Human RecQ Helicases in DNA Double-Strand Break Repair
 Huiming Lu
Huiming Lu Anthony J. Davis
Anthony J. Davis- Division of Molecular Radiation Biology, Department of Radiation Oncology, UT Southwestern Medical Center, Dallas, TX, United States
RecQ DNA helicases are a conserved protein family found in bacteria, fungus, plants, and animals. These helicases play important roles in multiple cellular functions, including DNA replication, transcription, DNA repair, and telomere maintenance. Humans have five RecQ helicases: RECQL1, Bloom syndrome protein (BLM), Werner syndrome helicase (WRN), RECQL4, and RECQL5. Defects in BLM and WRN cause autosomal disorders: Bloom syndrome (BS) and Werner syndrome (WS), respectively. Mutations in RECQL4 are associated with three genetic disorders, Rothmund–Thomson syndrome (RTS), Baller–Gerold syndrome (BGS), and RAPADILINO syndrome. Although no genetic disorders have been reported due to loss of RECQL1 or RECQL5, dysfunction of either gene is associated with tumorigenesis. Multiple genetically independent pathways have evolved that mediate the repair of DNA double-strand break (DSB), and RecQ helicases play pivotal roles in each of them. The importance of DSB repair is supported by the observations that defective DSB repair can cause chromosomal aberrations, genomic instability, senescence, or cell death, which ultimately can lead to premature aging, neurodegeneration, or tumorigenesis. In this review, we will introduce the human RecQ helicase family, describe in detail their roles in DSB repair, and provide relevance between the dysfunction of RecQ helicases and human diseases.
DNA Double-Strand Breaks and Repair Pathways
A number of elegant mechanisms have evolved that repair the vast number of DNA lesions an organism encounters each day. DNA repair mechanisms are described as guardians of the human genome because DNA is the template for the fundamental processes of replication and transcription, and preserving the integrity of genomic DNA ensures faithful propagation of genetic material and transmission to daughter cells (Hoeijmakers, 2009; Ciccia and Elledge, 2010; Tubbs and Nussenzweig, 2017). Arguably, the most important DNA repair mechanisms are those that repair DNA double-strand breaks (DSBs) (Ciccia and Elledge, 2010; Ceccaldi et al., 2016; Scully et al., 2019). DSBs are generated during endogenous events such as after the collapse of replication forks (Bouwman and Crosetto, 2018), SPO11-induced DSB formation during meiosis (Tock and Henderson, 2018), V(D)J (variable, diversity, and joining) recombination (Chi et al., 2020), and via reactive oxygen species generated during metabolism, as well as from various exogenous stresses which include ionizing radiation (IR) and cancer chemotherapeutic agents (Tubbs and Nussenzweig, 2017) (Figure 1). Unrepaired or misrepaired DSBs can cause chromosomal aberrations, genomic instability, senescence, or cell death, further leading to premature aging, neurodegeneration, or tumorigenesis (Figure 1) (White and Vijg, 2016; Tubbs and Nussenzweig, 2017; Taylor et al., 2019). To overcome severe consequences from DSBs, mammalian cells have evolved at least four pathways to repair this type of DNA lesion, termed non-homologous end joining (NHEJ), homologous recombination (HR), and the alternative end-joining pathways, microhomology-mediated end joining (MMEJ) and single-strand annealing (SSA) (Figure 2). In the following sections, we will give a brief overview of each DSB repair pathway.
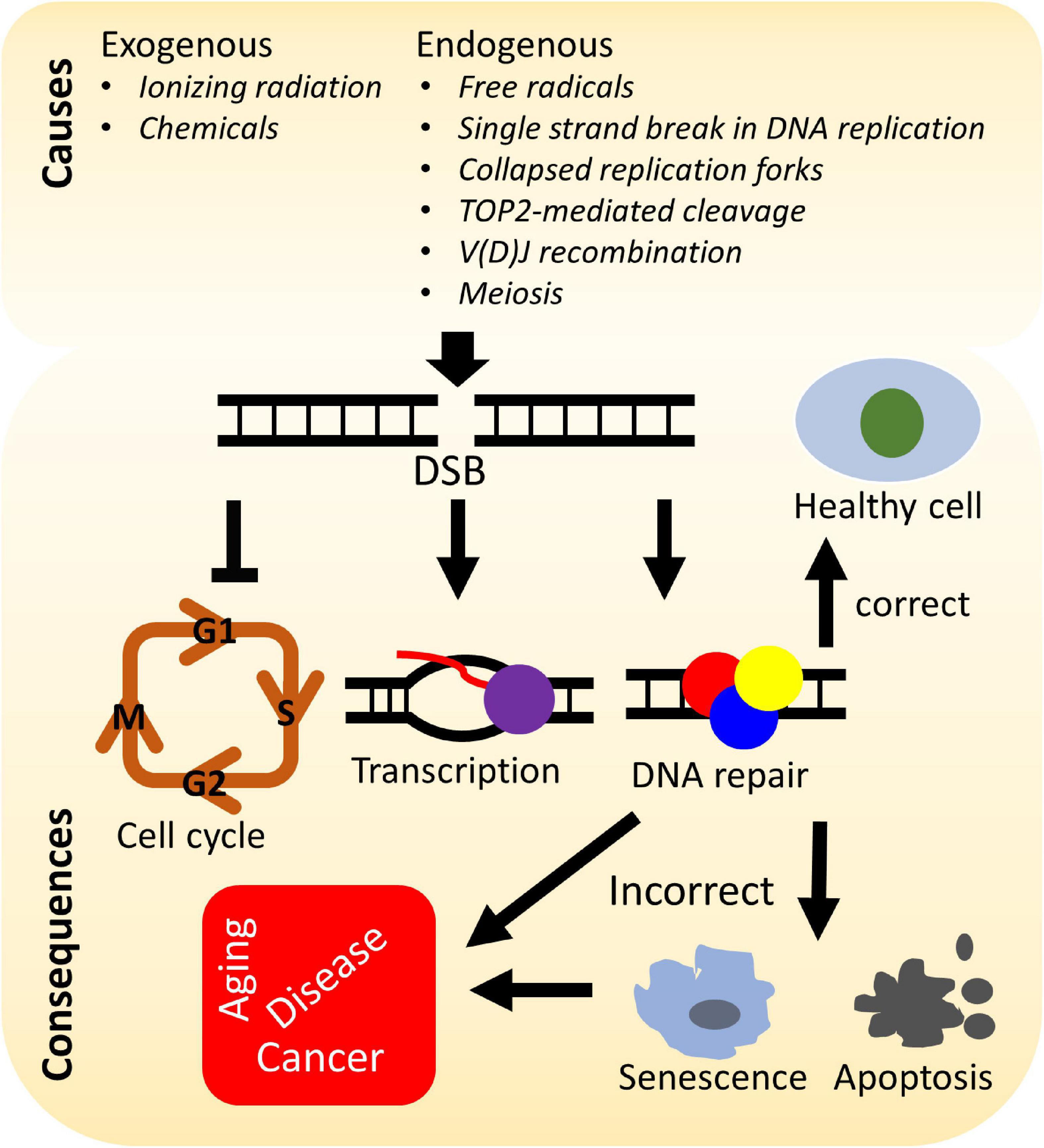
Figure 1. Causes and consequences of DNA double-strand breaks (DSBs). DSBs arise from various stresses by endogenous or exogenous factors and can lead to arrest of the cell cycle, transcription, activation of the DNA damage response, and repair of the DNA damage. Incorrectly repaired or unrepaired DSBs can result in cellular senescence, apoptosis, premature aging, genetic disorders, and/or tumorigenesis.
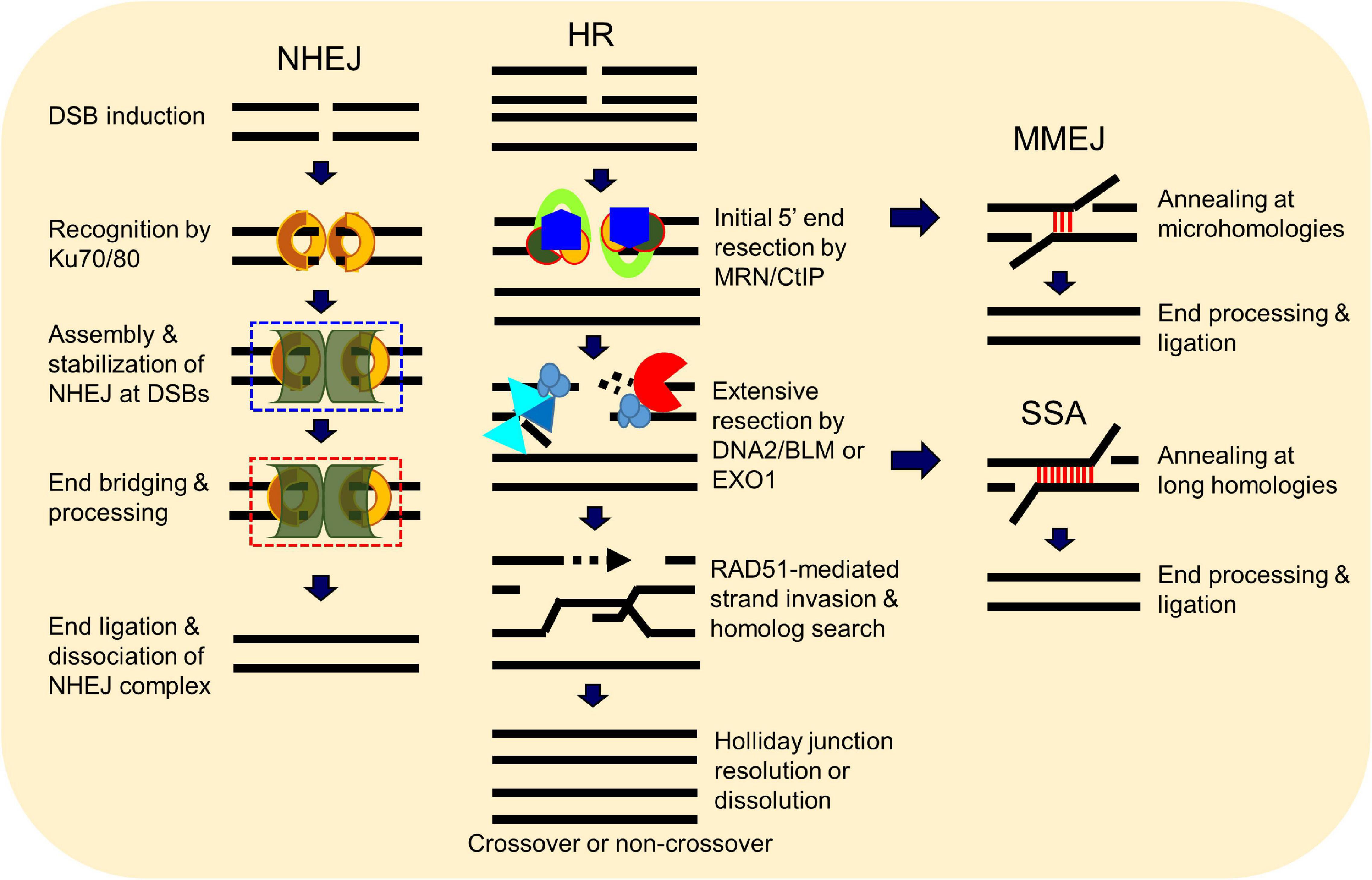
Figure 2. Double-strand break (DSB) repair pathways. DSBs in mammalian cells are able to be repaired by at least four pathways, including non-homologous end joining (NHEJ), homologous recombination (HR), microhomology-mediated end joining (MMEJ), and single-strand annealing (SSA). HR and NHEJ are the dominant DSB repair pathways in normal cells, but the two minor pathways SSA and MMEJ can occur under certain circumstances. Choice between these repair pathways is tightly regulated.
Non-homologous End Joining
Non-homologous end joining (NHEJ) is the major pathway responsible for the repair of two-ended DSBs generated by IR, restriction enzymes, and those intentionally generated for V(D)J and class switch recombination during T and B cell lymphocyte development (Davis and Chen, 2013; Davis et al., 2014; Scully et al., 2019). NHEJ is a flexible process that directs re-ligation of the broken DNA molecule in a template-independent manner and is active in all phases of the cell cycle (Davis et al., 2014; Pannunzio et al., 2018). Initiation of NHEJ occurs when the ring-shaped Ku heterodimer, composed of the Ku70 and Ku80 proteins, recognizes and binds to the DSB in a sequence-independent manner (Fell and Schild-Poulter, 2015). Once bound to the DSB ends, Ku then functions as a scaffold to recruit the NHEJ machinery to the damage site. In particular, Ku70/80 directly recruits DNA-dependent protein kinase catalytic subunit (DNA-PKcs) to the DNA ends to form the DNA-PK complex, resulting in the activation of DNA-PKcs kinase activity (Davis et al., 2014). DNA-PKcs promotes NHEJ and phosphorylates the histone H2AX and the chromatin remodeler KAP1 to promote chromatin relaxation proximal to the DSB to facilitate recruitment of the DNA damage response and repair machinery to the DNA damage site (Lu et al., 2019). If the ends of the DSB are not compatible for ligation, different enzymes are required, including those that resect DNA ends, fill in gaps, or remove blocking end groups, to process the DNA ends to allow ligation (Pannunzio et al., 2018). The terminal step in NHEJ is the ligation of the broken DNA ends by the DNA ligase IV(LIG4)/X-ray cross-complementing protein 4 (XRCC4) complex with the assistance of the XRCC4-like factor (XLF) (Ellenberger and Tomkinson, 2008). NHEJ is not intrinsically inaccurate, but the quality of end joining is dictated by the structure of the DSB ends as small insertions or deletions can be generated if end processing by nucleases and polymerases is required (Davis and Chen, 2013; Pannunzio et al., 2018; Scully et al., 2019).
Homologous Recombination
Homologous recombination (HR) requires a homologous DNA sequence to serve as a template for DNA synthesis-dependent repair. It is an accurate process as it employs DNA sequences homologous to/near the broken ends to drive repair, predominantly using the sister chromatid as a template for DSB repair rather than the homologous chromosome. As a sister chromatid is available after DNA replication, HR predominantly occurs in mid-S to the early G2 phase of the cell cycle (Ceccaldi et al., 2016; Hustedt and Durocher, 2016). HR is initiated by the MRE11/RAD50/NBS1 (MRN) complex in conjunction with CtIP via the endonuclease and 3′–5′ exonuclease activities of MRE11 (Lengsfeld et al., 2007; Sartori et al., 2007; Garcia et al., 2011; Cannavo and Cejka, 2014; Deshpande et al., 2020). The endonuclease activity of MRE11 generates a nick in the double-stranded DNA (dsDNA) near the DSB site, followed by its 3′–5′ exonuclease activity generating a short section of single-stranded DNA (ssDNA) next to the nick. This is followed by extensive resection by exonuclease 1 (EXO1) and/or the nuclease DNA2 with the RECQ helicase Bloom syndrome protein (BLM) to produce a long 3′ overhang (Mimitou and Symington, 2008; Nimonkar et al., 2011; Symington, 2014; Ronato et al., 2020). The resulting 3′ ssDNA is rapidly coated by the ssDNA-binding protein replication protein A (RPA), which is subsequently replaced by RAD51 via assistance by BRCA2 and DSS1 (Gudmundsdottir et al., 2004; Yang et al., 2005; Jensen et al., 2010; Thorslund et al., 2010; Zhao et al., 2015). RAD51 binds to ssDNA, forming a helical RAD51–ssDNA nucleoprotein filament that is capable of homology search and invasion of a homologous DNA sequence. If sufficient base pairing occurs between the invading RAD51-coated strand and the invaded DNA molecule, the non-base-paired strand of the invaded molecule is displaced in the form of a displacement loop (D-loop). A DNA polymerase extends the 3′-end of the invasion strand past the break using the invaded homologous strand as a template, followed by resolution of the Holliday junction from the extended D-loop by resolvases and dissolution by BLM-TOP3a-RMI1/2 complex, annealing, and ligation of the extended invasion strand to the other end of the DSB on the original DNA molecule (Wright et al., 2018; Scully et al., 2019).
