
A c.1775C > T Point Mutation of Sodium Channel Alfa Subunit Gene (SCN4A) in a Three-Generation Sardinian Family with Sodium Channel Myotonia
Abstract
Background:
The nondystrophic myotonias are rare muscle hyperexcitability disorders caused by gain-of-function mutations in the SCN4A gene or loss-of-function mutations in the CLCN1 gene. Clinically, they are characterized by myotonia, defined as delayed muscle relaxation after voluntary contraction, which leads to symptoms of muscle stiffness, pain, fatigue, and weakness. Diagnosis is based on history and examination findings, the presence of electrical myotonia on electromyography, and genetic confirmation.
Methods:
Next-generation sequencing including the CLCN1 and SCN4A genes was performed in patients with clinical neuromuscular disorders. Electromyography, Short Exercise Test, in vivo and in vitro electrophysiology, site-directed mutagenesis and heterologous expression were collected.
Results:
A heterozygous point mutation (c.1775C > T, p.Thr592Ile) of muscle voltage-gated sodium channel α subunit gene (SCN4A) has been identified in five female patients over three generations, in a family with non-dystrophic myotonia. The muscle stiffness and myotonia involve mainly the face and hands, but also affect walking and running, appearing early after birth and presenting a clear cold sensitivity. Very hot temperatures, menstruation and pregnancy also exacerbate the symptoms; muscle pain and a warm-up phenomenon are variable features. Neither paralytic attacks nor post-exercise weakness has been reported. Muscle hypertrophy with cramp-like pain and increased stiffness developed during pregnancy. The symptoms were controlled with both mexiletine and acetazolamide. The Short Exercise Test after muscle cooling revealed two different patterns, with moderate absolute changes of compound muscle action potential amplitude.
Conclusions:
The p.Thr592Ile mutation in the SCN4A gene identified in this Sardinian family was responsible of clinical phenotype of myotonia.
INTRODUCTION
Autosomal dominant point mutations in the gene coding for the α subunit of the muscle voltage-gated sodium channel (SCN4A gene) cause the phenotypes of paramyotonia congenita (PMC, MIM #168300), periodic paralysis (HYPP, MIM #170500; HOKPP2, MIM #613345), and sodium channel myotonias (SCM: PAM - potassium-aggravated myotonia, myotonia fluctuans and myotonia permanens, MIM #608390) [1, 2]. The main symptoms of these channelopathies, i.e. muscle stiffness and myotonia, episodic weakness and cold sensitivity, may variably combine to result in one of the three phenotypes; however, there are several reports of mutations causing mild, incomplete, or overlapping phenotypes, as well as unusual heat sensitivity or warm-up phenomenon, a feature typical of chloride channel (CLCN1 gene) myotonia [3–10] but increasingly seen in sodium channel myotonia. The electrophysiological behavior of mutated channels has been analyzed in detail for a number of mutations [11–15], disclosing impaired inactivation and/or enhanced activation as the main gating mechanisms causing the channel overactivity that determines hyperexcitability and muscle stiffness. The specific biophysical defect may also constitute a basis for a more selective therapeutic approach [16–18].
This study reports the clinical and electrophysiological phenotype of a point mutation in SCN4A gene (c.1775C > T, p.Thr592Ile) identified in five affected individuals of a three-generation family. In vitro functional characterization of the mutated channel was performed to confirm the pathogenicity of the mutation.
MATERIALS AND METHODS
Genetics
The genomic DNA of the proband was analyzed using a custom panel next generation sequencing (NGS) including CLCN1 (chromosome 7q34) and SCN4A genes (chromosome 17q23.3) as previously described [19, 20]. DNA libraries were prepared from 50 ng of genomic DNA by the HaloPlex Target Enrichment System (Agilent Technologies, Santa Clara, CA, USA), following the manufacturer’s instructions and were sequenced on the MiSeq Illumina (Illumina, San Diego, CA, USA). Validation of the variants identified by NGS in the proband and the segregation in available family members were performed by Sanger sequencing using the BigDye Terminator v3.1 Cycle Sequencing Kit on an Applied Biosystems 3130xl Genetic Analyzer (Thermo Fisher, Foster City, CA, USA). The results were analyzed with SeqScape v.3 software (Thermo Fisher Scientific, Monza, Italy) and compared with a reference wild-type sequence (GenBank accession numbers: CLCN1:NM_000083.3 and SCN4A:NM_000334.4). Public database of normal human variations, dbSNP (https://www.ncbi.nlm.nih.gov/snp/), 1000Genome (https://www.internationalgenome.org/1000-genomes-browsers), EVS (https://evs.gs.washington.edu/EVS/) and gnomAD databases (https://gnomad.broadinstitute.org/blog/), were used to filter the data to exclude variants with allele frequency higher than 1% .
The genomic DNA of the patient was also analyzed to exclude myotonic dystrophy types 1 and 2 (DM1 and DM2).
In vivo electrophysiology
EMG was recorded with concentric needles in resting abductor pollicis brevis muscles of patients I-1 and II-1 and extensor digitorum communis of patient II-1.
For the short Exercise Test (SET), the ulnar nerve was stimulated at the wrist and the compound muscle action potential (CMAP) was recorded from the immobilized abductor digiti minimi (ADM) according to the protocol by Fournier and collaborators [21, 22]. First, a baseline recording of the CMAP evoked by a single stimulus was performed in the resting ADM after cutaneous temperature stabilization. Thereafter, 10 seconds of maximal effort were followed by a single stimulus applied every 8–10 seconds for 50 seconds; this 1-minute short exercise test was repeated three times at room temperature, with hand skin temperature maintained at 32–33°C. The SET was then repeated in the contralateral hand after cooling it to 20°C with ice packs.
Low-frequency (3 Hz) repetitive nerve stimulation was performed in patient II-1.
The warm-up phenomenon was evaluated using the protocol described by Trip and collaborators [6]. For the face and hand-forearm muscles, the relaxation time was measured with a stopwatch after a 10-second maximal contraction of the orbicularis oculi or fist clenching. For the lower limbs, the patient was asked to rise from a chair, to walk around it and to sit again. This contraction-relaxation cycle was repeated ten times; the tenth relaxation times were compared to the first ones using the Wilcoxon signed-rank test for paired samples.
Site-directed mutagenesis and heterologous expression
The p.T592I mutation was introduced in the wild-type human SCN4A cDNA imbedded in the pRc/CMV plasmid, as previously described [16, 17]. Transient expression of WT or T592I sodium channels was obtained in HEK293T cells, by transfecting 0.2μg/mL of the corresponding pRC/CMC-SCN4A plasmid and 0.1μg/mL of the pCD8-IRES-hβ1 plasmid, which contains the sodium channel auxiliary β1 subunit and the CD8 receptor gene reporter. The cells were used 48–72 hours after transfection for patch-clamp experiments.
