Transcriptional Activation of Biosynthetic Gene Clusters in Filamentous Fungi
 László Mózsik
László Mózsik Riccardo Iacovelli
Riccardo Iacovelli Roel A. L. Bovenberg2,3
Roel A. L. Bovenberg2,3  Arnold J. M. Driessen
Arnold J. M. Driessen- 1Department of Molecular Microbiology, Groningen Biomolecular Sciences and Biotechnology Institute, University of Groningen, Groningen, Netherlands
- 2DSM Biotechnology Center, Delft, Netherlands
- 3Department of Synthetic Biology and Cell Engineering, Groningen Biomolecular Sciences and Biotechnology Institute, University of Groningen, Groningen, Netherlands
Filamentous fungi are highly productive cell factories, many of which are industrial producers of enzymes, organic acids, and secondary metabolites. The increasing number of sequenced fungal genomes revealed a vast and unexplored biosynthetic potential in the form of transcriptionally silent secondary metabolite biosynthetic gene clusters (BGCs). Various strategies have been carried out to explore and mine this untapped source of bioactive molecules, and with the advent of synthetic biology, novel applications, and tools have been developed for filamentous fungi. Here we summarize approaches aiming for the expression of endogenous or exogenous natural product BGCs, including synthetic transcription factors, assembly of artificial transcription units, gene cluster refactoring, fungal shuttle vectors, and platform strains.
Introduction
Secondary metabolites (SM), commonly referred to as natural products, are chemical substances that are produced by living organisms, often bearing distinctive pharmacological effects. The exploitation of microorganisms for generating these valuable products for our societies in an economical manner has a great history. Notably, the use of filamentous fungi in industrial biotechnology is well established. With the introduction of synthetic biology, new tools and alternative methods are provided to further aid the metabolic engineering and exploitation of fungal workhorses. Filamentous fungi (with key players from Aspergillus, Penicillium, Trichoderma, Fusarium, and Neurospora species) are highly efficient cell factories, often used industrially for the production of a diverse range of products such as proteins, enzymes, organic acids, and SMs (Meyer et al., 2020). SMs are not essential for the survival of the organism, but the production of these natural products often provides an evolutionary advantage. With the discovery of penicillin in 1928 produced by a mold identified as a Penicillium species, a new era started for industrial antibiotics production and the exploration and characterization of novel fungal SMs. This interest resulted in the discovery of not just antibacterial, but also antifungal (griseofulvin), cholesterol-lowering (lovastatin), immunosuppressant (cyclosporine), anticancer (paclitaxel), and food additive (carmine) compounds (Keller et al., 2005). Alongside the beneficial metabolites, fungi also produce SMs acting as toxins (e.g., aflatoxin, fumonisin, patulin), which negatively affect food, feed, livestock, and human health.
Filamentous fungi are known prolific producers of SMs. The fungal kingdom currently consists of around 120,000 identified species, but this number is estimated to represent only 3%–8% of the predicted number of existing species in our biosphere (Hawksworth and Lücking, 2017). Thanks to next-generation and third-generation sequencing technologies, in recent years the number of publicly available genomes has grown tremendously. As of this moment (early 2022), there are several thousand fungal genomes deposited in public databases, e.g., more than 2000 only on Mycocosm, a project maintained by the Joint Genome Initiative (JGI) (Grigoriev et al., 2014). The simultaneous development of automated genome mining tools such as antiSMASH (Blin et al., 2021) and other bioinformatics tools (Kautsar et al., 2019; Navarro-Muñoz et al., 2020) allowed researchers to identify a vast and unknown biosynthetic potential within the fungal kingdom in the form of SM encoding biosynthetic gene clusters (BGCs) (Figure 1A) (Mosunova et al., 2020). Bioinformatic analysis of 1,037 fungal genomes from the Ascomycota, Basidiomycota, and non-Dikarya revealed that the number of BGCs per genome significantly varies across fungal genomes (Robey et al., 2021). In the Ascomycota phylum Pezizomycotina genomes harbor on average 40 SM BGCs (25% of the genomes within this class possess >60 BGCs), however, this number is significantly lower in non-Dikarya (∼15 BGCs), in Basidiomycota (<10 BGCs) or non-Pezizomycotina Ascomycota genomes (∼5 BGCs) (Robey et al., 2021). Because of the richness and diversity of SM BGCs contained within their genomes, Pezizomycotina fungi are by far the most studied taxon in the field of SM discovery. Unfortunately, most of the BGCs encoded in their genomes are transcriptionally silent under laboratory cultivation conditions (Keller, 2019).
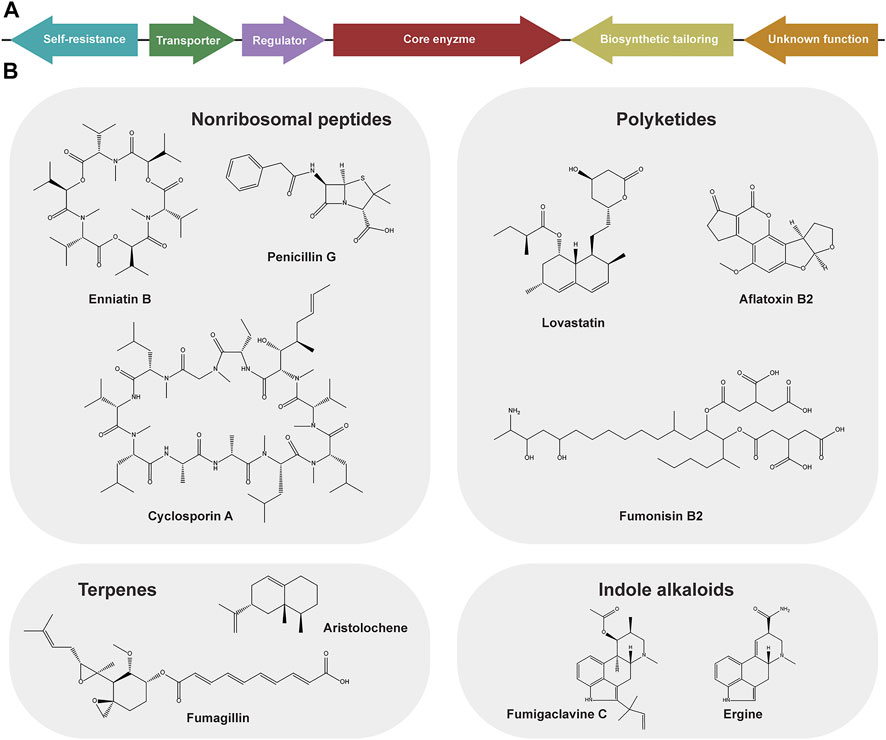
FIGURE 1. Schematic representation of a fungal biosynthetic gene cluster (BGCs) (A) and structurally different, representative members of nonribosomal peptides synthetase (NRPS), and polyketides synthase (PKS), terpenes synthase produced secondary metabolites and indole alkaloids from fungi (B).
Fungal SM BGCs can be activated via manipulation of cultivation conditions or by genetic modifications. Using different cultivation conditions or co-cultivation with other organisms (Bode et al., 2002; Oh et al., 2007) has led to successful examples of BGC activation, as we further discuss in Section. Replacement of the promoter driving the expression of local or global transcriptional regulators is a commonly used genome editing strategy for transcriptional activation, e.g., overexpression of transcriptional activators or knock-outs of transcriptional repressors, as well as manipulation of epigenetic modulators, which function as global chromatin regulators (Brakhage and Schroeckh, 2011). Traditional metabolic engineering methods combined with the implementation of the “clustered regularly interspaced short palindromic repeats” (CRISPR) technologies (Doudna and Charpentier, 2014) further accelerated strain construction and enabled more complex and sophisticated genetic engineering of filamentous fungi (Song et al., 2019). BGCs can be transcriptionally upregulated by conventional genome editing approaches, but thanks to the latest developments in synthetic biology, new attractive genetic tools have become available: synthetic transcription factors (STFs), artificial transcription units, fungal shuttle vectors, and various enhanced platform strains for heterologous expression. In the following sections we present recently developed tools and discuss how they compare to each other and to conventional metabolic engineering approaches.
Fungal Secondary Metabolite Biosynthetic Gene Clusters
SMs are low molecular weight, structurally heterogeneous compounds–synthesized by bacteria, fungi, and plants–which are not directly involved in the normal growth, development, or reproduction of the organism. SMs are synthesized from metabolic intermediates from primary metabolism. The produced SMs commonly provide a biological advantage to their producers, to thrive and survive in their environment, for instance in supporting the competition against other organisms (toxins and antimicrobials) and in the protection against harsh environments (pigments and iron-chelating siderophores), but SMs are also used for chemical signaling (Spiteller, 2015). Although many SMs have no known function, these compounds probably fulfill a role in complex communication networks in ecosystems, but so far it is just a human interpretation with limited experimental evidence.
The core skeleton of fungal SMs is produced by dedicated biosynthetic enzymes that belong to a few distinct families: nonribosomal peptide synthetases (NRPS), polyketide synthases (PKS), terpene synthases (TPS), dimethylallyltryptophan synthases (DMATS), or combinations thereof (Keller, 2019). NRPS and PKS are complex multi-modular megaenzymes that utilize a variety of amino acids and acyl-CoA monomers as substrates, respectively. TPS and DMATS are generally smaller and use a more limited set of substrates: the former use intermediates of the mevalonate pathway (IPP and DMAPP) as starter units for terpene synthesis (Keller, 2019); the latter utilize DMAPP to prenylate the amino acid tryptophan or other aromatic substrates (Kremer and Li, 2008). In all cases, the core scaffold is generally further modified by tailoring enzymes (oxidases, reductases, methyltransferases, cytochrome P450 monooxygenases, and others) whose genes are often found in the same BGC, ensuring a broad chemical diversity of the products (Figure 1B). Furthermore, these clusters frequently contain genes encoding transporters and regulatory proteins (Figure 1A). The size of a BGCs can span from a few kb to ∼100 kb incorporating as little as only two genes (valactamide BGC) (Clevenger et al., 2017) or up to ∼27 genes (aflatoxin BGC) (Caceres et al., 2020). Given that they are the most abundant in filamentous fungi—particularly in more commonly studied members of Pezyzomycotina—we will mainly discuss NRPS- and PKS-encoding BGCs in the remainder of this section.
NRPS enzymes synthesize a broad class of small peptides, typically 2–50 monomers from a wide variety of amino acids and their derivatives, as well as fatty acids and alpha hydroxy acids (Süssmuth and Mainz, 2017). These enzymes have a modular structure, where each module is responsible for the activation and coupling of a monomer to a growing peptide chain. A minimal NRPS module consists of an adenylation (A), condensation (C), and thiolation (T) domain (also called peptide-carrier protein). The A domain recognizes the monomer substrate and activates it as an (amino) acyl-AMP conjugate, which is subsequently transferred to the T domain via a transesterification reaction. The activated substrates/intermediates are then transported to the C domain, which is responsible for the formation of the peptide bond. Eventually, the final product is released by a terminal thioesterase domain (Fischbach and Walsh, 2006; Süssmuth and Mainz, 2017).
Polyketides represent the most abundant group of SMs. PKS enzymes utilize activated short-chain organic acids derived from primary metabolism, such as acetyl-, malonyl- or methylmalonyl-coenzyme A, for the biosynthesis of polyketides. The basic set of domains consists of an acyl carrier protein (ACP), a β-ketoacyl synthase (KS), and an acyltransferase domain (AT). The AT domain selects and loads both starter and extender monomers, while the KS domain catalyzes C-C bond formation between two adjacent substrates/intermediates. The ACP domain is responsible for storing and shuttling monomer substrates and synthesized intermediates during the elongation process. PKS enzymes are extremely diverse, many contain optional domains that introduce further chemical modifications, generating an incredible variety of products (Fischbach and Walsh, 2006; Robbins et al., 2016).
Molecules can be also constructed from hybrid NRPS-PKS assembly lines leading to mixed NRP-PK products, such as the bacterial bleomycin, rapamycin, epothilones, or the fungal fusarin C, pseurotin A, tenellin, and cytochalasin E. (Fischbach and Walsh, 2006; Boettger and Hertweck, 2013). NRP-PK hybrids can be synthesized by proteins containing domains and modules from both PKSs and NRPSs organized in the same polypeptide chain (tethered), but these enzymes can also be formed from individually expressed proteins in the BGC (non-tethered) (Miyanaga et al., 2018). In these hybrid systems, the different subunits need to communicate efficiently to coordinate the transport of substrates and intermediates across the hybrid system, and have to perform either C-C or C-N bond elongations at the corresponding PKS/NRPS interfaces (Fischbach and Walsh, 2006).
BGCs encoding NRPS and PKS clusters can readily be predicted and identified from genomic data by advanced bioinformatics algorithms, for example, using the conserved domains of the core enzymes. Such tools are the “Secondary Metabolite Unique Regions Finder” (SMURF) (Khaldi et al., 2010) or the “antibiotics and secondary metabolite analysis shell” (antiSMASH) (Blin et al., 2021). AntiSMASH is continuously updated since its release in 2011, incorporating several newly developed algorithms, e.g., searching for shared transcription factor (TF) binding sites in the promoter sequences [“Cluster Assignment by Islands of Sites” (CASSIS) (Wolf et al., 2016)]. Cross-referencing with public databases further aids the identification of uncharacterized BGCs. One example is the MIBiG (Minimum Information about a Biosynthetic Gene cluster) repository, which contains curated information in a unified format listing the BGC annotations and their molecular products (Kautsar et al., 2019). As these algorithms accept annotated DNA sequences as input, cluster predictions can be further advanced taking into account transcriptome data of the predicted BGC, assuming that the cluster can be brought to a transcriptionally activated state. Algorithms such as MIDDAS-M (motif-independent de novo detection algorithm for SM BGCs) aim to combine genomic data and transcriptomic data to predict coordinately regulated genes including fungal BGCs (Umemura et al., 2014). As homologues of clusters with known compounds can be easily identified, the use of databanks and algorithms can reduce the re-discovery rates or yield predictive information regarding the targeted BGCs. The discovery of a huge number of transcriptionally silent BGCs through bioinformatics fueled the interest in genome mining, and the interrogation of these unknown clusters by experimental identification (Nielsen et al., 2017; Caesar et al., 2020).
Regulation and Transcriptional Activation of Biosynthetic Gene Clusters
SM production is often regulated by a stimulus, and without it, the product of the BGC is not synthesized. When no known metabolite is connected to a BGC, the cluster is called “cryptic” or “orphan.” In most cases, BGCs react to various environmental stimuli, but often the connection between regulators and the stimuli is unknown. Under laboratory conditions, native environmental signals may not be present, rendering BGCs transcriptionally silent. Since cryptic BGCs appear to be silent under laboratory conditions (Keller, 2019), alternative strategies need to be employed to awaken these clusters and explore their biosynthetic potential.
Conventional methods for transcriptional activation of genes or even entire BGCs have been rapidly implemented in fungal biotechnology (Brakhage and Schroeckh, 2011; Lim et al., 2012). One of the strategies concerns the OSMAC (one strain many compounds) approach, which assumes that one strain is capable to produce numerous compounds, but different environmental or cultivation conditions regulate what subset of BGCs are activated (Bode et al., 2002). Indeed, modifications for cultivation parameters such as temperature, salinity, aeration and others, showed that Aspergillus ochraceus is capable of producing 15 compounds in addition to the previously known aspinonene (Bode et al., 2002). Co-culturing can also result in transcriptional activation of BGCs due to inter-species crosstalk (Oh et al., 2007; Bertrand et al., 2014; Netzker et al., 2018): co-cultivating A. nidulans with the soil-dwelling bacterium Streptomyces rapamycinicus resulted in the production of orsellinic acid (Schroeckh et al., 2009). Co-cultivation of A. fumigatus with the same bacterium resulted in the activation of the fumicycline BGC, which involved epigenetic regulation changes induced by the bacterium (König et al., 2013).
Global Regulators
Around half of the fungal BGCs do not harbor in-cluster regulators, and are only regulated by global transcription regulatory mechanisms (Keller, 2019). Global transcriptional regulators respond to environmental stimuli by coordinated up or downregulating of required gene sets, and the corresponding TFs to these signals have been identified in several cases: CreA responds to carbon levels; the velvet complex to light; AreA to nitrogen concentration; PacC to pH levels; and the CCAAT-binding complex to iron concentration (Macheleidt et al., 2016). These regulators act genome-wide on numerous genes, controlling morphological development, primary metabolism as well as SM production. Both overexpression (e.g., LaeA transcriptional activator of secondary metabolism) (Bok and Keller, 2004) and deletion of master regulators (e.g., McrA repressor protein) (Oakley et al., 2017) resulted in transcriptional activation of BGCs.
Chromatin-Mediated Regulation
Alterations in the structure of chromatin can result in global transcriptional regulatory effects (Collemare and Seidl, 2019). Histones are critical proteins responsible for the tight packing of DNA in the nucleus, creating the chromatin. Histones can undergo numerous modifications (acetylation, methylation, phosphorylation, uniquitination, sumoylation, and neddylation), and this can create less- or more accessible DNA segments (Pfannenstiel and Keller, 2019). Histone deacetylation results in a more closed chromatin structure causing transcriptional repression of affected genes. In contrast, histone acetylation can result in more accessible chromatin for regulator proteins and the transcription machinery, causing transcriptional activation. The structure of chromatin can be manipulated by using chemical agents or genetic modifications to achieve transcriptional regulation of genes of interest. For instance, chemical histone deacetylase (HDAC) inhibitors (e.g., suberoylanilide hydroxamic acid, trichostatin, and sodium butyrate) can be supplemented to the cultivation media to prevent histone deacetylation (Pfannenstiel and Keller, 2019). Such epigenetic perturbation may lead to a 100-fold up and downregulation of genes that are spread over the genome (Albright et al., 2015). Epigenome editing for transcriptional activation is also possible by genetic engineering of the regulation of the expression of histone acetyltransferase (HAT) or HDAC genes. In A. nidulans, downregulation of the gene rpdA that encodes a HDAC enzyme yielded similar results as observed with chemical HDAC inhibitors (Albright et al., 2015). Deletion of the hdaA (histone deacetylase A) in A. nidulans resulted in increased penicillin and sterigmatocystin production (Keats et al., 2007). In P. rubens [previously identified as P. chrysogenum (Houbraken et al., 2011)], deletion of a hdaA homolog positively affected the production of sorbicillins and roquefortine/meleagrin (Guzman-Chavez et al., 2018), and significantly downregulated the BGCs responsible for chrysogine and dihydroxynaptelene-melanin production (Ding et al., 2020).
