Expanding structural diversity of 5′-aminouridine moiety of sansanmycin via mutational biosynthesis
 Yuan Lu1
Yuan Lu1  Yihong Li1
Yihong Li1  Jiahui Fan1 Xingxing Li1
Jiahui Fan1 Xingxing Li1  Hongmin Sun1 Lifei Wang1 Xingli Han2,3,4,5 Yuting Zhu2,3,4
Hongmin Sun1 Lifei Wang1 Xingli Han2,3,4,5 Yuting Zhu2,3,4  Tianyu Zhang2,3,4,5 Yuanyuan Shi1*
Tianyu Zhang2,3,4,5 Yuanyuan Shi1*  Yunying Xie1*
Yunying Xie1*  Bin Hong1*
Bin Hong1*- 1CAMS Key Laboratory of Synthetic Biology for Drug Innovation and NHC Key Laboratory of Biotechnology of Antibiotics, Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China
- 2State Key Laboratory of Respiratory Disease, Guangzhou Institutes of Biomedicine and Health (GIBH), Chinese Academy of Sciences (CAS), Guangzhou, China
- 3China-New Zealand Joint Laboratory of Biomedicine and Health, Guangzhou Institutes of Biomedicine and Health (GIBH), Chinese Academy of Sciences (CAS), Guangzhou, China
- 4Guangdong-Hong Kong-Macau Joint Laboratory of Respiratory Infectious Diseases, Guangzhou, China
- 5University of Chinese Academy of Sciences (UCAS), Beijing, China
Sansanmycins represent a family of uridyl peptide antibiotics with antimicrobial activity specifically against Mycobacterium tuberculosis (including drug-resistant M. tuberculosis) and Pseudomonas aeruginosa. They target translocase I (MraY) to inhibit bacterial cell wall assembly. Given the unique mechanism of action, sansanmycin has emerged as a potential lead compound for developing new anti-tuberculosis drugs, while the 5′-aminouridine moiety plays a crucial role in the pharmacophore of sansanmycin. For expanding the structural diversity of the 5′-aminouridine moiety of sansanmycin through biosynthetic methods, we firstly demonstrated that SsaM and SsaK are responsible for the biosynthesis of the 5′-aminouridine moiety of sansanmycin in vivo. Using the ssaK deletion mutant (SS/KKO), we efficiently obtained a series of new analogues with modified 5′-aminouridine moieties through mutational biosynthesis. Based on molecular networking analysis of MS/MS, twenty-two new analogues (SS-KK-1 to -13 and SS-KK-A to -I) were identified. Among them, four new analogues (SS-KK-1 to -3 and SS-KK-C) were purified and bioassayed. SS-KK-2 showed better antibacterial activity against E. coli ΔtolC than the parent compound sansanmycin A. SS-KK-3 showed the same anti-TB activity as sansanmycin A against M. tuberculosis H37Rv as well as clinically isolated, drug-sensitive and multidrug-resistant M. tuberculosis strains. Furthermore, SS-KK-3 exhibited significantly improved structural stability compared to sansanmycin A. The results suggested that mutasynthesis is an effective and practical strategy for expanding the structural diversity of 5′-aminouridine moiety in sansanmycin.
1 Introduction
Tuberculosis (TB), an infectious disease caused by Mycobacterium tuberculosis, remains one of the most significant global health threats. The recently published “2022 Global Tuberculosis Report” (World Health Organization, 2022) points that TB is the second deadliest infectious disease following COVID-19. The COVID-19 pandemic has reversed the progress made against TB in recent years. Global TB deaths increased from 1.4 million in 2019 to 1.5 million in 2020 and further to 1.6 million in 2021. In the near future, TB may surpass COVID-19 again as the leading cause of death worldwide from a single infectious source as COVID-19 is gradually under control. The current approach to treating TB involves a 6-month course of quadruple therapy consisting of rifampicin (RIF), isoniazid (INH), ethambutol, and pyrazinamide (Zumla et al., 2013). While this treatment can effectively control drug-sensitive TB, it is ineffective against drug-resistant TB infections, which are becoming increasingly prevalent worldwide. Given the severity of the current TB situation and the propensity for drug resistance, the development of new drugs specifically targeting drug-resistant TB is urgently needed.
Sansanmycins (Xie et al., 2007), produced by Streptomyces sp. SS, belong to the family of uridyl peptide antibiotics (UPAs) including pacidamycins (Karwowski et al., 1989), napsamycins (Chatterjee et al., 1994), and mureidomycins (Inukai et al., 1989). They share a core scaffold of a 3′-deoxyuridine unit linked to a pseudo-tetra/pentapeptidyl backbone via an unusual enamide linkage. UPAs possess noteworthy antimicrobial activity against Mycobacterium tuberculosis (including drug-resistant Mycobacterium tuberculosis) (Shi et al., 2016) and Pseudomonas aeruginosa by targeting translocase I (MraY) to block bacterial cell wall assembly (Winn et al., 2010). As this unique mode of action has not been clinically targeted, UPA has emerged as a lead compound in the quest for new anti-TB drugs.
Extensive research has been conducted on UPAs’ biosynthesis logic and structural modification. UPAs are synthesized by a series of highly dissociated non-ribosomal peptide synthetases (NRPSs) (Zhang et al., 2010). Their assembly begins with the central building block 2,3-diaminobutyric acid (DABA), then the elongation of the peptide framework includes the attachment of the N-terminal amino acid (AA1) to the β-amino group of DABA, and the C-terminal ureidodipeptide (AA3 and AA4) linked to the α-amino group of DABA (Zhang et al., 2010; Zhang et al., 2011). The unusual ureidodipeptide assembly is achieved under the catalysis of a unique condensation enzyme PacN, resulting in the formation of a ureido-bond between AA3 and AA4 (Zhang et al., 2010; Ióca et al., 2021). The synthesis of 4′,5′-enamide-3′-deoxyuridine of UPA has been elucidated in vitro, which starts from uridine, and a flavin-dependent dehydrogenase Pac11 (i.e., PacK) catalyzes the oxidation of uridine to uridine-5′-aldehyde (Ragab et al., 2011). The aminotransferase Pac5 (i.e., PacE) catalyzes the transamination of the 5′-aldehyde group, and the dehydration reaction mediated by Pac13 (i.e., PacM) finally generates 4′,5′-enamide-3′-deoxyuridine (Ragab et al., 2011; Michailidou et al., 2017) (Supplementary Figure S1). Then under the catalysis of PacI, it is attached to pseudo-tetra backbone through an enamide bond, consequently producing UPAs (Zhang et al., 2011).
Previous structural modifications of UPAs mainly focused on substituting amino acid residues in peptide chains, such as obtaining C-terminally modified pacidamycin derivatives (Gruschow et al., 2009) and sansanmycin derivatives (Zhang et al., 2016) by precursor-directed biosynthesis. A series of new sansanmycin analogues with modification at N-terminal amino acid were obtained using mutational biosynthesis (Shi et al., 2016). Nevertheless, few analogue compounds showed significantly increase of the antibacterial activity. The 5′-aminouridine moiety in UPAs is believed to be essential for competing active site binding with the natural substrate UDP-N-acetylmuramoyl (UDP-MurNAc) pentapeptide of MraY (Walsh and Zhang, 2011). Recently, the MraY-inhibitor complex structure data show a common feature of the uridine binding pocket of naturally occurring nucleoside inhibitors including UPAs, and the residues that form the uridine binding pocket are likely involved in binding the natural substrate of MraY (Mashalidis et al., 2019). However, as an important part of pharmacophore, there have been no reported structural modifications of 5′-aminouridine through biosynthetic methods.
Currently, reported analogues with modified 5′-aminouridine are almost entirely obtained through chemical synthesis (Figure 1). Dihydropacidamycin D, the first analogue compound with modified 5′-aminouridine was obtained through the hydrogenation of C-4′ exocyclic olefin of pacidamycin D, and it showed comparable activity against P. aeruginosa to pacidamycin D (Boojamra et al., 2001) (Figures 1A–C,F), indicating that the hydrogenation of enamide linkage would not affect the activity of UPAs. In recent years, certain dihydrosansanmycin analogues with modified peptide chains have been reported to exhibit enhanced activity against M. tuberculosis (Tran et al., 2017; Tran et al., 2021) (Figures 1A–C, F). Notably, the stereochemistry at C-4′ of the ribose moiety in both dihydrosansanmycin and dihydropacidamycin has been suggested to be essential for antibacterial activity, with S-stereochemistry in analogues usually leading to loss or reduction of activity (Boojamra et al., 2001; Tran et al., 2017). The antibacterial activity of 3′-hydroxypacidamycin D against P. aeruginosa was comparable to that of pacidamycin D (Okamoto et al., 2012), while the MraY inhibition activity of 3′-hydroxymureidomycin A (Mashalidis et al., 2019) was also comparable to that of mureidomycin A (Inukai et al., 1993) (Figures 1A, D, F). These findings suggest that the introduction of a hydroxyl group at the 3′-position of the 5′-aminouridine moiety is well-tolerated for both MraY inhibition and antibacterial activity (Okamoto et al., 2012). In 2020, Niro et al. (Niro et al., 2020) reported a new hybrid structure, which consists of the peptide chain of sansanmycin B and uridine core moiety of 5′-deoxy muraymycin C4 (Figures 1A, E–F). Similar to UPA, the 5′-deoxy muraymycin C4 is also a uridine-derived nucleoside antibiotic, comprising a (6′S)-5′-deoxy-glycyluridine moiety, and demonstrates inhibitory activity against MraY (Koppermann et al., 2018). The muraymycin-sansanmycin B hybrid structure showed inhibition activity against MraY, but displayed no significant antibacterial activity against P. aeruginosa (Niro et al., 2020). This suggests that the hybrid structure resulting from the incorporation of uridine building blocks from other nucleoside antibiotics into UPAs may still exhibit inhibitory activity against MraY. Chemical synthesis of UPAs is challenging and low yielding, but it provides some insights into the structure-activity relationship of UPAs. What’s more, it indicates that making appropriate modifications to 5′-aminouridine can be further explored for new UPA derivatives to improve their activity and/or druggability.
