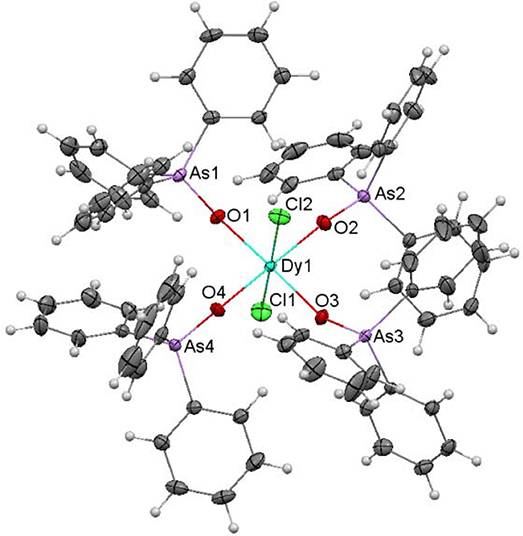
Figure 6. Ellipsoids diagram (50% probability) for [DyCl2(Ph3AsO)4]+ in 5. Only the Dy, As, Cl and donor O-atoms have been labeled, for clarity. Color code: Dy, light blue; As, violet; C, gray; Cl, green; H, light gray; O, red.
The asymmetric unit of 5·2.75CH3CH2OH·1.25H2O contains a [DyCl2(Ph3AsO)4]Cl molecule, and ethanol and water as solvates. The [DyCl2(Ph3AsO)4]+ cation shows the Dy(III) ion joined to four Ph3AsO ligands through their O-atom, and to two chloride ions. Thus, the dysprosium atom is in an O4Cl2 environment, with octahedral geometry. In this polyhedron, the chloride ions occupy the axial sites, what produces an elongation of the octahedron.
Although the octahedral geometry is not very common for lanthanide ions, given that they tend to achieve higher coordination numbers, the structure of [DyCl2(Ph3AsO)4]Cl is very similar to that previously described for other [LnX2(R3PO)4]X (X = Cl or Br) compounds (Glazier et al., 2004; Bowden et al., 2010; Platt, 2017), with distances and angles in the usual range, and that do not merit further consideration.
[Dy(OTf)2(MePh2PO)4]OTf·THF (6·THF)
The unit cell of 6·THF contains two crystallographically different but chemically equivalent [Dy(OTf)2(MePh2PO)4]+ cations, which will be called 6A and 6B, respectively, and CF3 counterions and THF as solvate. Each [Dy(OTf)2(MePh2PO)4]+ cation has an inversion center, located at the dysprosium atom. Besides, one of these [Dy(OTf)2(MePh2PO)4]+ cations, the one containing Dy2 (6B), has the coordinated triflate anions disordered over two sites (80 and 20% occupancy).
An ellipsoid diagram for one of the [Dy(MePh2PO)4(OTf)2]+ ions (6A) is shown in Figure 7 and main bonds distances and angles in Table S6.
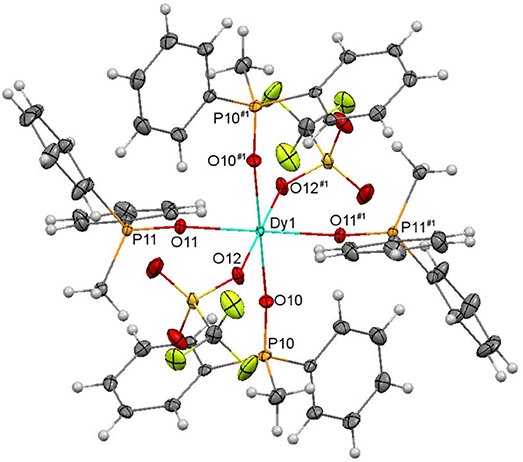
Figure 7. Ellipsoids diagram (50% probability) for [Dy(MePh2PO)4(OTf)2]+ in 6A. Only the Dy, P, and donor O-atoms have been labeled, for clarity. Color code: Dy, light blue; C, gray; F, fluorescent yellow; H, light gray; O, red; P, orange.
