
Abstract
The effect of carbonates on the formation of magnesium silicate hydrate phases (M-S-H) was investigated in paste experiments. At molar ratio Mg/Si = 1.5, M-S-H pastes were synthesised from silica fume and MgO or MgO/hydromagnesite in a sodium carbonate-rich environment (dissolved 1.7 g of Na2CO3 for 100 g of binder). Thermogravimetric analysis, X-ray diffraction, and 29Si MAS NMR data showed that M-S-H phases formed much faster in the presence of carbonates, which destabilised brucite. 23Na MAS NMR data, cation exchange capacity measurements, thermogravimetric analysis coupled with infrared exhaust gas analysis, and 13C MAS NMR data showed that traces of sodium and carbonates were sorbed on M-S-H. However, no evidence of the formation of Mg-carbonate phases was observed in crystalline or amorphous form. Thermodynamic modelling carried out with an updated database for Mg-phases indicated that M-S-H and hydromagnesite should be stable, in contrast to the observed partial destabilisation of hydromagnesite.
Similar content being viewed by others


Explore related subjects
Discover the latest articles and news from researchers in related subjects, suggested using machine learning.Avoid common mistakes on your manuscript.
1 Introduction
Magnesium-based binders manufactured from magnesium silicate-rich raw materials (MOMS = magnesium oxides derived from magnesium silicates) are potential low CO2 or even CO2 negative alternatives to Portland cement. The raw materials do not decarbonate naturally occurring carbonates as in the production of Portland-based cement, but use magnesium silicates or Mg containing brines as source materials [1]. MOMS production requires some processing to gain magnesium carbonates from magnesium silicates [2, 3]. Furthermore, the calcination temperature of such magnesium carbonates (≈800 °C) is significantly lower than the clinkering temperature of Portland cement [1]. Additionally, they have the potential to sequester at least a part of the CO2 released during this calcination in the form of stable magnesium carbonates and to use such material as constituent of binders, mortars and concretes [4]. Many different types of magnesium carbonate hydrates can potentially precipitate as detailed in Fig. 1: magnesite (MgCO3), magnesium carbonate monohydrate (MgCO3·H2O), barringtonite (MgCO3·2H2O), nesquehonite (MgCO3·3H2O), lansfordite (MgCO3·5H2O), magnesium carbonate hexahydrate (MgCO3·6H2O), hydromagnesite (Mg5(CO3)4(OH)2·4H2O), artinite (Mg2(CO3)(OH)2·3H2O), dypingite (Mg5(CO3)4(OH)2·5H2O) and amorphous magnesium carbonate hydrates. The kind of magnesium carbonate hydrate formed mainly depends on CO2 pressure, relative humidity, time, and temperature [5]. The development of the carbonatable MOMS cement is currently only in the research stage and remains far from industrial application [1,2,3,4].

MgO-CO2-H2O ternary plots based on the composition of the different magnesium carbonates and M-S-H phases (silica is only present in M-S-H)
Another use of the MOMS would be the hydration of reactive magnesia in the presence of a silicate source instead of carbonation of reactive magnesia. This hydration process results in precipitation of the magnesium silicate hydrate phase (M-S-H) [6], which can on the longterm generate high compressive strengths up to 50–80 MPa as measured on pastes [7] and concretes [8]. However, the precipitation of pure M-S-H from suspensions has been observed to be extremely slow [6, 9,10,11]. The addition of sodium hexametaphosphate (NaHMP) as a dispersant for M-S-H fastens the reaction and provides good mechanical properties after 28 days [7]. NaHMP acts as superplasticizer, thus enhancing the fluidity of the pastes, and the phosphate ions form complexes with dissolved magnesium ions increasing the Mg2+ concentration in solution which seems to enhance the formation of M-S-H-like phases [12,13,14]. However, at high loadings, the addition of NaHMP can decelerate the reaction [14]. NaHCO3 seems to also help to fasten the formation of M-S-H [15].
M-S-H phases are hydrated nano-crystalline phyllosilicates with variable Mg/Si ratios, usually between 0.8–1.3; the ratio Mg/H2O is relatively constant as detailed in Fig. 1. The M-S-H structure consists of mixed short layers T:O or T:O:T of silicate tetrahedral layers (T) and magnesia octahedral layers (O) [11, 16]. A large amount of hydroxyl groups is naturally present in the M-S-H structure, both Si–OH and Mg-OH [11, 16, 17]. When increasing the pH, the hydroxyl groups are deprotonated and the negative surface charges are compensated by exchangeable cations [18].
The precipitation of M-S-H, brucite and/or magnesium carbonate phases depends on the pH value, the availability of silicate and carbonate ions [3, 11, 19], although the specific conditions steering the precipitation kinetic remain unknown. This study aims to determine the stability of M-S-H versus the stability of different Mg-carbonate phases in the system MgO-SiO2-CO2-H2O, which has implications regarding the potential use of such systems as cementitious binders.
Therefore, in this study the phase assemblage formed upon the reaction of silica fume with magnesium oxide together with hydromagnesite (Mg5(CO3)4(OH)2·4H2O) and/or sodium carbonate (Na2CO3) solution was investigated at a Mg/Si of 1.5, to allow the formation of both M-S-H and magnesium carbonate hydrates.
2 Materials and methods
2.1 Raw materials
The X-ray fluorescence analyses of the raw materials are presented in Table 1. The MgO used for the experiments was obtained from Mg(OH)2 (95% reagent grade) burned at 600 °C for 2 h. A 50/50 wt. % mix of a commercial fumed silica (purity > 98 wt. % containing residual HCl [20]) and commercial silica fume (purity > 96 wt. %, see Table 1) were used as reactive silica sources in order to improve workability. Hydromagnesite material was commercially acquired and its composition is given in Table 1. The particle size distribution of the different Mg based phases is provided in Figure S 1 in the Electronic Supplementary Material (ESM).
2.2 Preparation of pastes
The samples were prepared at Mg/Si of 1.5 with MgO or MgO/hydromagnesite and SiO2 as summarized in Table 2. The sodium carbonate solution for the activation was prepared by dissolving 8.5 g of Na2CO3 (anhydrous, ≥ 99.8%) per litre. The experiments were carried out at 20 °C using a water/solid ratio (w/s) of 2 to ensure proper mixing and avoid the use of superplasticizer. The samples were mixed by hand for 30 s.
Directly after mixing, the pastes were cast in 12 mL PE-bottles, sealed and stored at 20 °C. After 3, 7, 28 and 182 days the pastes were crushed, and hydration was stopped by submerging the specimens for 15 min in isopropanol. The solid was filtered, rinsed with diethyl ether and dried for 15 min at 40 °C. Afterward the samples were ground by hand in an agate mortar (< 0.5 mm) and analysed by thermogravimetry (TGA), Fourier transform infrared (FT-IR) and X-ray diffraction (XRD) within the next 2 days; the remainders of the samples were stored in desiccators at a temperature of 20–23 °C and a relative humidity of ~ 34% (over-saturated CaCl2 solution) until further analyses by nuclear magnetic resonance spectroscopy (NMR), or cation exchange capacity (CEC).
