


Diabetes results from an inadequate mass of functional β-cells. Such inadequacy could result from loss of β-cells due to an immune assault or the inability to compensate for insulin resistance. Thus, mechanisms that regulate the number of β-cells will be key to understanding both the pathogenesis of diabetes and for developing therapies. In this study, we show that cell cycle regulator p27 plays a crucial role in establishing the number of β-cells formed before birth. We show that p27 accumulates in terminally differentiated β-cells during embryogenesis. Disabling p27 allows newly differentiated β-cells that are normally quiescent during embryogenesis to reenter the cell cycle and proliferate. As a consequence, excess β-cells are generated in the p27−/− mice, doubling their β-cell mass at birth. The early postnatal expansion of β-cell mass was unaffected in p27−/− mice, indicating that the main function of p27 is to maintain the quiescent state of newly differentiated β-cells generated during embryogenesis. The expanded β-cell mass was accompanied by increased insulin secretion; however, the p27−/− mice were glucose intolerant, as these mice were insulin insensitive. To assess the role of p27 to affect regeneration of β-cells in models of diabetes, p27−/− mice were injected with streptozotocin (STZ). In contrast to control mice that displayed elevated blood glucose levels, p27−/− mice showed decreased susceptibility to develop STZ-induced diabetes. Furthermore, β-cells retained the ability to reenter the cell cycle at a far greater frequency in p27−/− mice after developing STZ-induced diabetes compared with wild-type littermates. These data indicate that p27 is a key regulator in establishing β-cell mass and an important target for facilitating β-cell regeneration in therapies for diabetes.
During pancreatic organogenesis, endocrine cells are generated from a population of pancreatic progenitor cells (1–3). The endocrine cells that differentiate from progenitor cells are mitotically quiescent, and direct-lineage tracing analyses indicate that a population of progenitor cells persists throughout embryogenesis, which allows the differentiation of new endocrine cells (4,5). The early postnatal period is characterized by massive expansion of the β-cell mass as the endocrine pancreas adapts to metabolic needs. The number of β-cells during this period is governed by balancing endocrine cell growth and endocrine cell death (6–8). Recent studies from our laboratory have shown that high rates of β-cell proliferation account for most of this increase in β-cell mass (9). In addition, lineage analyses suggest that a significant portion of β-cells during adult life originate from preexisting β-cells (10). These studies have focused interest in the control of cell cycle regulation in β-cells, as it may be vital not only in determining the size of the endocrine pancreas but also as a means to regenerate β-cell mass.
The decision of terminally differentiated cells to replicate is often made at the interface of G0–G1 phase of the cell cycle (9). Mitotic stimulation can induce entry into the cell cycle in the G1 phase by assembly of d-type cyclin with Cdk4/6 (10,11). Cyclin D2 and CDK4 have been shown to be essential for the replication of β-cells (9,14). The activated cyclin d-Cdk4/6 complexes phosphorylate retinoblastoma (pRb) (rev. in 12,13). This initial pRb phosphorylation is followed by additional phosphorylation by cyclin E-Cdk2 complex. Once phosphorylated, pRb releases tethered transcription factor E2F and irreversibly commits the cell to progress through the cell cycle (rev. in 14). Thus, while cyclin D2-Cdk4 complex formation is clearly essential for β-cell proliferation, it alone is unlikely to be sufficient for committing a cell to divide.
Whether a β-cell would undergo a round of division or remain quiescent would depend on the balance between the cyclin D2-Cdk4 complex, which forms in response to mitotic signals, and cyclin kinase inhibitors (CKIs), which block the activity of the cyclin E-Cdk2 complex. Two groups of CKIs have been described (15). These include the Ink4 family, members of which specifically inhibit cyclin d-Cdk4/6 activity, and the CIP/KIP family, which includes p21, p27, and p57, which exhibit promiscuous CDK-inhibitory activity. What role do CKIs play in the differentiation of β-cells during embryogenesis and in the β-cell proliferation during postnatal period? We show here that newly differentiated β-cells during embryogenesis accumulate p27 and that disabling p27 in these cells allows these β-cells to divide. Consequently, we observed a significant increase in β-cell mass of p27−/− mice, which was accompanied by hyperinsulimia, consistent with recent studies that have reported that the deletion of p27 in mouse models of type 2 diabetes prevented hyperglycemia (16). We show that the increase in β-cell mass in the p27−/− mice was primarily due to newly formed β-cells that underwent extra rounds of division during embryogenesis. We propose that the cellular abundance of p27 is a critical determinant of whether a β-cell divides or remains quiescent and that p27 may play a crucial role in regulating β-cell proliferation in adapting to metabolic demands.