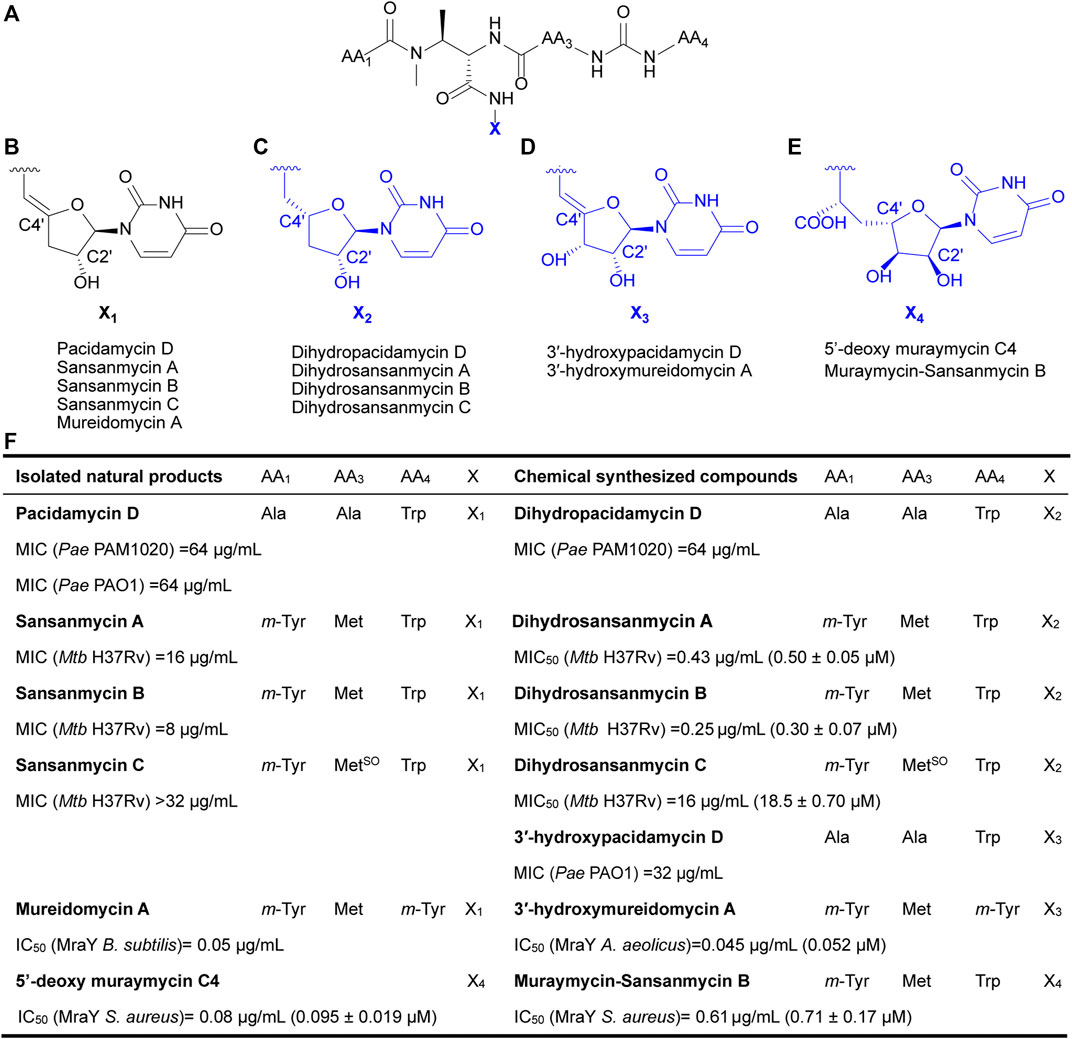
FIGURE 1. Chemical structures of natural UPAs (black) and synthetic analogues with modified 5′-aminouridine moiety (blue) and their corresponding inhibitory activity. (A) The pseudo-tetrapeptide backbone of UPAs. (B) The chemical structures of 5′-aminouridine of natural UPAs. (C–E) The chemical structures of modified 5′-aminouridine of synthetic analogues. (F) Detailed information on structure and inhibitory activities of natural UPAs and synthesized derivatives. Pae, Pseudomonas aeruginosa; Mtb, Mycobacterium tuberculosis; B. subtilis, Bacillus subtilis; S. aureus, Staphylococcus aureus; A. aeolicus, Aquifex aeolicus.
Here, we aimed to expand the structural diversity of sansanmycin and explore the structure-activity relationship of 5′-aminouridine moiety by mutational biosynthesis. Within sansanmycin biosynthetic gene cluster, SsaM, SsaK and SsaE share high homology with PacM, PacK and PacE, respectively, which have been previously confirmed to be responsible for the biosynthesis of the 5′-aminouridine of pacidamycin in vitro. In this study, through in vivo assays, we demonstrated that SsaM and SsaK are responsible for the biosynthesis of 5′-aminouridine in sansanmycin biosynthetic pathway, and confirmed SsaM acts as the dehydratase by the detection of a new hydrated derivative. On this basis, two 5′-aminouridine analogues were fed to the knockout strains of ssaK and ssaM, and ssaK knockout strain showed higher yield of derivatives. Twenty-two new derivatives were identified by the molecular networking of LC-MS/MS data. Among them, four monomers were purified and one of them were further confirmed by NMR. SS-KK-2 exhibited improved antibacterial activity against E. coli ΔtolC. SS-KK-3 showed the same activity against M. tuberculosis including clinically isolated, multidrug-resistant strains with improved structural stability.
2 Results
2.1 Blocking the biosynthesis of 4′,5′-enamide-3′-deoxyuridine
Upon scrutiny of the sansanmycin biosynthetic gene cluster, SsaM, SsaK, and SsaE were found to exhibit over 80% amino acid sequence identity with PacM, PacK, and PacE (Supplementary Figures S2–4), respectively, indicating their probable role in the biosynthesis of the 5′-aminouridine moiety in sansanmycin. To investigate the function of SsaM, SsaK, and SsaE in vivo, the corresponding encoding genes ssaM, ssaK, and ssaE were in-frame deleted from Streptomyces sp. SS by CRISPR/Cas9 using plasmid pKCcas9dO (He et al., 2015), which contains codon-optimized scocas9 encoding Streptococcus pyogenes Cas9 protein. Deletion plasmids pKC-M, pKC-K and pKC-E were constructed by replacing the sgRNA sequence and homology arm sequences in plasmid pKCcas9dO with the corresponding sequences of the genes to be knocked out, and were further introduced into Streptomyces sp. SS by conjugation (Figure 2A). Knock-out mutants SS/MKO, SS/KKO and SS/EKO of ssaM, ssaK, and ssaE were verified by PCR, and the results showed that the correct SS/MKO and SS/KKO were obtained (Supplementary Figures S5, 6). There was no correct SS/EKO obtained even after many efforts, which may be due to the off-target effect. The complete coding regions of ssaM and ssaK were cloned into the expression plasmid pL646 (Hong et al., 2007) derived from pSET152 (Bierman et al., 19922) under the control of a strong constitutive promoter, ermEp*, respectively. Then resulting plasmids pL-ssaM and pL-ssaK were introduced into SS/MKO and SS/KKO by conjugation, respectively, and the complementary strains SS/MKO/pL-ssaM and SS/KKO/pL-ssaK were obtained to exclude the possibility of polar effects in SS/MKO and SS/KKO.
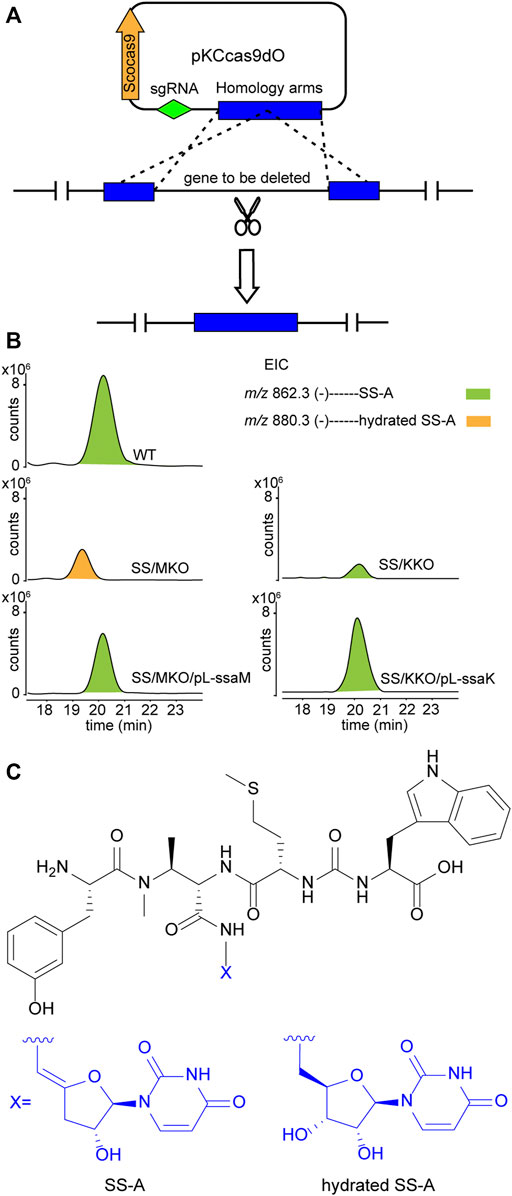
FIGURE 2. Effects of blocking 5′-aminouridine biosynthetic gene ssaM and ssaK on sansanmycin production. (A) Construction of mutant strains with deleted 5′-aminouridine biosynthetic genes. (B) LC-ESI-MS analyses of sansanmycin A (SS-A) and its derivatives in different strains. (C) Structures of SS-A and hydrated SS-A.
