Targeting the NLRP3 inflammasome and associated cytokines in scleroderma associated interstitial lung disease
 Samuel Woo
Samuel Woo Shifa Gandhi
Shifa Gandhi  Changwan Ryu
Changwan Ryu- Department of Internal Medicine, Yale School of Medicine, Section of Pulmonary, Critical Care and Sleep Medicine, New Haven, CT, United States
SSc-ILD (scleroderma associated interstitial lung disease) is a complex rheumatic disease characterized in part by immune dysregulation leading to the progressive fibrotic replacement of normal lung architecture. Because improved treatment options are sorely needed, additional study of the fibroproliferative mechanisms mediating this disease has the potential to accelerate development of novel therapies. The contribution of innate immunity is an emerging area of investigation in SSc-ILD as recent work has demonstrated the mechanistic and clinical significance of the NLRP3 inflammasome and its associated cytokines of TNFα (tumor necrosis factor alpha), IL-1β (interleukin-1 beta), and IL-18 in this disease. In this review, we will highlight novel pathophysiologic insights afforded by these studies and the potential of leveraging this complex biology for clinical benefit.
Introduction
SSc (scleroderma) is a complex rheumatic disease characterized by diffuse microvascular injury and immune dysregulation leading to the progressive fibrotic replacement of normal tissue architecture (Denton and Khanna, 2017). One of the leading causes of morbidity and mortality of this condition is the development of ILD (interstitial lung disease) (Volkmann and Fischer, 2021), where current therapeutic strategies include non-specific immunosuppressive drugs (Denton and Khanna, 2017) and anti-fibrotic agents (Distler et al., 2019) that are associated with heterogenous efficacy, high costs, and at times, intolerable side-effects (Rahaghi et al., 2023). Because better treatment options are sorely needed, additional study of the fibroproliferative mechanisms mediating this disease has the potential to accelerate development of novel therapies.
While the mechanisms by which autoimmunity engenders pathologic remodeling of the SSc lung remains poorly understood, aberrant adaptive immune responses and fibroblast activation are heavily implicated (Herzog et al., 2014). Although the contribution of innate immunity is less known in this context, its role in fibrogenesis has been an emerging area of investigation (Taroni et al., 2017). Innate immunity is activated by PRRs (pattern recognition receptors), which senses epitopes broadly conserved across pathogenic microbes, known as PAMPs (pathogen-associated molecular patterns), and endogenous ligands, known as DAMPs (danger-associated molecular patterns), that are released by stressed or activated cells and tissues (Ellson et al., 2014). When sensing excessive amounts of PAMPs or DAMPs, PRRs mediate activation of downstream inflammatory pathways (Li and Wu, 2021), including assembly of multimeric protein complexes known as inflammasomes, a caspase-1 activating platform (Franchi et al., 2009). While several PRRs are known to trigger inflammasome formation (Franchi et al., 2009; Kelley et al., 2019), the fibroproliferative role of the NLRP3 (NOD [nucleotide-binding oligomerization domain], LRR [leucine-rich repeat] containing proteins, and PYD-3 [pyrin domain-containing protein-3]) inflammasome has been of significant interest in SSc-ILD. Thus, the purpose of our review is to highlight recent work characterizing the mechanistic and clinical relevance of the NLRP3 inflammasome and its associated cytokines in this disease.
NLRP3 inflammasome
Initially described in 2002, NLRP3 is a sensor protein that along with the adaptor protein ASC (apoptosis-related spot-like protein containing CARD [carboxy-terminal caspase recruitment domain]) and the effector protein caspase-1 comprises the NLRP3 inflammasome (Swanson et al., 2019; Yang et al., 2019). Its activation first requires a priming step mediated by NF-κB (nuclear factor kappa light chain enhancer of activated B cells) signaling, which occurs via PAMP or DAMP activation of TLRs (Toll-like receptors) or NOD2 (nucleotide binding oligomerization domain containing 2), or through soluble mediators such as TNFα (tumor necrosis factor alpha) and IL-1β (interleukin-1 beta) (Swanson et al., 2019; Gritsenko et al., 2020). Priming serves to (1) upregulate transcription of inflammasome components of NLRP3, pro-IL-18, and pro-IL-1β, and (2) induce post-translational modifications of NLRP3 to stabilize its function (Swanson et al., 2019; Gritsenko et al., 2020). Following this step, NLRP3 forms oligomers through homotypic interactions via NACHT domains (NAIP [neuronal apoptosis inhibitor protein], CIITA [MHC class II transcription activator], HET-E [incompatibility locus protein from Podospora anserina] and TP1 [telomerase-associated protein]) (Damiano et al., 2004), which recruits ASC proteins via homotypic PYD–PYD interactions to nucleate helical ASC filament formation (Swanson et al., 2019). Multiple ASC filaments coalesce into a single macromolecule, known as an ASC speck, to recruit caspase-1 through CARD–CARD interactions to facilitate caspase-1 self-cleavage and activation (Swanson et al., 2019; Yang et al., 2019). Caspase-1 cleaves and activates IL-1β, IL-18 (Swanson et al., 2019; Li et al., 2020), and the membrane pore-forming protein GSDMD (gasdermin-D), where its N-terminal domain (GSDMDNterm) oligomerizes to create a cell membrane pore (Shi et al., 2015; Swanson et al., 2019). Though the exact mechanism of NLRP3 inflammasome activation remains elusive, many sources suggest that common second-messenger pathways link the multiple upstream signals to inflammasome activation, including potassium efflux, decrease in intracellular calcium, lysosomal disruption, mitochondrial dysfunction, and reactive oxygen species (Artlett and Thacker, 2015; Elliott and Sutterwala, 2015; Man and Kanneganti, 2015; He et al., 2016). Given the inflammatory properties inherent with NLRP3 inflammasome activation, this pathway has been interrogated in autoimmune conditions such as SSc-ILD.
Aberrant activation of the NLRP3 inflammasome has garnered significant interest for its potential pathogenic role in inflammatory conditions such as SSc-ILD. Genome-wide association studies have shown polymorphisms in the NLRP3 gene to be linked with the development of ILDs such as asbestosis (rs35829419) (Franko et al., 2020), coal workers pneumoconiosis (rs1539019) (Ji et al., 2012), and silicosis (rs1539019 and rs34298354) (Weng et al., 2015); while specific NLRP3 polymorphisms have yet to be identified in SSc-ILD, further study in this arena could identify convergent molecular mechanisms linking divergent disease states. However, translational work completed by Artlett and colleagues demonstrated the connection between NLRP3 inflammasome activation and SSc by showing that dermal fibroblasts from SSc patients exhibit increased expression of inflammasome components, and that experimentally induced caspase-1 inhibition of both dermal and lung SSc fibroblasts ameliorated collagen deposition, reduced IL-1β and IL-18 secretion, and decreased αSMA (alpha smooth muscle actin) expression (Artlett et al., 2011). Additionally, studies investigating the pathogenic contribution of chronic parvovirus B19 (B19V) infection in SSc revealed that monocytes derived from SSc patients express significantly elevated mRNA levels of NLRP3 than monocytes derived from healthy control subjects in the presence of B19V infection (Zakrzewska et al., 2019). Furthermore, mice deficient in NLRP3 (−/−) and ASC (−/−) were shown to be resistant to BLM (bleomycin) induced skin and lung fibrosis (Artlett et al., 2011). Additional work validated the finding of NLRP3 overexpression in SSc, including the association between skin thickness and NLRP3 expression (Martinez-Godinez et al., 2015). More recently, it was shown that miR-155 (microRNA-155) expression is indispensable for NLRP3 inflammasome mediated collagen production in SSc dermal and lung fibroblasts (Artlett et al., 2017; Henderson and O'Reilly, 2017). NLRP3−/− fibroblasts and those treated with caspase-1 inhibition had significantly decreased miR-155 expression and collagen synthesis, leading the authors to conclude that the NLRP3 inflammasome is required for miR-155 expression (Artlett et al., 2017; Henderson and O'Reilly, 2017). These discoveries not only advance our understanding of the pathophysiologic importance of the NLRP3 inflammasome in SSc, but also lend scientific rationale for targeting its inhibition.