In vitro electrophysiology
The whole-cell sodium currents were recorded at room temperature (∼22°C) in transfected HEK293T cells using the Axopatch 200B patch-clamp amplifier and Digidata 1550B AD/DA converter (Axon instruments). The pipette solution contained in mM: 120 CsF, 10 CsCl, 10 NaCl, 5 EGTA, and 5 Cs-HEPES (pH 7.2). The extracellular solution contained in mM: 150 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 5 Na-HEPES, and 5 mM glucose (pH 7.4). In these conditions, the pipette resistance was within 1.5 and 3.0 MΩ. The voltage clamp protocols are described in the Results section. Data were analyzed using pCLAMP 10.3 (Axon Instruments) and SigmaPlot 8.02 (Systat Software GmbH).
RESULTS
Clinical presentation
The proband, (patient III-1, Fig. 1A), was referred for neurologic examination during pregnancy at 32-year-old following a genetic counselling outpatient service as part of a beta-thalassemia prevention program. Since early infancy, the patient had complained of non-painful muscle stiffness that worsened with cold, fasting and during intermediate seasons, although very hot temperatures also caused some worsening; the stiffness involving limbs and face, was exacerbated during menstruation, and definitely increased during pregnancy. The patient did not complain of muscle pain, except for mild muscle soreness at times, which she described as being similar to that following muscle cramps. She reported a slight warm-up phenomenon and did not experience spontaneous or provoked paralytic attacks or weakness following muscle efforts. The patient’s shoulder girdle, arms and lower limbs muscles were hypertrophic, although this was in part attributable to her work as a masseuse in a beauty center; on examination, she had an athletic appearance and presented hand-grip, eyelid and percussion myotonia. Since she was pregnant, a potassium challenge was not performed. At the end of a normal pregnancy, she gave birth to a child bearing no symptoms of the disease; the proband’s mother, who was experienced in recognizing the myotonic symptoms in the daughter since her birth, correctly predicted that the baby would be healthy. When examined in the following years, the athletic appearance of the patient gradually waned. Nowadays the disease is stable, with somewhat fluctuating symptoms, as the patient can stop treatment for several days without complaining of disturbing myotonia.
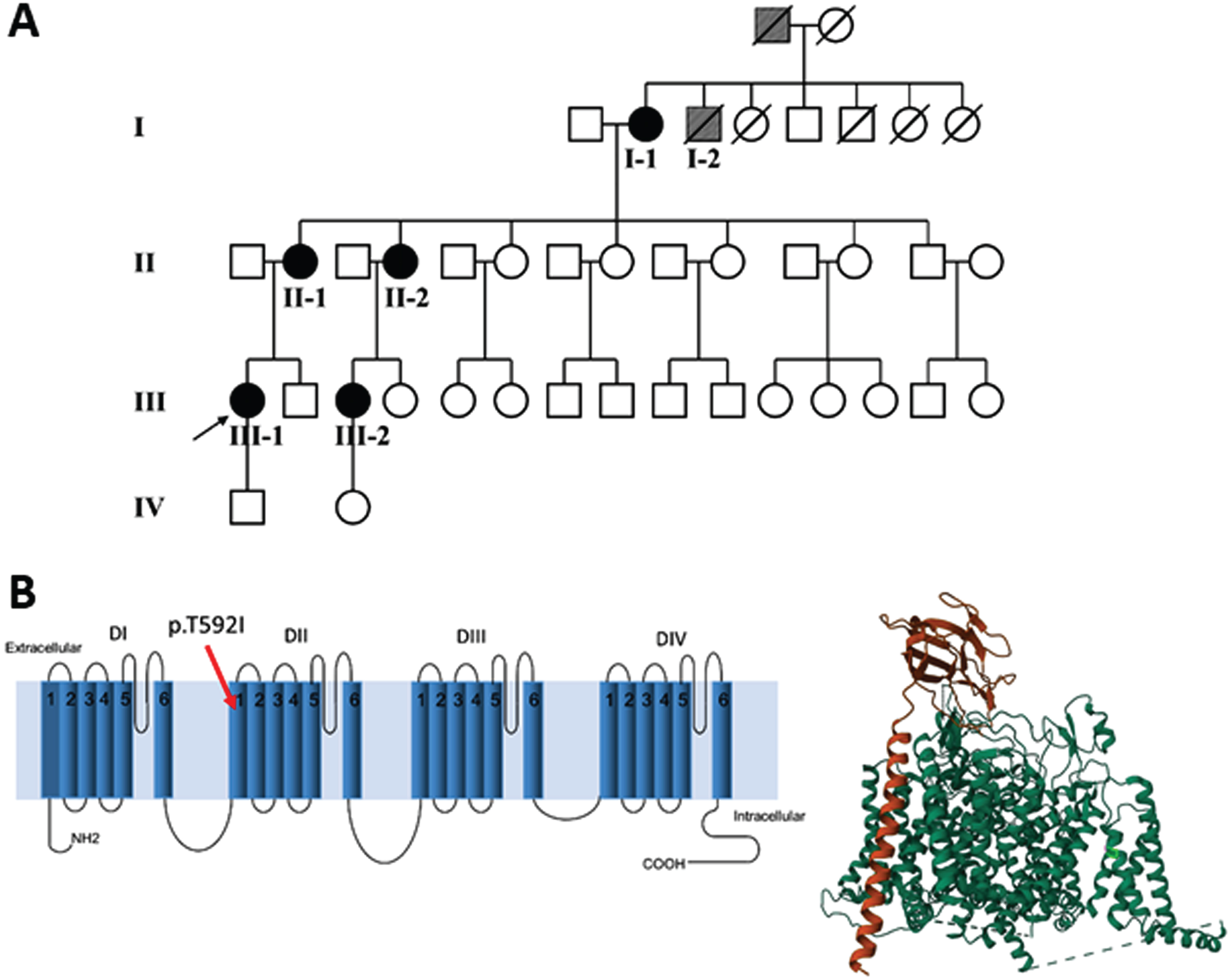
Fig. 1
(A) Family pedigree: patient I-2 and his father (grey symbols) are possible cases, based on the information provided. (B) Left panel. The mutation p.T592I identified in the affected patients is shown in the secondary structure of human Nav1.4. The position has been established using NextProt (https://www.nextprot.org/entry/NX_P35499/sequence). Right panel. The T592I mutation is shown in light green in the 3D plot of hNav1.4. The principal α subunit (green) of hNav1.4 and the auxiliary β1 subunit (orange) were obtained from RCSB Protein Data Bank (https://www.rcsb.org/).