In both [Dy(MePh2PO)4(OTf)2]+ cations, the DyIII ions are coordinated to four phosphine oxides and to two triflate ions, which act as monodentate ligands. This gives rise to an O6 octahedral environment about the dysprosium center. It should be noted that three lanthanoid complexes of empirical formula [Ln(OTf)2(Ph3PO)4](OTf)(Ln = Ce, Nd, Lu) have been previously reported, and in Ce and Nd complexes one of the triflate ligands acts as bidentate chelate and the other one as monodentate (Fawcett et al., 2002; Berthet et al., 2003), leading to coordination number 7, while in 6 both triflate donors act as monodentate. In fact, the structure of 6 is more similar to that of [Lu(OTf)2(Ph3PO)4](OTf) (Fawcett et al., 2002), where the metal ion is also in an octahedral environment. This seems to be related to the smaller size of DyIII compared with CeIII and NdIII that, in spite of the less sterically hindered phosphine oxide in 6 (MePh2PO) respect to the Ce and Nd complexes (Ph3PO), cannot allocate seven atoms in its surroundings.
The shortest Dy-O distances in both 6A and 6B cations correspond to Dy-Ophosphine oxide, the weakest bonds being the Dy-Otriflate ones, as expected in view of the poor ability of the triflate anions to act as ligands. Thus, the polyhedra are axially elongated. Nevertheless, it should be noted that while in 6A all the Dy-Ophosphine oxide distances are very close, in 6B the strength of these bonds differs significantly (Dy2-O21 distance of ca. 2.19 Å and Dy2-O20 distance of ca. 2.24 Å) and the Dy-Otriflate bond is a bit stronger in 6B than in 6A, showing a less elongated octahedron.
[Tb(hfa)3(Ph3PO)2] (7) and [Dy(hfa)3(Ph3PO)2] (8)
The crystal structures of both complexes are very similar, and they will be discussed together. The main difference between them is that the fluoride atoms of one CF3 moiety of one triflate ligand are disordered over two sites (56/44%) in 7, while this does not occur in 8. Ellipsoids diagrams for 7 and 8 are shown in Figure S2, Figure 8, respectively, and main bond distances and angles in Table S7.
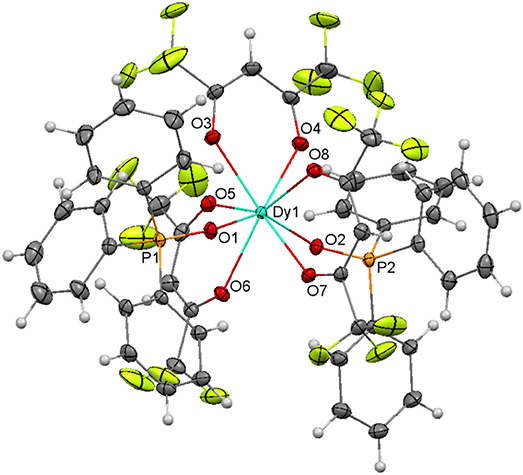
Figure 8. Ellipsoids diagram (50% probability) for 8. Only the Dy, P, and donor O-atoms have been labeled, for clarity. Color code: Dy, light blue; C, gray; F, fluorescent yellow; H, light gray; O, red; P, orange.
The structure of both compounds resembles that of [Eu(hfa)3(Ph3PO)2] (Hasegawa et al., 2013). Thus, in both complexes the lanthanoid ion is coordinated to three bidentate chelate hexafluoroacetylacetonate anions and to two phosphine oxides, giving rise to an O8 environment. Calculations of the degree of distortion of the MO8 environment with respect to an ideal eight-vertex polyhedron with the SHAPE software (Llunell et al., 2005, 2010; Ruiz-Martínez et al., 2008), indicates that the geometry is closer to square antiprism in both cases. In these polyhedra, all the distances and angles are in the expected range and do not merit further consideration.
Magnetic Properties
The direct current (dc) magnetic susceptibility studies of all the complexes (1-8) were recorded under a magnetic field of 1000 Oe in the temperature range 2–300 K. The plots of χMT vs. T for the complexes are shown in Figure S3. At room temperature (300 K), the χMT products for the terbium complexes 1 and 7 are in the range 11.81–12.37 cm3kmol−1 while for the dysprosium complexes 2-6 and 8 vary from 14.05 to 14.93 cm3kmol−1. All these values are similar to the expected ones for one isolated TbIII (4f8, 7F6) or DyIII (4f9, 6H15/2) ion of 11.82 and 14.17 cm3kmol−1, respectively.
