Mitochondrial DNA Mutations and its Role in the Genesis of Renal Diseases an Update
*Corresponding Author(s):
Rodolfo J Gordillo De AndaNephroimmunologist, Médica Sur Hospital, Ciudad De México, Mexico
Tel:+525 5568 6904,
Email:rodolfogordillo2000@yahoo.com
Abstract
Keywords
INTRODUCTION
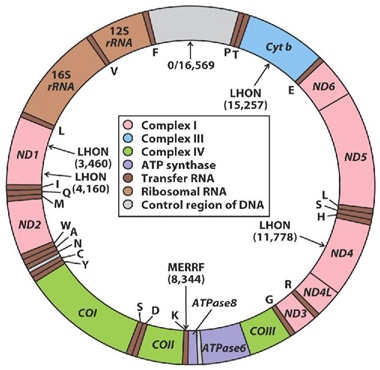
Figure 1: Mitochondrial DNA, showing its circular structure, constituted by two strands the outer a heavy and the inner a light strand, shows regions encoding for cytochrome b (Cyt B) and various subunits ofNADH-coenzyme Q reductase (ND), cytochrome c oxidase (COX), ATPasa, and ribosomal RNAs (rRNA) indicated.
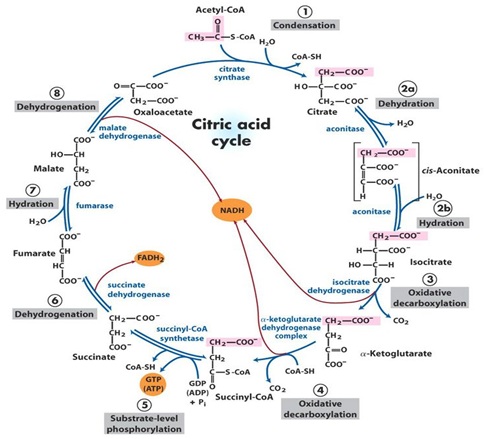
Human DNA was initially identified at the end of the 1860s by the swiss Chemistry Friederich-Miescher [4] and later two outstanding scientists, the rusian biochemistry PhiebusLevene and Erwin Chargaff conducted a series of reserch that revealed aditional details of the DNA molecule, Levene was the first to describe the order of the three components of a simple nucleotide (fosfate, sugar and base) and also was the first one to discover that the sugar in RNA was Ribosa and in DNA was Desoxiribosa [5], later the studies of Linus Pauling and Maurice Wilkins contributed but the later researcher due to a personal dispute with Professor Rosalind Franklin and without her permission in 1953 took the photography of the DNA molecule taken with X-Ray diffraction by her, and gave them to Watson, the photography of the DNA showed with clarity the helicoidal structure of the molecule, wich allowed Watson and Crick to deduce the structure of DNA in 1954 [6] without the help of theX-Ray diffraction photographies it would have been very difficult for Watson and Crick to have attained their discovery [6]. DNA has been totally sequenced recently by Vanter y cols [7] it is localized in the nucleous and orginized in structures denominated chromosoms; there are 46 chromosoms organized in 23 pairs, 22 of them are called autosoms and one pair are de sexual chromosoms: XX in the case of females and XY in the case of males [8] this is in reallity the real Human DNA (HDNA) wich contains our complete genetic code and contains 3.3 billions of bases pairs, the letters that encode the message for the sinthesis of specific proteins and, codifies aproximately 100,000 genes [9].
