Abstracts
Mascagnins A and B, two novel and unusual epicatechin and epiafzelechin derivatives bearing glucosylated phenyloctanoid units, along with quercetin-3-O-±-L-rhamnopyranosyl-(1→6)-²-Dglucopyranoside were isolated from the aerial parts of Mascagnia pubiflora(Malpighiaceae), a plant toxic to cattle. Their structures were established by a combination of 1D- and 2D-nuclear magnetic resonance spectroscopic techniques and mass spectrometry data.
Mascagnia pubiflora; Malpighiaceae; poisonous plant; phenyloctanoid catechins
Dois novos e incomuns derivados de epicatequina e epiafzelequina contendo unidades feniloctanóides glicosiladas e denominados mascagninas A e B foram isolados, juntamente com quercetina-3-O-±-L-ramnopiranosil-(1→6)-²-D-glucopiranosídeo, das partes aéreas de Mascagnia pubiflora(Malpighiaceae), uma planta tóxica para o gado. Suas estruturas foram determinadas pela combinação de espectroscopia de ressonância magnética nuclear (RMN) uni e bidimensionais e dados de espectroscopia de massas (MS).
ARTICLE
Two unusual epicatechin and epiafzelechin derivatives from Mascagnia pubiflora, a plant toxic to cattle
Walmir S. GarcezI,* * e-mail: wgarcez@nin.ufms.br ; Fernanda R. GarcezI; Deizeluci de F. P. ZanellaI, II; Lidilhone HamerskiI; Antonio G. FerreiraIII; Andersson BarisonIII; Alfredo R. AbotII
IDepartamento de Química Universidade Federal de Mato Grosso do Sul, 79070-900 Campo Grande-MS, Brazil
IIUniversidade Estadual de Mato Grosso do Sul, Unidade Universitária de Aquidauana, 79200-000 Aquidauana-MS, Brazil
IIIDepartamento de Química, Universidade Federal de São Carlos, 13560-970 São Carlos-SP, Brazil
ABSTRACT
Mascagnins A and B, two novel and unusual epicatechin and epiafzelechin derivatives bearing glucosylated phenyloctanoid units, along with quercetin-3-O-α-L-rhamnopyranosyl-(1→6)-β-Dglucopyranoside were isolated from the aerial parts of Mascagnia pubiflora(Malpighiaceae), a plant toxic to cattle. Their structures were established by a combination of 1D- and 2D-nuclear magnetic resonance spectroscopic techniques and mass spectrometry data.
Keywords:Mascagnia pubiflora, Malpighiaceae, poisonous plant, phenyloctanoid catechins
RESUMO
Dois novos e incomuns derivados de epicatequina e epiafzelequina contendo unidades feniloctanóides glicosiladas e denominados mascagninas A e B foram isolados, juntamente com quercetina-3-O-α-L-ramnopiranosil-(1→6)-β-D-glucopiranosídeo, das partes aéreas de Mascagnia pubiflora(Malpighiaceae), uma planta tóxica para o gado. Suas estruturas foram determinadas pela combinação de espectroscopia de ressonância magnética nuclear (RMN) uni e bidimensionais e dados de espectroscopia de massas (MS).
Introduction
In Brazil, poisoning by plants is one of the main causes of death to adult cattle and the number of plants which are considered to be toxic to ruminants is constantly increasing.1,2 Therefore, considerable losses due to cattle intoxication by ingestion of seeds, leaves or roots of toxic plants have become a significant problem in cattle producing areas. In the state of Mato Grosso do Sul, Brazil's greatest producer of pasture-herd cattle, M. pubiflora (Mascagnia pubiflora) (A. Juss.) Griseb., a liana popularly known as "cipó prata" and "corona" that belongs to the Malpighiaceae family, is the main causative agent of sudden bovine death as a consequence of heart attack, due to a peracute poisoning.2,3 The cardiotoxic activity of M. pubiflorawas studied by Saad et al.4 in 1970, and attempts to isolate and characterize the active principles pointed to the possibility of the presence of a biologically active glycoside.4 Considering the economic losses due to the toxic activity of M. pubifloraon grazing animals and the absence of a more detailed phytochemical study, we decided to investigate the chemical composition of the aerial parts of a specimen of M. pubifloragrowing in the "Pantanal" border of Mato Grosso do Sul. Herein, we report on the isolation of two novel and unusual epicatechin and epiafzelechin derivatives bearing glucosylated phenyloctanoid units, named mascagnins A and B, together with quercetin-3-Oα-L-rhamnopyranosyl-(1→6)-β-D-glucopyranoside.