The experimental χMT values remain basically constant (1, 2), decreases very slowly (6-8) or decrease a bit quickly (3-5·1.25H2O) upon cooling up to 50 K, and more rapidly below this temperature in all cases. This behavior appears to be mostly owing to the depopulation of the MJ sublevels of the lanthanoid ion, and possibly to very weak intermolecular interactions, which could be the cause of the more marked decrease in χMT at low temperature.
The variation of the magnetization with the field at 2 K for 1-8 (Figure S3) are akin in all cases, and shows a quite fast increase of the M/NμB value at low field, without attaining saturation at 5 T. The M/NμB value at the largest applied field of 5 T (in the 5.07–5.77 Ms/NμB range) is remarkably lower than the predictable one for isolated TbIII or DyIII ions (Ms/NμB = gjJ), which can be ascribed to the crystal-field effects that promote a significant magnetic anisotropy.
Alternating current (ac) magnetic susceptibility measurements were also done for microcrystalline samples of compounds 1-8, with the aim of knowing their low-temperature magnetic relaxation behavior. No one of the compounds show out-of-phase ac susceptibilities (χ″) peaks at a frequency of 1400 Hz at zero field, indicating the absence of SMM behavior. Nevertheless, it should be noted that, sometimes, application of an external static field (Ruiz et al., 2012), in order to eliminate the mixing of the ground ±Ms levels, can partially or completely suppress the quantum tunneling of the magnetization (QTM), allowing to observe the slow relaxation process through the real thermally activated energy barrier (U). Therefore, this approximation was tested with the aim of checking the quantum tunneling effects.
Accordingly, variable temperature alternating current (ac) susceptibilities were measured for all the compounds at 1400 Hz and under a small dc field of 1000 Oe. In these conditions, the χ” vs. T curve only shows a clear net peak for 2, 4, and 8 (Figure S4), but in the case of 4 the peak appears at 2.5 K at a frequency of 1400 Hz. Thus, in view of these results, given that the magnetic response of 4 seems too weak, variable frequency and temperature ac susceptibilities were measured only for 2 and 8 with the application of an experimental optimal dc field of 1000 Oe. As shown in Figure 9, in these conditions 2 and 8 show frequency and temperature-dependence of the out-of-phase susceptibility, with maxima for χ” below 5 and 5.8 K for 2 and 8, respectively.
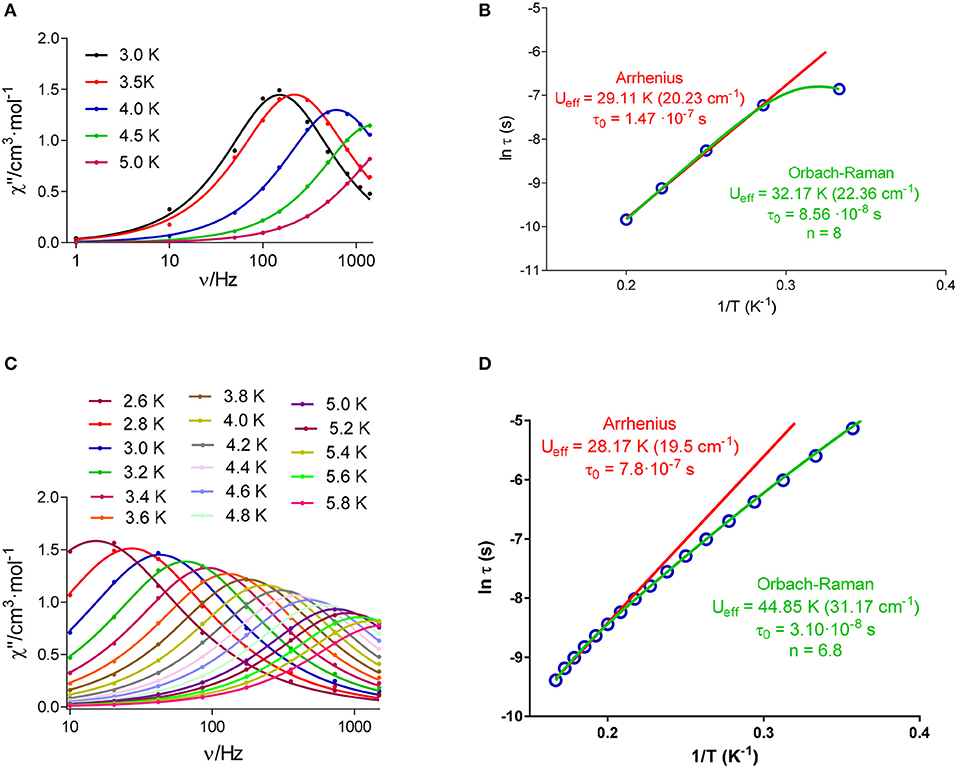
Figure 9. Frequency dependence of out-of-phase component of the ac susceptibility (χ”) under a dc applied field of 1000 Oe at different temperatures for 2 (A) and 8 (C); and Arrhenius plot for 2 (B) and 8 (D). The solid lines represent the best fit to simple Arrhenius or Orbach-Raman equation.
