Abstracts
The reactivity and diastereoselectivity of conjugate addition of different nitronates ions to (R)-carvone was systematically studied. The Michael adducts 2a-e were obtained in good yield and 3,2-cis-3,5-trans selectivity. The nitroadducts 2b,c were transformed via Nef reaction into (R)-carvone ketone derivatives 9,10 and nitroadducts 2b,d led to (R)-carvone alkylated derivatives 11,12, via a denitration reaction.
nitroalkyl anions; Michael addition; Nef reaction; tetrabutylammonium fluoride
A reatividade e diastereosseletividade da adição conjugada de íons nitronatos representativos à (R)-carvona foi estudada. Os adutos de Michael 2a-e foram obtidos em bom rendimento e boa 3,2-cis-3,5-trans-seletividade. Os nitroadutos 2b e 2c foram transformados via uma reação de Nef nos ceto-derivados 9 e 10 ao passo que uma reação de desnitração transformou 2b e 2d nos derivados alquilados da carvona 11 e 12.
ARTICLE
A new route to keto and alkyl derivatives of (R)-carvone via diastereoselective conjugate addition of nitronate ions
Jeronimo S. CostaII; Bruno S. FreireI; André L. S. MouraI; Vera L. Patrocinio Pereira* * e-mail: patrocinio@nppn.ufrj.br , I
INúcleo de Pesquisas de Produtos Naturais, Centro de Ciências da Saúde, Universidade Federal do Rio de Janeiro, Bloco H, Cidade Universitária, 21941-590 Rio de Janeiro-RJ, Brazil
IICentro Federal de Educação Tecnológica de Química, CEFETEQ, Rua Lúcio Tavares, 1045, 26530-060 Nilópolis-RJ, Brazil
ABSTRACT
The reactivity and diastereoselectivity of conjugate addition of different nitronates ions to (R)-carvone was systematically studied. The Michael adducts 2a-e were obtained in good yield and 3,2-cis-3,5-trans selectivity. The nitroadducts 2b,c were transformed via Nef reaction into (R)-carvone ketone derivatives 9,10 and nitroadducts 2b,d led to (R)-carvone alkylated derivatives 11,12, via a denitration reaction.
Keywords: nitroalkyl anions, Michael addition, Nef reaction, tetrabutylammonium fluoride
RESUMO
A reatividade e diastereosseletividade da adição conjugada de íons nitronatos representativos à (R)-carvona foi estudada. Os adutos de Michael 2a-e foram obtidos em bom rendimento e boa 3,2-cis-3,5-trans-seletividade. Os nitroadutos 2b e 2c foram transformados via uma reação de Nef nos ceto-derivados 9 e 10 ao passo que uma reação de desnitração transformou 2b e 2d nos derivados alquilados da carvona 11 e 12.
Introduction
Both enantiomers of carvone are commercially available at low cost and have been extensively used as chiral starting material in enantioselective syntheses of natural products with diverse biological activities.1-6 The functional richness of 1 allows different synthetic transformations such as conjugate additions, electrophilic additions to electron-rich double bond, frame rearrangement, a-alkylation, carbonyl 1,2-addition, etc. In the case of conjugate addition, different nucleophiles have been added onto electron deficient double bond of 1, for example alkyl Grignard reagents,7 organocopper reagents,8 cyanide anion,8 allyl organoindium reagent,9 thiophenolate,10 silyl enol ethers11 and alcoholate.12 The stereochemistry of the addition of these nucleophiles is preferentially trans to the isopropenyl group.7,8,10,13 Giving pursuit to our strategy to study the reactivity and diastereoselectivity of nitronate anions in nucleophilic additions to chiral nonracemic eletrophiles,14-17 we imagine to develop a new route for the stereocontrolled introduction of acyl and alkyl groups into the structure of naturally occuring (R)-carvone and in this manner provides an entry into various potentially important synthetic intermediates. Thus the keto and alkyl derivatives of (R)-Carvone (1) 9-12 could be synthesized via conjugate addition of representative nitroalkanes 3-8 to 1 followed by Nef and denitration reaction, respectively.