Alternative End-Joining Pathways
Non-homologous end joining and HR are the dominant DSB repair pathways, but there are two minor alternative end joining pathways called microhomology-mediated end joining (MMEJ) and single-strand annealing (SSA). MMEJ and SSA were initially identified in cells that were deficient in NHEJ and/or HR, and thus, both were believed to be strictly “backup pathways” (Fairman et al., 1992; Boulton and Jackson, 1996; Mason et al., 1996; Kabotyanski et al., 1998), but it has been found that, even in NHEJ- and HR-proficient cells, a small percentage of DSBs are repaired by these alternative pathways (Truong et al., 2013). Similar to NHEJ, the MMEJ and SSA pathways terminate with direct ligation of the two ends of the broken DNA, but MMEJ and SSA are distinct from NHEJ as they function completely independently of the Ku heterodimer and DNA-PKcs, require components of the HR machinery, and require longer tracts of microhomology to mediate repair (Sallmyr and Tomkinson, 2018). The factors and processes required for MMEJ and SSA are not well defined, but both initiate with the binding of PARP1 to the DSB ends and each requires DNA end resection (Bennardo et al., 2008; Xie et al., 2009; Lee-Theilen et al., 2011; Zhang and Jasin, 2011; Truong et al., 2013). Similar to HR, DNA end resection is initiated by the MRN–CtIP complex in MMEJ and SSA, but the enzymes required for long DNA end resection are not clearly defined, with speculation being that EXO1 and/or DNA2/BLM mediate this process (Sallmyr and Tomkinson, 2018). Each alternative end-joining pathway requires differing amounts of sequence homology to align the DNA. MMEJ requires a short region of complimentary sequence, 2–20 nucleotides, called microhomology sequences, whereas SSA needs >25 nucleotides of homologous sequence, which typically reside within tandem repeats (Bhargava et al., 2016). During SSA, the generated 3′ ssDNAs are annealed by RAD52 via alignment of homologous sequences, whereas multiple enzymes are responsive for end bridging and annealing in MMEJ, including the MRN complex, PARP1, and Polθ (Sallmyr and Tomkinson, 2018). Once aligned, the non-complementary sequences generate 3′ ssDNA overhangs that are removed by nucleases. Both MMEJ and SSA complete with gap filling and DNA ligation by DNA polymerases and DNA ligases, but the exact enzymes and mechanisms that drive these processes are not well defined (Sallmyr and Tomkinson, 2018; Patterson-Fortin and D’Andrea, 2020). The repair of DSBs by MMEJ and SSA are intrinsically mutagenic as they cause deletions and rearrangements, resulting in genomic instability (Sallmyr and Tomkinson, 2018; Patterson-Fortin and D’Andrea, 2020).
Human RecQ Helicases
Helicases are a ubiquitous family of molecular motors that unwind DNA, RNA, and DNA–RNA duplexes. They play essential roles in DNA and RNA metabolism, including DNA replication, transcription, DNA repair, translation, RNA maturation, ribosome synthesis, and splicing. A conserved helicase family is the RecQ family, which is named after the prototypical member found in Escherichia coli called RecQ (Cobb and Bjergbaek, 2006; Croteau et al., 2014). RecQ helicases possess 3′ to 5′ directionality and can unwind a variety of DNA structures including B-form DNA, forked DNA duplexes, D-loops, DNA junctions, and G-quadruplexes (Croteau et al., 2014). Furthermore, they promote the annealing of complementary ssDNAs and branch migration of Holliday junctions. Each RecQ helicase shares the highly conserved core helicase domain (DEAD/DEAH box, helicase conserved C-terminal domain), with the majority of the family members containing the RecQ C-terminal (RQC) domain, and the helicase and RNase D-like C-terminal (HRDC) domain is shared among members (Figure 3). An important function of RecQ helicases is that they are essential to maintain genome stability as they act at the interface between DNA replication, DNA recombination, DNA repair, telomere maintenance, and transcription (Bohr, 2008; Croteau et al., 2014; Urban et al., 2017). Besides RecQ, E. coli has another RecQ-like helicase, named RqlH, which does not appear in the K12 strain of E. coli but in many other strains (Russell and Mulvey, 2015). Two RecQ helicases, Sgs1 and Hrq1, have been identified in the budding yeast Saccharomyces cerevisiae as homologs to human BLM and RECQL4, respectively (Watt et al., 1996; Choi et al., 2013; Bochman et al., 2014; Rogers et al., 2017). Human cells have five distinct RecQ helicases, named RECQL1, BLM, Werner syndrome helicase (WRN), RECQL4, and RECQL5. All of them have the conserved helicase core domain, but only BLM and WRN possess the HRDC domain, and RECQL1, BLM, and WRN have the typical RQC domain (Figure 3). Notably, the human RecQ helicases play important functions in nearly all DNA repair pathways, in particular those required for the repair of DSBs (Hickson, 2003; Bohr, 2008; Croteau et al., 2014). The role of human RecQ helicases in DSB repair is supported by the observation that defects in these enzymes result in a number of distinct human genetic disorders, premature aging, and/or carcinogenesis, which may be driven due to defective DSB repair (Datta et al., 2020; Oshima et al., 2018). These RecQ helicases participate in multiple DSB repair pathways by physically and functionally interacting with key players in these pathways (Table 1). In this review, we will focus on the important roles of human RecQ helicases in DSB repair and maintenance of genome stability, as well as the biological relevance between these cellular functions and the mechanisms underlying RecQ-associated disorders and cancers.
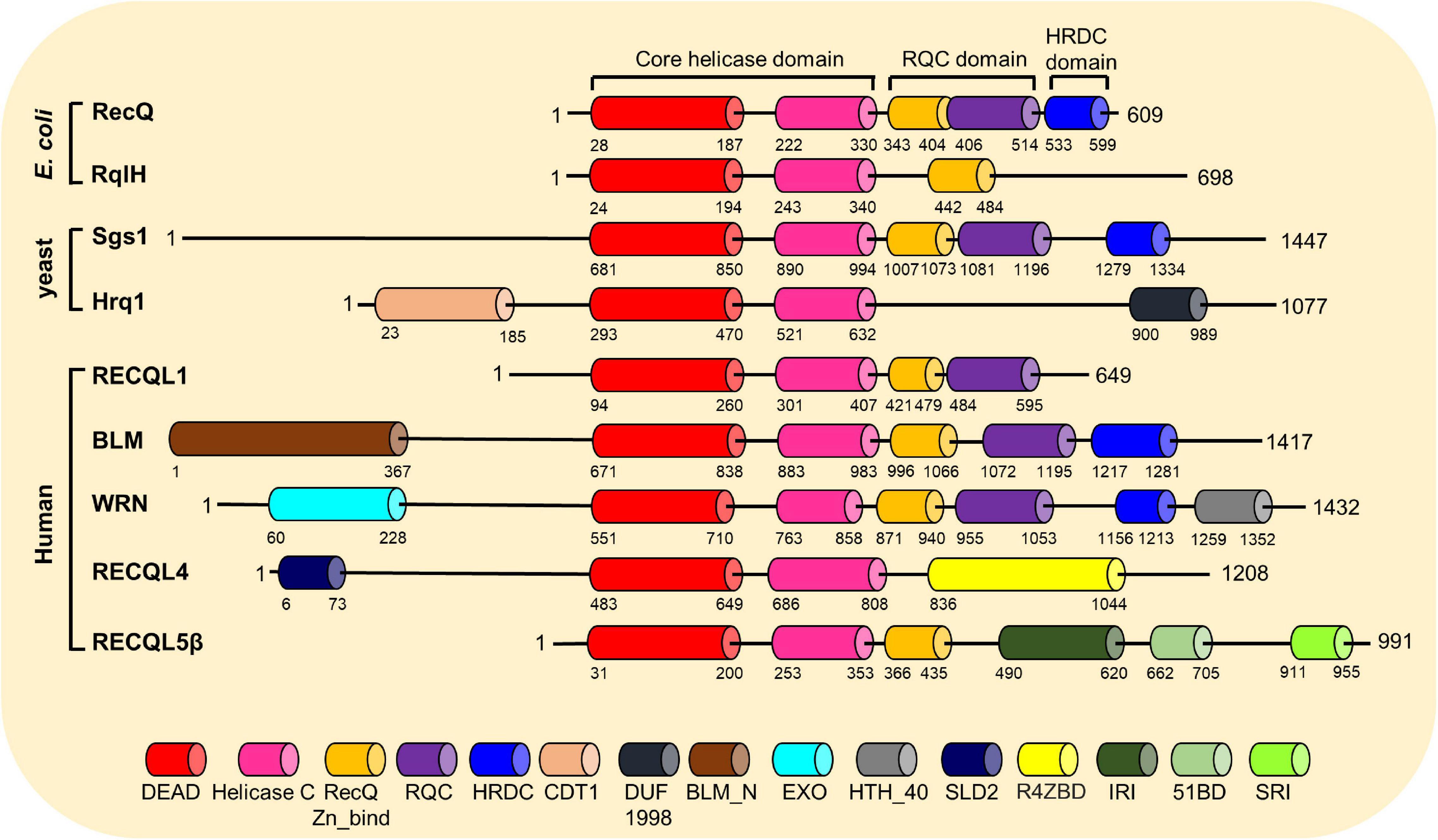
Figure 3. Conserved motifs and domains of RecQ helicases from Escherichia coli, budding yeast, and humans. The RecQ helicases’ motifs and domains are presented mainly based on the annotations by the GenomeNet Database (https://www.genome.jp/), with information from published literatures. DEAD, DEAD/DEAH box helicase; Helicase C, helicase conserved C-terminal motif; RecQ-Zn-Bind, RecQ Zn2+-binding motif; RQC, RecQ-C-terminal domain; HRDC, helicase and RNase D-like C-terminal domain; CDT1, DNA replication factor CDT1 like; DUF1998, domain of unknown function (DUF1998); BLM_N, N-terminal region of Bloom syndrome protein; EXO, DNA_POLA_EXO1, 3′–5′ exonuclease domain; HTH_40, helix-turn-helix domain; SLD2, DNA replication and checkpoint protein; R4ZBD, RECQL4-Zn2+-binding motif; IRI, internal RNAPII-interacting domain; 51BD, RAD51-binding domain; SRI, SET2-RPB1 interaction motif.
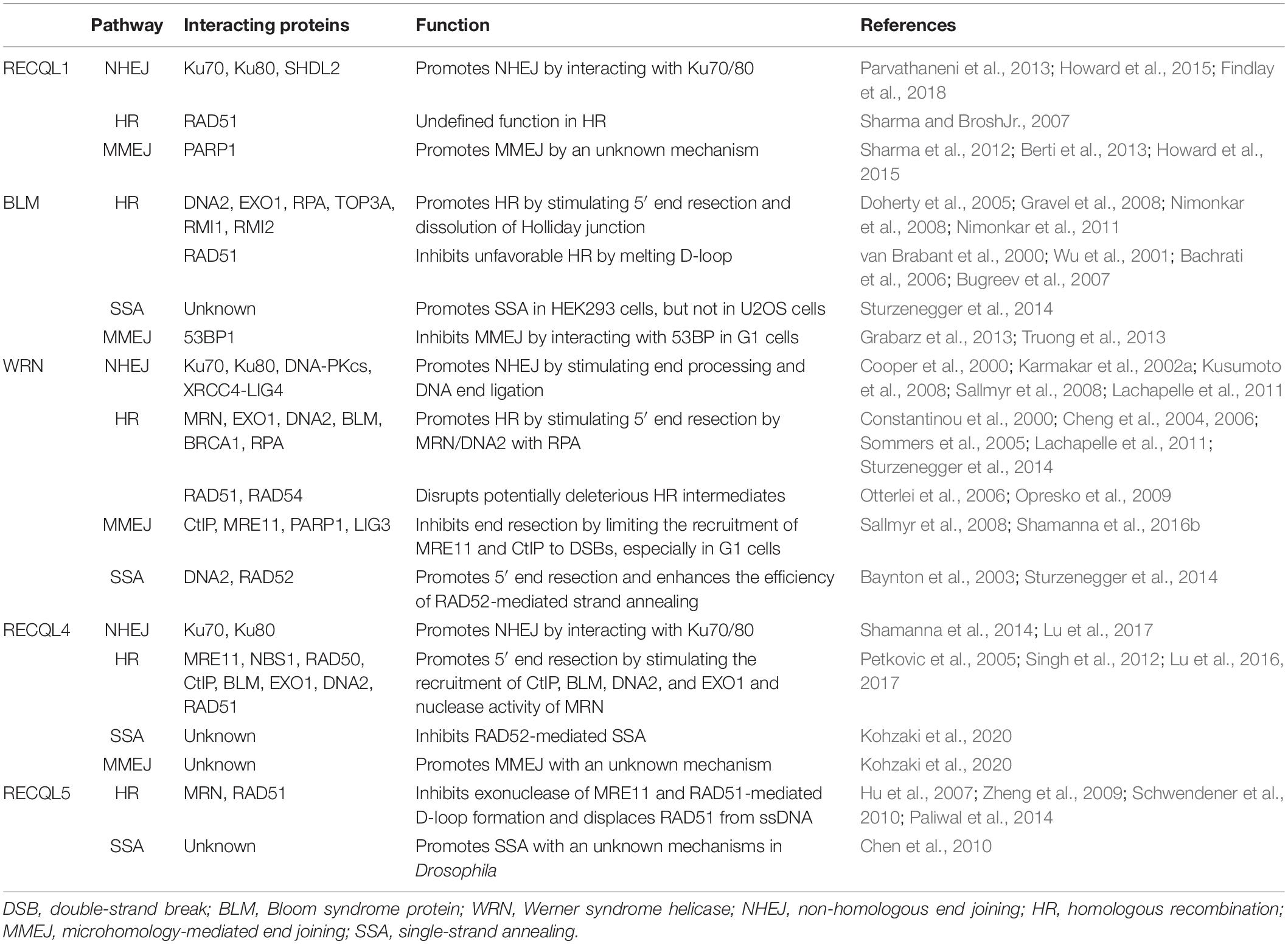
Table 1. RecQ helicase-interacting proteins in DSB repair pathways.
RECQL1
RECQL1, also known as RECQL and RECQ1, is the most abundant member of the five human RecQ helicases (Sharma and Brosh Jr., 2008). It is encoded by the RECQL1 gene, which is located at chromosome 12p12. RECQL1 consists of 649 amino acid residues, and it possesses the conserved core helicase domain, RecQ Zn2+ binding motif, and the RQC domain (Figure 3). RECQL1 can unwind 3′ overhang dsDNA, forked duplexes, 3′ or 5′ flap dsDNA, D-loops, bubble-structured dsDNA, and Holliday junctions, and its helicase activity is stimulated by RPA (Cui et al., 2003; Sharma et al., 2005). RECQL1 plays important roles in DNA repair, restart of stalled replication, and telomere maintenance (Sharma and Brosh Jr., 2007; Popuri et al., 2012a, 2014; Berti et al., 2013; Banerjee et al., 2015; Parvathaneni and Sharma, 2019).
RECQL1 and NHEJ
A role for RECQL1 in DSB repair was initially identified via the observation that Recql1 knockout mouse embryonic fibroblasts (MEFs) are highly sensitive to IR and that knockdown of RECQL1 in human cells results in increased cell death to IR and the topoisomerase 1 (TOP1) inhibitor camptothecin (CPT) (Sharma and Brosh Jr., 2007; Sharma et al., 2007). Interestingly, RECQL1 is phosphorylated following exposure to IR and accumulates at IR-damaged chromatin (Sharma and Brosh Jr., 2007), but the IR-induced phosphorylation site(s) and the kinase(s) mediating the RECQL1 phosphorylation have not been identified. A key piece of evidence supporting RECQL1 functions in NHEJ is that it directly interacts with the Ku heterodimer, and does so independently of DNA-PKcs, and that RECQL1 and Ku70/80 can simultaneously bind linearized plasmid DNA in vitro (Parvathaneni et al., 2013). Furthermore, RECQL1 can unwind Ku-bound DNA duplex in a manner that is dependent on intrinsic RECQL1 ATPase activity and RECQL1 modulates end joining in cell-free extracts (Parvathaneni et al., 2013). A reporter-based assay with small interfering RNA (siRNA) library targeting DNA damage response and repair proteins showed that RECQL1 siRNA treatment resulted in a loss of NHEJ efficiency by approximately 25% (Howard et al., 2015). However, exactly how RECQL1 directs end joining and its role in NHEJ in vivo are still undefined.
RECQL1 and HR
RECQL1 has also been implicated to play a role in HR, as the depletion of RECQL1 by siRNA resulted in increased sister chromatid exchanges in HeLa cells (LeRoy et al., 2005; Sharma and Brosh Jr., 2007). In addition, RECQL1 forms a complex with RAD51 (Sharma and Brosh Jr., 2007) and promotes the processing of Holliday junctions by promoting branch migration (LeRoy et al., 2005; Mazina et al., 2012), indicating that RECQL1 functions in resolving HR intermediates. However, knockdown of RECQL1 in U2OS cells did not significantly reduce HR efficiency, as assessed using a green fluorescent protein (GFP)-based reporter assay (Sharma et al., 2012). RECQL1, similar to RAD51, protects stalled replication forks from MRE11-dependent degradation (Berti et al., 2013; Mason et al., 2019), suggesting that RECQL1 may play an indirect role in the protection and/or repair of one-ended DSBs at replication forks and is not directly required for strand invasion or homology search in HR at two-ended DSBs. Further studies are required to define the mechanism of RECQL1 in the repression of sister chromatid exchanges and its role in HR.
RECQL1 and Cancers
The data support that RECQL1 is important for the maintenance of the genome as MEFs from Recql1 knockout mice show evaluated levels of chromosomal structural aberrations and aneuploidy, and loss of human RECQL1 also causes increased sister chromatid exchanges (LeRoy et al., 2005; Sharma and Brosh Jr., 2007; Sharma et al., 2007). Although no genetic disorders have been identified with mutations in RECQL1, germline mutations in RECQL1 are reported to be linked with an increased risk of breast cancer, suggesting that RECQL1 is a breast cancer susceptibility gene (Cybulski et al., 2015; Kwong et al., 2016; Sun J. et al., 2017; Sun et al., 2015; Tervasmaki et al., 2018). These accumulating clues suggest a tight association between RECQL1 dysfunction and the consequence of genome instability and cancer predisposition (Debnath and Sharma, 2020; Mojumdar, 2020).