This indicates that both complexes (2 and 8) are field-induced single ion magnets. In addition, it should be noticed that Figure 9 reveals that χ” goes to zero below the maxima at low temperature for 2, which suggest that the fast relaxation of the magnetization by a quantum tunneling mechanism has been completely suppressed by the application of the external magnetic field of 1000 Oe. Nevertheless, this does not occur for 8, where the QTM does not seem fully suppressed at low temperatures.
According to this, the experimental data for 2 (Figure 9B) were initially fitted to the Arrhenius equation (thermally activated Orbach relaxation process), and the best fit affords a small Ueff barrier of 29.11 K. But the experimental data deviate from the linear Arrhenius plot, which can be due to the simultaneous presence of different relaxation processes.
In keeping with this, the Cole-Cole plot for 2 (Figure 10) between 2.5 and 5 K gives parameters in the range 0.19–0.04, suggesting more than one relaxation process at the lowest temperature. Therefore, the experimental relaxation times were fitted to equation 1, which takes into account contributions from Orbach and Raman processes.
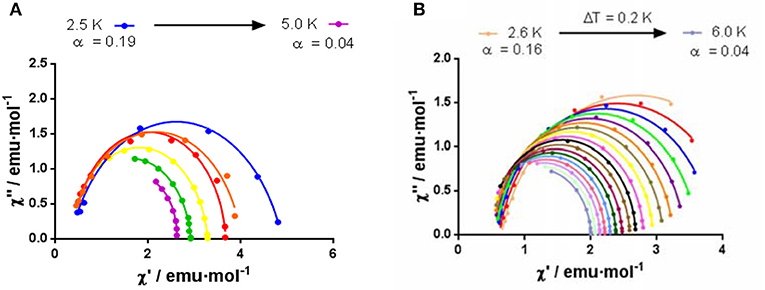
Figure 10. Cole–Cole plot in 1000 Oe dc field for 2 (A) and 8 (B).
The best fit with this equation (Figure 9B), which reproduces all the curve, leads to an Ueff barrier of 32.17 K.
In the case of 8, the best fit of the experimental relaxation times to the Arrhenius equation leads to an Ueff of 28.17 K. However, as in the case of 2, the experimental relaxation times deviate from linearity (Figure 9), indicating the possible existence of several relaxation processes. The Cole-Cole plot for 8 between 2.6 and 6 K (Figure 10), which renders parameters in the range 0.16–0.04, corroborate the presence of more than one relaxation process, especially in the low-temperature area. In view of this, the experimental data were also fitted to the equation 1. The best fit led to the parameters indicated in Figure 9D. Although for Kramers ion like DyIII an n value equal to 9 is expected (Abragam and Bleaney, 1970), values between 2 and 7 are also realistic when both acoustic and optical phonons are present (Singh and Shrivastava, 1979; Shrivastava, 1983).
At this point it is worth mentioning that the free ion electronic density of the DyIII ion has an oblate shape, which can be stabilized by an axial crystal field leading to an axial anisotropy in the ground state (MJ = ± 15/2). This bistable ground state favors the SMM behavior. The axial crystal field is relatively easy to attain in low-symmetry DyIII complexes, and this is the reason why this type of complexes usually shows slow relaxation of the magnetization and SMM behavior, either at zero applied dc field or in the presence of a magnetic field (when a significant QTM is operative).
The non-Kramers TbIII ion is also an oblate ion and therefore also requires an axial crystal field to reach an axial bistable ground state, and then to exhibit SMM behavior. However, to this end a rigorous axial symmetry must be maintained, which is not an easy task. Moreover, non-Kramers ions possess an intrinsic tunneling gap that promotes QTM at zero field. Therefore, the lack of SMM at zero field, and in the presence of a small field for complexes 1 and 7 is not unexpected.