2.3 Characterization techniques
2.3.1 X-ray diffraction analyses
X-ray diffraction analyses (XRD) were performed with a PANalytical X'Pert Pro MPD diffractometer in Bragg–Brentano configuration, equipped with a Cu X-ray source running at 45 mV and 40 mA and the X'Celerator detector. The samples were scanned from 5 to 75° 2θ.
2.3.2 Thermogravimetric analyses
Thermogravimetric analyses (TGA) were carried out using a Netzsch STA 449 F3 Jupiter TGA apparatus coupled with a Bruker Fourier-transform infrared (FT-IR) spectrometer for the analysis of the exhaust gases. Approximately 30 mg of each sample were heated from 30 to 980 °C with a heating rate of 20 K per minute in 150 μL alumina crucibles. The infrared absorbances of H2O and CO2 were integrated in the ranges of 1300—2000 cm−1 (O–H bending vibration in H2O molecules) and 2200—2450 cm−1 (C = O stretching vibration), respectively, and used as relative measures of H2O and CO2 contents in the exhaust gases.
2.3.3 Attenuated total reflectance Fourier Transformation-Infrared
Attenuated total reflectance (ATR) Fourier Transform-Infrared (FT-IR) spectra were recorded in the mid-region on a Bruker Tensor 27 FT-IR spectrometer between 600 and 4000 cm−1 with a resolution of 6 cm−1 by transmittance on small amounts of powder. Spectra were background corrected and scaled to ease comparison.
2.3.4 29 Si MAS NMR
The 29Si MAS NMR single pulse experiments were conducted on a Bruker Avance III 400 NMR spectrometer (Bruker BioSpin AG, Fällanden, Switzerland) using a 7 mm CP/MAS probe at 79.5 MHz applying the following parameters: 4500 Hz sample rotation rate, 7168 scans, 30° 29Si pulse of 2.5 μs, 30 s relaxation delays, RF field strength of 33.3 kHz during SPINAL64 proton decoupling. For representative samples, 29Si T1 relaxation times were evaluated by saturation recovery NMR experiments. For the samples containing M-S-H phases (samples "pure M-S-H" and "MS") relaxation times from 60–80 s were determined for the three narrow main resonances at -79, -86 and -92 ppm. Considering the parameters used to record the 29Si MAS NMR data, it is clear that the relative amounts of M-S-H phase determined by the line shape analysis of the Qn sites (see 3.2.) are systematically too small, but proportionally correct.
The 29Si NMR chemical shifts were referenced to the most intense resonance at − 2.3 ppm of an external sample of an octamethylsilsesquioxane (Aldrich No. 52,683–5) which was referenced to tetramethylsilane (TMS, δ29Si = 0.0 ppm), details are given in [21]. The observed 29Si NMR resonances were assigned using the Qn classification, where one Si tetrahedron is connected to n Si tetrahedral, where n varies from 0 to 4. The line shapes of the experimental data were analysed by non-linear least-square fits using the “DMFIT” software developed by Massiot et al. [22] as a linear combination of Gaussian and Lorentzian functions as detailed in Table S 1 in the ESM. The presence of unreacted silica was confirmed by the resonance Q4 at − 110 ppm.
2.3.5 13 C MAS NMR
Solid-state 13C CP-MAS NMR spectra were recorded on the same NMR instrument than for recording the 29Si MAS NMR data using a 7 mm CP-MAS probe. Weighed amounts of material (140–230 mg) were thoroughly packed into 7 mm zirconia rotors and to allow smoother sample rotation, Teflon inserts (3 mm thickness) were used. The filling heights were carefully chosen so that the Teflon spacer was in direct contact with the cap ensuring equivalent sample volumes for each rotor. The 13C CP-MAS NMR spectra were recorded at 100.6 MHz, using the following parameters: 6.0 μs 90° excitation pulse on 1H channel, 2 ms contact time with a ramp from 100 to 50% of power level on the proton channel, rotations rates of 4000 Hz, 5 s repetition time, 41 kHz SPINAL 64 proton decoupling was applied during acquisition. Appropriate numbers of scans (3072 up to 20,480) were recorded to yield reasonable signal-to-noise ratios. All absolute signal intensities of the individual samples were normalized by weight and by the number of scans with respect to the 13C CP MAS NMR spectrum of a sample containing pure hydromagnesite starting material (see 3.3).
The line shapes of the experimental data were analysed by non-linear least-square fits using the “DMFIT” software developed by Massiot et al. [22]. The individual spectra were analysed by line shape simulations applying i) Lorentzian shapes for the two resonances of hydromagnesite at 165.6 and 163.4 ppm (line widths of 80–100 Hz), respectively. ii) One additional Gaussian shape (line widths of 350–800 Hz) for the much broader signal at 167.1 ppm was needed to simulate the signal intensity in the foot of the narrow resonances.
The contents of hydromagnesite were determined from the relative proportions of the resonances of hydromagnesite in the individual 13C CP MAS NMR spectra and the absolute signal intensities (see above). Please note that a saturation transfer experiment obtained for the hydromagnesite NMR resonances observed at 165.6 and 163.4 ppm T1 relaxation times in the order of 140–170 s. This made it impossible to record quantitative single pulse 13C MAS NMR data for series of samples. Assuming that this compound's relaxation time and polarization transfer rates do not vary significantly from sample to sample, we therefore chose the indirect route via CP MAS NMR experiments to quantify hydromagnesite.
2.3.6 23 Na MAS NMR
The 23Na MAS NMR data were recorded at 105.9 MHz using a 4 mm CP/MAS probe applying the following parameters: 13′000 Hz sample rotation, 1024 scans, 20° pulses of 2.0\(\mu\) s, 1 s recycle delays, no 1H decoupling during acquisition. The 23Na chemical shifts were externally referenced to a 0.1 M solution of NaCl in D2O [23]. The line shapes were analysed by applying Lorentzian line shapes [22]. Please note that the 23Na MAS NMR resonances throughout were symmetrical and line shapes could be simulated by using Lorentzian shapes. All attempts to fit the 23Na NMR data with 2nd order quadrupolar broadened line shapes failed, indicating a high mobility of the sodium cations in the observed phases. The 23Na MAS NMR was recorded for the 6-month-old samples only. All absolute signal intensities of individual samples were evaluated from the absolute intensities over the full spectral range determined by the Bruker TopSpin software with respect to the signal intensity of the MS sample and normalized by the weight of the samples.