Results and Discussion
Dried and ground aerial parts of M. pubiflorawere extracted with EtOH. After removal of the solvent, the residue was subsequently partitioned between 1:1 MeOHH2O and CHCl3 and 1:1 MeOH-H2O and EtOAc. After a combination of column chromatography on reversed phase silica gel, gel filtration and reversed phase high performance liquid chromatography (HPLC) separations of the EtOAc phase, compounds 1(mascagnin A), 2(mascagnin B) and a known flavonoid diglycoside, quercetin-3-O-α-Lrhamnopyranosyl-(1→6)-β-D-glucopyranoside (3) were isolated.
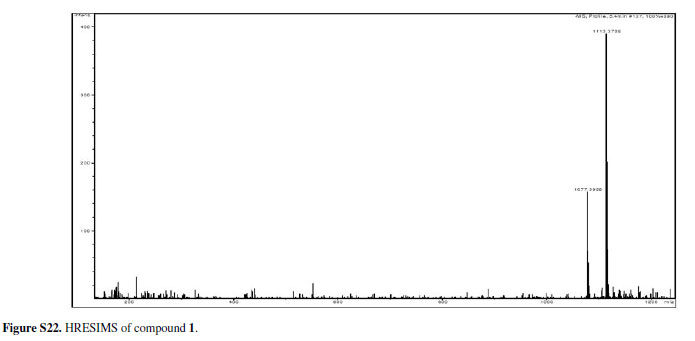
Compound 1was obtained as an amorphous powder and its molecular formula was suggested to be C55H70O24 on the basis of the quasi-molecular ion peak at m/z1113.3706 [M-H]- in the high resolution electron spray ionization mass spectrum (HRESIMS) and on NMR data. The molecular formula pointed to twenty-one degrees of unsaturation for the compound. Its IR spectrum showed absorption bands for hydroxyl (3410 cm-1) and aromatic (1617, 1510 cm-1) functionalities. The 1H and 13C NMR spectra of 1(Table 1) exhibited signals for fifteen aromatic quaternary carbons (nine oxygenated) and nine aromatic methine carbons, thus indicating the presence of four aromatic rings in its structure. In addition, signals attributable to carbons of aliphatic moieties were assigned to two methyls, nine methylenes, six methines (four oxymethines), and two oxygenated quaternary carbons. Additionally, twelve carbon signals corresponding to two sugar residues were also observed and were assigned to two D-glucopyranosyl units.5,6 The anomeric centers were each determined to have a β-configuration based on the coupling constant values of the α-oriented anomeric proton signals, which were observed as a two-proton doublet at δH 4.64 (J7.3 Hz).5,6 This, in turn, showed cross-peak correlations in the heteronuclear single quantum coherence (HSQC) spectrum with the signals at δ 104.5 and 104.8, accounting for the two anomeric carbons, whose chemical shifts around 100 ppm indicated the O-glucosidic nature of 1.5,6
Three of the four aromatic rings were found to be 1,2,4-trisubstituted, as ascertained by the connectivities observed in 1H-1H correlation spectroscopy (COSY) spectrum, in which three spin systems could be assigned by the signals at δ 6.57 (dd, J8.3 and 1.9 Hz), 7.00 (d, J8.3 Hz) and 6.62 (d, J1.9 Hz) [I]; δ 6.51 (dd, J8.3 and 1.9 Hz), 6.99 (d, J8.3 Hz) and 6.60 (d, J1.9 Hz) [II] and δ 6.75 (dd, J8.2 and 1.6 Hz), 6.78 (d, J8.2 Hz) and 6.96 (d, J1.6 Hz) [III]. In addition, the respective HSQC and heteronuclear multiple bond correlations (HMBC) along with the corresponding aromatic carbon resonances indicated the presence of one alkyl and two ortho-oxygenated substituents in each of the aromatic rings. The absence of other proton resonances in the aromatic region of the 1H NMR spectrum as well as the presence of six quaternary carbon signals ascribable to aromatic carbons in the 13C NMR spectrum defined the fourth aromatic ring in the structure of 1as a hexasubstituted one. The chemical shifts assigned to the carbons of this ring at δ 99.5, 105.3, 105.6, 150.9, 151.7 and 154.0 matched to an 1,3,5-trioxygenated aromatic ring bearing alkyl residues at the 2, 4 and 6 positions.