The proband’s mother (Patient II-1) at age 56 reported muscle stiffness and pain that worsened with cold and hot temperatures (e.g., on the beach during summer; the family lives in an area where in the summer temperatures can easily reach 35°C or more), as well during menstruation and pregnancy. Myotonia had been present since her early infancy and, after some falls, the patient was limited in her physical activities because of her mother’s caution rather than due to an actual impairment. She also reported exacerbations during fasting or after alcoholic drinks. Clinical myotonia was evident in the orbicularis oculi but absent in the hand muscles. Neither warm-up nor periodic paralysis was reported.
Patient III-2 is a 26-year-old woman, who presented symptoms similar to those of her cousin (Patient III-1). While at the time of the first examination her muscle bulk was not increased, recently during a pregnancy she experienced a marked worsening of the symptoms and an overt increase in muscle bulk. During pregnancy, muscle stiffness and cramp-like pain extended to involve the hands and abdominal muscles. Since she already had laboratory confirmation of the disease, she obtained a prenatal molecular genetics examination and, at the end of a normal pregnancy, she gave birth to a healthy baby.
Patient II-2 had symptoms and physical status similar to her sister (patient II-1); muscle stiffness involved her face, limbs and hand muscles and was more severe with cold, during pregnancy and, to a lesser extent, with hot temperatures. She did not report muscle pain, except for some cramps during pregnancy.
Since childhood, the proband’s 77-year-old grandmother (Patient I-1) was limited by muscle stiffness, particularly during quick movements. For this reason, in her youth, she avoided moving quickly due to the fear of falling. Her myotonia worsened both with cold and hot weather and could involve her face and tongue and affect swallowing.
From the anamnestic information, it was determined that two other family members, the father and a brother (I-2) of patient I-1, both deceased, were also possibly affected (Fig. 1A).
As a whole, the patients complained of a mild to moderate myotonic disorder that partially limited their physical activity but did not perturb daily activities. Myotonia was visible since birth and involved the lower limbs, hands and face. It worsened with cold and very hot temperatures, with muscle pain varying from a mild soreness to a cramp-like aching. The patients were uncertain about whether they experienced a clear-cut warm-up phenomenon in their lower limbs or hands, but two of them reported it to be present to some extent. Pregnancy and menstruation exacerbated the symptoms. No patient ever had a paralytic attack or reported significant post-exercise weakness; no paradoxical worsening of myotonia was reported.
Four patients were initially treated with mexiletine 200 mg tid, except during pregnancy, but all of them had gastric intolerance and were eventually switched to acetazolamide 250 to 500 mg/day. According to their assessment, both drugs were effective, but mexiletine was slightly superior in attenuating symptoms. More recently, patient III-2 has started treatment, first with acetazolamide and later with mexiletine, but stopped both drugs because of side effects; further trials with class I anti-arrhythmic or anti-epileptic drugs are planned.
Genetic testing