2.3.7 pH measurements
Leaching experiments for the aged samples were carried out to estimate the pH values of the pore solutions following the procedure outlined in [24]. Accordingly, 1.5 g of powder were blended with 1 g of distilled water 15 min, 3 h and 24 h, respectively, prior to pH measurement. The pH measurements were carried in the supernatant of the suspension at 23 °C out using a Thermo Scientific™ Orion™ PerpHecT™ ROSS™ Combination pH Micro Electrode. The calibration was done with built-in calibration using pH 7, 10, and 12 standard solutions.
2.3.8 Effective cation exchange capacity
The effective cation exchange capacity (CEC) was measured on stopped pastes using 5 mL of cobalt hexamine solution (Sigma-Aldrich, assay: 99%, 3 g/L in deionized water) and an equilibration time of 30 min. The pH of the CEC solution was adapted with KOH to the pH of the leached solutions to avoid any destabilisation or structural change in the M-(A-)S–H as detailed in [25]. After the exchange, the suspensions were filtered and the Co3+ remaining in solution was measured by colorimetry (absorption band at 473 nm) using a UNI-CAM UV visible spectrometer. The total Co3+ adsorbed and thus effective CEC is calculated from the difference in the cobalt hexamine concentration from the original solution (coloured) and from the leachate (less coloured). The composition of the liquid phase was analysed by ion chromatography (IC) immediately after filtration. The dissolved concentrations of Mg, Na, and K in solutions diluted by factors 10, 100 were quantified using a Dionex DP serie ICS-3000 ion chromatography system. This method was previously used for M-S-H and M-A-S–H [17, 18, 26] to quantify the surface charges based on the measured content of exchangeable cations per g of solid.
2.4 Thermodynamic modelling
Thermodynamic modelling of the experiments was carried out using the Gibbs free energy minimization program GEMS [27, 28]. GEMS is a broad-purpose geochemical modelling code which computes equilibrium phase assemblage and speciation in a complex chemical system from its total bulk elemental composition. The thermodynamic data for aqueous species as well as for the amorphous SiO2, and brucite were taken from the PSI-GEMS and Cemdata18 thermodynamic database [29, 30]. The thermodynamic data selected for the magnesium carbonate phases were from [31,32,33] as detailed in [19] completed with newer detailed studies [34, 35], where available. A summary of the data used is given in Table 3.
3 Results & discussions
3.1 Accelerated formation of M-S-H in sodium carbonate solution
Figure 2a shows the TG curves of hydrated MS pastes at different curing times together with the one of a 2-year-old synthetic M-S-H with Mg/Si = 1.4 from [17]. For pastes older than 3 days, the dehydration peak centred at ~ 110 °C shifts to ~ 130 °C, indicating the presence of more physically bound water in the solid, which is characteristic for the formation of M-S-H [17]. The prominent peak centred at 400 °C is typical for the dehydroxylation of brucite [10, 11, 20]. With increasing hydration time, the intensity of the brucite peak decreases, while a more pronounced weight loss is observed between 250 and 650 °C corresponding to hydroxyl groups in M-S-H [11, 17]. In addition, the physically bound water content increases over time from 12 wt. % after 7 days to 17 wt. % after 6 months, due to the higher content of M-S-H in the samples. The content of brucite quantified by TGA decreases steadily over time: from 46 mol% of the total Mg after 3 days, to 22 mol% after 7 days, 2 mol% after 28 and < 0.1 mol% after 182 days, indicating the full reaction of Mg(OH)2 to M-S-H. For comparison the synthetic M-S-H 1.4 sample still contained brucite, about ~ 23 mol% of the total Mg after 3.3 years of equilibration time [17]. No clear weight loss due to carbonates is observed, although a slightly increased weight loss at around 100 °C is observed for the MS samples compared to the plain M-S-H (see the TGA-FTIR results in Figure S 3 in the ESM). This weight loss occurs at low temperature compared to the temperature region where magnesium carbonates loose water [19, 39] indicating rather sorbed CO32− on the solid than directly incorporated in a solid (as discussed below).

a Thermogravimetric analysis (TGA) and b X‐ray diffraction (XRD) patterns of MS pastes with Na2CO3 solutions after different hydration times. Light blue rectangles correspond to M-S-H broad humps. *Synthetic M-S-H 1.4 aged of 2 years from [20] is added for comparison
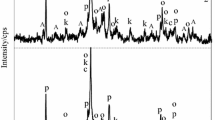
The XRD patterns confirm the TGA data. The prominent MgO reflections of the unhydrated mix are very small after 3 days and completely disappear within 7 days (Fig. 2b), indicating the full reaction of the MgO into hydration products. The XRD patterns from day 3 onwards confirm the presence of brucite with characteristic reflections at 18.6, 32.7, 38.0, 50.9, 58.7, 62.0°2θ CuKα [6, 40] and of M-S-H with broad humps at 19.7, 26.7, 35.0, and 59.9°2θ CuKα [11, 16]. The intensity of the hump at 26.7°2θ CuKα, characteristic of the layer to layer distance in M-S-H, seems higher in the MS paste compared to the pure M-S-H with Mg/Si 1.4. In addition, the hump is shifted to lower 2θ CuKα, indicating rather a T:O structure (sepiolite-like) compared to the synthetic M-S-H 1.4, where a T:O:T structure (talc-like) would be expected [17]. The XRD patterns (Fig. 2b) confirm the total consumption of brucite and MgO within 182 days and thus a significant impact of the Na2CO3 solution on kinetic and solid phase composition.
Figure 3a displays the FT-IR spectra of the MS samples after 3, 7, 28 and 182 days compared to the FT-IR spectra of the unhydrated mix and of the synthetic M-S-H 1.4 from [20]. In the unhydrated sample, the characteristic bands of amorphous silica are contained in the large band centered at ~ 1050 cm−1 with a shoulder at 1200 cm−1 and in an additional band at 805 cm−1. With the hydration, the band at 1650 cm−1 characteristic of free water (bending of the H2O molecule) is visible together with some carbonate bands at 1300 and 1550 cm−1. After 3 days, the unreacted silica is still present based on the bands at ~ 1110 cm−1, 1060 cm−1 and 805 cm−1. Remarkably, the band at 990 cm−1 characteristic of Si–O stretching in M-S-H is clearly visible as well. With time, the bands characteristic of the unreacted silica decrease in favour of the band of the M-S-H. After 182 days, the FT-IR region between 2000 and 800 cm−1 of the MS sample looks similar to the one in the FT-IR spectrum of the M-S-H 1.4 from [20]. Only the shoulders at 1060 and 890 cm−1 are better visible.