Secondary metabolites of SS/MKO and SS/KKO, as well as the complementary strains SS/MKO/pL-ssaM and SS/KKO/pL-ssaK were analyzed by LC-MS/MS, with wild-type strain as control. In SS/MKO, sansanmycin A (SS-A), the major component compound of wild-type strain, was eliminated and instead a new hydrated sansanmycin A (hydrated SS-A) was detected (Figures 2B, C, Supplementary Figure S7). The production of hydrated SS-A confirmed that SsaM acts as the dehydratase responsible for biosynthesizing the 5′-aminouridine of sansanmycins in vivo. Mutant SS/KKO still produced trace amount of SS-A (Figure 2B), potentially due to its function being partially complemented by other enzymes in the production strain. Production of sansanmycins could be restored in both complementary strains (Figure 2B). All of above results demonstrated that SsaM and SsaK were responsible for the biosynthesis of 5′-aminouridine in sansanmycin biosynthetic pathway in vivo.
2.2 Production of new sansanmycin analogues by mutational biosynthesis
To explore the possibility of mutational biosynthesis on modification of 5′-aminouridine moiety, we intended to feed uridine analogues to the knock-out mutants. Besides the natural amide linkage between 5′-aminouridine and tetra/pentapeptides, PacI could also catalyze the formation of an oxoester in the in vitro assays for its relaxed substrate specificities (Zhang et al., 2011). Therefore, a series of commercially accessible uridine analogues (Supplementary Figure S8) were firstly fed to SS/KKO. However, no new derivatives were detected in the LC-MS/MS analysis, potentially due to the low efficiency of SsaI (the homologue of PacI) in catalyzing the formation of ester bonds in vivo. Then 5′-aminouridine analogues were taken into our consideration and only two commercial compounds were available, 2′-deoxy-5′-aminouridine (1) and 5-methyl-2′-deoxy-5′-aminouridine (2) both with the R-configuration of the chiral C-4' (Figure 3A). The two 5′-aminouridine analogues 1 and 2 (final concentration of 3 mM) were respectively fed to both SS/MKO and SS/KKO mutants. The fermentation broths were analyzed by extracting ion chromatograms of target compounds derived from SS-A biosynthetic pathway. It was found that two peaks (Figure 3B), with identical MS/MS fragmentations (Figure 4C), appeared in each of the two cultures fed with 1 and 2. The new derivatives generated from feeding substrate 1 were named SS-KK-1 and SS-KK-2, while those produced from feeding substrate 2 were named SS-KK-A and SS-KK-B.
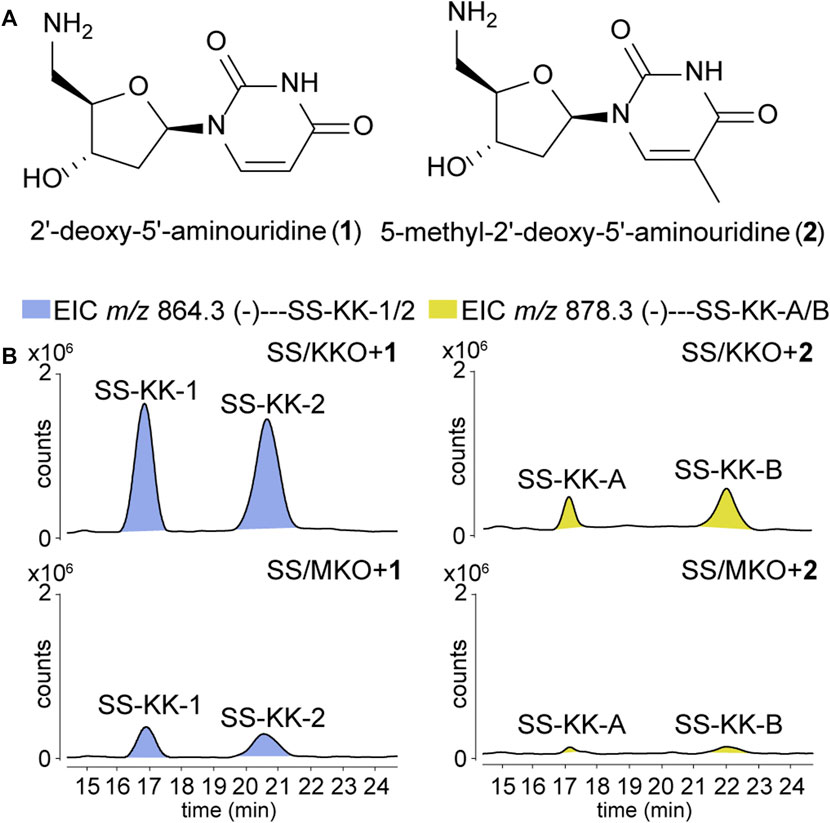
FIGURE 3. Production of new sansanmycin analogues with 5′-aminouridine modification by mutational biosynthesis. (A) Structures of 2′-deoxy-5′-aminouridine (1) and 5-methyl-2′-deoxy-5′-aminouridine (2). (B) LC-ESI-MS analyses of fermentations fed with different 5′-aminouridine analogues 1 and 2.
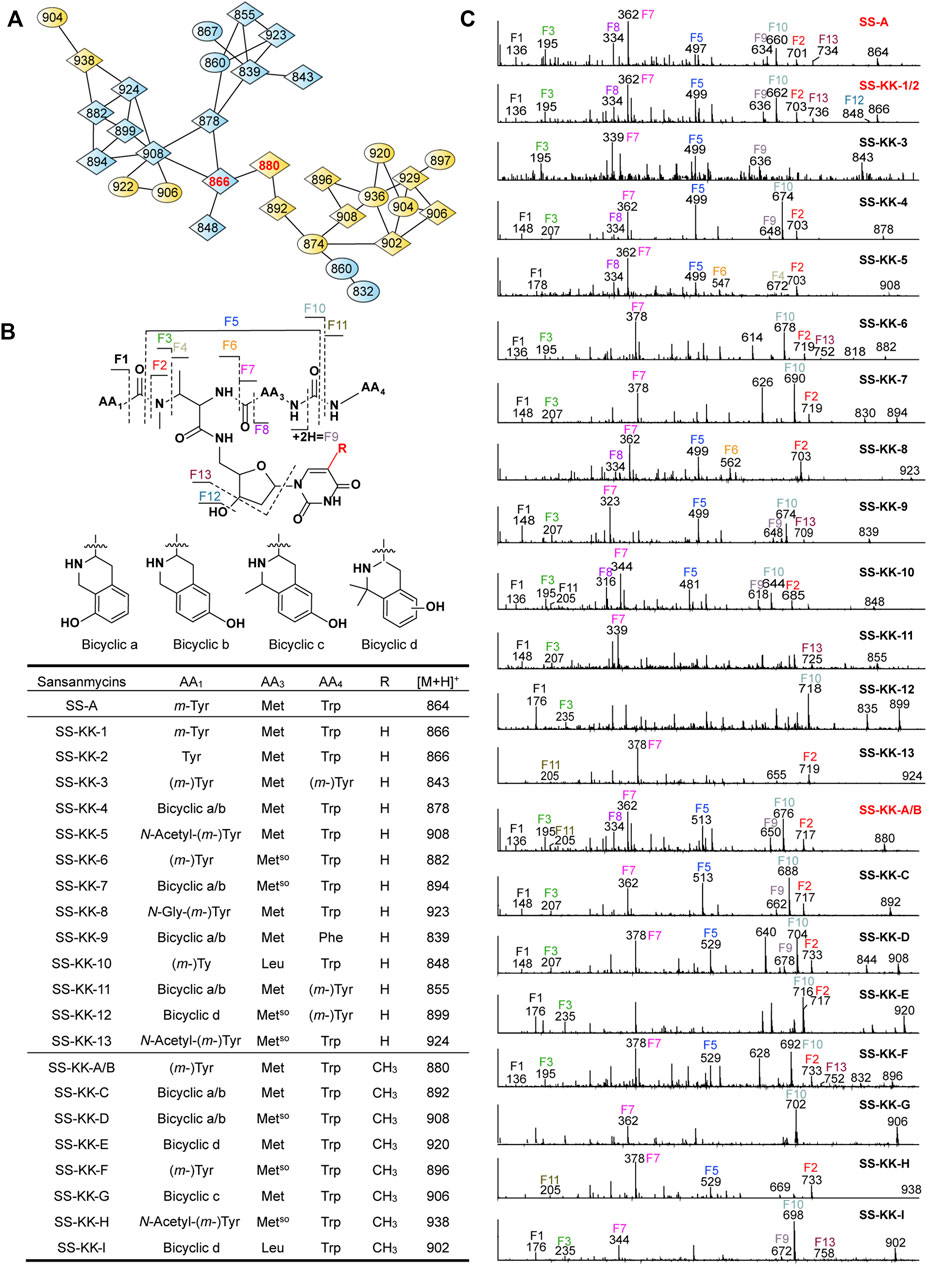
FIGURE 4. Molecular networking directed discovery of new sansanmycin analogues. (A) Molecular network consisting of all parent ions detected by LC–MS/MS in the extract crude of SS/KKO when fed with 1 or 2. A constellation for potential sansanmycins was picked out using SS-KK-1/2 and SS-KK-A/B (highlight in red) as a probe from the whole molecular network (Supplementary Figure S9) and amplified for displaying. The derivatives produced by feeding SS/KKO with 1 are labeled in blue, while the derivatives produced by feeding SS/KKO with 2 are labeled in yellow. All identified sansanmycin analogues are represented as nodes with diamond symbols. (B) The tentative structures of SS-KK-1 to SS-KK-13 and SS-KK-A to SS-KK-I. (C) MS/MS analysis for corresponding compounds. The diagnostic fragments are indicated.
