Abstract
Nitric oxide (NO), a free radical gas produced endogenously from the amino acid L-arginine by NO synthase (NOS), has important functions in modulating vasopressin and oxytocin secretion from the hypothalamo-neurohypophyseal system. NO production is stimulated during increased functional activity of magnocellular neurons, in parallel with plastic changes of the supraoptic nucleus (SON) and paraventricular nucleus. Electrophysiological data recorded from the SON of hypothalamic slices indicate that NO inhibits firing of phasic and non-phasic neurons, while L-NAME, an NOS inhibitor, increases their activity. Results from measurement of neurohypophyseal hormones are more variable. Overall, however, it appears that NO, tonically produced in the forebrain, inhibits vasopressin and oxytocin secretion during normovolemic, isosmotic conditions. During osmotic stimulation, dehydration, hypovolemia and hemorrhage, as well as high plasma levels of angiotensin II, NO inhibition of vasopressin neurons is removed, while that of oxytocin neurons is enhanced. This produces a preferential release of vasopressin over oxytocin important for correction of fluid imbalance. During late pregnancy and throughout lactation, fluid homeostasis is altered and expression of NOS in the SON is down- and up-regulated, respectively, in parallel with plastic changes of the magnocellular system. NO inhibition of magnocellular neurons involves GABA and prostaglandin synthesis and the signal-transduction mechanism is independent of the cGMP-pathway. Plasma hormone levels are unaffected by icv 1H-[1, 2, 4]oxadiazolo-[4,3-a]quinoxalin-1-one (a soluble guanylyl cyclase inhibitor) or 8-Br-cGMP administered to conscious rats. Moreover, cGMP does not increase in homogenates of the neural lobe and in microdialysates of the SON when NO synthesis is enhanced during osmotic stimulation. Among alternative signal-transduction pathways, nitrosylation of target proteins affecting activity of ion channels is considered.
L-NAME; Vasopressin; Oxytocin; cGMP; ODQ; Microdialysis
Braz J Med Biol Res, April 2004, Volume 37(4) 441-450 (Review)
Nitric oxide modulation of the hypothalamo-neurohypophyseal system
M. Kadekaro
Division of Neurosurgery, The University of Texas Medical Branch at Galveston, Galveston, TX, USA
 Text
Text
References
Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes
Abstract
Nitric oxide (NO), a free radical gas produced endogenously from the amino acid L-arginine by NO synthase (NOS), has important functions in modulating vasopressin and oxytocin secretion from the hypothalamo-neurohypophyseal system. NO production is stimulated during increased functional activity of magnocellular neurons, in parallel with plastic changes of the supraoptic nucleus (SON) and paraventricular nucleus. Electrophysiological data recorded from the SON of hypothalamic slices indicate that NO inhibits firing of phasic and non-phasic neurons, while L-NAME, an NOS inhibitor, increases their activity. Results from measurement of neurohypophyseal hormones are more variable. Overall, however, it appears that NO, tonically produced in the forebrain, inhibits vasopressin and oxytocin secretion during normovolemic, isosmotic conditions. During osmotic stimulation, dehydration, hypovolemia and hemorrhage, as well as high plasma levels of angiotensin II, NO inhibition of vasopressin neurons is removed, while that of oxytocin neurons is enhanced. This produces a preferential release of vasopressin over oxytocin important for correction of fluid imbalance. During late pregnancy and throughout lactation, fluid homeostasis is altered and expression of NOS in the SON is down- and up-regulated, respectively, in parallel with plastic changes of the magnocellular system. NO inhibition of magnocellular neurons involves GABA and prostaglandin synthesis and the signal-transduction mechanism is independent of the cGMP-pathway. Plasma hormone levels are unaffected by icv 1H-[1, 2, 4]oxadiazolo-[4,3-a]quinoxalin-1-one (a soluble guanylyl cyclase inhibitor) or 8-Br-cGMP administered to conscious rats. Moreover, cGMP does not increase in homogenates of the neural lobe and in microdialysates of the SON when NO synthesis is enhanced during osmotic stimulation. Among alternative signal-transduction pathways, nitrosylation of target proteins affecting activity of ion channels is considered.
Key words: L-NAME, Vasopressin, Oxytocin, cGMP, ODQ, Microdialysis

Introduction
Since the discovery that nitric oxide (NO), a lipophilic gas synthesized by NO synthase (NOS) from the amino acid L-arginine, is endogenously produced not only in peripheral organs but also in the central nervous system, considerable evidence has accumulated implicating this molecular messenger in a diversity of biological functions. For example, NO is involved in the regulation of blood pressure and blood flow, plasticity of the nervous system, ingestive behaviors, immunoresponses, and secretion of anterior and posterior pituitary hormones (1-3). Of particular interest to this review is the role of NO in the modulation of vasopressin and oxytocin secretion from the hypothalamo-neurohypophyseal system.