About 1,500 millions of years ago a pro-eukarioteamitochondrial cell [10] engulfed an anaerobic alfa-proto-bacteria member of the sub-division of Rickettsias, a group of intracellular bacteria probably an Archezoaprimitiva, this interaction resulted in a symbiotic relation for both; the remanents of this bacterial endosymbiosis are known as the Mitochondrion, they are of monophylicorigen [10] and appeared both in aerobic and in anaerobic bacterias [11]. In the eukariotic organisms the Mithochondrion evolve into different organels like the mitochondrias, the hidrogenosoms and the mitososms [12] that diverge among them in a very important ways, because one of the outstandig function of the mitochondria is the ATP synthesis dependent on the oxidative phosphorylation that has not been observed in the rest of the organeles derived from the mitochondrion. Therefore the hidrogenosoms uptakes ADP from the sourounding medium and excrete equimolar quantities of ATP. ATP synthesis in this organels is done by the phosphorylation at the sustrate level through the catalytic convertion of Succinil-Coenzyme A to Succinate by the enzyme succinate-thiokinase [13]. In the mitosoms ATP is synthetized trough the phosphorilation at the sustrate level but inside the cytosol without a direct involvement of the mitososms [14], because of these differences it can be concluded that the energetic metabolism is not the unifying fact among the mitochondria and the rest of organeles derived from the mitochondrion. Theseorganeles do share among them ATP dependant chaperon molecules (Cpn60) and/or mitochondrial heat shock proteins like mtSP70, as well as ATP/ADP transporters, but what it seems to be in reality the unifying line among them are the iron sulphataded proteins (FES). This FES [15] have important function in the electron transference during the enzymatic catalysis and in the metabolic regulation, moreover this proteins have an universal distribution in proeukariote and eukariote organisms and the current evidences suggest that these proteins have a central role for the establisment and maintenance of the original mitochondrial endosymbiosis. Among these proteins one that is highligth is nominated Rli-1 [16] with universal distribution in eukariote organisms an in the organeles derived from the mitochondrion and it is involve in the maturation of both the ribosomal RNA and transference RNA and therefore it is of outsanding importance.
The nuclear DNA controls the transcription activity of the mitochondrial DNA trough regulator proteins like the Mitochondrial Transcriptional Factor (MTFA) dependant of nuclear DNA. It is clear that the mitochondria is a so complex organele that it requires more than 37 genetic products for its function; in fact 850 polipetides codify by hDNA are required for its function, aproximately 75 are structural components of the respiratory complex and at least another 20 are required to maintain their structure and function. Mitochondria are diveded in 4 principal components: the External Mitochondrial Membrane (EMM), the Internal Mitochondrail Membrane (IMM), the Intermediate Mitochondiral Space (IMS) and the matrix localized in the inside (organelecytoplasm) [2].
The five complexes of the respiratory chain/system OXPHOS are: complex I (NADH ubiquinonaoxireductase), complex II (Succinate-ubquinonaoxireductase), complex III (Ubiquinol-cytochrome c oxidoreductase), complex IV (cytochrome c oxidase) and complex V (ATP sinthase) are localized in the IMM, there are also two electron transporters: the ubicuinona localized in the IMM and the cytochrome C in the EMM [17]. Beside that the Mitochondrial DNA works subjet under a double genetic control (nuclear and mitocondrial) there are another unique four findings for the behaivor of this organele that are important to know and comprehend to understand the mitochondrial functions.
Instead of the nuclear DNA where there exists only one pair of chromosoms in each cell, there are thousands copies of mitochonrial DNA and aproximately 5 copies per mitochondia. The división of the mitochondria and the replication of mitochondrial DNA take place independently of the cell cycle. After the cell división the mitochondrias and its DNAm are randomely distributed among the daughter´s cells (mitotic segregation).
The number of organels among the different cells is variable and depends primarily on the energetic requirements of that linage cells; that is why the fibroblasts contain a few hundreds of mitochondrias while the neurons can contain thousands and the cardiomiocytes ten thousands of mtochondrias, this shows that mitochondrias do not follow a genetic mendelian pattern but in fact they obey laws according with the genetic-energetic requirements of the specific cell lineage [18]. The mitochondrial DNA is inherited to the human offspring exclusively by the maternal line [19] due to the fact that the Father´s mitochondrias are present in the flagelous of the spermatozoids and once the spermatozoid penetrates the ovule it loses its flagelous and with it their mitochondrias (Figure 3). This fact allows establishing with great precisión the genetic line to whom we belong for hundreds or thousands of generations prior to us and by this way give us information about or must remotorigens. In the mayority of cases, the mitochondrial DNA copies are identical among each other condition called homoplasmia. During the celular division, the mitochondrias are inherited randomly to the daughter cells (19).