The NGS revealed a previously unreported heterozygous point mutation (c.1775C > T) in exon 11 of the SCN4A gene, which codes for p.Thr592Ile located in the transmembrane S1 segment of domain II of the Nav1.4 sodium channel (Fig. 1B). This mutation is not included in mutation databases [23, 24]; dbSNP (https://www.ncbi.nlm.nih.gov/snp/), 1000Genome (https://www.internationalgenome.org/1000-genomes-browsers), EVS (https://evs.gs.washington.edu/EVS/) and gnomAD databases (https://gnomad.broadinstitute.org/blog/), but was reported on ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/variation) as “uncertain significance variant”, but without functional evidence or citations. The pathogenicity of the variant was further evaluated in silico using UMD-Predictor [25], PROVEAN [26], Mutation Taster [27], PolyPhen-2 [28] and VarSome [29]. All the mentioned tools predicted the variant as pathogenic (UMD-Predictor), deleterious (PROVEAN), disease-causing (Mutation Taster), probably damaging (PolyPhen2) and pathogenic strong (VarSome). We submitted the p.The592Ile variant to the Leiden Open Variation Database (DB-ID: SCN4A_000288).
The variant p.Thr592Ile was found in all the five related patients and was absent in one asymptomatic family member (a brother of patients II-1 and II-2), in 100 unrelated sex- and age-matched healthy Italian blood volunteers (200 alleles) and in more than 300 patients affected by non-dystrophic myotonia investigated in our laboratory, suggesting that this variant might be responsible for the disease phenotype.
In vivo electrophysiology
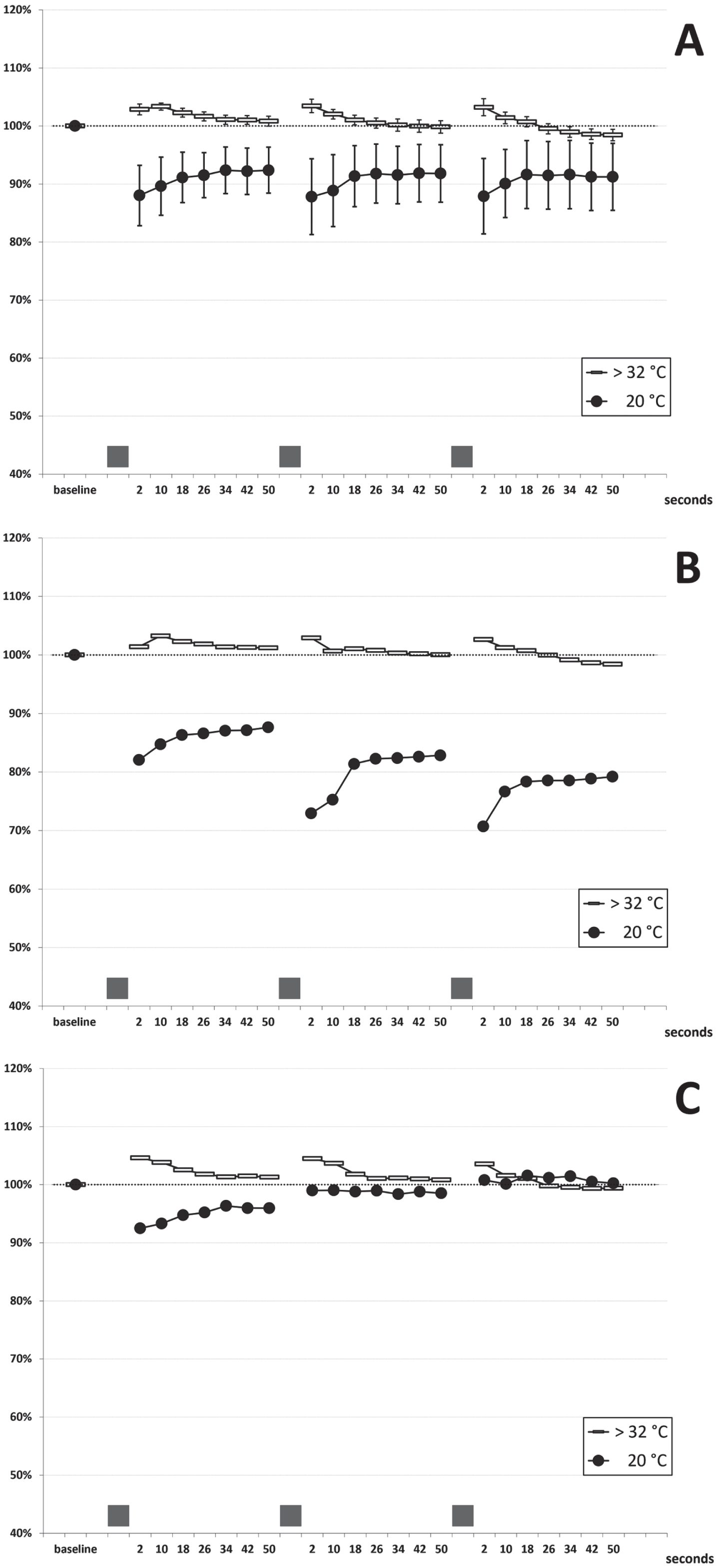
In patients I-1 and II-1, the needle EMG examination of abductor pollicis brevis (both patients) and extensor digitorum communis (patient II-1) showed typical myotonic runs of positive waves. Neither fasciculations nor spontaneous denervation activity were recorded. The short exercise test (SET) was performed at normal temperature (hand skin temperature > 32°C). A normal pattern was observed in all the patients, consisting of a slight amplitude increase (mean: +3%) within the first 10 seconds after exercise, a return to pre-exercise values in the following 40 seconds, and no cumulative changes at the end of the three repetitions (see cumulated data of 11 examinations on five subjects in Fig. 2A). This pattern was consistent across patients and in the follow-up examinations performed over three years. After cooling to 20°C, exercise caused a transient amplitude decrease that partially recovered within 50-seconds (Fig. 2A); there was some test-retest variability in a given subject when examined several months apart (data not shown). Two different patterns could be identified in the five patients: in patients II-1, II-2 and III-2, a slight gradual amplitude decrease followed by partial recovery was observed (Fig. 2B); on the contrary, in patients I-1 and III-1, a mild progressive amplitude increase occurred with the three repeated contractions (Fig. 2C). However, extreme amplitude change values stood between –35% and +9% at any time point, and four out of five patients always presented an increase or decrease of the CMAP amplitude within±20%, which have been considered normal limits in previous studies [22, 30, 31]. No post-exercise myotonic potentials (PEMP) [21, 22] were recorded, either at room temperature or after cooling. Repetitive (3 Hz) nerve stimulation revealed no decrement or late potentials.
Fig. 2
Short Exercise Test. (A) cumulated data (mean±SEM bars): 11 examinations in five patients at room temperature (room T; hand skin temperature >32°C) and after cooling (cold; hand skin temperature 20°C) (B) findings in patients II-1, II-2 and III-2 (C) findings in patients III-1 and I-1 The grey bars on the x-axis represent the three 10-seconds maximal efforts.
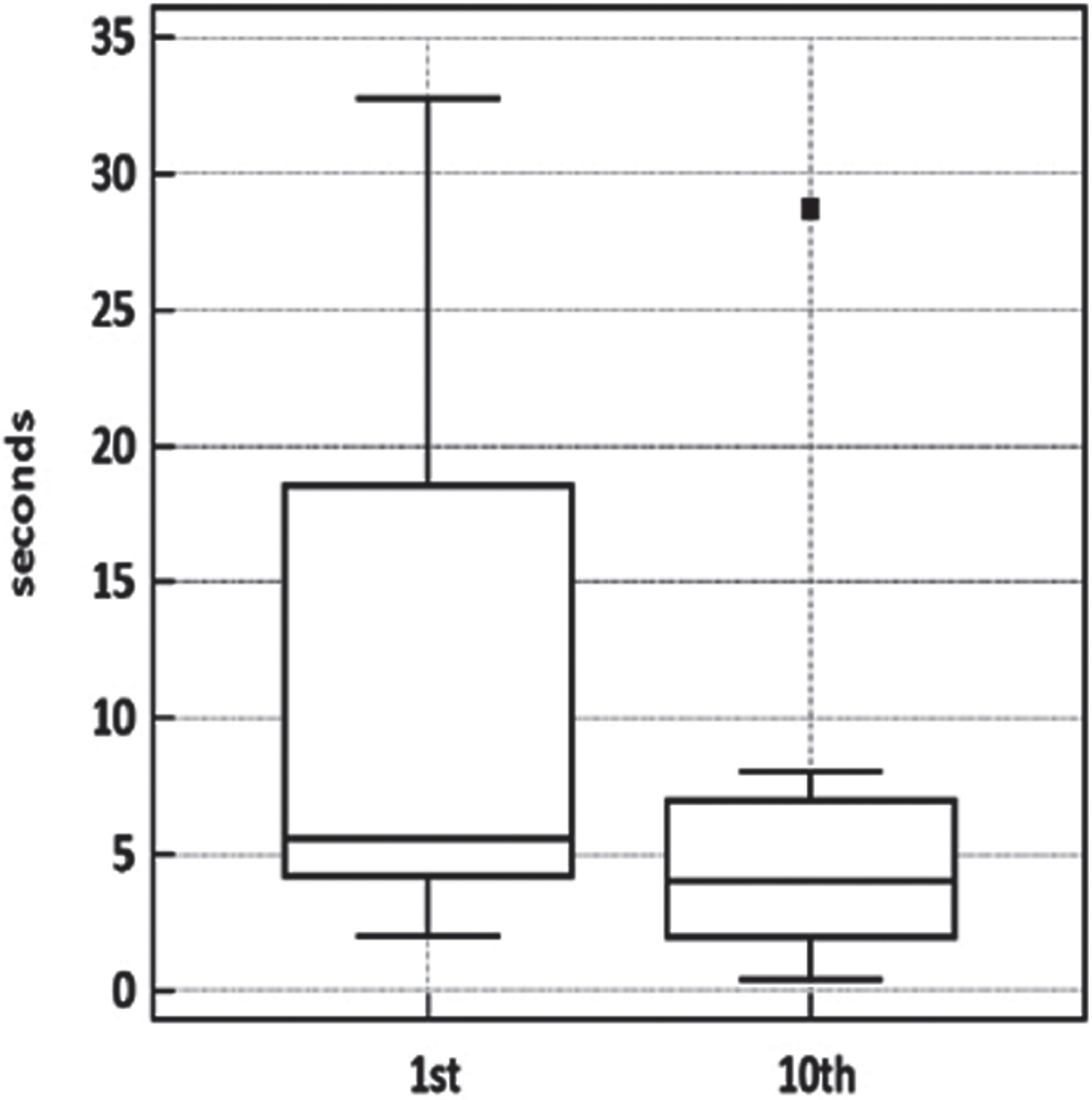
Muscle relaxation times were clearly affected by exercise in the orbicularis oculi and, less consistently, in the forearm-hand muscles, which presented a high variability across subjects (Table 1 and Fig. 3). No definite warm-up could be demonstrated in the lower limb muscles. Despite the contribution of the lower limb figures to this small dataset, the tenth-to-first trial difference was significant (p = 0.0039, Wilcoxon matched-pairs signed-rank test).