Another spin system detected in the 1H-1H COSY spectrum consisted of two broad singlets at δH 4.19 (1H) and 4.68 (1H), one double doublet at δ 2.95 (1H, J16.7 and 4.0 Hz) and one broad doublet at δ 2.82 (1H, J16.7 Hz). In the HSQC spectrum, cross-peaks were observed between the last two signals and the carbon signal at δ 29.5 as well as between the two broad singlets at δ 4.19 and 4.68 and the carbon signals at δ 67.1 and 79.9, respectively. This information, along with the presence of the aforementioned hexasubstituted aromatic ring and the spin system III, which in turn showed correlations in the HSQC spectrum with the methine carbons at δ 119.3, 116.4 and 115.1, respectively, were in accordance with the presence of an epicatechin residue in the structure of 1bearing alkyl side chains at carbons C-6 and C-8 of ring A.7,8 The remaining carbon resonances in the aliphatic region of the 13C NMR and distortionless enhancement by polarization transfer (DEPT) spectra suggested two C8 carbon-containing chains, each composed of four sp3 methylenes in the region of δ 31.6 to 39.7, two sp3 methines, one being oxymethinic (δ 70.0/70.8, δ 24.9), one tertiary methyl group (δ 28.5) and one sp3 quaternary carbon indicative of the presence of a hemiacetal functionality (δ 98.8/ 98.9). An interesting feature of the 13C NMR spectrum of 1 was the appearance of these sets of signals, which showed very similar chemical shifts and the same multiplicities in the DEPT spectrum, the same being observed for the signals ascribed to the carbons of the two remaining 1,2,4-trisubstituted aromatic rings as well as the signals of their corresponding hydrogens (the above-mentioned spin systems I and II). This led to the assumption that 1 had identical groups, which formed two disjoint spin-systems, linked to ring A of the epicatechin unit. On the basis of correlations discernible from 1H-1H COSY, HSQC and HMBC experiments, they were characterized as being two glucosylated phenyloctanoid units, as shown in Figure 1. A cross-peak between the anomeric proton of the two glucosyl moieties at δ 4.64 and the oxygenated carbon of the two trisubstituted aromatic rings at δ 145.9 (attributed to C-12a) and δ 144.7 (attributed to C-12b) in the HMBC spectrum revealed the linkage between these two groups. The HMBC connectivities between H-14a (δ 6.57) and C-12a and between H-14b (δ 6.51) and C-12b provided further support for these assignments. Likewise, the correlations between the carbon signals at δ 139.4 (C-9a) and 139.6 (C-9b) with the proton signals ascribed to, respectively, H-8a (δ 2.46) and H-8b (δ 2.30 and 2.57) established the linkage between the aromatic rings and the benzylic methylene group of the two aliphatic eight-carbon chains. The signals of these methylene protons, in turn, were found to have long-range correlations with the signal of the corresponding vicinal carbons (C-7a and C-7b, d 39.7). The remaining degrees of unsaturation suggested by the molecular formula of 1 indicated that the eight-carbon moieties must have one ring system each. This information, together with the above-mentioned structural fragments, and the evidence of substitution at C-6 and C-8 positions of the epicatechin unit, were strongly indicative that the carbons of these alkyl side chains formed, along with C-6/C-8 and the oxygen atoms at C-5/C-7 positions of the epicatechin core, a six-membered hemiacetal bearing a methyl group bonded to the hemiacetal carbon. Accordingly, the attachment of C-1a/C-1b to C-6/C-8 of the epicatechin skeleton was deduced unambiguously from the correlations between H-1a/H-1b (δ 3.40/3.42) and C-6/C-8 (δ 105.3/105.6) in the HMBC spectrum. Furthermore, the cross-peaks between the methylene carbon signals at d 35.6/35.4 (C-2a/C2b) and the proton signals of H-1a and H-1b, as well as between the hemiacetal carbon signals C-3a and C-3b at δ 98.9 and 98.8, respectively, and the methyl singlets at δH 1.47/1.52 (H-4a and H-4b, respectively) corroborated the aforementioned suggestion. The methylene carbon signals observed at δ 36.8 and those assignable to hydroxylbearing methine carbons at δ 70.8/70.0 accounted for the remaining carbons of the side chains, namely C-5a/C-5b and C-6a/C-6b, respectively, which showed connectivities in the HSQC spectrum with the signals attributed to H-5a/H-5b (δ 1.60; 1.88 /1.64; 1.53) and H-6a/H-6b (δ 3.53/3.57), respectively. Cross-peaks between H-6a/H-6b (δ 3.53/3.57) and H-7a/H-7b (δ 1.47) in the 1H-1H COSY spectrum along with two-bond C-H correlations observed between H-1a/H-1b (δ 3.40/3.42) and C-5a/C-5b (δ 36.8) in the HMBC spectrum were in accordance with these assignments. The stereochemistry at C-6a and C-6b could not be elucidated while the stereochemical relationships of H-1a/H-1b with the methyl groups C-4a/C-4b were determined to be syn, as revealed by analysis of the nuclear Overhauser enhancement spectroscopy (NOESY) spectrum. Further evidence of structure 1was given by additional two- and three-bond correlations discernible in the HMBC spectrum (Table 1).