Bloom Syndrome Protein (BLM)
BLM helicase, encoded by the BLM gene that is located on chromosome 15q26.1, is a multifunctional protein consisting of 1,417 amino acids. BLM possesses a conserved helicase domain, the RQC domain, and the HRDC domain (Figure 3). BLM can unwind Y-structured dsDNA, bubble-like dsDNA, G-quadruplex, and Holliday junction and also disrupts mobile D-loops (Mohaghegh et al., 2001; Bachrati et al., 2006). The HRDC domain is important for BLM to direct the annealing of DNA strands and the dissolution of double Holliday junctions, but not for unwinding DNA substrates (Cheok et al., 2005; Wu et al., 2005; Gyimesi et al., 2012). An HRDC-dependent conformational change coupled with ATP hydrolysis/DNA translocation cycle promotes BLM to process complex DNA structures (Newman et al., 2015). The N-terminal domain of BLM is unique and conserved in vertebrates, and it is believed to regulate the oligomerization of BLM (Beresten et al., 1999; Janscak et al., 2003; Xu et al., 2012; Shi et al., 2017). BLM participates in different pathways required for DNA metabolism, which it achieves by interacting with many proteins via either its N-terminal domain or undefined C-terminal region (Croteau et al., 2014). BLM resolves complex secondary structures such as G-quadruplexes and hairpins during DNA replication and transcription and at telomeres (Schawalder et al., 2003; Johnson et al., 2010; Barefield and Karlseder, 2012; Grierson et al., 2012, 2013; Drosopoulos et al., 2015). BLM can also stabilize stalled replication forks and promote the restart of the stalled forks (Ralf et al., 2006; Pan et al., 2017). BLM functions in multiple DNA repair pathways, with its most-defined role in HR.
BLM and HR
Bloom syndrome protein plays a multifaceted role in HR as it is required for the early phase of the pathway (DNA end resection) as well as one of the terminal steps (dissolution of Holliday junctions) (Croteau et al., 2014). Consistent with BLM playing a role at the early and later steps in HR, GFP-tagged BLM accumulates at laser-generated DSBs a few seconds after induction of damage, and it stays at the damage site for hours (Karmakar et al., 2006; Singh et al., 2010). As mentioned above, HR initiates with 5′ DNA end resection by the MRN complex with CtIP and then the 3′ ssDNA is extended by further resection by EXO1 and/or this DNA2/BLM complex. During this step, BLM unwinds dsDNA after binding to 3′ ssDNA to facilitate the endonuclease activity of DNA2, resulting in the generation of an extended 3′ ssDNA (Gravel et al., 2008; Nimonkar et al., 2011). During this process, RPA interacts with BLM and promotes its helicase activity (BroshJr., Li et al., 2000; Doherty et al., 2005; Nimonkar et al., 2011; Soniat et al., 2019; Qin et al., 2020). A recent study reported that CtIP interacts with BLM and stimulates its helicase activity, further promoting DNA2/BLM-mediated extensive resection (Daley et al., 2017). Interestingly, in addition to working with DNA2, another biochemical study found that BLM, but not other RecQ helicases, promotes the nuclease activity of EXO1 on dsDNA (Nimonkar et al., 2008).
Resection-generated 3′ ssDNA is used by RAD51 for strand invasion and exchange with intact sister chromatid, resulting in the formation of a D-loop. BLM can displace the invading strand from the D-loop and thus can disrupt the RAD51–ssDNA filaments, which explains why BLM has also been termed an “anti-recombination” protein, which is a feature observed in Bloom syndrome (BS) patient cells (van Brabant et al., 2000; Wu et al., 2001; Bachrati et al., 2006; Bugreev et al., 2007). The role of BLM as an anti-recombinase is dependent on its helicase activity, but does not require association with DNA topoisomerase IIIα (Patel et al., 2017). BLM stimulates the strand exchange of active ATP-bound RAD51 filaments, but dismantles inactive ADP-bound filaments (Bugreev et al., 2007; Bugreev et al., 2009; Kikuchi et al., 2009), indicating that BLM may not be an actual anti-recombinase but is required to inspect nascent D-loops in order to drive proper HR (Manthei and Keck, 2013; Xue et al., 2019). Another important role of BLM in HR is to process double Holliday junction structures along with topoisomerase IIIα, RMI1, and RMI2, generating only non-crossover recombinant products, which is termed as “dissolution of the Holliday junction” (Wu and Hickson, 2003; Raynard et al., 2006; Wu et al., 2006; Singh et al., 2008; Xu et al., 2008).
BLM and Alternative End-Joining Pathways
Although BLM possesses a strong ssDNA annealing activity and is crucial for extensive DNA end resection, its role in the alternative end-joining DSB repair pathways is unclear. Using a GFP-based reporter assay, depletion of BLM by siRNA reduces SSA in HEK293 cells, but not in U2OS cells (Sturzenegger et al., 2014). In contrast, depletion of BLM by short hairpin RNA (shRNA) leads to a significant increase in MMEJ in U2OS cells (Truong et al., 2013). Consistently, another study showed that BLM inhibits MMEJ and long-range CtIP/MRE11-dependent deletions by interacting with 53BP1 in G1 cells, suggesting that BLM plays a role in DSB repair pathway choice (Grabarz et al., 2013).
BLM and Diseases
Mutations in BLM lead to a rare autosomal-recessive genetic disorder, Bloom syndrome (BS; OMIM#210900). BS is characterized by growth deficiency, insulin resistance, immune deficiency, photosensitive skin changes, and increased risk for diabetes, as well as high risk of cancer predisposition at a young age (Cunniff et al., 2017; de Renty and Ellis, 2017). Unfortunately, BS patients have a short life span (less than 30 years), and the major cause of death is cancer (Cunniff et al., 2017; de Renty and Ellis, 2017). In addition to BS, heterozygous deleterious mutations in BLM increase the risk of breast cancer (Thompson et al., 2012), prostate cancer (Ledet et al., 2020), and colorectal cancer (Gruber et al., 2002; Cleary et al., 2003; Baris et al., 2007; de Voer et al., 2015). The cells from BS patients display chromosomal instability, which is characterized by elevated rates of chromatid gaps, breaks, sister chromatid exchanges, and quadriradials (Ellis et al., 1995; German et al., 2007). In support of this, an increased frequency of sister chromatid exchanges of >10-fold is used for the standard diagnosis of BS (Chaganti et al., 1974).
Werner Syndrome Helicase (WRN)
Werner syndrome helicase (WRN) is encoded by the WRN gene, which is located at chromosome 8p11-12 (Goto et al., 1992; Yu et al., 1996; Goto et al., 1997). WRN is a 1,432-amino acid-long protein with multiple functional domains, including 3′–5′ exonuclease, conserved helicase domain, RQC domain, and HRDC domain (Croteau et al., 2014; Oshima et al., 2017). The 3′–5′ exonuclease domain in the N-terminal region of WRN is required for multiple DNA repair pathways, and this domain can resolve various DNA substrates, such as fork-shaped duplex, flap-structured dsDNA, D-loops, bubble-structured duplex, Holliday junctions, and G-quadruplexes (Croteau et al., 2014; Oshima et al., 2017). WRN efficiently unwinds duplex DNA with 3′ or 5′ ssDNA tails that are >10 nucleotides long, as well as unwinding duplex DNA duplex with shorter 3′ ssDNA tails (BroshJr., Waheed and Sommers, 2002). The helicase activity of WRN is stimulated by a number of proteins, including RPA, the Ku heterodimer, MRN complex, and the telomere protein TRF2 (Cheng et al., 2004; Sommers et al., 2005; BroshJr., Opresko and Bohr, 2006). Interestingly, a recent study showed that the binding of multiple RPAs super boosts the unwinding activity of WRN so that this helicase can unidirectionally unwind a duplex with a size of >1 kb (Lee et al., 2018). The conserved RQC domain is critical for the substrate-specific DNA binding of WRN to initiate unwinding (Kitano et al., 2010; Tadokoro et al., 2012) and is also required for the ability of WRN to localize at telomere regions, but not at other genomic sites, after oxidative stress (Sun L. et al., 2017). The HRDC domain of WRN plays a role in DNA binding (von Kobbe et al., 2003; Kitano et al., 2007) and is important for the recruitment of WRN protein to DSBs (Lan et al., 2005). In addition, a small region between the RQC and HRDC domains promotes the ability of WRN to execute ssDNA annealing activity and oligomerization (Muftuoglu et al., 2008). The nuclear localization signal is located in the C-terminal region of WRN, and mutations in these sequence result in the translocation of WRN to the nucleolus and account for a significant portion of the mutations that drive the pathogenesis of Werner syndrome (WS) (Matsumoto et al., 1997; Suzuki et al., 2001). Each of these activities of WRN allows it to play a role in multiple DNA-associated metabolisms, including DNA replication, recombination and repair, telomere maintenance, and transcription (Oshima et al., 2017; Shamanna et al., 2017; Mukherjee et al., 2018).
Recruitment of WRN to DSB
The WRN plays various roles in NHEJ, HR, MMEJ, and SSA and does so by interacting with key participants in these pathways (Croteau et al., 2014; Shamanna et al., 2017). Cells isolated from WS patients or those with WRN knockdown are sensitive to DSB-inducing agents, including IR, CPT, etoposide, and chromium (Yannone et al., 2001; Imamura et al., 2002; Zecevic et al., 2009; Ammazzalorso et al., 2010). WRN quickly accumulates at laser-induced DSBs and is also retained at the damage sites for many hours (Lan et al., 2005; Singh et al., 2010). The recruitment of WRN to DSBs requires the presence of the HRDC domain, and its recruitment is independent of DSB sensors, such as PARP1, Ku80, DNA-PKcs, NBS1, and histone H2AX (Lan et al., 2005). In line with WRN’s involvement in multiple DSB repair pathways, the recruitment of this RecQ helicase to DSBs occurs in the G1, S, and G2 phases of the cell cycle (Shamanna et al., 2016b).
WRN and NHEJ
The involvement of WRN in NHEJ is supported by the observations that the depletion of WRN results in a reduced in vitro ligation of DNA substrates with either blunt or sticky ends, as well as a decrease in NHEJ as monitored by an in vivo NHEJ GFP reporter assay (Chen et al., 2003; Shamanna et al., 2016b). WRN physically and functionally interacts with multiple members of the NHEJ machinery, including the Ku heterodimer, DNA-PKcs, and the XRCC4–LIG4 complex (Cooper et al., 2000; Karmakar et al., 2002a; Kusumoto et al., 2008; Sallmyr et al., 2008; Lachapelle et al., 2011). WRN directly interacts with both subunits of the Ku70/80 heterodimer, which stimulates its exonuclease activity (Cooper et al., 2000; Li and Comai, 2000; Orren et al., 2001). Interestingly, Ku enables WRN to digest DNA containing 8-oxoadenine and 8-oxoguanine modifications, lesions that block the exonuclease activity of WRN in the absence of Ku (Orren et al., 2001). Two putative Ku-binding motifs are located in the N-terminus and C-terminus of WRN, and the interaction between WRN and Ku facilitates the nucleolytic processing of ends (Karmakar et al., 2002b). A recent study reported that these two Ku-binding motifs of WRN function cooperatively to bind the Ku heterodimer and that the N-terminal Ku-binding motif mediates Ku-dependent stimulation of WRN exonuclease activity, promoting DSB repair (Grundy et al., 2016). DNA-PKcs, phosphorylates WRN at Ser440 and Ser467, and regulates the enzymatic activities of WRN (Yannone et al., 2001; Karmakar et al., 2002a; Kusumoto-Matsuo et al., 2014). The XRCC4-LIG4 complex also interacts with WRN and stimulates its exonuclease activity, but not helicase activity, to generate DNA ends suitable for XRCC4-LIG4-mediated ligation (Kusumoto et al., 2008). Another report indicates that WRN is involved in NHEJ indirectly by upregulating the transcription level of the key NHEJ factor XLF (Liu et al., 2014). Together, the data show that the enzymatic activities of WRN, in particular DNA end processing by the exonuclease activity of WRN, promote NHEJ.
WRN and HR
In addition to NHEJ, WRN promotes HR-mediated DSB repair in multiple ways (Saintigny et al., 2002; Chen et al., 2003). WRN participates in DNA end resection during the early stages of HR. WRN interacts with MRE11 and NBS1, which is enhanced by IR, and results in the promotion of WRN helicase activity (Cheng et al., 2004). WRN also physically interacts with DNA2 and promotes extensive DNA end resection in an RPA-dependent matter (Sturzenegger et al., 2014; Pinto et al., 2016). Moreover, during the late S/G2 and M phases of the cell cycle, cyclin-dependent kinase 1 (CDK1) phosphorylates WRN to promote the long-range end resection by DNA2 at replication-associated DSBs, which stimulates HR and the recovery of collapsed replication forks and promotes the maintenance of chromosome stability (Palermo et al., 2016). Specifically, CDK1-dependent phosphorylation of WRN occurs at Ser1133, and this phosphorylation is required for the WRN–MRE11 interaction and promotes MRE11 foci formation at CPT-induced DSBs (Palermo et al., 2016). Moreover, BRCA1 also directly interacts with WRN and stimulates both the helicase and exonuclease activities of WRN, which likely promotes DNA end resection (Cheng et al., 2006). WRN processes intermediates during HR as it promotes the ATP-dependent translocation of Holliday junctions (Constantinou et al., 2000), disrupts mobile D-loop by promoting branch migration, and degrades the invading strand both prior to and after release from the D-loop (Opresko et al., 2009). WRN has also been found to interact with the following HR proteins: RAD51, RAD54, and RAD52 (Baynton et al., 2003; Otterlei et al., 2006; Lachapelle et al., 2011). In addition, the BRCA1/BARD1 complex interacts with WRN in vivo and stimulates WRN helicase activity toward forked and Holliday junction substrates (Cheng et al., 2006). RAD52 modulates WRN activity and inhibits its ability to unwind four-way Holliday junctions (Baynton et al., 2003). Collectively, WRN plays important roles in multiple steps during HR.
WRN and Alternative End-Joining Pathways
As mentioned above, resected dsDNA with 3′ ssDNA overhangs can be used for HR, MMEJ, and SSA, which is initiated by MRN with the assistance of CtIP (Ronato et al., 2020). Interestingly, WRN actively inhibits DNA end resection by limiting the recruitment of MRE11 and CtIP to DSBs in the G1 phase of the cell cycle (Shamanna et al., 2016b). Accordingly, elevated MMEJ was observed in both WS and WRN-deficient cells, leading to telomere fusions (Shamanna et al., 2016b). In agreement with this finding, limiting MMEJ by depleting CtIP suppresses telomere fusions in WRN-deficient cells (Shamanna et al., 2016b).
WRN interacts with the key SSA player RAD52 and enhances the efficiency of RAD52-mediated strand annealing between non-duplex DNA and homologous sequences contained within a double-stranded plasmid (Baynton et al., 2003). The C-terminal region of WRN may be required for SSA (Muftuoglu et al., 2008). WRN deletion by siRNA causes a 25–50% reduction of SSA-mediated DSB repair in two human cell lines, indicating that WRN promotes SSA (Sturzenegger et al., 2014). However, many questions remain with regard to WRN’s role in SSA, including whether it helps mediate the long resection required for SSA and whether WRN is required for the RAD52-dependent annealing of ssDNA to drive SSA.
WRN and Diseases
Mutations in WRN cause the autosomal-recessive disorder Werner syndrome (MIM #277700), which is a segmental progeria (Oshima et al., 2017; Shamanna et al., 2017; Lebel and MonnatJr., 2018). The average life span of WS patients is 54 years, and the major death causes of WS patients are cancer and myocardial infarction (Huang et al., 2006; Goto et al., 2013; Oshima et al., 2017). WS confers a strong predisposition to a diversity of neoplasia, and two thirds of these neoplasia are collectively thyroid neoplasms, malignant melanoma, meningioma, soft tissue sarcomas, leukemia and pre-leukemic conditions of the bone marrow, and primary bone neoplasms (Goto et al., 2013; Lauper et al., 2013). The elevated risk of these neoplasms ranges from 8.9-fold for thyroid epithelial neoplasms to 53.5-fold for melanoma compared to the normal population (Lauper et al., 2013). Meningioma frequently occurs in WS patients (Tsurubuchi et al., 2008; Huang et al., 2018; Pattankar et al., 2020), and this is likely a consequence of a reduced WRN expression due to the elevated methylation of the WRN promoter (Li et al., 2015). In addition, WRN mutations are also associated with other tumors, such as breast cancer (Romanowicz et al., 2017), oral squamous cell carcinoma (Kuribayashi et al., 2019), and colorectal cancer (Lee et al., 2017; Zhunussova et al., 2019; Zimmer et al., 2020). The cells from WS patients display an increased rate of somatic mutations, chromosome losses, deletions, and genomic rearrangements (Subino et al., 1982; Fukuchi et al., 1989; Oshima et al., 2002; Friedrich et al., 2010).