Table 1
Warm-up phenomenon in four patients, examined with the protocol of Trip and coll. (6); orbicularis oculi and handgrip relaxation time (seconds) and time to walk around the chair (seconds) in the first and tenth trials. Wilcoxon matched-pairs signed-ranks test p = 0.0039 (total time in 10th vs 1st trial)
| Patient | Trial | II-1 | II-2 | III-1 | III-2 | Mean(median) | |
| eyelid | 1st | 30.0 | 5.0 | 29.0 | 32.7 | 24.2(29.5) | NS |
| 10th | 8.0 | 2.1 | 7.0 | 28.7 | 11.5(7.5) | ||
| hand | 1st | 2.0 | 3.5 | 8.0 | 4.7 | 4.6(4.1) 1.4 | NS |
| 10th | 2.0 | 1.3 | 2.0 | 0.4 | 1.4(1.7) 5.5 | ||
| legs | 1st | 6.8 | 3.8 | 5.4 | 5.8 | 5.5(5.6) | NS |
| 10th | 6.9 | 3.8 | 4.3 | 4.6 | 4.9(4.5) | ||
| total time | 1st | 38.8 | 12.3 | 42.4 | 43.2 | 34.2(40.6) | **p = 0.0039 |
| 10th | 16.9 | 7.2 | 13.3 | 33.7 | 17.8(15.1) |
Fig. 3
Warm-up testing in four patients. Boxplot of warm-up test data. Wilcoxon signed-rank test for paired data p < 0.0039.
In vitro electrophysiology
Sodium currents generated by p.T592I mutant in transfected HEK cells were similar to wild type hNav1.4 currents (Fig. 4A), showing rapid activation followed by complete inactivation. The exponential fit of sodium current decay between –40 and +20 mV indicates no difference in the rate of entry into fast inactivation (Fig. 4B). The current-voltage relationships were similar (Fig. 4C). The average maximal current was –4208±588 pA (n = 26) for T592I and –3638±594 (n = 15) for WT (not significant with unpaired Student’s t test). The voltage of maximal current was –32.8±1.9 (n = 26) and –31.3±1.2 mV for T592I and WT, respectively (not significant). Yet, T592I currents were activated at more negative voltages, which was confirmed by a significant, ∼5-mV negative shift of the voltage-dependence of activation (Fig. 4D, Table 2). The voltage dependence of fast inactivation was unaltered by the mutation (Fig. 4D, Table 2). The likelihood to open was greater for T592I compared to wild-type, especially at negative voltages, as evidenced by the window currents determined by the intersection of activation and fast inactivation voltage dependences (Fig. 4E, Table 2). The voltage dependence of slow inactivation was unaffected by the mutation (Fig. 4F, Table 2).
Fig. 4
Functional characterization of WT and T592I Nav1.4 channel variants expressed in HEK283T cells. (A) Representative sodium current traces for T592I and WT. Sodium currents were elicited according the protocol shown inset. Interval between each pulse was 10 s. (B) Time constants for sodium channel entry in fast inactivation calculated from current decay monoexponential fit. (C) Normalized current-voltage (I-V) relationships were drawn from the sodium current traces recorded as shown in A. (D) Voltage dependence of activation and fast inactivation. For activation, the conductance was calculated from I-V relationships using the equation gNa = INa/(V –ENa), where gNa is the sodium conductance, INa is the peak sodium current, V is the membrane voltage, and ENa is the electrochemical gradient at equilibrium for sodium ions calculated from Nernst equation (ENa = +68.4 mV). The relationships (in green and black) were fitted to the Boltzmann equation gNa/gNa,max = 1/{1 + exp.((V-aV50)/Ka)}, where aV50 is the half-maximum activation voltage and Ka is the slope factor. Voltage dependence of fast inactivation was studied using a two-pulse voltage clamp protocol shown inset. The relationships (in blue and red), showing the normalized peak current amplitude recorded during the second pulse versus the first pulse membrane potential, were fitted to the Boltzmann equation INa/INa,max = 1 / {1 + exp.((V-fV50)/Kf)}, where fV50 is the half-maximum inactivation voltage, and Kf is the slope factor. (E) Window current resulting from the overlap of activation and fast inactivation relationships. (F) The voltage dependence of slow inactivation was studied using a three-pulse voltage clamp protocol. The normalized peak current amplitude during the third pulse was plotted versus the membrane potential of the first pulse. The relationships were fitted to the Boltzmann equation INa/INa,max = IR + (1 - IR)/{1 + exp.((V-sV50)/Ks)}, where sV50 is the half-maximum inactivation voltage, Ks is the slope factor, and IR is the steady fraction of non-inactivating channels. Data points in the relationships are mean±SEM from n cells. The fit parameter values are given in Table 2.