The Kidney is a highly vascularized organ because it recives 25% of the cardiac output per minute, aproximately 125 ml/min wich equals to a volume of filtered blood of 180 liters per day. Taking into account that the adult bloodvolume is about 6 liters, it filters that amount about 30 times a day. With this high flow you can understand that the kidney filtrates a great variety of substances, some toxic to the body but others not that require to be recovered from the urine by different mechanisms; an example is the sodium, we know that its normal serum concentration is around 140 mmol/l, so during a day an amount of 25 200 mmol/day are filtered, but the kidney reabsorbs 99% in their different segments and primaraly in the proximal tubule and excrete less tan 1%, lets say if the kidney reabsorbs 99.4% (24,948 mmol of sodium ) the losses will equal 0.6% or 151.2 mmol, that amount excreted is called the Fractional Excretion of S'odium (FENA), if FENA is greater than 1 %( 252 mmol) it would indicate acute renal failure. To acomplish the reabsortion of this valuable filtered elements the kidney counts with different methods for reabsortion among them; there is the paracellular transport that takes place in the adyacent portions of the cells in the tight junctions; and there is also the tanscellular transport, altough some of these mechanisms involve a facilitated transport trough a concentration gradient and an advantageous pH gradient, the mayority requires an active trasporter with ATP consumption, for these reasons the epithelium of the renal tubules in its luminal side include a great number of mitochondrias to provide the required energy (Figure 4). Therefore the reduction or dysfunction of this organeles produces severe hydro-electrolytic alterations.
Mutations on mitochondrial DNA with neruromuscular clinical presentation are well known and recognized among them is the MELAS syndrome with mitochondrial encephalopathy, lactic acidosis and cerebrovascular stroke; the MERRF síndrome with myoclonic epilepsy with red twisted fibers; the medula and pancreas Pearson syndrome and the Kearn-Sayre syndrome, the Leber hereditary optic neuritis and the Leber plus syndrome in wich there is also degeneration of the basal ganglia and dystonia with a variety of parkinsonism that do respond with Levodopa treatment. The Leigh syndrome is a mortal neurodegenerative disease with subcortical brain lesions [21]. Renal diseases can ocurrer in these four syndromes, they show up as nephrotic syndrome and, patients with a puntualmutation in m3243A>G manifiest as Fanconi syndrome in the mitochondrial Pearson and Kearns-Sayre syndromes.
The presence of multisystemic diseases in childhood should sugest the exitance of mitochondrial defects especially if they are associated with metabolic acidosis and renal tubular defects. One has to recall that the renal tubulo-intersticial disease with poliuria and hyposthenuria are a very common manifestation of mitochondrial diseases affecting the proximal renal tubule. More than a hundread of mitochondrial diseases inherited with mendelian character have beeen reported, many of them are associated with nephropaties [22].
Clinical expression of mitochondrial renal cytophaties:
1. Tubulophaties: In the renal proximal tubule is where thake place more than the 90% of reabsortion of the necessary elements for the body that are filtrated trough the glomeruli and this activity requires must of the time to spend energy (ATP) what explains the elevated number of mitochondrias at this level.
Proximal Tubulophaties:
a. Toni Debré Fanconi syndome: With glucosuria, aminoaciduria, phosphaturia proteinuria, uricosuria and kaluresis.
b. Proximal Tubular Acidosis: With Bicarbonaturia and hypercalciuria.
c. Tubulointersticialnephrophaty: With hypostenuria, poliuria, hypernatriuresis and nicturia
d. Bartter syndrome: With metabolic alcalosis, hypokalemia, hiperreninemia, and hyperaldosteronism without arterial hypertension.
2. Glomerulophaties: Nephrotic syndrome with Focal Semental Sclerosis resistant to corticosteroids therapy.
3. End Stage Renal disease, with elvation of serum creatinine over 1.3 mg/dl, BUN elevation, hyperphospahemia, hypocalcemia, hyperkalemia, and anemia, hyponatremia of variable degree, hyperparathyroidsm and anemia.
The vast mayority of patients with renal findings have extra-renal symptoms like muscular findings as myophaties, muscle pain, myclonus; and also show ocular findings like diplopía, palpebral ptosis, restrictions of ocular movments, pigmentary retinophaty, also course with several neurological findings like psicomotor delay development, seizures, neurosensorial deafness, optic atrophy, myclonus, pheriphericneurophaty, dementia and also show cerebro-vascular events and cardiac findings as blokage of different degrees, disrritmias, hypertrohic concentric cardiomiopathy. Also the endocrine glands may be involved showing as diabetes mellitus, hypoparathyroidsm and growth hormone deficiency [23].