RECQL4
RECQL4, located at chromosome 8q24.3, encodes a 1,208-amino acid protein, which possesses the highly conserved 3′ to 5′ helicase domain, but does not have the typical RQC and HRDC domains (Figure 3). A recent study identified a novel C-terminal domain containing a Zn2+-binding site and two distinct winged-helix domains, which are not involved in canonical DNA binding or helicase activity (Kaiser et al., 2017). The N-terminus of RECQL4 contains multiple functional domains, including both nuclear and mitochondrial targeting sequences (Burks et al., 2007; Croteau et al., 2012a). Furthermore, an SLD2-like domain is located in the N-terminus of RECQL4, and it is important for DNA replication (Sangrithi et al., 2005; Xu et al., 2009). Many proteins interact with the N-terminal region of RECQ4 (Croteau et al., 2012b). In addition, functional phosphorylation and acetylation events have also been identified in this region (Dietschy et al., 2009; Lu et al., 2017). Consequently, the N-terminus of RECQL4 is essential for cell viability and for the cellular response to IR (Abe et al., 2011; Lu et al., 2017). The C-terminal region of RECQL4 is not well characterized, but is important for cells to survive IR (Kohzaki et al., 2012). The helicase activity of RECQL4 can unwind forked duplexes, D-loops, and bubble structures, but not duplex DNA or Holliday junctions (Rossi et al., 2010). Interestingly, a recent study identified an unwinding activity of RECQL4 and its yeast homolog Hrq1 on G-quadruplexes (Rogers et al., 2017). RECQL4 has a strong ssDNA annealing activity, which may mask the detection of unwound products (Rossi et al., 2010). The helicase activity is important for the role of RECQL4 in DNA repair and prevention of cellular senescence (Lu et al., 2014, 2016). Overall, RECQL4 is involved in various cellular functions, including DNA replication (Sangrithi et al., 2005), multiple repair pathways (Fan and Luo, 2008; Schurman et al., 2009; Singh et al., 2010; Duan et al., 2020), preservation of the mitochondrial genome (Croteau et al., 2012a), and telomere maintenance (Ghosh et al., 2012).
Recruitment of RECQL4 to DSBs
RECQL4-depleted human cells and fibroblasts from Rothmund–Thomson syndrome (RTS) patients are sensitive to IR and other DSB-inducing chemicals, and these cells accumulate unrepaired DSBs, as indicated by elevated γH2AX and 53BP1 foci (Jin et al., 2008; Singh et al., 2010; Kohzaki et al., 2012; Shamanna et al., 2014; Lu et al., 2014, 2016). In Xenopus egg extracts, RECQL4 accumulates on chromatin containing EcoRI-induced DSBs in vitro, which is dependent on RPA, DNA-PK, and ataxia telangiectasia mutated (ATM), but not on DNA replication or RAD51 (Kumata et al., 2007). In human cells, GFP-tagged RECQL4 is recruited immediately to laser−induced DSBs and dissociates from DSBs much more quickly than WRN and BLM, suggesting that RECQL4 only functions at the early time points in DSB repair (Singh et al., 2010; Lu et al., 2016). The unique N−terminus between amino acids 363–492 is the region required for the recruitment of RECQL4 to DSBs (Singh et al., 2010). Moreover, the accumulation of RECQL4 at laser-induced DSBs occurs in both G1 and S/G2 cells (Lu et al., 2017). Inhibition of ATM or knockdown of BLM or WRN does not significantly alter the dynamics of RECQL4 to laser-induced DSBs in human cells (Singh et al., 2010; Lu et al., 2017). The accumulation of RECQL4 at DSBs is stimulated by both phosphorylation at Ser89 and Ser251 by CDK1 and CDK2 and ubiquitination by the DDB1-CUL4A E3 ubiquitin ligase (Lu et al., 2017). However, the ubiquitination sites on RECQL4 by DDB1-CUL4A E3 Ligase have not been identified.
RECQL4 and NHEJ
A role for RECQL4 in NHEJ was first indicated when it was found that RECQL4 binds to restriction enzyme-generated DSBs near the Ku70-binding site and that RECQL4 is required for the repair of these DSBs in Xenopus extracts (Kumata et al., 2007). Depletion of RECQL4 reduced the end-joining activity on DNA substrates with either cohesive or non-cohesive ends in vitro and also decreased the end-joining activity of a GFP reporter plasmid in vivo, providing strong evidence that RECQL4 functions in NHEJ in human cells (Shamanna et al., 2014). Further investigations showed that RECQL4 forms a complex with the Ku70/Ku80 heterodimer through its N-terminal domain and stimulates the DNA binding of Ku70/Ku80 to a blunt-ended dsDNA substrate (Shamanna et al., 2014). Interestingly, the interaction of RECQL4 with the Ku complex is enhanced in the G1 phase of the cell cycle compared to that in S/G2 cells, indicating that RECQL4 promotes NHEJ in G1 cells (Lu et al., 2017).
RECQL4 and HR
As an important protein in the assembly of DNA replication machinery, RECQL4 is highly expressed during the S phase, the cell cycle phase when HR is the dominant DSB repair pathway, and thus it was predicted that it would also play a role in HR (Sangrithi et al., 2005; Im et al., 2009; Xu et al., 2009; Singh et al., 2012). The initial study suggesting RECQL4 functions in HR reported that it forms a complex with RAD51 following treatment with etoposide (Petkovic et al., 2005). Its role was further elucidated when it was found that the depletion of RECQL4 by siRNAs caused a dramatic loss of HR efficiency, as monitored by a GFP reporter assay, and that RECQL4 is required for DNA end resection (Lu et al., 2016). Specifically, MRE1 regulates the recruitment of RECQL4 to DSBs and RECQL4 promotes the nuclease activity of MRE11 in vitro. In addition, RECQL4 also forms a complex with CtIP via its N-terminal domain and promotes the recruitment of CtIP to MRN at DSBs. Furthermore, the helicase activity of RECQL4 promotes DNA end processing and HR, and this process is regulated by CDK1/2-mediated phosphorylation (Lu et al., 2017). Specifically, CDK1 and CDK2 phosphorylate RECQL4 on serines 89 and 251, and this facilitates the interaction between MRE11 and RECQL4 as well as RECQL4 recruitment to DSBs (Lu et al., 2017). Interestingly, RECQL4 promotes and coordinates NHEJ and HR in a cell cycle-dependent manner (Lu et al., 2017). RECQL4 preferably interacts with Ku70 to promote NHEJ in G1 when the overall CDK activity is low. During the S/G2 phases, RECQL4 is phosphorylated by CDK1 and CDK2, which promotes the RECQL4–MRN interaction to stimulate HR. These findings indicate a role for RECQL4 in the regulation of DSB repair pathway choice.
RECQL4 and Alternative End-Joining Pathways
A study recently discovered a role for RECQL4 in DSB repair pathway choice between MMEJ and SSA (Kohzaki et al., 2020). RECQL4ΔC HCT116 cells (lacking the C-terminal domain) exhibit increased SSA activity and decreased MMEJ activity, and ectopic expression of RECQL4 increased HR and MMEJ but repressed SSA (Kohzaki et al., 2020). Knockdown of RAD52 inhibits SSA in RECQL4ΔC HCT116 cells, but does not influence HR and MMEJ (Kohzaki et al., 2020). The involvement of RECQL4 in the initial end resection by MRN/CtIP may account for the pro-MMEJ role of RECQL4 since a short resected ssDNA is required for MMEJ. However, it is unclear how RECQL4 functions in repressing RAD52-mediated SSA.
RECQL4 and Diseases
Mutations in RECQL4 are associated with three rare autosomal-recessive disorders: Rothmund–Thomson syndrome (RTS; OMIM #268400), RAPADILINO (RAPA; OMIM #266280), and Baller–Gerold syndrome (BGS; OMIM #218600) (Siitonen et al., 2009; Larizza et al., 2010, 2013). RTS is accompanied with an increased risk of malignant tumor osteosarcoma in childhood (Wang et al., 2003; Larizza et al., 2010; Lu et al., 2020). RAPADILINO patients develop lymphoma and osteosarcoma (Siitonen et al., 2009), while a BGS patient was reported with lymphoma (Debeljak et al., 2009). In addition, germline mutations of RECQL4 have also been identified to associate with several cancers, including prostate cancer (Paulo et al., 2018), colorectal cancer (Zhunussova et al., 2019), choroid plexus papilloma (Taher et al., 2019), and melanoma (Bodelon et al., 2012; Aoude et al., 2020). Chromosome aberrations, mainly mosaic trisomies and isochromosomes, frequently occur in RTS patient cells, leading to the cancer predisposition of these patients (Larizza et al., 2006). In line with this finding in human RTS cells, defective sister chromatid cohesion, aneuploidy, and cancer predisposition were observed in the cells from an RTS mouse model (Mann et al., 2005). Therefore, RECQL4 is crucial to maintain the integrity of the genome.
RECQL5
RECQL5, encoded by the RECQL5 gene at chromosome 17q25, exists in three isoforms generated by alternative splicing: RECQL5α, RECQL5β, and RECQL5γ. RECQL5α and RECQL5γ consist of 410 and 435 amino acids, respectively, while RECQL5β contains 991 amino acids (Figure 3) (Sekelsky et al., 1999; Shimamoto et al., 2000). All three isoforms have the core helicase domain, while only RECQL5β has helicase activity in vitro (Shimamoto et al., 2000). Additionally, RECQL5β (referred to as RECQL5 in the following text) has a Zn2+-binding motif and a unique C-terminal region harboring multiple specific protein interaction domains: a RAD51-binding domain, internal RNAPII-interacting (IRI) domain, Set2-Rpb1-interacting (SRI) domain, RNA polymerase I (RNAPI)-binding domain, and a proliferating cell nuclear antigen (PCNA)-interacting protein (PIP) motif (Andrs et al., 2020). The Zn2+-binding motif is essential for helicase activity and DNA binding (Ren et al., 2008). RECQL5 has intrinsic ssDNA strand annealing activity, which is inhibited by ATP (Garcia et al., 2004; Ren et al., 2008). However, a recent study showed that RECQL5 possesses a stronger annealing activity on long or small duplexed substrates compared to the other RecQ helicases, which is not inhibited by the presence of ATP (Khadka et al., 2016). RECQL5 efficiently catalyzes the annealing of RNA to DNA in vitro in the presence or absence of ATP (Khadka et al., 2016). However, the cellular function of this activity for RECQL5 is yet to be delineated.
Recruitment of RECQL5 to DSBs
RECQL5-depleted cells accumulate persistent IR-induced 53BP1 foci, implicating a role for RECQL5 in DSB repair (Popuri et al., 2012b). RECQL5 is recruited quickly to laser-induced DSBs and remains for a shorter duration than BLM and WRN, but persists longer than RECQL4 (Zheng et al., 2009; Popuri et al., 2012b). Both the helicase and KIX domains are required for DNA damage recognition and the stable association of RECQL5 to DSB sites (Popuri et al., 2012b). The recruitment of RECQL5 requires MRE11, but not the exonuclease activity of MRE11, and its recruitment to DSB is independent of RNA polymerase II, BLM, WRN, ATM, MDC1, and CtIP (Zheng et al., 2009; Popuri et al., 2012b). RECQL5 interacts with Poly(ADP-ribose) (PAR) and Poly(ADP-ribose) polymerase 1 (PARP1), and PARylation by PARP1 stimulates the recruitment of RECQL5-GFP to laser-induced DSBs (Khadka et al., 2015).
RECQL5 and HR
Deletion of RECQL5 increases HR in MEFs (Hu et al., 2007). Consistent with this, it was found that RECQL5 binds to RAD51, inhibits RAD51-mediated D-loop formation, and displaces RAD51 from ssDNA in assistance with ATP hydrolysis and RPA (Hu et al., 2007; Schwendener et al., 2010). Another study reported that RECQL5 counteracts the inhibitory effect of RAD51 on RAD52-mediated DNA annealing with its ATPase and RAD51-binding activity (Paliwal et al., 2014). Furthermore, RECQL5 deficiency causes an increased occupancy of RAD51 at DSBs and evaluated sister chromatid exchange when the Holliday junction dissolution pathway is inactivated or a high load of DNA damage is generated in the cell (Paliwal et al., 2014). Together, these findings suggest that RECQL5 functions during the postsynaptic phase of synthesis-dependent strand annealing to prevent the formation of aberrant RAD51 filaments on the extended invading strand and inappropriate HR events (Hu et al., 2007; Schwendener et al., 2010; Paliwal et al., 2014). RECQL5 forms a constitutive complex with MRN and specifically inhibits the exonuclease activity of MRE11 (Zheng et al., 2009), but it is unclear whether this activity plays a role in the ability of RECQL5 to suppress HR. Interestingly, it was reported that RECQL5 promotes HR-mediated repair with non-crossover products in a direct repeat GFP (DR-GFP) reporter assay in both U2OS and HEK293 cells (Paliwal et al., 2014; Khadka et al., 2015). RECQL5 deficiency in Drosophila causes sensitivity to IR and DSBs induced by the I-SceI endonuclease and impairs SSA-mediated DSB repair (Chen et al., 2010), but it is unknown whether RECQL5 modulates SSA in mammalian cells.
RECQL5 and Cancers
The downregulation of RECQ5 leads to a transcription-dependent chromosome fragmentation during S phase of the cell cycle as well as the accumulation of chromosomal rearrangements with the breakpoints located in genes and common fragile sites, indicating the importance of this helicase in maintaining genome integrity (Li et al., 2011; Saponaro et al., 2014). Although defects in RECQL5 have not been associated with human genetic disorders, mutations in RECQL5 have been associated with tumorigenesis, including breast cancer (He et al., 2014), osteosarcoma (Zhi et al., 2014; Dong et al., 2015), NUT midline carcinoma (Stirnweiss et al., 2017), head and neck cancer (Das et al., 2018), and hereditary diffuse gastric cancer (Fewings et al., 2018).
Perspective Remarks
A significant number of clues have uncovered the roles that the RecQ helicases play in DSB repair and the maintenance of genome stability. Furthermore, the evidence has started to reveal how defects in the RecQ helicases drive specific genetic disorders and carcinogenesis. However, many questions still remain, including: (1) a clear role for each RecQ helicase’s enzymatic activity in DSB repair is yet to be demonstrated; (2) it is still unclear how BLM, WRN, and RECQL4 cooperate in 5′ DNA end resection during HR; (3) RECQL4 interacts with Ku70/80 heterodimers and is required for NHEJ, but its role in this pathway is unknown; (4) the role of RecQ helicases in MMEJ and SSA are also limited; (5) does RECQL5 play a role in RNA-mediated DSB repair; (6) do the RecQ helicases play a direct or passive role in DSB repair pathway choice; (7) what role does posttranslational modifications play in regulating the RecQ helicases; and (8) can RecQ helicases be targeted for specific cancer therapies. Among these questions, investigating the posttranslational modifications of RecQ helicases may shed light on many unknown mechanisms, including the enzymatic roles of RecQ helicases in specific steps of DSB repair pathways, coordination between the RecQ helicases during specific repair processes, and the role(s) of RecQ helicases in DSB repair pathway choice. Thanks to the great advance of mass spectrometry, hundreds of posttranslational modifications, including phosphorylation, acetylation, ubiquitination, etc., have been identified on RecQ helicases1 (Hornbeck et al., 2012, 2015, 2019). However, the biological functions of these modifications are yet to be elucidated. Hence, it would be promising to investigate posttranslational modifications of RecQ helicases with the combination of spatial and temporal factors as well as stresses. Recently, WRN was identified as a synthetic lethal vulnerability in cancers with microsatellite instability (Chan et al., 2019; van Wietmarschen et al., 2020). Interestingly, CPT treatment leads to the degradation of WRN (Shamanna et al., 2016a; Li et al., 2020), suggesting that cancers with microsatellite instability may be a candidate target for CPT treatment to drive WRN-specific vulnerable tumors. These findings together demonstrate WRN as an important genome guardian to prevent diseases and as a potential drug target. Collectively, these and other unanswered questions drive us and others to understand the role of each RecQ helicase in DSB repair, how defects drive human disorders, and how these enzymes may be targets for therapy.
Author Contributions
HL and AD conceived the concept and wrote the manuscript together. Both authors contributed to the article and approved the submitted version.
Funding
This research was supported by grants from the National Institutes of Health CA092584, CA162804, and GM04725 to AD and by an Initiative Seed Grant for Career Development from the Department of Radiation Oncology at UT Southwestern Medical Center to HL.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
Abe, T., Yoshimura, A., Hosono, Y., Tada, S., Seki, M., and Enomoto, T. (2011). The N-terminal region of RECQL4 lacking the Helicase domain is both essential and sufficient for the viability of vertebrate cells. Role of the N-terminal region of RECQL4 in cells. Biochim. Biophys. Acta 1813, 473–479. doi: 10.1016/j.bbamcr.2011.01.001
Ammazzalorso, F., Pirzio, L. M., Bignami, M., Franchitto, A., and Pichierri, P. (2010). ATR and ATM differently regulate WRN to prevent DSBs at stalled replication forks and promote replication fork recovery. EMBO J. 29, 3156–3169. doi: 10.1038/emboj.2010.205
Andrs, M., Hasanova, Z., Oravetzova, A., Dobrovolna, J., and Janscak, P. (2020). RECQ5: a mysterious Helicase at the interface of DNA replication and transcription. Genes 11:232. doi: 10.3390/genes11020232
Aoude, L. G., Bonazzi, V. F., Brosda, S., Patel, K., Koufariotis, L. T., Oey, H., et al. (2020). Pathogenic germline variants are associated with poor survival in stage III/IV melanoma patients. Sci. Rep. 10:17687.