Early preclinical studies targeting the NLRP3 inflammasome identified the sulfonylurea glyburide as a potential inhibiting agent as in vitro studies showed a reduction in caspase-1 activation and IL-1β and IL-18 secretion in ATP-treated, lipopolysaccharide-sensitized macrophages (Lamkanfi et al., 2009). MCC950 (also known as CRID-3 or CP-456,773) is a disulfonylurea compound similar to glyburide that has been shown to specifically bind to NLRP3 and prevent inflammasome activation, interrupting IL-1β, IL-1α, and IL-18 secretion in a myriad of preclinical work (Coll et al., 2015; Primiano et al., 2016; van der Heijden et al., 2017; van Hout et al., 2017; Perera et al., 2018; Coll et al., 2019; Tapia-Abellan et al., 2019; Vande Walle et al., 2019; Corcoran et al., 2021). While clinical evaluation of MCC950 led to its discontinuation due to hepatotoxicity (Chen et al., 2021), these early efforts provided the scientific premise for other small-molecule NLRP3 inhibitors that are in various phases of clinical trials, including dapansutrile, a specific NLRP3 inhibitor (Marchetti et al., 2018; Sanchez-Fernandez et al., 2019) that has been shown to be safe in heart failure (Wohlford et al., 2020) and gout (Kluck et al., 2020). Other small molecule NLRP3 inhibitors under investigation include IFM2427, inzomelid, somalix, IZD334, and NT-0167 (El-Sharkawy et al., 2020; Chen et al., 2021); their relevance and benefit to SSc-ILD will require additional translational and clinical evaluation.
Along with direct antagonism of the NLRP3 inflammasome, targeting soluble mediators related to this pathway has also been an area of active study. As stated above, TNFα and IL-1β mediate priming of the inflammasome, and its activation result in the production of IL-1β and IL-18. Moreover, integrative analysis of RNA sequencing studies of peripheral blood and lungs from SSc patients has further demonstrated the pathophysiologic significance of these cytokines (Kobayashi et al., 2021). Single cell RNA sequencing of lung tissue from SSc-ILD subjects revealed a subpopulation of monocytes (termed FCN1hi) that highly expressed, among others, genes related to TNF, IL1B, and IL1R2 (Valenzi et al., 2019; Kobayashi et al., 2021). Thus, in the next section of this review, we will discuss the fibroproliferative contribution of TNFα, IL-1β, and IL-18 in SSc-ILD.
Tumor necrosis factor alpha
TNFα is a pleiotropic pro-inflammatory cytokine produced by various stromal and immune cells that is initially expressed as a transmembrane precursor protein and undergoes cleavage by TACE (TNFα-converting enzyme) to release soluble TNFα (Black et al., 1997; Moha et al., 2002). Both transmembrane and soluble TNFα binds to TNFR1 (TNF receptor 1), while only its transmembrane form is able to recognize TNFR2 (TNF receptor 2) (Vandenabeele et al., 1995). TNFR1 is ubiquitously expressed and contains a conserved death domain that facilitates recruitment of the adaptor protein TRADD (TNFR1-associated death domain), triggering activation of four potential signaling complexes (Hsu et al., 1995). Complex I forms when TNF binds to TNFR1, leading to a conformation change in its cytoplasmic domain, leading to recruitment of key mediators that include TRADD, RIPK1 (receptor-interacting serine/threonine-protein kinase 1), TRAF2 (TNFR-associated factor 2), cIAP1/2 (cellular inhibitor of apoptosis protein 1 or 2), and LUBAC (linear ubiquitin chain assembly complex); these interactions mediate downstream events critical for canonical NF-κB and MAPK (mitogen-activated protein kinases) signaling that promote tissue and cell inflammation, survival, and proliferation (Baud and Karin, 2001; Micheau and Tschopp, 2003; Brenner et al., 2015). The formation of complexes IIa and IIb (known as apoptosomes) also involve TNF-TNFR1-TRADD-RIPK1 interactions, but also include recruitment of FADD (Fas-associated protein with death domain) and pro-caspase 8 to induce cytoplasmic apoptotic signaling; complex IIb also requires activation of RIPK3 (Cain et al., 1999; Wang et al., 2008; Brenner et al., 2015). Complex IIc (known as a necrosome) requires TNF-TNFR1-TRADD-RIPK1-RIPK3 interactions that mediate activation of MLKL (mixed lineage kinase domain-like protein) to initiate cellular necroptosis (Cho et al., 2009; Brenner et al., 2015). Meanwhile, TNFR2, expressed exclusively by immune and endothelial cells, lacks the death domain present in TNFR1, and alternatively recruits TRAF1 and TRAF2 to form Complex 1, leading to activation of NF-κB and MAPK pathways (Faustman and Davis, 2010; Brenner et al., 2015). Not surprisingly, TNFα signaling has been extensively evaluated in inflammatory conditions such as SSc-ILD.
Early translational studies with this cytokine demonstrated its critical role in experimental models of pulmonary fibrosis as TNFα was shown to modulate expression of TGFβ in various cells in the lungs (Warshamana et al., 2001; Sullivan et al., 2005; Sullivan et al., 2009). In elegant work completed by Sullivan and colleagues, they showed that both transcriptional and post-transcriptional modifications in TGFβ expression in mouse lung fibroblasts are induced by TNFα (Sullivan et al., 2005; Sullivan et al., 2009). In rodent models of lung fibrosis, mice exposed to BLM displayed increased expression of TNFα that was associated with TGFβ levels (Ortiz et al., 1998; Brass et al., 1999; Hou et al., 2018); moreover, adenoviral mediated TNFα overexpression in otherwise normal rat lungs resulted in upregulation of TGFβ1 and accumulation of αSMA expressing myofibroblasts (Sime et al., 1998). Additionally, in various murine models, genetic (Liu et al., 1998) and pharmacologic (Phan and Kunkel, 1992; Piguet et al., 1993) knockdown of TNFα and its receptor ameliorated chemically-induced lung fibrosis in a TGFβ dependent manner, suggesting a synergistic association between TNFα and TGFβ-mediated fibrosis. In a direct connection to the SSc disease state, high levels of TNFα have been detected in the BAL (bronchoalveolar lavage) (Bolster et al., 1997; Pantelidis et al., 2001) of SSc-ILD patients. Moreover, relative to alveolar macrophages (AMs) derived from SSc patients without pulmonary fibrosis, AMs from fibrotic SSc lungs displayed enhanced secretion of this cytokine (Pantelidis et al., 2001). However, other reports implicate an antagonistic relationship as chronic overexpression of TNFα not only protected mice from BLM-induced lung fibrosis (Fujita et al., 2003), but also accelerated resolution of this pathology through a reduction in profibrotic lung macrophages (Redente et al., 2014). This constellation of findings demonstrates a paradigm wherein TNFα exerts pathogenic or protective roles in pulmonary fibrosis that depends on cell-specific and temporal cues that would benefit from further evaluation.
Despite these findings, TNFα antagonism has been clinically therapeutic in inflammatory diseases such as RA (rheumatoid arthritis), ankylosing spondylitis, inflammatory bowel disease, and psoriasis, as well as various off-label indications (Haraoui, 2005; Jang et al., 2021). Not unexpectedly, this treatment strategy has been associated with exacerbating underlying ILD or resulting in de novo pneumonitis (Tengstrand et al., 2005; Andrew et al., 2006; Perez-Alvarez et al., 2011; Tamao et al., 2014), which has tempered enthusiasm for this agent in SSc-ILD. However, given that the blood of SSc patients is enriched for this cytokine (Pehlivan et al., 2012), particularly among those with lung disease (Murdaca et al., 2014), as well as the availability of several FDA-approved TNFα inhibitors, the indication for these agents in SSc-ILD have been explored. In an open-label pilot trial for diffuse cutaneous SSc, infliximab, a recombinant chimeric mouse/human monoclonal antibody, showed no significant improvement in MRSS (modified Rodnan skin scores); while there was potential for stabilization of skin disease, a high number of transfusion reactions limited use of this agent in this population (Denton et al., 2009). The role of infliximab remains unknown in SSc-ILD as there were no reports of its effect on lung function (Bosello et al., 2005; Denton et al., 2009); however, studies in sarcoidosis, another multi-system ILD, suggest that it has the potential to improve lung function (Baughman et al., 2006) and ameliorate multi-organ disease (Russell et al., 2013). Etanercept, a human TNF-receptor p75 Fc fusion protein that binds TNFα, showed promise in decreasing SSc associated inflammatory synovitis and was safely tolerated (Gordon et al., 2007). Although its role in SSc-ILD is largely unknown, in the same study, there was a no significant change in lung function (Gordon et al., 2007), and studies with this agent in sarcoidosis also demonstrated no change in lung function (Utz et al., 2003). Other FDA approved TNFα monoclonal antibodies include adalimumab and golimumab (Haraoui, 2005; Jang et al., 2021). While these agents have yet to be studied in SSc, they have been evaluated in sarcoidosis, where adalimumab has shown promise in improving lung function (Kamphuis et al., 2011; Milman et al., 2012; Sweiss et al., 2014), while golimumab demonstrated no benefit (Judson et al., 2014). Thus, despite the inflammatory nature of SSc-ILD, antagonism of TNFα signaling has demonstrated mixed results.