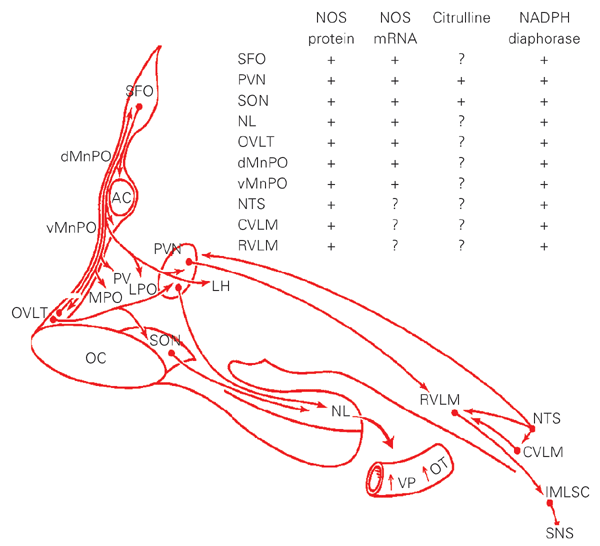
Vasopressin and oxytocin are nonapeptide hormones synthesized in magnocellular neurons of the supraoptic and paraventricular nuclei (SON and PVN, respectively), transported to the pituitary neural lobe, where they are stored and released into the circulation in response to increased neuronal activity. The SON and PVN receive inputs from the forebrain osmosensitive network in the lamina terminalis to regulate secretion of hormones. Although both hypothalamic nuclei are themselves osmosensitive, they necessitate inputs from the network for their normal functional activity. The forebrain network (Figure 1) encompasses the circumventricular organs, subfornical organ and organum vasculosum laminae terminalis, and the median preoptic nucleus. In turn, this network has anatomical connections with the nucleus tractus solitarius and caudal and rostral ventrolateral medulla in the hindbrain. The activity of these structures is influenced by inputs from central and peripheral osmoreceptors, volume and baroreceptors and circulating angiotensin II (ANG II) to regulate secretion of vasopressin and oxytocin into plasma. It is now evident that NO has an important modulatory role in the secretion of neurohypophyseal hormones. Immunohistochemical and in situ hybridization studies have shown that the enzyme NOS (4-6) and its mRNA (7), citrulline (8), the co-product of NO synthesis, as well as NADPH-diaphorase activity (9), a histochemical marker of NOS, are expressed in forebrain and hindbrain osmoregulatory networks. These findings, as well as results from microdialysis studies (10), indicate that NO is produced at these sites, thus having a potential to modulate the activity of these structures, either directly or indirectly by influencing neurotransmitter release. Release of neurotransmitters within the circuit regulates secretion of vasopressin and oxytocin into plasma. Expression of NOS and its mRNA (11-14), as well as NADPH-diaphorase activity (9,15) increases throughout the entire hypothalamo-neurohypophyseal system when functional activity of magnocellular neurons is elevated during acute and chronic osmotic stimulation, dehydration by water deprivation and hypovolemia. These results indicate that NO production is enhanced during disturbances of fluid balance, presumably to meet the increasing demand for NO modulation of the magnocellular system. This enhanced action of NO occurs in parallel with morphological changes in the hypothalamo-neurohypophyseal system that are characterized by retraction of glia cells interposed among magnocellular neurons, resulting in formation of new synaptic connections that facilitate hormone release (16). Importantly, linkage between NO and plasticity of the nervous system has been demonstrated in the hippocampus, cerebellum (1) and neurohypophysis (17).
Sagittal view of a simplified schematic diagram of brain structures that generate nitric oxide (NO; inserted table) and their neural circuitry regulating body fluid homeostasis. The presence of NO synthase (NOS) (4-6) and its mRNA (7), citrulline (8), the co-product of NO synthesis, as well as NADPH-diaphorase activity (9), a histochemical marker of NOS, are expressed throughout the structures in the lamina terminalis, hypothalamo-neurohypophyseal system, as well as hindbrain. AC = anterior commissure; CVLM = caudal ventrolateral medulla; dMnPO = dorsal median preoptic nucleus; IMLSC = intermedio-lateral column of the spinal cord; LH = lateral hypothalamus; LPO = lateral preoptic area; MPO = medial preoptic area; NL = neural lobe; NTS = nucleus tractus solitarius; OC = optic chiasma; OVLT = organum vasculosum laminae terminalis; OT = oxytocin; PV = periventricular nucleus; PVN = paraventricular nucleus; RVLM = rostral ventrolateral medulla; SFO = subfornical organ; SNS = sympathetic nervous system; SON, supraoptic nucleus; vMnPO = ventral median preoptic nucleus; VP = vasopressin. Reproduced with permission from Ref. 3.
NO's influence on electrical activity of magnocellular neurons
Electrophysiological and neuroendocrine studies also demonstrate that NO modulates the activity of the magnocellular system. Specifically, electrophysiological evidence demonstrates that NO inhibits the activity of magnocellular neurons. Firing of both phasic (vasopressin) and non-phasic (oxytocin) neurons recorded from the SON in slices of rat hypothalamus in vitro is inhibited by sodium nitroprusside (SNP), a spontaneous releaser of NO (18). The response is prevented by hemoglobin, a scavenger of NO. Conversely, neuronal activity is enhanced by Nw-nitro-L-arginine methyl ester (L-NAME), an inhibitor of NOS. These results indicate that NO has an inhibitory influence on both vasopressin and oxytocin neurons. The effect may be direct, as well as indirect, by modulating release of neurotransmitters such as gamma-aminobutyric acid (GABA) and glutamate.