FT-IR spectra of the a) MS pastes and b) H-MS pastes over time compared to pure M-S-H 1.4 from [20]
Figure 4 shows the 29Si MAS NMR spectra of the MS pastes after 28 and 182 days. For comparison, 29Si MAS NMR data of pure M-S-H [20] and M-S-H synthesized in the presence of NaOH [18] are added. In the 29Si MAS NMR spectra of pure M-S-H phases, the signals at − 78.5 ppm can be assigned to Q1, at − 85 ppm to Q2 and the broader resonances between − 91.5 and − 97 ppm to Q3 in tetrahedral silicates [11, 17]. The deconvolutions of the spectra are detailed in Figure S 4 in the ESM and the numerical data are summarized in Table 4.

In the MS paste, the presence of Q1 at − 78.4 ppm, Q2 at − 85.1 ppm and Q3 at − 91.7 ppm indicates the presence of M-S-H. The weak Q4 resonance at − 110 ppm is attributed to the presence of traces of amorphous silica after 28 days, while the absence of this resonance after 182 indicates that all amorphous silica had been reacted. Unlike our previous work where three different Q3 environments were observed in pure M-S-H [20], only one main Q3 at − 92 ppm with a weak shoulder at − 95.6 ppm is identified in the present study. This is also observed for the M-S-H synthesized in presence of NaHMP [14] or sodium hydroxide [18], but not in the presence of NaNO3. This could indicate a rearrangement of Mg resulting in structure containing less T:O:T layers but more T:O layers [18].
A better fit is obtained with an additional resonance at − 83.2 ppm; this shoulder can be attributed to Q2Na and indicates some sodium in the vicinity of silica, resulting in a lower shift [18] as sodium shields the Si signal stronger than magnesium. Only relatively small amounts of these Q2Na species are determined here (< 10%, see Table 3), while ~ 15% were determined in 100 mM NaOH [18]. The content of Q1 is slightly higher in this study compared to pure M-S-H from [20] but comparable to samples with NaOH [18]. This indicates that sodium (hydroxide and carbonate) depolymerizes the silicate network, similar to the depolymerisation of silica by NaOH in C-S–H [41,42,43].
3.2 Effect of hydromagnesite as a partial replacement of MgO
The TGA of pure hydromagnesite (see Figure S 2 in the ESM) shows three different weight losses, a first one at about 270 °C due to the loss of water and second and third at 440° and at ~ 520 °C due to the loss of H2O, OH and CO2 groups in hydromagnesite [19]. TG curves and for the H-MS pastes, i.e. where half of the MgO content was replaced by hydromagnesite, are shown in Fig. 5a. In the unhydrated blend the second and third weight losses appear together at ~ 480 °C (as observed previously in [19]), while in the hydrating pastes the three peaks are well separated. The specific weight loss of hydromagnesite occurring at 440 °C (Figure S 2 in the ESM) is difficult to differentiate from the weight loss associated to brucite (~ 400 °C), as the presence of both brucite and hydromagnesite results in a broad water loss between 400 and 450 °C. The final shift after 182 days of this weight loss to higher temperature (centred ~ 440 °C) indicates the full reaction of brucite into M-S-H and the presence of hydromagnesite only.

a Thermogravimetric analysis (TGA) and b X‐ray diffraction (XRD) patterns of H-MS samples after different hydration times. Light blue areas correspond to the M-S-H broad reflections
At later ages, similar to the observations for the MS pastes (see Fig. 2), the dehydration peak centred at ~ 100 °C and a small continuous dehydroxylation weight loss between 250 and 650 °C. This information indicates the presence of physically bound water and some hydroxyl groups, both characteristic for M-S-H. With increasing hydration time, the weight losses at 270, 440 and 520 °C decrease (Fig. 5a), indicating the consumption of hydromagnesite presumably resulting in the formation of M-S-H. The quantification of hydromagnesite and the TGA-FTIR description are detailed in Sect. 3.3.
The XRD patterns (Fig. 5b) confirm the presence of M-S-H and hydromagnesite [44] in each sample. Traces of brucite are also observed with reflections at 18.6, 32.7, 38.0, 50.9°2θ CuKα at early age, but are absent after 182 days. The broad humps observed in the XRD patterns (light blue highlighted areas, Fig. 5b) are characteristic of M-S-H and become better defined, more narrow, and more intense with time.
According to the 29Si-NMR data, traces of amorphous silica are still present in the H-MS sample (Fig. 4) after 28 days (~ 12%, Table 4), but have completely reacted after 182 days. For the MS samples, a reasonable result is obtained by line shape analysis with only one Q3 resonance at ~ − 91.7 ppm, while the 29Si NMR data for the H-MS samples could only be simulated with an additional resonance at − 95.6 ppm (28 and 182 days hydration time, Figure S 4 in the ESM), similar to the observation in plain M-S-H. The additional resonance at − 95.6 ppm seems to indicate that the M-S-H structure in the H-MS sample contains some T:O:T layers in addition to the T:O layers.
Figure 3b displays the FT-IR spectra for the H-MS samples over time. Hydromagnesite is visible by the two characteristic bands at 1420 and 1480 cm−1 in the unhydrated and hydrated samples confirming his presence in the samples. Similar to the MS samples, the large amount of unreacted silica is observed together with M-S-H at early ages, and the spectrum of the final product is comparable to the ones of the MS sample and of the synthetic M-S-H.
To conclude, TGA, XRD, FT-IR, and 29Si NMR data show the complete reaction of the MgO, the brucite and the SiO2 to M-S-H after 182 days, while some hydromagnesite persisted.
3.3 Carbonate phases (13C CP MAS NMR data and TGA)
In the 13C CP MAS NMR spectra, all resonances are observed in the narrow chemical shift range from 160 to 175 ppm, typical for carboxyl and carbonate compounds (Fig. 6). The spectrum of pure hydromagnesite shows two narrow resonances at 165.6 and 163.4 ppm as already reported by [45]. These signals are observed in the 13C MAS NMR spectra of the H-MS sample prepared with hydromagnesite as starting material (Fig. 6b). Additionally, in the spectra of the aged sample, broad signals can be recognized at the base of the resonances of hydromagnesite.

Region of carboxylate species of 13C CP MAS NMR spectra of 182 days old samples prepared without (MS) and with hydromagnesite (H-MS) as starting material compared to the spectrum of pure hydromagnesite
The individual spectra were analysed by line shape simulations (details are given in Figure S 5 and Figure S 6 in the ESM). The results of the deconvolutions of the 13C NMR data are summarized in Table 5 and selected spectra including the simulated signals are shown in Figure S 5 in the ESM. The samples MS and H-MS show broad 13C MAS NMR resonances in the region of 167 ppm, which have been assigned to the sorption of carbonate on the solid, following the suggestion of [46], as even in freshly calcined MgO a weak resonance at 167 ppm can be observed, which grows over time when the material is exposed to ambient conditions (Figure S 6 in the ESM). Note that no other signal which could potentially be attributed to further carbonated phases is present. This together with the XRD (see 3.1.), confirmed that no or only small amounts of nano-crystalline/amorphous magnesium carbonates seem to be present.