Results and Discussion
The Table 1 shows the results obtained in the conjugate addition of nitroalkanes 3-8 to 1 using mostly TBAF.3H2O as base.
Our results show that TBAF is an efficient promoter of the conjugate addition of 3-7 to 1 (entries 1-7). Others bases such as triethylamine, amberlyst-A21 or KF supported on alumine were not effective. Secundary nitroalkane 8 was essentially not reactive, even under severe reaction conditions (entries 8,9). The addition of the primary nitroalkanes 4 and 5 required excess of reagent (5 equiv.) or longer reaction time for efficient transformation to the desired adducts (entries 2-4). The diastereoselectivity was substantially increased by use of higher concentration of base and longer reaction time (entries 5-7), possibly due to epimerization of the stereocenter at a-carbonyl position18 (compare entries 2-4 with 5-7). No stereocontrol in the nitromethine stereocenter could be obtained due to easy epimerization of this sterereocenter in basic reaction media.15,16 The transformation of the nitroadducts obtained in synthetic useful alkylated and keto-derivatives of 1 was also accomplished. Thus, the nitroadducts 2b,c could be transformed into corresponding ketone derivatives 9, 10 via a Nef reaction19-21 mediated by OxoneTM in good yield and good 3,2-cis-3,5-trans-selectivity, (d.e. 80%), Scheme 1.
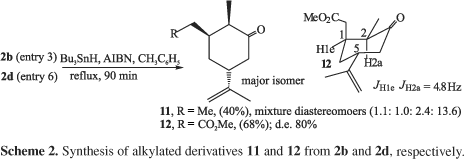
Similarly, the nitroadducts 2b, 2d were easily denitrated22 to corresponding alkylated derivatives 11 and 12 (Scheme 2).
The unambiguous stereochemical assignment to the two new stereocenters formed in the conjugate addition (excluding the nitromethine stereocenter) could be done by chemical correlation with diastereomers of 1218 and by analysis of the spin coupling constant between H-2 and H-3 proton (JH1e H2a 4.8Hz) in the 1H NMR spectrum obtained, see Scheme 2. This valor is in agreement with a cis relationship among these hydrogens.18 The synthesis of 12 confirm the trans kinetical controled addition of nitronate ions to (R)-carvone (1)7,8,10,13 and the epimerization of a carbonyl stereocenter in the conditions employed.18
Conclusions
The transformation of the obtained nitroadducts into alkylated derivatives of carvone complements methodologies of alkylation with the advantage of employing less demanding reaction conditions and low cost reagents. Additionally, the successful Nef reaction allowed the polarity inversion at the nitromethine center, enabling the direct introduction of different acyl groups in the in carvone which might be less practical via other methodologies. The highly functionalised chiral cyclohexanones 10-12 can be view as excellent starting compounds for the total synthesis of enantiomerically pure natural products.1-6
Experimental
TBAF.3H2O solid, nitromethane (3), nitroethane (4), 1-nitropropane (5), 2-nitropropane (8), THF, benzene, AIBN, n-Bu3SnH and amberlyst-A21 were commercially available (Aldrich, Across or Merck) and were used as purchased. Methyl nitroacetate (6),23 1,1-dimetoxi-3-nitropropane (7)24 were prepared according to literature procedures. 1H NMR and 13C NMR spectra were recorded on Gemini-200 (200 MHz) Varian Instruments or on Brucker avance DRX (400 MHz) with TMS as internal reference. The coupling constant (J) is in Hertz (Hz). The IR spectra were recorded on a Nicolet Magna-IR-760 spectrometer and only the principal bands are reported. The analyses by GC-mass was realized on Shimadzu GC/MS-QP 500.