The molecular weight of SS-KK-1 and SS-KK-2 showed 2 Da larger than that of SS-A, while the molecular weights of 1 is 2 Da larger than that of original 5′-aminouridine moiety, suggesting that 5′-aminouridine of SS-KK-1 and SS-KK-2 were replaced by 1. This hypothesis was supported by MS/MS spectra (Figure 4C), in which the characteristic fragment ions containing 5′-aminouridine portion, including F5, F6, F12, F13, were 2 Da larger than that of SS-A, and other characteristic fragment ions were consistent with those of SS-A. The F1 ion at m/z 136, corresponding to immonium ions of meta-tyrosine or tyrosine (Tyr), together with the F2 ion at m/z 703, corresponding to the loss of an m-Tyr or Tyr residue from N-terminus, inferred that SS-KK-1 and SS-KK-2 hold an m-Tyr or Tyr at AA1 position as found in SS-A (Xie et al., 2007) and SS MX-2 (Shi et al., 2016) respectively.
The molecular weight of 2 was 14 Da larger than that of 1. Similarly, the quasi molecular of SS-KK-A/B were also 14 Da larger than that of SS-KK-1/2, indicating that the fed precursor 2 was incorporated into the structures of SS-KK-A/B. This inference was supported by the corresponding tandem mass spectrum fragments, in which the characteristic fragment ions containing 5′-aminouridine moiety, including F2, F5, F9, F10, were found to be 14 Da larger than that of SS-KK-1/2, while other characteristic fragment ions were consistent with those of SS-KK-1/2 (Figure 4C). Therefore, it is speculated that SS-KK-A/B are similar to SS-KK-1/2 except for the replacement of 5′-aminouridine with 2.
The production of SS-KK-1/2 and SS-KK-A/B indicates that feeding 5′-aminouridine analogues to knockout strains SS/MKO and SS/KKO can afford new derivatives. Among them, SS/KKO showed a higher yield of new derivatives (Figure 3), indicating it is a better host for expanding the structural diversity of 5′-aminouridine by mutational biosynthesis. The above results suggest the potential of using mutational biosynthesis for production of 5′-aminouridine-modified sansanmycin derivatives.
2.3 New sansanmycin analogues discovered by molecular networking
The method of tandem mass-based molecular networking was previously established for rapid identification of sansanmycin analogues (Jiang et al., 2018). Here, we applied this method for analyzing sansanmycin derivatives produced by the above two cultures fed with precursors 1 and 2, respectively. Through this analysis, secondary metabolites showing similar fragmentation patterns in tandem mass spectra were grouped into multiple sub-networks (Supplementary Figure S9). Using SS-KK-1/2 and SS-KK-A/B as probes, a sub-network consisting of over 30 nodes was discovered in the molecular networks of extracts from the two cultures (Figure 4A; Supplementary Figure S9). The visualization of metabolites from the two cultures in different colors allowed us to easily distinguish the source of the molecules (Figure 4A; Supplementary Figure S9). Further manual analysis of the MS/MS data corresponding to these molecules reveals that the uridine moiety of the molecules from the culture fed with precursor 1 have altered from the original 5′-aminouridine moiety to 1. Similarly, the molecules from the culture fed with precursor 2 are found to bear the unit of 2 in place of the original 5′-aminouridine moiety in sansanmycin (detailed analyses are shown in Supplementary Results).
By closer inspection of their MS/MS spectra, we found that, in addition to the alterations in the 5′-aminouridine, the amino acids comprising the pseudopeptide chain also exhibit diversity. Due to the susceptibility of these molecules to cleavage at peptide bonds, analysis of their MS/MS data can provide a clear inference of the types and positions of amino acids in the pseudopeptide chain (Figure 4 and Supplementary Results). For instance, the fragment losses for M-F10 indicate the molecular weight of the C-terminal amino acid residue (AA4). A mass loss of 204 Da for M-F10 (SS-KK-1/2, SS-KK-4, SS-KK-6, SS-KK-7, SS-KK-10, SS-KK-A/B to SS-KK-G and SS-KK-I) suggests the presence of Trp at AA4. Similarly, a shortage of 165 Da indicates that Phe is at AA4 (SS-KK-9), while 181 Da (SS-KK-12) points to m-Tyr/Tyr ((m-)Tyr) in the place of AA4. Therefore, by analyzing the MS/MS fragments and comparing them with those of known sansanmycins (Jiang et al., 2018), the structures of additional eighteen new sansanmycin analogues (SS-KK-3 to SS-KK-13 and SS-KK-C to SS-KK-I) were tentatively deduced (Figure 4 and Supplementary Results).
Due to the limited production of many sansanmycin analogues, their structures can only be deduced through MS/MS data. The molecular networking approach, based on MS/MS data, provides valuable clues towards discovering more 5′-aminouridine-modified sansanmycin derivatives.
2.4 Isolation and structural determination of new sansanmycin analogues
To further characterize the structures deduced by MS/MS analysis of sansanmycin derivatives and determine their antimicrobial activity, SS/KKO fermentations fed with 1 and 2 were scaled up. The main components were isolated as previously described (Xie et al., 2007; Xie et al., 2014), and then further purified by semi-preparative HPLC to give SS-KK-1 (2.1 mg), SS-KK-2 (0.8 mg), SS-KK-3 (0.9 mg) and SS-KK-C (0.4 mg). The purity of these compound monomers was determined by HPLC (Supplementary Figures S10–13). Subsequently, the 1D and 2D NMR spectra of SS-KK-1 were collected in DMSO-d6 to aid in structural elucidation. However, due to the limited quantity of SS-KK-2, SS-KK-3 and SS-KK-C, their structures could not be further confirmed by NMR methods.
The NMR spectra in DMSO-d6 for SS-KK-1 were very similar to those of SS-A (Xie et al., 2007), except the signals from sugar moiety. Further analysis of these different signals disclosed that the fed 2-deoxy-5′-aminouridine in SS-KK-1 (δH 6.05 for H-sugar-1, 2.02 for H-sugar-2, 3.99 for H-sugar-3, 3.66 for H-sugar-4, 3.21 for H-sugar-5) was in place of 5′-aminouridine in SS-A, which was further confirmed by the sequential 1H–1H COSY correlations of H-sugar-1/H-sugar-2/H-sugar-3/H-sugar-4/H-sugar-5. Interpretation of the 1D and 2D NMR spectra (Supplementary Figures S14–19 and Supplementary Table S1) confirmed the proposed structure which holds an m-Tyr at the N-terminus.
Based on above MS/MS analysis, SS-KK-1 and SS-KK-2 produced the same fragmentations and was expected to hold an m-Tyr or Tyr at N-terminus, respectively. As m-Tyr was confirmed in the SS-KK-1, Tyr was inferred at the N-terminus of SS-KK-2.
2.5 Antibacterial activity and stability of new sansanmycin analogues
The antibacterial activity of sansanmycin analogues SS-KK-1, SS-KK-2, SS-KK-3, and SS-KK-C was tested against a range of bacteria (Table 1), including both Gram-negative and Gram-positive bacteria, as well as standard strain of M. tuberculosis H37Rv, and clinically isolated strains of M. tuberculosis S17, M6600, M3551 and M9483. Among them, M6600, M3551, and M9483 are multidrug-resistant strains, resistant to INH at 0.1 μg/mL and RIF at 1 μg/mL. Unexpectedly, both SS-KK-1 and SS-KK-C lost their antibacterial activity against P. aeruginosa, M. tuberculosis, and even E. coli ΔtolC. SS-KK-3 showed the same antibacterial activity as SS-A against E. coli ΔtolC and M. tuberculosis strains including H37Rv and clinically isolated, drug-sensitive and multidrug-resistant strains. Interestingly, although SS-KK-2 lost its inhibitory activity against P. aeruginosa, it showed the best inhibition activity against E. coli ΔtolC compared with SS-KK-3 and SS-A. The bioactivity of our newly obtained SS-KK-2 and SS-KK-3, which do not contain the 2′-OH group, suggests that it is not essential for their activity.
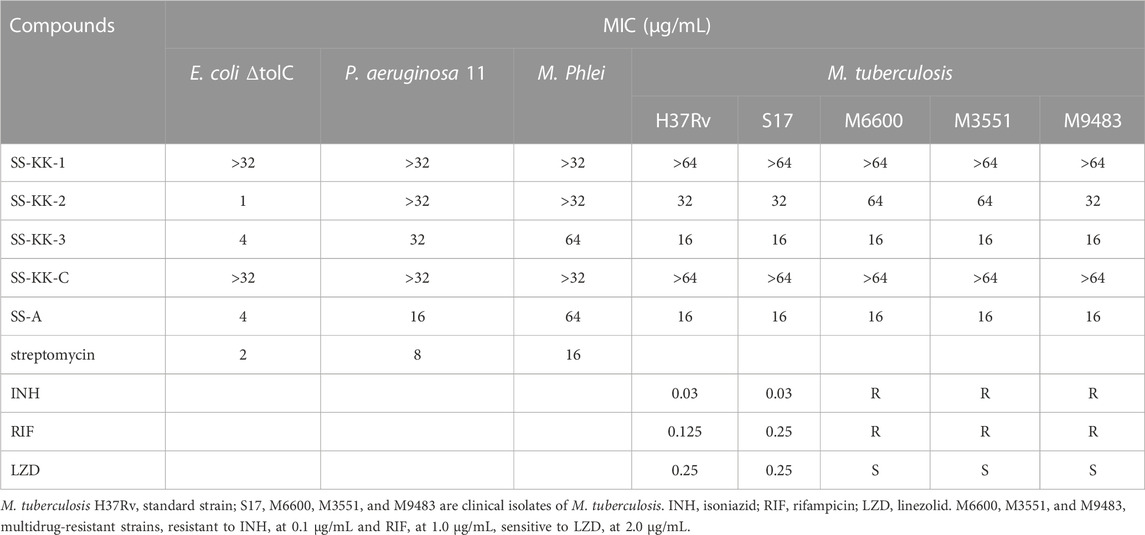
TABLE 1. Activities of sansanmycin analogues.