Many synaptic functions of NO are linked to N-methyl-D-aspartate (NMDA)-type glutamate receptors (1). This selectivity is possible because of the compartmentalization of NOS with NMDA receptors at certain synaptic sites. At these sites, the modular protein-protein motif or PDZ domain of NOS, implicated in the signal transduction mechanism, links the synthase to protein complexes (19). Significantly, NOS and glutamate receptors are widely distributed in the hypothalamo-neurohypophyseal system (20) and circumventricular organs (21). Additionally, GABAergic innervation of magnocellular neurons is abundant (22) and presynaptic activation of glutamate receptors modulates release of GABA in these neurons (23). Moreover, Bains and Ferguson (24) have demonstrated that activation of NMDA receptors in the PVN, in addition to producing post-synaptic excitatory effects, also induces inhibitory synaptic activity in magnocellular neurons. This is due to an increase in activity of GABAergic neurons in response to enhanced production of NO induced by stimulation of NMDA receptors. The effect is blocked by L-NAME and bicuculline and mimicked by applications of NO to the bath. SNP also reduces the depolarization of supraoptic neurons elicited by NMDA in vitro (25). Moreover, S-nitroso-N-acetylpenicillamine (SNAP), a donor of NO, increases the frequency of spontaneous inhibitory post-synaptic current (IPSC) from supraoptic neurons in slice preparations, without affecting its amplitude (26). This indicates that NO potentiates IPSCs via a pre-synaptic mechanism, possibly through NO's action on GABA terminals. On the other hand, Stern and Ludwig (27) have shown that SNP increases not only the frequency, but also the amplitude of miniature IPSCs of vasopressin and oxytocin neurons, an indication that NO acts at presynaptic, as well as postsynaptic sites.
Using an in situ preparation from urethane-anesthetized rats, Srisawat et al. (28) demonstrated that the spontaneous activity of vasopressin and oxytocin neurons in the SON, antidromically identified as projecting to the posterior pituitary, is inhibited by enhancing NO action with SNP retrodialyzed onto the nucleus. In contrast, firing rates of magnocellular neurons increase during local administration of N w-nitro-L-arginine (L-NNA), another inhibitor of NOS, indicating that NO tonically produced in vivo is inhibitory. In lactating rats anesthetized with urethane, the high-frequency activity of oxytocin neurons of the SON during suckling is also inhibited when NO action is enhanced by intracerebroventricular (icv) administration of SNP, whereas bursting activity is increased by an NOS inhibitor (29).
Collectively, the pharmacological data from in vitro and in vivo studies show that NO inhibits the electrical activity of vasopressin and oxytocin neurons.
NO and neurohypophyseal hormones
Basal condition
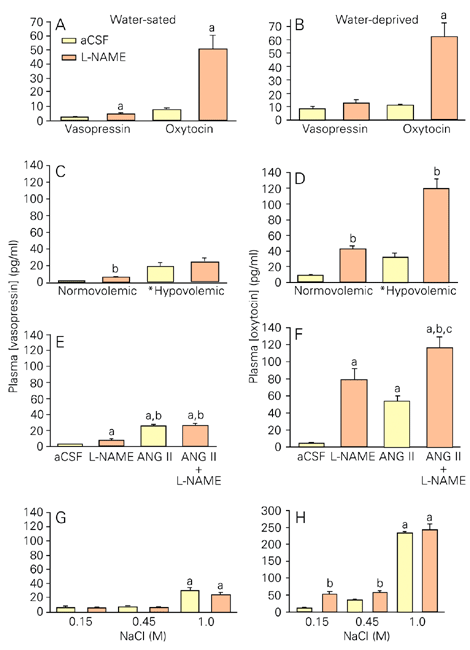
Although electrophysiological data show unequivocally that NO has an inhibitory influence on magnocellular neurons, studies using the measurement of plasma levels of vasopressin and oxytocin have provided some incongruent results, particularly with regard to NO's effect on basal levels of circulating hormones. This, most likely, relates to varied experimental protocols and animal species, anesthetic, route of drug administration and doses of drugs. Our studies (3) have demonstrated that when central production of NO is reduced with L-NAME administered icv to conscious rats in normal hydration, plasma levels of vasopressin and oxytocin become elevated (Figure 2A). Similar results are observed when the availability of NO is reduced by the NO scavenger, carboxy PTIO (Summy-Long JY and Bui V, unpublished data). This is an important comparison to interpret NO's effects on hormone secretion because it has been shown that, in addition to inhibiting NOS activity, L-NAME also has antimuscarinic effects (30). Thus, our studies indicate that NO is tonically produced in the forebrain during euvolemic isosmotic conditions to inhibit secretion of both hormones from the magnocellular system. In agreement with our results, Chiu and Reid (31) and Goyer et al. (32) have shown that L-NAME administered intravenously (iv) to normovolemic conscious rabbits increases plasma levels of vasopressin, even in the presence of elevated blood pressure. In humans, however, Chiodera et al. (33) have reported that iv administration of L-NAME does not elevate basal levels of neurohypophyseal hormones. In view of our findings demonstrating an elevation in vasopressin and oxytocin levels within 2 min after L-NAME (icv), it is possible that this effect was missed in their studies measuring the hormone at 10-min intervals following drug treatment. Srisawat et al. (28), however, have shown that L-NAME or L-NNA injected intraperitoneally does not modify basal plasma concentration of oxytocin in rats anesthetized with urethane, a finding incongruent with the reported increased firing rates of magnocellular neurons observed in their studies. Conversely, others have reported that in rats anesthetized with pentobarbital, L-NAME decreases vasopressin levels (34). To complicate matters further, Yamaguchi and Hama (35) demonstrated that SNP given icv to conscious rats does not modify plasma vasopressin levels. On the other hand, when SNP was infused into the anteroventral third ventricle area, vasopressin levels increased despite the proximity of the icv infusion to this anatomical site. In contrast to these findings, Ota et al. (36) reported that SNAP (icv), a drug that spontaneously forms NO, increases vasopressin release in conscious rats 5 min after its administration.