RESEARCH DESIGN AND METHODS
Animal breeding and genotyping.
Targeted disruption of the p27 allele has been described previously (17). Mice lacking p27 gene function were obtained from crosses of p27+/− animals in order to generate heterozygous and wild-type littermates. The day of birth was designated postnatal day 0. Embryos isolated from these mice were considered to be 0.5 days of gestation at 12:00 p.m. on the day the plugs were detected. DNA extracted from tails was used for PCR-based genotyping using standard methods. Primers used for genotyping were: p27WT-F 5′ TCAAACGTGAGAGTGTCTAACGC3′, p27WTR5′AGGGGCTTATGATTCTGAAAGTCG3′, and p27NullR5′ATATTGCTGAAGAGCTTGGCGG-3′. Pancreatic tissue was dissected in cold PBS, fixed in 4% formaldehyde for 4 h to overnight, followed by dehydration in grades of ethanol, and stored at −20°C until processed for paraffin embedding.
Immuohistochemistry.
The pancreas was oriented during the paraffin embedding process such that sections were cut along the head-tail axis. Sections were deparaffinized in toluene, rehydrated in grades of alcohol, and washed in H2O. All slides were subject to antigen retrieval protocols using Antigen Unmasking Buffer (Vector Labs). After antigen unmasking, the slides were cooled to room temperature, permeablized in 0.2% Triton X-100/Tris-buffered solution for 20 min, and blocked with 0.2% Tween 20/3% IgG-free BSA/Tris-buffered solution. Primary antibodies were diluted in the blocking solution at the following dilutions: mouse anti-glucagon 1:1,000 (Sigma); guinea pig anti-insulin 1:500 (Dako); rabbit anti-phosphohistone H3 1:200 (Upstate); Islet1 (Isl1) 1:50 (Developmental hybridoma); Neurogenin3 (Ngn3) 1:100 (gift of Maike Sanders); and mouse anti-p27 1:200 (Santa Cruz). Donkey- and goat-derived secondary antibodies conjugated to fluorescein isothiocyanate and Cy3 were diluted 1:500 (The Jackson Laboratories). All slides were mounted with Vectashield (Vector) or Permount (Fisher). Fluorescent slides were viewed using a Leica DM6000 microscope and images acquired using Openlab software.
5-bromo-2′-deoxyuridine incorporation.
Thymidine analog 5-bromo-2′-deoxyuridine (BrdU; 0.025 g/g body wt) was injected intraperitoneally before harvesting the pancreas as indicated. Pancreas were isolated and processed for histology as described above. Mouse anti-BrdU antibody/nuclease solution (Amersham/Pharmacia) was applied for 1 h at room temperature.
β-Cell mass analysis.
The pancreas was trimmed of all nonpancreatic tissue, weighed, and processed for histology. The pancreas was embedded in paraffin such that longitudinal sections from tail to head were obtained. Sections were deparaffinized in toluene and incubated in 0.3% hydrogen peroxide in ethanol for 30 min. Sections were prepared for immunohistochemistry as described above. A montage of the whole pancreatic section was created using Openlab (Improvision) and ImageJ (National Institutes of Health) software on a Leica DM6000. Five representative sections that spanned the width of the pancreas were used in the analysis of β-cell mass. The relative cross-sectional area of β-cells was determined by quantification of the cross-sectional area occupied by β-cells. Each section was analyzed to estimate β-cell and total tissue area. The β-cell mass per pancreas was estimated as the product of the cross-sectional area of β-cells/total tissue and the weight of the pancreas.