Bachrati, C. Z., Borts, R. H., and Hickson, I. D. (2006). Mobile D-loops are a preferred substrate for the Bloom’s syndrome Helicase. Nucleic Acids Res. 34, 2269–2279. doi: 10.1093/nar/gkl258
Banerjee, T., Sommers, J. A., Huang, J., Seidman, M. M., and Brosh, R. M. Jr. (2015). Catalytic strand separation by RECQ1 is required for RPA-mediated response to replication stress. Curr. Biol. 25, 2830–2838. doi: 10.1016/j.cub.2015.09.026
Barefield, C., and Karlseder, J. (2012). The BLM Helicase contributes to telomere maintenance through processing of late-replicating intermediate structures. Nucleic Acids Res. 40, 7358–7367. doi: 10.1093/nar/gks407
Baris, H. N., Kedar, I., Halpern, G. J., Shohat, T., Magal, N., Ludman, M. D., et al. (2007). Prevalence of breast and colorectal cancer in Ashkenazi Jewish carriers of Fanconi anemia and Bloom syndrome. Isr. Med. Assoc. J. 9, 847–850.
Baynton, K., Otterlei, M., Bjoras, M., von Kobbe, C., Bohr, V. A., and Seeberg, E. (2003). WRN interacts physically and functionally with the recombination mediator protein RAD52. J. Biol. Chem. 278, 36476–36486. doi: 10.1074/jbc.m303885200
Bennardo, N., Cheng, A., Huang, N., and Stark, J. M. (2008). Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 4:e1000110. doi: 10.1371/journal.pgen.1000110
Beresten, S. F., Stan, R., van Brabant, A. J., Ye, T., Naureckiene, S., and Ellis, N. A. (1999). Purification of overexpressed hexahistidine-tagged BLM N431 as oligomeric complexes. Protein Expr. Purif. 17, 239–248. doi: 10.1006/prep.1999.1135
Berti, M., Ray Chaudhuri, A., Thangavel, S., Gomathinayagam, S., Kenig, S., Vujanovic, M., et al. (2013). Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat. Struct. Mol. Biol. 20, 347–354. doi: 10.1038/nsmb.2501
Bhargava, R., Onyango, D. O., and Stark, J. M. (2016). Regulation of single-strand annealing and its role in genome maintenance. Trends Genet. 32, 566–575. doi: 10.1016/j.tig.2016.06.007
Bochman, M. L., Paeschke, K., Chan, A., and Zakian, V. A. (2014). Hrq1, a homolog of the human RecQ4 Helicase, acts catalytically and structurally to promote genome integrity. Cell Rep. 6, 346–356. doi: 10.1016/j.celrep.2013.12.037
Bodelon, C., Pfeiffer, R. M., Bollati, V., Debbache, J., Calista, D., Ghiorzo, P., et al. (2012). On the interplay of telomeres, nevi and the risk of melanoma. PLoS One 7:e52466. doi: 10.1371/journal.pone.0052466
Bohr, V. A. (2008). Rising from the RecQ-age: the role of human RecQ Helicases in genome maintenance. Trends Biochem. Sci. 33, 609–620. doi: 10.1016/j.tibs.2008.09.003
Boulton, S. J., and Jackson, S. P. (1996). Saccharomyces cerevisiae Ku70 potentiates illegitimate DNA double-strand break repair and serves as a barrier to error-prone DNA repair pathways. EMBO J. 15, 5093–5103. doi: 10.1002/j.1460-2075.1996.tb00890.x
Bouwman, B. A. M., and Crosetto, N. (2018). Endogenous DNA double-strand breaks during DNA transactions: emerging insights and methods for genome-wide profiling. Genes 9:632. doi: 10.3390/genes9120632
Brosh, R. M. Jr., Li, J. L., Kenny, M. K., Karow, J. K., Cooper, M. P., Kureekattil, R. P., et al. (2000). Replication protein A physically interacts with the Bloom’s syndrome protein and stimulates its Helicase activity. J. Biol. Chem. 275, 23500–23508. doi: 10.1074/jbc.m001557200
Brosh, R. M. Jr., Opresko, P. L., and Bohr, V. A. (2006). Enzymatic mechanism of the WRN Helicase/nuclease. Methods Enzymol. 409, 52–85. doi: 10.1016/s0076-6879(05)09004-x
Brosh, R. M. Jr., Waheed, J., and Sommers, J. A. (2002). Biochemical characterization of the DNA substrate specificity of Werner syndrome Helicase. J. Biol. Chem. 277, 23236–23245. doi: 10.1074/jbc.m111446200
Bugreev, D. V., Mazina, O. M., and Mazin, A. V. (2009). Bloom syndrome Helicase stimulates RAD51 DNA strand exchange activity through a novel mechanism. J. Biol. Chem. 284, 26349–26359. doi: 10.1074/jbc.m109.029371
Bugreev, D. V., Yu, X., Egelman, E. H., and Mazin, A. V. (2007). Novel pro- and anti-recombination activities of the Bloom’s syndrome Helicase. Genes Dev. 21, 3085–3094. doi: 10.1101/gad.1609007
Burks, L. M., Yin, J., and Plon, S. E. (2007). Nuclear import and retention domains in the amino terminus of RECQL4. Gene 391, 26–38. doi: 10.1016/j.gene.2006.11.019
Cannavo, E., and Cejka, P. (2014). Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature 514, 122–125. doi: 10.1038/nature13771
Ceccaldi, R., Rondinelli, B., and D’Andrea, A. D. (2016). Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 26, 52–64. doi: 10.1016/j.tcb.2015.07.009
Chaganti, R. S., Schonberg, S., and German, J. (1974). A manyfold increase in sister chromatid exchanges in Bloom’s syndrome lymphocytes. Proc. Natl. Acad. Sci. U.S.A. 71, 4508–4512. doi: 10.1073/pnas.71.11.4508
Chan, E. M., Shibue, T., McFarland, J. M., Gaeta, B., Ghandi, M., Dumont, N., et al. (2019). WRN Helicase is a synthetic lethal target in microsatellite unstable cancers. Nature 568, 551–556.
Chen, L., Huang, S., Lee, L., Davalos, A., Schiestl, R. H., Campisi, J., et al. (2003). WRN, the protein deficient in Werner syndrome, plays a critical structural role in optimizing DNA repair. Aging Cell 2, 191–199. doi: 10.1046/j.1474-9728.2003.00052.x
Chen, Y., Dui, W., Yu, Z., Li, C., Ma, J., and Jiao, R. (2010). Drosophila RecQ5 is required for efficient SSA repair and suppression of LOH in vivo. Protein Cell 1, 478–490. doi: 10.1007/s13238-010-0058-2
Cheng, W. H., Kusumoto, R., Opresko, P. L., Sui, X., Huang, S., Nicolette, M. L., et al. (2006). Collaboration of Werner syndrome protein and BRCA1 in cellular responses to DNA interstrand cross-links. Nucleic Acids Res. 34, 2751–2760. doi: 10.1093/nar/gkl362
Cheng, W. H., von Kobbe, C., Opresko, P. L., Arthur, L. M., Komatsu, K., Seidman, M. M., et al. (2004). Linkage between Werner syndrome protein and the Mre11 complex via Nbs1. J. Biol. Chem. 279, 21169–21176. doi: 10.1074/jbc.m312770200
Cheok, C. F., Wu, L., Garcia, P. L., Janscak, P., and Hickson, I. D. (2005). The Bloom’s syndrome Helicase promotes the annealing of complementary single-stranded DNA. Nucleic Acids Res. 33, 3932–3941. doi: 10.1093/nar/gki712
Chi, X. Y., Li, Y., and Qiu, X. Y. (2020). V(D)J recombination, somatic hypermutation and class switch recombination of immunoglobulins: mechanism and regulation. Immunology 160, 233–247. doi: 10.1111/imm.13176
Choi, D. H., Lee, R., Kwon, S. H., and Bae, S. H. (2013). Hrq1 functions independently of Sgs1 to preserve genome integrity in Saccharomyces cerevisiae. J. Microbiol. 51, 105–112. doi: 10.1007/s12275-013-3048-2
Ciccia, A., and Elledge, S. J. (2010). The DNA damage response: making it safe to play with knives. Mol. Cell 40, 179–204. doi: 10.1016/j.molcel.2010.09.019
Cleary, S. P., Zhang, W., Di Nicola, N., Aronson, M., Aube, J., Steinman, A., et al. (2003). Heterozygosity for the BLM(Ash) mutation and cancer risk. Cancer Res. 63, 1769–1771.
Cobb, J. A., and Bjergbaek, L. (2006). RecQ Helicases: lessons from model organisms. Nucleic Acids Res. 34, 4106–4114. doi: 10.1093/nar/gkl557
Constantinou, A., Tarsounas, M., Karow, J. K., Brosh, R. M., Bohr, V. A., Hickson, I. D., et al. (2000). Werner’s syndrome protein (WRN) migrates Holliday junctions and co-localizes with RPA upon replication arrest. EMBO Rep. 1, 80–84. doi: 10.1093/embo-reports/kvd004
Cooper, M. P., Machwe, A., Orren, D. K., Brosh, R. M., Ramsden, D., and Bohr, V. A. (2000). Ku complex interacts with and stimulates the Werner protein. Genes Dev. 14, 907–912.
Croteau, D. L., Popuri, V., Opresko, P. L., and Bohr, V. A. (2014). Human RecQ Helicases in DNA repair, recombination, and replication. Annu. Rev. Biochem. 83, 519–552. doi: 10.1146/annurev-biochem-060713-035428
Croteau, D. L., Rossi, M. L., Canugovi, C., Tian, J., Sykora, P., Ramamoorthy, M., et al. (2012a). RECQL4 localizes to mitochondria and preserves mitochondrial DNA integrity. Aging Cell 11, 456–466. doi: 10.1111/j.1474-9726.2012.00803.x
Croteau, D. L., Singh, D. K., Hoh Ferrarelli, L., Lu, H., and Bohr, V. A. (2012b). RECQL4 in genomic instability and aging. Trends Genet. 28, 624–631. doi: 10.1016/j.tig.2012.08.003
Cui, S., Klima, R., Ochem, A., Arosio, D., Falaschi, A., and Vindigni, A. (2003). Characterization of the DNA-unwinding activity of human RECQ1, a Helicase specifically stimulated by human replication protein A. J. Biol. Chem. 278, 1424–1432. doi: 10.1074/jbc.m209407200
Cunniff, C., Bassetti, J. A., and Ellis, N. A. (2017). Bloom’s syndrome: clinical spectrum, molecular pathogenesis, and cancer predisposition. Mol. Syndromol. 8, 4–23. doi: 10.1159/000452082
Cybulski, C., Carrot-Zhang, J., Kluzniak, W., Rivera, B., Kashyap, A., Wokolorczyk, D., et al. (2015). Germline RECQL mutations are associated with breast cancer susceptibility. Nat. Genet. 47, 643–646. doi: 10.1038/ng.3284
Daley, J. M., Jimenez-Sainz, J., Wang, W., Miller, A. S., Xue, X., Nguyen, K. A., et al. (2017). Enhancement of BLM-DNA2-mediated long-range DNA end resection by CtIP. Cell Rep. 21, 324–332. doi: 10.1016/j.celrep.2017.09.048
Das, R., Kundu, S., Laskar, S., Choudhury, Y., and Ghosh, S. K. (2018). Assessment of DNA repair susceptibility genes identified by whole exome sequencing in head and neck cancer. DNA Repair. 66–67, 50–63. doi: 10.1016/j.dnarep.2018.04.005
Datta, A., Dhar, S., Awate, S., and Brosh, R. M. Jr. (2020). Synthetic Lethal Interactions of RECQ Helicases. Trends Cancer 7, 146–161. doi: 10.1016/j.trecan.2020.09.001
Davis, A. J., Chen, B. P., and Chen, D. J. (2014). DNA-PK: a dynamic enzyme in a versatile DSB repair pathway. DNA Repair. 17, 21–29. doi: 10.1016/j.dnarep.2014.02.020
Davis, A. J., and Chen, D. J. (2013). DNA double strand break repair via non-homologous end-joining. Transl. Cancer Res. 2, 130–143.
de Renty, C., and Ellis, N. A. (2017). Bloom’s syndrome: why not premature aging?: a comparison of the BLM and WRN Helicases. Age. Res. Rev. 33, 36–51.
de Voer, R. M., Hahn, M. M., Mensenkamp, A. R., Hoischen, A., Gilissen, C., Henkes, A., et al. (2015). Deleterious germline BLM mutations and the risk for early-onset colorectal cancer. Sci. Rep. 5:14060.
Debeljak, M., Zver, A., and Jazbec, J. (2009). A patient with Baller-gerold syndrome and midline NK/T lymphoma. Am. J. Med. Genet. A 149A, 755–759. doi: 10.1002/ajmg.a.32736
Debnath, S., and Sharma, S. (2020). RECQ1 Helicase in genomic stability and cancer. Genes 11:622. doi: 10.3390/genes11060622
Deshpande, R. A., Myler, L. R., Soniat, M. M., Makharashvili, N., Lee, L., Lees-Miller, S. P., et al. (2020). DNA-dependent protein kinase promotes DNA end processing by MRN and CtIP. Sci. Adv. 6:eaay0922. doi: 10.1126/sciadv.aay0922
Dietschy, T., Shevelev, I., Pena-Diaz, J., Huhn, D., Kuenzle, S., Mak, R., et al. (2009). p300-mediated acetylation of the Rothmund-Thomson-syndrome gene product RECQL4 regulates its subcellular localization. J. Cell Sci. 122, 1258–1267. doi: 10.1242/jcs.037747
Doherty, K. M., Sommers, J. A., Gray, M. D., Lee, J. W., von Kobbe, C., Thoma, N. H., et al. (2005). Physical and functional mapping of the replication protein a interaction domain of the werner and bloom syndrome Helicases. J. Biol. Chem. 280, 29494–29505. doi: 10.1074/jbc.m500653200
Dong, Y. Z., Huang, Y. X., and Lu, T. (2015). Single nucleotide polymorphism in the RECQL5 gene increased osteosarcoma susceptibility in a Chinese Han population. Genet. Mol. Res. 14, 1899–1902. doi: 10.4238/2015.march.13.18
Drosopoulos, W. C., Kosiyatrakul, S. T., and Schildkraut, C. L. (2015). BLM Helicase facilitates telomere replication during leading strand synthesis of telomeres. J. Cell Biol. 210, 191–208. doi: 10.1083/jcb.201410061
Duan, S., Han, X., Akbari, M., Croteau, D. L., Rasmussen, L. J., and Bohr, V. A. (2020). Interaction between RECQL4 and OGG1 promotes repair of oxidative base lesion 8-oxoG and is regulated by SIRT1 deacetylase. Nucleic Acids Res. 48, 6530–6546. doi: 10.1093/nar/gkaa392
Ellenberger, T., and Tomkinson, A. E. (2008). Eukaryotic DNA ligases: structural and functional insights. Annu. Rev. Biochem. 77, 313–338. doi: 10.1146/annurev.biochem.77.061306.123941
Ellis, N. A., Groden, J., Ye, T. Z., Straughen, J., Lennon, D. J., Ciocci, S., et al. (1995). The Bloom’s syndrome gene product is homologous to RecQ Helicases. Cell 83, 655–666. doi: 10.1016/0092-8674(95)90105-1
Fairman, M. P., Johnson, A. P., and Thacker, J. (1992). Multiple components are involved in the efficient joining of double stranded DNA breaks in human cell extracts. Nucleic Acids Res. 20, 4145–4152. doi: 10.1093/nar/20.16.4145
Fan, W., and Luo, J. (2008). RecQ4 facilitates UV light-induced DNA damage repair through interaction with nucleotide excision repair factor xeroderma pigmentosum group A (XPA). J. Biol. Chem. 283, 29037–29044. doi: 10.1074/jbc.m801928200
Fell, V. L., and Schild-Poulter, C. (2015). The Ku heterodimer: function in DNA repair and beyond. Mutat. Res. Rev. Mutat. Res. 763, 15–29. doi: 10.1016/j.mrrev.2014.06.002
Fewings, E., Larionov, A., Redman, J., Goldgraben, M. A., Scarth, J., Richardson, S., et al. (2018). Germline pathogenic variants in PALB2 and other cancer-predisposing genes in families with hereditary diffuse gastric cancer without CDH1 mutation: a whole-exome sequencing study. Lancet Gastroenterol. Hepatol. 3, 489–498. doi: 10.1016/s2468-1253(18)30079-7
Findlay, S., Heath, J., Luo, V. M., Malina, A., Morin, T., Coulombe, Y., et al. (2018). SHLD2/FAM35A co-operates with REV7 to coordinate DNA double-strand break repair pathway choice. EMBO J. 37:e100158.