Results from in vitro studies are also contradictory. Using an isolated neural lobe preparation of the pituitary gland, Lutz-Bucher and Koch (37) have shown that L-NAME and free ferrous hemoglobin (a scavenger of NO) produce a transient, but significant increase in oxytocin and vasopressin. In contrast, syndnonimine-1 (SIN-1), a donor of NO, dampens spontaneous release of these neuropeptides. On the other hand, Raber and Bloom (38) have reported that SNP increases basal vasopressin release from hypothalamic slices.
As mentioned previously, differences in experimental design, dose and time responses to the drugs most likely contribute to these data documenting inhibition, activation or no change in the basal release of neurohypophyseal hormones in response to NO.
A,B, Effects of L-NAME (250 µg/5 µl; icv) or artificial cerebrospinal fluid (aCSF; 5 µl) on plasma vasopressin and oxytocin levels in water-sated (A) and 24-h water-deprived rats (B). Animals were decapitated 2 min later. aP < 0.05 vs aCSF. C, D, Effects of L-NAME (icv) injected 5 min after hemorrhage (20% of total blood volume) or at corresponding times in normovolemic rats on plasma hormones. Animals were decapitated 2 min later. For vasopressin (C), volume effect: *hypovolemic > normovolemic, P < 0.01; drug effect P = 0.07. Bonferroni multiple comparisons: aCSF, *hypovolemic > normovolemic, P < 0.01; L-NAME, *hypovolemic > normovolemic, P < 0.01; normovolemic, bL-NAME > aCSF, P < 0.01; hypovolemic, aCSF = L-NAME, P > 0.05. For oxytocin (D): volemic state x drug effect, P = 0.01; aCSF: *hypovolemic > normovolemic, P < 0.05; L-NAME: *hypovolemic > normovolemic, P < 0.01; normovolemic: bL-NAME > aCSF, P < 0.01; hypovolemic: bL-NAME > aCSF, P < 0.01. E, F, Effects of L-NAME (icv) alone or in combination with ANG II on plasma hormones. Animals were decapitated 90 s later. For vasopressin (E): aP < 0.05 vs aCSF; bP < 0.05 vs L-NAME. For oxytocin (F): aP < 0.05 vs aCSF; bP < 0.05 vs L-NAME; cP < 0.05 vs ANG II. G, H, Effects of L-NAME (icv) administered 30 min after subcutaneous injection of various concentrations of NaCl solutions on plasma hormones. Animals were decapitated 5 min later. For vasopressin and oxytocin: salt effect, aP < 0.01 vs all others; drug effect, bP < 0.01 L-NAME > aCSF. Differences among groups were determined by two-way ANOVA and the Newman-Keuls t-test. Reproduced with permission from Ref. 3.
Stimulated condition
The modulatory role of NO when activity of the hypothalamo-neurohypophyseal system is increased is more consistent and less controversial. In hypothalamic explants stimulated with high concentration of KCl, Yasin et al. (39) have demonstrated that enhancing NO's action with SNP, SIN-1 and L-arginine attenuates vasopressin release.
In in vivo studies, we have shown (3) that L-NAME administered icv during dehydration induced by water deprivation (a condition that depletes volumes from intracellular and intravascular compartments) produces a selective increase in plasma levels of oxytocin, but not vasopressin (Figure 2B). Thus, during dehydration the inhibitory action of NO on vasopressin neurons is removed, while that on oxytocin neurons is enhanced, a phenomenon resulting in a preferential release of vasopressin over oxytocin. This mechanism is independent of opiate receptors but dependent upon production of prostaglandins, inasmuch as indomethacin, an inhibitor of cyclooxygenase, prevents the response (3).
When intracellular volume is decreased during moderate osmotic stimulation with 0.45 M NaCl administered subcutaneously (sc), centrally administered L-NAME (icv) further increases plasma levels of oxytocin, but not of vasopressin (Figure 2G,H). In response to a stronger hypertonic solution (1.0 M NaCl, sc), however, L-NAME no longer enhances the already high levels of oxytocin. This indicates that, under extreme hypertonic conditions, NO inhibition of oxytocin neurons is also removed, resulting in elevated levels of the hormone, presumably needed to promote natriuresis (40). Srisawat et al. (28) also reported that L-NAME or L-NNA administered after hypertonic saline stimulation further increases the already elevated plasma concentration of oxytocin, while SNP (icv) significantly reduces it. Additionally, Goyer et al. (32) have shown that when NOS activity is inhibited during osmotic stimulation vasopressin secretion remains unchanged. Thus, these results show that decreasing production of NO with NOS inhibitors during osmotic stimulation further increases oxytocin, but not vasopressin levels.
During hypovolemia, a similar pattern of NO inhibition of magnocellular neurons occurs. For example, when intravascular volume is decreased during hemorrhage, independent of change in tonicity (3), L-NAME (icv) preferentially increases plasma oxytocin, but not vasopressin (Figure 2C,D). In accordance with these results, Chiu and Reid (31) have reported that iv administration of L-NAME to hypovolemic rabbits does not further elevate plasma levels of vasopressin.
Hypovolemia and hemorrhage are potent activators of the renal renin-angiotensin system. ANG II produced in response to these physiological conditions activates ANG II receptors in the subfornical organ and organum vasculosum laminae terminalis (41) and thereby promotes release of neurohypophyseal hormones. Similar to these effects observed during hypovolemia, NO also modulates hormone secretion in response to ANG II, such that when administered in conjunction with ANG II, L-NAME enhances oxytocin, but not vasopressin secretion (Figure 2E,F).