Glucose tolerance tests, plasma insulin levels, and streptozotocin treatment.
Following a 16-h fast, baseline blood glucose levels (milligrams per deciliter) were measured in tail-vein blood from mice using a OneTouch Ultra glucose meter (LifeScan, Milpitas, CA). Glucose (2 mg dextrose/g body mass) in sterile PBS was injected intraperitoneally and blood glucose measured 15, 30, 60, and 120 min after injection. Glucose tolerance tests were performed between 10 and 12 weeks after birth. To measure plasma insulin levels, ∼40 μl blood was collected from the tail vein before, and 30 min after, interperitoneal injection with glucose (3 mg/g body mass). Blood samples were centrifuged, and serum was used to measure insulin concentrations with an enzyme-linked immunosorbent assay Rat/Mouse Insulin kit (Linco Research). Insulin tolerance tests were performed on animals between 10 and 12 weeks of age. In animals that were fasted for 6 h, 1 mU/g body mass of human insulin diluted in PBS was injected interperitoneally. Blood glucose levels were measured immediately before injection and every 20 min thereafter for the 1st h after injection. Data are represented as means of n = 5 p27 mutants and n = 4 age-matched wild-type mice. p27−/− mice and wild-type littermates (aged 12 weeks; n = 3) were given a single interperitoneal bolus injection of 90 mg/kg streptozotocin (STZ; Sigma-Aldrich) freshly prepared in citrate buffer (pH 4.5). Control p27−/− and wild-type littermates (n = 3) received a sham injection of vehicle alone. Glucose was monitored weekly for 3 weeks after the STZ injection. After 3 weeks, the pancreas was collected and processed for histology.
Statistical analysis.
All data were expressed as means ± SE. The statistical significance of differences was measured by unpaired Student’s t test. A P value <0.05 indicated statistical significance.
RESULTS
As understanding the role of cell cycle proteins in the specification of endocrine cells from pancreatic progenitors is essential to developing methods of β-cell regeneration, we began by analyzing the role of p27 during pancreatic development. The major mechanism for regulating p27 levels is thought to be posttranslational proteolytic degradation, as the mRNA levels of p27 are constant throughout the cell cycle (18,19). The cellular distribution of p27 protein was examined by performing immunohistochemistry on pancreatic sections from various embryonic stages. The p27 protein was confined to the nucleus of epithelial cells and absent from the mesenchyme during early stages of pancreatic bud formation. Costaining with endocrine hormones at embryonic day 11.5 revealed that p27 was present in the nucleus of glucagon-expressing cells (Fig. 1A). To assess whether p27 is expressed in other endocrine cells, we examined the pancreatic epithelium for the expression of insulin and p27 at embryonic day 14.5. At this stage, p27 expression was detected in the nucleus of all the newly formed β-cells (Fig. 1B). Expression of p27 was not detected in cells stained with exocrine marker, carboxypeptidase A, indicating that p27-expressing cells were restricted to the endocrine pancreas throughout embryogenesis (data not shown). To assess when p27 protein accumulates during endocrine cell differentiation, we compared its expression with two transcription factors that are necessary for endocrine differentiation and mark temporally different stages of differentiation. Ngn3 is the earliest marker expressed in pancreatic progenitors that are committed to an endocrine cell fate, while Isl1 is expressed later in the differentiation process and marks differentiated endocrine cells before hormone activation (20,21). Costaining of p27 with these markers revealed that all p27-expressing cells did not overlapped with with Ngn3-expressing progenitors (Fig. 1C) but did costain with all Isl1-expressing cells (Fig. 1d–F). These analyses suggest that during embryonic development, differentiated endocrine cells accumulate p27 after the pancreatic progenitors have committed to the endocrine cell fate.