The pediatricians and pediatric pephrologist have to be aware of these class of diseases, new discovered of seven defects in the biosynthesis of coenzyme Q10 in three of these COQ2, PDSS2 and COQ6 have an association with a prominent renal phenotype and show up as a corticosteroid resistant nephrotic syndrome with variable association with multisystemic findings as neurosensorial deafness, epilepsy, ataxia and syndromes alike cerebro vascular events. Many progress to End Stage Renal Diseases and require renal replacement function procedures like dyalisis either Hemo or peritoneal or Kidney transplantation, nevertheless as it has been pointed out by several researchers that the presymptomatic therapy with Coenzima Q10 in high dosages can prevent the progression of the renal disease and save of the neurological symptoms [24].
To establish the diagnosis of mitochondrial mutations it is very helpfull the familiar background, in the particular patient the presence of multisystemic disease afecting other organs highly aerobics: brain, liver, muscle, the presence of metabolic acidosis with elevation of lactate and piruvate help to establish the diagnosis but these alterations are not always present. In the kidney the presence of corticoresistant nephrotic syndrome, Fanconi syndrome, poliuria with incapacity to concentrate the urine and the presence of túbulo-intersticial findings in the renal biopsy and or focal segmental sclerosis and the finding in the ephitelium of the renal proximal tubule evuidence of mitochondrial dismorphogenesis or its presence in the muscle biopsy help to establish the diagnosis. Up to date more than a 100 mitochondrial diseases heredited in a mendelian way are known, those who affect primarialy the kidney appear on table 1 [25].
|
Molecular defects |
Gen(es) |
Renal Afection |
Other Clinical Findings |
|
Maintainace of |
RRM2B, DGUPK, TK2 |
Proximal Tubulopathy |
s. of Mitochondrial DNA depletion |
|
Mitochondrial DNA |
SUCLA2, MPV17 |
3 principal phenotypes: hepato-cerebralmyopathic andencéfalomyopathic |
|
|
TranslationMitochondrial(aminoacylation) |
SARS2 |
Tubulointerstiscial salt looser disease and with Hypomagnesemia |
Pulmonary hypertension |
|
Mitochondrial Ribosomas |
MRPS22 |
Tubulopathy |
HypertrophicCardiomiopathy and Encephalopathy |
|
TranslationMitochondrial (elongation) |
TFSM |
Tubulopathy |
Intrauterine growth delay, hepatic failure and Hipotonicity |
|
Assembly of Complex I |
NDUFAF2 |
Renal Tubular Acidosis |
Leigh Syndrome |
|
Assembley of Complex III |
BCSIL |
Proximal TubulophatyEncephalopahtyand Hepatic failure |
|
|
Assambley of Complex IV |
COX10, SURF 1 |
Renal Tubulopathy and distal tubular Acidosis(SURF 1) |
Leigh Syndrome |
|
Assambley of Complex V |
TMEM70 |
Proximal Tubulopathy |
HypertrophicCardiomiopathy |
|
Co-Enzyme Q10 |
PDSS2, COQ2,COQ6, COQ9 |
RSNS, Tubulopathy |
Seizures, Ataxia, neurosensorialdeafeness, multisystemic disease |
TREATMENT
The Therapy consists of the following mesurament.
General Mesuraments
a) Prevention: Prevent the use of commonly use medication that interfiere with the respiratory chain and can cause acute liver failure like valproic acid and barbiturates, prevent the use of comon antibiotics that can alter mitochondrial protein synthesis like tetracyclines and cloranfenicol; also prevent the use of biguanaides and steroids.
b) Infections and extenuated excercise should be avoid because they can excacerbate the lactic acidosis that has to be treated with a low infusión of sodium bicarbonate and good hydration.
c) Suplementation: In cases of Complex III deficiency it can improve with Vitamin K3 suplements (40-60 mg/day) and Coenzyme Q10(80-300mg/day) both show that is early use besides improving the neurological symptoms can have a beneficial effect in the prognostic of renal function. Proton aceptors, Carnitine and Vitamin C are partially effective though there effects are minimal. Use of Citrate solution, potasium, phosphorous, Vitamin-D and fluids may be required for patients with renal tubulophaties with poliuria, renal tubular-acidosis and Fanconi syndrome that produce those deficiencies [27].