Friedrich, K., Lee, L., Leistritz, D. F., Nurnberg, G., Saha, B., Hisama, F. M., et al. (2010). WRN mutations in Werner syndrome patients: genomic rearrangements, unusual intronic mutations and ethnic-specific alterations. Hum. Genet. 128, 103–111. doi: 10.1007/s00439-010-0832-5
Fukuchi, K., Martin, G. M., and Monnat, R. J. Jr. (1989). Mutator phenotype of Werner syndrome is characterized by extensive deletions. Proc. Natl. Acad. Sci. U.S.A. 86, 5893–5897. doi: 10.1073/pnas.86.15.5893
Garcia, P. L., Liu, Y., Jiricny, J., West, S. C., and Janscak, P. (2004). Human RECQ5beta, a protein with DNA Helicase and strand-annealing activities in a single polypeptide. EMBO J. 23, 2882–2891. doi: 10.1038/sj.emboj.7600301
Garcia, V., Phelps, S. E., Gray, S., and Neale, M. J. (2011). Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature 479, 241–244. doi: 10.1038/nature10515
German, J., Sanz, M. M., Ciocci, S., Ye, T. Z., and Ellis, N. A. (2007). Syndrome-causing mutations of the BLM gene in persons in the Bloom’s syndrome registry. Hum. Mutat. 28, 743–753. doi: 10.1002/humu.20501
Ghosh, A. K., Rossi, M. L., Singh, D. K., Dunn, C., Ramamoorthy, M., Croteau, D. L., et al. (2012). RECQL4, the protein mutated in Rothmund-Thomson syndrome, functions in telomere maintenance. J. Biol. Chem. 287, 196–209. doi: 10.1074/jbc.m111.295063
Goto, M., Imamura, O., Kuromitsu, J., Matsumoto, T., Yamabe, Y., Tokutake, Y., et al. (1997). Analysis of helicase gene mutations in Japanese Werner’s syndrome patients. Hum. Genet. 99, 191–193. doi: 10.1007/s004390050336
Goto, M., Ishikawa, Y., Sugimoto, M., and Furuichi, Y. (2013). Werner syndrome: a changing pattern of clinical manifestations in Japan (1917~2008). Biosci. Trends 7, 13–22.
Goto, M., Rubenstein, M., Weber, J., Woods, K., and Drayna, D. (1992). Genetic linkage of Werner’s syndrome to five markers on chromosome 8. Nature 355, 735–738. doi: 10.1038/355735a0
Grabarz, A., Guirouilh-Barbat, J., Barascu, A., Pennarun, G., Genet, D., Rass, E., et al. (2013). A role for BLM in double-strand break repair pathway choice: prevention of CtIP/Mre11-mediated alternative nonhomologous end-joining. Cell Rep. 5, 21–28. doi: 10.1016/j.celrep.2013.08.034
Gravel, S., Chapman, J. R., Magill, C., and Jackson, S. P. (2008). DNA Helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 22, 2767–2772. doi: 10.1101/gad.503108
Grierson, P. M., Acharya, S., and Groden, J. (2013). Collaborating functions of BLM and DNA topoisomerase I in regulating human rDNA transcription. Mutat. Res. 743–744, 89–96. doi: 10.1016/j.mrfmmm.2012.12.002
Grierson, P. M., Lillard, K., Behbehani, G. K., Combs, K. A., Bhattacharyya, S., Acharya, S., et al. (2012). BLM Helicase facilitates RNA polymerase I-mediated ribosomal RNA transcription. Hum. Mol. Genet. 21, 1172–1183. doi: 10.1093/hmg/ddr545
Gruber, S. B., Ellis, N. A., Scott, K. K., Almog, R., Kolachana, P., Bonner, J. D., et al. (2002). BLM heterozygosity and the risk of colorectal cancer. Science 297:2013. doi: 10.1126/science.1074399
Grundy, G. J., Rulten, S. L., Arribas-Bosacoma, R., Davidson, K., Kozik, Z., Oliver, A. W., et al. (2016). The Ku-binding motif is a conserved module for recruitment and stimulation of non-homologous end-joining proteins. Nat. Commun. 7:11242.
Gudmundsdottir, K., Lord, C. J., Witt, E., Tutt, A. N., and Ashworth, A. (2004). DSS1 is required for RAD51 focus formation and genomic stability in mammalian cells. EMBO Rep. 5, 989–993. doi: 10.1038/sj.embor.7400255
Gyimesi, M., Harami, G. M., Sarlos, K., Hazai, E., Bikadi, Z., and Kovacs, M. (2012). Complex activities of the human Bloom’s syndrome Helicase are encoded in a core region comprising the RecA and Zn-binding domains. Nucleic Acids Res. 40, 3952–3963. doi: 10.1093/nar/gks008
He, Y. J., Qiao, Z. Y., Gao, B., Zhang, X. H., and Wen, Y. Y. (2014). Association between RECQL5 genetic polymorphisms and susceptibility to breast cancer. Tumour Biol. 35, 12201–12204. doi: 10.1007/s13277-014-2528-2
Hickson, I. D. (2003). RecQ Helicases: caretakers of the genome. Nat. Rev. Cancer 3, 169–178. doi: 10.1038/nrc1012
Hornbeck, P. V., Kornhauser, J. M., Latham, V., Murray, B., Nandhikonda, V., Nord, A., et al. (2019). 15 years of PhosphoSitePlus(R): integrating post-translationally modified sites, disease variants and isoforms. Nucleic Acids Res. 47, D433–D441.
Hornbeck, P. V., Kornhauser, J. M., Tkachev, S., Zhang, B., Skrzypek, E., Murray, B., et al. (2012). PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 40, D261–D270.
Hornbeck, P. V., Zhang, B., Murray, B., Kornhauser, J. M., Latham, V., and Skrzypek, E. (2015). PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 43, D512–D520.
Howard, S. M., Yanez, D. A., and Stark, J. M. (2015). DNA damage response factors from diverse pathways, including DNA crosslink repair, mediate alternative end joining. PLoS Genet. 11:e1004943. doi: 10.1371/journal.pgen.1004943
Hu, Y., Raynard, S., Sehorn, M. G., Lu, X., Bussen, W., Zheng, L., et al. (2007). RECQL5/Recql5 Helicase regulates homologous recombination and suppresses tumor formation via disruption of Rad51 presynaptic filaments. Genes Dev. 21, 3073–3084. doi: 10.1101/gad.1609107
Huang, G., Feng, J., Hao, S., Li, D., Wang, K., Wang, L., et al. (2018). CASP8, XRCC1, WRN, NF2, and BRIP1 polymorphisms analysis shows their genetic susceptibility for meningioma risk and the association with tumor-related phenotype in a Chinese population. World Neurosurg. 114, e883–e891.
Huang, S., Lee, L., Hanson, N. B., Lenaerts, C., Hoehn, H., Poot, M., et al. (2006). The spectrum of WRN mutations in Werner syndrome patients. Hum. Mutat. 27, 558–567.
Hustedt, N., and Durocher, D. (2016). The control of DNA repair by the cell cycle. Nat. Cell Biol. 19, 1–9.
Im, J. S., Ki, S. H., Farina, A., Jung, D. S., Hurwitz, J., and Lee, J. K. (2009). Assembly of the Cdc45-Mcm2-7-GINS complex in human cells requires the Ctf4/And-1, RecQL4, and Mcm10 proteins. Proc. Natl. Acad. Sci. U.S.A. 106, 15628–15632. doi: 10.1073/pnas.0908039106
Imamura, O., Fujita, K., Itoh, C., Takeda, S., Furuichi, Y., and Matsumoto, T. (2002). Werner and Bloom Helicases are involved in DNA repair in a complementary fashion. Oncogene 21, 954–963. doi: 10.1038/sj.onc.1205143
Janscak, P., Garcia, P. L., Hamburger, F., Makuta, Y., Shiraishi, K., Imai, Y., et al. (2003). Characterization and mutational analysis of the RecQ core of the bloom syndrome protein. J. Mol. Biol. 330, 29–42. doi: 10.1016/s0022-2836(03)00534-5
Jensen, R. B., Carreira, A., and Kowalczykowski, S. C. (2010). Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 467, 678–683. doi: 10.1038/nature09399
Jin, W., Liu, H., Zhang, Y., Otta, S. K., Plon, S. E., and Wang, L. L. (2008). Sensitivity of RECQL4-deficient fibroblasts from Rothmund-Thomson syndrome patients to genotoxic agents. Hum. Genet. 123, 643–653. doi: 10.1007/s00439-008-0518-4
Johnson, J. E., Cao, K., Ryvkin, P., Wang, L. S., and Johnson, F. B. (2010). Altered gene expression in the Werner and Bloom syndromes is associated with sequences having G-quadruplex forming potential. Nucleic Acids Res. 38, 1114–1122. doi: 10.1093/nar/gkp1103
Kabotyanski, E. B., Gomelsky, L., Han, J. O., Stamato, T. D., and Roth, D. B. (1998). Double-strand break repair in Ku86- and XRCC4-deficient cells. Nucleic Acids Res. 26, 5333–5342. doi: 10.1093/nar/26.23.5333
Kaiser, S., Sauer, F., and Kisker, C. (2017). The structural and functional characterization of human RecQ4 reveals insights into its Helicase mechanism. Nat. Commun. 8:15907.
Karmakar, P., Piotrowski, J., Brosh, R. M. Jr., Sommers, J. A., Miller, S. P., Cheng, W. H., et al. (2002a). Werner protein is a target of DNA-dependent protein kinase in vivo and in vitro, and its catalytic activities are regulated by phosphorylation. J. Biol. Chem. 277, 18291–18302. doi: 10.1074/jbc.m111523200
Karmakar, P., Snowden, C. M., Ramsden, D. A., and Bohr, V. A. (2002b). Ku heterodimer binds to both ends of the Werner protein and functional interaction occurs at the Werner N-terminus. Nucleic Acids Res. 30, 3583–3591. doi: 10.1093/nar/gkf482
Karmakar, P., Seki, M., Kanamori, M., Hashiguchi, K., Ohtsuki, M., Murata, E., et al. (2006). BLM is an early responder to DNA double-strand breaks. Biochem. Biophys. Res. Commun. 348, 62–69. doi: 10.1016/j.bbrc.2006.07.037
Khadka, P., Croteau, D. L., and Bohr, V. A. (2016). RECQL5 has unique strand annealing properties relative to the other human RecQ Helicase proteins. DNA Repair. 37, 53–66. doi: 10.1016/j.dnarep.2015.11.005
Khadka, P., Hsu, J. K., Veith, S., Tadokoro, T., Shamanna, R. A., Mangerich, A., et al. (2015). Differential and concordant roles for Poly(ADP-Ribose) Polymerase 1 and Poly(ADP-Ribose) in Regulating WRN and RECQL5 Activities. Mol. Cell Biol. 35, 3974–3989. doi: 10.1128/mcb.00427-15
Kikuchi, K., Abdel-Aziz, H. I., Taniguchi, Y., Yamazoe, M., Takeda, S., and Hirota, K. (2009). Bloom DNA Helicase facilitates homologous recombination between diverged homologous sequences. J. Biol. Chem. 284, 26360–26367. doi: 10.1074/jbc.m109.029348
Kitano, K., Kim, S. Y., and Hakoshima, T. (2010). Structural basis for DNA strand separation by the unconventional winged-helix domain of RecQ Helicase WRN. Structure 18, 177–187. doi: 10.1016/j.str.2009.12.011
Kitano, K., Yoshihara, N., and Hakoshima, T. (2007). Crystal structure of the HRDC domain of human Werner syndrome protein, WRN. J. Biol. Chem. 282, 2717–2728. doi: 10.1074/jbc.m610142200
Kohzaki, M., Chiourea, M., Versini, G., Adachi, N., Takeda, S., Gagos, S., et al. (2012). The Helicase domain and C-terminus of human RecQL4 facilitate replication elongation on DNA templates damaged by ionizing radiation. Carcinogenesis 33, 1203–1210. doi: 10.1093/carcin/bgs149
Kohzaki, M., Ootsuyama, A., Sun, L., Moritake, T., and Okazaki, R. (2020). Human RECQL4 represses the RAD52-mediated single-strand annealing pathway after ionizing radiation or cisplatin treatment. Int. J. Cancer 146, 3098–3113. doi: 10.1002/ijc.32670
Kumata, Y., Tada, S., Yamanada, Y., Tsuyama, T., Kobayashi, T., Dong, Y. P., et al. (2007). Possible involvement of RecQL4 in the repair of double-strand DNA breaks in Xenopus egg extracts. Biochim. Biophys. Acta 1773, 556–564. doi: 10.1016/j.bbamcr.2007.01.005
Kuribayashi, N., Uchida, D., Hamasaki, Y., and Kawamata, H. (2019). Oral squamous cell carcinoma arising in a patient with Werner syndrome. Int. J. Oral. Maxillofac. Surg. 48, 1394–1397. doi: 10.1016/j.ijom.2019.06.005
Kusumoto, R., Dawut, L., Marchetti, C., Wan Lee, J., Vindigni, A., Ramsden, D., et al. (2008). Werner protein cooperates with the XRCC4-DNA ligase IV complex in end-processing. Biochemistry 47, 7548–7556. doi: 10.1021/bi702325t
Kusumoto-Matsuo, R., Ghosh, D., Karmakar, P., May, A., Ramsden, D., and Bohr, V. A. (2014). Serines 440 and 467 in the Werner syndrome protein are phosphorylated by DNA-PK and affects its dynamics in response to DNA double strand breaks. Aging 6, 70–81. doi: 10.18632/aging.100629
Kwong, A., Shin, V. Y., Cheuk, I. W. Y., Chen, J., Au, C. H., Ho, D. N., et al. (2016). Germline RECQL mutations in high risk Chinese breast cancer patients. Breast Cancer Res. Treat. 157, 211–215. doi: 10.1007/s10549-016-3784-1
Lachapelle, S., Gagne, J. P., Garand, C., Desbiens, M., Coulombe, Y., Bohr, V. A., et al. (2011). Proteome-wide identification of WRN-interacting proteins in untreated and nuclease-treated samples. J. Proteome Res. 10, 1216–1227. doi: 10.1021/pr100990s
Lan, L., Nakajima, S., Komatsu, K., Nussenzweig, A., Shimamoto, A., Oshima, J., et al. (2005). Accumulation of Werner protein at DNA double-strand breaks in human cells. J. Cell Sci. 118, 4153–4162. doi: 10.1242/jcs.02544
Larizza, L., Magnani, I., and Roversi, G. (2006). Rothmund-Thomson syndrome and RECQL4 defect: splitting and lumping. Cancer Lett. 232, 107–120. doi: 10.1016/j.canlet.2005.07.042
Larizza, L., Roversi, G., and Verloes, A. (2013). Clinical utility gene card for: Rothmund-Thomson syndrome. Eur. J. Hum. Genet. 21:260.
Larizza, L., Roversi, G., and Volpi, L. (2010). Rothmund-Thomson syndrome. Orphanet. J. Rare Dis. 5:2.
Lauper, J. M., Krause, A., Vaughan, T. L., and Monnat, R. J. Jr. (2013). Spectrum and risk of neoplasia in Werner syndrome: a systematic review. PLoS One 8:e59709. doi: 10.1371/journal.pone.0059709
Lebel, M., and Monnat, R. J. Jr. (2018). Werner syndrome (WRN) gene variants and their association with altered function and age-associated diseases. Age. Res. Rev 41, 82–97. doi: 10.1016/j.arr.2017.11.003
Ledet, E. M., Antonarakis, E. S., Isaacs, W. B., Lotan, T. L., Pritchard, C., and Sartor, A. O. (2020). Germline BLM mutations and metastatic prostate cancer. Prostate 80, 235–237. doi: 10.1002/pros.23924
Lee, J. H., Kim, S. S., Kim, M. S., Yoo, N. J., and Lee, S. H. (2017). WRN, the Werner syndrome gene, exhibits frameshift mutations in gastric and colorectal cancers. Pathol. Oncol. Res. 23, 451–452. doi: 10.1007/s12253-016-0173-3
Lee, M., Shin, S., Uhm, H., Hong, H., Kirk, J., Hyun, K., et al. (2018). Multiple RPAs make WRN syndrome protein a superhelicase. Nucleic Acids Res. 46, 4689–4698. doi: 10.1093/nar/gky272
Lee-Theilen, M., Matthews, A. J., Kelly, D., Zheng, S., and Chaudhuri, J. (2011). CtIP promotes microhomology-mediated alternative end joining during class-switch recombination. Nat. Struct. Mol. Biol. 18, 75–79. doi: 10.1038/nsmb.1942
Lengsfeld, B. M., Rattray, A. J., Bhaskara, V., Ghirlando, R., and Paull, T. T. (2007). Sae2 is an endonuclease that processes hairpin DNA cooperatively with the Mre11/Rad50/Xrs2 complex. Mol. Cell 28, 638–651. doi: 10.1016/j.molcel.2007.11.001
LeRoy, G., Carroll, R., Kyin, S., Seki, M., and Cole, M. D. (2005). Identification of RecQL1 as a Holliday junction processing enzyme in human cell lines. Nucleic Acids Res. 33, 6251–6257. doi: 10.1093/nar/gki929
Li, B., and Comai, L. (2000). Functional interaction between Ku and the werner syndrome protein in DNA end processing. J. Biol. Chem. 275, 28349–28352. doi: 10.1074/jbc.c000289200
Li, M., Liu, B., Yi, J., Yang, Y., Wang, J., Zhu, W. G., et al. (2020). MIB1-mediated degradation of WRN promotes cellular senescence in response to camptothecin treatment. FASEB J. 34, 11488–11497. doi: 10.1096/fj.202000268rrr
Li, M., Xu, X., and Liu, Y. (2011). The SET2-RPB1 interaction domain of human RECQ5 is important for transcription-associated genome stability. Mol. Cell Biol. 31, 2090–2099. doi: 10.1128/mcb.01137-10
Li, P., Hao, S., Bi, Z., Zhang, J., Wu, Z., and Ren, X. (2015). Methylation of Werner syndrome protein is associated with the occurrence and development of invasive meningioma via the regulation of Myc and p53 expression. Exp. Ther. Med. 10, 498–502. doi: 10.3892/etm.2015.2519
Liu, D., Deng, X., Yuan, C., Chen, L., Cong, Y., and Xu, X. (2014). Werner syndrome protein positively regulates XRCC4-like factor transcription. Mol. Med. Rep. 9, 1648–1652. doi: 10.3892/mmr.2014.2030
Lu, H., Fang, E. F., Sykora, P., Kulikowicz, T., Zhang, Y., Becker, K. G., et al. (2014). Senescence induced by RECQL4 dysfunction contributes to Rothmund-Thomson syndrome features in mice. Cell Death Dis. 5:e1226. doi: 10.1038/cddis.2014.168
Lu, H., Saha, J., Beckmann, P. J., Hendrickson, E. A., and Davis, A. J. (2019). DNA-PKcs promotes chromatin decondensation to facilitate initiation of the DNA damage response. Nucleic Acids Res. 47, 9467–9479. doi: 10.1093/nar/gkz694
Lu, H., Shamanna, R. A., de Freitas, J. K., Okur, M., Khadka, P., Kulikowicz, T., et al. (2017). Cell cycle-dependent phosphorylation regulates RECQL4 pathway choice and ubiquitination in DNA double-strand break repair. Nat. Commun. 8:2039.