To explore the relationship of p27 accumulation to cell cycle regulation in endocrine cells, we injected pregnant dams with bromodeoxyuridine (BrdU) and analyzed the embryos for the expression of p27. No overlap of p27 expression was observed with BrdU after a 1-h pulse, indicating that p27-expression cells were not in S-phase (Fig. 1H). As the timing of p27 accumulation in endocrine cells may reflect changes in their cell cycle characteristics, we assessed whether differentiated endocrine cells could incorporate BrdU. Endocrine cells expressing Isl1 or glucagon (Fig. 1I and J) did not incorporate BrdU at embryonic day 12.5. These results suggested that the newly generated endocrine cells during embryogenesis are quiescent. The accumulation of p27 in these differentiated cells coincided with the acquisition of a postmitotic state for newly differentiated endocrine cells during embryogenesis.
To determine whether p27 accumulation in endocrine cells was involved in maintaining the quiescent state, we analyzed the cell cycle characteristics of endocrine cells in p27−/− embryos. We assessed whether the absence of p27 affected the quiescent state of endocrine cells in p27−/− embryos by examining for the incorporation of BrdU. Glucagon-expressing cells are the first endocrine cells to differentiate during development, and these cells did not incorporate BrdU in day 11.5 wild-type embryos (Fig. 2A). By contrast, a number of glucagon-expressing cells within the pancreatic epithelium of the p27−/− littermates incorporated BrdU (Fig. 2B). Similarly, analysis of pancreatic sections from embryonic day 14.5 p27−/− embryos showed a number of insulin-expressing cells that incorporate BrdU in contrast to wild-type littermates in which insulin-expressing cells did not incorporate BrdU (Fig. 2C and D). Analysis of pancreas from embryonic day 15.5 embryos also showed a dramatic increase in the number of endocrine cells in the pancreatic epithelium of p27−/− embryos compared with wild-type littermates (Fig. 2E–G). The increase in the number of endocrine cells in p27−/− embryos was likely due to the ability of terminally differentiated endocrine cells to transition from quiescence to proliferation. No change in endocrine cell size was detected in p27−/− embryos. The increase in the number of endocrine cells, along with the expression analysis of p27 described above, suggested that p27 more likely played a role in regulating the number of endocrine cells generated during development rather than had direct involvement in the differentiation of endocrine cells per se.
To quantify the increase in β-cells, we analyzed the β-cell mass of p27−/− pups and their wild-type littermates. At birth, the p27−/− pups displayed a twofold increase in β-cell mass when compared with wild-type littermates (Fig. 3A–C). The increase in β-cell mass in p27−/− pups was specific to the endocrine pancreas and occurred despite the fact that body mass and pancreas mass were similar to the wild-type littermates (Fig. 2D and E). To assess whether p27 played a role in the early postnatal expansion of β-cells, we measured β-cell mass in 3-week-old mice. The β-cell mass in the pancreas from 3-week-old p27−/− mice was twofold greater than in wild-type littermates. However, the proportional increase in β-cell mass during this 3-week period was similar in p27−/− and wild-type littermate pancreas, which suggested that the growth of β-cells during this early postnatal period was unaffected by the loss of p27. To confirm this observation, we assessed β-cell proliferation by costaining phosphorylated histone H3 with insulin (which marks cells in both G2 and M phases). No difference in the frequency of proliferating β-cells was observed in p27−/− mice and their wild-type littermates (Fig. 2F).
To determine if there were metabolic effects in the p27−/− mice due to increased β-cell mass, we measured the ability to metabolize glucose in both wild-type and p27−/− mice. The p27−/− mice had fasting blood glucose levels that were slightly elevated when compared with the wild-type mice (P < 0.05) (Fig. 4). Following intraperitoneal glucose, p27−/− mice showed a decreased ability to clear glucose from the blood after injection. This indicated that the p27−/− mice were glucose intolerant. After overnight fasting, the serum insulin levels were elevated in p27−/− mice compared with wild-type littermates and increased after a glucose challenge in both p27−/− and wild-type littermates (Fig. 5A). The increased serum insulin levels coupled with mild impaired glucose tolerance indicated changes in insulin sensitivity in the p27−/− mice. We carried out an insulin tolerance test, in which blood glucose was monitored after intraperitoneal insulin injection. The p27−/− mice displayed impairment in insulin responsiveness relative to their wild-type littermates (P < 0.05) (Fig. 5B).