In summary, all the spectral data provided the basis to determine the structure 1for mascagnin A, an unusual epicatechin derivative, which has not been previously reported. Compound 2, to which was assigned the trivial name mascagnin B, displayed a quasi-molecular ion peak at m/z1097.3313 [M-H]-in the HRESIMS spectrum, which was sixteen mass units lower than that of 1and in agreement with the molecular formula C55H70O23. The 1H and 13C NMR spectra of 2are closely comparable to those of 1(Table 2). However, the signals of ring B of the epicatechin moiety were not present, suggesting that 2differed from 1only in the nature of the flavan-3-ol nucleus. Accordingly, the presence of a typical pair of AA'BB' doublets at δH 7.32 (2H, J8.2 Hz, H-2' and H-6') and δH 6.80 (2H, J8.2 Hz, H-3' and H-5') was clearly indicative of a para-hydroxy substituted ring B in the structure of 2. This could also be deduced from the carbon resonances at δC 131.8 (C-1'), 128.9 (C-2',C-6'), 115.9 (C-3', C-5') and 157.9 (C-4') and from the connectivities observed between the proton and carbon signals of ring B in the HSQC and HMBC spectra (Table 2). These data can be accounted for in 2if the epicatechin nucleus of 1 is replaced by a flavan-3-ol moiety of the epiafzelechintype. The spectral data reported in the literature for epiafzelechin supported this assumption.7,9 The remaining 1H and 13C NMR assignments of mascagnin B were based on the information afforded by DEPT, HSQC and HMBC experiments and also by comparison with the data obtained for 1(Table 2). Thus, the structure of 2, a novel epiafzelechin derivative, was established as that depicted in Figure 1.
The biogenetic origin of the phenyloctanoid residues found in the unique epicatechin and epiafzelechin derivatives 1and 2can be postulated as arising from chain extension of a C6-C3 unit with three malonyl-Co A units, followed by hydrolysis and decarboxylation of the terminal thioester function of the polyketide moiety.
The 1H and 13C NMR spectral data of compound 3(Figure 1) were found to be in good agreement with those of a di-O-glycosylflavonol of the rutinoside-type which was identified as the known quercetin-3-O-α-L-rhamnopyranosyl(1→6)-β-D-glucopyranoside (rutin). 5,10
In spite of the wide occurrence of epicatechin and epiafzelechin as plant constituents, the isolation of flavan3-ol derivatives bearing glucosylated phenyloctanoid units from M. pubiflorain the present work is noteworthy, since only two records are found in the literature on the chemical constituents of members of the genus Mascagnia: the aforementioned preliminary chemical study of M. pubiflora4 and the reported isolation of two naphtho-γ pyrone glycosides from M. rigida.11
Experimental
General experimental procedures
Optical rotations were determined on a Perkin Elmer 341 polarimeter (Na filter, λ = 589 nm). Infrared (IR) spectra were recorded as KBr pellets on a Bomem-Hartmann & Braun FT IR spectrometer. 1H and 13C 1D and 2D NMR spectra were obtained in CD3OD on Bruker DPX-300 and Bruker DRX 400 spectrometers operating at 300.13 MHz (1H)/75.47 MHz (13C) and 400.35 MHz (1H)/100.10 MHz (13C), respectively. Standard pulse sequences were used for homo-and heteronuclear correlation experiments. HRESIMS spectra were acquired in negative ion mode on an UltrOTOF-Q instrument (Bruker Daltonics). Column chromatography procedures were performed on silica gel 60 RP-18 (230-400 mesh) and Sephadex LH-20. Reversed phase semi-preparative HPLC separations were performed with a Shimadzu LC-6AD pump, using a RP-18, 21.6 x 250 mm, 5 µm particle size, Phenomenex Luna column, with a flow rate of 14 mL min-1, monitoring at 254 nm with Shimadzu Detector.