Interleukin-1 beta
IL-1β is a cytokine mainly produced by myeloid cells in its inactive form, pro-IL-1β (Dinarello, 2011). It is cleaved intracellularly by caspase 1, and extracellularly by serine proteases that result in its activation (Lamkanfi, 2011; Bode et al., 2012). IL-1β binds to its receptor, IL-1R1, which heterodimerizes with the accessory protein IL-1RAcP (IL-1 receptor accessory protein) to transmit signaling via intracellular activation of adaptor proteins such as MyD88 (myeloid differentiation primary response gene-88), leading to the production of NF-κB and MAPK (Dinarello, 2009; Van Gorp et al., 2019; Chauhan et al., 2020). IL-1β signaling results in the production of additional pro-inflammatory cytokines and chemokines such as TNFα, IL-6, IL-8, MCP1 (monocyte chemoattractant protein 1), CXCL1 (chemokine ligand 1), and MIP2 (macrophage inflammatory protein 2) (Chauhan et al., 2020). IL-1β signaling also drives amplification and polarization of CD4+ T-cells toward a Th1 and Th17 phenotype and promotes differentiation of antigen-specific CD8+ T-cells (Ben-Sasson et al., 2009; Santarlasci et al., 2013; Chauhan et al., 2020). As an effector of both innate and adaptive immune responses, the study of IL-1β can lend novel immunopathogenic insight into inflammatory conditions such as SSc-ILD.
Somewhat surprisingly, studies of IL-1β signaling have led to conflicting findings. Initial translational efforts found that excessive concentrations of IL-1β were present in the lungs, blood, and skin of SSc patients (Hussein et al., 2005). In experimental models of lung fibrosis, mice treated with BLM displayed increased IL-1β production, inflammation, remodeling, and fibrosis in a manner dependent on IL-1R1/MyD88 signaling (Gasse et al., 2007). These findings were significantly reduced in IL-1R1-and MyD88-deficient mice, suggesting the indispensable nature of endogenous IL-1β in pulmonary inflammation and fibrosis (Gasse et al., 2007). However, these findings are tempered by recent reports showing that IL-1 receptor inhibition can exacerbate pulmonary fibrosis; employing a murine model of SSc through the overexpression of Fra-2 (fos-related antigen-2), mice treated with an IL-1 inhibitor demonstrated worse lung function, enhanced Th2 inflammation, and greater numbers of pro-fibrotic, alternatively activated macrophages (Birnhuber et al., 2019). Similar to that of TNFα, these data demonstrate the context-dependent nature of IL-1β that warrants additional investigation.
Nonetheless, re-purposing FDA-approved IL-1β antagonists has been considered for this and other autoimmune diseases. Anakinra is a recombinant human IL-1Ra that competitively inhibits IL-1α and IL-1β interaction with IL-1R1 and is currently indicated for the treatment of refractory RA and for CAPS (Cryopyrin Associated Periodic Syndrome) (Cavalli and Dinarello, 2015). Its safety and efficacy have yet to be explored in SSc. Canakinumab is a human monoclonal anti-IL-1β antibody that has demonstrated efficacy in treating multiple autoimmune conditions (De Benedetti et al., 2018) as well atherosclerotic diseases in the CANTOS Trial (Ridker et al., 2012; Ridker et al., 2017a). Interestingly, additional analysis of the CANTOS cohort revealed a significant reduction in incident lung cancer and lung cancer mortality (Ridker et al., 2017b); these findings suggest that this agent is able to modulate processes in the lung, making it a promising candidate for treating SSc-ILD. Lastly, rilonacept is a soluble IL-1 trap that binds IL-1α and IL-1β; it has been approved for the treatment of CAPS (Hoffman et al., 2012) and has demonstrated safety and efficacy in other inflammatory disorders (Krause et al., 2012). Moreover, in a small cohort of SSc patients (n = 19), this drug, while demonstrating an acceptable safety and tolerability profile, did not demonstrate biologic (based on gene expression) or clinical (based on MRSS) efficacy (Manter et al., 2018). Ultimately, further work is required to determine the clinical benefit of IL-1β inhibition in SSc-ILD.
Interleukin-18
Along with IL-1β, NLRP3 inflammasome activation mediates production of IL-18, formerly known as IFN-γ (interferon-gamma) inducing factor. This cytokine is produced as pro-IL-18 by a number of cells, including macrophages and dendritic cells, and as with IL-1β, requires cleavage by caspase-1 to become biologically active (Arend et al., 2008; Dinarello et al., 2013). The IL-18 receptor, comprised of IL-18Rα and IL-18Rβ subunits, forms a high-affinity heterodimer expressed on various immune, endothelial, and smooth muscle cells, and is modulated by various other cytokines (Arend et al., 2008). Similar to the IL-1 receptor, IL-18 signal transduction proceeds via several intracellular adaptor molecules, including MyD88, IRAK1 (interleukin 1 receptor associated kinase 1), and TRAF6 to activate MAPK, NF-κB, and JNK (c-Jun N-terminal kinase) (Arend et al., 2008; Dinarello et al., 2013; Xu et al., 2019; Yasuda et al., 2019). IL-18 stimulates production of GM-CSF (granulocyte-monocyte colony-stimulating factor), TNFα, and IL-1β, and mediates both innate and adaptive immune responses (Arend et al., 2008). In the presence of IL-12 and IL-15, IL-18 can induce IFN-γ production by various T-cells and favors Th1 differentiation, a pro-inflammatory state; in the absence of IL-12 and IL-15, IL-18 signaling favors Th2 differentiation and promotes fibrosis (Dinarello et al., 2013; Kaplanski, 2018). Much like IL-1β, IL-18 has pleotropic effects in immune activation, suggesting a potential contribution in inflammatory conditions such as SSc-ILD.
Similar to work done with TNFα and IL-1β, studies of IL-18 have also led to contradictory results in SSc-ILD. In the serum of patients with SSc, increased concentrations of IL-18 were identified, and these levels negatively correlated with lung function, suggesting a role in mediating lung disease (Lin et al., 2019). However, both profibrotic and anti-fibrotic properties of IL-18 have been illustrated. In experimentally-induced pulmonary fibrosis, mice subjected to BLM administration exhibited increased concentrations of IL-18 in the lung (Oku et al., 2008). Additionally, novel work investigating the myeloid-specific contribution of autophagy in lung injury showed that mice deficient in the key autophagy gene Atg7 develop spontaneous lung inflammation that was predominantly mediated by IL-18 from constitutive inflammasome activation (Abdel Fattah et al., 2015). Here, the authors showed that IL-18 antagonism inhibited the recruitment of lymphocytes and neutrophils in the lungs of these mice, suggesting a critical role for myeloid-specific IL-18 (Abdel Fattah et al., 2015). However, another study showed that mice deficient in IL-18 exhibited greater lung injury and weight loss in response to BLM administration (Nakatani-Okuda et al., 2005). In that same study, while therapeutic administration of IL-18 had no effect on BLM-induced lung injury, prophylactic IL-18 treatment prior to BLM administration ameliorated findings of lung injury (Nakatani-Okuda et al., 2005). The pleotropic effects of IL-18 altering the balance between Th1 and Th2 responses is likely contributing to this conundrum (Kim et al., 2010), and further study is sorely needed to unravel this complex biology.