The adult p27−/− mice displayed a twofold greater β-cell mass compared with wild-type littermates (Fig. 6A). Unlike the early postnatal period, the frequency of mitotic β-cells is very low (<0.1%), although the frequency of mitotic β-cells in adult p27−/− pancreas was slightly higher than their wild-type littermates (Fig. 6G). We hypothesized that β-cells from p27−/− mice have a greater propensity to proliferate, which could have a competitive advantage during the regeneration process. To test this, we used a STZ model, which leads to the selective loss of β-cells (22). p27−/− and wild-type littermates were subject to a single STZ dose to induce partial loss of β-cells, and blood glucose was monitored for 3 weeks. Wild-type mice displayed elevated blood glucose and developed STZ-induced diabetes within 2 weeks (Fig. 6B). By contrast, p27−/− littermates’ blood glucose levels did not rise dramatically and remained significantly lower than STZ-injected littermates. Neither the p27−/− nor wild-type littermates injected with vehicle alone developed hyperglycemia. Analysis of pancreas 3 weeks after STZ injections showed substantial loss of β-cells in the pancreas from both p27−/− and wild-type littermates (Fig. 6E and F). However, quantification of the frequency of mitotic β-cells revealed that β-cells from p27−/− mice proliferated at greater frequency than the wild-type littermates (Fig. 6G–I).
DISCUSSION
During embryogenesis, β-cells arise from differentiation of pancreatic progenitor cells. These newly differentiated β-cells are mitotically quiescent and do not proliferate during embryogenesis. We show here that the cell cycle regulator p27 accumulates in differentiated β-cells. In the absence of p27, the differentiated β-cells are no longer quiescent and reenter the cell cycle to proliferate. This proliferation of β-cells in p27−/− mice during embryogenesis results in increased β-cell mass at birth when compared with wild-type littermates. We suggest that the accumulation of p27 in β-cells, which differentiate during embryogenesis, prevents reentry into the cell cycle. Thus, p27 functions to maintain the quiescent state of the newly differentiated β-cells during embryogenesis. This function of p27 is essential in establishing the total number of β-cell within the pancreas.
In contrast to embryogenesis, the proliferation of the terminally differentiated β-cells during early postnatal period accounts for a massive increase in β-cell mass. Endocrine cells that incorporate BrdU are first observed as early as embryonic day 17.5, indicating that at the late stages of embryogenesis, endocrine cells transition to allow replication. This does not coincide with changes in the levels of p27, but, instead, cyclin D2 levels are upregulated, allowing cells to reenter the cell cycle (23). This scenario is consistent with our observation that the proportional increase in β-cell mass in the 3-week period after birth was the same in both p27−/− mice and their wild-type littermates. Furthermore, quantification of proliferating β-cells shows the same frequency of mitotic β-cells in p27−/− mice and their wild-type littermates during this period. These analyses indicate that the increased β-cell mass observed in the adult p27−/− mice was primarily due to the excess β-cells generated during embryogenesis. Our focus here has been on the role of p27 in cell cycle regulation; however, epigenetic mechanism can operate in the pancreas to regulate p27 transcription, which thereby regulate β-cell growth (24).
The increased β-cell mass in p27−/− pancreas correlated with the increased insulin content (data not shown), suggesting that the excess β-cells generated during embryogenesis were functional. However, having excess functional β-cells does not necessarily lead to increased insulin secretion, unless there is an increased metabolic demand (25). The enhanced insulin secretion in p27−/− mice was due to insulin insensitivity, as suggested by the insulin tolerance test. These observations are consistent with a recent study (26) that demonstrated adipocyte hyperplasia in p27−/− mice, which is likely to lead to insulin insensitivity. Together, these data suggest that the loss of p27 resulted in two opposing metabolic phenotypes: adipocyte hyperplasia, which could cause insulin insensitivity, and an expanded β-cell mass, which could increase insulin secretion in response to metabolic needs (27). We propose that as a result of these opposing metabolic phenotypes, the p27−/− mice generated by conventional gene targeting approaches display relatively mild metabolic phenotype. We predict that tissue-specific loss of p27 would lead to more severe metabolic consequences. Conditional knockout of p27 in either β-cells or adipocytes would resolve the role of p27 in these different tissues to contribute to glucose metabolism.