Cluster Specific Regulators
Around half of the predicted BGCs harbor genes encoding TFs, which are often transcriptional activators of the complete cluster (Lyu et al., 2020). These regulators bind to the corresponding recognition sequence in the promoters of the genes in the BGC. As promoter replacement is a good strategy to override the native regulation of a transcriptionally silent gene, replacing the promoter of the in-cluster TF can result in cluster-specific activation. Although overexpression of cluster specific TFs has led to the production of aspyridones, asperfuranone, and emodin derivatives in several Aspergilli (Bergmann et al., 2007; Chiang et al., 2009; Lyu et al., 2020), a systematic promoter replacement approach in A. nidulans showed that only 3 out of 17 overexpressed cluster specific TFs effectively led to the production of an obtainable amount of SMs (Ahuja et al., 2012). Although inducers and protein-protein interactions affect the activity of the TF, it is currently unknown what other mechanism(s) are required for complete BGC activation alongside the overexpression of an in-cluster TF. Cross-talk between different cluster specific TFs have also been described in A. nidulans, as the overexpression of the ScpR TF (from the fellutamide BGC) caused upregulation of the in-cluster inpA and inpB NRPS genes, as well as the asperfuranone BGC, located on a different chromosome (Sebastian et al., 2010). Since the AflR (aflatoxin transcriptional activator) recognition sequence can be found in most of the promoters in the sterigmatocystin and aflatoxin cluster, this TF was shown to be able to regulate positively both BGCs, as well as some genes outside of these BGCs (Brodhagen and Keller, 2006; Price et al., 2006).
In-cluster SM BGC transcriptional repressors with DNA-binding capacity have so far not been discovered in filamentous fungi. Rather, repressor proteins interacting with transcriptional activators is a more common mechanism. For example, the primary metabolism BGC responsible for the quinic acid degradation in N. crassa is controlled by a transcriptional activator/repressor regulator pair (qa-1F/qa-1S) (Giles et al., 1991). Similarly, in A. niger the repression of galacturonic acid utilization pathway is modulated by a regulator pair (Gaar/GaaX) (Niu et al., 2017). It is believed, that these in-cluster repressors are responsible for keeping the positive transcriptional regulator inactive in the absence of an inducer (Giles et al., 1991; Niu et al., 2017). The sorbicillin SM BGC in P. rubens harbors an activator/repressor pair as well, and the metabolites of the cluster are acting as autoinducers for the pathway (Guzmán-Chávez et al., 2017). Overexpression of the transcriptional repressor (sorR2) results in transcriptional suppression, while deletion of sorR2 results in early-stage transcriptional activation of the sorbicillin BGC, but with hardly any sorbicillin production (Guzmán-Chávez et al., 2017). Although promoter replacement, gene deletion or complete BGC refactoring in the native host leads to direct transcriptional activation of the gene of interest, but these methods require editing the genome of the fungus.
Fungal Genome Editing
Precise and flexible genome editing is key for efficient engineering of the fungal host. Targeted gene manipulation in wild type filamentous fungal species is challenging due to the relatively low rates of homologous recombination (HR) and high rate of random integration of the transformed DNA. Targeting efficiency to the desired location is relatively low in Aspergillus and Penicillium species (0.1%–5.0%) (da Silva Ferreira et al., 2006; Snoek et al., 2009), and it differs by the organism and targeted locus. The fungal homologs of the human ku70/ku80 genes encode a protein complex functioning in the non-homologous end-joining (NHEJ) DNA repair pathway, which favors random integration of the transformed donor DNA. Deletion of either of these genes is highly advantageous for fungal strains that are employed for precise genome editing and HR-mediated DNA delivery. Inactivation of either of the fungal homolog of the human ku70/ku80 genes drastically decreases or eliminates the functionality of the NHEJ DNA repair pathway and highly increases the efficiency of targeted DNA delivery through HR (Pöggeler and Kück, 2006; Takahasi et al., 2006; Snoek et al., 2009; Carvalho et al., 2010). With the advent of CRISPR-based tools the genome editing efficiency was further increased, in some filamentous fungal strains reaching more than 90% (Pohl et al., 2016; Schuster et al., 2016; Nødvig et al., 2018; Song et al., 2018).
A commonly used alternative to engineer fungal hosts is the Agrobacterium-mediated transformation, particularly employed when little or no genetic tools are available for that host. A. tumefacians is a gram-negative plant pathogen that has been shown to be capable of transferring its T-DNA (transfer-DNA, used by the bacteria to infect plants) into the genome of several filamentous fungi. This made it possible to achieve successful DNA delivery in fungal hosts that cannot be transformed with traditional methods (de Groot et al., 1998; Idnurm et al., 2017). The system is commonly used for random integration of one single copy of a gene of interest, but it can also be used for targeted genome editing in NHEJ-deficient hosts via homologous recombination using long homologous flanking sequences (>1000 bp) (Idnurm et al., 2017).
Before the global application of CRISPR-based genetic tools, zinc finger nucleases (ZFNs) and transcription activator-like effector (TALE) nucleases were established for locus-specific genome engineering applications. Engineering the DNA binding domain (DBD) of these nucleases allows targeting specific genomic loci. ZFNs are artificial restriction enzymes and are typically generated by fusing DBSs with the FokI nuclease domain. DNA targeting is provided by fusing together three to six DNA-binding zinc-finger proteins, each of which is capable to recognize a specific 3 bp DNA sequence. Although ZFNs are relatively small proteins, which are easy to deliver to the host, their targeting efficiency is rather weak and the relatively high levels of off-target effects may lead to cytotoxicity (Ramalingam et al., 2011). The next generation of targeted DNA editing was the discovery of the transcription activator-like effector nuclease (TALEN) elements, which are acting as TFs in the species of Xanthomonas. The DBD builds up from 33 to 34 amino acid long tandem repeats, which determines the targeted DNA sequence. These repeats can be altered to recognize one specific nucleotide and by combining these repeats in sequential order, the protein can be targeted to any DNA sequence (preceded by a thymine or cytosine base). Direct fusion of the TALE DBD and with restriction endonuclease (FokI) domain created guidable TALENs, meanwhile fusions to transcription activation domains (ADs) to created STFs for targeted transcriptional regulation (TALE-TFs) (Gao et al., 2014).
CRISPR-Mediated Genome Editing
The CRISPR systems and their CRISPR-associated (Cas) proteins have recently been repurposed for transcriptional gene regulation in eukaryotes (Gilbert et al., 2013; Perez-Pinera et al., 2013), where they can be utilized as components of STFs. Native Cas proteins provide a self-defense mechanism against bacteriophage virus infection in prokaryotes. When these organisms encounter the virus for the first time, they embed small viral sequences in their genome, which are later transcribed into small RNAs molecules. These RNAs form complexes with the Cas proteins, which are now able to recognize and cleave the complementary nucleic acid sequence in the viral genome at any next infection event, effectively eliminating the intruder (Rodolphe et al., 2007). Cas proteins commonly cleave double or single stranded DNA, but RNA-cleaving Cas proteins have also been identified (Knott and Doudna, 2018). Repurposing and utilizing these systems for targeted genome editing has revolutionized precise genome engineering in various organisms. In these two component CRISPR systems, the Cas protein is guided by a CRISPR RNA (crRNA) to a target specific locus for nucleic acid cleavage. For the commonly used Cas9 systems, specificity is delivered on a single guide RNA (sgRNA) complex that encodes both the short trans-activating CRISPR RNA (tracrRNA) and crRNA transcripts. The tracrRNA forms the stem loops that anchor the endonuclease protein while the crRNA is the actual targeting sequence. In contrast, the other commonly applied Cas12a (Cpf1) nuclease is capable to processes its own crRNAs from pre-crRNA, and does not need a tracrRNA (Fonfara et al., 2016).
With the Cas9 system, the genomic locus is targeted by a sequence-specific 17–20 nucleotide crRNA which is complementary to its genomic target (protospacer), that must be followed by a protospacer adjacent motif (PAM). This PAM sequence is recognized at the DNA level by the protein and is unique for different Cas proteins. These unique PAM sequences limit the number of sequences that one can target since they show minimal flexibility for different nucleotides: For example, the PAM sequence recognized by the commonly used Streptococcus pyogenes Cas9 (SpCas9) is 5′-NGG-3′ (to a lesser extent non-canonical NAG and NGA are also recognized, where N is any nucleotide) located downstream the protospacer, meanwhile the Cas12a nuclease recognizes 5′-TTTV-3′ (where V can be G, C or A) sequences located upstream of a typically 20–24 nucleotide long protospacer (Pickar-Oliver and Gersbach, 2019). Careful design of the crRNA is therefore essential to avoid off-target CRISPR effects, as the nuclease complex is capable to bind to highly similar sequences (Fu et al., 2013), which represents another limitation of this system.
In recent years, highly efficient CRISPR-based genome editing tools have been developed and established for several organisms, such as bacteria, yeast, and human cells (Wang et al., 2016). CRISPR/Cas9-based genome editing in filamentous fungi has been established for several organisms including A. fumigatus, A. oryzae, Neurospora crassa, Pyricularia oryzae, Trichoderma reesei, Ustilago maydis, and P. rubens (Song et al., 2019). CRISPR elements can be delivered as ribonucleoproteins preassembled in vitro, or as genetic elements that are expressed by the host. The AMA1 (autonomous maintenance in Aspergillus) sequence from A. nidulans supports autonomous vector replication in several filamentous fungal species (Fierro et al., 1996; Aleksenko and Clutterbuck, 1997), and thus vectors encoding this sequence have been extensively used for gene delivery and expression purposes, as well as delivering CRISPR components (Fuller et al., 2015; Nødvig et al., 2015; Pohl et al., 2016; Salazar-Cerezo et al., 2020; Iacovelli et al., 2021b). Single vector-based CRISPR/Cas9 genome editing systems have been previously developed for several filamentous fungal species (Song et al., 2019). Recently, a similar system was developed based on the nuclease Cas12a (Cpf1) for Aspergilli (Vanegas et al., 2019).
Fungal Synthetic Biology Tools
Synthetic biology has revolutionized metabolic engineering with tools–created by repurposing or redesigning biological systems found in nature–enabling the exploitation of industrial microorganisms at whole new levels. Since synthetic biology strives to engineer highly predictable and controllable genetic systems, genetic circuits are often constructed in a standardized and preferably modular fashion. The modularity of various DNA parts encoding genetic elements allows rapid assembly of novel, more predictable genetic circuits, like logic gates, and genetic switches. Inducible or synthetic transcriptional regulators (activators and repressors) can be used to enable fine-tuning of gene expression or controlling entire pathways. Synthetic biology-based tools have been established in several model bacterial and eukaryotic systems, and recently also in filamentous fungi where they are still relatively underdeveloped compared to more common hosts.
Modular Assemblies
The Design-Build-Test-Learn (DBTL) cycle of synthetic biology represents a systematic and efficient workflow for the optimization of biological systems for speciffic functionalities (e.g., strain improvement). Complex genetic systems can be constructed in a modular manner with desired features–synthetic regulatory tools and rewired expression of biosynthetic pathways–enabling an affordable genetic engineering of biological systems. Synthetic transcription units can be rapidly assembled by cloning methods supporting the assembly of multiple DNA fragments [e.g., Gibson Assembly (Gibson et al., 2009) or USER Cloning (Geu-Flores et al., 2007)], or high-throughput, modular cloning methods, such as Golden Gate cloning-based (Engler et al., 2009) Modular Cloning (Weber et al., 2011), and GoldenBraid (Sarrion-Perdigones et al., 2011) assemblies. With these methods, genetic parts (promoters, coding sequences, and terminators) can be arranged into transcription units, where the building blocks are interchangeable within the same synthetic biology language, but their order and orientation are commonly predetermined. Collections of such DNA building blocks (toolkits) have been established for bacteria (Moore et al., 2016), yeasts (Lee et al., 2015; Obst et al., 2017), plants (Engler et al., 2014), mammalian host cell lines (Martella et al., 2017) and also for filamentous fungi (Sarkari et al., 2017; Hernanz-Koers et al., 2018; Mózsik et al., 2021b; Dahlmann et al., 2021). These toolkits can provide backbone vectors to facilitate modular assembly and fungal delivery, or pre-assembled vectors containing various genetic elements, suitable for generic applications or specific needs. The deposition of such toolkits containing ready-to-use, established DNA parts or modules, highly accelerates the biological DBTL cycle for synthetic biology applications. A major repository for genetic parts is Addgene, a free online database that facilitates the exchange of genetic material between laboratories around the world.
Artificial Promoters and Synthetic Transcription Factors
Eukaryotic promoters are complex DNA structures responsible for recruiting transcriptional regulatory elements (transcriptional regulators). The simplest functioning unit of the promoter—often called minimal or core promoter (CP)—is incorporating the transcription start site (TSS) and is required to initiate transcription of the gene of interest. CPs contain specific DNA elements that the RNA polymerase II requires to initiate transcription (Michael, 1998). In eukaryotes, CP sequences are highly diverse: many motifs can be present such as the TATA, CCAAT boxes, the B recognition element, and the initiator element (Juven-Gershon and Kadonaga, 2010). Several regulatory TF-binding sequences are located upstream of the CP sequence, where they recruit transcriptional activator or repressor proteins, hence, modulating the transcription of the gene. The length of CPs is not well defined in filamentous fungi, but these sequences are located roughly 140–200 bp upstream of the starting codon (Rantasalo et al., 2018; Mózsik et al., 2019). The precise identification of upstream regulatory DNA sequences and CPs is essential for the engineering of functional synthetic promoters.
Synthetic gene expression systems can be used for the production of metabolites or proteins of interest, and have been established in numerous filamentous fungal species (Vogt et al., 2005; Rantasalo et al., 2018; Mózsik et al., 2019). Such orthogonal systems do not rely on the regulatory system of the host, but instead depend on hybrid or synthetic TFs, composed of different DNA-bindings (DBDs) and transcriptional effector domains, and on synthetic promoters. DBDs target and bind to unique upstream activating sequences (UASs) in the promoter region, effectively regulating gene expression. If the TFs are inducible and/or repressible upon the addition of small molecules, these systems can be used as genetic switches as well. STFs have been repurposed from different prokaryotic, eukaryotic, or viral transcriptional regulators and have been shown to be functional in several hosts including yeast and filamentous fungi. Using such synthetic transcriptional regulators, activation or repression of genes can be achieved in a controlled manner. Transcription can be fine-tuned for each gene individually by changing different elements of the system. Synthetic promoters created by fusing specific UAS and CPs, or by integrating UAS elements into native promoters, can be used to rewire the native transcriptional regulation system of the genes of interest. Synthetic promoters bring the promise of a pre-defined, fine-tuned, and metabolism-independent expression for multiple individual genes. Synthetic promoters in combination with an inducer-dependent STF can allow further tuning of gene expression in a gene dosage and/or inducer concentration dependent manner (Meyer et al., 2011). Such refactoring would allow the overexpression of entire BGCs bypassing the need for established strong promoters for each gene of the cluster, since the number of such promoters is limited for filamentous fungal hosts. Functional STFs have been successfully introduced in Aspergilli (Vogt et al., 2005; Wanka et al., 2016; Grau et al., 2018; Rantasalo et al., 2018), P. rubens (Mózsik et al., 2019), T. reesei (Derntl et al., 2019), and Ustilago maydis (Zarnack et al., 2006).
Many STFs have been constructed to regulate genes in primary metabolism (Gao et al., 2017; Zhang et al., 2018; Han et al., 2020; Kun et al., 2021; Yamashita et al., 2021). Fusing the DNA-binding domain of the CreA/Cre1 (carbon catabolite repression) transcription factor to the complete Xyr1 transcription factor (Xylanase Regulator 1) resulted in enhanced cellulase production in a CreA/Cre1 deficient T. reesei strain grown on glucose (Zhang et al., 2017). Fusing the DBD of the Xyr1 with the regulator domain of Ypr2 (transcriptional activator of the sorbicillinoid SM BGC) resulted in high expression of xylanases and cellulases in T. reesei nearly independently from the carbon source used (Derntl et al., 2019). Replacing the regulatory domain of a weak in-cluster transcriptional TF with a highly active activator domain (AD) can lead to activation of target SM BGCs, without the integration of additional synthetic promoter elements. When the DNA-binding domain of the transcriptional activator (AlnR) from the asperlin BGC was fused to the regulatory domain of the transcriptionally highly active asperfuranone TF (AfoA), it led to the production of asperlins in A. nidulans (Grau et al., 2018). In these works, the DBD in the STF retained its capability to bind to its native operator sequences in the promoters, while the newly fused activator domain provided transcriptional activation of the genes.
Although STFs (also called altered, artificial, or hybrid TFs) have been studied for more than 30 years (often using the Gal4 TF as a model from Saccharomyces cerevisiae) (Ma and Ptashne, 1987; Corton, 1989; Hach et al., 2000), there is limited information about how these domain fusions should be engineered to avoid creating nonfunctional STFs. Cluster-specific TFs commonly consist of a Zn(II)2Cys6 (C6 zinc) DBD and a transcriptional regulator domain. DBDs often contain at least one structural motif that recognizes and bind to double- or single-stranded DNA sequences. Generally, DBDs can be further divided into sub regions: the zinc finger, the linker region, and a coiled-coil element. Numerous Gal4-family TFs contain a coiled coil between the linker and the regulator domain, which is possibly responsible for mediating protein-protein interactions or homodimer formation before binding to DNA (Hach et al., 2000). While structural changes in the zinc-finger motif, the linker region or the coiled-coil regions negatively affect the functionality of the TF, the regions between these coiled-coil sequences and the regulator domains are often non-essential for retaining activity (Ma and Ptashne, 1987; Corton, 1989; Marmorstein and Harrison, 1994).
The tetracycline-inducible (TET) expression system has been originally developed for mammalian cells (Gossen and Bujard, 1992), and later adopted for other eukaryotic systems, as well as for Aspergilli and U. maydis (Vogt et al., 2005; Zarnack et al., 2006; Meyer et al., 2011). Within the endogenous tetracycline-resistance system in Gram-negative bacteria, the TetR transcriptional repressor represses the expression of the tetracycline transporter gene (TetA) by binding to the TetO (or “tetracycline response element” TRE) operator sequences in the promoter. In the presence of the antibiotic tetracycline, TetR will bind the compound and be released from TetO, enabling expression of the transporter gene which eventually provides self-resistance (Orth et al., 2000). This repressor was engineered into an activator in the Tetracycline-Off (Tet-Off) system, where a tetracycline-controlled STF, the tTA (TetR-VP16 fusion) provides inducible repression. Transcriptional repression can be achieved by feeding tetracycline (or its synthetic derivative doxycycline) to the medium, which binds to the synthetic activator (tTA), thus preventing binding to the TetO sequences and the expression of the gene of interest. Several copies of the TetO sequences are inserted upstream of a weak or transcriptionally silent minimal (core) promoter for transcriptional regulation of the gene of interest. As tetracycline and doxycycline have relatively short half-lives, these chemicals need to be added to the medium repeatedly to maintain transcriptional repression, and in their absence transcriptional activation occurs, as the tTA binds to the TetO sequences.