Despite these conflicting in vivo data, IL-18 inhibition is currently under investigation for therapeutic benefit. Tadekinig alfa, a recombinant human IL-18 binding protein, was shown to be effective in treating adult onset Still’s disease (Gabay et al., 2018; Kiltz et al., 2020) and NLRC4 associated macrophage activation syndrome (Canna et al., 2017). GSK1070806, a monoclonal IL-18 antibody, was shown to be safely tolerated (Mistry et al., 2014) and is currently being evaluated in a phase 1b clinical trial for atopic dermatitis (NCT04975438) and phase 2 clinical trial for moderate to severe Crohn’s disease (NCT03681067). Additional work will be needed to determine whether IL-18 is promoting disease or protecting the lung in SSc-ILD.
Conclusion
This concludes our review of NLRP3 inflammasome signaling and associated cytokines of TNFα, IL-1β, and IL-18 as novel therapeutic targets in SSc-ILD; these findings are summarized in Table 1 and depicted in Figure 1. Study of this complex biology requires further investigation to fully characterize the context-dependent roles of this signaling pathway and soluble mediators in protecting against or promoting pulmonary fibrosis in SSc-ILD. Addressing such knowledge gaps require improved modeling systems that better recapitulate human disease; rodent models exhibit relatively swift progression of lung fibrosis, fail to reproduce lung histopathology, and resolve fibrosis with discontinuation of the pro-fibrotic agent (Herzog et al., 2014). Thus, while animal modeling is viewed as a useful tool for the in vivo study of lung disease that would be unethical and impractical in humans, their value is limited. Work complemented by studies of primary human cells and tissues, as well as functional studies of explanted cells and organs, have been instrumental in replicating the salient features of the healthy and diseased adult mammalian lung (Herzog et al., 2014). Translational studies integrating traditional in vivo models with state-of-the-art ex vivo mimetics will have more direct relevance to the human disease state. Such work has the potential to yield insight into novel pathophysiologic mechanisms while catalyzing new treatment approaches, including repurposing old and developing new drugs, in this intractable condition.

TABLE 1. Antagonists and their mechanism of action.
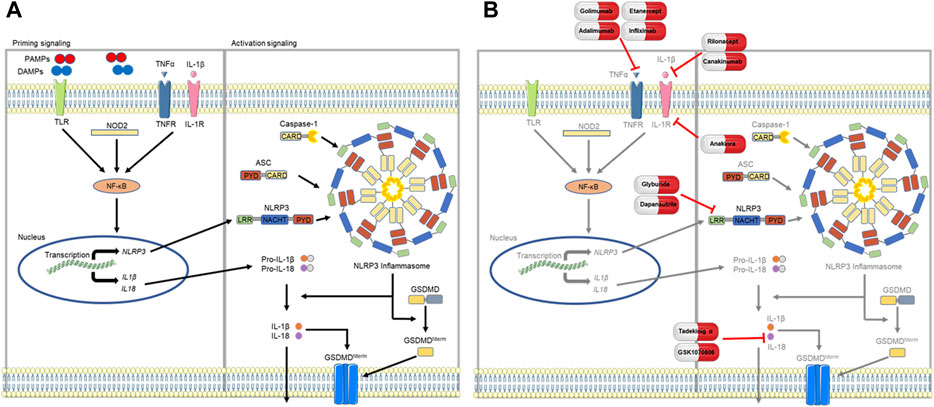
FIGURE 1. Proposed model of NLRP3 inflammasome signaling. (A) Assembly of the NLRP3 inflammasome first requires a priming step that is mediated by NF-κB (nuclear factor kappa light chain enhancer of activated B cells) signaling, which occurs via PAMP (pathogen associated molecular pattern) or DAMP (damage associated molecular pattern) activation of TLRs (toll-like receptors) or NOD2 (nucleotide binding oligomerization domain containing 2), or through soluble mediators such as TNFα (tumor necrosis factor alpha) and IL-1β (interleukin-1 beta). Priming upregulates transcription of inflammasome components of NLRP3, pro-IL-18, and pro-IL-1β. Following this step, components of the NLRP3 inflammasome coalesce to form the ASC speck and subsequently the inflammasome, activating caspase-1 to cleave pro-IL-1β and pro-IL-18 into their respective products, IL-1β and IL-18. (B) Antagonism of the NLRP3 inflammasome or mediating cytokines (TNFα, IL-1β, and IL-18) have shown potential as novel therapeutic targets in SSc-ILD. Parts of the figure were drawn by using pictures from Servier Medical Art. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/).
Author contributions
SW: Writing–original draft, Writing–review and editing. SG: Writing–review and editing, Conceptualization, Supervision. AG: Writing–original draft, Writing–review and editing. TS: Conceptualization, Supervision, Writing–review and editing. CL: Writing–review and editing, Writing–original draft. CR: Conceptualization, Supervision, Writing–review and editing, Funding acquisition, Visualization, Writing–original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. SG was supported by T32HL007778-21. AG was supported by T32HL007778-21. CR was supported by K08HL151970-01 and the Cystic Fibrosis Foundation.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abdel Fattah, E., Bhattacharya, A., Herron, A., Safdar, Z., and Eissa, N. T. (2015). Critical role for IL-18 in spontaneous lung inflammation caused by autophagy deficiency. J. Immunol. 194 (11), 5407–5416. doi:10.4049/jimmunol.1402277
Andrew, J. K. O., Chilvers, E. R., Somerville, M. F., Lim, A. Y. N., Lane, S. E., Crisp, A. J., et al. (2006). Pulmonary complications of infliximab therapy in patients with rheumatoid arthritis. J. Rheumatology 33 (3), 622–628.
Arend, W. P., Palmer, G., and Gabay, C. (2008). IL-1, IL-18, and IL-33 families of cytokines. Immunol. Rev. 223, 20–38. doi:10.1111/j.1600-065X.2008.00624.x
Artlett, C. M., Sassi-Gaha, S., Hope, J. L., Feghali-Bostwick, C. A., and Katsikis, P. D. (2017). Mir-155 is overexpressed in systemic sclerosis fibroblasts and is required for NLRP3 inflammasome-mediated collagen synthesis during fibrosis. Arthritis Res. Ther. 19 (1), 144. doi:10.1186/s13075-017-1331-z
Artlett, C. M., Sassi-Gaha, S., Rieger, J. L., Boesteanu, A. C., Feghali-Bostwick, C. A., and Katsikis, P. D. (2011). The inflammasome activating caspase 1 mediates fibrosis and myofibroblast differentiation in systemic sclerosis. Arthritis Rheum. 63 (11), 3563–3574. doi:10.1002/art.30568
Artlett, C. M., and Thacker, J. D. (2015). Molecular activation of the NLRP3 inflammasome in fibrosis: common threads linking divergent fibrogenic diseases. Antioxid. Redox Signal 22 (13), 1162–1175. doi:10.1089/ars.2014.6148
Baud, V., and Karin, M. (2001). Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 11 (9), 372–377. doi:10.1016/s0962-8924(01)02064-5
Baughman, R. P., Drent, M., Kavuru, M., Judson, M. A., Costabel, U., du Bois, R., et al. (2006). Infliximab therapy in patients with chronic sarcoidosis and pulmonary involvement. Am. J. Respir. Crit. Care Med. 174 (7), 795–802. doi:10.1164/rccm.200603-402OC
Ben-Sasson, S. Z., Hu-Li, J., Quiel, J., Cauchetaux, S., Ratner, M., Shapira, I., et al. (2009). IL-1 acts directly on CD4 T cells to enhance their antigen-driven expansion and differentiation. Proc. Natl. Acad. Sci. U. S. A. 106 (17), 7119–7124. doi:10.1073/pnas.0902745106
Birnhuber, A., Crnkovic, S., Biasin, V., Marsh, L. M., Odler, B., Sahu-Osen, A., et al. (2019). IL-1 receptor blockade skews inflammation towards Th2 in a mouse model of systemic sclerosis. Eur. Respir. J. 54 (3), 1900154. doi:10.1183/13993003.00154-2019
Black, R. A., Rauch, C. T., Kozlosky, C. J., Peschon, J. J., Slack, J. L., Wolfson, M. F., et al. (1997). A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 385 (6618), 729–733. doi:10.1038/385729a0