General procedure-preparation of the nitroadducts 2a-e
(2R,3R,5R)-5-isopropenyl-2-methyl-3-nitromethyl-cyclohexanone 2a. To a solution of 1 (1.0 g, 6.6 mmol) in 20 mL THF, under stirring, was added 3 (0.5 mL, 7.3 mmol) followed a solution of TBAF.3H2O (0.6 g, 2 mmol) in 5 mL THF. The mixture was stirred at room temperature for 12 hours, then washed with H2O (50.0 mL), extracted with CH2Cl2 (3 ´ 50.0 mL), the organic phase was dried over Na2SO4 and the solvent was removed in vacuum. The residue was purified by column chromatography on silica gel (Hexane/EtOAc 80:20) yielding 1.2 g (88%) of a mixture of diastereoisomers 2a (6.2 : 17.9 : 8. 4 : 1.0) as yellow oil. IR (neat) nmax/cm-1: 2974; 2939; 2882; 1713; 1645; 1552; 1452; 1435; 1382; 1220; 898. 13C NMR (50 MHz, CDCl3), major isomer: d 209.9; 145.5; 112.7; 78.7; 46.7; 43.6; 39.7; 38.7; 30.1; 21.6; 12.7. MS (70eV): m/z (%): M+. 211 (3); 150 (6); 107 (20); 67 (52); 41 (100).
Nitroadduct 2d, mixture of diastereoisomers (10.0 : 7.5 : 4.5 : 1.0). IR (neat) nmax/cm-1: 2972; 2920; 2884; 1756; 1713; 1645; 1561; 1439; 1370; 1217. 13C NMR (50 MHz, CDCl3), spectroscopy data for the major isomer: d 209.5; 163.8; 145.4; 112.9; 88.6; 53.5; 46.2; 43.9; 43.7; 40.6; 27.7; 21.5; 12.3. MS (70eV): m/z (%): M+. 269 (9); 222 (2); 163 (10); 150 (20); 121 (33); 107 (36); 95 (49); 67 (100); 55 (63).
(2R,3R,5R)-3-acetyl-5-isopropenyl-2-methyl-cyclohexanone 9a+9b
To a solution of 2b, (entry 3, table 1; 0.22g, 1 mmol) in MeOH (6 mL), was added 8 mL of a 0.5 mol L-1 solution of Na2HPO4 whose solvent is a 1 mol L-1 solution of NaOH. After 1 h, a solution of OxoneTM (0.61 g, 1 mmol) in 3 mL of water was added to the stirred suspension. The resulting mixture was stirred at room temperature for 1 h, then acidified with a 10% solution of HCl and extracted with CH2Cl2. The combined organic layers were washed with brine, dried with anhydrous Na2SO4 and evaporated under reduced pressure. The residue was subjected to column chromatography on silica gel (Hexane/EtOAc 95:5) providing 0.14 g of 9a+9b (yield 70%, 9a:9b, 90:10, d.e. 80%) as a colorless oil. IR (neat) nmax/cm-1: 2973, 2937, 2881, 1713,1646, 897. 1H NMR (200 MHz, CDCl3) for the major isomer: d 4.79 (d, 2H, J 8.1); 2.00-2.80 (m, 4H); 2.25 (s, 3H); 1.77 (s, 3H); 1.10-1.70 (m, 3H); 0.97 (d, 3H, J 6.2, 3H). 13C NMR (50 MHz, CDCl3): d 210.9; 209.9; 146.4; 110.5; 57.2; 46.2; 45.3; 33.7; 30.0; 20.0; 16.6; 12.1. MS (70eV): m/z (%): M+ 194 (2); 151( M+. - COCH3, 8 ); 109 (17); 55 (51); 43 ([CH3CO]+, 100).