The fact that, among the DyIII complexes, only compounds 2, 4, and 8 exhibit slow relaxation of the magnetization can be explained by analyzing the Dy-O distances and their distribution in the DyIII coordination sphere, as these factors play an essential role in dictating the SMM behavior. The required axial crystal field can be reached when the donors atoms with the largest electron densities (generally those involved in the shortest Dy-donor distances) are located above and below the equatorial plane, thus diminishing the repulsive interactions between the donor atoms of the ligands and the oblate charge cloud of the DyIII ion. Nevertheless, the crystal field can also be generated when a Dy-donor distance is significantly shorter that the other Dy-donor distances. Taking into account these considerations, complexes 5 and 6, which exhibit an elongated octahedral geometry, with the four shortest Dy-O distances in the equatorial plane, present an equatorial crystal field. Therefore, the fact that these complexes do not present SMM behavior is not surprising. In the case of 2, the three Dy-Ophosphine oxide distances, with a near T disposition, are significantly shorter than the other ones. Nevertheless, two of them (Dy-O1 and Dy-O2) are slightly but significantly shorter that the other one. This disposition leads to a feeble axial crystal field that favors slow relaxation of the magnetization. The fact that at zero-field this compound does not show any peak in the ac measurements can be due to the fast QTM promoted transverse anisotropy, as well as to intermolecular and hyperfine interactions. For complex 4, the shortest Dy-O distances are also those involving the oxygen atoms of the arsine-oxide ligands. These distances are approximately 0.2 and 0.1 Å shorter than those of the coordinated nitrate anions and ethanol molecule, respectively. This disposition of the shortest Dy-O distances is similar to that of 2 and therefore the appearance of field induced slow relaxation in 4 is not unexpected. Finally, for compound 8, the two shortest Dy-O bond distances are those involving the Ph3PO ligands, which are at opposite sides of the DyIII ions, with a O1-Dy-O2 angle of 143.63(4)°. This distribution of the phosphine ligands around the DyIII ions generates a clear axial crystal field that is responsible of the SMM behavior observed for this compound.
Conclusions
The stoichiometry of the Dy and Tb complexes derived from R3XO (R3 = Ph or MePh2, X = P or As) does not seem to rely on the nature of the X atom, nor in the molar ratio of the lanthanoid salt:R3XO employed. Nevertheless, this stoichiometry highly depends on the nature of the lanthanoid salt, as well as on the election of the solvent of the reaction.
Comparison of the results obtained herein with previously published data allows concluding that the cone angle of the phosphine or arsine, as well as the degree of steric hindrance of the auxiliary donors also play an important role in the stoichiometry of the isolated compounds. Thus, in our case, it was not possible to obtain pentagonal bipyramidal complexes. In this way, nona-coordinated complexes of different stoichiometry ([M(NO3)3(Ph3PO)3] (M = Tb, 1; Dy, 2) or [Dy(NO3)3(EtOH)(Ph3XO)2] (X = P, 3; As, 4) are isolated by mixing the lanthanoid salt and Ph3XO in 1:2 molar ratio, just varying the solvent of the reaction. Hexa-coordinated complexes [DyCl2(Ph3AsO)4]Cl (5) and [Dy(OTf)2(MePh2PO)4]OTf (6; OTf = triflate) are also obtained from 1:2 molar ratios of salt and phosphine oxide, while octa-coordinated [M(hfa)3(Ph3PO)2] (M = Tb, 7; Dy, 8) compounds are formed when the lanthanoid precursor and the phosphine oxide are mixed in 1:1 molar ratio.
The magnetic characterization of all the complexes show that only 2, 4, and 8 present slow relaxation of the magnetization under an external dc field of 1000 Oe. This indicates that, between oblate TbIII and DyIII, the Kramers dysprosium ion increases the effective barrier for reversal of the magnetization with respect to non-Kramers terbium ion, as frequently observed, provided that the relaxation of the magnetization is seen for the dysprosium complexes 2, 4, and 8 but not for the homologous terbium compounds 1 and 7. Besides, the arsine oxide also favors the SMM behavior compared with the phosphine oxide, given that 4 shows slow relaxation of the magnetization at low temperature (< 2.5 K) while 3 lacks this relaxation. In addition, the replacement of a phosphine oxide by an ethanol molecule decreases the ability of the complex to act as a SIM, since 2 is a field induced SIM, with an Ueff barrier of 32.17 K, while 3 does not show any evidence of SMM behavior. Furthermore, [Dy(hfa)3(Ph3PO)2] is also a weak induced SIM, with Ueff of 44.85 K. The field induced slow relaxation of the magnetization in complexes 2, 4, and 8, as well as the absence of slow relaxation in complexes 5 and 6 has been justified by using the simple prolate-oblate electrostatic model and the distribution of the shortest Dy-O distances in the DyIII coordination sphere.