Poor stability is one of the factors hindering the development of sansanmycin analogues into drug candidates. Thus, we conducted stability experiments followed the method reported previously (Shi et al., 2016) on the two active sansanmycin analogues, SS-KK-2 and SS-KK-3 (Figure 5). These experiments were carried out at room temperature with the parent compound SS-A as a control. When subjected to these conditions, the residual quantities of SS-A were depleted below 70% after 9 days of incubation, whereas SS-KK-2 showed a slightly higher level of stability than SS-A. SS-KK-3 exhibited minimal degradation and substantially greater stability than both SS-A and SS-KK-2. As SS-KK-3 displayed antimicrobial activity comparable to that of SS-A, it is a more promising lead compound for anti-TB drug development.
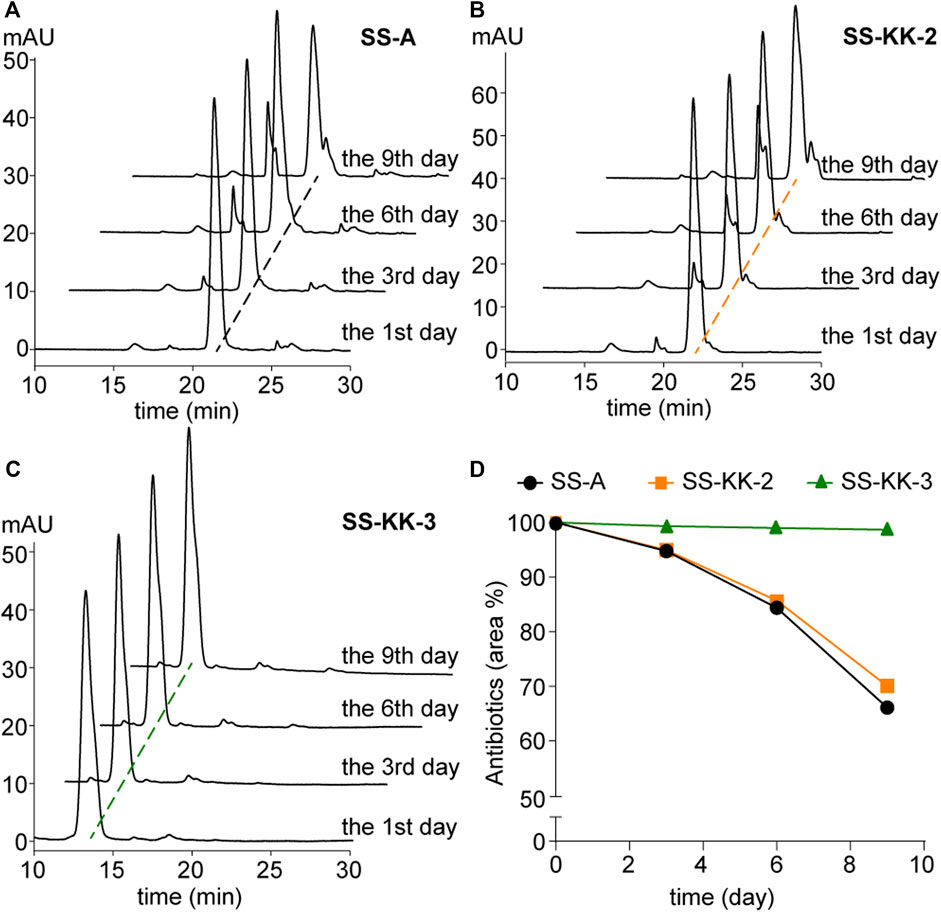
FIGURE 5. Stability of SS-A, SS-KK-2 and SS-KK-3. (A–C) HPLC analysis of SS-A, SS-KK-2 and SS-KK-3 at indicated days in KH2PO4 buffer (pH 6.0) at room temperature. (D) The changes of the level of SS-A, SS-KK-2 and SS-KK-3 over time. All samples were analyzed by HPLC and quantified according to the areas of peaks. Each sample underwent three parallel repeats. SS-A, black line; SS-KK-2, orange line; SS-KK-3, green line.
3 Discussion
To expand the structural diversity of 5′-aminouridine moiety of sansanmycin through biosynthetic methods, the function of ssaM and ssaK as biosynthetic genes for 5′-aminouridine moiety of sansanmycins was firstly validated in vivo. The production of hydrated SS-A in the ssaM-deletion mutant confirmed the role of ssaM as a dehydratase. Unfortunately, due to the low yield of hydrated SS-A, the pure compound could not be obtained for activity assay. Preliminary attempt of feeding easily accessible uridine analogues containing 5′-OH in vivo, which can form ester bonds with tetra/pentapeptides catalyzed by PacI in vitro (Zhang et al., 2011), did not result in the expected derivatives by mutational biosynthesis. This could be due to either the varying efficiency of SsaI, which is the homologue of PacI, between in vivo and in vitro conditions, or to the larger substrate tolerance of PacI compared to SsaI. As expected, 5′-aminouridine analogues were shown to be readily incorporated into sansanmycin biosynthetic pathway in SS/KKO and SS/MKO mutant, with higher effectiveness in SS/KKO mutant. To the best of our knowledge, it is the first report to modify 5′-aminouridine of UPAs and obtained a range of 5′-aminouridine-modified derivatives through mutational biosynthesis, which confirms that SS/KKO obtained in this study is a good host for enhancing the structural diversity of 5′-aminouridine of UPAs. In contrast to tedious and challenging chemical synthesis, employing SS/KKO to increase the structural diversity of 5′-aminouridine via mutasynthesis is more efficient and effective.
he 5′-aminouridine moiety of UPAs, as present in other classes of naturally occurring nucleoside inhibitors such as muraymycin, caprazamycin and capuramycin, resembles the uridine moiety of UDP-MurNAc pentapeptide, the natural substrate of MraY. Recent structural biological data of MraY-inhibitor complex also showed a common feature of the uridine binding pocket of naturally occurring nucleoside inhibitors including caprazamycin, capuramycin, and mureidomycin (Mashalidis et al., 2019). The chemically synthesized dihydropacidamycin D resulting from reduction of the chemically unstable 4′-exo double bond of ribosyl moiety showed similar antibacterial activity to that of pacidamycin D (Boojamra et al., 2001). Lemoine et al. further synthesized a series of dihydropacidamycin D derivatives by chemically modifying the uracil ring of 5′-aminouridine, however, none of these derivatives were capable of inhibiting MraY, highlighting the crucial role of uracil for UPAs’ activity (Lemoine et al., 2002). These early discoveries were also consistent with recent structural biological data of MraY-inhibitor complex, which revealed that the uridine pocket of MraY has a certain level of spatial tolerance. Moreover, compared to the ribosyl moiety, the modifications in the uracil moiety are more likely to interfere with the interactions between the residues forming the uridine pocket of MraY, leading to the loss of inhibitory effect (Mashalidis et al., 2019). In this study, SS-KK-C, an analogue obtained from the culture fed precursor 2 with a methyl modification on uracil, also lost its antibacterial activity against P. aeruginosa, M. tuberculosis, and even E. coli ΔtolC.
Previous chemically synthesized 5′-aminouridine-modified derivatives have always contained a 2′-OH on the nucleoside ribosyl group (Figure 1) (Inukai et al., 1993; Boojamra et al., 2001; Okamoto et al., 2012; Tran et al., 2017; Mashalidis et al., 2019; Niro et al., 2020; Tran et al., 2021). By generating a series of 5′-aminouridine analogues that lacks 2′-OH for the first time, this study demonstrated that the presence of 2′-OH is dispensable for the bioactivity of UPAs. In previous reports, the R-stereochemistry at the C-4′ position of dihydropacidamycin has been considered as playing a significant role in maintaining antibacterial activity (Boojamra et al., 2001; Tran et al., 2017). However, we noticed that SS-KK-1 with the R-stereochemistry at the C-4′ position of the ribose attached to the same tetrapeptide backbone as sansanmycin A completely losses antibacterial activity. Further inspection of the structure of uridine unit disclosed that although the new dihydro UPA derivatives obtained in this study share the R-stereochemistry at the C-4′ position of the ribose with the chemically synthesized dihydro UPAs reported previously (Boojamra et al., 2001; Tran et al., 2017), their relative configurations differ. The chemically synthesized dihydro UPAs bear an α-l-ribose configuration, while the dihydro UPA derivatives in this study show a β-d-ribose configuration. It appears that the role of stereochemistry of C-4′ position required further evaluation using more UPA derivatives with structurally diverse uridine.
As uridine moiety is an important part of pharmacophore, further research should be conducted on the structure-activity relationship of the nucleoside part of UPAs. Here we show that mutational biosynthesis offers an efficient and accessible method to expand the structural diversity of 5′-aminouridine moiety of sansanmycin. In the future, by synthesizing structurally varied 5′-aminouridine analogues for mutasynthesis in SS/KKO, the diversity of UPA structures may be further increased, which would provide more valuable insights into the structure-activity relationship of the nucleoside part in UPAs. Additionally, utilizing the biosynthetic method to obtain UPAs with different 5′-aminouridine modifications may be combined with genetically manipulated biosynthesis on the pseudopeptide parts in vivo, which will significantly enhance the diversity of UPAs and hold promise for obtaining UPAs with improved activity and/or druggability.