Lu, H., Shamanna, R. A., Keijzers, G., Anand, R., Rasmussen, L. J., Cejka, P., et al. (2016). RECQL4 promotes DNA end resection in repair of DNA double-strand breaks. Cell Rep. 16, 161–173. doi: 10.1016/j.celrep.2016.05.079
Lu, L., Jin, W., and Wang, L. L. (2020). RECQ DNA Helicases and Osteosarcoma. Adv. Exp. Med. Biol. 1258, 37–54.
Mann, M. B., Hodges, C. A., Barnes, E., Vogel, H., Hassold, T. J., and Luo, G. (2005). Defective sister-chromatid cohesion, aneuploidy and cancer predisposition in a mouse model of type II Rothmund-Thomson syndrome. Hum. Mol. Genet. 14, 813–825. doi: 10.1093/hmg/ddi075
Manthei, K. A., and Keck, J. L. (2013). The BLM dissolvasome in DNA replication and repair. Cell Mol. Life Sci. 70, 4067–4084. doi: 10.1007/s00018-013-1325-1
Mason, J. M., Chan, Y. L., Weichselbaum, R. W., and Bishop, D. K. (2019). Non-enzymatic roles of human RAD51 at stalled replication forks. Nat. Commun. 10:4410.
Mason, R. M., Thacker, J., and Fairman, M. P. (1996). The joining of non-complementary DNA double-strand breaks by mammalian extracts. Nucleic Acids Res. 24, 4946–4953. doi: 10.1093/nar/24.24.4946
Matsumoto, T., Shimamoto, A., Goto, M., and Furuichi, Y. (1997). Impaired nuclear localization of defective DNA Helicases in Werner’s syndrome. Nat. Genet. 16, 335–336. doi: 10.1038/ng0897-335
Mazina, O. M., Rossi, M. J., Deakyne, J. S., Huang, F., and Mazin, A. V. (2012). Polarity and bypass of DNA heterology during branch migration of Holliday junctions by human RAD54, BLM, and RECQ1 proteins. J. Biol. Chem. 287, 11820–11832. doi: 10.1074/jbc.m112.341347
Mimitou, E. P., and Symington, L. S. (2008). Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 455, 770–774. doi: 10.1038/nature07312
Mohaghegh, P., Karow, J. K., Brosh, R. M. Jr., Bohr, V. A., and Hickson, I. D. (2001). The Bloom’s and Werner’s syndrome proteins are DNA structure-specific Helicases. Nucleic Acids Res. 29, 2843–2849. doi: 10.1093/nar/29.13.2843
Mojumdar, A. (2020). Mutations in conserved functional domains of human RecQ Helicases are associated with diseases and cancer: a review. Biophys. Chem. 265:106433. doi: 10.1016/j.bpc.2020.106433
Muftuoglu, M., Kulikowicz, T., Beck, G., Lee, J. W., Piotrowski, J., and Bohr, V. A. (2008). Intrinsic ssDNA annealing activity in the C-terminal region of WRN. Biochemistry 47, 10247–10254. doi: 10.1021/bi800807n
Mukherjee, S., Sinha, D., Bhattacharya, S., Srinivasan, K., Abdisalaam, S., and Asaithamby, A. (2018). Werner syndrome protein and DNA replication. Int. J. Mol. Sci. 19:3442. doi: 10.3390/ijms19113442
Newman, J. A., Savitsky, P., Allerston, C. K., Bizard, A. H., Ozer, O., Sarlos, K., et al. (2015). Crystal structure of the Bloom’s syndrome Helicase indicates a role for the HRDC domain in conformational changes. Nucleic Acids Res. 43, 5221–5235. doi: 10.1093/nar/gkv373
Nimonkar, A. V., Genschel, J., Kinoshita, E., Polaczek, P., Campbell, J. L., Wyman, C., et al. (2011). BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 25, 350–362. doi: 10.1101/gad.2003811
Nimonkar, A. V., Ozsoy, A. Z., Genschel, J., Modrich, P., and Kowalczykowski, S. C. (2008). Human exonuclease 1 and BLM Helicase interact to resect DNA and initiate DNA repair. Proc. Natl. Acad. Sci. U.S.A. 105, 16906–16911. doi: 10.1073/pnas.0809380105
Opresko, P. L., Sowd, G., and Wang, H. (2009). The Werner syndrome Helicase/Exonuclease processes mobile D-loops through branch migration and degradation. PLoS One 4:e4825. doi: 10.1371/journal.pone.0004825
Orren, D. K., Machwe, A., Karmakar, P., Piotrowski, J., Cooper, M. P., and Bohr, V. A. (2001). A functional interaction of Ku with Werner exonuclease facilitates digestion of damaged DNA. Nucleic Acids Res. 29, 1926–1934. doi: 10.1093/nar/29.9.1926
Oshima, J., Huang, S., Pae, C., Campisi, J., and Schiestl, R. H. (2002). Lack of WRN results in extensive deletion at nonhomologous joining ends. Cancer Res. 62, 547–551.
Oshima, J., Kato, H., Maezawa, Y., and Yokote, K. (2018). RECQ Helicase disease and related progeroid syndromes: RECQ2018 meeting. Mech. Age. Dev. 173, 80–83. doi: 10.1016/j.mad.2018.05.002
Oshima, J., Sidorova, J. M., and Monnat, R. J. Jr. (2017). Werner syndrome: Clinical features, pathogenesis and potential therapeutic interventions. Age. Res. Rev. 33, 105–114. doi: 10.1016/j.arr.2016.03.002
Otterlei, M., Bruheim, P., Ahn, B., Bussen, W., Karmakar, P., Baynton, K., et al. (2006). Werner syndrome protein participates in a complex with RAD51, RAD54, RAD54B and ATR in response to ICL-induced replication arrest. J. Cell Sci. 119, 5137–5146. doi: 10.1242/jcs.03291
Palermo, V., Rinalducci, S., Sanchez, M., Grillini, F., Sommers, J. A., and Brosh, R. M. Jr., et al. (2016). CDK1 phosphorylates WRN at collapsed replication forks. Nat. Commun. 7:12880.
Paliwal, S., Kanagaraj, R., Sturzenegger, A., Burdova, K., and Janscak, P. (2014). Human RECQ5 Helicase promotes repair of DNA double-strand breaks by synthesis-dependent strand annealing. Nucleic Acids Res. 42, 2380–2390. doi: 10.1093/nar/gkt1263
Pan, X., Drosopoulos, W. C., Sethi, L., Madireddy, A., Schildkraut, C. L., and Zhang, D. (2017). FANCM, BRCA1, and BLM cooperatively resolve the replication stress at the ALT telomeres. Proc. Natl. Acad. Sci. U.S.A. 114, E5940–E5949.
Pannunzio, N. R., Watanabe, G., and Lieber, M. R. (2018). Nonhomologous DNA end-joining for repair of DNA double-strand breaks. J. Biol. Chem. 293, 10512–10523. doi: 10.1074/jbc.tm117.000374
Parvathaneni, S., and Sharma, S. (2019). The DNA repair Helicase RECQ1 has a checkpoint-dependent role in mediating DNA damage responses induced by gemcitabine. J. Biol. Chem. 294, 15330–15345. doi: 10.1074/jbc.ra119.008420
Parvathaneni, S., Stortchevoi, A., Sommers, J. A., Brosh, R. M. Jr., and Sharma, S. (2013). Human RECQ1 interacts with Ku70/80 and modulates DNA end-joining of double-strand breaks. PLoS One 8:e62481. doi: 10.1371/journal.pone.0062481
Patel, D. S., Misenko, S. M., Her, J., and Bunting, S. F. (2017). BLM Helicase regulates DNA repair by counteracting RAD51 loading at DNA double-strand break sites. J. Cell Biol. 216, 3521–3534. doi: 10.1083/jcb.201703144
Pattankar, S., Churi, O., and Misra, B. K. (2020). Meningioma in a patient with Werner syndrome. Neurol. India 68, 483–486. doi: 10.4103/0028-3886.284350
Patterson-Fortin, J., and D’Andrea, A. D. (2020). Exploiting the microhomology-mediated end-joining pathway in cancer therapy. Cancer Res. 80, 72.
Paulo, P., Maia, S., Pinto, C., Pinto, P., Monteiro, A., Peixoto, A., et al. (2018). Targeted next generation sequencing identifies functionally deleterious germline mutations in novel genes in early-onset/familial prostate cancer. PLoS Genet. 14:e1007355. doi: 10.1371/journal.pgen.1007355
Petkovic, M., Dietschy, T., Freire, R., Jiao, R., and Stagljar, I. (2005). The human Rothmund-Thomson syndrome gene product, RECQL4, localizes to distinct nuclear foci that coincide with proteins involved in the maintenance of genome stability. J. Cell Sci. 118, 4261–4269. doi: 10.1242/jcs.02556
Pinto, C., Kasaciunaite, K., Seidel, R., and Cejka, P. (2016). Human DNA2 possesses a cryptic DNA unwinding activity that functionally integrates with BLM or WRN Helicases. eLife 5:e18574.
Popuri, V., Croteau, D. L., Brosh, R. M. Jr., and Bohr, V. A. (2012a). RECQ1 is required for cellular resistance to replication stress and catalyzes strand exchange on stalled replication fork structures. Cell Cycle 11, 4252–4265. doi: 10.4161/cc.22581
Popuri, V., Ramamoorthy, M., Tadokoro, T., Singh, D. K., Karmakar, P., Croteau, D. L., et al. (2012b). Recruitment and retention dynamics of RECQL5 at DNA double strand break sites. DNA Repair. 11, 624–635. doi: 10.1016/j.dnarep.2012.05.001
Popuri, V., Hsu, J., Khadka, P., Horvath, K., Liu, Y., Croteau, D. L., et al. (2014). Human RECQL1 participates in telomere maintenance. Nucleic Acids Res. 42, 5671–5688. doi: 10.1093/nar/gku200
Qin, Z., Bi, L., Hou, X. M., Zhang, S., Zhang, X., Lu, Y., et al. (2020). Human RPA activates BLM’s bidirectional DNA unwinding from a nick. eLife 9:e54098.
Ralf, C., Hickson, I. D., and Wu, L. (2006). The Bloom’s syndrome Helicase can promote the regression of a model replication fork. J. Biol. Chem. 281, 22839–22846. doi: 10.1074/jbc.m604268200
Raynard, S., Bussen, W., and Sung, P. (2006). A double holliday junction dissolvasome comprising BLM, topoisomerase IIIalpha, and BLAP75. J. Biol. Chem. 281, 13861–13864. doi: 10.1074/jbc.c600051200
Ren, H., Dou, S. X., Zhang, X. D., Wang, P. Y., Kanagaraj, R., Liu, J. L., et al. (2008). The zinc-binding motif of human RECQ5beta suppresses the intrinsic strand-annealing activity of its DExH Helicase domain and is essential for the Helicase activity of the enzyme. Biochem. J. 412, 425–433. doi: 10.1042/bj20071150
Rogers, C. M., Wang, J. C., Noguchi, H., Imasaki, T., Takagi, Y., and Bochman, M. L. (2017). Yeast Hrq1 shares structural and functional homology with the disease-linked human RecQ4 Helicase. Nucleic Acids Res. 45, 5217–5230. doi: 10.1093/nar/gkx151
Romanowicz, H., Pyziak, L., Jablonski, F., Brys, M., Forma, E., and Smolarz, B. (2017). Analysis of DNA repair genes polymorphisms in breast cancer. Pathol. Oncol. Res. 23, 117–123. doi: 10.1007/s12253-016-0110-5
Ronato, D. A., Mersaoui, S. Y., Busatto, F. F., Affar, E. B., Richard, S., and Masson, J. Y. (2020). Limiting the DNA double-strand break resectosome for genome protection. Trends Biochem. Sci. 45, 779–793. doi: 10.1016/j.tibs.2020.05.003
Rossi, M. L., Ghosh, A. K., Kulikowicz, T., Croteau, D. L., and Bohr, V. A. (2010). Conserved Helicase domain of human RecQ4 is required for strand annealing-independent DNA unwinding. DNA Repair. 9, 796–804. doi: 10.1016/j.dnarep.2010.04.003
Russell, C. W., and Mulvey, M. A. (2015). The extraintestinal pathogenic Escherichia coli factor RqlI constrains the genotoxic effects of the RecQ-Like Helicase RqlH. PLoS Pathog. 11:e1005317. doi: 10.1371/journal.ppat.1005317
Saintigny, Y., Makienko, K., Swanson, C., Emond, M. J., and Monnat, R. J. Jr. (2002). Homologous recombination resolution defect in werner syndrome. Mol. Cell Biol. 22, 6971–6978. doi: 10.1128/mcb.22.20.6971-6978.2002
Sallmyr, A., and Tomkinson, A. E. (2018). Repair of DNA double-strand breaks by mammalian alternative end-joining pathways. J. Biol. Chem. 293, 10536–10546. doi: 10.1074/jbc.tm117.000375
Sallmyr, A., Tomkinson, A. E., and Rassool, F. V. (2008). Up-regulation of WRN and DNA ligase IIIalpha in chronic myeloid leukemia: consequences for the repair of DNA double-strand breaks. Blood 112, 1413–1423. doi: 10.1182/blood-2007-07-104257
Sangrithi, M. N., Bernal, J. A., Madine, M., Philpott, A., Lee, J., Dunphy, W. G., et al. (2005). Initiation of DNA replication requires the RECQL4 protein mutated in Rothmund-Thomson syndrome. Cell 121, 887–898. doi: 10.1016/j.cell.2005.05.015
Saponaro, M., Kantidakis, T., Mitter, R., Kelly, G. P., Heron, M., Williams, H., et al. (2014). RECQL5 controls transcript elongation and suppresses genome instability associated with transcription stress. Cell 157, 1037–1049. doi: 10.1016/j.cell.2014.03.048
Sartori, A. A., Lukas, C., Coates, J., Mistrik, M., Fu, S., Bartek, J., et al. (2007). Human CtIP promotes DNA end resection. Nature 450, 509–514. doi: 10.1038/nature06337
Schawalder, J., Paric, E., and Neff, N. F. (2003). Telomere and ribosomal DNA repeats are chromosomal targets of the bloom syndrome DNA Helicase. BMC Cell Biol. 4:15. doi: 10.1186/1471-2121-4-15
Schurman, S. H., Hedayati, M., Wang, Z., Singh, D. K., Speina, E., Zhang, Y., et al. (2009). Direct and indirect roles of RECQL4 in modulating base excision repair capacity. Hum. Mol. Genet. 18, 3470–3483. doi: 10.1093/hmg/ddp291
Schwendener, S., Raynard, S., Paliwal, S., Cheng, A., Kanagaraj, R., Shevelev, I., et al. (2010). Physical interaction of RECQ5 Helicase with RAD51 facilitates its anti-recombinase activity. J. Biol. Chem. 285, 15739–15745. doi: 10.1074/jbc.m110.110478
Scully, R., Panday, A., Elango, R., and Willis, N. A. (2019). DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 20, 698–714. doi: 10.1038/s41580-019-0152-0
Sekelsky, J. J., Brodsky, M. H., Rubin, G. M., and Hawley, R. S. (1999). Drosophila and human RecQ5 exist in different isoforms generated by alternative splicing. Nucleic Acids Res. 27, 3762–3769. doi: 10.1093/nar/27.18.3762
Shamanna, R. A., Croteau, D. L., Lee, J. H., and Bohr, V. A. (2017). Recent advances in understanding Werner syndrome. F1000Research 6:1779. doi: 10.12688/f1000research.12110.1
Shamanna, R. A., Lu, H., Croteau, D. L., Arora, A., Agarwal, D., Ball, G., et al. (2016a). Camptothecin targets WRN protein: mechanism and relevance in clinical breast cancer. Oncotarget 7, 13269–13284. doi: 10.18632/oncotarget.7906
Shamanna, R. A., Lu, H., de Freitas, J. K., Tian, J., Croteau, D. L., and Bohr, V. A. (2016b). WRN regulates pathway choice between classical and alternative non-homologous end joining. Na.t Commun. 7:13785.