Author Contributions
JC-V and JS-M did the experimental work to prepare, crystallize and characterize the samples. AMG-D carried out the XRD analyses. JMH and EC made the magnetic measurements, interpreted some of the magnetic results and participate in the writing. MF designed and supervised the work, interpreted some of the magnetic data and wrote the draft.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Financial support from Ministerio de Economía y Competitividad (MINECO, CTQ2014-56312-P), Junta de Andalucía (FQM-195 and Project of excellence P11-FQM-7756) and the University of Granada are grateful acknowledged. JC-V acknowledges Xunta de Galicia for his Ph.D. fellowship.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2018.00420/full#supplementary-material
References
Abragam, A., and Bleaney, B. (1970). Electron Paramagnetic Resonance of Transition Ions. Oxford, UK: Clarendon Press.
Berthet, J.-C., Nierlich, M., and Ephritikhine, M. (2003). A comparison of analogous 4f- and 5f-element compounds: syntheses and crystal structures of triphenylphosphine oxide complexes of lanthanide(III) and uranium(III) triflates and iodides [MX2(OPPh3)4][X] (X = OTf and M = Ce or U; X = I and M = Nd, Ce, La, U). Polyhedron 22, 3475–3482. doi: 10.1016/j.poly.2003.09.008
Blessing, R. H. (1995). An empirical correction for absorption anisotropy. Acta Crystallogr. A. 51, 33–38. doi: 10.1107/S0108767394005726
Bowden, A., Coles, S. J., Pitak, M. B., and Platt, A. W. G. (2014). Complexes of lanthanide nitrates with tri-isopropylphosphine oxide. Polyhedron 68, 258–264. doi: 10.1016/j.poly.2013.10.028
Bowden, A., Horton, P. N., and Platt, A. W. G. (2011). Lanthanide nitrate complexes of tri-isobutylphosphine oxide: solid state and CD2Cl2 solution structures. Inorg. Chem. 50, 2553–2561. doi: 10.1021/ic102385p
Bowden, A., Platt, A. W. G., Singh, K., and Townsend, R. (2010). Complexes of triphenylphosphine oxide with lanthanide bromides. Inorg. Chim. Acta 363, 243–249. doi: 10.1016/j.ica.2009.08.016
Bowden, A., Singh, K., and Platt, A. W. G. (2012). Lanthanide nitrate complexes with triphenylphosphine oxide. Solid state and solution properties. Polyhedron 42, 30–35. doi: 10.1016/j.poly.2012.04.021
Chen, Y.-C., Liu, J.-L., Lan, Y., Zhong, Z.-Q., Mansikkam-ki, A., Ungur, L., et al. (2017). Dynamic magnetic and optical insight into a high performance pentagonal bipyramidal DyIII single-ion magnet. Chem. Eur. J. 23, 5708–5715. doi: 10.1002/chem.201606029
Chen, Y.-C., Liu, J.-L., Ungur, L., Liu, J., Li, Q.-W., Wang, L.-F., et al. (2016).Symmetry-supported magnetic blocking at 20 K in pentagonal bipyramidal Dy(III) single-ion magnets. J. Am. Chem. Soc. 138, 2829–2837. doi: 10.1021/jacs.5b13584
Cousins, D. R., and Hart, F. A. (1967). Complexes of triphenylphosphine with lanthanide and yttrium nitrates. J. Inorg. Nucl. Chem. 29, 1745–1757. doi: 10.1016/0022-1902(67)80218-5
Ding, Y.-S., Chilton, N. F., Winpenny, R. E. P., and Zheng, Y.-Z. (2016). On approaching the limit of molecular magnetic anisotropy: a near-perfect pentagonal bipyramidal dysprosium(III) single-molecule magnet. Angew Chem Int Ed Engl. 55, 16071–16074. doi: 10.1002/anie.201609685