Bode, J. G., Albrecht, U., Häussinger, D., Heinrich, P. C., and Schaper, F. (2012). Hepatic acute phase proteins-regulation by IL-6- and IL-1-type cytokines involving STAT3 and its crosstalk with NF-κB-dependent signaling. Eur. J. Cell Biol. 91 (6-7), 496–505. doi:10.1016/j.ejcb.2011.09.008
Bolster, M. B., Ludwicka, A., Sutherland, S. E., Strange, C., and Silver, R. M. (1997). Cytokine concentrations in bronchoalveolar lavage fluid of patients with systemic sclerosis. Arthritis & Rheumatism 40 (4), 743–751. doi:10.1002/art.1780400422
Bosello, S., De Santis, M., Tolusso, B., Zoli, A., and Ferraccioli, G. (2005). Tumor necrosis factor-alpha inhibitor therapy in erosive polyarthritis secondary to systemic sclerosis. Ann. Intern. Med. 143 (12), 918–920. doi:10.7326/0003-4819-143-12-200512200-00019
Brass, D. M., Hoyle, G. W., Poovey, H. G., Liu, J. Y., and Brody, A. R. (1999). Reduced tumor necrosis factor-alpha and transforming growth factor-beta1 expression in the lungs of inbred mice that fail to develop fibroproliferative lesions consequent to asbestos exposure. Am. J. Pathol. 154 (3), 853–862. doi:10.1016/s0002-9440(10)65332-1
Brenner, D., Blaser, H., and Mak, T. W. (2015). Regulation of tumour necrosis factor signalling: live or let die. Nat. Rev. Immunol. 15 (6), 362–374. doi:10.1038/nri3834
Cain, K., Brown, D. G., Langlais, C., and Cohen, G. M. (1999). Caspase activation involves the formation of the aposome, a large (approximately 700 kDa) caspase-activating complex. J. Biol. Chem. 274 (32), 22686–22692. doi:10.1074/jbc.274.32.22686
Canna, S. W., Girard, C., Malle, L., de Jesus, A., Romberg, N., Kelsen, J., et al. (2017). Life-threatening NLRC4-associated hyperinflammation successfully treated with IL-18 inhibition. J. Allergy Clin. Immunol. 139 (5), 1698–1701. doi:10.1016/j.jaci.2016.10.022
Cavalli, G., and Dinarello, C. A. (2015). Treating rheumatological diseases and co-morbidities with interleukin-1 blocking therapies. Rheumatol. Oxf. 54 (12), 2134–2144. doi:10.1093/rheumatology/kev269
Chauhan, D., Vande Walle, L., and Lamkanfi, M. (2020). Therapeutic modulation of inflammasome pathways. Immunol. Rev. 297 (1), 123–138. doi:10.1111/imr.12908
Chen, Q. L., Yin, H. R., He, Q. Y., and Wang, Y. (2021). Targeting the NLRP3 inflammasome as new therapeutic avenue for inflammatory bowel disease. Biomed. Pharmacother. 138, 111442. doi:10.1016/j.biopha.2021.111442
Cho, Y. S., Challa, S., Moquin, D., Genga, R., Ray, T. D., Guildford, M., et al. (2009). Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137 (6), 1112–1123. doi:10.1016/j.cell.2009.05.037
Coll, R. C., Hill, J. R., Day, C. J., Zamoshnikova, A., Boucher, D., Massey, N. L., et al. (2019). MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat. Chem. Biol. 15 (6), 556–559. doi:10.1038/s41589-019-0277-7
Coll, R. C., Robertson, A. A. B., Chae, J. J., Higgins, S. C., Muñoz-Planillo, R., Inserra, M. C., et al. (2015). A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 21 (3), 248–255. doi:10.1038/nm.3806
Corcoran, S. E., Halai, R., and Cooper, M. A. (2021). Pharmacological inhibition of the nod-like receptor family pyrin domain containing 3 inflammasome with MCC950. Pharmacol. Rev. 73 (3), 968–1000. doi:10.1124/pharmrev.120.000171
Damiano, J. S., Oliveira, V., Welsh, K., and Reed, J. C. (2004). Heterotypic interactions among NACHT domains: implications for regulation of innate immune responses. Biochem. J. 381 (1), 213–219. doi:10.1042/BJ20031506
De Benedetti, F., Gattorno, M., Anton, J., Ben-Chetrit, E., Frenkel, J., Hoffman, H. M., et al. (2018). Canakinumab for the treatment of autoinflammatory recurrent fever syndromes. N. Engl. J. Med. 378 (20), 1908–1919. doi:10.1056/NEJMoa1706314
Denton, C. P., Engelhart, M., Tvede, N., Wilson, H., Khan, K., Shiwen, X., et al. (2009). An open-label pilot study of infliximab therapy in diffuse cutaneous systemic sclerosis. Ann. Rheumatic Dis. 68 (9), 1433–1439. doi:10.1136/ard.2008.096123
Denton, C. P., and Khanna, D. (2017). Systemic sclerosis. Lancet 390 (10103), 1685–1699. doi:10.1016/S0140-6736(17)30933-9
Dinarello, C. A. (2011). A clinical perspective of IL-1β as the gatekeeper of inflammation. Eur. J. Immunol. 41 (5), 1203–1217. doi:10.1002/eji.201141550
Dinarello, C. A. (2009). Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 27, 519–550. doi:10.1146/annurev.immunol.021908.132612
Dinarello, C. A., Novick, D., Kim, S., and Kaplanski, G. (2013). Interleukin-18 and IL-18 binding protein. Front. Immunol. 4, 289. doi:10.3389/fimmu.2013.00289
Distler, O., Highland, K. B., Gahlemann, M., Azuma, A., Fischer, A., Mayes, M. D., et al. (2019). Nintedanib for systemic sclerosis-associated interstitial lung disease. N. Engl. J. Med. 380 (26), 2518–2528. doi:10.1056/NEJMoa1903076
El-Sharkawy, L. Y., Brough, D., and Freeman, S. (2020). Inhibiting the NLRP3 inflammasome. Molecules 25 (23), 5533. doi:10.3390/molecules25235533
Elliott, E. I., and Sutterwala, F. S. (2015). Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol. Rev. 265 (1), 35–52. doi:10.1111/imr.12286
Ellson, C. D., Dunmore, R., Hogaboam, C. M., Sleeman, M. A., and Murray, L. A. (2014). Danger-associated molecular patterns and danger signals in idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 51 (2), 163–168. doi:10.1165/rcmb.2013-0366TR
Faustman, D., and Davis, M. (2010). TNF receptor 2 pathway: drug target for autoimmune diseases. Nat. Rev. Drug Discov. 9 (6), 482–493. doi:10.1038/nrd3030
Franchi, L., Eigenbrod, T., Muñoz-Planillo, R., and Nuñez, G. (2009). The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 10 (3), 241–247. doi:10.1038/ni.1703
Franko, A., Goricar, K., Kovac, V., Dodic-Fikfak, M., and Dolzan, V. (2020). NLRP3 and CARD8 polymorphisms influence risk for asbestos-related diseases. J. Med. Biochem. 39 (1), 91–99. doi:10.2478/jomb-2019-0025
Fujita, M., Shannon, J. M., Morikawa, O., Gauldie, J., Hara, N., and Mason, R. J. (2003). Overexpression of tumor necrosis factor-alpha diminishes pulmonary fibrosis induced by bleomycin or transforming growth factor-beta. Am. J. Respir. Cell Mol. Biol. 29 (6), 669–676. doi:10.1165/rcmb.2002-0046OC
Gabay, C., Fautrel, B., Rech, J., Spertini, F., Feist, E., Kötter, I., et al. (2018). Open-label, multicentre, dose-escalating phase II clinical trial on the safety and efficacy of tadekinig alfa (IL-18BP) in adult-onset Still's disease. Ann. Rheum. Dis. 77 (6), 840–847. doi:10.1136/annrheumdis-2017-212608
Gasse, P., Mary, C., Guenon, I., Noulin, N., Charron, S., Schnyder-Candrian, S., et al. (2007). IL-1R1/MyD88 signaling and the inflammasome are essential in pulmonary inflammation and fibrosis in mice. J. Clin. investigation 117 (12), 3786–3799. doi:10.1172/JCI32285
Gordon, K. L., Hummers, L. K., Woods, A., and Wigley, F. M. (2007). Efficacy and safety of etanercept in the treatment of scleroderma-associated joint disease. J. Rheumatology 34 (7), 1636–1637.