Shamanna, R. A., Singh, D. K., Lu, H., Mirey, G., Keijzers, G., Salles, B., et al. (2014). RECQ Helicase RECQL4 participates in non-homologous end joining and interacts with the Ku complex. Carcinogenesis 35, 2415–2424. doi: 10.1093/carcin/bgu137
Sharma, S., and Brosh, R. M. Jr. (2007). Human RECQ1 is a DNA damage responsive protein required for genotoxic stress resistance and suppression of sister chromatid exchanges. PLoS One 2:e1297. doi: 10.1371/journal.pone.0001297
Sharma, S., and Brosh, R. M. Jr. (2008). Unique and important consequences of RECQ1 deficiency in mammalian cells. Cell Cycle 7, 989–1000. doi: 10.4161/cc.7.8.5707
Sharma, S., Phatak, P., Stortchevoi, A., Jasin, M., and Larocque, J. R. (2012). RECQ1 plays a distinct role in cellular response to oxidative DNA damage. DNA Repair. 11, 537–549. doi: 10.1016/j.dnarep.2012.04.003
Sharma, S., Sommers, J. A., Choudhary, S., Faulkner, J. K., Cui, S., Andreoli, L., et al. (2005). Biochemical analysis of the DNA unwinding and strand annealing activities catalyzed by human RECQ1. J. Biol. Chem. 280, 28072–28084. doi: 10.1074/jbc.m500264200
Sharma, S., Stumpo, D. J., Balajee, A. S., Bock, C. B., Lansdorp, P. M., and Brosh, R. M. Jr., et al. (2007). RECQL, a member of the RecQ family of DNA Helicases, suppresses chromosomal instability. Mol. Cell Biol. 27, 1784–1794. doi: 10.1128/mcb.01620-06
Shi, J., Chen, W. F., Zhang, B., Fan, S. H., Ai, X., Liu, N. N., et al. (2017). A helical bundle in the N-terminal domain of the BLM Helicase mediates dimer and potentially hexamer formation. J. Biol. Chem. 292, 5909–5920. doi: 10.1074/jbc.m116.761510
Shimamoto, A., Nishikawa, K., Kitao, S., and Furuichi, Y. (2000). Human RecQ5beta, a large isomer of RecQ5 DNA Helicase, localizes in the nucleoplasm and interacts with topoisomerases 3alpha and 3beta. Nucleic Acids Res. 28, 1647–1655. doi: 10.1093/nar/28.7.1647
Siitonen, H. A., Sotkasiira, J., Biervliet, M., Benmansour, A., Capri, Y., Cormier-Daire, V., et al. (2009). The mutation spectrum in RECQL4 diseases. Eur. J. Hum. Genet. 17, 151–158. doi: 10.1038/ejhg.2008.154
Singh, D. K., Karmakar, P., Aamann, M., Schurman, S. H., May, A., Croteau, D. L., et al. (2010). The involvement of human RECQL4 in DNA double-strand break repair. Aging Cell 9, 358–371. doi: 10.1111/j.1474-9726.2010.00562.x
Singh, D. K., Popuri, V., Kulikowicz, T., Shevelev, I., Ghosh, A. K., Ramamoorthy, M., et al. (2012). The human RecQ Helicases BLM and RECQL4 cooperate to preserve genome stability. Nucleic Acids Res. 40, 6632–6648. doi: 10.1093/nar/gks349
Singh, T. R., Ali, A. M., Busygina, V., Raynard, S., Fan, Q., Du, C. H., et al. (2008). BLAP18/RMI2, a novel OB-fold-containing protein, is an essential component of the Bloom Helicase-double Holliday junction dissolvasome. Genes Dev. 22, 2856–2868. doi: 10.1101/gad.1725108
Sommers, J. A., Sharma, S., Doherty, K. M., Karmakar, P., Yang, Q., Kenny, M. K., et al. (2005). p53 modulates RPA-dependent and RPA-independent WRN Helicase activity. Cancer Res. 65, 1223–1233. doi: 10.1158/0008-5472.can-03-0231
Soniat, M. M., Myler, L. R., Kuo, H. C., Paull, T. T., and Finkelstein, I. J. (2019). RPA phosphorylation inhibits DNA resection. Mol. Cell 75, 145–153.e145.
Stirnweiss, A., Oommen, J., Kotecha, R. S., Kees, U. R., and Beesley, A. H. (2017). Molecular-genetic profiling and high-throughput in vitro drug screening in NUT midline carcinoma-an aggressive and fatal disease. Oncotarget 8, 112313–112329. doi: 10.18632/oncotarget.22862
Sturzenegger, A., Burdova, K., Kanagaraj, R., Levikova, M., Pinto, C., Cejka, P., et al. (2014). DNA2 cooperates with the WRN and BLM RecQ helicases to mediate long-range DNA end resection in human cells. J. Biol. Chem. 289, 27314–27326. doi: 10.1074/jbc.m114.578823
Subino, S., Fiori, P. L., Lubinu, G., Scappaticci, S., and Cerimele, D. (1982). Immunofluorescence of the tubulin system in human skin fibroblasts in Werner’s syndrome, Kaposi sarcoma, and psoriasis. Arch. Dermatol. Res. 272, 143–145. doi: 10.1007/bf00510405
Sun, J., Meng, H., Yao, L., Lv, M., Bai, J., Zhang, J., et al. (2017). Germline mutations in cancer susceptibility genes in a large series of unselected breast cancer patients. Clin. Cancer Res. 23, 6113–6119. doi: 10.1158/1078-0432.ccr-16-3227
Sun, L., Nakajima, S., Teng, Y., Chen, H., Yang, L., Chen, X., et al. (2017). WRN is recruited to damaged telomeres via its RQC domain and tankyrase1-mediated poly-ADP-ribosylation of TRF1. Nucleic Acids Res. 45, 3844–3859. doi: 10.1093/nar/gkx065
Sun, J., Wang, Y., Xia, Y., Xu, Y., Ouyang, T., Li, J., et al. (2015). Mutations in RECQL gene are associated with predisposition to breast cancer. PLoS Genet. 11:e1005228. doi: 10.1371/journal.pgen.1005228
Suzuki, T., Shiratori, M., Furuichi, Y., and Matsumoto, T. (2001). Diverged nuclear localization of Werner Helicase in human and mouse cells. Oncogene 20, 2551–2558. doi: 10.1038/sj.onc.1204344
Symington, L. S. (2014). End resection at double-strand breaks: mechanism and regulation. Cold Spring Harb. Perspect. Biol. 6:a016436. doi: 10.1101/cshperspect.a016436
Tadokoro, T., Kulikowicz, T., Dawut, L., Croteau, D. L., and Bohr, V. A. (2012). DNA binding residues in the RQC domain of Werner protein are critical for its catalytic activities. Aging 4, 417–429. doi: 10.18632/aging.100463
Taher, M. M., Hassan, A. A., Saeed, M., Jastania, R. A., Nageeti, T. H., Alkhalidi, H., et al. (2019). Next generation DNA sequencing of atypical choroid plexus papilloma of brain: identification of novel mutations in a female patient by Ion Proton. Oncol. Lett. 18, 5063–5076.
Taylor, A. M. R., Rothblum-Oviatt, C., Ellis, N. A., Hickson, I. D., Meyer, S., Crawford, T. O., et al. (2019). Chromosome instability syndromes. Nat. Rev. Dis. Primers 5:64.
Tervasmaki, A., Mantere, T., Hartikainen, J. M., Kauppila, S., Lee, H. M., Koivuluoma, S., et al. (2018). Rare missense mutations in RECQL and POLG associate with inherited predisposition to breast cancer. Int. J. Cancer 142, 2286–2292. doi: 10.1002/ijc.31259
Thompson, E. R., Doyle, M. A., Ryland, G. L., Rowley, S. M., Choong, D. Y., Tothill, R. W., et al. (2012). Exome sequencing identifies rare deleterious mutations in DNA repair genes FANCC and BLM as potential breast cancer susceptibility alleles. PLoS Genet. 8:e1002894. doi: 10.1371/journal.pgen.1002894
Thorslund, T., McIlwraith, M. J., Compton, S. A., Lekomtsev, S., Petronczki, M., Griffith, J. D., et al. (2010). The breast cancer tumor suppressor BRCA2 promotes the specific targeting of RAD51 to single-stranded DNA. Nat. Struct. Mol. Biol. 17, 1263–1265. doi: 10.1038/nsmb.1905
Tock, A. J., and Henderson, I. R. (2018). Hotspots for initiation of meiotic recombination. Front. Genet. 9:521. doi: 10.3389/fgene.2018.00521
Truong, L. N., Li, Y., Shi, L. Z., Hwang, P. Y., He, J., Wang, H., et al. (2013). Microhomology-mediated end joining and homologous recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 110, 7720–7725. doi: 10.1073/pnas.1213431110
Tsurubuchi, T., Yamamoto, T., Tsukada, Y., Matsuda, M., Nakai, K., and Matsumura, A. (2008). Meningioma associated with Werner syndrome–case report. Neurol. Med. Chir. 48, 470–473.
Tubbs, A., and Nussenzweig, A. (2017). Endogenous DNA damage as a source of genomic instability in cancer. Cell 168, 644–656. doi: 10.1016/j.cell.2017.01.002
Urban, V., Dobrovolna, J., and Janscak, P. (2017). Distinct functions of human RecQ helicases during DNA replication. Biophys. Chem. 225, 20–26. doi: 10.1016/j.bpc.2016.11.005
van Brabant, A. J., Ye, T., Sanz, M., German, I. J., Ellis, N. A., and Holloman, W. K. (2000). Binding and melting of D-loops by the Bloom syndrome Helicase. Biochemistry 39, 14617–14625. doi: 10.1021/bi0018640
van Wietmarschen, N., Sridharan, S., Nathan, W. J., Tubbs, A., Chan, E. M., Callen, E., et al. (2020). Repeat expansions confer WRN dependence in microsatellite-unstable cancers. Nature 586, 292–298. doi: 10.1038/s41586-020-2769-8
von Kobbe, C., Thoma, N. H., Czyzewski, B. K., Pavletich, N. P., and Bohr, V. A. (2003). Werner syndrome protein contains three structure-specific DNA binding domains. J. Biol. Chem. 278, 52997–53006. doi: 10.1074/jbc.m308338200
Wang, L. L., Gannavarapu, A., Kozinetz, C. A., Levy, M. L., Lewis, R. A., Chintagumpala, M. M., et al. (2003). Association between osteosarcoma and deleterious mutations in the RECQL4 gene in Rothmund-Thomson syndrome. J. Natl. Cancer Inst. 95, 669–674. doi: 10.1093/jnci/95.9.669
Watt, P. M., Hickson, I. D., Borts, R. H., and Louis, E. J. (1996). SGS1, a homologue of the Bloom’s and Werner’s syndrome genes, is required for maintenance of genome stability in Saccharomyces cerevisiae. Genetics 144, 935–945. doi: 10.1093/genetics/144.3.935
White, R. R., and Vijg, J. (2016). Do DNA double-strand breaks drive aging? Mol. Cell 63, 729–738. doi: 10.1016/j.molcel.2016.08.004
Wright, W. D., Shah, S. S., and Heyer, W. D. (2018). Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 293, 10524–10535.
Wu, L., Bachrati, C. Z., Ou, J., Xu, C., Yin, J., Chang, M., et al. (2006). BLAP75/RMI1 promotes the BLM-dependent dissolution of homologous recombination intermediates. Proc. Natl. Acad. Sci. U.S.A. 103, 4068–4073. doi: 10.1073/pnas.0508295103
Wu, L., Chan, K. L., Ralf, C., Bernstein, D. A., Garcia, P. L., Bohr, V. A., et al. (2005). The HRDC domain of BLM is required for the dissolution of double Holliday junctions. EMBO J. 24, 2679–2687. doi: 10.1038/sj.emboj.7600740
Wu, L., Davies, S. L., Levitt, N. C., and Hickson, I. D. (2001). Potential role for the BLM Helicase in recombinational repair via a conserved interaction with RAD51. J. Biol. Chem. 276, 19375–19381. doi: 10.1074/jbc.m009471200
Wu, L., and Hickson, I. D. (2003). The Bloom’s syndrome Helicase suppresses crossing over during homologous recombination. Nature 426, 870–874. doi: 10.1038/nature02253
Xie, A., Kwok, A., and Scully, R. (2009). Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat. Struct. Mol. Biol. 16, 814–818. doi: 10.1038/nsmb.1640
Xu, D., Guo, R., Sobeck, A., Bachrati, C. Z., Yang, J., Enomoto, T., et al. (2008). RMI, a new OB-fold complex essential for Bloom syndrome protein to maintain genome stability. Genes Dev. 22, 2843–2855. doi: 10.1101/gad.1708608
Xu, X., Rochette, P. J., Feyissa, E. A., Su, T. V., and Liu, Y. (2009). MCM10 mediates RECQ4 association with MCM2-7 Helicase complex during DNA replication. EMBO J. 28, 3005–3014. doi: 10.1038/emboj.2009.235
Xu, Y. N., Bazeille, N., Ding, X. Y., Lu, X. M., Wang, P. Y., Bugnard, E., et al. (2012). Multimeric BLM is dissociated upon ATP hydrolysis and functions as monomers in resolving DNA structures. Nucleic Acids Res. 40, 9802–9814. doi: 10.1093/nar/gks728
Xue, C., Daley, J. M., Xue, X., Steinfeld, J., Kwon, Y., Sung, P., et al. (2019). Single-molecule visualization of human BLM Helicase as it acts upon double- and single-stranded DNA substrates. Nucleic Acids Res. 47, 11225–11237. doi: 10.1093/nar/gkz810
Yang, H., Li, Q., Fan, J., Holloman, W. K., and Pavletich, N. P. (2005). The BRCA2 homologue Brh2 nucleates RAD51 filament formation at a dsDNA-ssDNA junction. Nature 433, 653–657. doi: 10.1038/nature03234
Yannone, S. M., Roy, S., Chan, D. W., Murphy, M. B., Huang, S., Campisi, J., et al. (2001). Werner syndrome protein is regulated and phosphorylated by DNA-dependent protein kinase. J. Biol. Chem. 276, 38242–38248. doi: 10.1074/jbc.m101913200
Yu, C. E., Oshima, J., Fu, Y. H., Wijsman, E. M., Hisama, F., Alisch, R., et al. (1996). Positional cloning of the Werner’s syndrome gene. Science 272, 258–262.
Zecevic, A., Menard, H., Gurel, V., Hagan, E., DeCaro, R., and Zhitkovich, A. (2009). WRN Helicase promotes repair of DNA double-strand breaks caused by aberrant mismatch repair of chromium-DNA adducts. Cell Cycle 8, 2769–2778. doi: 10.4161/cc.8.17.9410
Zhang, Y., and Jasin, M. (2011). An essential role for CtIP in chromosomal translocation formation through an alternative end-joining pathway. Nat. Struct. Mol. Biol. 18, 80–84. doi: 10.1038/nsmb.1940
Zhao, W., Vaithiyalingam, S., San Filippo, J., Maranon, D. G., Jimenez-Sainz, J., Fontenay, G. V., et al. (2015). Promotion of BRCA2-dependent homologous recombination by DSS1 via RPA targeting and Dna Mimicry. Mol. Cell 59, 176–187. doi: 10.1016/j.molcel.2015.05.032
Zheng, L., Kanagaraj, R., Mihaljevic, B., Schwendener, S., Sartori, A. A., Gerrits, B., et al. (2009). MRE11 complex links RECQ5 Helicase to sites of DNA damage. Nucleic Acids Res. 37, 2645–2657. doi: 10.1093/nar/gkp147
Zhi, L. Q., Ma, W., Zhang, H., Zeng, S. X., and Chen, B. (2014). Association of RECQL5 gene polymorphisms and osteosarcoma in a Chinese Han population. Tumour Biol. 35, 3255–3259. doi: 10.1007/s13277-013-1425-4
Zhunussova, G., Afonin, G., Abdikerim, S., Jumanov, A., Perfilyeva, A., Kaidarova, D., et al. (2019). Mutation spectrum of cancer-associated genes in patients with early onset of colorectal cancer. Front. Oncol. 9:673. doi: 10.3389/fonc.2019.00673
Keywords: RecQ helicase, RECQL1, BLM, WRN, RECQL4, RECQL5, DNA double-strand break repair, genome stability
Citation: Lu H and Davis AJ (2021) Human RecQ Helicases in DNA Double-Strand Break Repair. Front. Cell Dev. Biol. 9:640755. doi: 10.3389/fcell.2021.640755
Received: 12 December 2020; Accepted: 29 January 2021;
Published: 25 February 2021.
Edited by:
Binfeng Lu, University of Pittsburgh, United StatesReviewed by:
Sudha Sharma, Howard University, United StatesMatthew Bochman, Indiana University, United States
Copyright © 2021 Lu and Davis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huiming Lu, huiming.lu@utsouthwestern.edu; Anthony J. Davis, anthony.davis@utsouthwestern.edu