Our main objective here was to elucidate the role of p27 in the pancreatic β-cell and evaluate its potential in regenerative therapies. The most immediate application of this work is the potential to target p27 in ways that could be beneficial for cell-based therapies in which the extent of cell proliferation could have potential benefit, given the current need to expand β-cells for islet transplantation. We propose that islets in which p27 is disabled could provide a competitive advantage upon transplantation and would markedly improve islet engraftment. Much less immediate is a therapy for diabetes that uses suppression of p27 activity to induce division and restore β-cell mass. Any such therapy would depend on the ability to reactivate p27 in order to halt proliferation and to do so with the proper amount of β-cell mass to restore blood glucose homeostatis.

p27 accumulates in mitotically quiescent, terminally differentiated endocrine cell. A: Immunofluorescence staining shows that p27 (red) is expressed in glucagon-expressing cells (green) at embryonic day 11.5. B: p27 (red) expression in insulin cells (green) at embryonic day 14.5. C: Endocrine precursor cells, marked by Ngn3 (green), do not express p27 (red) at embryonic day 13.5. D–F: Costaining reveals p27 (red) is expressed in all committed endocrine cells, as marked by the transcription factor Isl1 (green) at embryonic day 12.5; nuclei are marked by 4,6-diamidino-2-phenylindole (DAPI) (blue). G: p27 staining was not observed in the pancreas derived from a p27−/− embryo, at embryonic day 14.5 H: p27-expressing cells do not incorporate BrdU (green) at embryonic day 12.5. H and I: Newly differentiated endocrine cells are postmitotic. Isl1 (red) and glucagon-expressing cells (red) do not incorporate BrdU (green) at embryonic day 11.5.
p27 accumulates in mitotically quiescent, terminally differentiated endocrine cell. A: Immunofluorescence staining shows that p27 (red) is expressed in glucagon-expressing cells (green) at embryonic day 11.5. B: p27 (red) expression in insulin cells (green) at embryonic day 14.5. C: Endocrine precursor cells, marked by Ngn3 (green), do not express p27 (red) at embryonic day 13.5. D–F: Costaining reveals p27 (red) is expressed in all committed endocrine cells, as marked by the transcription factor Isl1 (green) at embryonic day 12.5; nuclei are marked by 4,6-diamidino-2-phenylindole (DAPI) (blue). G: p27 staining was not observed in the pancreas derived from a p27−/− embryo, at embryonic day 14.5 H: p27-expressing cells do not incorporate BrdU (green) at embryonic day 12.5. H and I: Newly differentiated endocrine cells are postmitotic. Isl1 (red) and glucagon-expressing cells (red) do not incorporate BrdU (green) at embryonic day 11.5.

In the absence of p27, terminally differentiated endocrine cells proliferate. A: In wild-type (WT) animals, glucagon cells (green) do not incorporate BrdU (red). B: Glucagon cells (green) in p27−/− embryos show aberrant incorporation of BrdU (red) at embryonic day 11.5. C: At embryonic day 14.5, insulin cells (green) do not incorporate BrdU (red). D: In p27−/− embryos, insulin cells (green) incorporate BrdU (red). E: Typical numbers of glucagon (green) and insulin (red) cells in embryonic day 15.5 wild-type embryos. F: p27−/− embryos show increased numbers of classically postmitotic endocrine cell types. Insulin (red) and glucagon (green) at embryonic day 14.5. G: Quantification of the glucagon-expressing cell area in wild-type and embryonic day 15.5 p27−/− pancreas.