Gritsenko, A., Green, J. P., Brough, D., and Lopez-Castejon, G. (2020). Mechanisms of NLRP3 priming in inflammaging and age related diseases. Cytokine & Growth Factor Rev. 55, 15–25. doi:10.1016/j.cytogfr.2020.08.003
Haraoui, B., Differentiating the efficacy of the tumor necrosis factor inhibitors. Seminars Arthritis Rheumatism, 2005. 34(5): p. 7–11. doi:10.1016/j.semarthrit.2005.01.003
He, Y., Hara, H., and Nunez, G. (2016). Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem. Sci. 41 (12), 1012–1021. doi:10.1016/j.tibs.2016.09.002
Henderson, J., and O'Reilly, S. (2017). Inflammasome lights up in systemic sclerosis. Arthritis Res. Ther. 19 (1), 205. doi:10.1186/s13075-017-1420-z
Herzog, E. L., Mathur, A., Tager, A. M., Feghali-Bostwick, C., Schneider, F., and Varga, J. (2014). Review: interstitial lung disease associated with systemic sclerosis and idiopathic pulmonary fibrosis: how similar and distinct? Arthritis Rheumatol. 66 (8), 1967–1978. doi:10.1002/art.38702
Hoffman, H. M., Throne, M. L., Amar, N. J., Cartwright, R. C., Kivitz, A. J., Soo, Y., et al. (2012). Long-term efficacy and safety profile of rilonacept in the treatment of cryopryin-associated periodic syndromes: results of a 72-week open-label extension study. Clin. Ther. 34 (10), 2091–2103. doi:10.1016/j.clinthera.2012.09.009
Hou, J., Ma, T., Cao, H., Chen, Y., Wang, C., Chen, X., et al. (2018). TNF-α-induced NF-κB activation promotes myofibroblast differentiation of LR-MSCs and exacerbates bleomycin-induced pulmonary fibrosis. J. Cell Physiol. 233 (3), 2409–2419. doi:10.1002/jcp.26112
Hsu, H., Xiong, J., and Goeddel, D. V. (1995). The TNF receptor 1-associated protein TRADD signals cell death and NF-κB activation. Cell 81 (4), 495–504. doi:10.1016/0092-8674(95)90070-5
Hussein, M. R., Hassan, H. I., Hofny, E. R. M., Elkholy, M., Fatehy, N. A., Abd Elmoniem, A. E. A., et al. (2005). Alterations of mononuclear inflammatory cells, CD4/CD8+ T cells, interleukin 1beta, and tumour necrosis factor alpha in the bronchoalveolar lavage fluid, peripheral blood, and skin of patients with systemic sclerosis. J. Clin. pathology 58 (2), 178–184. doi:10.1136/jcp.2004.019224
Jang, D.-i., Lee, A. H., Shin, H. Y., Song, H. R., Park, J. H., Kang, T. B., et al. (2021). The role of tumor necrosis factor alpha (TNF-α) in autoimmune disease and current TNF-α inhibitors in therapeutics. Int. J. Mol. Sci. 22, 2719. doi:10.3390/ijms22052719
Ji, X., Hou, Z., Wang, T., Jin, K., Fan, J., Luo, C., et al. (2012). Polymorphisms in inflammasome genes and risk of coal workers' pneumoconiosis in a Chinese population. PLoS One 7 (10), e47949. doi:10.1371/journal.pone.0047949
Judson, M. A., Baughman, R. P., Costabel, U., Drent, M., Gibson, K. F., Raghu, G., et al. (2014). Safety and efficacy of ustekinumab or golimumab in patients with chronic sarcoidosis. Eur. Respir. J. 44 (5), 1296–1307. doi:10.1183/09031936.00000914
Kamphuis, L. S., Lam-Tse, W. K., Dik, W. A., van Daele, P. L., van Biezen, P., Kwekkeboom, D. J., et al. (2011). Efficacy of adalimumab in chronically active and symptomatic patients with sarcoidosis. Am. J. Respir. Crit. Care Med. 184 (10), 1214–1216. doi:10.1164/ajrccm.184.10.1214
Kaplanski, G. (2018). Interleukin-18: biological properties and role in disease pathogenesis. Immunol. Rev. 281 (1), 138–153. doi:10.1111/imr.12616
Kelley, N., Jeltema, D., Duan, Y., and He, Y. (2019). The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 20 (13), 3328. doi:10.3390/ijms20133328
Kiltz, U., Kiefer, D., Braun, J., Schiffrin, E. J., Girard-Guyonvarc'h, C., and Gabay, C. (2020). Prolonged treatment with Tadekinig alfa in adult-onset Still's disease. Ann. Rheum. Dis. 79 (1), e10. doi:10.1136/annrheumdis-2018-214496
Kim, H. J., Song, S. B., Choi, J. M., Kim, K. M., Cho, B. K., Cho, D. H., et al. (2010). IL-18 downregulates collagen production in human dermal fibroblasts via the ERK pathway. J. Investigative Dermatology 130 (3), 706–715. doi:10.1038/jid.2009.302
Kluck, V., Jansen, T. L. T. A., Janssen, M., Comarniceanu, A., Efdé, M., Tengesdal, I. W., et al. (2020). Dapansutrile, an oral selective NLRP3 inflammasome inhibitor, for treatment of gout flares: an open-label, dose-adaptive, proof-of-concept, phase 2a trial. Lancet Rheumatol. 2 (5), e270–e280. doi:10.1016/s2665-9913(20)30065-5
Kobayashi, S., Nagafuchi, Y., Okubo, M., Sugimori, Y., Shirai, H., Hatano, H., et al. (2021). Integrated bulk and single-cell RNA-sequencing identified disease-relevant monocytes and a gene network module underlying systemic sclerosis. J. Autoimmun. 116, 102547. doi:10.1016/j.jaut.2020.102547
Krause, K., Weller, K., Stefaniak, R., Wittkowski, H., Altrichter, S., Siebenhaar, F., et al. (2012). Efficacy and safety of the interleukin-1 antagonist rilonacept in schnitzler syndrome: an open-label study. Allergy 67 (7), 943–950. doi:10.1111/j.1398-9995.2012.02843.x
Lamkanfi, M. (2011). Emerging inflammasome effector mechanisms. Nat. Rev. Immunol. 11 (3), 213–220. doi:10.1038/nri2936
Lamkanfi, M., Mueller, J. L., Vitari, A. C., Misaghi, S., Fedorova, A., Deshayes, K., et al. (2009). Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J. Cell Biol. 187 (1), 61–70. doi:10.1083/jcb.200903124
Li, D., and Wu, M. (2021). Pattern recognition receptors in health and diseases. Signal Transduct. Target. Ther. 6 (1), 291. doi:10.1038/s41392-021-00687-0
Li, Z., Guo, J., and Bi, L. (2020). Role of the NLRP3 inflammasome in autoimmune diseases. Biomed. Pharmacother. 130, 110542. doi:10.1016/j.biopha.2020.110542
Lin, E., Vincent, F. B., Sahhar, J., Ngian, G. S., Kandane-Rathnayake, R., Mende, R., et al. (2019). Analysis of serum interleukin(IL)-1α, IL-1β and IL-18 in patients with systemic sclerosis. Clin. Transl. Immunol. 8 (4), e1045. doi:10.1002/cti2.1045
Liu, J. Y., Brass, D. M., Hoyle, G. W., and Brody, A. R. (1998). TNF-alpha receptor knockout mice are protected from the fibroproliferative effects of inhaled asbestos fibers. Am. J. Pathol. 153 (6), 1839–1847. doi:10.1016/s0002-9440(10)65698-2
Man, S. M., and Kanneganti, T. D. (2015). Regulation of inflammasome activation. Immunol. Rev. 265 (1), 6–21. doi:10.1111/imr.12296
Mantero, J. C., Kishore, N., Ziemek, J., Stifano, G., Zammitti, C., Khanna, D., et al. (2018). Randomised, double-blind, placebo-controlled trial of IL1-trap, rilonacept, in systemic sclerosis. A phase I/II biomarker trial. Clin. Exp. Rheumatol. 36 (4): p. 146–149.
Marchetti, C., Swartzwelter, B., Gamboni, F., Neff, C. P., Richter, K., Azam, T., et al. (2018). OLT1177, a beta-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc. Natl. Acad. Sci. U. S. A. 115 (7), E1530–E1539. doi:10.1073/pnas.1716095115
Martinez-Godinez, M. A., Cruz-Domínguez, M. P., Jara, L. J., Domínguez-López, A., Jarillo-Luna, R. A., Vera-Lastra, O., et al. (2015). Expression of NLRP3 inflammasome, cytokines and vascular mediators in the skin of systemic sclerosis patients. Isr. Med. Assoc. J. 17 (1), 5–10.