As detailed above (see 3.1.), the H2O and CO2 losses in TGA occur over a large temperature range from 30 to 1000 °C. The FTIR analysis of the exhausted gas of the TGA was used to understand the nature of the weight losses for the different samples. An example of deconvolution of the TGA-FTIR is presented in Figure S 3 in the ESM and the repartition CO2/H2O exhausted is summarized in Table S2 in the ESM.
Most of the CO2 losses are related to the presence of hydromagnesite in the samples, but some of the CO2 also starts to be lost at low temperatures (< 200 °C). This low weight loss at temperature can probably be related to the presence of sorbed CO32− and HCO3− as observed by 13C NMR. The amount of hydromagnesite is estimated from the repartition hydromagnesite/total for the carbonate species from the 13C NMR data (Table 5) and the estimated amount of CO2 in the samples from TGA and expressed per 100 g of dry paste. Since the 13C NMR data were obtained by CP-MAS experiments, the amounts of carbonate species determined by line shape analysis (Table 5) are not quantitative but yield only relative amounts. Assuming that the relaxation time and polarization transfer rates of hydromagnesite do not vary significantly from sample to sample, we therefore chose an indirect route via CP MAS NMR experiments to quantify the absolute amounts of hydromagnesite. The absolute signal intensities of the individual 13C NMR data sets were normalized by weight and by the number of scans with respect to the 13C CP MAS NMR spectrum of the sample containing pure hydromagnesite starting material (Table 5). Taking into account the relative signal intensity of the hydromagnesite species in relation to the total signal intensity in the 13C NMR spectra (Table 5) the absolute amounts of hydromagnesite per 100 g of dry paste (for the H-MS sample) were calculated.
The results obtained from 13C CP MAS NMR show that ~ 74 ± 5% of the hydromagnesite has reacted after 6 months in the H-MS sample, while from the FTIR data extracted from the TGA-FTIR data (Table S2 in the ESM) a reaction of 66 ± 5% was estimated. These two estimations are in the range of error of the two techniques (measurement errors and deviation of the measurements).
3.4 pH values and sodium uptake
In the leached solutions, pH values of 9.8 and 9.7 at 23 °C were determined for the MS and H-MS, respectively, after 182 days. These low pH values confirm that brucite is unstable, while M-S-H formation is favoured [11, 47], as brucite is stabilised only above pH 10.5.
The CEC values obtained by colorimetry for the two samples are about 49 meq/100 g (Fig. 7), corresponding to 0.07 charge per silicon, while the cations dissolved during the CEC measurements are about 70–78 meq/100 g. The ions released during the CEC measurements showed that approximately 50 meq/100 g are due to the presence of Mg2+, while the excess charge is due to the presence of Na+ near the surface. Indeed, the sodium measured matches the total amount of sodium (molar Natot/Si = 0.042 ± 0.004) present in the samples due to the addition of 1.7 g Na2CO3 per 100 g unreacted binder (Table 6). The lower amount of CEC obtained by colorimetry compared to the amount of ions released appears to be related to Na+ in the vicinity but further away of the stern layer of the M-S-H, in contrast to M-S-H in NaNO3 or NaOH solution, where Na+ is directly sorbed at the surface of the silicate sheet [18]. This assignment is also supported by the 23Na MAS NMR spectra of the 182-day-samples displayed in Figure S 7 in the ESM, which are narrow and symmetric, indicating the presence of fully hydrated sodium (Na(H2O)xy+) in solution. The 23Na NMR resonances at − 2.7 ± 1 ppm are relatively narrow (\(\Delta v_{\frac{1}{2}}\) 1/2 ≈ 370 Hz). In comparison, partially hydrated sodium (Na(H2O)x+) sorbed at the cation exchange surface sites of M-S-H have a broader resonance at − 5.8 ± 1 ppm (\(\Delta v_{\frac{1}{2}}\) ≈ 630 Hz) (see Figure S 8 in the ESM or [21]). Hence, the sodium is indeed present but is further away of the solid, i.e. in the edge of the diffuse layer.

CEC from the colorimetric measurements and the cations released in solution 483 from the MS and H-MS samples (error of the CEC measured by colorimetry is reflected by the size of the circle). The surplus of Na+ indicates the presence of ions near the surface
In the presence of NaOH or NaNO3, the CEC of M-S-H contains both magnesium and sodium, although the Na+ is mainly present due to the very low concentration of the divalent cation Mg2+ [18]. In contrast, for MS and H-MS samples, mainly Mg2+ is responsible for CEC, although the initial sodium concentration is with 160 mM quite high. This is due to the presence of carbonate in the solution, which increases the concentration of Mg in solution by complexing with it as detailed in Fig. 8. Indeed, at pH 9.7–9.9, Mg2+ and Mg(CO3)0 represent ~ 45% and ~ 54%, respectively, of the total dissolved Mg (Fig. 8b).

a Calculated brucite solubility curves in pure water and in carbonated solution (CO32− = 80 mmol/L) and b different Mg species associated to the solubility of brucite in carbonated solution (CO32− = 80 mmol/L). Mg(CO3)@ corresponds to the dissolved complex
The exchangeable Mg2+ measured by IC corresponds to the CEC measured by colorimetry: 0.035 Mg exchangeable per Si. This is higher than the 0.030 Mgexchangeable per Si obtained for M-S-H with Mg/Si = 1.2 [17, 18] and confirms the increasing trend of CEC with increasing Mg/Si in M-S-H as observed in Fig. 9. Finally, the CECs measured here are relatively low compared to the CECs (CEC > 150 meq/100 g at pH ~ 12.5) of M-S-H synthesized in the presence of NaOH, mainly due to the lower pH in the MS and H-MS samples, which limits the dehydroxylation of the silanol groups [18].

3.5 Discussion of the solid phase composition
The composition of the hydrates formed was calculated by mass balance from the initial content of SiO2 and MgO, deducing the amount of SiO2 and hydromagnesite present estimated from 29Si MAS NMR and TGA coupled with the 13C NMR data. The total ratios in the solid phase are presented in Table 6 and Fig. 9.