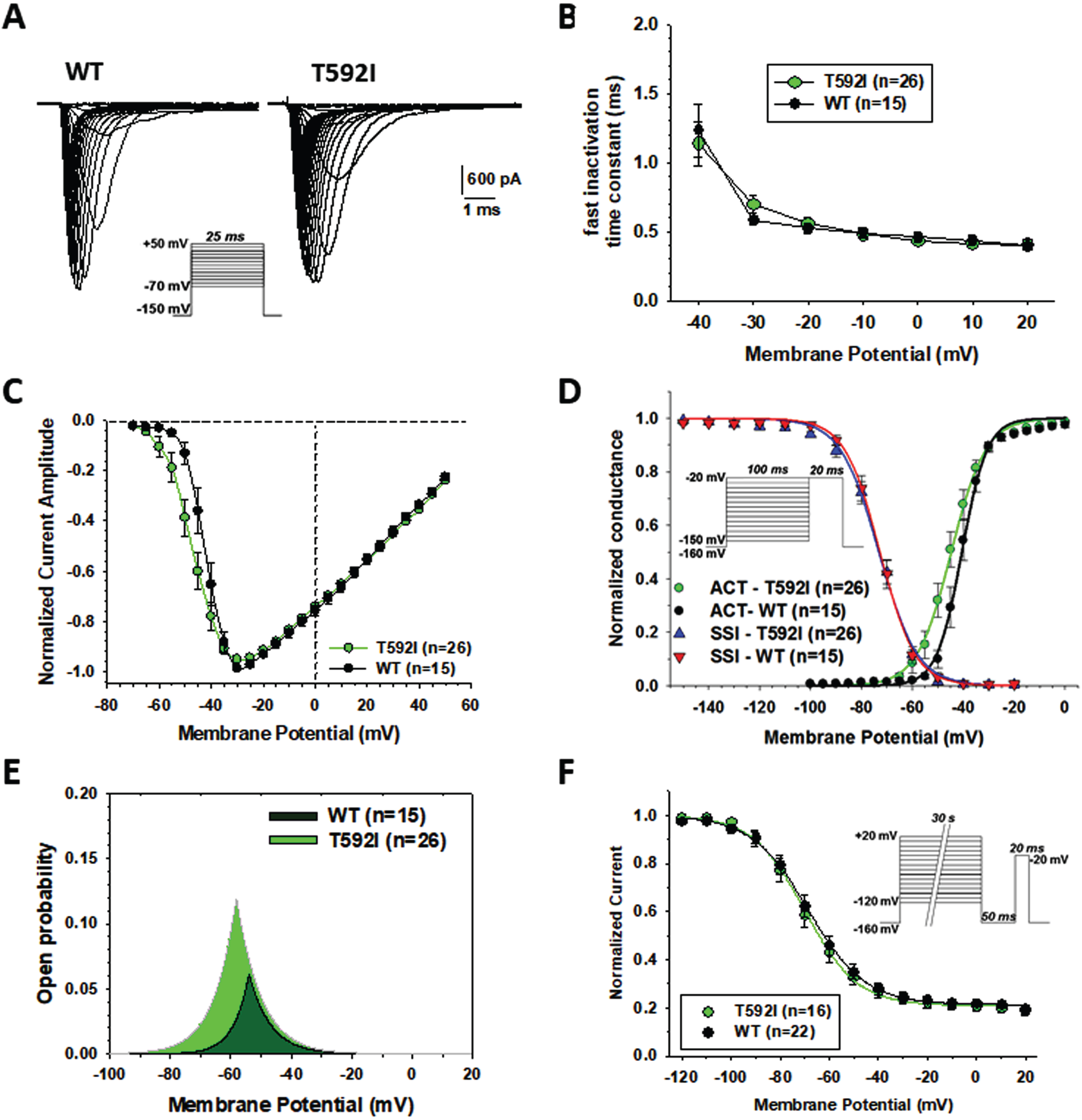
Table 2
Parameters of WT and T592I Nav1.4 channel variants
| Parameter | p.T592I | Wild-type | |
| Activation voltage-dependence | aV50 (mV) | –45.5±1.5 (n = 26) * | –40.9±1.0 (n = 15) |
| Ka (mV) | 3.8±0.2 | 3.4±0.4 | |
| Fast inactivation voltage- dependence | fV50 (mV) | –73.3±1.4 (n = 26) | –72.6±1.6 (n = 15) |
| Kf (mV) | –6.1±0.3 | –5.7±0.3 | |
| Open probability | Window current | 0.64±0.09 (n = 26) * | 0.34±0.07 (n = 15) |
| Slow inactivation voltage-dependence | sV50 (mV) | –69.7±2.5 (n = 16) | –68.7±2.6 (n = 22) |
| Ks (mV) | –8.6±0.6 | –9.3±0.6 | |
| IR | 0.21±0.02 | 0.21±0.01 |
The fit parameters were calculated as shown in Fig. 5, and reported as mean±S.E.M. from n cells. Statistical analysis was performed using unpaired Student’s t test. Significant differences were found between T592I and WT for activation voltage dependence midpoint ( *p < 0.05) and window current ( *p < 0.02). The aV50, fV50, and sV50 are the half-maximum potentials; Ka, Kf, and Ks are the slope factors; IR is the steady fraction of non-inactivating channels.
DISCUSSION
The clinical picture in this family is consistent with SCM, a non-dystrophic myotonia mostly affecting the face and hand muscles but also involving the lower limbs and abdominal or pharyngeal muscles, without any episodic paralysis or post-exercise weakness. Temperature sensitivity is obvious, since cold and very hot temperatures can worsen the symptoms. Several other physiological or external factors, such as pregnancy, menstruation, fasting and alcohol intake, can also increase muscle stiffness. Particularly, pregnancy caused a severe limb muscles stiffness extending to the abdominal muscles (patient III-2) and led to an overt muscle hypertrophy in both younger patients (patients III-1 and III-2). Warm-up phenomenon was a variable feature, reported by three out of five patients, and involving different muscle groups to a variable degree (Table 1); no patient reported paradoxical myotonia, thus excluding paramyotonia congenita and suggesting a SCM diagnosis.
The SET conducted at normal hand temperature produced a normal pattern (Fournier pattern III), which has also been reported for other SCM mutations (A715T, I1310N, V445N, S804N, V1293I) [22]. After cooling the hand to 20°C, the repetition of the exercises produced more variable patterns, resembling those of A715T and I1310N mutations (Fig. 4D of ref. 23) and G1306A/V mutation (Fig. 4E of ref. 22). These changes however were mild and restricted to the –20% – +10% range in all but 1 test in 1 patient, as reported in several pathogenic SCN4A mutations [22].
The SCN4A sequencing disclosed a previously unreported point mutation c.1775C > T in exon 11, corresponding to Thr592Ile substitution. This region codes for the transmembrane S1 segment of domain II of the NaV1.4 sodium channel (DII-S1, Fig. 1B). The mutation was present in all five of the symptomatic patients and is predicted pathogenic by most software. It is noteworthy that the majority of reported pathogenic SCN4A mutations [32–34] are located in the S4–S6 segments of the four domains, often in domain IV or the intra- or extra-cytoplasmic loops. The S1–S3 segments of domain II seem to be an uncommon site for pathogenic mutations. To our knowledge, only p.I588V from domain II segment 1, found in a patient with myotonia and episodes of weakness, has been functionally characterized [35]. The mutation induced a hyperpolarization of shift of activation voltage dependence. In addition, a variant at position 591 (p.N591K) has been reported in the Ensembl database (www.ensembl.org) as deleterious/probably damaging, suggesting that this region might have an effect on the normal physiology of the channel. Interestingly, we found that T592I induced a left shift of Nav1.4 activation voltage dependence resulting in larger window currents, similarly to I558V. The interaction between the S4–S6 and S1–S3 segments could be relevant for the correct functioning of the channel. Indeed, the crystal structure of the bacterial NavAb channel revealed that the S1 and S4 segments are very close to each other [36], and the mutation p.I141V (domain I, segment 1) was demonstrated to stabilize the open conformation of the channel by modifying the interactions between the S1 and S4 segments [37]. Thus the biophysical defect identified in T592I likely accounts for the myotonia phenotype. Such a defect was also recently observed in another mutation K1302R associated with myotonia [13].