Plant material
The aerial parts of M. pubiflorawere collected in Aquidauana, Mato Grosso do Sul, Brazil, in August 2006. The plant material was identified by Dr. Arnildo Pott from the Empresa Brasileira de Pesquisa Agropecuária, EMBRAPA-CPAO, Campo Grande, MS, Brazil, where a voucher specimen (No. 5218) is deposited.
Extraction and isolation of chemical constituents
Dried and ground aerial parts of M. pubiflora(466.0 g) were extracted at room temperature with EtOH. After concentration in vacuo, the residue obtained from the EtOH extract was suspended in 1:1 MeOH-H2O and subsequently partitioned with CHCl3 and EtOAc to give the corresponding CHCl3 (19.6 g) and EtOAc (14.5 g) phases. Part of the EtOAc soluble portion (5.0 g) was fractionated on a RP-18 silica gel column eluted successively with stepwise gradients of H2O-MeOH (9.5:0.5, 4:1, 7:3, 3:2, 2:3, 1:4 and MeOH) to yield seven fractions (A→G). Fraction C (667.4 mg, 7:3 H2O-MeOH) was further chromatographed on a Sephadex LH-20 column using 3:2 EtOAc-MeOH as eluent to give two hundred and fifty fractions of 10 mL each. Fractions were grouped on the basis of their TLC profiles into twenty-two fractions (F1→F22). Compound 3(14.5 mg) was obtained from F16 (64.8 mg), after semi-preparative HPLC (1.5:8.5 CH3CN-H2O). A portion (200.0 mg) of fraction D (434.9 mg, 3:2 H2O-MeOH) was further purified by semi-preparative HPLC (1:4 CH3CN-H2O) to yield 1 (30.2 mg) and 2 (8.5 mg).
Mascagnin A (1)
Amorphous powder; [α]D23: +11.43º (c0.17, MeOH); IR (KBr) ν max/cm-1: 3410, 2935, 1617, 1510, 1197, 1123, 1072; 1H and 13C NMR data: see Table 1; HRESIMS m/z1113.3706 [M-H]-(calcd for C55H69O24, 1113.4177).
Mascagnin B (2)
Amorphous powder; [α]D23: +12.32º (c0.28, MeOH); IR (KBr) ν max/cm-1: 3421, 2932, 1614, 1506, 1124, 1073; 1H and 13C NMR data: see Table 2; HRESIMS m/z1097.3313 [M-H]-(calcd. for C55H69O23, 1097.4228).
Acknowledgements
The authors are grateful to FUNDECT-MS and CPq-PROPP-UFMS for their financial support and to FUNDECT-MS and the CNPq for scholarships. Dr.Arnildo Pott (EMBRAPA-CPAO, Campo Grande, MS, Brazil, present address CGMS Herbarium, Universidade Federal de Mato Grosso do Sul, Campo Grande, MS, Brazil) is acknowledged for his assistance in the identification of the plant material. Thanks are also due to Dr. Norberto P. Lopes (Faculdade de Ciências Farmacêuticas, USP, Ribeirão Preto, Brazil) for the HRESIMS measurements.
Supplementary Information
1H NMR, 13C NMR, 1H-1H COSY, HSQC, HMBC and/ or HRESIMS spectra of compounds 1-3are available free of charge at http://jbcs.org.br, as PDF file.
References
1. Riet-Correa, F.; Medeiros, R. M. T.; Pesq. Vet. Bras.2001, 21, 38.
2. Tokarnia, C. H.; Döbereiner, J.; Peixoto, P. V.; Toxicon2002, 40, 1635.
3. Tokarnia, C. H.; Döbereiner, J.; Pesq. Agropec. Bras.1973, 8, 61; Afonso, E.; Pott, A.; Plantas no Pantanal Tóxicas para Bovinos, 5ª ed., EMBRAPA: Brasília, 2001.
4. Saad, A. D.; Andrade, S. O.; Aguiar, A. A.; An. Acad. Bras. Ciênc.1970, 42(Supl.), 235.
5. Agrawal, P. K.; Thakur, R. S.; Bansal, M. C. In Carbon-13 NMR of Flavonoids; Agrawal P. K., ed.; Elsevier: Amsterdam, 1989, ch. 6.
6. Agrawal, P. K.; Phytochemistry1992, 31, 3307.
7. Agrawal, P. K.; Thakur, R. S.; Bansal, M. C. In Carbon-13 NMR of Flavonoids;Agrawal P. K., ed. Elsevier: Amsterdam, 1989, ch. 8.
8. Abe, Y.; Shoji, T.; Kawahara, N; Kamakura, H., Kanda, T.; Goda, Y.; Tetrahedron Lett.2008, 49, 6413.
9. Kashiwada, Y.; Morita, M.; Nonaka, G.; Nishioka, I.; Chem. Pharm. Bull.1990, 38, 856.
10. Beck, M. A.; Häberlein, H.; Phytochemistry1999, 50, 329.
11. Nascimento, M. S.; Habermehl, G. G.; Fitoterapia1995, 66, 539.
Received: November 12, 2008
Web Release Date: April 17, 2009
FAPESP helped in meeting the publication costs of this article.
Supplementary Information