Using the further engineered Tetracycline-On (Tet-On) system, gene activation can be achieved in a concentration-dependent manner by feeding the inducer and using the reverse tetracycline-controlled transcriptional activator (rtTA, mutated TetR-VP16 fusion) as the STF. In the presence of tetracycline (or doxycycline), the affinity of this rtTA STF towards the TetO sequences increases, therefore enhancing the transcription of the gene of interest downstream. Unfortunately, the rtTA retains some binding affinity to its TetO sequences in the absence of the inducer, leading to leaky transcription. Thus, an advanced version of rtTA (rtTA2S-M2, TetR-3xVP16) was designed, showing increased specificity, stability, and inducibility using doxycycline without leaky expression (Urlinger et al., 2000). This Tet-On system was applied with A. fumigatus for inducible expression of the gene of interest using seven copies of the TetO sequence upstream a short 175 bp CP sequence of the commonly used gpdA promoter (Vogt et al., 2005). The system was established in A. niger using fluorescent reporters, and was applied for the production of fructose-6-phosphate amidotransferase (Wanka et al., 2016) and biologically active fungal cyclodepsipeptides (Enniatin B, Beauvericin, Bassianolide) on a grams-per-liter scale (Boecker et al., 2018).
The bacterial Bm3R1-based STF (Bm3R1-DBD-VP16) was shown to be functional for transcriptional activation in yeasts as well as in A. niger and T. reesei (Rantasalo et al., 2018). This STF was delivered to different fungal hosts harboring several copies of the BS-UAS, enhancing the transcription capacity of various, native and non-native CPs to control gene expression in fungi. These results showed that, although CPs function differently among hosts, universally functional CPs which operate both in filamentous fungi and yeast hosts can be designed. Some of these synthetic promoters even performed better than commonly used native “strong” promoters. As the native TFs have no known inducers, controlling or inducing the transcription in this system is not established.
The transcriptional activator and repressor of the quinic acid metabolism from Neurospora crassa (Giles et al., 1985) have been implemented as a binary expression system for Drosophila melanogaster and mammalian cell lines, known as the “Q-system” (Potter et al., 2010). This system has controllable features as in the native host the repression of qa-1F by the qa-1S transcriptional repressor can be relieved by feeding with quinic acid, resulting in inducible activation. In the earliest example of an engineered Q-system, a STF was constructed by fusing the DBD of the qa-1F transcriptional activator to the GAL4 AD. This DBD binds to its corresponding recognition sequences upstream of the targeted promoter (called QARE QA response element or QUAS Q-System UAS) (Giles et al., 1985; Potter et al., 2010). The Q-system was later also adapted and established for mammalian cells, Caenorhabditis elegans, zebrafish, and malaria mosquitos (Riabinina et al., 2016). Based on the Q-system transcriptional activator (qa-1F), a STF using the VP16 AD (qa-1F-DBD-VP16-GFP) has been constructed in P. rubens, where the strength of the Q-system STF device showed scalability by using different CPs, by increasing the expression levels of the STF or the number of UAS elements (1, 5 or 11) upstream of the CP (Mózsik et al., 2019). The system was capable to produce expression levels ranging from hardly detectable to a level similar to that of highly expressed native genes. These synthetic expression devices were validated using fluorescent reporters while the application potential was confirmed by synthetically controlling the expression of the penicillin BGC. The development of such a system further increased the number of genetic regulation tools available for filamentous fungi.
CRISPR-Based Transcriptional Regulation
Mutations in the nickase domain(s) of the CRISPR protein eliminate its nuclease activity while retaining the capability of the protein to bind to the DNA. Such “nuclease-dead” CRISPR proteins (dCas) are engineered from Cas9 by introducing point-mutations in the RuvC and HNH nuclease domains (in dCas9m2 from S. pyogenes these are the D10A and H840A, respectively). Similarly, point mutations are introduced in the RuvC-like domain of Cas12a to generate its corresponding dCas variant (E993A in dCas12a from Acidaminococcus sp.) (Yamano et al., 2017). These proteins can be fused to ADs and used as STFs (CRISPRa, activation), thereby recruiting a transcriptional regulator to the promoter of the gene of interest. Since dCas proteins can still bind tightly to their target sequences, they can be guided to regions upstream of a gene of interest where they form a “road-block” for the transcriptional machinery, resulting in transcriptional repression (CRISPRi, interference). Taking advantage of the guidable DNA-binding capability, these dCas proteins can be applied for various other applications depending on the delivered modulator, e.g., targeted DNA modifications (e.g., methylation), transcriptional regulation, fluorescent imaging can be achieved (Pickar-Oliver and Gersbach, 2019). Inactivated Cas proteins can be used to deliver transcriptional regulator domains to the promoter of the gene of interest by direct fusion of regulatory domains, or repetitive peptide epitopes that recruit multiple copies of antibody-fused regulators (SunTag), or by using MS2 RNA stem-loops in the sequence of the tracrRNA to recruit MS2-tagged regulators (Synergistic Activation Mediator “SAM” system) (Konermann et al., 2015; Chavez et al., 2016).
The protospacer sequence is crucial in CRISPRi/CRISPRa applications for targeted repression and activation, respectively. CRISPRi has been successfully adapted to several bacterial and eukaryotic hosts for targeted gene repression (Farzadfard et al., 2013; Gilbert et al., 2013, 2014; Qi et al., 2013; Rock et al., 2017; Sato’o et al., 2018; Román et al., 2019; Lauren et al., 2022; Tan et al., 2022). Targeting in close distance to the TSS of the gene of interest with this system leads to successful downregulation, presumably by blocking transcriptional initiation or elongation. In prokaryotic CRISPRi applications, the bare dCas9 without any fused regulator domain is already capable of achieving significant repression. It is believed that the binding of dCas9 can hinder the binding of positive enhancers or the mediator complex for transcriptional elongation. In eukaryotes, the levels of repression achieved by using dCas9 alone are low, but can be enhanced by fusing repressor domains such as KRAB (Krüppel associated box) or Mxi1 (a histone deacetylation mediator) (Qi et al., 2013). The efficiency of CRISPRi-based repression differs depending on several factors including the type of fused effector, off- and on-target effects of the CRISPR protein, the distance of the protospacer from the TSS, and the chromatin state of the target genomic region (Smith et al., 2016). Presumably, the native transcription levels of the target genes and the presence of regulatory protein binding sequences in close proximity of the CRISPR complex also affect the degree of repression. In mammalian cell lines, the dCas9-KRAB fusion provides repression when targeted in the range of −50 to +300 bp relative to the TSS of a gene, with the highest efficiency of ∼100-fold repression in the −50 to +100 bp region (Gilbert et al., 2014). In S. cerevisiae, the dCas9-Mxi1 fusion resulted in a maximal ∼10-fold repression when targeted to the −200 to +1 region relative to the TSS, but this reduced efficiency could be explained by the mode of repression employed by Mxi1, which mediates DNA deacetylation (Smith et al., 2016). These experiments also highlight how nucleosome occupancy and chromatin accessibility can affect crRNA efficiency. The level of repression can be further increased by deploying multiple sgRNAs in combination with the Cas9 systems (Farzadfard et al., 2013) which can be achieved by using sgRNA-arrays in combination with self-cleaving sequences (Hammerhead and HDV ribozymes and tRNA) (Gao and Zhao, 2014; Zhang Y. et al., 2019) or exogenous nucleases and their cleavage factor recognition sequences (Csy4 nuclease) (Ferreira et al., 2018).
CRISPR-based transcriptional activation systems commonly use the VP16 AD or variants thereof where the VP16 is arranged in tandem repeats (VP64, VP160). This regulatory domain originates from herpes simplex virus, but it was shown to function in various organisms (Sadowski et al., 1988; Gossen and Bujard, 1992; Vogt et al., 2005; Rantasalo et al., 2018; Mózsik et al., 2019). VP64 was also combined with two other potent transcriptional activators to generate the VPR (VP64-p65-Rta) tripartite activator domain, which has been shown to be superior compared to other activator domains tested in human, mouse, and fly cell lines as well as in the yeast S. cerevisiae (Chavez et al., 2015). The Cas-VPR fusion system has been successfully adopted for filamentous fungi, and established for A. nidulans (Roux et al., 2020), and P. rubens (Mózsik et al., 2021a). In A. nidulans, the dCas9-VPR and dCas12a-VPR activators were expressed from an episomal vector and were guided to the transcriptionally silent elcA promoter of the PKS gene of the elsinochrome BGC from Parastagonospora nodorum, which was fused to an mCherry fluorescent reporter gene. After transcriptional activation of elcA was validated using fluorescence microscopy, the system was used to overexpress individual genes of the native microperfuranone BGC in A. nidulans, which resulted in enhanced production of microperfuranone and the identification of dehydromicroperfuranone (Roux et al., 2020). In P. rubens, a vector-based dCas9-VPR system was used to activate the transcriptionally silent, native P. rubens macrophorin BGC by activating the promoter of the transcriptional activator of the cluster (Mózsik et al., 2021a). This CRISPR activator system was validated using a transcriptionally silent CP (Mózsik et al., 2019) driving a DsRed fluorescent reporter (Mózsik et al., 2021a). Cas12a is natively able to process its own crRNAs from an array of pre-crRNAs, while Cas9 requires additional engineering for the delivery of multiple crRNAs (e.g., individual sgRNA transcription units, self-cleaving ribozyme sequences, or Csy4 endoribonuclease cleaving) (Ferreira et al., 2018). Since targeting the same promoter with multiple crRNAs shows synergistic effects in various eukaryotic CRISPRa applications (McCarty et al., 2020), dCas12a systems are superior compared to dCas9 for gene regulation purposes.
In mammalian cell lines, CRISPRa seems to be the most effective in the range of 400–50 bp upstream of the TSS (Gilbert et al., 2014). Since the genes of cryptic BGCs in filamentous fungi are often transcriptionally silent, the TSSs are not known. In this case, crRNAs can be designed to target regions close to the predicted TSS or to the start codon of the gene of interest. Both in A. nidulans and P. rubens, this approach was successfully used to achieve transcriptional activation using individual sgRNAs guiding the dCas9-VPR activator to 162–190 bp (PelcA) or 106–170 bp (PpenDE) and 68–73 bp (PmacR) region upstream of the start codon, respectively (Roux et al., 2020; Mózsik et al., 2021a). Next to the general rules to identify CRISPR protospacer candidates (selecting predicted high on-target and low off-target binding efficiency, and avoiding strong secondary RNA structures), regulatory DNA elements in the targeted promoter, as well as local chromatin organization should be considered when designing crRNA sequences.
When designing CRISPRa strategies, particular attention should be paid to prevent undesired blockages to the transcription complex. Targeting in close proximity upstream from the TSS seems favorable, but the CRISPR complex should not be too close to create physical hindrance for the transcription complex formation, and it should also bind outside of known enhancer or transcriptional regulatory elements (TATA or CCAAT box) in the promoter. Without precise knowledge of the regulatory elements in the sequence of the targeted promoter, empirical testing of crRNAs will remain necessary. In the extent of transcriptional activation achieved with CRISPRa, upregulation is dependent on the effect of native regulatory proteins as well as the native transcription level. When the CRISPRa system is correctly positioned, transcriptionally silent genes can be drastically upregulated, while enhanced activation of transcriptionally active genes is generally marginal (Strezoska et al., 2020). Problems of incorrectly positioned CRISPR guides could be potentially solved by deploying multiple spacers to the same promoter if the chosen CRISPRa/i system supports multiplexing. To conclude, with careful design CRISPRa can be applied as a targeted transcriptional activation tool for SM discovery, bypassing the need for laborious genome editing efforts.
CRISPR-Based Chromatin Remodeling
As discussed before, the chromatin landscape plays an important role in transcriptional regulation in filamentous fungi (Bok et al., 2009; Collemare and Seidl, 2019; Pfannenstiel and Keller, 2019). Since prokaryotic Cas proteins are not suited to cope with such obstacles as nucleosomes, it is expected that nucleosome-bound DNA hinders CRISPR activity. As CRISPR-based genome editing only involves a one-time event, and as the organization of chromatin is continuously changing, it is hypothesized that these spontaneous remodeling events contribute to the efficacy of CRISPR-based editing and its widespread success and applicability in eukaryotic organisms (Isaac et al., 2016). In contrast, for achieving potent CRISPRa/i transcriptional regulation at the promoter region, a persistent binding of the regulator is likely needed, which can be negatively affected by the chromatin state.
Nucleosome maps for fungal genomes are essentially undescribed. Since the chromatin organization can change depending on the cultivation conditions, it is advised to perform mapping in the same conditions as the CRISPRa/i application is planned to be executed. Fungal nucleosome maps could potentially help to identify genomic regions obscured by nucleosomes and therefore less accessible to the transcriptional complex, as well as nucleosome-free DNA regions, which are more favorable targets for CRISPR-based applications. Nucleosome mapping has been applied for A. nidulans to facilitate the design of efficient protospacers for dCas9-VPR. Indeed, targeting the nucleosome-free region of a bidirectional promoter in a cryptic BGC with a single sgRNA resulted in significant transcriptional activation of genes up and downstream of the spacer sequence (Schüller et al., 2020). Further, by targeting multiple protospacers (nucleosome-free and nucleosome-bound) synergistic activation effects were observed. For targeted chromatin remodeling, a fusion of dCas9 with the core domain of the human acetyltransferase p300 (dCas9-p300Core) has been successfully employed in mammalian cells to target enhancers regions upstream of the promoter of interest. This targeted acetylation resulted in increased expression of the downstream genes (Hilton et al., 2015). Recently, the dCas9-p300Core system has been employed in A. niger, where three different genes were targeted individually and successfully upregulated (Li et al., 2021).
Biosynthetic Gene Cluster Refactoring
Expression of all the relevant genes of a BGC with constitutive, inducible, or synthetic promoters—thus involving major refactoring and cloning efforts—is an effective approach to characterize cryptic BGCs. Although using filamentous fungal hosts has numerous advantages (as will be discussed later), tools and expression platforms are still underdeveloped compared to other well-established species such as S. cerevisiae.
The promoter replacement technology is commonly used for the overexpression of a gene of interest. Selected promoters are capable of a high transcription rate under the employed cultivation conditions. Strong promoters are often selected by using transcriptome data analysis or empirical testing. Usually, these are strong constitutive promoters responsible for the transcription of housekeeping genes or other genes that are highly expressed in vivo, or show inducibility (Kluge et al., 2018) [e.g., gpdA (ANIA_08041), glaA (An03g06550), pcbC (Pc21g21380), 40S-rps8 (An0465), tef1 (ANIA_04218)]. Inducible promoters are found in a similar manner, but employing a well-defined chemical (alcohols, antibiotics, hormones, or carbon sources) as a potential inducer that can be added in various amounts to repress or enhance gene expression levels (Kluge et al., 2018).
Unfortunately, individual replacement of all native promoters in a large BGC with strong promoters is an elaborate and time-consuming task, further complicated by the limited availability of well characterized fungal promoters. Nonetheless, such extensive refactoring can still be attempted with a filamentous fungal host that shows a high HR rate that facilitates recombination of DNA fragments in vivo (Chiang et al., 2020; Pohl et al., 2020). Single promoter replacement is much more practical when it is employed to overexpress an in-cluster regulator, which in turn results in complete BGC activation with minimal engineering efforts (Bromann et al., 2012; Zabala et al., 2012). Alternatively, prior to the fungal transformation the target BGC can be pre-assembled with the chosen promoters and terminators using advanced cloning methods or hosts with high HR rate, such as S. cerevisiae (Kim et al., 2010). For example, the 25 kb long geodin BGC from A. terreus was delivered into A. nidulans after pre-assembly using USER fusion from 8 PCR products containing the 13 native genes, and at the same time replacing and overexpressing the transcriptional activator of the cluster (Nielsen et al., 2013). Alternatively, such large genomic segments can be captured on fungal artificial chromosomes (FACs), as discussed in the following section (Bok et al., 2015). Since fungal promoters for overexpression approaches are limited, and not every BGC contains a specific transcriptional activator to overexpress, alternative solutions are needed. Although the decreasing prices of DNA synthesis could revolutionize BGC screening by making the synthesis of entire clusters affordable, the current price levels only allow for the synthesis of smaller DNA fragments. Polycistronic expression of multiple genes has been successfully applied in filamentous fungi using only one established promoter and one terminator (Unkles et al., 2014), and this could be a potential alternative for BGC refactoring. Synthetic promoters with orthogonal STF-based regulation (discussed earlier) could be used for a scaled, tunable or coordinated expression of refactored BGCs. Such systems can be delivered via genomic engineering of the native host, or by using shuttle vectors and suitable heterologous hosts.
Fungal Shuttle Vectors
Next to methods that require introducing genetic parts permanently into the genome of the host organism, vector-based, genome-editing-free alternatives are also available for gene expression in filamentous fungi. Fungal shuttle vectors allow the pre-assembly of genes of interest or complete BGCs in well-established model organisms like Escherichia coli or S. cerevisiae, thereby facilitating rapid cloning and subsequent delivery to the desired expression host. Since the isolation and identification of the AMA1 replicator sequence from A. nidulans (Gems et al., 1991), vectors bearing this sequence were shown to self-replicate in species within the genera Aspergillus, Penicillium, Giberella (Aleksenko and Clutterbuck, 1997) as well as in Trichoderma reseei (Kubodera et al., 2002), Lecanicillium (Ishidoh et al., 2014), and Paecilomyces variotii (Seekles et al., 2021). Telomeric sequences have also been reported to promote replication (and often integration) in various filamentous fungi like A. nidulans (Kistler and Benny, 1992), Fusarium oxysporum (Powell and Kistler, 1990), and Chrysosporium lucknowense (Verdoes et al., 2007). All of these vectors can be used efficiently for rapid assembly and delivery of transcription units expressing the gene(s) of interest into the host organism. The copy number of vectors maintained within the host differs by fungal species, and it is also influenced by the strength of the selection marker or the cultivation conditions. Aspergillus strains were shown to maintain numerous copies of AMA1 vectors in one nucleus (Fierro et al., 1996). Since they do not integrate in the genome, these vectors are easily lost without marker selection pressure (Aleksenko and Clutterbuck, 1997), which allows easy recycling of the same vector-based system.