Micheau, O., and Tschopp, J. (2003). Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114 (2), 181–190. doi:10.1016/s0092-8674(03)00521-x
Milman, N., Graudal, N., Loft, A., Mortensen, J., Larsen, J., and Baslund, B. (2012). Effect of the TNF-alpha inhibitor adalimumab in patients with recalcitrant sarcoidosis: A prospective observational study using FDG-PET. Clin. Respir. J. 6 (4), 238–247. doi:10.1111/j.1752-699X.2011.00276.x
Mistry, P., Reid, J., Pouliquen, I., McHugh, S., Abberley, L., DeWall, S., et al. (2014). Safety, tolerability, pharmacokinetics, and pharmacodynamics of single-dose antiinterleukin- 18 mAb GSK1070806 in healthy and obese subjects. Int. J. Clin. Pharmacol. Ther. 52 (10), 867–879. doi:10.5414/CP202087
Mohan, M. J., Seaton, T., Mitchell, J., Howe, A., Blackburn, K., Burkhart, W., et al. (2002). The tumor necrosis factor-alpha converting enzyme (TACE): A unique metalloproteinase with highly defined substrate selectivity. Biochemistry 41 (30), 9462–9469. doi:10.1021/bi0260132
Murdaca, G., Spanò, F., Contatore, M., Guastalla, A., and Puppo, F. (2014). Potential use of TNF-α inhibitors in systemic sclerosis. Immunotherapy 6 (3), 283–289. doi:10.2217/imt.13.173
Nakatani-Okuda, A., Ueda, H., Kashiwamura, S. I., Sekiyama, A., Kubota, A., Fujita, Y., et al. (2005). Protection against bleomycin-induced lung injury by IL-18 in mice. Am. J. Physiology-Lung Cell. Mol. Physiology 289 (2), L280–L287. doi:10.1152/ajplung.00380.2004
Oku, H., Shimizu, T., Kawabata, T., Nagira, M., Hikita, I., Ueyama, A., et al. (2008). Antifibrotic action of pirfenidone and prednisolone: different effects on pulmonary cytokines and growth factors in bleomycin-induced murine pulmonary fibrosis. Eur. J. Pharmacol. 590 (1), 400–408. doi:10.1016/j.ejphar.2008.06.046
Ortiz, L. A., Lasky, J., Hamilton, R. F., Holian, A., Hoyle, G. W., Banks, W., et al. (1998). Expression of TNF and the necessity of TNF receptors in bleomycin-induced lung injury in mice. Exp. Lung Res. 24 (6), 721–743. doi:10.3109/01902149809099592
Pantelidis, P., McGrath, D. S., Southcott, A. M., Black, C. M., and du Bois, R. M. (2001). Tumour necrosis factor-alpha production in fibrosing alveolitis is macrophage subset specific. Respir. Res. 2 (6), 365–372. doi:10.1186/rr87
Pehlivan, Y., Onat, A. M., Ceylan, N., Turkbeyler, I. H., Buyukhatipoglu, H., Comez, G., et al. (2012). Serum leptin, resistin and TNF-α levels in patients with systemic sclerosis: the role of adipokines in scleroderma. Int. J. Rheumatic Dis. 15 (4), 374–379. doi:10.1111/j.1756-185X.2012.01755.x
Perera, A. P., Fernando, R., Shinde, T., Gundamaraju, R., Southam, B., Sohal, S. S., et al. (2018). MCC950, a specific small molecule inhibitor of NLRP3 inflammasome attenuates colonic inflammation in spontaneous colitis mice. Sci. Rep. 8 (1), 8618. doi:10.1038/s41598-018-26775-w
Perez-Alvarez, R., Perez-de-Lis, M., Diaz-Lagares, C., Pego-Reigosa, J. M., Retamozo, S., Bove, A., et al. (2011). Interstitial lung disease induced or exacerbated by TNF-targeted therapies: analysis of 122 cases. Semin. Arthritis Rheum. 41 (2), 256–264. doi:10.1016/j.semarthrit.2010.11.002
Phan, S. H., and Kunkel, S. L. (1992). Lung cytokine production in bleomycin-induced pulmonary fibrosis. Exp. Lung Res. 18 (1), 29–43. doi:10.3109/01902149209020649
Piguet, P. F., Ribaux, C., Karpuz, V., Grau, G. E., and Kapanci, Y. (1993). Expression and localization of tumor necrosis factor-alpha and its mRNA in idiopathic pulmonary fibrosis. Am. J. Pathol. 143 (3), 651–655.
Primiano, M. J., Lefker, B. A., Bowman, M. R., Bree, A. G., Hubeau, C., Bonin, P. D., et al. (2016). Efficacy and pharmacology of the NLRP3 inflammasome inhibitor CP-456,773 (CRID3) in murine models of dermal and pulmonary inflammation. J. Immunol. 197 (6), 2421–2433. doi:10.4049/jimmunol.1600035
Rahaghi, F. F., Hsu, V. M., Kaner, R. J., Mayes, M. D., Rosas, I. O., Saggar, R., et al. (2023). Expert consensus on the management of systemic sclerosis-associated interstitial lung disease. Respir. Res. 24 (1), 6. doi:10.1186/s12931-022-02292-3
Redente, E. F., Keith, R. C., Janssen, W., Henson, P. M., Ortiz, L. A., Downey, G. P., et al. (2014). Tumor necrosis factor-alpha accelerates the resolution of established pulmonary fibrosis in mice by targeting profibrotic lung macrophages. Am. J. Respir. Cell Mol. Biol. 50 (4), 825–837. doi:10.1165/rcmb.2013-0386OC
Ridker, P. M., Everett, B. M., Thuren, T., MacFadyen, J. G., Chang, W. H., Ballantyne, C., et al. (2017a). Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 377 (12), 1119–1131. doi:10.1056/nejmoa1707914
Ridker, P. M., Howard, C. P., Walter, V., Everett, B., Libby, P., Hensen, J., et al. (2012). Effects of interleukin-1β inhibition with canakinumab on hemoglobin A1c, lipids, C-reactive protein, interleukin-6, and fibrinogen: A phase IIb randomized, placebo-controlled trial. Circulation 126 (23), 2739–2748. doi:10.1161/CIRCULATIONAHA.112.122556
Ridker, P. M., MacFadyen, J. G., Thuren, T., Everett, B. M., Libby, P., Glynn, R. J., et al. (2017b). Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 390 (10105), 1833–1842. doi:10.1016/S0140-6736(17)32247-X
Russell, E., Luk, F., Manocha, S., Ho, T., O'Connor, C., and Hussain, H. (2013). Long term follow-up of infliximab efficacy in pulmonary and extra-pulmonary sarcoidosis refractory to conventional therapy. Semin. Arthritis Rheum. 43 (1), 119–124. doi:10.1016/j.semarthrit.2012.10.008
Sanchez-Fernandez, A., Skouras, D. B., Dinarello, C. A., and López-Vales, R. (2019). OLT1177 (dapansutrile), a selective NLRP3 inflammasome inhibitor, ameliorates experimental autoimmune encephalomyelitis pathogenesis. Front. Immunol. 10, 2578. doi:10.3389/fimmu.2019.02578
Santarlasci, V., Cosmi, L., Maggi, L., Liotta, F., and Annunziato, F. (2013). IL-1 and T Helper immune responses. Front. Immunol. 4, 182. doi:10.3389/fimmu.2013.00182
Shi, J., Zhao, Y., Wang, K., Shi, X., Wang, Y., Huang, H., et al. (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526 (7575), 660–665. doi:10.1038/nature15514
Sime, P. J., Marr, R. A., Gauldie, D., Xing, Z., Hewlett, B. R., Graham, F. L., et al. (1998). Transfer of tumor necrosis factor-alpha to rat lung induces severe pulmonary inflammation and patchy interstitial fibrogenesis with induction of transforming growth factor-beta1 and myofibroblasts. Am. J. Pathol. 153 (3), 825–832. doi:10.1016/s0002-9440(10)65624-6
Sullivan, D. E., Ferris, M., Nguyen, H., Abboud, E., and Brody, A. R. (2009). TNF-alpha induces TGF-beta1 expression in lung fibroblasts at the transcriptional level via AP-1 activation. J. Cell Mol. Med. 13 (8), 1866–1876. doi:10.1111/j.1582-4934.2009.00647.x
Sullivan, D. E., Ferris, M., Pociask, D., and Brody, A. R. (2005). Tumor necrosis factor-alpha induces transforming growth factor-beta1 expression in lung fibroblasts through the extracellular signal-regulated kinase pathway. Am. J. Respir. Cell Mol. Biol. 32 (4), 342–349. doi:10.1165/rcmb.2004-0288OC
Swanson, K. V., Deng, M., and Ting, J. P. (2019). The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 19 (8), 477–489. doi:10.1038/s41577-019-0165-0
Sweiss, N. J., Noth, I., Mirsaeidi, M., Zhang, W., Naureckas, E. T., Hogarth, D. K., et al. (2014). Efficacy results of a 52-week trial of adalimumab in the treatment of refractory sarcoidosis. Sarcoidosis Vasc. Diffuse Lung Dis. 31 (1), 46–54.