DECLARATIONS
CONSENT PUBLICATION
A written informed consent for genetic analysis and for EMG was obtained from the proband and her relatives, as required by the Ethical Committee of the Foundation Neurological Institute Carlo Besta and by the Ospedale Binaghi, in accordance with the Helsinki Declaration.
DATASETS/DATA AVAILABILITY STATEMENT
The genetic, electromyography, Short Exercise Test, and in vitro electrophysiology datasets generated and/or analyzed during the current study are available from the corresponding author, Paolo Tacconi and Jean-François Desaphy authors, respectively, on reasonable request.
FUNDING
This study was partially supported by University of Bari (Horizon Europe Seeds project “Medineuropa” to JFD) and Italian Ministry of Health (RRC 2024).
CONFLICT OF INTEREST
The authors have no conflict of interest to report. Lorenzo Maggi is a member of the ERN-NMD. Jean-François Desaphy and Lorenzo Maggi are members of the Molecular Therapeutic Board in Neurological Channelopathies of the ERN-EpiCARE, ERN-NMD, and ERN-RND.
AUTHOR’S CONTRIBUTIONS
All authors contributed to data acquisition and analysis. P.T., R. B., and J-F.D. wrote the manuscript.
ACKNOWLEDGMENTS
We are grateful to the patients for their cooperation. This paper is dedicated to Pia Bernasconi, for her great commitment to scientific research.
REFERENCES
[1] | Maggi L , Bonanno S , Altamura C , Desaphy JF . Ion Channel Gene Mutations Causing Skeletal Muscle Disorders: Pathomechanisms and Opportunities for Therapy. Cells. (2021) ;10: (6):1521. doi:10.3390/cells10061521. |
[2] | Matthews E , Holmes S , Fialho D . Skeletal muscle channelopathies: A guide to diagnosis and management. Pract Neurol. (2021) ;21: (3):196–204. doi:10.1136/practneurol-2020-002576. |
[3] | Ricker K , Moxley RT 3rd , Heine R , Lehmann-Horn F . Myotonia fluctuans. A third type of muscle sodium channel disease. Arch Neurol. (1994) ;51: :1095–102. |
[4] | Sugiura Y , Aoki T , Sugiyama Y , Hida C , Ogata M , Yamamoto T . Temperature-sensitive sodium channelopathy with heat-induced myotonia and cold-induced paralysis. Neurology. (2000) ;54: :2179–81. |
[5] | Colding-Jorgensen E , Duno M , Vissing J . Autosomal dominant monosymptomatic myotonia permanens. Neurology. (2006) ;67: :153–5. |
[6] | Trip J , Faber CG , van Engelen BGM , Drost G . Warm-up phenomenon in myotonia associated with the V445M sodium channel mutation. J Neurol. (2007) ;254: :257–8. |
[7] | Webb J , Cannon SC . Cold-induced defects of sodium channel gating in atypical periodic paralysis plus myotonia. Neurology. (2008) ;70: :755–61. |
[8] | Stunnenberg BC , Ginjaar HB , Trip J , Faber CG , van Engelen BG , Drost G . Isolated eyelid closure myotonia in two families with sodium channel myotonia. Neurogenetics. (2010) ;11: :257–60. |
[9] | Saleem R , Setty G , Khan A , Farrell D , Hussain N . Phenotypic heterogeneity in skeletal muscle sodium channelopathies: A case report and literature review. J Pediatr Neurosci. (2013) ;8: :138–40. |
[10] | Stunnenberg BC , LoRusso S , Arnold WD , Barohn RJ , Cannon SC , Fontaine B , Griggs RC , Hanna MG , Matthews E , Meola G , Sansone VA , Trivedi JR , van Engelen BGM , Vicart S , Statland JM . Guidelines on clinical presentation and management of nondystrophic myotonias. Muscle Nerve. (2020) ;62: (4):430–44. doi:10.1002/mus.26887. |
[11] | Cannon SC . Sodium Channelopathies of Skeletal Muscle. Handb Exp Pharmacol. (2018) ;246: :309–30. doi:10.1007/164_2017_52. |
[12] | Cannon SC . Channelopathies of skeletal muscle excitability. Compr Physiol. (2015) ;5: (2):761–90. doi:10.1002/cphy.c140062. |
[13] | Vacchiano V , Brugnoni R , Campanale C , Imbrici P , Dinoi G , Canioni E , Laghetti P , Saltarella I , Altamura C , Maggi L , Liguori R , Donadio V , Desaphy JF . Coexistence of SCN4A and CLCN1 mutations in a family with atypical myotonic features: A clinical and functional study. Exp Neurol. (2023) ;362: :114342. doi:10.1016/j.expneurol.2023.114342. |
[14] | Ke Q , Zhao Y , Li Y , Ye J , Tang S , He F , Ji F , Dai X , Ni J , Li Y , Griggs RC , Cheng X . Clinical comparison and functional study of the L703P: A recurrent mutation in human SCN4A that causes sodium channel myotonia. Neuromuscul Disord. (2022) ;32: (10):811–9. doi:10.1016/j.nmd.2022.08. |
[15] | Huang CW , Lai HJ , Lin PC , Lee MJ . Changes of Resurgent Na+ Currents in the Nav1. 4 Channel Resulting from an SCN4A Mutation Contributing to Sodium Channel Myotonia. Int J Mol Sci. (2020) ;21: (7):2593. doi:10.3390/ijms21072593. |
[16] | Desaphy JF , Carbonara R , D’Amico A , Modoni A , Roussel J , Imbrici P , Pagliarani S , Lucchiari S , Lo Monaco M , Conte Camerino D . Translational approach to address therapy in myotonia permanens due to a new SCN4A mutation. Neurology. (2016) ;86: (22):2100–8. doi:10.1212/WNL.0000000000002721. |
[17] | Farinato A , Altamura C , Imbrici P , Maggi L , Bernasconi P , Mantegazza R , Pasquali L , Siciliano G , Lo Monaco M , Vial C , Sternberg D , Carratù MR , Conte D , Desaphy JF . Pharmacogenetics of myotonic hNav1. 4 sodium channel variants situated near the fast inactivation gate. Pharmacol Res. (2019) ;141: :224–35. doi:10.1016/j.phrs.2019.01.004. |
[18] | Desaphy JF , Altamura C , Vicart S , Fontaine B . Targeted Therapies for Skeletal Muscle Ion Channelopathies: Systematic Review and Steps Towards Precision Medicine. J Neuromuscul Dis. (2021) ;8: (3):357–81. doi:10.3233/JND-200582. |