In an impressive study, fungal artificial chromosomes based on AMA1 shuttle vectors have been used to capture the entire genome of A. terreus and to successfully clone all of its native BGCs in A. nidulans, resulting in the discovery of the astechrome biosynthetic machineries (Bok et al., 2015). The same approach was used to clone and overexpress 56 BGCs from other Aspergillus species in A. nidulans, resulting in the discovery of 15 novel metabolites (Clevenger et al., 2017). Although these shuttle vectors contained the BGCs with their native promoters, most of these compounds were not produced in the native hosts. Activation of these cryptic BGCs could be due to the presence of multiple copies of the vectors, or to the absence of native repressing factors such as epigenetic repression. Using fungal shuttle vectors in combination with modular cloning technologies and other well characterized advanced DNA assembly tools allows rapid refactoring and validation of multi-gene expression cassettes as well as synthetic metabolic pathways (Sarkari et al., 2017; Mózsik et al., 2021b).
Biosynthetic Gene Cluster Expression Using Polycistronic mRNA
To allow simultaneous expression of multiple genes from one established fungal promoter, and to avoid tedious promoter replacement of all genes in a BGC, the cluster can be reconstructed using so-called “Stop-Carry On,” or ribosomal “skipping,” sequences between genes cloned in a sequential organization. Viral 2A peptides have been shown to promote ribosomal skipping during translation from polycistronic mRNA (Sharma et al., 2012). Since its discovery, this method has been widely applied in eukaryotes for multiple protein delivery from a single transcript. The 2A peptide sequences have been used to express the three genes of the penicillin BGC from one polycistronic mRNA in A. nidulans (Unkles et al., 2014). As the same promoter is driving the expression of all the genes of the BGC an equimolar production of each enzyme might be expected, which can lead to imbalances in the pathway and accumulation of toxic intermediates (Hoefgen et al., 2018). Some technical limitations with P2-based BGC expression are potential enzyme activity problems created by the remnants of the 2A peptide sequence at the C-termini of the proteins, validation that all genes from the transcript are effectively translated, and tedious vector construction time (Hoefgen et al., 2018). To solve the first issue, the additional amino acids can be removed introducing Tobacco Etch Virus (TEV) endopeptidase recognition sequences and co-expressing this peptidase in the host, and a seamless cloning step has been utilized to clone the genes of the BGC and label them with P2 and TEV recognition sequences (Hoefgen et al., 2018). To solve the second issue, it is possible to incorporate a split fluorescent reporter to ensure that the first and last genes are correctly translated. This advanced 2A-based expression system was applied for the heterologous expression of the austinoid BGC (∼13 kb) from A. calidoustus and the psilocybin BGC (∼7.4 kb) from Psilocybe cubensis in A. nidulans (Hoefgen et al., 2018).
Filamentous Fungi as Platforms for the Heterologous Production of Secondary Metabolites
Because the great majority of fungi cannot be cultivated under laboratory conditions, model host strains are required for the heterologous expression of fungal BGCs and product identification. While common organisms as E. coli and S. cerevisiae have been successfully used for this purpose in some cases (Gao et al., 2010; Haynes et al., 2011; Davison et al., 2012; Bond et al., 2016; Zhang J. J. et al., 2019; Ma et al., 2021), filamentous fungi are much more suitable hosts for BGC expression for several reasons. Firstly introns do not strictly need to be removed when cloning a putative biosynthetic gene of interest, since filamentous fungi are more likely able to process them accurately during splicing, yielding the correct mRNA. Naturally, the chances of correct splicing are higher when cloning genes from organisms that are more closely related to the host of choice (Karnaukhova et al., 2007). The chances of a successful expression are further increased by a more ideal codon usage (Su et al., 2012). Secondly, fungi are more likely to produce the building blocks utilized by biosynthetic enzymes for secondary metabolite biosynthesis, because they are naturally wired for such processes. For the same reason, these hosts possess a plethora of accessory enzymes that are required for the correct functioning of the BGCs enzymes, such as phosphopantetheinyl transferases (PPTases), redox partners for P450s, prenyltransferases, and other enzymes (Keller, 2019). Additionally, hosts such as A. niger, A. oryzae are classified as GRAS (generally recognized as safe) organisms, and therefore they are suitable for the industrial production of compounds destined to be used in humans. The most commonly employed hosts for the characterization of heterologous BGCs are A. nidulans and A. oryzae (Anyaogu and Mortensen, 2015; Meng et al., 2021; Meyer, 2021), but other species have been successfully developed into platform strains, as showcased in Table 1. In the following section, we will discuss the most relevant examples and highlight their major features.
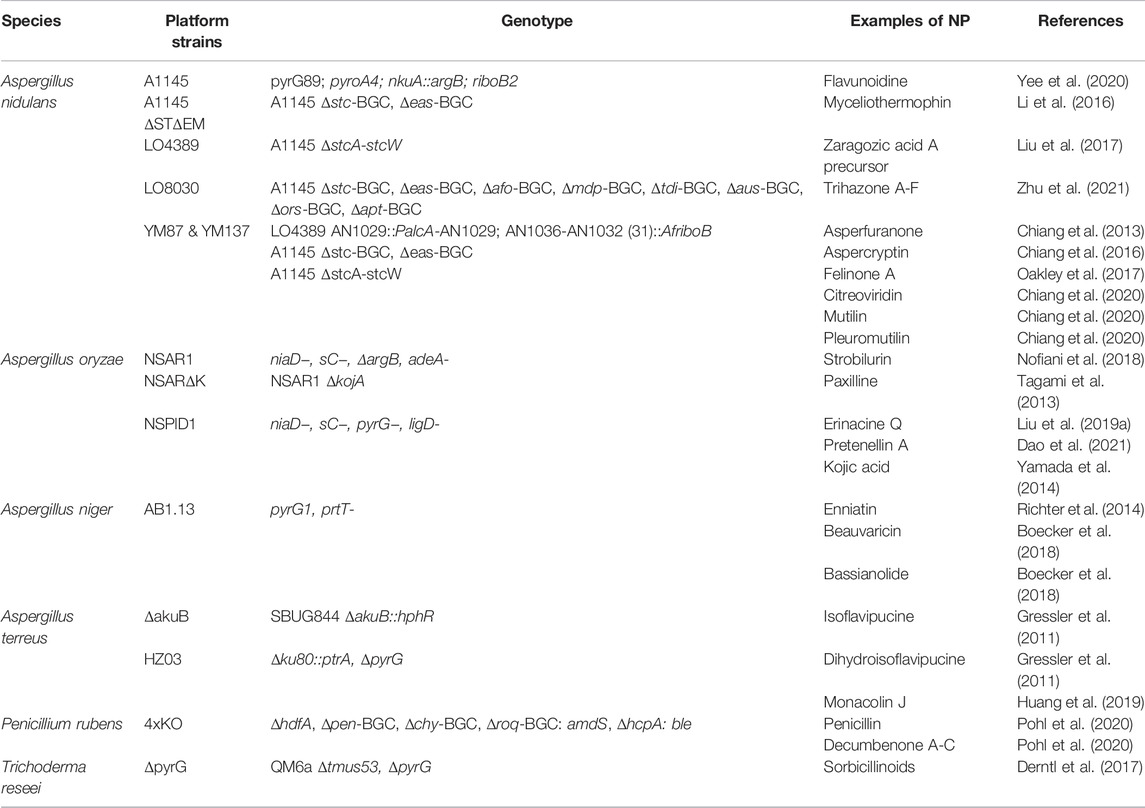
TABLE 1. Examples of fungal expression platforms for the production of natural products and the characterization of biosynthetic gene clusters.
The filamentous fungus most widely used as heterologous host is by far A. nidulans. This species has been used for decades to study important cellular processes such as recombination, DNA repair, and chromatin regulation (Morris, 1976; Chaveroche, 2000; Osmani and Mirabito, 2004), because it can be easily manipulated and cultivated in the lab. This has led to the development of several platform strains that have been engineered to characterize and overexpress heterologous genes and, ultimately, produce natural products. The most interesting platform strains, showcased in Table 1, are derived from a triple auxotrophic strain called A1145 (Nayak et al., 2006). This strain also carries a deletion of the nkuA gene (homolog of human ku70) that renders NHEJ DNA repair less favorable, facilitating precise genomic integration of heterologous genes via HR. A1145 has been successfully used to elucidate a diverse range of biosynthetic pathways belonging to all major classes of SMs (Li et al., 2016; Hai and Tang, 2018; Liu L. et al., 2019; Yee et al., 2020). Despite these attractive features, A. nidulans also has a major downside. It is a prolific producer of SMs (Anyaogu and Mortensen, 2015; Meng et al., 2021) that typically yields a crowded chromatographic background, which can render the identification of new metabolites a cumbersome task. To partially overcome this problem, strains with a cleaner SM background have been developed. In one instance, the BGCs responsible for the biosynthesis of sterigmatocystin and emericellamids, two major classes of compounds produced by A. nidulans, were deleted. This allowed the identification of an important intermediate of the cholesterol-lowering compound zaragozic acid A in an engineered strain, previously overshadowed by the host metabolites (Liu et al., 2017). A similar strain—LO4389—where only the sterigmatocystin pathway was deleted, was used for the identification of 6 polyketides from A. terreus along with the complete reconstitution of the A. terreus asperfuranone pathway (Chiang et al., 2013), underlining once again the potential of such cleaner platform strains. Following the success of such trials, the same authors reported the construction of strain LO8030 where eight of the most highly expressed BGCs were deleted, resulting in a considerable reduction of the genome size (Chiang et al., 2016). Despite that, the strain showed no significant defects in growth. Additionally, not only did the strain offer a minimal background, but it also benefitted from a higher availability of SM precursors, as demonstrated by the synthesis and detection of the previously undiscovered metabolite aspercryptin (Chiang et al., 2016). Recently, LO4389 was used to construct two new strains that possess genetic features that are especially advantageous for the expression of entire biosynthetic pathways. In these strains, up to 6 or 7 genes of interest (GOIs) can be placed under the control of the native regulatory elements of the asperfuranone pathway, whose genes have been removed, while the inducible promoter PalcA controls the major TF regulating the BGC pathway. This elegant approach allows for the induction of entire heterologous BGCs upon the addition of methyl ethyl ketone, and its potential was demonstrated by the successful production of citreoviridin, mutilin, and pleuromutilin (Chiang et al., 2020).
Another common choice as a cell factory for the identification and production of SMs is A. oryzae. This fungus plays an important role in food manufacturing in Asia, where it is widely used for the production of alcoholic beverages and fermented products such as soy sauce and miso. A. oryzae contains numerous BGCs in its genome, many of which are also found in the toxins-producing species A. flavus, which is a prolific producer of natural products. In fact, the species are so closely related that it is believed that A. oryzae is actually a product of the domestication of A. flavus (Payne et al., 2006). However, A. oryzae produces only a few endogenous SMs, which makes it a perfect host for heterologous production (Anyaogu and Mortensen, 2015). The most common platform strain is the quadruple auxotroph NSAR1 (Jin et al., 2004) which offers great versatility and does not require the need of expensive additives for the selection of the transformants. NSAR1 has been used successfully by many researchers to elucidate diverse biosynthetic pathways (Tagami et al., 2013; Nofiani et al., 2018; Liu C. et al., 2019; Han et al., 2021). Despite the fact that its use is relatively more recent than A. nidulans, an extensive toolkit is now available to transform A. oryzae and express heterologous genes (Pahirulzaman et al., 2012; Lazarus et al., 2014). Of particular interest is a system of vectors—pTYargB/niaD/adeA/sC-eGFPac—that carry an inducible expression cassette (under control of the amyB promoter and terminator) and three constitutive cassettes. There are four versions of these vectors, each carrying a different selection marker, that can be co-transformed to allow the overexpression of up to 16 heterologous genes in the recipient NSAR1 strain (Lazarus et al., 2014). Although A. oryzae already offers a clean SM background, it produces a relatively abundant compound called kojic acid. The presence of kojic acid can complicate the purification of metabolites of interest and interfere with structural characterization. Deletion of the gene kojA, that encodes a crucial oxidoreductase for the synthesis of kojic acid, resulted in an astoundingly clean SM background and easy detection of the metabolite of interest (Dao et al., 2021). Another platform strain that merits attention is the triple auxotroph NSlPD1 (Maruyama and Kitamoto, 2008). The advantage of this particular strain over NSAR1 and its derivative strains is the deletion of the ligD gene, which facilitates HR-mediated genetic engineering (analogously to the deletion of nkuA in A. nidulans). NSlPD1 was successfully engineered to generate a strain that produces higher titers of kojic acid and uses cellulose as starting material (Yamada et al., 2014).
Two other well-known Aspergilli that have been explored as hosts for the biosynthesis of natural products are A. niger and A. terreus. The former has been used for the efficient production of the depsipeptides enniatin (Richter et al., 2014), beauvericin, and bassianolide (Boecker et al., 2018). These compounds show high insecticidal activity (Grove and Pople, 1980) and some have been proposed as potential candidates for the treatment of HIV infections (Shin et al., 2009). A. terreus is less commonly used as a heterologous host, but its tremendous natural capabilities as a producer of SMs (Huang et al., 2021) suggest that it could represent a worthy alternative to other fungal species, especially for the production of polyketides. In fact, this species is widely used for the industrial production of lovastatin (Boruta and Bizukojc, 2017), an essential pharmaceutical, that is, used to treat high blood cholesterol. In recent years, platform strains of A. terreus have been used to elucidate the biosynthetic pathway of the mycotoxin flavipucine (Gressler et al., 2011), and to generate a high-performance strain capable of producing high titers of monacolin J, a key precursor to the synthesis of semi-synthetic statins (Huang et al., 2019).
Another important fungal workhorse for industrial applications is P. rubens, which is used for the production of penicillin, cephalosporin, and other β-lactam antibiotics. To increase the titers of penicillin produced, early strains of P. rubens have been subjected to decades of random mutagenesis and selection processes, collectively known as the classical strain improvement program. This has also led to increased capabilities to grow in submerged cultivation conditions and in defined media, features that are very desirable in the industry (Harris et al., 2009; Guzmán-Chávez et al., 2018; Iacovelli et al., 2021a). The CSI program also led to a major reduction of the expression and/or mutational inactivation of non-penicillin BGCs. One of the industrial penicillin producer strains was recently engineered to abolish production of β-lactam antibiotics, with the assumption that this strain would retain its metabolic capabilities while offering a cleaner background. Indeed, this particular strain was used to heterologously express the compactin BGC from P. citrinum, together with an engineered cytochrome P450 from Amycolatopsis orientalis. The newly engineered strain was able to catalyze the final hydroxylation step of compactin to the cholesterol-lowering drug pravastatin, with an impressive yield of more than 6 g/L in pilot scale fermentations (McLean et al., 2015). This idea was further explored even more recently, when the same β-lactam-deficient strain was used as a template to generate a quadruple deletion strain in which alongside the penicillin BGC three major BGCs (chrysogine, fungisporin, and roquefortine) were removed, resulting in a considerably clean SM background, ideal for natural product production (Pohl et al., 2020). As proof of concept, the SM-deficient platform strain was used to reintroduce the penicillin BGC, resulting in restored production of the antibiotic, and to overexpress an endogenous PKS (PKS17), leading to the production of YWA1, a common precursor to fungal pigments. Additionally, the strain was used for the successful reconstitution of the calbistrin BGC from Penicillium decumbens, which resulted in the heterologous production of decumbenone A, B, and C (Pohl et al., 2020). This strain is also devoid of the hdfA gene (homolog of human ku70) and is therefore suitable for HR-mediated genetic engineering. These results highlight the P. rubens 4xKO strain as a valuable option for SM research.
Another filamentous fungus worth mentioning is T. reesei. For decades, this organism has been used in industry for its astonishing ability to produce cellulolytic enzymes such as cellulases and hemicellulases (Bischof et al., 2016), but it never attracted natural product researchers, probably due to the broad availability of other hosts and a lack of well-developed synthetic biology tools. Recently, a strain which carries deletions for the genes tmus53 (ligD homolog) and pyrG was engineered (Steiger et al., 2011). Analogously to other fungal hosts, the Δtmus53 deletion facilitates HR-mediated targeted gene integration, while ΔpyrG allows easy selection through complementation of uracil/uridine auxotrophy. These features raise interest for T. reesei as a potential natural product producing host. In fact, this particular strain has already been utilized to investigate the endogenous biosynthetic pathway of sorbicillinoids (Derntl et al., 2017). What could set this platform apart and make it a concrete option for industrial applications is its natural ability to degrade and thrive on cellulosic material, which could lead to the production of natural products starting from biomass material, such as agricultural waste.
Undoubtedly, the fungal platforms discussed above provide a rich choice for researchers who want to investigate unknown biosynthetic pathways or produce industrially relevant metabolites. Nevertheless, despite the broad availability of hosts, not all species might be capable to produce the desired natural product, and even when they are, it is very likely that one species performs better than another in terms of yield. This is difficult to predict and engineering more species at once to optimize production can be extremely time-consuming as well as costly. To reduce the workload and facilitate simultaneous cloning and screening of more hosts, a group of researchers recently developed the first multispecies fungal platform for heterologous gene expression (Jarczynska et al., 2020). The first version of this system, called DIVERSIFY, is based upon four Aspergillus species: A. nidulans, A. oryzae, A. niger, and A. aculeatus. Each individual species was first engineered to contain in the genome a “common synthetic gene integration site” (COSI), which encodes a reporter gene for white/blue selection placed under the control of a constitutive promoter. Additionally, the COSI contains two 500 bp sequences flanking the reporter cassette that can be used for HR-mediated target gene replacement, whereby GOIs can be easily inserted and overexpressed. Since the COSI is equal in each recipient strain, only one integration cassette has to be designed and built. As a proof of concept, the DIVERSIFY platform has been successfully used to overexpress a fluorescent reporter (mRFP) and cellobiohydrolase, and for the production of 6-MSA, a model polyketide (Jarczynska et al., 2020). Because many of the synthetic biology tools developed for fungi are readily adaptable to other species, this platform can be expanded with other hosts in the future.
Clearly, filamentous fungi are powerful instruments for the elucidation of biosynthetic pathways and the production of SMs. In many cases, though, the desired compounds are produced at very low yields which are unsuitable for commercial applications. The development of a great array of hosts, each with specific benefits, as well as multispecies platforms that allow fast and simultaneous screening of several fungi with reduced workloads, will offer researchers the tools to readily optimize the production yield of metabolites of interest. Ultimately, this is necessary to engineer efficient fungal cell factories that are ready to be employed at an industrial scale.