Fawcett, J., Platt, A. W. G., and Russell, D. R. (2002). Structures and catalytic properties of triphenylphosphine oxide complexes of scandium and lanthanide triflates. Polyhedron 21, 287–293. doi: 10.1016/S0277-5387(01)00987-1
Glazier, M. J., Levason, W., Matthews, M. L., Thornton, P. L., and Webster, M. (2004). Synthesis, properties and solution speciation of lanthanide chloride complexes of triphenylphosphine oxide.Inorg. Chim. Acta 357, 1083–1091. doi: 10.1016/j.ica.2003.09.015
Goodwin, C. A. P., Ortu, F., Reta, D., Chilton, N. F., and Mills, D. P. (2017). Molecular magnetic hysteresis at 60 kelvin in dysprosocenium. Nature 548, 439–442. doi: 10.1038/nature23447
Guo, F.-S., Day, B. M., Chen, Y.-C., Tong, M.-L., Mansikkam-ki, A., and Layfield, R. A. (2017). A dysprosium metallocene single-molecule magnet functioning at the axial limit. Angew. Chem. Int. Ed. 56, 11445–11449. doi: 10.1002/anie.201705426
Hasegawa, Y., Ohkubo, T., Nakanishi, T., Kobayashi, A., Kato, M.;, Seki, T., et al. (2013). Effect of ligand polarization on asymmetric structural formation for strongly luminescent lanthanide complexes. Eur. J. Inorg. Chem. 5911–5918. doi: 10.1002/ejic.201301102
Hunter, A. P., Lees, A. M. J., and Platt, A. W. G. (2007). Synthesis, structures and mass spectrometry of lanthanide nitrate complexes with tricyclohexylphosphine oxide. Polyhedron 26, 4865–4876. doi: 10.1016/j.poly.2007.06.023
Ishikawa, N., Sugita, M., Ishikawa, T., Koshihara, S., and Kaizu, Y. (2003). Lanthanide double-decker complexes functioning as magnets at the single-molecular level. J. Am. Chem. Soc. 125, 8694–8695. doi: 10.1021/ja029629nv
Ishikawa, N., Sugita, M., and Wernsdorfer, W. (2005). Quantum tunneling of magnetization in lanthanide single-molecule magnets: bis(phthalocyaninato)terbium and bis(phthalocyaninato)dysprosium anions. Angew Chem. Int. Ed. Engl. 44, 2931–2935. doi: 10.1002/anie.200462638
Johnston, D. H., and Shiver, D. F. (1993). Vibrational study of the trifluoromethanesulfonate anion: unambiguous assignment of the asymmetric stretching modes. Inorg. Chem. 32, 1045–1047. doi: 10.1021/ic00058a050
Krause, L., Herbst-Irmer, R., Sheldrick, G. M., and Stalke, D. (2015). Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Cryst. 48, 3–10. doi: 10.1107/S1600576714022985
Lees, A. M. J., and Platt, A. W. G. (2014). Complexes of lanthanide chlorides with tricyclohexylphosphine oxide. the single crystal X-ray structures and solution properties of pentagonal bipyramidal complexes [Ln(H2O)5(Cy3PO)2]3+;·Cy3PO·3Cl– Ln = Dy, Er. Polyhedron 67, 368–372. doi: 10.1016/j.poly.2013.09.026
Lees, A. M. J., and Platt, A. W. G. (2015). Inner and outer sphere coordination of tricyclohexylphosphine oxide with lanthanide bromides. Polyhedron 91, 110–119. doi: 10.1016/j.poly.2015.02.008
Levason, W., Patel, B., Popham, M. L. C., Reid, G., and Webster, M. (2001). Scandium, yttrium and lanthanum nitrate complexes of tertiary arsine oxides: synthesis and multinuclear spectroscopic studies. X-raystructuresof [M(Me3AsO)6](NO3)3 (M = Sc or Y), [Sc(Ph3AsO)3(NO3)2]NO3, [M″(Ph3AsO)4(NO3)2]NO3 (M″ = Y or La) and [La(Ph3AsO)2(EtOH)(NO3)3]. Polyhedron 20, 2711–2720. doi: 10.1016/S0277-5387(01)00889-0
Llunell, M., Casanova, D., Cirera, J., Bofill, J. M., Alemany, P., Alvarez, S., et al. (2005). SHAPE v1.1b, Barcelona.