d) Dislipidemia when severe has to be controled because high blood concentrations of free fatty acids can enter the cell and the mitocondriastraspassing their internal membrane incresing their concentrations in this compartament and due to the absence of AcylCoAsyntethasa for large chain free fatty acids they can not be directed to the β oxidative pathway which results in and increased Lipid Perioxidation with lipotoxicity and damage to the mitochondrias, here the treatment besides diet with reduction in the content of mono and polyinsaturated fats has to include PPARγ agonists using any of them in the appropiate dosages [28].
e) Antioxidantes: Like omega-3 plolyinsaturated fats, the N-acetilcisteine and alopurinol.
f) Thiazolinedions: Is a PPARγ useful in patients with adquiredmitocondrial dysfunction due to its anti-inflamatories properties and other inmunological effects known currently, they inhibit the production of Tumor necrosis factor alfa, as well as the Nuclear Factor kappa Beta, they also inhibit the attraction and migration of macrophges through the inhibition of macrophge Migration Protein-1 (MP-1), with all thes effects they reduce inflamation and fibrosis and at the renal level they stimulate the Nephrin gene expression a protein crucial in the renal filter that prevents proteinuria and consequentely the progression of renal disease [29].
g) Sirtruins and Resveratrol:A new strategy that has demostrated to prolong life span in all the animal species is the caloric restriction [30] but is a complicated task to get to the target and can produce proteins and vitamins deficiencies; on the other hand the Sirtruins family in particular Sirtruin-1 act as desacetylation enzymes of Histones and other proteins regulators of the DNA transcription including HIF-2α COX2, PGC-α, Smad3, Smad7, the tumuralsupressor p53, FOXO3, FOXO4, NK-κβand induce Nitric Oxide Synthesis (NOS), all of them are related with the biogénesis and the mitochondrial function, besides they reduce the oxidative stress, fibrosis and apoptosis [31]. The resveratrol founded in the red grapes and strawberries, cranberries, blueberries, raspberries and red wines (wich is an inductor of Sirtruin-1) improves the mitochondrial function and the lipid concentrations, keep and mantain the PGC-1α protecting at the renl level the integrity of the podocytes and therefore the renal filter. Other new Sirtruin-1 agonist have been described like the mononuclide precursor of NAD+ or the riboside that incresase metabolism [32].
Genetic Therapy
CONCLUSION
REFERENCES
- Balk J, Pierik AJ, Netz DJ, Mühlenhoff U, Lill R (2004) The hydrogenase-like Nar1p is essential for maturation of cytosolic and nuclear iron-sulphur proteins. EMBO 23: 2105-2115.
- Chial H, Craig J(2008) mtDNA and Mitochondrial Diseases. Nature Education 1:217.
- Ingman M, Kaessmann H, Pääbo S, Gyllensten U (2000) Mitochondrial genome variation and the origin of modern humans. Nature 408: 708-713.
- Francessco Emma, Enrico Bertini (2012) Leionardo Salviati and Giovanni Montini: Renal Involvement in mitochondrial cytophaties, PediatrNephrol 27: 539-550.
- McFarlandR, Taylor RW, Turbull DM (2007) Mitochondrial diseases- its iumpact, etiology and pathology. Curr Top Dev Biol 77: 113-155.
- Chargaff E (1950) Chemical specificity of nucleic acids and mechanims of their enzimatic degradation. experientia 6: 201-209.
- http:www.biography.com/people/rosalind-franklin-9301344.
- Wilkins MH, Strokes AR, Wilson HR (1953) Molecular structure of deoxypentose nucleic acid. Nature.421: 398-400.
- Venter JC, Mark D Adams, Eugene W Myers, Peter W Li, Richard J Mural, et al.,(2001) The sequence of the human genoma. Science 291: 1304-1351.
- Michael W Gray, Gertraud Burger, Franz Lang B (1999) Science 283: 1476-1481.
- International Human Genome Sequencing Consortium(2001) Nature 409, 860Mark van der Giezen and Jorge Tovar(2005);Degenerate Mitochondria, EMBO 6: 525-530.
- Maechler P, Wolheim CB (2011) Mitochondrial function in normal and diabetic cells. Nature 13: 807-812.
- Allen JF (2003) The function of genomes in bioenergetic organelles. Philos Trans R Soc B Biol Sci 358:19-38.