Concluding Remarks
Thanks to the rapidly expanding number of filamentous fungal genome sequences and the advanced bioinformatics tools that are now available, it has become obvious that filamentous fungi represent an untapped reservoir of natural products. Each genome contains a high number of BGCs for which the product has not been identified, and many of these BGCs are transcriptionally silent under laboratory conditions. The products of these clusters can be unearthed with the combined efforts of bioinformatics, chemistry, and synthetic biology, to reveal new chemistries and biological activities of interest. A major challenge, however, remains prioritizing these BGCs for their potential value since the bioinformatics tools available at the moment cannot reliably predict the resulting products. Hence, much relies on laborious empirical testing, whereby many BGCs have to be expressed to screen for bioactive compounds. Here, we have discussed conventional tools and the development of new synthetic biology tools that aid in the transcriptional activation of silent BGCs in filamentous fungi, therefore offering new approaches for compound discovery (Table 2 and Figure 2).
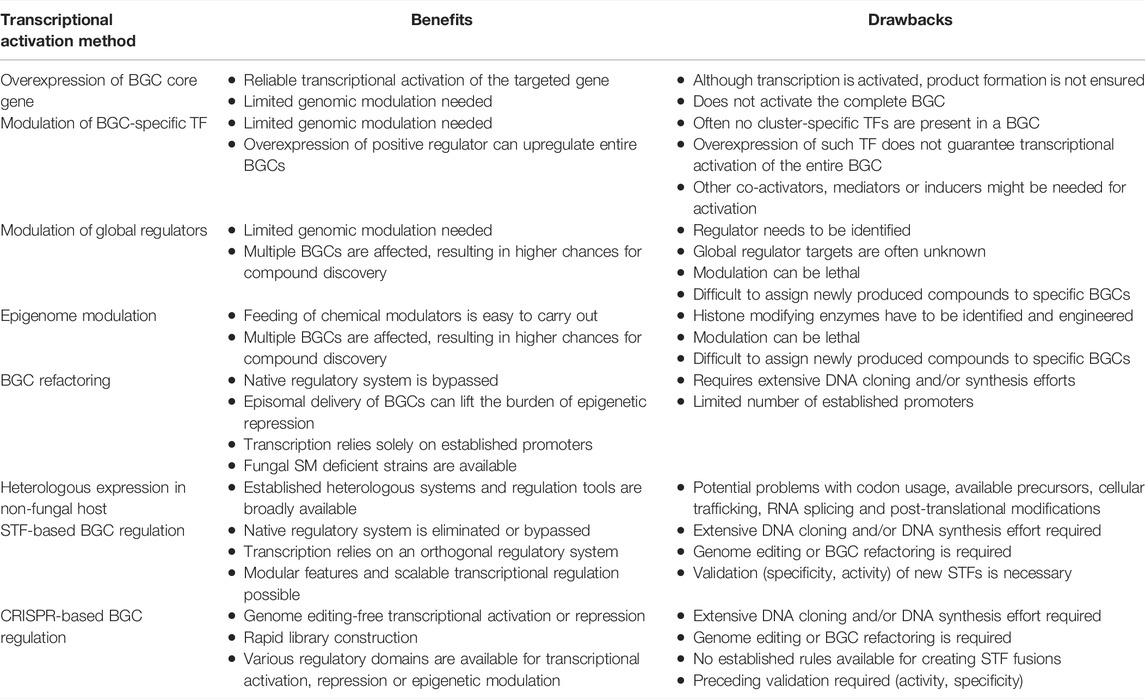
TABLE 2. Transcriptional activation tools and methods for fungal biosynthetic gene clusters.
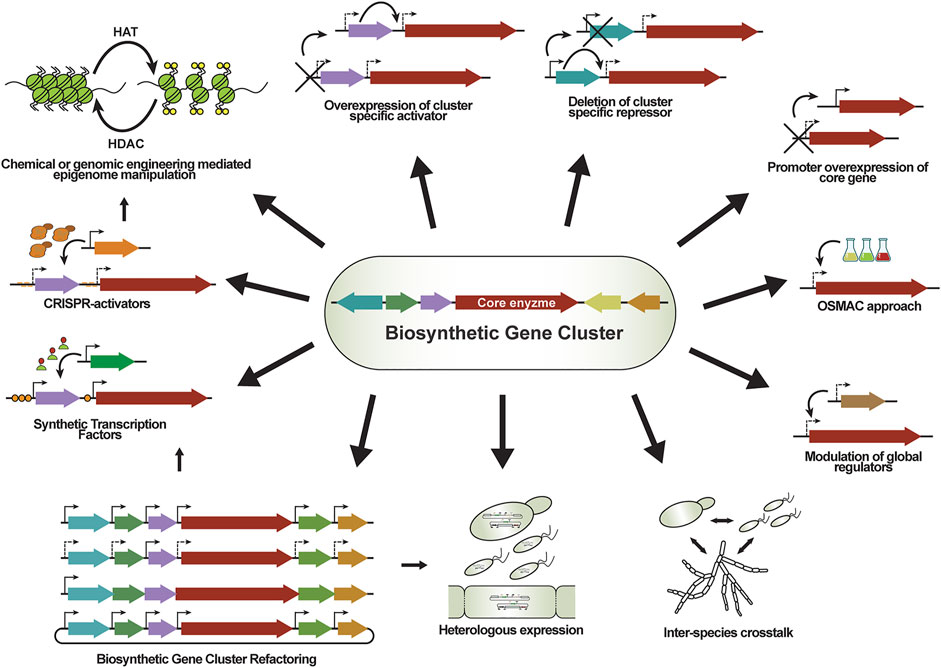
FIGURE 2. Strategies for transcriptional activation for fungal biosynthetic gene clusters. Dashed arrows indicate native, solid arrows indicate engineered (strong or inducible) promoters.
Modulating global regulatory systems does not require prior knowledge regarding the cluster-specific regulation mechanism of a given BGCs, and although numerous genes can be affected when using this approach, transcriptional activation of BGCs of interest and subsequent production of new metabolites are not guaranteed. Instead, overriding the native regulatory system of the cluster can result in a more direct transcriptional activation. Such approaches involve partial or complete BGC refactoring, and require a deep level of understanding about the number of genes in the BGC, and/or its cluster specific regulators. Refactoring approaches can take place within a host which supports the assembly of numerous DNA fragments, often achieved via homologous recombination or advanced synthetic cloning methods. The only approach that ensures transcriptional activation of an entire BGC is promoter replacement of all its genes, but this is generally a laborious and cumbersome task. Furthermore, the number of established strong fungal promoters and fungal selection markers is still limited, and this represents a bottleneck towards rapid or consecutive genomic modifications. An alternative to serial replacement of promoters could be the utilization of promoters from BGCs which are transcriptionally active in the selected host, or synthetic promoters containing regulatory elements for orthogonal regulatory systems such as STFs. The more recent CRISPRa systems further increase the number of available activator tools adding a new layer of control, since such systems do not require BGC refactoring anymore.
Since genetic manipulation and precise engineering of a non-model or wild type fungal strains is challenging—mainly due to the low rate of HR and difficulties to grow these organisms in laboratory conditions—the most versatile method is to express BGCs in an established heterologous host. Ideal host strains are those in which it is easy to perform genetic manipulation, that are convenient to cultivate at different scales, for which compatible genetic tools are available, and convenient downstream processing steps have been established such as rapid compound screening and a clean metabolite spectrum. A combination of advanced transcriptional activation tools and established expression hosts can ensure reliable, targeted transcriptional activation and efficient methods for compound identification.
Although new host strains and tools are continuously being developed for the characterization of cryptic BGCs in filamentous fungi, high-throughput BGC screening remains a major challenge. Automatized engineering of protoplasted filamentous fungi using microtiter plates and robotic liquid handling robots have been successfully established (Kuivanen et al., 2019), as well as fully-automated microscale bioreactor cultivations (Jansen et al., 2021), but working with BGC-coding DNA is demanding. The large size, the numerous genes, and the costs for total cluster DNA synthesis are limiting factors for rapid assembly and screening of numerous BGCs.
In the future, the combination of emerging genetic tools, tailored heterologous hosts with high metabolic capacity, and automated systems, will facilitate the development of highly efficient, targeted, multiplexing-compatible transcriptional activation applications for novel natural product discovery. This, coupled with the development of bioinformatics tools that are able to prioritize the most valuable BGCs within genomic sequences, will revolutionize the field and eliminate time-consuming and costly wet lab procedures, while starkly increasing the chances of identifying novel and potent bioactive compounds.
Author Contributions
LM and RI wrote the manuscript with the critical feedback of AJMD and RALB. LM and AJMD conceived the manuscript. AD supervised the manuscript. RALB co-supervised the manuscript.
Funding
LM received funding from the European Union’s Horizon 2020 Research and Innovation Programme under the Marie Skłodowska-Curie grant agreement No. 713482 (ALERT Program).
Conflict of Interest
Author RALB was employed by the company DSM Biotechnology Center.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbrevations
AD, activation domain; CP, core promoter; DBD, DNA-binding domain; STF, synthetic transcription factor; UAS, upstream activating sequence
References
Ahuja, M., Chiang, Y.-M., Chang, S.-L., Praseuth, M. B., Entwistle, R., and Sanchez, J. F. (2012). Illuminating the Diversity of Aromatic Polyketide Synthases in Aspergillus nidulans. J. Am. Chem. Soc. 134, 8212–8221. doi:10.1021/ja3016395
Albright, J. C., Henke, M. T., Soukup, A. A., McClure, R. A., Thomson, R. J., Keller, N. P., et al. (2015). Large-Scale Metabolomics Reveals a Complex Response of Aspergillus nidulans to Epigenetic Perturbation. ACS Chem. Biol. 10, 1535–1541. doi:10.1021/acschembio.5b00025
Aleksenko, A., and Clutterbuck, A. J. (1997). Autonomous Plasmid Replication in Aspergillus nidulans:AMA1 and MATE Elements. Fungal Genet. Biol. 21, 373–387. doi:10.1006/fgbi.1997.0980
Anyaogu, D. C., and Mortensen, U. H. (2015). Heterologous Production of Fungal Secondary Metabolites in Aspergilli. Front. Microbiol. 6, 1–6. doi:10.3389/fmicb.2015.00077
Bergmann, S., Schümann, J., Scherlach, K., Lange, C., Brakhage, A. A., and Hertweck, C. (2007). Genomics-driven Discovery of PKS-NRPS Hybrid Metabolites from Aspergillus nidulans. Nat. Chem. Biol. 3, 213–217. doi:10.1038/nchembio869
Bertrand, S., Bohni, N., Schnee, S., Schumpp, O., Gindro, K., and Wolfender, J.-L. (2014). Metabolite Induction via Microorganism Co-culture: A Potential Way to Enhance Chemical Diversity for Drug Discovery. Biotechnol. Adv. 32, 1180–1204. doi:10.1016/j.biotechadv.2014.03.001
Bischof, R. H., Ramoni, J., and Seiboth, B. (2016). Cellulases and beyond: The First 70 Years of the Enzyme Producer Trichoderma Reesei. Microb. Cell Fact. 15, 1–13. doi:10.1186/s12934-016-0507-6
Blin, K., Shaw, S., Kloosterman, A. M., Charlop-Powers, Z., van Wezel, G. P., Medema, M. H., et al. (2021). antiSMASH 6.0: Improving Cluster Detection and Comparison Capabilities. Nucleic Acids Res. 49, W29–W35. doi:10.1093/nar/gkab335
Bode, H. B., Bethe, B., Höfs, R., and Zeeck, A. (2002). Big Effects from Small Changes: Possible Ways to Explore Nature’s Chemical Diversity. ChemBioChem 3, 619–627. doi:10.1002/1439-7633(20020703)3:7%3C619::AID-CBIC619%3E3.0.CO10.1002/1439-7633(20020703)3:7<619::aid-cbic619>3.0.co;2-9
Boecker, S., Grätz, S., Kerwat, D., Adam, L., Schirmer, D., Richter, L., et al. (2018). Aspergillus niger Is a Superior Expression Host for the Production of Bioactive Fungal Cyclodepsipeptides. Fungal Biol. Biotechnol. 5, 1–14. doi:10.1186/s40694-018-0048-3
Boettger, D., and Hertweck, C. (2013). Molecular Diversity Sculpted by Fungal PKS–NRPS Hybrids. ChemBioChem 14, 28–42. doi:10.1002/cbic.201200624
Bok, J. W., Chiang, Y.-M., Szewczyk, E., Reyes-Dominguez, Y., Davidson, A. D., Sanchez, J. F., et al. (2009). Chromatin-level Regulation of Biosynthetic Gene Clusters. Nat. Chem. Biol. 5, 462–464. doi:10.1038/nchembio.177
Bok, J. W., and Keller, N. P. (2004). LaeA, a Regulator of Secondary Metabolism in Aspergillus Spp. Eukaryot. Cell 3, 527–535. doi:10.1128/EC.3.2.527-535.2004
Bok, J. W., Ye, R., Clevenger, K. D., Mead, D., Wagner, M., Krerowicz, A., et al. (2015). Fungal Artificial Chromosomes for Mining of the Fungal Secondary Metabolome. BMC Genomics 16, 343. doi:10.1186/s12864-015-1561-x
Bond, C., Tang, Y., and Li, L. (2016). Saccharomyces cerevisiae as a Tool for Mining, Studying and Engineering Fungal Polyketide Synthases. Fungal Genet. Biol. 89, 52–61. doi:10.1016/j.fgb.2016.01.005
Boruta, T., and Bizukojc, M. (2017). Production of Lovastatin and Itaconic Acid by Aspergillus terreus: a Comparative Perspective. World J. Microbiol. Biotechnol. 33, 1–12. doi:10.1007/s11274-017-2206-9
Brakhage, A. A., and Schroeckh, V. (2011). Fungal Secondary Metabolites - Strategies to Activate Silent Gene Clusters. Fungal Genet. Biol. 48, 15–22. doi:10.1016/j.fgb.2010.04.004
Brodhagen, M., and Keller, N. P. (2006). Signalling Pathways Connecting Mycotoxin Production and Sporulation. Mol. Plant Pathol. 7, 285–301. doi:10.1111/j.1364-3703.2006.00338.x
Bromann, K., Toivari, M., Viljanen, K., Vuoristo, A., Ruohonen, L., and Nakari-Setälä, T. (2012). Identification and Characterization of a Novel Diterpene Gene Cluster in Aspergillus nidulans. PLoS One 7, 1–14. doi:10.1371/journal.pone.0035450
Caceres, I., Al Khoury, A., El Khoury, R., Lorber, S., Oswald, P., Khoury, A. E., et al. (2020). Aflatoxin Biosynthesis and Genetic Regulation: A Review. Toxins 12. doi:10.3390/toxins12030150
Caesar, L. K., Kelleher, N. L., and Keller, N. P. (2020). In the Fungus where it Happens: History and Future Propelling Aspergillus nidulans as the Archetype of Natural Products Research. Fungal Genet. Biol. 144, 103477. doi:10.1016/j.fgb.2020.103477
Carvalho, N. D. S. P., Arentshorst, M., Jin Kwon, M., Meyer, V., and Ram, A. F. J. (2010). Expanding the Ku70 Toolbox for Filamentous Fungi: Establishment of Complementation Vectors and Recipient Strains for Advanced Gene Analyses. Appl. Microbiol. Biotechnol. 87, 1463–1473. doi:10.1007/s00253-010-2588-1
Chaveroche, M.-K. (2000). A Rapid Method for Efficient Gene Replacement in the Filamentous Fungus Aspergillus nidulans. Nucleic Acids Res. 28, 97e–97. doi:10.1093/nar/28.22.e97
Chavez, A., Scheiman, J., Vora, S., Pruitt, B. W., Tuttle, M., P R Iyer, E., et al. (2015). Highly Efficient Cas9-Mediated Transcriptional Programming. Nat. Methods 12, 326–328. doi:10.1038/nmeth.3312
Chavez, A., Tuttle, M., Pruitt, B. W., Ewen-Campen, B., Chari, R., Ter-Ovanesyan, D., et al. (2016). Comparison of Cas9 Activators in Multiple Species. Nat. Methods 13, 563–567. doi:10.1038/nmeth.3871
Chiang, Y.-M., Ahuja, M., Oakley, C. E., Entwistle, R., Asokan, A., Zutz, C., et al. (2016). Development of Genetic Dereplication Strains in Aspergillus nidulans Results in the Discovery of Aspercryptin. Angew. Chem. Int. Ed. 55, 1662–1665. doi:10.1002/anie.201507097
Chiang, Y.-M., Lin, T.-S., Chang, S.-L., Ahn, G., and Wang, C. C. C. (2020). An Aspergillus nidulans Platform for the Complete Cluster Refactoring and Total Biosynthesis of Fungal Natural Products. ACS Synth. Biol. 3. doi:10.1021/acssynbio.0c00536
Chiang, Y.-M., Szewczyk, E., Davidson, A. D., Keller, N., Oakley, B. R., and Wang, C. C. C. (2009). A Gene Cluster Containing Two Fungal Polyketide Synthases Encodes the Biosynthetic Pathway for a Polyketide, Asperfuranone, in Aspergillus nidulans. J. Am. Chem. Soc. 131, 2965–2970. doi:10.1021/ja8088185
Chiang, Y. M., Oakley, C. E., Ahuja, M., Entwistle, R., Schultz, A., Chang, S. L., et al. (2013). An Efficient System for Heterologous Expression of Secondary Metabolite Genes in Aspergillus nidulans. J. Am. Chem. Soc. 135, 7720–7731. doi:10.1021/ja401945a
Clevenger, K. D., Bok, J. W., Ye, R., Miley, G. P., Verdan, M. H., Velk, T., et al. (2017). A Scalable Platform to Identify Fungal Secondary Metabolites and Their Gene Clusters. Nat. Chem. Biol. 13, 895–901. doi:10.1038/nchembio.2408
Collemare, J., and Seidl, M. F. (2019). Chromatin-dependent Regulation of Secondary Metabolite Biosynthesis in Fungi: Is the Picture Complete? FEMS Microbiol. Rev. 43, 591–607. doi:10.1093/femsre/fuz018
Corton, J. C. (1989). Altering DNA-Binding Specificity of GAL4 Requires Sequences Adjacent to the Zinc Finger. Nature 340, 724–727. doi:10.1038/340724a0
da Silva Ferreira, M. E., Kress, M. R. V. Z., Savoldi, M., Goldman, M. H. S., Härtl, A., Heinekamp, T., et al. (2006). The akuB KU80 Mutant Deficient for Nonhomologous End Joining Is a Powerful Tool for Analyzing Pathogenicity in Aspergillus fumigatus. Eukaryot. Cell 5, 207–211. doi:10.1128/EC.5.1.207-211.2006
Dahlmann, T. A., Terfehr, D., Becker, K., and Teichert, I. (2021). Golden Gate Vectors for Efficient Gene Fusion and Gene Deletion in Diverse Filamentous Fungi. Curr. Genet. 67, 317–330. doi:10.1007/s00294-020-01143-2
Dao, T. T., de Mattos-Shipley, K. M. J., Prosser, I. M., Williams, K., Zacharova, M. K., Lazarus, C. M., et al. (2021). Cleaning the Cellular Factory–Deletion of McrA in Aspergillus oryzae NSAR1 and the Generation of a Novel Kojic Acid Deficient Strain for Cleaner Heterologous Production of Secondary Metabolites. Front. Fungal Biol. 2, 1–12. doi:10.3389/ffunb.2021.632542
Davison, J., Al Fahad, A., Cai, M., Song, Z., Yehia, S. Y., Lazarus, C. M., et al. (2012). Genetic, Molecular, and Biochemical Basis of Fungal Tropolone Biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 109, 7642–7647. doi:10.1073/pnas.1201469109