[19] | Brugnoni R , Maggi L , Canioni E , Verde F , Gallone A , Ariatti A , et al. Next-generation sequencing application to investigate skeletal muscle channelopathies in a large cohort of Italian patients. Neuromuscul Disord. (2021) ;31: (4):336–47. doi:10.1016/j.nmd.2020.12.003. |
[20] | Maggi L , Ravaglia S , Farinato A , Brugnoni R , Altamura C , Imbrici P , Camerino DC , Padovani A , Mantegazza R , Bernasconi P , Desaphy JF , Filosto M . Coexistence of CLCN1 and SCN4A mutations in one family suffering from myotonia. Neurogenetics. (2017) ;18: (4):219–25. doi:10.1007/s10048-017-0525-5. |
[21] | Fournier E , Arzel M , Sternberg D , Vicart S , Laforet P , Eymard B , Willer JC , Tabti N , Fontaine B . Electromyography guides toward subgroups of mutations in muscle channelopathies. Ann Neurol. (2004) ;56: (5):650–61. doi:10.1002/ana.20241. |
[22] | Fournier E , Viala K , Gervais H , Sternberg D , Arzel-Hézode M , Laforêt P , Eymard B , Tabti N , Willer JC , Vial C , Fontaine B . Cold extends electromyography distinction between ion channel mutations causing myotonia. Ann Neurol. (2006) ;60: (3):356–65. doi:10.1002/ana.20905. |
[23] | Fokkema IF , Taschner PE , Schaafsma GC , Celli J , Laros JF , den Dunnen JT . LOVD v.2.0: The next generation in gene variant databases. Hum Mutat. (2011) ;32: (5):557–63. doi:10.1002/humu.21438. (see also http://www.lovd.nl/3.0/home). |
[24] | Stenson PD , Mort M , Ball EV , Evans K , Hayden M , Heywood S , Hussain M , Phillips AD , Cooper DN . The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet. (2017) ;136: (6):665–77. doi:10.1007/s00439-017-1779-6. (see also http://www.hgmd.cf.ac.uk/ac/index.php). |
[25] | Salgado D , Desvignes JP , Rai G , Blanchard A , Miltgen M , Pinard A , Lévy N , Collod-Béroud G , Béroud C . UMD-Predictor: A High-Throughput Sequencing Compliant System for Pathogenicity Prediction of any Human cDNA Substitution. Hum Mutat. (2016) ;37: (5):439–46. doi:10.1002/humu.22965. (see also https://umd-predictor.genomnis.com/mutation). |
[26] | Choi Y , Sims GE , Murphy S , Miller JR , Chan AP . Predicting the functional effect of amino acid substitutions and indels. PLoS One. (2012) ;7: (10):e46688. doi:10.1371/journal.pone.0046688. |
[27] | Schwarz JM , Cooper DN , Schuelke M , Seelow D . MutationTaster2: Mutation prediction for the deep-sequencing age. Nat Methods. (2014) ;11: (4):361–2. doi:10.1038/nmeth.2890. |
[28] | Adzhubei IA , Schmidt S , Peshkin L , Ramensky VE , Gerasimova A , Bork P , Kondrashov AS , Sunyaev SR . A method and server for predicting damaging missense mutations. Nat Methods. (2010) ;7: (4):248–9. doi:10.1038/nmeth0410-248. |
[29] | Richards S , Aziz N , Bale S , Bick D , Das S , Gastier-Foster J , Grody WW , Hegde M , Lyon E , Spector E , Voelkerding K , Rehm HL ACMG Laboratory Quality Assurance Committee Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) ;17: (5):405–24. doi:10.1038/gim.2015.30. |
[30] | Kuntzer T , Michel P . Muscle membrane polarisation after provocative tests, and after cooling: The normal CMAP changes to be expected. Clin Neurophysiol. (2004) ;115: (6):1457–63. doi:10.1016/j.clinph.2004.01.004. |
[31] | Tan SV , Matthews E , Barber M , Burge JA , Rajakulendran S , Fialho D , Sud R , Haworth A , Koltzenburg M , Hanna MG . Refined exercise testing can aid DNA-based diagnosis in muscle channelopathies. Ann Neurol. (2011) ;69: (2):328–40. doi:10.1002/ana.22238. |
[32] | Maggi L , Brugnoni R , Canioni E , Tonin P , Saletti V , Sola P , Piccinelli SC , Colleoni L , Ferrigno P , Pini A , Masson R , Manganelli F , Lietti D , Vercelli L , Ricci G , Bruno C , Tasca G , Pizzuti A , Padovani A , Fusco C , Pegoraro E , Ruggiero L , Ravaglia S , Siciliano G , Morandi L , Dubbioso R , Mongini T , Filosto M , Tramacere I , Mantegazza R , Bernasconi P . Clinical and Molecular Spectrum of Myotonia and Periodic Paralyses Associated With Mutations in SCN4A in a Large Cohort of Italian Patients. Front Neurol. (2020) ;11: :646. doi:10.3389/fneur.2020.00646. |
[33] | Brunklaus A , Feng T , Brünger T , Perez-Palma E , Heyne H , Matthews E , Semsarian C , Symonds JD , Zuberi SM , Lal D , Schorge S . Gene variant effects across sodium channelopathies predict function and guide precision therapy. Brain. (2022) ;145: (12):4275–86. doi:10.1093/brain/awac006. |
[34] | Sun J , Luo S , Suetterlin KJ , Song J , Huang J , Zhu W , Xi J , Zhou L , Lu J , Lu J , Zhao C , Hanna MG , Männikkö R , Matthews E , Qiao K . Clinical and genetic spectrum of a Chinese cohort with SCN4A gene mutations. Neuromuscul Disord. (2021) ;31: (9):829–38. doi:10.1016/j.nmd.2021.03.014. |
[35] | Corrochano S , Männikkö R , Joyce PI , McGoldrick P , Wettstein J , Lassi G , Raja Rayan DL , Blanco G , Quinn C , Liavas A , Lionikas A , Amior N , Dick J , Healy EG , Stewart M , Carter S , Hutchinson M , Bentley L , Fratta P , Cortese A , Cox R , Brown SD , Tucci V , Wackerhage H , Amato AA , Greensmith L , Koltzenburg M , Hanna MG , Acevedo-Arozena A . Novel mutations in human and mouse SCN4A implicate AMPK in myotonia and periodic paralysis. Brain. (2014) ;137: (Pt 12):3171–85. doi:10.1093/brain/awu292. |
[36] | Payandeh J , Scheuer T , Zheng N , Catterall WA . The crystal structure of a voltage-gated sodium channel. Nature. (2011) ;475: (7356):353–8. doi:10.1038/nature10238. |
[37] | Amarouch MY , Kasimova MA , Tarek M , Abriel H . Functional interaction between S1 and S4 segments in voltage-gated sodium channels revealed by human channelopathies. Channels (Austin). (2014) ;8: (5):414–20. doi:10.4161/19336950.2014.958922. |