In the absence of p27, terminally differentiated endocrine cells proliferate. A: In wild-type (WT) animals, glucagon cells (green) do not incorporate BrdU (red). B: Glucagon cells (green) in p27−/− embryos show aberrant incorporation of BrdU (red) at embryonic day 11.5. C: At embryonic day 14.5, insulin cells (green) do not incorporate BrdU (red). D: In p27−/− embryos, insulin cells (green) incorporate BrdU (red). E: Typical numbers of glucagon (green) and insulin (red) cells in embryonic day 15.5 wild-type embryos. F: p27−/− embryos show increased numbers of classically postmitotic endocrine cell types. Insulin (red) and glucagon (green) at embryonic day 14.5. G: Quantification of the glucagon-expressing cell area in wild-type and embryonic day 15.5 p27−/− pancreas.

Increased β-cell mass is established during embryogenesis and maintained during postnatal development. A: Immunofluorescence shows representative pictures of wild-type pancreas at postnatal day 21, stained for insulin (green) and glucagon (red). B: p27−/− pancreas have increased numbers of endocrine cells (insulin, green; glucagon, red). C: β-Cell mass quantification at postnatal days 0 (P0) and 21 (P21) (n = 3 per genotype, per age-group). D and E: Pancreas mass (D) and body mass (E) were not significantly different between wild-type and p27−/− animals at postnatal days 0 or 21 (n = 10 per genotype, per age-group). F: β-Cell replication measured by expression of phosphorylated histone H3 (PPH3) immunostaining at postnatal day 21 (n = 3 per genotype; P = 0.38). Data are shown as means ± SE. *P < 0.05; **P < 0.01.
Increased β-cell mass is established during embryogenesis and maintained during postnatal development. A: Immunofluorescence shows representative pictures of wild-type pancreas at postnatal day 21, stained for insulin (green) and glucagon (red). B: p27−/− pancreas have increased numbers of endocrine cells (insulin, green; glucagon, red). C: β-Cell mass quantification at postnatal days 0 (P0) and 21 (P21) (n = 3 per genotype, per age-group). D and E: Pancreas mass (D) and body mass (E) were not significantly different between wild-type and p27−/− animals at postnatal days 0 or 21 (n = 10 per genotype, per age-group). F: β-Cell replication measured by expression of phosphorylated histone H3 (PPH3) immunostaining at postnatal day 21 (n = 3 per genotype; P = 0.38). Data are shown as means ± SE. *P < 0.05; **P < 0.01.
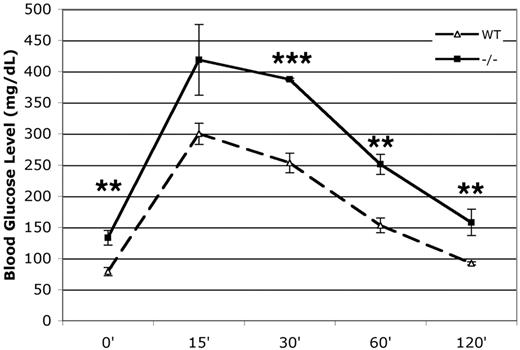
p27−/− are glucose intolerant. Glucose tolerance tests were performed after an injection of 2 g/kg glucose in 10- to 12-week-old overnight-fasted mice (n = 5 per genotype; data are expressed as means ± SE). *P < 0.05; **P < 0.01; ***P < 0.005.
p27−/− are glucose intolerant. Glucose tolerance tests were performed after an injection of 2 g/kg glucose in 10- to 12-week-old overnight-fasted mice (n = 5 per genotype; data are expressed as means ± SE). *P < 0.05; **P < 0.01; ***P < 0.005.