The formation of M-S-H is observed already after 3 days, while the content of brucite decreases drastically within the first 28 days of hydration. After 182 days neither amorphous silica nor brucite are observed indicating an M-S-H with Mg/Si ~ 1.5 (mol/mol) in the MS sample and Mg/Si ~ 1.4 in the H-MS sample (see Table 6 and Fig. 9). This is somewhat higher than the maximum Mg/Si of 1.3 observed for synthetic M-S-H described in the literature [11, 17]. M-S-H with Mg/Si ratios of ~ 1 or even lower are reported when sodium metasilicate and Mg(NO3)2·6H2O are used as starting materials [6, 9, 10, 40, 48, 49]. A wider range of Mg/Si ratios from 0.8—1.4 was obtained when MgO and SiO2 were used as starting materials in combination with longer equilibrium times and/or higher temperatures [11, 16, 17, 50]. The maximum Mg/Si in synthetic M-S-H reported in literature is 1.4 but is obtained together with brucite after 3.3 years of equilibration time [17]. However, due to the very slow formation of M-S-H, it could be possible that after even longer reaction times more magnesium could be present in M-S-H as in natural occurring phyllosilicates such as antigorite or chrysotile a Mg/Si of 1.5 is present. M-(A-)S-H observed as deterioration product of PC in the context of seawater or groundwater interaction actually showed higher Mg/Si, which was attributed to the possible formation of brucite or hydrotalcite [51, 52]. Although no evidence for such a phase was found, a potential additional amorphous magnesium carbonate hydrate, as observed in MgO-CO2-H2O systems by [19], could have formed contributing to the observed high Mg/Si in the solid phase. Alternatively a change in solution composition or reaction kinetics could also have contributed to the high Mg/Si.
Most studies under laboratory conditions, i.e. in inert atmosphere, showed that M-S-H formation from MgO and silica fume is very slow [6, 9,10,11, 17, 20]. The rapid dissolution of MgO and the precipitation of brucite occur prior to the dissolution of silica fume [20]. Subsequently, in the absence of carbonate solution, the magnesium hydroxide dissolves only slowly in the presence of M-S-H, which was attributed to the presence of dissolved silicon decreasing the brucite dissolution rate [20].
The 8.5 g/L sodium carbonate in solution results in a (calculated) pH value of 11.3 in the mixing solution. This pH value is expected to decrease to around pH 9 because of M-S-H formation. As the Mg concentration decreases with higher pH (Fig. 8) due to brucite precipitation, it is expected that brucite would dissolve slower at higher pH values and M-S-H would form slower. However, in the presence of carbonate ions, brucite dissolution is faster due to the formation of carbonate surface complexes on brucite, which promote brucite dissolution rate [53]. In addition, the formation of MgHCO3+ and MgCO30 complexes increases the magnesium concentrations in the presence of brucite (Fig. 8). The faster brucite reaction, together with the increased total Mg concentrations could favour the formation of M-S-H with a Mg/Si of 1.5 as no evidence for the formation of a significant amount of amorphous magnesium carbonate hydrate together with M-S-H has been observed.
3.6 Thermodynamic modelling
Figure 10 displays the phase contents of the anhydrous binder and of the hydrated binder as predicted by thermodynamic modelling. The main modelled hydrated phases are M-S-H and hydromagnesite. Note that although magnesite and artinite are thermodynamically more stable than the other Mg-carbonate phases, their formations were prevented in the modelling, as their formation seems to be kinetically hindered. Similarly, Winnefeld et al. [19] modelled artinite precipitation, but observed an amorphous magnesium carbonate hydrate experimentally in a MgO-CO2-H2O chemical system.

a initial mix and b thermodynamically modelled changes in the MgO-SiO2-CO2-H2O system (magnesite and artinite precipitations were prohibited)
Compared to the experiments, where the MS sample contained only M-S-H (~ 62 cm3/100 g of paste) and the H-MS sample contained 93 vol % M-S-H and 7 vol % of hydromagnesite (~ 54 and 4 cm3/100 g of paste respectively), the formation of some hydromagnesite is modelled in the MS sample, but not in the H-MS samples. Experimentally, the destabilisation of hydromagnesite results in the incorporation of additional magnesia in the M-S-H, but most of the carbonate was not found in the solids (13C MAS NMR or TGA) indicating the presence of dissolved CO2 mainly in solution. Independent whether a potential degassing of CO2 to the atmosphere was considered or not in the thermodynamic calculations, hydromagnesite was stable in the modelling. In contrast to [19], where amorphous magnesium carbonate hydrate was clearly observed experimentally, no amorphous magnesium carbonate hydrate could be observed in the present study, indicating that the additional bicarbonate remained in solution or was present as magnesium carbonate complexes at the surface of the solids.
The Mg/Si modelled is also presented in Fig. 10b. With the addition of hydromagnesite the Mg/Si decreases and reaches 0.9, indicating that the present model underestimates Mg content in M-S-H.
For the MS sample, the modelled volume (Fig. 10) is similar to the volume estimated from the experimentally observed phase composition (Table S 4 in the ESM), 61 and 62 cm3/100 g of paste, respectively. In contrast, the H-MS sample's modelled volume is 53 cm3 per 100 g of dry binder, considerably lower than the experimentally observed volume of about 58 cm3 per 100 g, as less hydromagnesite but more M-S-H is observed experimentally. The experimental and modeled H-MS volume are slightly lower than for the MS sample.
The compressive and flexural strengths of the two samples were followed from 3 to 91 days on mortar samples with water/solid (w/s) = 1 (details given in the ESM). The MS and H-MS mortars show similar compressive strengths over time, 4.1 ± 0.3 MPa after 3 days, 7.7 ± 0.3 MPa after 7 days, 11.0 ± 0.7 MPa after 14 days, 11.2 ± 1.0 MPa after 28 days and 12.8 ± 0.3 MPa after 91 days, and similar flexural strengths about at 1.1 ± 0.2 MPa and 2.5 ± 0.2 MPa after 3 and 91 days respectively. The strengths for the MS mortars are rather low compared to the literature, but this is mainly related to the high w/s ratio used in this study, which was not optimized for high strength development. The calculated porosity in the H-MS sample with 70% of hydromagnesite reacted slightly increases compared to the MS sample from 55 to 59% (Table S 4 in the ESM) but this is not reflected in the mechanical properties.
4 Conclusions
M-S-H forms faster when sodium carbonate is present as brucite dissolution is accelerated. The presence of bicarbonate accelerates brucite dissolution due to the formation of magnesium carbonate complexes similarly to magnesium phosphate complexes [12]. M-S-H is the main hydration product in the samples studied, with a high Mg/Si ~ 1.4—1.5 and a T:O structure similar to antigorite/chrysotile minerals. The high percentage of Q1 and Q2 (about 55–65% of the tetrahedral silicate) indicates the presence of very small coherent regions only. The XRD data confirm a nano-crystalline character of the M-S-H formed, indicating that although sodium carbonate accelerates the formation of M-S-H, its structure is comparable to synthetic M-S-H formed in the absence of carbonates. The 23Na MAS NMR data and the cation exchange capacity measurements show that sodium is present in the M-S-H phase, but rather near the M-S-H silicate layers than within its structure. The negative surface charge (cation exchange capacity, CEC) is compensated by Mg ions, as the presence of carbonates increases Mg concentrations at pH 9.5–10. The water content and the CEC observed in the M-S-H increase with the increasing Mg/Si, and for the M-S-H with Mg/Si of 1.4–1.5 a H2O/Si of about 2.3–2.7 and a CEC (Mgexch/Si) of 0.035 was observed, which follows the trends observed for synthetic M-S-H [17, 20].