- Ho Joong Sung, Wenzhe Ma, Ping-yuan Wang, James Hynes, Tomas C. O’Riordan, Christian A. Combs, J. Phillip McCoy Jr, Fred Bunz, Ju-Gyenong Kang Paul, M. Hwang. (2010). Nature: Communications 1:1-8
- Abrahamsen MS, Templeton TJ, Enomoto S, Abrahante JE, Zhu G, et al. (2004) Complete genome sequence of the apicomplexan, Cryptosporidium parvum. (2004), Complete genoma sequence of the apicomplexan, Cryptosporiudium parvum. Science 304: 441-445.
- Wallance DC, Fan W, Procaccio V (2010) Mitochondrial Energetics and Therapeutics. Annu Rev Pathol 5: 297-348.
- Takasi T, Thomas L (2008) Quality control of mitochondria: protection against neurodegeneration and ageing. EMBO; 27: 306-314.
- McBride HM, Neuspiel M, Wasiak S (2006) Mitochondria: more than just a powerhouse.. Curr Biol 16: 551-560.
- Ema F, Enrico B, Leonardo S, Giovani M (2012) Renal involvement in mitochondrial cytopathies. Pediatr Nephrol 27: 539-550.
- Barratt CLR, Kay V, Oxenham SK (2009) The human spermatozoon - a stripped down but refined machine. J Biol 8: 63-66.
- Schon EA, Manfredi G (2003) Neuronal Degeneration and mitochondrial dysfunction. J Clin Inves 111: 303-312.
- Russell JF, Fu YH, Ptá?ek LJ (2013) Episodic neurologic disorders: syndromes, genes, and mechanisms. Annu. Annu Rev Neurosci 36: 25-50.
- Che R, Yuan Y, Huang S, Zhang A (2014) Mitochondrial dysfunction in the pathophysiology of renal diseases.Am J Physiol Renal Physiol 306: 367-378.
- Armstrong JS (2007) Mitochondrial Medicine: Pharmacological targeting of mitochondria in disease. British J Pharmacol 151: 1154-1165.
- Emma F, Bertini E, Salviati L, Montini G (2012): Renal involvement in mitochondrial cytopathies. Pediatr Nephrol 27: 539-550.
- Rahman S, Hall AM (2013) Mitochondrial diease-an important cause of end-stage renal failure. Pediatr Nephrol 28: 357-361.
- Kao MP, Ang DS, Gandy SJ, Nadir MA, Houston JG, et al. (2011) Allopurinol benefits left ventricular mass and endotelial dysfunction inchronic kidney disease. J Am Soc Nephrol 22: 1382-1389.
- Ambros V (2004) The Function of animal microRNAs. Nature 431: 350-355.
- Hesselink MKC, Mensink M, Schrauwen P (2007) Lipotoxicity and Mitochondrial Dysfunction in Type 2 Diabetes. Inmun, Endoc. & Metab. Agents in Med. Chem 7: 3-17.
- Sarafidis PA, Stafylas PC, Georgianos PI, Saratzis AN, Lasaridis AN (2010) Effect of Thiazolinedions on albuminuria and proteinuria in diabetes: a meta-analysis. Am J Kidney Dis 55: 835-847.
- Timmers S, Konings E, Bilet L, Houtkooper RH, van de Weijer T, et. al (2011) Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab 14: 612-622.
- Kitada M, Kume S, Takeda-Watanabe A, Kanasaki K, Koya D (2013) Sirtruins and renal diseases: relationship with ageing and diabetic nephropathy Clin Sci (Lond) 124: 153-164.
- Mai A, Valente S, Meade S, Carafa V, Tardugno M, et al. (2009) Study of 1,4-dihydropyridine structural scaffold: discovery of novel sirtuin activators and inhibitors. J Med Chem 52: 5496-5504.
- Bandiera S, Ruberg S, girard M, Cagnard N, Hanein S, et al. (2011) Nuclear outsourcing of RNA interference components to mictochondria. Plos One 6: 20746.
Citation: Gordillo de Anda RJ (2019) Mitochondrial DNA Mutations and its Rol in the Genesis of Renal Diseases an Update. J Nephrol Renal Ther 5: 021
Copyright: © 2019 Rodolfo J Gordillo de Anda, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.