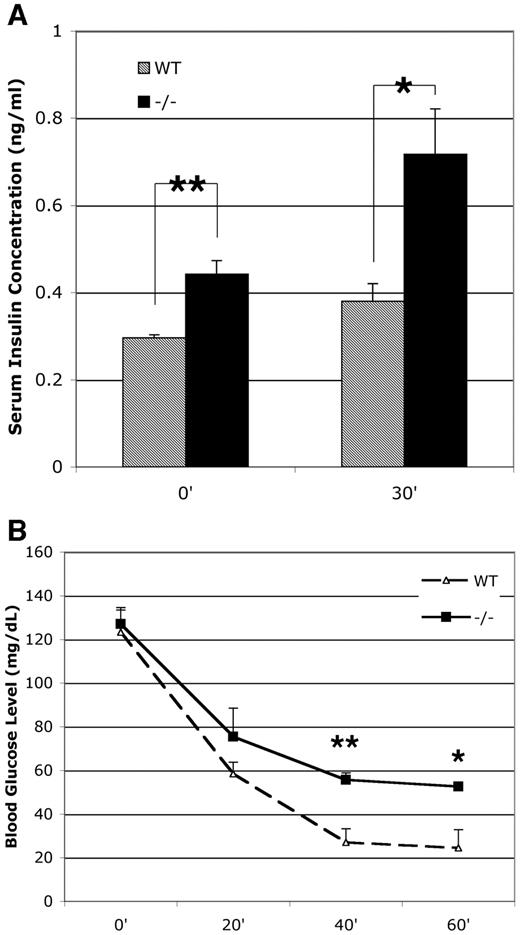
Elevated plasma insulin levels and decreased insulin tolerance is indicative of decreased insulin sensitivity. A: Serum insulin levels were measured after an injection of 3 g/kg glucose in 10- to 12-week-old overnight-fasted mice. B: Insulin tolerance tests were performed after an injection of 1 IU/kg insulin in animals fasted for 6 h (n = 3 per genotype; data are expressed as means ± SE). *P < 0.05; **P < 0.01.
Elevated plasma insulin levels and decreased insulin tolerance is indicative of decreased insulin sensitivity. A: Serum insulin levels were measured after an injection of 3 g/kg glucose in 10- to 12-week-old overnight-fasted mice. B: Insulin tolerance tests were performed after an injection of 1 IU/kg insulin in animals fasted for 6 h (n = 3 per genotype; data are expressed as means ± SE). *P < 0.05; **P < 0.01.

p27−/− mice have decreased susceptibility to develop STZ-induced diabetes due to enhanced β-cell proliferation. A: β-Cell mass in 16-week-old mice. B: Random-fed blood glucose levels of wild-type and p27−/− animals injected with 90 mg/kg STZ or control citrate buffer (n = 3 per genotype and condition; data are expressed as means ± SE). C and D: Islets from wild-type (WT) (C) and p27−/− (D) mice injected with control citrate buffer mice. Islets from 15-week-old wild-type (E) and p27−/− (F) mice injected with 90 mg/kg STZ at 12 weeks of age. G and H: Representative islets from wild-type (G) and p27−/− (H) mice injected with 90 mg/kg STZ and stained for phosphorylated histone H3 (PHH3; red) and insulin (green). I: β-Cell proliferation as measured by phosphorylated histone H3 staining in animals injected with 90 mg/kg STZ or control citrate buffer. *P < 0.05.
p27−/− mice have decreased susceptibility to develop STZ-induced diabetes due to enhanced β-cell proliferation. A: β-Cell mass in 16-week-old mice. B: Random-fed blood glucose levels of wild-type and p27−/− animals injected with 90 mg/kg STZ or control citrate buffer (n = 3 per genotype and condition; data are expressed as means ± SE). C and D: Islets from wild-type (WT) (C) and p27−/− (D) mice injected with control citrate buffer mice. Islets from 15-week-old wild-type (E) and p27−/− (F) mice injected with 90 mg/kg STZ at 12 weeks of age. G and H: Representative islets from wild-type (G) and p27−/− (H) mice injected with 90 mg/kg STZ and stained for phosphorylated histone H3 (PHH3; red) and insulin (green). I: β-Cell proliferation as measured by phosphorylated histone H3 staining in animals injected with 90 mg/kg STZ or control citrate buffer. *P < 0.05.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Article Information
This work was supported by the National Institutes of Health Grant R01 DK-6876 and the Larry Hillblom and Juvenile Diabetes Research Foundations (to A.B.). S.G. is supported by Ruth L. Kirschstein National Research Service Award GM07185.
We received superb technical help from Rosemary Soliz. We are grateful to James Roberts for p27−/− mice, Maike Sander for the NGN3 antibody, and Peter Butler for helpful discussions. The antibodies to Islet1, developed by T. Jessell, were obtained from the Developmental Studies Hybridoma Bank.