de Groot, M. J. A., Bundock, P., Hooykaas, P. J. J., and Beijersbergen, A. G. M. (1998). Agrobacterium Tumefaciens-Mediated Transformation of Filamentous Fungi. Nat. Biotechnol. 16, 839–842. doi:10.1038/nbt0998-839
Derntl, C., Guzmán-Chávez, F., Mello-de-Sousa, T. M., Busse, H. J., Driessen, A. J. M., Mach, R. L., et al. (2017). In Vivo study of the Sorbicillinoid Gene Cluster in Trichoderma Reesei. Front. Microbiol. 8, 1–12. doi:10.3389/fmicb.2017.02037
Derntl, C., Mach, R. L., and Mach-Aigner, A. R. (2019). Fusion Transcription Factors for Strong, Constitutive Expression of Cellulases and Xylanases in Trichoderma Reesei. Biotechnol. Biofuels 12, 231. doi:10.1186/s13068-019-1575-8
Ding, Z., Zhou, H., Wang, X., Huang, H., Wang, H., Zhang, R., et al. (2020). Deletion of the Histone Deacetylase HdaA in Endophytic Fungus Penicillium chrysogenum Fes1701 Induces the Complex Response of Multiple Bioactive Secondary Metabolite Production and Relevant Gene Cluster Expression. Mol 25. doi:10.3390/molecules25163657
Doudna, A. J., and Charpentier, E. (2014). The New Frontier of Genome Engineering with CRISPR-Cas9. Sci. (80-. ) 346, 1258096. doi:10.1126/science.1258096
Engler, C., Gruetzner, R., Kandzia, R., and Marillonnet, S. (2009). Golden Gate Shuffling: A One-Pot DNA Shuffling Method Based on Type Ils Restriction Enzymes. PLoS One 4. doi:10.1371/journal.pone.0005553
Engler, C., Youles, M., Gruetzner, R., Ehnert, T.-M., Werner, S., Jones, J. D. G., et al. (2014). A Golden Gate Modular Cloning Toolbox for Plants. ACS Synth. Biol. 3, 839–843. doi:10.1021/sb4001504
Farzadfard, F., Perli, S. D., and Lu, T. K. (2013). Tunable and Multifunctional Eukaryotic Transcription Factors Based on CRISPR/Cas. ACS Synth. Biol. 2, 604–613. doi:10.1021/sb400081r
Ferreira, R., Skrekas, C., Nielsen, J., and David, F. (2018). Multiplexed CRISPR/Cas9 Genome Editing and Gene Regulation Using Csy4 in Saccharomyces cerevisiae. ACS Synth. Biol. 7, 10–15. doi:10.1021/acssynbio.7b00259
Fierro, F., Kosalková, K., Gutiérrez, S., and Martín, J. F. (1996). Autonomously Replicating Plasmids Carrying the AMA1 Region in Penicillium chrysogenum. Curr. Genet. 29, 482–489. doi:10.1007/BF02221518
Fischbach, M. A., and Walsh, C. T. (2006). Assembly-Line Enzymology for Polyketide and Nonribosomal Peptide Antibiotics: Logic, Machinery, and Mechanisms. Chem. Rev. 106, 3468–3496. doi:10.1021/cr0503097
Fonfara, I., Richter, H., Bratovič, M., Le Rhun, A., and Charpentier, E. (2016). The CRISPR-Associated DNA-Cleaving Enzyme Cpf1 Also Processes Precursor CRISPR RNA. Nature 532, 517–521. doi:10.1038/nature17945
Fu, Y., Foden, J. A., Khayter, C., Maeder, M. L., Reyon, D., Joung, J. K., et al. (2013). High-frequency Off-Target Mutagenesis Induced by CRISPR-Cas Nucleases in Human Cells. Nat. Biotechnol. 31, 822–826. doi:10.1038/nbt.2623
Fuller, K. K., Chen, S., Loros, J. J., and Dunlap, J. C. (2015). Development of the CRISPR/Cas9 System for Targeted Gene Disruption in Aspergillus fumigatus. Eukaryot. Cell 14, 1073–1080. doi:10.1128/EC.00107-15
Gao, L., Xia, C., Xu, J., Li, Z., Yu, L., Liu, G., et al. (2017). Constitutive Expression of Chimeric Transcription Factors Enables Cellulase Synthesis under Non-inducing Conditions in Penicillium oxalicum. Biotechnol. J. 12, 1700119. doi:10.1002/biot.201700119
Gao, X., Tsang, J. C. H., Gaba, F., Wu, D., Lu, L., and Liu, P. (2014). Comparison of TALE Designer Transcription Factors and the CRISPR/dCas9 in Regulation of Gene Expression by Targeting Enhancers. Nucleic Acids Res. 42, e155. doi:10.1093/nar/gku836
Gao, X., Wang, P., and Tang, Y. (2010). Engineered Polyketide Biosynthesis and Biocatalysis in Escherichia coli. Appl. Microbiol. Biotechnol. 88, 1233–1242. doi:10.1007/s00253-010-2860-4
Gao, Y., and Zhao, Y. (2014). Self‐processing of Ribozyme‐flanked RNAs into Guide RNAs In Vitro and In Vivo for CRISPR‐mediated Genome Editing. J. Integr. Plant Biol. 56, 343–349. doi:10.1111/jipb.12152
Gems, D., Johnstone, I. L., and Clutterbuck, A. J. (1991). An Autonomously Replicating Plasmid Transforms Aspergillus nidulans at High Frequency. Gene 98, 61–67. doi:10.1016/0378-1119(91)90104-J
Geu-Flores, F., Nour-Eldin, H. H., Nielsen, M. T., and Halkier, B. A. (2007). USER Fusion: a Rapid and Efficient Method for Simultaneous Fusion and Cloning of Multiple PCR Products. Nucleic Acids Res. 35, e55. doi:10.1093/nar/gkm106
Gibson, D. G., Young, L., Chuang, R. Y., Venter, J. C., Hutchison, C. A., and Smith, H. O. (2009). Enzymatic Assembly of DNA Molecules up to Several Hundred Kilobases. Nat. Methods 6, 343–345. doi:10.1038/nmeth.1318
Gilbert, L. A., Horlbeck, M. A., Adamson, B., Villalta, J. E., Chen, Y., Whitehead, E. H., et al. (2014). Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 159, 647–661. doi:10.1016/j.cell.2014.09.029
Gilbert, L. A., Larson, M. H., Morsut, L., Liu, Z., Brar, G. A., Torres, S. E., et al. (2013). CRISPR-Mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes. Cell 154, 442–451. doi:10.1016/j.cell.2013.06.044
Giles, N. H., Case, M. E., Baum, J., Geever, R., Huiet, L., Patel, V., et al. (1985). Gene Organization and Regulation in the Qa (Quinic Acid) Gene Cluster of Neurospora Crassa. Microbiol. Rev. 49, 338–358. doi:10.1128/mr.49.3.338-358.1985
Giles, N. H., Geever, R. F., Asch, D. K., Avalos, J., and Case, M. E. (1991). Organization and Regulation of the Qa (Quinic Acid) Genes in Neurospora Crassa and Other Fungi. J. Hered. 82, 1–7. doi:10.1093/jhered/82.1.1
Gossen, M., and Bujard, H. (1992). Tight Control of Gene Expression in Mammalian Cells by Tetracycline-Responsive Promoters. Proc. Natl. Acad. Sci. 89, 5547–5551. doi:10.1073/pnas.89.12.5547
Grau, M. F., Entwistle, R., Chiang, Y.-M., Ahuja, M., Oakley, C. E., Akashi, T., et al. (2018). Hybrid Transcription Factor Engineering Activates the Silent Secondary Metabolite Gene Cluster for (+)-Asperlin in Aspergillus nidulans. ACS Chem. Biol. 13, 3193–3205. doi:10.1021/acschembio.8b00679
Gressler, M., Zaehle, C., Scherlach, K., Hertweck, C., and Brock, M. (2011). Multifactorial Induction of an Orphan PKS-NRPS Gene Cluster in Aspergillus terreus. Chem. Biol. 18, 198–209. doi:10.1016/j.chembiol.2010.12.011
Grigoriev, I. V., Nikitin, R., Haridas, S., Kuo, A., Ohm, R., Otillar, R., et al. (2014). MycoCosm Portal: Gearing up for 1000 Fungal Genomes. Nucleic Acids Res. 42, D699–D704. doi:10.1093/nar/gkt1183
Grove, J. F., and Pople, M. (1980). The Insecticidal Activity of Beauvericin and the Enniatin Complex. Mycopathologia 70, 103–105. doi:10.1007/BF00443075
Guzmán-Chávez, F., Salo, O., Nygård, Y., Lankhorst, P. P., Bovenberg, R. A. L., and Driessen, A. J. M. (2017). Mechanism and Regulation of Sorbicillin Biosynthesis by Penicillium chrysogenum. Microb. Biotechnol. 10, 958–968. doi:10.1111/1751-7915.12736
Guzman-Chavez, F., Salo, O., Samol, M., Ries, M., Kuipers, J., Bovenberg, R. A. L., et al. (2018). Deregulation of Secondary Metabolism in a Histone Deacetylase Mutant of Penicillium chrysogenum. Microbiologyopen 7, e00598. doi:10.1002/mbo3.598
Guzmán-Chávez, F., Zwahlen, R. D., Bovenberg, R. A. L., and Driessen, A. J. M. (2018). Engineering of the Filamentous Fungus Penicillium Chrysogenumas Cell Factory for Natural Products. Front. Microbiol. 9. doi:10.3389/fmicb.2018.02768
Hach, A., Hon, T., and Zhang, L. (2000). The Coiled Coil Dimerization Element of the Yeast Transcriptional Activator Hap1, a Gal4 Family Member, Is Dispensable for DNA Binding but Differentially Affects Transcriptional Activation. J. Biol. Chem. 275, 248–254. doi:10.1074/jbc.275.1.248
Hai, Y., and Tang, Y. (2018). Biosynthesis of Long-Chain N-Acyl Amide by a Truncated Polyketide Synthase-Nonribosomal Peptide Synthetase Hybrid Megasynthase in Fungi. J. Am. Chem. Soc. 140, 1271–1274. doi:10.1021/jacs.7b13350
Han, L., Liu, K., Ma, W., Jiang, Y., Hou, S., Tan, Y., et al. (2020). Redesigning Transcription Factor Cre1 for Alleviating Carbon Catabolite Repression in Trichoderma Reesei. Synth. Syst. Biotechnol. 5, 230–235. doi:10.1016/j.synbio.2020.07.002
Han, Y. B., Bai, W., Ding, C. X., Liang, J., Wu, S. H., and Tan, R. X. (2021). Intertwined Biosynthesis of Skyrin and Rugulosin A Underlies the Formation of Cage-Structured Bisanthraquinones. J. Am. Chem. Soc. 143, 14218–14226. doi:10.1021/jacs.1c05421
Harris, D. M., van der Krogt, Z. A., Klaassen, P., Raamsdonk, L. M., Hage, S., van den Berg, M. A., et al. (2009). Exploring and Dissecting Genome-wide Gene Expression Responses of Penicillium chrysogenum to Phenylacetic Acid Consumption and penicillinG Production. BMC Genomics 10, 75. doi:10.1186/1471-2164-10-75
Hawksworth, D. L., and Lücking, R. (2017-2016). Fungal Diversity Revisited: 2.2 to 3.8 Million Species. Microbiol. Spectr. 5. doi:10.1128/microbiolspec.FUNK-005210.1128/microbiolspec.FUNK-0052-2016
Haynes, S. W., Ames, B. D., Gao, X., Tang, Y., and Walsh, C. T. (2011). Unraveling Terminal C-Domain-Mediated Condensation in Fungal Biosynthesis of Imidazoindolone Metabolites. Biochemistry 50, 5668–5679. doi:10.1021/bi2004922
Hernanz-Koers, M., Gandía, M., Garrigues, S., Manzanares, P., Yenush, L., Orzaez, D., et al. (2018). FungalBraid: A GoldenBraid-Based Modular Cloning Platform for the Assembly and Exchange of DNA Elements Tailored to Fungal Synthetic Biology. Fungal Genet. Biol. 116, 51–61. doi:10.1016/j.fgb.2018.04.010
Hilton, I. B., D’Ippolito, A. M., Vockley, C. M., Thakore, P. I., Crawford, G. E., Reddy, T. E., et al. (2015). Epigenome Editing by a CRISPR-Cas9-Based Acetyltransferase Activates Genes from Promoters and Enhancers. Nat. Biotechnol. 33, 510–517. doi:10.1038/nbt.3199
Hoefgen, S., Lin, J., Fricke, J., Stroe, M. C., Mattern, D. J., Kufs, J. E., et al. (2018). Facile Assembly and Fluorescence-Based Screening Method for Heterologous Expression of Biosynthetic Pathways in Fungi. Metab. Eng. 48, 44–51. doi:10.1016/j.ymben.2018.05.014
Houbraken, J., Frisvad, J. C., and Samson, R. A. (2011). Fleming’s Penicillin Producing Strain Is Not Penicillium chrysogenum but P. rubens. IMA Fungus 2, 87–95. doi:10.5598/imafungus.2011.02.01.12
Huang, X., Men, P., Tang, S., and Lu, X. (2021). Aspergillus terreus as an Industrial Filamentous Fungus for Pharmaceutical Biotechnology. Curr. Opin. Biotechnol. 69, 273–280. doi:10.1016/j.copbio.2021.02.004
Huang, X., Tang, S., Zheng, L., Teng, Y., Yang, Y., Zhu, J., et al. (2019). Construction of an Efficient and Robust Aspergillus terreus Cell Factory for Monacolin J Production Monacolin J Production. ACS Synth. Biol. 8. doi:10.1021/acssynbio.8b00489
Iacovelli, R., Bovenberg, R. A. L., and Driessen, A. J. M. (2021a). Nonribosomal Peptide Synthetases and Their Biotechnological Potential in Penicillium rubens. J. Ind. Microbiol. Biotechnol. 48. doi:10.1093/jimb/kuab045
Iacovelli, R., Mózsik, L., Bovenberg, R. A. L., and Driessen, A. J. M. (2021b). Identification of a Conserved N-Terminal Domain in the First Module of ACV Synthetases. Microbiologyopen 10, e1145. doi:10.1002/mbo3.1145
Idnurm, A., Bailey, A. M., Cairns, T. C., Elliott, C. E., Foster, G. D., Ianiri, G., et al. (2017). A Silver Bullet in a Golden Age of Functional Genomics: the Impact of Agrobacterium-Mediated Transformation of Fungi. Fungal Biol. Biotechnol. 4, 6. doi:10.1186/s40694-017-0035-0
Isaac, R. S., Jiang, F., Doudna, J. A., Lim, W. A., Narlikar, G. J., and Almeida, R. (2016). Nucleosome Breathing and Remodeling Constrain CRISPR-Cas9 Function. Elife 5, e13450. doi:10.7554/eLife.13450
Ishidoh, K., Kinoshita, H., Ihara, F., and Nihira, T. (2014). Efficient and Versatile Transformation Systems in Entomopathogenic Fungus Lecanicillium Species. Curr. Genet. 60, 99–108. doi:10.1007/s00294-013-0399-5
Jansen, R., Küsters, K., Morschett, H., Wiechert, W., and Oldiges, M. (2021). A Fully Automated Pipeline for the Dynamic At‐line Morphology Analysis of Microscale Aspergillus Cultivation. Fungal Biol. Biotechnol. 8, 2. doi:10.1186/s40694-021-00109-4
Jarczynska, D., Rendsvig, J. K. H., Pagels, N., Viana, V. R., Nødvig, C. S., Kirchner, F. H., et al. (2020). DIVERSIFY: A Fungal Multispecies Gene Expression Platform. ACS Synth. Biol. 10. doi:10.1021/acssynbio.0c00587
Jin, F. J., Maruyama, J. I., Juvvadi, P. R., Arioka, M., and Kitamoto, K. (2004). Development of a Novel Quadruple Auxotrophic Host Transformation System by argB Gene Disruption Using adeA Gene and Exploiting Adenine Auxotrophy in Aspergillus oryzae. FEMS Microbiol. Lett. 239, 79–85. doi:10.1016/j.femsle.2004.08.025
Juven-Gershon, T., and Kadonaga, J. T. (2010). Regulation of Gene Expression via the Core Promoter and the Basal Transcriptional Machinery. Dev. Biol. 339, 225–229. doi:10.1016/j.ydbio.2009.08.009
Karnaukhova, E., Ophir, Y., Trinh, L., Dalal, N., Punt, P. J., Golding, B., et al. (2007). Expression of Human α1-proteinase Inhibitor in Aspergillus niger. Microb. Cell Fact. 6, 1–10. doi:10.1186/1475-2859-6-34
Kautsar, S. A., Blin, K., Shaw, S., Navarro-Muñoz, J. C., Terlouw, B. R., van der Hooft, J. J. J., et al. (2019). MIBiG 2.0: a Repository for Biosynthetic Gene Clusters of Known Function. Nucleic Acids Res. 48, D454–D458. doi:10.1093/nar/gkz882
Keats, S. E., Woo, B. J., Martin, T., Johannes, G., Stefan, G., and Keller, N. P. (2007). Histone Deacetylase Activity Regulates Chemical Diversity in Aspergillus. Eukaryot. Cell 6, 1656–1664. doi:10.1128/EC.00186-07
Keller, N. P. (2019). Fungal Secondary Metabolism: Regulation, Function and Drug Discovery. Nat. Rev. Microbiol. 17, 167–180. doi:10.1038/s41579-018-0121-1
Keller, N. P., Turner, G., and Bennett, J. W. (2005). Fungal Secondary Metabolism - from Biochemistry to Genomics. Nat. Rev. Microbiol. 3, 937–947. doi:10.1038/nrmicro1286
Khaldi, N., Seifuddin, F. T., Turner, G., Haft, D., Nierman, W. C., Wolfe, K. H., et al. (2010). SMURF: Genomic Mapping of Fungal Secondary Metabolite Clusters. Fungal Genet. Biol. 47, 736–741. doi:10.1016/j.fgb.2010.06.003
Kim, J. H., Feng, Z., Bauer, J. D., Kallifidas, D., Calle, P. Y., and Brady, S. F. (2010). Cloning Large Natural Product Gene Clusters from the Environment: Piecing Environmental DNA Gene Clusters Back Together with TAR. Biopolymers 93, 833–844. doi:10.1002/bip.21450
Kistler, H. C., and Benny, U. (1992). Autonomously Replicating Plasmids and Chromosome Rearrangement during Transformation of Nectria Haematococca. Gene 117, 81–89. doi:10.1016/0378-1119(92)90493-9