4 Conclusion
This study confirmed the functions of SsaM and SsaK in the biosynthesis of 5′-aminouridine of sansanmycin through gene deletion and complementation experiments. Based on this, mutational biosynthesis enriched the diversity of nucleoside moiety of sansanmycins by applying two 5′-aminouridine analogues, especially in ssaK knockout strain. Combining manual analysis and molecular networking of MS/MS data, twenty-two new 5′-aminouridine-modified sansanmycin derivatives were identified, among which four were purified and their antibacterial activities were determined. Of note, SS-KK-3 exhibited anti-mycobacterial activity comparable to that of sansanmycin A, even to clinically isolated multidrug-resistant strains of M. tuberculosis. Furthermore, SS-KK-3 exhibited substantial improvements in stability compared to sansanmycin A. The enhanced druggability of this compound may spur further research into the development of new anti-TB drug candidates acting against clinically unexplored target MraY.
5 Materials and methods
5.1 Strains, plasmids and growth conditions
To propagate and transform the sansanmycin-producing strain Streptomyces sp. SS, obtained from China Pharmaceutical Culture Collection (CPCC 200442), the strain was grown on solid S5 medium (Wang et al., 2009) at 28°C for sporulation and in liquid fermentation medium (Xie et al., 2007) for sansanmycin production. Mannitol soya flour (MS) agar medium (Kieser et al., 2000) was used for conjugation, and liquid phage medium (Korn et al., 1978) was used for genomic DNA isolation. Escherichia coli DH5α was used for general cloning experiments (Sambrook, 2001), while E. coli ET12567/pUZ8002 (Kieser et al., 2000) was used for conjugal transfer following established protocols. E. coli strains were incubated in Luria–Bertani medium (LB) (Sambrook, 2001) at 37°C. Strains were incubated with apramycin (Am, 50 μg/mL), kanamycin (Km, 25 μg/mL), and chloramphenicol (Cm, 25 μg/mL) when required. The antimicrobial activity of the compounds was tested against E. coli ΔtolC mutant, P. aeruginosa 11, M. phlei, and M. tuberculosis. All strains and plasmids used in this study see Table 2.
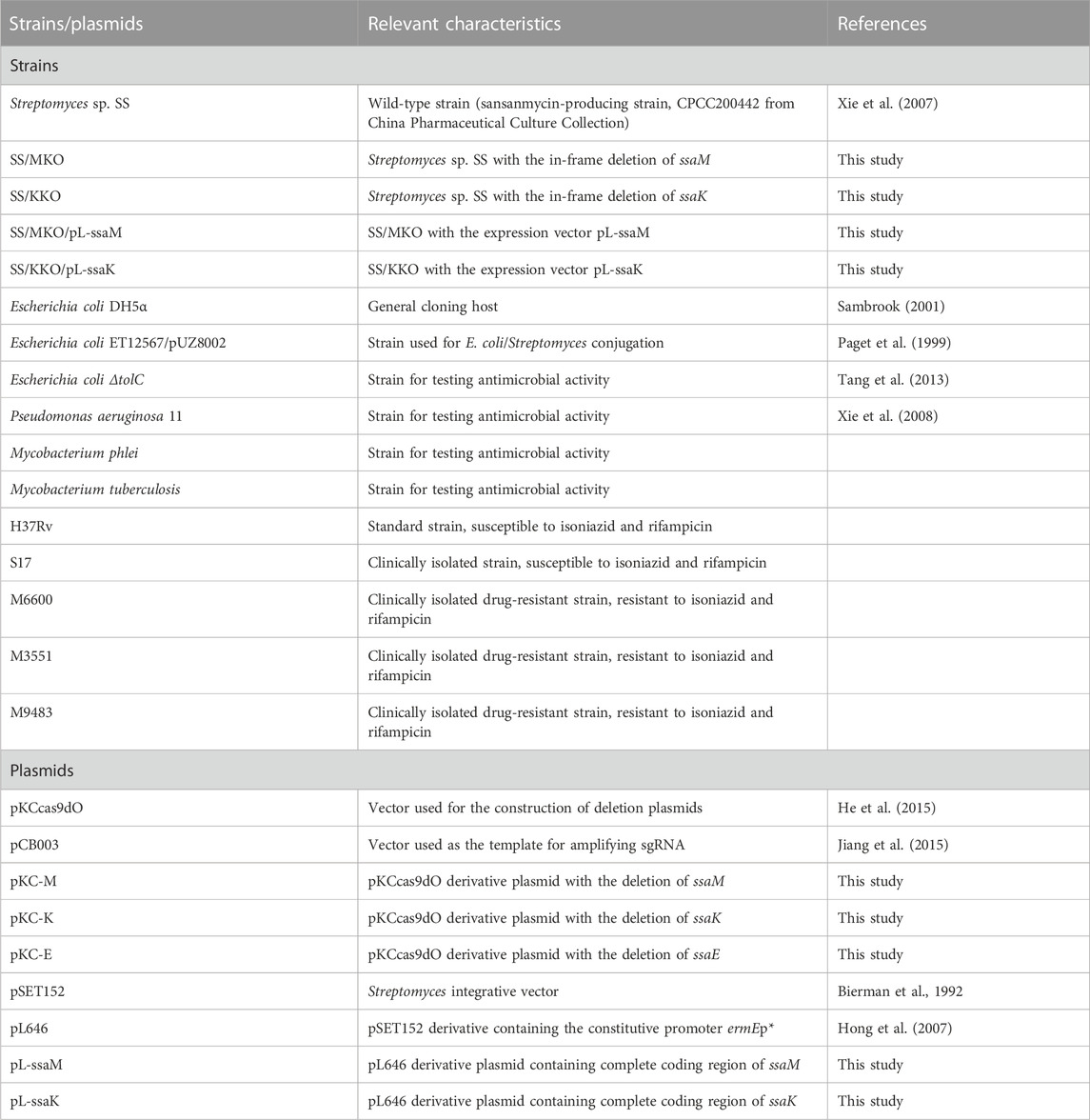
TABLE 2. Strains and plasmids used in this study.
5.2 Construction of Streptomyces sp. SS ssaM and ssaK mutant
The mutant strains ΔssaM or ΔssaK were generated by CRISPR/Cas9-mediated deletion. The mutant strain ΔssaE was also attempted to be constructed using the same method, but the correct strain was not obtained in the end. The pKCcas9dO (He et al., 2015) which comprises a codon-optimized cas9 for Streptomyces was used for gene deletion. To design the sgRNA targeting sequences, the gene sequences of interest were firstly submitted on the website http://www.rgenome.net/cas-designer/, and the output result is a series of sgRNA targeting sequences containing 20 bp located inside the gene of interest. Then, the off-target rate of sgRNA targeting sequences is assessed using CasOT (Xiao et al., 2014) to find sgRNA sequences that can be used to knock out the gene of interest. The sgRNA module plasmid pCB003 was used for amplifying M-sgRNA, K-sgRNA, and E-sgRNA. M-sgRNA, K-sgRNA, and E-sgRNA fragments were amplified with specific forward primers M-sg/K-sg/E-sg (Supplementary Table S2) which contain sgRNA targeting sequences of ssaM/ssaK/ssaE, and general reverse primers P2 (Supplementary Table S2), respectively. Homologous arm flanking ssaM, ssaK and ssaE were amplified from Streptomyces sp. SS genomic DNA by PCR with specific primer pairs MP3/MP4 and MP5/MP6, KP3/KP4 and KP5/KP6, and EP3/EP4 and EP5/EP6 respectively. For the deletion plasmid of ssaM, the fragment containing M-sgRNA and homologous arms were assembled by overlapping PCR with primer pair M-sg/MP6, then the fragment was cloned into SpeI-HindIII sites of pKCcas9dO to obtain the pKC-M. For the deletion plasmids of ssaK and ssaE, the fragment containing K-sgRNA or E-sgRNA and corresponding homologous arms were assembled into NdeI-BamHI sites of pL646 (Hong et al., 2007) with NEBuilder HiFi DNA Assembly Cloning Kit (New England Biolabs, United States) to obtain the pL-KO and pL-EO, then the fragment obtained by pL-KO and pL-EO digested with SpeI/HindIII was cloned into SpeI-HindIII sites of pKCcas9dO to obtain the pKC-K and pKC-E. The constructed plasmids were introduced into Streptomyces sp. SS by conjugation from E. coli ET12567/pUZ8002. The resulting mutant strains were designated SS/MKO and SS/KKO, but SS/EKO was not obtained.
5.3 Construction of complementary strains
Complementation plasmids for mutant strains ΔssaM or ΔssaK were constructed using pL646 (Hong et al., 2007) as the vector. The ssaM and ssaK genes were amplified from the genomic DNA of S. sp. SS using ssaM-F/ssaM-R and ssaK-F/ssaK-R primers (Supplementary Table S2), respectively. These genes contained NdeI and BamHI restriction sites and were cloned into the T-vector prior to sequencing. After correctly sequencing the ssaM and ssaK genes, they were ligated into the NdeI and BamHI restriction sites of the pL646 (Hong et al., 2007) vector resulting in the plasmids pL-ssaM and pL-ssaK. The recombined plasmids were validated by conjugation transfer into their corresponding knockout strains resulting in complemented strains SS/MKO/pL-ssaM and SS/KKO/pL-ssaK.
5.4 Analysis of production of sansanmycins
The methods for fermentation, isolation, and high-pressure liquid chromatography (HPLC) analysis of sansanmycins were conducted in accordance with previously published procedures (Xie et al., 2007; Xie et al., 2014). Cultivation of Streptomyces sp. SS and its derivatives proceeded in a 100 mL volume of liquid fermentation medium at 28°C under agitation at 200 rpm. After 2 days of culture, a 5% seed culture was transferred into new fermentation medium and maintained at 28°C for an additional 5 days under agitation at 200 rpm. During the feeding experiment, exogenous substrate (uridine analogue-1 to -12 (Supplementary Figure S8) and 5′-aminouridine analogues 1/2 (Figure 3A)) was added to the fermentation medium at a final concentration of 3 mM. The fermented broth was clarified by centrifugation at 4 °C for 20 min at 4,000 rpm, and the supernatant was harvested and subjected to HPLC-MS analysis after extraction with a Sep-Pack C18 cartridge. HPLC-MS analysis was performed on an Agilent 6,410 (Agilent Technologies, United States) equipped with XBridge® C18 column (4.6 × 150 mm, 3.5 μm, Waters, Dublin, Ireland), using a gradient mobile phase that consisted of solvent A (water +0.1% w/v ammonium acetate) and solvent B (100% MeOH). The gradient started from 80:20 solvent A: solvent B and was changed to 40:60 solvent A: solvent B within 30 min. The flow rate was set at 0.8 mL/min.