The thermogravimetric analysis coupled with infrared exhaust gas analysis, XRD and 13C MAS NMR data did not indicate the possible formation of Mg-carbonate phases in crystalline or amorphous form.
It was experimentally observed that hydromagnesite reacts to M-S-H, although it remained unclear where the carbonate was bound. In contrast, thermodynamic modelling indicated that crystalline hydromagnesite would react to artinite (which was not observed) or else would be stable under the experimental conditions. This discrepancy could be due to i) mismatched thermodynamic data for hydromagnesite or ii) M-S-H thermodynamic data and/or iii) the presence of a small amount of amorphous magnesium carbonate hydrate that could not be detected by the methods used.
Last, the mechanical properties of the mortar samples containing mainly M-S-H or a mixture of M-S-H/hydromagnesite were relatively similar with compressive and flexural strengths of about 13 and 2.5 MPa after 91 days at water/solid (w/s) = 1. However, the w/s was not optimized, and much higher values could be achieved if the w/s value would be significantly reduced by optimizing kind and content of superplasticizer.
In conclusion, this study shows that M-S-H with Mg/Si = 1.5 forms more rapidly and in higher proportions when carbonates are used as activators/accelerators in MgO-SiO2-CO2-H2O systems. Under these conditions, experimentally M-S-H appears to be more stable than crystalline hydrated Mg-carbonate phases, which were not observed to form. Furthermore, the hydromagnesite in the mix partially dissolves to preferentially form M-S-H in this specific system.
References
Gartner E, Sui T (2018) Alternative cement clinkers. Cem Concr Res 114:27–39
Scott A, Oze C, Shah V, Yang N, Shanks B, Cheeseman C, Marshall A, Watson M (2021) Transformation of abundant magnesium silicate minerals for enhanced CO2 sequestration. Commun Earth Environ 2:1–6
Gartner E, Gimenez M, Meyer V, Pisch A (2014) Novel atmospheric pressure approach to the mineral capture of co2 from industrial point sources, In: Thirteenth annual conference on carbon capture, utilization and storage. Pittsburgh, Pennsylvania
Evans SM (2010) Novacem–carbon negative cement to transform the construction industry, Novacem slide deck presentation. Cambridge, Dec, 4 (2009)
Chaka AM, Felmy AR (2014) Ab initio thermodynamic model for magnesium carbonates and hydrates. J Phys Chem A 118:7469–7488
Zhang T, Cheeseman C, Vandeperre L (2011) Development of low pH cement systems forming magnesium silicate hydrate (MSH). Cem Concr Res 41:439–442
Zhang T, Vandeperre LJ, Cheeseman CR (2014) Formation of magnesium silicate hydrate (MSH) cement pastes using sodium hexametaphosphate. Cem Concr Res 65:8–14
Sonat C, Teo W, Unluer C (2018) Performance and microstructure of MgO-SiO2 concrete under different environments. Constr Build Mater 184:549–564
Li Z, Zhang T, Hu J, Tang Y, Niu Y, Wei J, Yu Q (2014) Characterization of reaction products and reaction process of MgO–SiO2–H2O system at room temperature. Constr Build Mater 61:252
Szczerba J, Prorok R, Śnieżek E, Madej D, Maślona K (2013) Influence of time and temperature on ageing and phases synthesis in the MgO–SiO2–H2O system. Thermochim Acta 567:57–64
Nied D, Enemark-Rasmussen K, L’Hopital E, Skibsted J, Lothenbach B (2016) Properties of magnesium silicate hydrates (MSH). Cem Concr Res 79:323–332
Tonelli M, Martini F, Milanesi A, Calucci L, Geppi M, Borsacchi S, Ridi F (2019) Effect of phosphate additives on the hydration process of magnesium silicate cements. J Therm Anal Calorim 138:3311–3321
Jia Y, Wang B, Wu Z, Han J, Zhang T, Vandeperre LJ, Cheeseman CR (2016) Role of sodium hexametaphosphate in MgO/SiO2 cement pastes. Cem Concr Res 89:63–71
Walling SA, Kinoshita H, Bernal SA, Collier NC, Provis JL (2015) Structure and properties of binder gels formed in the system Mg(OH)2-SiO2-H2O for immobilisation of Magnox sludge. Dalton Trans 44:8126–8137
Zhao H, Hanein T, Li N, Alotaibi A, Li A, Walling S (2019) Kinoshita H, Acceleration of MSH gel formation through the addition of alkali carbonates, In: Proceedings of the 15th international congress on the chemistry of cement (ICCC 2019), Prague
Roosz C, Grangeon S, Blanc P, Montouillout V, Lothenbach B, Henocq P, Giffaut E, Vieillard P, Gaboreau S (2015) Crystal structure of magnesium silicate hydrates (MSH): the relation with 2: 1 Mg–Si phyllosilicates. Cem Concr Res 73:228–237
Bernard E, Lothenbach B, Chlique C, Wyrzykowski M, Dauzères A, Pochard I, Cau-Dit-Coumes C (2019) Characterization of magnesium silicate hydrate (M-S-H). Cem Concr Res 116:309–330
Bernard E, Lothenbach B, Pochard I, Cau-Dit-Coumes C (2019) Alkali binding by magnesium silicate hydrates. J Am Ceram Soc 102:6322–6336
Winnefeld F, Epifania E, Montagnaro F, Gartner EM (2019) Further studies of the hydration of MgO-hydromagnesite blends. Cem Concr Res 126:105912
Bernard E, Lothenbach B, Rentsch D, Pochard I, Dauzères A (2017) Formation of magnesium silicate hydrates (M-S-H). Physics and Chemistry of the Earth, Parts A/B/C 99:142–157
Bernard E, Lothenbach B, Rentsch D (2021) Influence of sodium nitrate on the phases formed in the MgO-Al2O3-SiO2-H2O system. Mater Des 198:109391
Massiot D, Fayon F, Capron M, King I, Le Calvé S, Alonso B, Durand JO, Bujoli B, Gan Z, Hoatson G (2002) Modelling one-and two-dimensional solid-state NMR spectra. Magn Reson Chem 40:70–76
Harris RK, Becker ED, de Menezes SMC, Goodfellow R, Granger P (2001) NMR nomenclature. Nuclear spin properties and conventions for chemical shifts (IUPAC Recommendations 2001). Pure Appl Chem 73:1795–1818
Plusquellec G, Geiker M, Lindgård J, Duchesne J, Fournier B, De Weerdt K (2017) Determination of the pH and the free alkali metal content in the pore solution of concrete: review and experimental comparison. Cem Concr Res 96:13–26
Bernard E, Yan Y, Lothenbach B (2021) Effective cation exchange capacity of calcium silicate hydrates (CSH). Cem Concr Res 143:106393
Bernard E, Lothenbach B, Cau-Dit-Coumes C, Pochard I, Rentsch D (2020) Aluminum incorporation into magnesium silicate hydrate (M-S-H). Cem Concr Res 128:105931
Kulik D, Wagner T, Dmytrieva SV, Kosakowski G, Hingerl F, Chudnenko KV, Berner U (2013) GEM-Selektor geochemical modeling package: revised algorithm and GEMS3K numerical kernel for coupled simulation codes. Computat Geochem 17:1–24
Wagner T, Kulik DA, Hingerl FF, Dmytrieva SV (2012) GEM-Selektor geochemical modeling package: TSolMod library and data interface for multicomponent phase models. Can Mineral 50:1173–1195
Thoenen T, Hummel W, Berner U, Curti E (2014) The PSI/Nagra chemical thermodynamic database 12/07, PSI report 14–04, Villigen PSI, Switzerland
Lothenbach B, Kulik DA, Matschei T, Balonis M, Baquerizo L, Dilnesa B, Miron GD, Myers RJ (2019) Cemdata18: a chemical thermodynamic database for hydrated Portland cements and alkali-activated materials. Cem Concr Res 115:472–506
Helgeson HC (1978) Summary and critique of the thermodynamic properties of rock-forming minerals. Am J Sci 278:1–229
Johnson JW, Oelkers EH, Helgeson HC (1992) SUPCRT92: A software package for calculating the standard molal thermodynamic properties of minerals, gases, aqueous species, and reactions from 1 to 5000 bar and 0 to 1000 C. Comput Geosci 18:899–947
Robie RA, Hemingway BS, Thermodynamic properties of minerals and related substances at 298.15 K and 1 bar (105 Pascals) pressure and at higher temperatures, US Government Printing Office1995.