Figure S1 - Click to enlarge

Figure S2 - Click to enlarge

Figure S3 - Click to enlarge

Figure S4 - Click to enlarge

Figure S5 - Click to enlarge

Figure S6 - Click to enlarge

Figure S7 - Click to enlarge

Figure S8 - Click to enlarge

Figure S9 - Click to enlarge

Figure S10 - Click to enlarge

Figure S11 - Click to enlarge

Figure S12 - Click to enlarge

Figure S13 - Click to enlarge

Figure S14 - Click to enlarge

Figure S15 - Click to enlarge

Figure S16 - Click to enlarge

Figure S17 - Click to enlarge

Figure S18 - Click to enlarge

Figure S19 - Click to enlarge

Figure S20 - Click to enlarge

Figure S21 - Click to enlarge

Figure S22 - Click to enlarge

Figure S23 - Click to enlarge

Figure S24 - Click to enlarge

Figure S25 - Click to enlarge

Figure S26 - Click to enlarge

Figure S27 - Click to enlarge

Figure S28 - Click to enlarge

Figure S29 - Click to enlarge

Figure S30 - Click to enlarge

Figure S31 - Click to enlarge

Figure S32 - Click to enlarge

Figure S33 - Click to enlarge

Figure S34 - Click to enlarge

Figure S35 - Click to enlarge

Figure S36 - Click to enlarge

Figure S37 - Click to enlarge

Figure S38 - Click to enlarge

Figure S39 - Click to enlarge

Figure S40 - Click to enlarge

Figure S41 - Click to enlarge

Figure S42 - Click to enlarge

Figure S43 - Click to enlarge
- 1. Riet-Correa, F.; Medeiros, R. M. T.; Pesq. Vet. Bras.2001, 21, 38.
- 2. Tokarnia, C. H.; Döbereiner, J.; Peixoto, P. V.; Toxicon2002, 40, 1635.
- 3. Tokarnia, C. H.; Döbereiner, J.; Pesq. Agropec. Bras.1973, 8, 61;
- Afonso, E.; Pott, A.; Plantas no Pantanal Tóxicas para Bovinos, 5Ş ed., EMBRAPA: Brasília, 2001.
- 4. Saad, A. D.; Andrade, S. O.; Aguiar, A. A.; An. Acad. Bras. Ciênc.1970, 42(Supl.), 235.
- 5. Agrawal, P. K.; Thakur, R. S.; Bansal, M. C. In Carbon-13 NMR of Flavonoids; Agrawal P. K., ed.; Elsevier: Amsterdam, 1989, ch. 6.
- 6. Agrawal, P. K.; Phytochemistry1992, 31, 3307.
- 7. Agrawal, P. K.; Thakur, R. S.; Bansal, M. C. In Carbon-13 NMR of Flavonoids;Agrawal P. K., ed. Elsevier: Amsterdam, 1989, ch. 8.
- 8. Abe, Y.; Shoji, T.; Kawahara, N; Kamakura, H., Kanda, T.; Goda, Y.; Tetrahedron Lett.2008, 49, 6413.
- 9. Kashiwada, Y.; Morita, M.; Nonaka, G.; Nishioka, I.; Chem. Pharm. Bull.1990, 38, 856.
- 10. Beck, M. A.; Häberlein, H.; Phytochemistry1999, 50, 329.
- 11. Nascimento, M. S.; Habermehl, G. G.; Fitoterapia1995, 66, 539.
Publication Dates
-
Publication in this collection
10 June 2009 -
Date of issue
2009
History
-
Accepted
17 Apr 2009 -
Received
12 Nov 2008