5.5 Tandem mass-based molecular networking
For analyzing sansanmycins through LC-MS/MS, the fermentation broth was enriched using macroporous absorbent resin D4006 (Nankai University Fine Chemical Experiment Factory, Tianjin, China) column. After elution with acetone aqueous solutions of various concentrations, sansanmycins were obtained in 20% and 30% solutions respectively. The crude extract consisting of sansanmycins was acquired after removing the acetone through rotary evaporation. LC-ESI(+)MS/MS data was collected on a Waters ACQUITY UPLC H-Class system (Milford, MA, United States) with a Waters Xevo G2-XS QTof detector (Manchester, United Kingdom). ACQUITY UPLC CSHTM C18 column (1.7 μm, 2.1 × 100 mm, Waters) was employed during UPLC analysis carried out at a flow rate of 0.3 mL/min. Solvent A consisted of 0.1% (w/v) formic acid in water, while Solvent B contained 0.1% (w/v) formic acid in acetonitrile. Over 10 min, mobile phase B increased gradually from 10% to 40%. Mass spectral data were acquired in continuum mode using the fast DDA function. The obtained raw data was converted into.mzML format before undergoing further analysis via the molecular networking tool on the GNPS (global natural product social molecular networking) website (https://gnps.ucsd.edu/ProteoSAFe/static/gnps-splash.jsp). Various parameters defaulted and precursor ion mass was set to 1.00 Da with an MS/MS fragment ion tolerance of 0.02 Da. Finally, results were downloaded and visualized by employing Cytoscape 3.9.1.
5.6 Purification and characterization of sansanmycin analogues
Separation and purification of sansanmycin analogues followed the method reported previously (Xie et al., 2014) with some modifications.
Fermentation supernatants were subjected to macroporous absorbent resin D4006 column chromatography. Sansanmycins were eluted in 20% and 30% acetone aqueous solutions and then separately passed through DEAE-Sephadex A-25 chromatograph (GE Healthcare, United States), utilizing Tris-HCl (20 mM, pH 8.5) plus varying concentrations of NaCl, while being monitored by HPLC-UV. The effluent consisting of target compounds was collected and subjected to further purification using preparative HPLC, conducted on a SHIMADZU LC-20 HPLC machine equipped with a diode array detector (DAD). A XBridge® prep C18 column (5 μm, 250 × 10 mm, Waters) was used during preparative HPLC. NMR data from the purified samples were acquired employing Bruker spectrometers and DMSO-d6 as solvent.
SS-KK-1: white amorphous powder; UV (MeOH) λmax (log ε) 216 (4.77), 265 (4.26) nm [α]20D −21.05 (c0.19, MeOH).
5.7 Antibacterial assay
To determine the inhibition activity of sansanmycin analogues against M. tuberculosis H37Rv and clinically isolated strains, a measurement was conducted by the microplate Alamar blue assay, following the method described by (Zhang et al., 2013). The analogues were first dissolved in dimethyl sulfoxide (DMSO) and then serially diluted to final concentrations (μg/mL) ranging from 64 to 0.125 before being tested with 7H9 broth supplemented. INH, RIF and linezolid (LZD) were used as controls for M. tuberculosis H37Rv and clinical strains isolated by previous reported method (Hameed et al., 2022). DMSO served as a negative control in these experiments. Minimum inhibitory concentration (MIC) is defined as the lowest concentration of samples that prevents the conversion of the blue color into pink color. Triplicate wells per drug concentration were used.
The microdilution assay recommended by the Clinical and Laboratory Standards Institute (CLSI, 2009, formerly NCCLS) was employed to determine the MICs of other bacterial strains. Strains were cultured in Mueller-Hinton broth (MHB) (Weissauer-Condon et al., 1987) and the final bacterial suspension (in MHB medium) was adjusted to 106 cells/ml. Dilutions of test compounds were substituted with MHB medium as described above. Then, triplicate 100 μL transfers of serial dilutions were placed into 96-well plates, after which 100 μL of bacterial suspension was added to each well. MIC is defined as the lowest concentration of samples that restrains the growth of the test organism detected visually following incubation for 10 h at 37°C. Streptomycin served as a positive control.
5.8 Stability determination of sansanmycin analogues
The stability assessment of sansanmycin A and its analogues was conducted using a previously reported method (Shi et al., 2016). Sansanmycins were dissolved in 0.05 M KH2PO4 buffer (with pH adjusted to 6.0 using NaOH) and incubated at 28 °C for 9 days with testing conducted every 3 days. Each sample underwent three parallel repeats. All samples were analyzed by HPLC and quantified by peak area analysis.
Data availability statement
The GNPS data of this study are available from the corresponding author upon request.
Author contributions
YLu: Investigation, Data curation, Methodology, Writing–original draft. YLi: Methodology, Writing–original draft. JF: Methodology, Investigation, Writing–original draft. XL: Methodology, Writing–original draft, Funding acquisition. HS: Methodology, Writing–original draft. LW: Methodology, Writing–original draft. XH: Writing–original draft, Data curation, Investigation. YZ: Writing–original draft, Data curation. TZ: Methodology, Data curation, Writing–review and editing. YS: Data curation, Methodology, Writing–review and editing. YX: Data curation, Methodology, Writing–review and editing. BH: Writing–review and editing, Conceptualization, Funding acquisition, Investigation, Project administration, Supervision.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Key Research and Development Program of China (2018YFA0902000, 2021YFA1300904), National Natural Science Foundation of China (82273832, 32270106, 82104046, 81973372), and the CAMS Innovation Fund for Medical Sciences (2021-I2M-1-029).
Acknowledgments
We thank Bertolt Gust (Pharmaceutical Institute, University of Tuebingen, Germany) for kindly providing Escherichia coli ΔtolC mutant strain.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2023.1278601/full#supplementary-material
References
Bierman, M., Logan, R., O’Brien, K., Seno, E. T., Rao, R. N., and Schoner, B. E. (1992). Plasmid cloning vectors for the conjugal transfer of DNA from Escherichia coli to Streptomyces spp. Gene 116, 43–49. doi:10.1016/0378-1119(92)90627-2
Boojamra, C. G., Lemoine, R. C., Lee, J. C., Léger, R., Stein, K. A., Vernier, N. G., et al. (2001). Stereochemical elucidation and total synthesis of dihydropacidamycin D, a semisynthetic pacidamycin. J. Am. Chem. Soc. 123, 870–874. doi:10.1021/ja003292c
Chatterjee, S., Nadkarni, S. R., Vijayakumar, E. K., Patel, M. V., Ganguli, B. N., Fehlhaber, H. W., et al. (1994). Napsamycins, new Pseudomonas active antibiotics of the mureidomycin family from Streptomyces sp. HIL Y-82,11372. J. Antibiot. 47, 595–598. doi:10.7164/antibiotics.47.595
CLSI (2009). “Methods for Dilution antimicrobial susceptibility tests for bacteria that grow aerobically: approved standard—8th Edition,” in CLSI document M07-A8 (Wayne: Clinical and laboratory standards institute).
Gruschow, S., Rackham, E. J., Elkins, B., Newill, P. L., Hill, L. M., and Goss, R. J. (2009). New pacidamycin antibiotics through precursor-directed biosynthesis. Chembiochem 10, 355–360. doi:10.1002/cbic.200800575
Hameed, H. M. A., Fang, C., Liu, Z., Ju, Y., Han, X., Gao, Y., et al. (2022). Characterization of genetic variants associated with rifampicin resistance level in Mycobacterium tuberculosis clinical isolates collected in Guangzhou chest hospital, China. Infect. Drug. Resist. 15, 5655–5666. doi:10.2147/idr.s375869
He, H., Zheng, G., Jiang, W., Hu, H., and Lu, Y. (2015). One-step high-efficiency CRISPR/Cas9-mediated genome editing in Streptomyces. Acta. Biochim. Biophys. Sin. 47, 231–243. doi:10.1093/abbs/gmv007
Hong, B., Phornphisutthimas, S., Tilley, E., Baumberg, S., and Mcdowall, K. J. (2007). Streptomycin production by Streptomyces griseus can be modulated by a mechanism not associated with change in the adpA component of the A-factor cascade. Biotechnol. Lett. 29, 57–64. doi:10.1007/s10529-006-9216-2
Inukai, M., Isono, F., Takahashi, S., Enokita, R., Sakaida, Y., and Haneishi, T. (1989). Mureidomycins A-D, novel peptidylnucleoside antibiotics with spheroplast forming activity. I. Taxonomy, fermentation, isolation and physico-chemical properties. J. Antibiot. 42, 662–666. doi:10.7164/antibiotics.42.662
Inukai, M., Isono, F., and Takatsuki, A. (1993). Selective inhibition of the bacterial translocase reaction in peptidoglycan synthesis by mureidomycins. Antimicrob. Agents. Chemother. 37, 980–983. doi:10.1128/AAC.37.5.980
Ióca, L. P., Dai, Y., Kunakom, S., Diaz-Espinosa, J., Krunic, A., Crnkovic, C. M., et al. (2021). A family of nonribosomal peptides modulate collective behavior in Pseudovibrio bacteria isolated from marine sponges. Angew. Chem. Int. Ed. Engl. 60, 15891–15898. doi:10.1002/anie.202017320
Jiang, Y., Chen, B., Duan, C., Sun, B., Yang, J., Yang, S., et al. (2015). Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 System. Appl. Environ. Microbiol. 81, 2506–2514. doi:10.1128/AEM.04023-14
Jiang, Z. B., Ren, W. C., Shi, Y. Y., Li, X. X., Lei, X., Fan, J. H., et al. (2018). Structure-based manual screening and automatic networking for systematically exploring sansanmycin analogues using high performance liquid chromatography tandem mass spectroscopy. J. Pharm. Biomed. Anal. 158, 94–105. doi:10.1016/j.jpba.2018.05.024
Karwowski, J. P., Jackson, M., Theriault, R. J., Chen, R. H., Barlow, G. J., and Maus, M. L. (1989). Pacidamycins, a novel series of antibiotics with anti-Pseudomonas aeruginosa activity. I. Taxonomy of the producing organism and fermentation. J. Antibiot. 42, 506–511. doi:10.7164/antibiotics.42.506
Kieser, T., Bibb, M. J., Buttner, M. J., Chater, K. F., Hopwood, D. A., Charter, K., et al. (2000). Practical Streptomyces genetics. Norwich: John Innes Foundation.