Llunell, M., Casanova, D., Cirera, J., Alemany, P., and Alvarez, S. (2010). SHAPE: Program for the Stereochemical Analysis of Molecular Fragments by Means of Continuous Shape Measures and Associated tools. University of Barcelona (Barcelona).
Lu, J., Guo, M., and Tang, J. (2017). Recent developments in lanthanide single-molecule magnets. Chem. Asian J. 12, 2772–2779. doi: 10.1002/asia.201701032
Platt, A. W. G. (2017). Lanthanide phosphine oxide complexes. Coord. Chem. Rev. 340, 62–78. doi: 10.1016/j.ccr.2016.09.012
Richardson, M. F., Wagner, W. F., and Sands, D. E. (1968). Rare-earth trishexafluoroacetylacetonates and related compounds. J. Inorg. Nucl. Chem. 30, 1275–1289. doi: 10.1016/0022-1902(68)80557-3
Rinehart, J. D., and Long, J. R. (2011). Exploiting single-ion anisotropy in the design of f-element single-molecule magnets. Chem. Sci. 2, 2078–2085. doi: 10.1039/c1sc00513h
Ruiz, J., Mota, A. J., Rodríguez-Diéguez, A., Titos, S., Herrera, J. M., Ruiz, E., et al. (2012). Field and dilution effects on the slow relaxation of a luminescent DyO9 low-symmetry single-ion magnet. Chem. Commun. 48, 7916–7918. doi: 10.1039/C2CC32518G
Ruiz-Martínez, A., Casanova, D., and Alvarez, S. (2008). Polyhedral structures with an odd number of vertices: nine-coordinate metal compounds. Chem. Eur. J. 14, 1291-1303. doi: 10.1002/chem.200701137
Sheldrick, G. M. (2008). A short history of SHELX. Acta Crystallogr. A 64, 112–122. doi: 10.1107/S0108767307043930
Sheldrick, G. M. (2015a). SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. A 71, 3–8. doi: 10.1107/S2053273314026370
Sheldrick, G. M. (2015b). Crystal structure refinement with SHELXL. Acta Cryst. 71, 3–8. doi: 10.1107/S2053229614024218
Shiddiq, M., Komijani, D., Duan, Y., Gaita-Ariño, A., Coronado, E., and Hill, S. (2016). Enhancing coherence in molecular spin qubits via atomic clock transitions. Nature 531, 348–351. doi: 10.1038/nature16984
Shrivastava, K. N. (1983). Theory of spin–lattice relaxation. Phys. Status Solidi B 117, 437–458. doi: 10.1002/pssb.2221170202
Singh, A., and Shrivastava, K. N. (1979). Optical-acoustic two-phonon relaxation in spin systems. Phys. Status Solidi B 95, 273–277. doi: 10.1002/pssb.2220950131
Tolman, C. A. (1977). Steric effects of phosphorus ligands in organometallic chemistry and homogeneous catalysis. Chem. Rev. 77, 313–348. doi: 10.1021/cr60307a002
Ungur, L., and Chibotaru, L. F. (2011). Magnetic anisotropy in the excited states of low symmetry lanthanide complexes. Phys. Chem. Chem. Phys. 13, 20086–20090. doi: 10.1039/C1CP22689D
Woodruff, D. N., Winpenny, R. E. P., and Layfield, R. A. (2013). Lanthanide single-molecule magnets. Chem. Rev. 113, 5110–5148. doi: 10.1021/cr400018q
Keywords: terbium, dysprosium, triphenylarsine oxide, phosphine oxide, single ion magnet
Citation: Fondo M, Corredoira-Vázquez J, García-Deibe AM, Sanmartín-Matalobos J, Herrera JM and Colacio E (2018) Field-Induced Single Molecule Magnets of Phosphine- and Arsine-Oxides. Front. Chem. 6:420. doi: 10.3389/fchem.2018.00420
Received: 29 June 2018; Accepted: 24 August 2018;
Published: 12 September 2018.
Edited by:
Carlos Lodeiro, Universidade Nova de Lisboa, PortugalReviewed by:
Guo-Hong Tao, Sichuan University, ChinaSukhendu Mandal, Indian Institute of Science Education and Research, Thiruvananthapuram, India
Copyright © 2018 Fondo, Corredoira-Vázquez, García-Deibe, Sanmartín-Matalobos, Herrera and Colacio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matilde Fondo, matilde.fondo@usc.es