Collectively, results from electrophysiological data and the majority of neuroendocrine studies indicate that NO is produced tonically during conditions of normal hydration to inhibit secretion of both vasopressin and oxytocin. When intracellular and intravascular volumes decrease, or plasma ANG II levels increase, however, NO inhibition of vasopressin secretion is removed while that of oxytocin secretion is enhanced to promote preferential release of vasopressin. This phenomenon is physiologically significant for correction of fluid imbalance.
Osmoregulation of the magnocellular system during pregnancy and lactation
Changes in reproductive state result in plastic changes in supraoptic oxytocinergic neurons (42) and profound alterations in body fluid homeostasis (43). Specifically, late in gestation blood volume expands to about twice that of non-pregnant rats and plasma osmolality decreases. This adaptive mechanism, teleologically significant in anticipation of blood losses during delivery, is a result of a decrease in the apparent osmotic threshold for release of vasopressin and oxytocin from the magnocellular system, without a change in the sensitivity of the osmoreflex (43). In parallel with these changes, during late pregnancy expression of NOS mRNA in the SON is down-regulated (28,44). Thus, in late-pregnant rats pretreatment with L-NNA, another inhibitor of NOS, does not further elevate plasma oxytocin levels after osmotic stimulation, in sharp contrast with the response in virgin rats (28). Throughout lactation, however, the apparent osmotic threshold for vasopressin and oxytocin release returns to the non-pregnant level, but the sensitivity of the hypothalamo-neurohypophyseal system becomes attenuated in response to stimuli unrelated to milk ejection reflex (43). Expression of NOS mRNA becomes up-regulated not only in the SON, but also in the lamina terminalis (7). This may be responsible for the reduction of sensitivity of the forebrain circuit to osmotic stimuli at a time when oxytocin is in high demand for the milk-ejection reflex (45).
NO and signal-transduction pathway
The signal-transduction pathway involved in the modulatory action of NO on the hypothalamo-neurohypophyseal system has not been fully elucidated. A prominent physiological receptor for NO is soluble guanylyl cyclase (sGC) (46). As described from research on the cerebellum, NO diffuses into nearby neurons and glia, where it binds to sGC and activates the enzyme, elevating intracellular levels of cyclic GMP (cGMP) (46). cGMP either inhibits or stimulates cellular function by several mechanisms, such as regulating protein kinase or phosphodiesterase activities and ion channels. It is possible that this signaling pathway is similarly utilized in magnocellular neurons because, in addition to expressing NOS, cells in the SON and PVN are rich in the a1 and ß1 subunits of sGC (47). Moreover, Yang and Hatton (48) have shown that cGMP enhances dye coupling of supraoptic neurons, a plastic phenomenon also demonstrated with arginine, the substrate for NOS, and SNP, a donor of NO. Despite these findings, however, our studies do not support the postulate that cGMP is involved in the signal-transduction mechanism of NO's action on magnocellular neuroendocrine neurons. This was demonstrated by the following findings from our laboratory (10): 1) basal plasma concentrations of vasopressin and oxytocin do not become elevated when sGC activity is inhibited with 1H-[1, 2, 4]oxadiazolo-[4,3-a] quinoxalin-1-one (ODQ) administered icv to conscious rats; 2) administration of a membrane-permeable analog of cGMP (8-Br-cGMP; icv) to conscious dehydrated rats does not modify plasma hormone levels; 3) activity of NOS increases significantly in homogenates of the pituitary neural lobe of rats drinking 2% NaCl as the sole source of fluids for 4 days without any change in sGC activity or production of cGMP, and 4) the activity of NOS increases in microdialysates of the SON during osmotic stimulation in conscious and pentobarbital-anesthetized rats without changes in cGMP production.
In support of our findings, Ozaki et al. (26) observed that SNAP, a donor of NO, increases the frequency of inhibitory postsynaptic potentials in hypothalamic slices containing the SON by a mechanism independent of cGMP. Similarly, Cui et al. (25) have demonstrated that the inhibitory effects of NO on NMDA-induced depolarization of membrane potentials in SON neurons of hypothalamic slices are independent of cGMP. In an immunocytochemical study, Briski (49) reported that in response to acute glucose deprivation (a stimulus that enhances the activity of vasopressin and oxytocin neurons), pretreatment with a high dose of ODQ (icv) does not change the number of Fos-positive vasopressin neurons. In contrast, animals treated with SIN-1, an NO donor, showed a decrease in the number of Fos-positive vasopressin neurons. While these experiments indicate that cGMP is not involved in NO's actions on magnocellular neurons, in the same study Briski showed that ODQ administered in conjunction with SIN-1 prevents the action of this NO donor. This finding is difficult to interpret. Nonetheless, results of the majority of studies indicate that NO modulates magnocellular neurons by mechanism(s) other than the NO-cGMP pathway.