Koppermann, S., Cui, Z., Fischer, P. D., Wang, X., Ludwig, J., Thorson, J. S., et al. (2018). Insights into the target interaction of naturally occurring muraymycin nucleoside antibiotics. ChemMedChem 13, 779–784. doi:10.1002/cmdc.201700793
Korn, F., Weingrtner, B., and Kutzner, H. J. (1978). “A study of twenty actinophages: morphology, serological relationship and host range,” in Genetics of the actinomycetales. Editors E. Freerksen, I. Tarnok, and H. Thumin (New York: Gustav Fischer Verlag Press), 251–270.
Lemoine, R. C., Magon, A., and Hecker, S. J. (2002). Synthesis of base-modified dihydropacidamycins. Bioorg. Med. Chem. Lett. 12, 1121–1123. doi:10.1016/s0960-894x(02)00100-2
Mashalidis, E. H., Kaeser, B., Terasawa, Y., Katsuyama, A., Kwon, D. Y., Lee, K., et al. (2019). Chemical logic of MraY inhibition by antibacterial nucleoside natural products. Nat. Commun. 10, 2917. doi:10.1038/s41467-019-10957-9
Michailidou, F., Chung, C. W., Brown, M. J. B., Bent, A. F., Naismith, J. H., Leavens, W. J., et al. (2017). Pac13 is a Small, Monomeric dehydratase that mediates the formation of the 3'-deoxy nucleoside of pacidamycins. Angew. Chem. Int. Ed. Engl. 56, 12492–12497. doi:10.1002/anie.201705639
Niro, G., Weck, S. C., and Ducho, C. (2020). Merging natural products: muraymycin-sansanmycin hybrid structures as novel scaffolds for potential antibacterial agents. Chemistry 26, 16875–16887. doi:10.1002/chem.202003387
Okamoto, K., Sakagami, M., Feng, F., Togame, H., Takemoto, H., Ichikawa, S., et al. (2012). Total synthesis and biological evaluation of pacidamycin D and its 3'-hydroxy analogue. J. Org. Chem. 77, 1367–1377. doi:10.1021/jo202159q
Paget, M. S., Chamberlin, L., Atrih, A., Foster, S. J., and Buttner, M. J. (1999). Evidence that the extracytoplasmic function sigma factor ςE is required for normal cell wall structure in Streptomyces coelicolor A3(2). J. Bacteriol. 181, 204–211. doi:10.1128/JB.181.1.204-211.1999
Ragab, A. E., Gruschow, S., Tromans, D. R., and Goss, R. J. (2011). Biogenesis of the unique 4',5'-dehydronucleoside of the uridyl peptide antibiotic pacidamycin. J. Am. Chem. Soc. 133, 15288–15291. doi:10.1021/ja206163j
Sambrook, J. R. D. (2001). Molecular cloning: a laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
Shi, Y., Jiang, Z., Lei, X., Zhang, N., Cai, Q., Li, Q., et al. (2016). Improving the N-terminal diversity of sansanmycin through mutasynthesis. Microb. Cell. Fact. 15, 77. doi:10.1186/s12934-016-0471-1
Tang, X., Gross, M., Xie, Y., Kulik, A., and Gust, B. (2013). Identification of mureidomycin analogues and functional analysis of an N-acetyltransferase in napsamycin biosynthesis. Chembiochem 14, 2248–2255. doi:10.1002/cbic.201300287
Tran, A. T., Watson, E. E., Pujari, V., Conroy, T., Dowman, L. J., Giltrap, A. M., et al. (2017). Sansanmycin natural product analogues as potent and selective anti-mycobacterials that inhibit lipid I biosynthesis. Nat. Commun. 8, 14414. doi:10.1038/ncomms14414
Tran, W., Kusay, A. S., Hawkins, P. M. E., Cheung, C. Y., Nagalingam, G., Pujari, V., et al. (2021). Synthetic sansanmycin analogues as potent Mycobacterium tuberculosis translocase I inhibitors. J. Med. Chem. 64, 17326–17345. doi:10.1021/acs.jmedchem.1c01407
Walsh, C. T., and Zhang, W. (2011). Chemical logic and enzymatic machinery for biological assembly of peptidyl nucleoside antibiotics. ACS Chem. Biol. 6, 1000–1007. doi:10.1021/cb200284p
Wang, L., Hu, Y., Zhang, Y., Wang, S., Cui, Z., Bao, Y., et al. (2009). Role of sgcR3 in positive regulation of enediyne antibiotic C-1027 production of Streptomyces globisporus C-1027. Bmc. Microbiol. 9, 14. doi:10.1186/1471-2180-9-14
Weissauer-Condon, C., Engels, I., and Daschner, F. D. (1987). In vitro activity of four new quinolones in Mueller-Hinton broth and peritoneal dialysis fluid. Eur. J. Clin. Microbiol. 6, 324–326. doi:10.1007/BF02017630
Winn, M., Goss, R. J., Kimura, K., and Bugg, T. D. (2010). Antimicrobial nucleoside antibiotics targeting cell wall assembly: recent advances in structure-function studies and nucleoside biosynthesis. Nat. Prod. Rep. 27, 279–304. doi:10.1039/b816215h
World Health Organization (2022). Global tuberculosis report 2022. Available at: https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2022 (Accessed October 27, 2022).
Xiao, A., Cheng, Z., Kong, L., Zhu, Z., Lin, S., Gao, G., et al. (2014). CasOT: a genome-wide Cas9/gRNA off-target searching tool. Bioinformatics 30, 1180–1182. doi:10.1093/bioinformatics/btt764
Xie, Y., Cai, Q., Ren, H., Wang, L., Xu, H., Hong, B., et al. (2014). NRPS substrate promiscuity leads to more potent antitubercular sansanmycin analogues. J. Nat. Prod. 77, 1744–1748. doi:10.1021/np5001494
Xie, Y., Chen, R., Si, S., Sun, C., and Xu, H. (2007). A new nucleosidyl-peptide antibiotic, sansanmycin. J. Antibiot. 60, 158–161. doi:10.1038/ja.2007.16
Xie, Y., Xu, H., Si, S., Sun, C., and Chen, R. (2008). Sansanmycins B and C, new components of sansanmycins. J. Antibiot. 61, 237–240. doi:10.1038/ja.2008.34
Zhang, N., Liu, L., Shan, G., Cai, Q., Lei, X., Hong, B., et al. (2016). Precursor-directed biosynthesis of new sansanmycin analogs bearing para-substituted-phenylalanines with high yields. J. Antibiot. 69, 765–768. doi:10.1038/ja.2016.2
Zhang, W., Ntai, I., Bolla, M. L., Malcolmson, S. J., Kahne, D., Kelleher, N. L., et al. (2011). Nine enzymes are required for assembly of the pacidamycin group of peptidyl nucleoside antibiotics. J. Am. Chem. Soc. 133, 5240–5243. doi:10.1021/ja2011109
Zhang, W., Ostash, B., and Walsh, C. T. (2010). Identification of the biosynthetic gene cluster for the pacidamycin group of peptidyl nucleoside antibiotics. Proc. Natl. Acad. Sci. U.S.A. 107, 16828–16833. doi:10.1073/pnas.1011557107
Zhang, Y. J., Reddy, M. C., Ioerger, T. R., Rothchild, A. C., Dartois, V., Schuster, B. M., et al. (2013). Tryptophan biosynthesis protects mycobacteria from CD4 T-cell-mediated killing. Cell 155, 1296–1308. doi:10.1016/j.cell.2013.10.045
Keywords: sansanmycin, 5′-aminouridine, structural diversity, mutational biosynthesis, new analogues
Citation: Lu Y, Li Y, Fan J, Li X, Sun H, Wang L, Han X, Zhu Y, Zhang T, Shi Y, Xie Y and Hong B (2023) Expanding structural diversity of 5′-aminouridine moiety of sansanmycin via mutational biosynthesis. Front. Bioeng. Biotechnol. 11:1278601. doi: 10.3389/fbioe.2023.1278601
Received: 16 August 2023; Accepted: 18 October 2023;
Published: 30 October 2023.
Edited by:
Dipesh Dhakal, University of Florida, United StatesReviewed by:
Chin-Yuan Chang, National Chiao Tung University, TaiwanCuauhtemoc Licona-Cassani, Monterrey Institute of Technology and Higher Education (ITESM), Mexico
Copyright © 2023 Lu, Li, Fan, Li, Sun, Wang, Han, Zhu, Zhang, Shi, Xie and Hong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bin Hong, binhong69@hotmail.com, hongbin@imb.pumc.edu.cn; Yunying Xie, xieyy@imb.pumc.edu.cn; Yuanyuan Shi, luckshiyuanyuan@163.com