Harrison AL, Mavromatis V, Oelkers EH, Bénézeth P (2019) Solubility of the hydrated Mg-carbonates nesquehonite and dypingite from 5 to 35° C: Implications for CO2 storage and the relative stability of Mg-carbonates. Chem Geol 504:123–135
Gautier Q, Bénézeth P, Mavromatis V, Schott J (2014) Hydromagnesite solubility product and growth kinetics in aqueous solution from 25 to 75 C. Geochim Cosmochim Acta 138:1–20
Hill R, Canterford J, Moyle F (1982) New data for lansfordite. Miner Mag 46:453–457
Königsberger E, Königsberger L-C, Gamsjäger H (1999) Low-temperature thermodynamic model for the system Na2CO3− MgCO3− CaCO3− H2O. Geochim Cosmochim Acta 63:3105–3119
Raade G (1970) Dypingite, a new hydrous basic carbonate of magnesium, from Norway. Am Miner J Earth Planet Mater 55:1457–1465
Hollingbery L, Hull TR (2012) The thermal decomposition of natural mixtures of huntite and hydromagnesite. Thermochim Acta 528:45–52
Brew DRM, Glasser FP (2005) Synthesis and characterisation of magnesium silicate hydrate gels. Cem Concr Res 35:85–98
Lothenbach B, Nonat A (2015) Calcium silicate hydrates: solid and liquid phase composition. Cem Concr Res 78:57–70
L’Hôpital E, Lothenbach B, Scrivener K, Kulik D (2016) Alkali uptake in calcium alumina silicate hydrate (CASH). Cem Concr Res 85:122–136
Yan Y, Yang S-Y, Miron GD, Collings IE, L’Hôpital E, Skibsted J, Winnefeld F, Scrivener K, Lothenbach B (2022) Effect of alkali hydroxide on calcium silicate hydrate (CSH). Cem Concr Res 151:106636
Akao M, Iwai S (1977) The hydrogen bonding of hydromagnesite, acta crystallographica section b: structural crystallography and crystal. Chemistry 33:1273–1275
Moore JK, Surface JA, Brenner A, Skemer P, Conradi MS, Hayes SE (2015) Quantitative identification of metastable magnesium carbonate minerals by solid-state 13C NMR spectroscopy. Environ Sci Technol 49:657–664
Ishihara S, Sahoo P, Deguchi K, Ohki S, Tansho M, Shimizu T, Labuta J, Hill JP, Ariga K, Watanabe K (2013) Dynamic breathing of CO2 by hydrotalcite. J Am Chem Soc 135:18040–18043
Bernard E, Lothenbach B, Le Goff F, Pochard I, Dauzères A (2017) Effect of magnesium on calcium silicate hydrate (C-S-H). Cem Concr Res 97:61–72
Brew D, Glasser F (2005) The magnesia–silica gel phase in slag cements: alkali (K, Cs) sorption potential of synthetic gels. Cem Concr Res 35:77–83
d’Espinose de Lacaillerie J-B, Kermarec M, Clause O (1995) 29Si NMR observation of an amorphous magnesium silicate formed during impregnation of silica with Mg(II) in aqueous solution. J Phys Chem 99(47):17273–17281
Kalousek GL, Mui D (1954) Studies on formation and recrystallization of intermediate reaction products in the system magnesia-silica-water. J Am Ceram Soc 37:38–42
Kobayashi M, Takahashi K, Kawabata Y (2021) Physicochemical properties of the Portland cement-based mortar exposed to deep seafloor conditions at a depth of 1680 m. Cem Concr Res 142:106335
Jenni A, Mäder U, Lerouge C, Gaboreau S, Schwyn B (2014) In situ interaction between different concretes and Opalinus clay. Phys Cheme Earth Parts A/B/C 70:71–83
Pokrovsky OS, Schott J, Castillo A (2005) Kinetics of brucite dissolution at 25 C in the presence of organic and inorganic ligands and divalent metals. Geochim Cosmochim Acta 69:905–918
Acknowledgements
The authors would like to thank Luigi Brunetti, Boris Ingold, Dr. Josef Kaufmann (Empa) and Dr. Marcus Yio (Imperial College) for the TGA and the density measurements. The financial support of the Swiss National Science Foundation (SNSF) for Ellina Bernard’s Postdoc mobility under the grant P400P2_194345 is gratefully acknowledged. The NMR hardware was partially granted by the Swiss National Science Foundation (SNSF, grant no. 206021_150638/1).
Funding
Open Access funding provided by Lib4RI – Library for the Research Institutes within the ETH Domain: Eawag, Empa, PSI & WSL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no affiliation with any organization with a direct or indirect financial interest in the subject matter discussed in the manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bernard, E., Lothenbach, B., Rentsch, D. et al. Effect of carbonates on the formation of magnesium silicate hydrates. Mater Struct 55, 183 (2022). https://doi.org/10.1617/s11527-022-02018-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1617/s11527-022-02018-3