Kluge, J., Terfehr, D., and Kück, U. (2018). Inducible Promoters and Functional Genomic Approaches for the Genetic Engineering of Filamentous Fungi. Appl. Microbiol. Biotechnol. 102, 6357–6372. doi:10.1007/s00253-018-9115-1
Knott, G. J., and Doudna, J. A. (2018). CRISPR-cas Guides the Future of Genetic Engineering. Science 361, 866–869. doi:10.1126/science.aat5011
Konermann, S., Brigham, M. D., Trevino, A. E., Joung, J., Abudayyeh, O. O., Barcena, C., et al. (2015). Genome-scale Transcriptional Activation by an Engineered CRISPR-Cas9 Complex. Nature 517, 583–588. doi:10.1038/nature14136
König, C. C., Scherlach, K., Schroeckh, V., Horn, F., Nietzsche, S., Brakhage, A. A., et al. (2013). Bacterium Induces Cryptic Meroterpenoid Pathway in the Pathogenic Fungus Aspergillus fumigatus. ChemBioChem 14, 938–942. doi:10.1002/cbic.201300070
Kremer, A., and Li, S.-M. (2008). Potential of a 7-dimethylallyltryptophan Synthase as a Tool for Production of Prenylated Indole Derivatives. Appl. Microbiol. Biotechnol. 79, 951–961. doi:10.1007/s00253-008-1505-3
Kubodera, T., Yamashita, N., and Nishimura, A. (2002). Transformation of Aspergillus sp. and Trichoderma reesei Using the Pyrithiamine Resistance Gene ( ptrA ) of Aspergillus oryzae. Biosci. Biotechnol. Biochem. 66, 404–406. doi:10.1271/bbb.66.404
Kuivanen, J., Korja, V., Holmström, S., and Richard, P. (2019). Development of microtiter plate scale CRISPR/Cas9 transformation method for Aspergillus niger based on In Vitro assembled ribonucleoprotein complexes. Fungal Biol. Biotechnol. 6, 3. doi:10.1186/s40694-019-0066-9
Kun, R. S., Garrigues, S., Di Falco, M., Tsang, A., and de Vries, R. P. (2021). The chimeric GaaR-XlnR transcription factor induces pectinolytic activities in the presence of D-xylose in Aspergillus niger. Appl. Microbiol. Biotechnol. 105, 5553–5564. doi:10.1007/s00253-021-11428-2
Lauren, W., Jehoshua, S., Deeva, U., Yannic, P., Alejandro, C., Shapiro, S. R., et al. (2022). A CRISPR Interference Platform for Efficient Genetic Repression in Candida albicans. mSphere 4, e00002–19. doi:10.1128/mSphere.00002-19
Lazarus, C. M., Williams, K., and Bailey, A. M. (2014). Reconstructing fungal natural product biosynthetic pathways. Nat. Prod. Rep. 31, 1339–1347. doi:10.1039/c4np00084f
Lee, M. E., DeLoache, W. C., Cervantes, B., and Dueber, J. E. (2015). A Highly Characterized Yeast Toolkit for Modular, Multipart Assembly. ACS Synth. Biol. 4, 975–986. doi:10.1021/sb500366v
Li, L., Yu, P., Tang, M. C., Zou, Y., Gao, S. S., Hung, Y. S., et al. (2016). Biochemical Characterization of a Eukaryotic Decalin-Forming Diels-Alderase. J. Am. Chem. Soc. 138, 15837–15840. doi:10.1021/jacs.6b10452
Li, X., Huang, L., Pan, L., Wang, B., and Pan, L. (2021). CRISPR/dCas9-mediated epigenetic modification reveals differential regulation of histone acetylation on Aspergillus niger secondary metabolite. Microbiol. Res. 245, 126694. doi:10.1016/j.micres.2020.126694
Lim, F. Y., Sanchez, J. F., Wang, C. C. C., and Keller, N. P. (2012). “Toward awakening cryptic secondary metabolite gene clusters in filamentous fungi,” in Methods in Enzymology. doi:10.1016/B978-0-12-404634-4.00015-2
Liu, C., Minami, A., Ozaki, T., Wu, J., Kawagishi, H., Maruyama, J., et al. (2019a). Efficient Reconstitution of Basidiomycota Diterpene Erinacine Gene Cluster in Ascomycota Host Aspergillus oryzae Based on Genomic DNA Sequences. J. Am. Chem. Soc. 141, 15519–15523. doi:10.1021/jacs.9b08935
Liu, L., Tang, M. C., and Tang, Y. (2019b). Fungal highly reducing polyketide synthases biosynthesize salicylaldehydes that are precursors to epoxycyclohexenol natural products. J. Am. Chem. Soc. 141, 19538–19541. doi:10.1021/jacs.9b09669
Liu, N., Hung, Y. S., Gao, S. S., Hang, L., Zou, Y., Chooi, Y. H., et al. (2017). Identification and Heterologous Production of a Benzoyl-Primed Tricarboxylic Acid Polyketide Intermediate from the Zaragozic Acid A Biosynthetic Pathway. Org. Lett. 19, 3560–3563. doi:10.1021/acs.orglett.7b01534
Lyu, H.-N., Liu, H.-W., Keller, N. P., and Yin, W.-B. (2020). Harnessing diverse transcriptional regulators for natural product discovery in fungi. Nat. Prod. Rep. 37, 6–16. doi:10.1039/C8NP00027A
Ma, J., and Ptashne, M. (1987). Deletion analysis of GAL4 defines two transcriptional activating segments. Cell 48, 847–853. doi:10.1016/0092-8674(87)90081-X
Ma, K., Zhang, Y., Guo, C., Yang, Y., Han, J., Yu, B., et al. (2021). Reconstitution of biosynthetic pathway for mushroom-derived cyathane diterpenes in yeast and generation of new “non-natural” analogues. Acta Pharm. Sin. B 11, 2945–2956. doi:10.1016/j.apsb.2021.04.014
Macheleidt, J., Mattern, D. J., Fischer, J., Netzker, T., Weber, J., Schroeckh, V., et al. (2016). Regulation and Role of Fungal Secondary Metabolites. Annu. Rev. Genet. 50, 371–392. doi:10.1146/annurev-genet-120215-035203
Marmorstein, R., and Harrison, S. C. (1994). Crystal structure of a PPR1-DNA complex: DNA recognition by proteins containing a Zn2Cys6 binuclear cluster. Genes Dev. 8, 2504–2512. doi:10.1101/gad.8.20.2504
Martella, A., Matjusaitis, M., Auxillos, J., Pollard, S. M., and Cai, Y. (2017). EMMA: An Extensible Mammalian Modular Assembly Toolkit for the Rapid Design and Production of Diverse Expression Vectors. ACS Synth. Biol. 6, 1380–1392. doi:10.1021/acssynbio.7b00016
Maruyama, J. I., and Kitamoto, K. (2008). Multiple gene disruptions by marker recycling with highly efficient gene-targeting background (ΔligD) in Aspergillus oryzae. Biotechnol. Lett. 30, 1811–1817. doi:10.1007/s10529-008-9763-9
McCarty, N. S., Graham, A. E., Studená, L., and Ledesma-Amaro, R. (2020). Multiplexed CRISPR technologies for gene editing and transcriptional regulation. Nat. Commun. 11, 1281. doi:10.1038/s41467-020-15053-x
McLean, K. J., Hans, M., Meijrink, B., van Scheppingen, W. B., Vollebregt, A., Tee, K. L., et al. (2015). Single-step fermentative production of the cholesterol-lowering drug pravastatin via reprogramming of Penicillium chrysogenum. Proc. Natl. Acad. Sci. 112, 2847–2852. doi:10.1073/pnas.1419028112
Meng, X., Fang, Y., Ding, M., Zhang, Y., Jia, K., Li, Z., et al. (202110786). Developing fungal heterologous expression platforms to explore and improve the production of natural products from fungal biodiversity. Biotechnol. Adv. doi:10.1016/j.biotechadv.2021.107866
Meyer, V., Basenko, E. Y., Benz, J. P., Braus, G. H., Caddick, M. X., Csukai, M., et al. (2020). Growing a circular economy with fungal biotechnology: a white paper. Fungal Biol. Biotechnol. 7, 5. doi:10.1186/s40694-020-00095-z
Meyer, V. (2021). “Metabolic Engineering of Filamentous Fungi,” in Metabolic Engineering (Wiley), 765–801. doi:10.1002/9783527823468.ch20
Meyer, V., Wanka, F., van Gent, J., Arentshorst, M., van den Hondel, C. A. M. J. J., and Ram, A. F. J. (2011). Fungal Gene Expression on Demand: an Inducible, Tunable, and Metabolism-Independent Expression System for Aspergillus niger. Appl. Environ. Microbiol. 77, 2975–2983. doi:10.1128/AEM.02740-10
Michael, H. (1998). Molecular Genetics of the RNA Polymerase II General Transcriptional Machinery. Microbiol. Mol. Biol. Rev. 62, 465–503. doi:10.1128/MMBR.62.2.465-503.1998
Miyanaga, A., Kudo, F., and Eguchi, T. (2018). Protein–protein interactions in polyketide synthase–nonribosomal peptide synthetase hybrid assembly lines. Nat. Prod. Rep. 35, 1185–1209. doi:10.1039/C8NP00022K
Moore, S. J., Lai, H.-E., Kelwick, R. J. R., Chee, S. M., Bell, D. J., Polizzi, K. M., et al. (2016). EcoFlex: A Multifunctional MoClo Kit for E. coli Synthetic Biology. ACS Synth. Biol. 5, 1059–1069. doi:10.1021/acssynbio.6b00031
Morris, N. R. (1976). Nucleosome structure in Aspergillus nidulans. Cell 8, 357–363. doi:10.1016/0092-8674(76)90147-1
Mosunova, O., Navarro-Muñoz, J. C., and Collemare, J. (2020). “The Biosynthesis of Fungal Secondary Metabolites: From Fundamentals to Biotechnological Applications,” in Reference Module in Life Sciences (Elsevier), 1–19. doi:10.1016/B978-0-12-809633-8.21072-8
Mózsik, L., Büttel, Z., Bovenberg, R. A. L., Driessen, A. J. M., and Nygård, Y. (2019). Synthetic control devices for gene regulation in Penicillium chrysogenum. Microb. Cell Fact. 18. doi:10.1186/s12934-019-1253-3
Mózsik, L., Hoekzema, M., de Kok, N. A. W., Bovenberg, R. A. L., Nygård, Y., and Driessen, A. J. M. (2021a). CRISPR-based transcriptional activation tool for silent genes in filamentous fungi. Sci. Rep. 11, 1118. doi:10.1038/s41598-020-80864-3
Mózsik, L., Pohl, C., Meyer, V., Bovenberg, R. A. L., Nygård, Y., and Driessen, A. J. M. (2021b). Modular Synthetic Biology Toolkit for Filamentous Fungi. ACS Synth. Biol. doi:10.1021/acssynbio.1c00260
Navarro-Muñoz, J. C., Selem-Mojica, N., Mullowney, M. W., Kautsar, S. A., Tryon, J. H., Parkinson, E. I., et al. (2020). A computational framework to explore large-scale biosynthetic diversity. Nat. Chem. Biol. 16, 60–68. doi:10.1038/s41589-019-0400-9
Nayak, T., Szewczyk, E., Oakley, C. E., Osmani, A., Ukil, L., Murray, S. L., et al. (2006). A Versatile and Efficient Gene-Targeting System for Aspergillus nidulans. Genetics 172, 1557–1566. doi:10.1534/genetics.105.052563
Netzker, T., Flak, M., Krespach, M. K. C., Stroe, M. C., Weber, J., Schroeckh, V., et al. (2018). Microbial interactions trigger the production of antibiotics. Curr. Opin. Microbiol. 45, 117–123. doi:10.1016/j.mib.2018.04.002
Nielsen, J. C., Grijseels, S., Prigent, S., Ji, B., Dainat, J., Nielsen, K. F., et al. (2017). Global analysis of biosynthetic gene clusters reveals vast potential of secondary metabolite production in Penicillium species. Nat. Microbiol. 2. doi:10.1038/nmicrobiol.2017.44
Nielsen, M. T., Nielsen, J. B., Anyaogu, D. C., Holm, D. K., Nielsen, K. F., Larsen, T. O., et al. (2013). Heterologous Reconstitution of the Intact Geodin Gene Cluster in Aspergillus nidulans through a Simple and Versatile PCR Based Approach. PLoS One 8, e72871. doi:10.1371/journal.pone.0072871
Niu, J., Alazi, E., Reid, I. D., Arentshorst, M., Punt, P. J., Visser, J., et al. (2017). An Evolutionarily Conserved Transcriptional Activator-Repressor Module Controls Expression of Genes for D-Galacturonic Acid Utilization in Aspergillus niger. Genetics 205, 169–183. doi:10.1534/genetics.116.194050
Nødvig, C. S., Hoof, J. B., Kogle, M. E., Jarczynska, Z. D., Lehmbeck, J., Klitgaard, D. K., et al. (2018). Efficient oligo nucleotide mediated CRISPR-Cas9 gene editing in Aspergilli. Fungal Genet. Biol. 115, 78–89. doi:10.1016/j.fgb.2018.01.004
Nødvig, C. S., Nielsen, J. B., Kogle, M. E., and Mortensen, U. H. (2015). A CRISPR-Cas9 system for genetic engineering of filamentous fungi. PLoS One 10. doi:10.1371/journal.pone.0133085
Nofiani, R., de Mattos-Shipley, K., Lebe, K. E., Han, L.-C., Iqbal, Z., Bailey, A. M., et al. (2018). Strobilurin biosynthesis in Basidiomycete fungi. Nat. Commun. 9, 3940. doi:10.1038/s41467-018-06202-4
Oakley, C. E., Ahuja, M., Sun, W.-W., Entwistle, R., Akashi, T., Yaegashi, J., et al. (2017). Discovery of McrA, a master regulator of Aspergillus secondary metabolism. Mol. Microbiol. 103, 347–365. doi:10.1111/mmi.13562
Obst, U., Lu, T. K., and Sieber, V. (2017). A Modular Toolkit for Generating Pichia pastoris Secretion Libraries. ACS Synth. Biol. 6, 1016–1025. doi:10.1021/acssynbio.6b00337
Oh, D.-C., Kauffman, C. A., Jensen, P. R., and Fenical, W. (2007). Induced Production of Emericellamides A and B from the Marine-Derived Fungus Emericella sp. in Competing Co-culture. J. Nat. Prod. 70, 515–520. doi:10.1021/np060381f
Orth, P., Schnappinger, D., Hillen, W., Saenger, W., and Hinrichs, W. (2000). Structural basis of gene regulation by the tetracycline inducible Tet repressor–operator system. Nat. Struct. Biol. 7, 215–219. doi:10.1038/73324
Osmani, S. A., and Mirabito, P. M. (2004). The early impact of genetics on our understanding of cell cycle regulation in Aspergillus nidulans. Fungal Genet. Biol. 41, 401–410. doi:10.1016/j.fgb.2003.11.009
Pahirulzaman, K. A. K., Williams, K., and Lazarus, C. M. (2012). A Toolkit for Heterologous Expression of Metabolic Pathways in Aspergillus Oryzae. 1st ed. Elsevier. doi:10.1016/B978-0-12-404634-4.00012-7
Payne, G. A., Nierman, W. C., Wortman, J. R., Pritchard, B. L., Brown, D., Dean, R. A., et al. (2006). Whole genome comparison of Aspergillus flavus and A. oryzae. Med. Mycol. 44, 9–11. doi:10.1080/13693780600835716
Perez-Pinera, P., Kocak, D. D., Vockley, C. M., Adler, A. F., Kabadi, A. M., Polstein, L. R., et al. (2013). RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat. Methods 10, 973–976. doi:10.1038/nmeth.2600
Pfannenstiel, B. T., and Keller, N. P. (2019). On top of biosynthetic gene clusters: How epigenetic machinery influences secondary metabolism in fungi. Biotechnol. Adv. 37, 107345. doi:10.1016/j.biotechadv.2019.02.001
Pickar-Oliver, A., and Gersbach, C. A. (2019). The next generation of CRISPR–Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 20, 490–507. doi:10.1038/s41580-019-0131-5
Pöggeler, S., and Kück, U. (2006). Highly efficient generation of signal transduction knockout mutants using a fungal strain deficient in the mammalian ku70 ortholog. Gene 378, 1–10. doi:10.1016/j.gene.2006.03.020
Pohl, C., Kiel, J. A. K. W., Driessen, A. J. M., Bovenberg, R. A. L., and Nygård, Y. (2016). CRISPR/Cas9 Based Genome Editing of Penicillium chrysogenum. ACS Synth. Biol. 5, 754–764. doi:10.1021/acssynbio.6b00082
Pohl, C., Polli, F., Schütze, T., Viggiano, A., Mózsik, L., Jung, S., et al. (2020). A Penicillium rubens platform strain for secondary metabolite production. Sci. Rep. 10, 7630. doi:10.1038/s41598-020-64893-6
Potter, C. J., Tasic, B., Russler, E. V., Liang, L., and Luo, L. (2010). The Q System: A Repressible Binary System for Transgene Expression, Lineage Tracing, and Mosaic Analysis. Cell 141, 536–548. doi:10.1016/j.cell.2010.02.025
Powell, W. A., and Kistler, H. C. (1990). In Vivo rearrangement of foreign DNA by Fusarium oxysporum produces linear self-replicating plasmids. J. Bacteriol. 172, 3163–3171. doi:10.1128/jb.172.6.3163-3171.1990
Price, M. S., Yu, J., Nierman, W. C., Kim, H. S., Pritchard, B., Jacobus, C. A., et al. (2006). The aflatoxin pathway regulator AflR induces gene transcription inside and outside of the aflatoxin biosynthetic cluster. FEMS Microbiol. Lett. 255, 275–279. doi:10.1111/j.1574-6968.2005.00084.x