Recently, emerging research has demonstrated that NO may react directly with thiol groups in cell membrane proteins to exert its biologic actions (50). For example, NO oxidizes vicinal thiol groups to form disulfide (51). This oxidation reaction appears to occur via formation of an intermediate nitrosothiol to increase or decrease the activity of proteins such as p21RAS (52), the olfactory cyclic nucleotide-gated channel (53), glyceraldehyde-3-phosphate dehydrogenase (54), caspase-3 (55), and ryanodine receptor (56). A similar mechanism appears to mediate NO inhibition of ion flux through the NMDA receptor (apparently through cysteine residues in the NR1 and NR2A subunits that are particularly sensitive to S-nitrosylation), with the possible formation of disulfide linkage. Dithiothreitol, a disulfide reducing agent that decomposes nitrosothiol bonds, enhances NMDA receptor activity. In contrast, 5,5 dithio-bis-2-nitrobenzioic acid, an oxidizing agent that oxidizes free thiol groups and forms disulfide bonds, decreases NMDA receptor function (57). Interestingly, in our studies (Kadekaro M, Terrell ML, Bui V and Summy-Long JY, unpublished data), we have observed that dithiothreitol administered icv to conscious rats in normal hydration increases plasma levels of oxytocin and vasopressin, a result similar to the effects of L-NAME that is also prevented by indomethacin, an inhibitor of prostaglandin synthesis.
Inasmuch as S-nitrosylation of proteins is a reversible phenomenon, it has been postulated that it may function as a post-translational modification mechanism analogous to phosphorylation or acetylation to regulate several physiological processes (58). In recent years, Ahern et al. (59) have demonstrated that NO activates large conductance Ca2+-activated K+ channels (also called BK or Maxi-K channels) of the axon terminals in the posterior pituitary, by a cGMP-independent mechanism. In neurons, BK channels participates in repolarization and fast after-hyperpolarization of action potential, affecting the amount of neurotransmitter release (60) and, therefore, the excitability of neurons. Activation of BK channels by NO in the terminals of magnocellular neurons in the posterior pituitary depresses the excitability of the terminals. This is likely to inhibit impulse activity that could explain the inhibitory action of NO on hormone secretion. Further studies using quantitative immunohistochemistry, molecular biology techniques and mass spectrometry are needed to clarify further the signal-transduction molecule mediating NO's action on the hypothalamo-neurohypophyseal system.
Acknowledgments
The author thanks Dr. J.Y. Summy-Long (Department of Pharmacology, Pennsylvania State University) and Mary Lee Terrell (Department of Internal Medicine, University of Michigan Health Systems) for comments on the manuscript, and Diana Crowell for editorial assistance.
Address for correspondence: M. Kadekaro, University of Texas Medical Branch, 301 University Blvd. Galveston, TX 77555-0517 USA. Fax: +1-409-772-6352. E-mail: mkutyna@utmb.edu
Presented at the XVII Annual Meeting of the Federação de Sociedades de Biologia Experimental, Salvador, BA, Brazil, August 28-31, 2002. Research supported by grants from the National Institutes of Health 2RO1-NS23055. Received August 11, 2003. Accepted February 3, 2004.
- 1. Garthwaite J & Boulton CL (1995). Nitric oxide signaling in the central nervous system. Annual Review of Physiology, 57: 683-706.
- 2. Nelson RJ, Kriegsfeld LJ, Dawson VL & Dawson TM (1997). Effects of nitric oxide on neuroendocrine function and behavior. Frontiers in Neuroendocrinology, 18: 463-491.
- 3. Kadekaro M & Summy-Long JY (2000). Centrally produced nitric oxide and the regulation of body fluid and blood pressure homeostases. Clinical and Experimental Pharmacology and Physiology, 27: 450-459.
- 4. Bredt DS, Glatt CE, Hwang PM, Fotuhi M, Dawson TM & Snyder SH (1991). Nitric oxide synthase protein and mRNA are discretely localized in neuronal populations of the mammalian CNS together with NADPH diaphorase. Neuron, 7: 615-624.
- 5. Wang H & Morris JF (1996). Constitutive nitric oxide synthase in hypothalami of normal and hereditary diabetes insipidus rats and mice: role of nitric oxide in osmotic regulation and its mechanism. Endocrinology, 137: 1745-1751.
- 6. Yamada K, Emson P & Hokfelt T (1996). Immunohistochemical mapping of nitric oxide synthase in the rat hypothalamus and colocalization with neuropeptides. Journal of Chemical Neuroanatomy, 10: 295-316.
- 7. Luckman SM, Huckett L, Bicknell RJ, Voisin DL & Herbison AE (1997). Up-regulation of nitric oxide synthase messenger RNA in an integrated forebrain circuit involved in oxytocin secretion. Neuroscience, 77: 37-48.
- 8. Pasqualotto BA, Hope BT & Vincent SR (1991). Citrulline in the rat brain: immunohistochemistry and coexistence with NADPH-diaphorase. Neuroscience Letters, 128: 155-160.
- 9. Pow DV (1992). NADPH-diaphorase activity (nitric oxide synthase) staining in the rat supraoptic nucleus is activity-dependent: possible functional implications. Journal of Neuroendocrinology, 4: 377-380.
- 10. Terrell ML, Salas N, Bui V, Summy-Long JY & Kadekaro M (2004). NO inhibition of the magnocellular neuroendocrine system in rats is independent of cGMP signaling pathway. Experimental Neurology, 184: 846-856.
- 11. Kadowaki K, Kishimoto J, Leng G & Emson PC (1994). Up-regulation of nitric oxide synthase (NOS) gene expression together with NOS activity in the rat hypothalamo-hypophyseal system after chronic salt loading: Evidence of neuromodulatory role of nitric oxide in arginine vasopressin and oxytocin secretion. Endocrinology, 134: 1011-1017.