Tamao, N., Ando, K., Kaneko, N., Takahashi, K., and Motojima, S. (2014). Potential risk of TNF inhibitors on the progression of interstitial lung disease in patients with rheumatoid arthritis. BMJ Open 4 (8), e005615. doi:10.1136/bmjopen-2014-005615
Tapia-Abellan, A., Angosto-Bazarra, D., Martínez-Banaclocha, H., de Torre-Minguela, C., Cerón-Carrasco, J. P., Pérez-Sánchez, H., et al. (2019). MCC950 closes the active conformation of NLRP3 to an inactive state. Nat. Chem. Biol. 15 (6), 560–564. doi:10.1038/s41589-019-0278-6
Taroni, J. N., Greene, C. S., Martyanov, V., Wood, T. A., Christmann, R. B., Farber, H. W., et al. (2017). A novel multi-network approach reveals tissue-specific cellular modulators of fibrosis in systemic sclerosis. Genome Med. 9 (1), 27. doi:10.1186/s13073-017-0417-1
Tengstrand, B., Ernestam, S., Engvall, I. L., Rydvald, Y., and Hafström, I. (2005). TNF blockade in rheumatoid arthritis can cause severe fibrosing alveolitis. Six case reports. Lakartidningen. 102(49): p. 3788–3790.
Utz, J. P., Limper, A. H., Kalra, S., Specks, U., Scott, J. P., Vuk-Pavlovic, Z., et al. (2003). Etanercept for the treatment of stage II and III progressive pulmonary sarcoidosis. Chest 124 (1), 177–185. doi:10.1378/chest.124.1.177
Valenzi, E., Bulik, M., Tabib, T., Morse, C., Sembrat, J., Trejo Bittar, H., et al. (2019). Single-cell analysis reveals fibroblast heterogeneity and myofibroblasts in systemic sclerosis-associated interstitial lung disease. Ann. Rheum. Dis. 78 (10), 1379–1387. doi:10.1136/annrheumdis-2018-214865
van der Heijden, T., Kritikou, E., Venema, W., van Duijn, J., van Santbrink, P. J., Slütter, B., et al. (2017). NLRP3 inflammasome inhibition by MCC950 reduces atherosclerotic lesion development in apolipoprotein E-deficient mice-brief report. Arterioscler. Thromb. Vasc. Biol. 37 (8), 1457–1461. doi:10.1161/ATVBAHA.117.309575
Van Gorp, H., Van Opdenbosch, N., and Lamkanfi, M. (2019). Inflammasome-dependent cytokines at the crossroads of health and autoinflammatory disease. Cold Spring Harb. Perspect. Biol. 11 (1), a028563. doi:10.1101/cshperspect.a028563
van Hout, G. P., Bosch, L., Ellenbroek, G. H. J. M., de Haan, J. J., van Solinge, W. W., Cooper, M. A., et al. (2017). The selective NLRP3-inflammasome inhibitor MCC950 reduces infarct size and preserves cardiac function in a pig model of myocardial infarction. Eur. Heart J. 38 (11), 828–836. doi:10.1093/eurheartj/ehw247
Vande Walle, L., Stowe, I. B., Šácha, P., Lee, B. L., Demon, D., Fossoul, A., et al. (2019). MCC950/CRID3 potently targets the NACHT domain of wild-type NLRP3 but not disease-associated mutants for inflammasome inhibition. PLoS Biol. 17 (9), e3000354. doi:10.1371/journal.pbio.3000354
Vandenabeele, P., Declercq, W., Vanhaesebroeck, B., Grooten, J., and Fiers, W. (1995). Both TNF receptors are required for TNF-mediated induction of apoptosis in PC60 cells. J. Immunol. 154 (6), 2904–2913. doi:10.4049/jimmunol.154.6.2904
Volkmann, E. R., and Fischer, A. (2021). Update on morbidity and mortality in systemic sclerosis-related interstitial lung disease. J. Scleroderma Relat. Disord. 6 (1), 11–20. doi:10.1177/2397198320915042
Wang, L., Du, F., and Wang, X. (2008). TNF-alpha induces two distinct caspase-8 activation pathways. Cell 133 (4), 693–703. doi:10.1016/j.cell.2008.03.036
Warshamana, G. S., Corti, M., and Brody, A. R. (2001). TNF-Alpha, PDGF, and TGF-beta(1) expression by primary mouse bronchiolar-alveolar epithelial and mesenchymal cells: tnf-alpha induces TGF-beta(1). Exp. Mol. Pathology 71 (1), 13–33. doi:10.1006/exmp.2001.2376
Weng, S., Wang, L., Rong, Y., Liu, Y., Wang, X., Guan, H., et al. (2015). Effects of the interactions between dust exposure and genetic polymorphisms in Nalp3, caspase-1, and IL-1β on the risk of silicosis: A case-control study. PLOS ONE 10 (10), e0140952. doi:10.1371/journal.pone.0140952
Wohlford, G. F., Van Tassell, B. W., Billingsley, H. E., Kadariya, D., Canada, J. M., Carbone, S., et al. (2020). Phase 1B, randomized, double-blinded, dose escalation, single-center, repeat dose safety and pharmacodynamics study of the oral NLRP3 inhibitor dapansutrile in subjects with NYHA II-III systolic heart failure. J. Cardiovasc Pharmacol. 77 (1), 49–60. doi:10.1097/FJC.0000000000000931
Xu, D., Mu, R., and Wei, X. (2019). The roles of IL-1 family cytokines in the pathogenesis of systemic sclerosis. Front. Immunol. 10, 2025. doi:10.3389/fimmu.2019.02025
Yang, Y., Wang, H., Kouadir, M., Song, H., and Shi, F. (2019). Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 10 (2), 128. doi:10.1038/s41419-019-1413-8
Yasuda, K., Nakanishi, K., and Tsutsui, H. (2019). Interleukin-18 in health and disease. Int. J. Mol. Sci. 20 (3), 649. doi:10.3390/ijms20030649
Zakrzewska, K., Arvia, R., Torcia, M. G., Clemente, A. M., Tanturli, M., Castronovo, G., et al. (2019). Effects of parvovirus B19 in vitro infection on monocytes from patients with systemic sclerosis: enhanced inflammatory pathways by caspase-1 activation and cytokine production. J. Investigative Dermatology 139 (10), 2125–2133. doi:10.1016/j.jid.2019.03.1144
Keywords: scleroderma associated interstitial lung disease, NLRP3, tumor necrosis factor, alpha, IL-1 beta, IL-18
Citation: Woo S, Gandhi S, Ghincea A, Saber T, Lee CJ and Ryu C (2023) Targeting the NLRP3 inflammasome and associated cytokines in scleroderma associated interstitial lung disease. Front. Cell Dev. Biol. 11:1254904. doi: 10.3389/fcell.2023.1254904
Received: 07 July 2023; Accepted: 19 September 2023;
Published: 02 October 2023.
Edited by:
Chunheng Mo, Sichuan University, ChinaReviewed by:
Mark Mellett, University Hospital Zürich, SwitzerlandSarang Tartey, IGM Biosciences, United States
Copyright © 2023 Woo, Gandhi, Ghincea, Saber, Lee and Ryu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Changwan Ryu, changwan.ryu@yale.edu