Qi, L. S., Larson, M. H., Gilbert, L. A., Doudna, J. A., Weissman, J. S., Arkin, A. P., et al. (2013). Repurposing CRISPR as an RNA-Guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183. doi:10.1016/j.cell.2013.02.022
Ramalingam, S., Kandavelou, K., Rajenderan, R., and Chandrasegaran, S. (2011). Creating Designed Zinc-Finger Nucleases with Minimal Cytotoxicity. J. Mol. Biol. 405, 630–641. doi:10.1016/j.jmb.2010.10.043
Rantasalo, A., Landowski, C. P., Kuivanen, J., Korppoo, A., Reuter, L., Koivistoinen, O., et al. (2018). A universal gene expression system for fungi. Nucleic Acids Res. 46, e111. doi:10.1093/nar/gky558
Riabinina, O., Task, D., Marr, E., Lin, C.-C., Alford, R., O’Brochta, D. A., et al. (2016). Organization of olfactory centres in the malaria mosquito Anopheles gambiae. Nat. Commun. 7, 13010. doi:10.1038/ncomms13010
Richter, L., Wanka, F., Boecker, S., Storm, D., Kurt, T., Vural, Ö., et al. (2014). Engineering of Aspergillus niger for the production of secondary metabolites. Fungal Biol. Biotechnol. 1, 1–13. doi:10.1186/s40694-014-0004-9
Robbins, T., Liu, Y.-C., Cane, D. E., and Khosla, C. (2016). Structure and mechanism of assembly line polyketide synthases. Curr. Opin. Struct. Biol. 41, 10–18. doi:10.1016/j.sbi.2016.05.009
Robey, M. T., Caesar, L. K., Drott, M. T., Keller, N. P., and Kelleher, N. L. (2021). An interpreted atlas of biosynthetic gene clusters from 1,000 fungal genomes. Proc. Natl. Acad. Sci. U. S. A. 118, e2020230118. doi:10.1073/pnas.2020230118
Rock, J. M., Hopkins, F. F., Chavez, A., Diallo, M., Chase, M. R., Gerrick, E. R., et al. (2017). Programmable transcriptional repression in mycobacteria using an orthogonal CRISPR interference platform. Nat. Microbiol. 2, 16274. doi:10.1038/nmicrobiol.2016.274
Rodolphe, B., Christophe, F., Hélène, D., Melissa, R., Patrick, B., Sylvain, M., et al. (2007). CRISPR Provides Acquired Resistance Against Viruses in Prokaryotes. Sci. (80-. ) 315, 1709–1712. doi:10.1126/science.1138140
Román, E., Coman, I., Prieto, D., Alonso-Monge, R., and Pla, J. (2019). Implementation of a CRISPR-Based System for Gene Regulation in Candida albicans. mSphere 4. doi:10.1128/msphere.00001-19
Roux, I., Woodcraft, C., Hu, J., Wolters, R., Gilchrist, C. L. M., and Chooi, Y.-H. (2020). CRISPR-Mediated Activation of Biosynthetic Gene Clusters for Bioactive Molecule Discovery in Filamentous Fungi. ACS Synth. Biol. doi:10.1021/acssynbio.0c00197
Sadowski, I., Ma, J., Triezenberg, S., and Ptashne, M. (1988). GAL4-VP16 is an unusually potent transcriptional activator. Nature 335, 563–564. doi:10.1038/335563a0
Salazar-Cerezo, S., Kun, R. S., de Vries, R. P., and Garrigues, S. (2020). CRISPR/Cas9 technology enables the development of the filamentous ascomycete fungus Penicillium subrubescens as a new industrial enzyme producer. Enzyme Microb. Technol. 133, 109463. doi:10.1016/j.enzmictec.2019.109463
Sarkari, P., Marx, H., Blumhoff, M. L., Mattanovich, D., Sauer, M., and Steiger, M. G. (2017). An efficient tool for metabolic pathway construction and gene integration for Aspergillus niger. Bioresour. Technol. 245, 1327–1333. doi:10.1016/j.biortech.2017.05.004
Sarrion-Perdigones, A., Falconi, E. E., Zandalinas, S. I., Juárez, P., Fernández-del-Carmen, A., Granell, A., et al. (2011). GoldenBraid: An Iterative Cloning System for Standardized Assembly of Reusable Genetic Modules. PLoS One 6, e21622. doi:10.1371/journal.pone.0021622
Sato’o, Y., Hisatsune, J., Yu, L., Sakuma, T., Yamamoto, T., and Sugai, M. (2018). Tailor-made gene silencing of Staphylococcus aureus clinical isolates by CRISPR interference. PLoS One 13, e0185987. doi:10.1371/journal.pone.0185987
Schroeckh, V., Scherlach, K., Nützmann, H.-W., Shelest, E., Schmidt-Heck, W., Schuemann, J., et al. (2009). Intimate bacterial–fungal interaction triggers biosynthesis of archetypal polyketides in Aspergillus nidulans. Proc. Natl. Acad. Sci. 106, 14558–14563. doi:10.1073/pnas.0901870106
Schüller, A., Wolansky, L., Berger, H., Studt, L., Gacek-Matthews, A., Sulyok, M., et al. (2020). A novel fungal gene regulation system based on inducible VPR-dCas9 and nucleosome map-guided sgRNA positioning. Appl. Microbiol. Biotechnol. 104, 9801–9822. doi:10.1007/s00253-020-10900-9
Schuster, M., Schweizer, G., Reissmann, S., and Kahmann, R. (2016). Genome editing in Ustilago maydis using the CRISPR–Cas system. Fungal Genet. Biol. 89, 3–9. doi:10.1016/j.fgb.2015.09.001
Sebastian, B., Funk, N. A., Kirstin, S., Volker, S., Ekaterina, S., Uwe, H., et al. (2010). Activation of a Silent Fungal Polyketide Biosynthesis Pathway through Regulatory Cross Talk with a Cryptic Nonribosomal Peptide Synthetase Gene Cluster. Appl. Environ. Microbiol. 76, 8143–8149. doi:10.1128/AEM.00683-10
Seekles, S. J., Teunisse, P. P. P., Punt, M., van den Brule, T., Dijksterhuis, J., Houbraken, J., et al. (2021). Preservation stress resistance of melanin deficient conidia from Paecilomyces variotii and Penicillium roqueforti mutants generated via CRISPR/Cas9 genome editing. Fungal Biol. Biotechnol. 8, 4. doi:10.1186/s40694-021-00111-w
Sharma, P., Yan, F., Doronina, V. A., Escuin-Ordinas, H., Ryan, M. D., and Brown, J. D. (2012). 2A peptides provide distinct solutions to driving stop-carry on translational recoding. Nucleic Acids Res. 40, 3143–3151. doi:10.1093/nar/gkr1176
Shin, C. G., An, D. G., Song, H. H., and Lee, C. (2009). Beauvericin and enniatins H, i and MK1688 are new potent inhibitors of human immunodeficiency virus type-1 integrase. J. Antibiot. (Tokyo) 62, 687–690. doi:10.1038/ja.2009.102
Smith, J. D., Suresh, S., Schlecht, U., Wu, M., Wagih, O., Peltz, G., et al. (2016). Quantitative CRISPR interference screens in yeast identify chemical-genetic interactions and new rules for guide RNA design. Genome Biol. 17, 45. doi:10.1186/s13059-016-0900-9
Snoek, I. S. I., van der Krogt, Z. A., Touw, H., Kerkman, R., Pronk, J. T., Bovenberg, R. A. L., et al. (2009). Construction of an hdfA Penicillium chrysogenum strain impaired in non-homologous end-joining and analysis of its potential for functional analysis studies. Fungal Genet. Biol. 46, 418–426. doi:10.1016/j.fgb.2009.02.008
Song, L., Ouedraogo, J.-P., Kolbusz, M., Nguyen, T. T. M., and Tsang, A. (2018). Efficient genome editing using tRNA promoter-driven CRISPR/Cas9 gRNA in Aspergillus niger. PLoS One 13, e0202868. doi:10.1371/journal.pone.0202868
Song, R., Zhai, Q., Sun, L., Huang, E., Zhang, Y., Zhu, Y., et al. (2019). CRISPR/Cas9 genome editing technology in filamentous fungi: progress and perspective. Appl. Microbiol. Biotechnol. 103, 6919–6932. doi:10.1007/s00253-019-10007-w
Spiteller, P. (2015). Chemical ecology of fungi. Nat. Prod. Rep. 32, 971–993. doi:10.1039/C4NP00166D
Steiger, M. G., Vitikainen, M., Uskonen, P., Brunner, K., Adam, G., Pakula, T., et al. (2011). Transformation System for Hypocrea jecorina ( Trichoderma reesei ) That Favors Homologous Integration and Employs Reusable Bidirectionally Selectable Markers. Appl. Environ. Microbiol. 77, 114–121. doi:10.1128/AEM.02100-10
Strezoska, Ž., Dickerson, S. M., Maksimova, E., Chou, E., Gross, M. M., Hemphill, K., et al. (2020). CRISPR-mediated transcriptional activation with synthetic guide RNA. J. Biotechnol. 319, 25–35. doi:10.1016/j.jbiotec.2020.05.005
Su, X., Schmitz, G., Zhang, M., Mackie, R. I., and Cann, I. K. O. (2012). “Heterologous Gene Expression in Filamentous Fungi,” in Advances in Applied Microbiology (Elsevier), 1–61. doi:10.1016/B978-0-12-394382-8.00001-0
Süssmuth, R. D., and Mainz, A. (2017). Nonribosomal Peptide Synthesis—Principles and Prospects. Angew. Chem. Int. Ed. 56, 3770–3821. doi:10.1002/anie.201609079
Tagami, K., Liu, C., Minami, A., Noike, M., Isaka, T., Fueki, S., et al. (2013). Reconstitution of biosynthetic machinery for indole-diterpene paxilline in Aspergillus oryzae. J. Am. Chem. Soc. 135, 1260–1263. doi:10.1021/ja3116636
Takahasi, T., Masuda, T., and Koyama, Y. (2006). Identification and Analysis of Ku70 and Ku80 Homologs in the koji Molds Aspergillus sojae and Aspergillus oryzae. Biosci. Biotechnol. Biochem. 70, 135–143. doi:10.1271/bbb.70.135
Tan, S. Z., Reisch, C. R., Prather, K. L. J., and O’Toole, G. (2022). A Robust CRISPR Interference Gene Repression System in Pseudomonas. J. Bacteriol. 200, e00575–17. doi:10.1128/JB.00575-17
Umemura, M., Koike, H., Nagano, N., Ishii, T., Kawano, J., Yamane, N., et al. (2014). MIDDAS-M: Motif-Independent De Novo Detection of Secondary Metabolite Gene Clusters through the Integration of Genome Sequencing and Transcriptome Data. PLoS One 8, e84028. doi:10.1371/journal.pone.0084028
Unkles, S. E., Valiante, V., Mattern, D. J., and Brakhage, A. A. (2014). Synthetic Biology Tools for Bioprospecting of Natural Products in Eukaryotes. Chem. Biol. 21, 502–508. doi:10.1016/j.chembiol.2014.02.010
Urlinger, S., Baron, U., Thellmann, M., Hasan, M. T., Bujard, H., and Hillen, W. (2000). Exploring the sequence space for tetracycline-dependent transcriptional activators: Novel mutations yield expanded range and sensitivity. Proc. Natl. Acad. Sci. 97, 7963–7968. doi:10.1073/pnas.130192197
Vanegas, K. G., Jarczynska, Z. D., Strucko, T., and Mortensen, U. H. (2019). Cpf1 enables fast and efficient genome editing in Aspergilli. Fungal Biol. Biotechnol. 6, 1–10. doi:10.1186/s40694-019-0069-6
Verdoes, J. C., Punt, P. J., Burlingame, R., Bartels, J., Dijk, R., Slump, E., et al. (2007). ORIGINAL RESEARCH: A dedicated vector for efficient library construction and high throughput screening in the hyphal fungus Chrysosporium lucknowense. Ind. Biotechnol. 3, 48–57. doi:10.1089/ind.2007.3.048
Vogt, K., Bhabhra, R., Rhodes, J. C., and Askew, D. S. (2005). Doxycycline-regulated gene expression in the opportunistic fungal pathogen Aspergillus fumigatus. BMC Microbiol. 5, 1. doi:10.1186/1471-2180-5-1
Wang, H., La Russa, M., and Qi, L. S. (2016). CRISPR/Cas9 in Genome Editing and Beyond. Annu. Rev. Biochem. 85, 227–264. doi:10.1146/annurev-biochem-060815-014607
Wanka, F., Cairns, T., Boecker, S., Berens, C., Happel, A., Zheng, X., et al. (2016). Tet-on, or Tet-off, that is the question: Advanced conditional gene expression in Aspergillus. Fungal Genet. Biol. 89, 72–83. doi:10.1016/j.fgb.2015.11.003
Weber, E., Engler, C., Gruetzner, R., Werner, S., and Marillonnet, S. (2011). A Modular Cloning System for Standardized Assembly of Multigene Constructs. PLoS One 6, e16765. doi:10.1371/journal.pone.0016765
Wolf, T., Shelest, V., Nath, N., and Shelest, E. (2016). CASSIS and SMIPS: promoter-based prediction of secondary metabolite gene clusters in eukaryotic genomes. Bioinformatics 32, 1138–1143. doi:10.1093/bioinformatics/btv713
Yamada, R., Yoshie, T., Wakai, S., Asai-Nakashima, N., Okazaki, F., Ogino, C., et al. (2014). Aspergillus oryzae-based cell factory for direct kojic acid production from cellulose. Microb. Cell Fact. 13, 1–8. doi:10.1186/1475-2859-13-71
Yamano, T., Zetsche, B., Ishitani, R., Zhang, F., Nishimasu, H., and Nureki, O. (2017). Structural Basis for the Canonical and Non-canonical PAM Recognition by CRISPR-Cpf1. Mol. Cell 67, 633–645. e3. doi:10.1016/j.molcel.2017.06.035
Yamashita, M., Tsujikami, M., Murata, S., Kobayashi, T., Shimizu, M., and Kato, M. (2021). Artificial AmyR::XlnR transcription factor induces α-amylase production in response to non-edible xylan-containing hemicellulosic biomass. Enzyme Microb. Technol. 145, 109762. doi:10.1016/j.enzmictec.2021.109762
Yee, D. A., Kakule, T. B., Cheng, W., Chen, M., Chong, C. T. Y., Hai, Y., et al. (2020). Genome Mining of Alkaloidal Terpenoids from a Hybrid Terpene and Nonribosomal Peptide Biosynthetic Pathway. J. Am. Chem. Soc. 142, 710–714. doi:10.1021/jacs.9b13046
Zabala, A. O., Xu, W., Chooi, Y.-H., and Tang, Y. (2012). Characterization of a Silent Azaphilone Gene Cluster from Aspergillus niger ATCC 1015 Reveals a Hydroxylation-Mediated Pyran-Ring Formation. Chem. Biol. 19, 1049–1059. doi:10.1016/j.chembiol.2012.07.004
Zarnack, K., Maurer, S., Kaffarnik, F., Ladendorf, O., Brachmann, A., Kämper, J., et al. (2006). Tetracycline-regulated gene expression in the pathogen Ustilago maydis. Fungal Genet. Biol. 43, 727–738. doi:10.1016/j.fgb.2006.05.006
Zhang, J. J., Tang, X., and Moore, B. S. (2019a). Genetic platforms for heterologous expression of microbial natural products. Nat. Prod. Rep. 36, 1313–1332. doi:10.1039/c9np00025a
Zhang, J., Zhang, G., Wang, W., Wang, W., and Wei, D. (2018). Enhanced cellulase production in Trichoderma reesei RUT C30 via constitution of minimal transcriptional activators. Microb. Cell Fact. 17, 75. doi:10.1186/s12934-018-0926-7
Zhang, X., Li, Y., Zhao, X., and Bai, F. (2017). Constitutive cellulase production from glucose using the recombinant Trichoderma reesei strain overexpressing an artificial transcription activator. Bioresour. Technol. 223, 317–322. doi:10.1016/j.biortech.2016.10.083
Zhang, Y., Wang, J., Wang, Z., Zhang, Y., Shi, S., Nielsen, J., et al. (2019b). A gRNA-tRNA array for CRISPR-Cas9 based rapid multiplexed genome editing in Saccharomyces cerevisiae. Nat. Commun. 10, 1–10. doi:10.1038/s41467-019-09005-3
Keywords: secondary metabilites, biosynthetic gene cluster, synthetic biology, synthetic transcriptional regulators, fungal platform strains
Citation: Mózsik L, Iacovelli R, Bovenberg RAL and Driessen AJM (2022) Transcriptional Activation of Biosynthetic Gene Clusters in Filamentous Fungi. Front. Bioeng. Biotechnol. 10:901037. doi: 10.3389/fbioe.2022.901037
Received: 21 March 2022; Accepted: 17 June 2022;
Published: 15 July 2022.
Edited by:
A. Pedro Gonçalves, National Cheng Kung University, TaiwanReviewed by:
Mario Andrea Marchisio, Tianjin University, ChinaKai Blin, Technical University of Denmark, Denmark
Andy Bailey, University of Bristol, United Kingdom
Wan Abd Al Qadr Imad Wan Mohtar, University of Malaya, Malaysia
Copyright © 2022 Mózsik, Iacovelli, Bovenberg and Driessen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Arnold J. M. Driessen, a.j.m.driessen@rug.nl
†Present address: László Mózsik, Department Molecular Microbiology and Biotechnology, Institute of Biology, Leiden University, Leiden, Netherlands; Riccardo Iacovelli, Department of Chemical and Pharmaceutical Biology, Groningen Research Institute of Pharmacy, University of Groningen, Groningen, Netherlands