- 12. Ueta Y, Levy A, Chowdrey HS & Lightman SL (1995). Water deprivation in the rat induces nitric oxide synthase (NOS) gene expression in the hypothalamic paraventricular and supraoptic nuclei. Neuroscience Research, 23: 317-319.
- 13. Villar MJ, Ceccatelli S, Rönnquist M & Hökfelt T (1994). Nitric oxide synthase increases in hypothalamic magnocellular neurons after salt loading in the rat. An immunohistochemical and in situ hybridization study. Brain Research, 644: 273-281.
- 14. Ueta Y, Levy A, Lightman SL, Hara Y, Seino R, Nomura M, Shibuya I, Hattori Y & Yamashita H (1998). Hypovolemia upregulates the expression of nitric oxide synthase gene in the paraventricular and supraoptic nuclei of rats. Brain Research, 790: 25-32.
- 15. Sagar SM & Ferriero DM (1987). NADPH diaphorase activity in the posterior pituitary: relation to neuronal function. Brain Research, 400: 348-352.
- 16. Hatton GI (1997). Function-related plasticity in hypothalamus. American Review of Neuroscience, 20: 375-397.
- 17. Beagley GH & Cobbett P (1997). Inhibition of nitric oxide synthase induces ultrastructural changes in the neurohypophysis of dehydrated rats. Neuroscience Letters, 222: 143-146.
- 18. Liu QS, Jia YS & Ju G (1997). Nitric oxide inhibits neuronal activity in the supraoptic nucleus of the rat hypothalamic slices. Brain Research Bulletin, 43: 121-125.
- 19. Brenman JE, Chao DS, Gee SH et al. (1996). Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and a-1 syntrophin mediated by PDZ domains. Cell, 84: 757-767.
- 20. Meeker RB, Swanson DJ, Greenwood RS & Hayward JN (1993). Quantitative mapping of glutamate presynaptic terminals in the supraoptic nucleus and surrounding hypothalamus. Brain Research, 600: 112-122.
- 21. Brann DW (1995). Glutamate: a major excitatory transmitter in neuroendocrine regulation. Neuroendocrinology, 61: 213-225.
- 22. Theodosis DT, Paul L & Tappaz ML (1986). Immunocytochemical analysis of the GABAergic innervation of oxytocin- and vasopressin-secreting neurons in the rat supraoptic nucleus. Neuroscience, 19: 207-222.
- 23. Schrader LA & Tasker JG (1997). Presynaptic modulation by metabotrophic glutamate receptors of excitatory and inhibitory synaptic inputs to hypothalamic magnocellular neurons. Journal of Neurophysiology, 77: 527-536.
- 24. Bains JS & Ferguson AV (1997). Nitric oxide regulates NMDA-driven GABAergic inputs to type I neurons of the rat paraventricular nucleus. Journal of Physiology, 499: 733-746.
- 25. Cui LN, Inenaga K, Nagatoma T & Yamashita H (1994). Sodium nitroprusside modulates NMDA response in the rat supraoptic neurons in vitro Brain Research Bulletin, 35: 253-260.
- 26. Ozaki M, Shibuya I, Kabashima N, Isse T, Noguchi J, Ueta Y, Inoue Y, Shigematsu A & Yamashita H (2000). Preferential potentiation by nitric oxide of spontaneous inhibitory postsynaptic currents in rat supraoptic neurons. Journal of Neuroendocrinology, 12: 273-281.
- 27. Stern JE & Ludwig M (2001). NO inhibits supraoptic oxytocin and vasopressin neurons via activation of GABAergic synaptic inputs. American Journal of Physiology, 280: R1815-R1822.
- 28. Srisawat R, Ludwig M, Bull PM, Douglas JJ, Russell JA & Leng G (2000). Nitric oxide and the oxytocin system in pregnancy. Journal of Neuroscience, 20: 6721-6727.
- 29. Okere CO, Wang Y-F, Higuchi T, Negoro H, Okutani F, Takahashi S & Murata T (1996). The effect of systemic and central nitric oxide administration on milk availability in lactating rats. NeuroReport, 8: 243-247.
- 30. Buxton IL, Cheek DJ, Eckman D, Westfall DP, Sanders KM & Keef KD (1993). NG-nitro L-arginine methyl ester and other alkyl esters of arginine are muscarinic receptor antagonists. Circulatory Research, 72: 387-395.
- 31. Chiu T & Reid IA (1995). Effect of inhibition of nitric oxide synthesis on the cardiovascular and endocrine responses to hemorrhage in conscious rabbits. Hypertension Research, 18: 55-61.
- 32. Goyer M, Bui H, Chou L, Evans J, Kiel LC & Reid IA (1994). Effect of inhibition of nitric oxide synthesis on vasopressin secretion in conscious rabbits. American Journal of Physiology, 266: H822-H828.
- 33. Chiodera P, Volpi R & Coiro V (1994). Inhibitory control of nitric oxide on the arginine-vasopressin and oxytocin response to hypoglycae-mia in normal men. NeuroReport, 5: 1822-1824.
- 34. Cao L, Sun X & Shen E (1996). Nitric oxide stimulates both the basal and reflex release of vasopressin in anesthetized rats. Neuroscience Letters, 221: 49-52.
- 35. Yamaguchi K & Hama H (2003). A study on the mechanism by which sodium nitroprusside, a nitric oxide donor, applied to the anteroventral third ventricular region provokes facilitation of vasopressin secretion in conscious rats. Brain Research, 968: 35-43.
- 36. Ota M, Crofton JT, Festavan GT & Share L (1993). Evidence that nitric oxide can act centrally to stimulate vasopressin release. Neuroendocrinology, 57: 955-959.
- 37. Lutz-Bucher B & Koch B (1994). Evidence for an inhibitory effect of nitric oxides on neuropeptides secretion from isolated neural lobe of the pituitary gland. Neuroscience Letters, 165: 48-50.
- 38. Raber J & Bloom FE (1994). IL-2 induces vasopressin release from the hypothalamus and the amygdala: role of nitric oxide-mediated signaling. Journal of Neuroscience, 14: 6187-6195.
- 39. Yasin S, Costa A, Trainer P, Windle R, Forsling ML & Grossman A (1993). Nitric oxide modulates the release of vasopressin from rat hypothalamic explants. Endocrinology, 133: 1466-1469.
- 40. Verbalis JG, Mangione MP & Stricker EM (1991). Oxytocin produces natriuresis in rats at physiological plasma concentrations. Endocrinology, 128: 1317-1322.
- 41. Johnson AK, Cunningham JT & Thunhorst RL (1996). Integrative role of the lamina terminalis in the regulation of cardiovascular and body fluid homeostasis. Clinical and Experimental Pharmacology and Physiology, 23: 183-199.
- 42. Theodosis DT & Poulain DA (1984). Evidence for structural plasticity in the supraoptic nucleus of the rat hypothalamus in relation to gestation and lactation. Neuroscience, 11: 183-193.
- 43. Koehler EM, McLemore GL, Tang W & Summy-Long JY (1993). Osmoregulation of the magnocellular system during pregnancy and lactation. American Journal of Physiology, 264: R555-R560.
- 44. Okere CO & Higuchi T (1996). Down-regulation of endogenous nitric oxide synthase in late-pregnancy and parturition in the rat hypothalamic magnocellular neurons and neurohypophysis. Neuroscience Letters, 220: 133-136.
- 45. Summy-Long JY, Gestl S, Terrell ML, Wolz G & Kadekaro M (1997). Osmoregulation of the magnocellular neuroendocrine system during lactation. American Journal of Physiology, 272: R275-R288.
- 46. Southam E & Garthwaite J (1993). The nitric oxide-cyclic GMP signaling pathway in rat brain. Neuropharmacology, 32: 1267-1277.
- 47. Furuyama T, Inagaki S & Takagi H (1993). Localizations of a1 and ß1 subunits of soluble guanylate cyclase in the rat brain. Molecular Brain Research, 20: 335-344.
- 48. Yang QZ & Hatton GI (1999). Nitric oxide via cGMP-dependent mechanisms increases dye coupling and excitability of rat supraoptic neurons. Journal of Neuroscience, 19: 4270-4279.
- 49. Briski KP (1999). Pharmacological manipulation of central nitric oxide/guanylate cyclase activity alters Fos expression by rat hypothalamic vasopressinergic neurons during acute glucose deprivation. Journal of Chemical Neuroanatomy, 17: 13-19.
- 50. Butler AR, Flitney FW & Williams DL (1995). NO, nitrosonium ions, nitroxide ions, nitrosothiols, and iron-nitrosyls in biology: a chemist's perspective. Trends in Pharmacological Sciences, 16: 18-22.
- 51. Pryor W, Church DF, Govindan C & Crank G (1982). Oxidation of thiols by nitric oxide and nitrogen dioxide: Synthetic utility and toxicological implications. Journal of Organic Chemistry, 47: 156-159.
- 52. Lander HM, Ogiste JS, Pearce SF, Levi R & Novogrodsky R (1995). Nitric oxide-stimulated guanine nucleotide exchange one p21 ras. Journal of Biological Chemistry, 270: 7017-7020.
- 53. Broillet MC & Firestein S (1996). Direct activation of the olfactory cyclic nucleotide-gated channel through modification of sulfhydryl groups by NO compounds. Neuron, 16: 377-385.
- 54. Molina y Vedia L, McDonald B, Reep B, Brune B, Di Silvio M, Billiar TR & Lapetina EG (1992). Nitric oxide-induced S-nitrosylation of glyceraldehyde-3-phosphate dehydrogenase inhibits enzymatic activity and increases endogenous ADP-ribosylation. Journal of Biological Chemistry, 267: 24929-24932.
- 55. Tenneti L, D'Emilia DM & Lipton SA (1997). Suppression of neuronal apoptosis by S-nitrosylation of caspases. Neuroscience Letters, 236: 139-142.
- 56. Xu L, Eu JP, Meissner G & Stamler JS (1998). Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science, 279: 234-237.
- 57. Aizenman E, Lipton SA & Loring RH (1989). Selective modulation of NMDA responses by reduction and oxidation. Neuron, 2: 1257-1263.
- 58. Stamler JS, Lamas S & Fang FC (2001). Nitrosylation. The prototypic redox-based signaling mechanism. Cell, 106: 675-683.
- 59. Ahern GP, Hsu SF & Jackson MB (1999). Direct actions of nitric oxide on rat neurohypophyseal K+ channels. Journal of Physiology, 520: 165-176.
- 60. Kaczorowki GJ, Knaus HG, Leonard RJ, McManus OB & Garcia ML (1996). High-conductance calcium-activated potassium channels; structure, pharmacology, and function. Journal of Bioenergetics and Biomembranes, 28: 255-267.
Correspondence and Footnotes
Publication Dates
-
Publication in this collection
22 Apr 2004 -
Date of issue
Apr 2004
History
-
Accepted
03 Feb 2004 -
Received
11 Aug 2003