Denitration of 2b and 2d to 11 and 12, respectively-typical procedure
Methyl 2-((1S,2R,5R)-5-isopropenyl-2-methyl-3-oxo-cyclohexyl)acetate 12. A mixture of 2d (entry 6, Table 1) (0.72 g, 2.68 mmol), Bu3SnH (3.9 g, 13.4 mmol) and AIBN (0.36 g, 2.2 mmol) in toluene (5 mL) was heated at 90 ºC for 90 min. The reaction mixture was subjected to column chromatography on silica gel (Hexane/EtOAc 96:4) to give 0.24 g (yield 68%) of 12 as colorless oil (d.e. 80%). IR (neat) nmax/cm-1: 2971; 2952; 2938; 2880; 1737; 1712; 1645; 1437; 1378; 1249; 1220; 1163; 896. 1H NMR (400 MHz, C6D6): d 1.02 (d, 3H, J 6.8); 1.34 (ddd, 1H, J 13.8, 9.5, 4.1); 1.63 (br s, 3H); 1.8-2.0 (m, 3H), 2.13 (dd, 1H, J 14.3, 5.9); 2.26 (dd, 1H, J 14.3, 3.4); 2.30-2.38 (m, 2H); 2.55 (1H, ddd, J 14.4, 4.8, 1.7); 3.4 (s, 3H); 4.85 (br s, 2H).13C NMR (100 MHz, C6D6): d 13.7; 23.6; 33.3; 37.6; 39.3; 41.4; 44.8; 51.8; 51.9; 113.1; 147.4; 173.1; 210.8. MS (70eV): m/z (%): M+ 224 (7); 193 (7); 150 (100); 151 (54); 109 (87); 67 (97).
(2R, 3R, 5R)-3-ethyl-5-isopropenyl-2-methyl-cyclohexanone 11. Data for the major isomer from mixture of diastereoisomers (1.1 : 1.0 : 2.4 : 13.6). IR (neat) nmax/cm-1: 2966; 2935; 2875; 1712; 1645; 1445; 1377; 891. 1H NMR (200MHz, CDCl3): d 4.76 (m, 2H); 0.86-2.45 (m, 12H); 1.74 (brs, 3H); 1.04 (d, 3H, J 6.6). 13C NMR (50MHz, CDCl3); spectroscopy data for the major isomer: d 212.9; 147.6; 109.4; 48.6; 46.5; 45.3; 45.0; 35.6; 26.1; 20.2; 11.2; 9.9. MS (70eV): m/z (%): M+. 180 (9); 165 (6); 151 ( M+. -Et, 3 ); 139 (3); 109 (44); 55 (78); 41 (100).
Acknowledgments
The authors thank FAPERJ (Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro) for financial support and Professor Alessandro B. C. Simas for text revision.
Received: March 6, 2006
Published on the web: August 1, 2006
- 1. Ho, T. L.; Enantioselective Syhthesis, Natural Products from Chiral Terpenes, John Wiley & Sons, Inc.: New York, 1992, pp. 123-183.
- 2. Jansen, B. J. M.; de Groot, A.; Nat. Prod. Rep 1991, 8, 309.
- 3. Jansen, B. J. M.; de Groot, A.; Nat. Prod. Rep. 1991, 8, 319.
- 4. González, M. A.; Ghosh, S.; Rivas F.; Fisher, D.; Theodorakis, E. A.; Tetrahedron Lett. 2004, 45, 5039.
- 5. Pogrebnoi, S.; Sarabèr, F. C. E.; Jansen, B. J. M.; de Groot, A.; Tetrahedron 2006, 62, 1743.
- 6. Srikrishna, A.; Ramasastry, S. S.V.; Tetrahedron Lett. 2006, 47, 335.
- 7. Jenniskens, L. H. D.; de Groot, A.; Tetrahedron 1998, 54, 5617.
- 8. Verstegen-Haaksma, A. A.; Swarts, H. J.; Jansen, B. J. M.; De Groot, A.; Tetrahedron 1994, 50, 10073.
- 9. Lee, P. H.; Anh, H.; Lee, K.; Sung, S.; Kim, S.; Tetrahedron Lett 2001, 42, 37.
- 10. Solladie, G.; Hutt, J.; Bull. Soc. Chim. Fr. 1986, 4, 643.
- 11. Sarabèr, F. C. E.; Dratch, S.; Bosselaar, G.; Jansen, B. J. M.; de Groot, A.; Tetrahedron 2006, 62,1717.
- 12. Osuka, A.; Taka-oka, K.; Suzuki, H.; Chem. Lett. 1984, 271.
- 13. Permutter, P.; Conjugate Addition Reactions in Organic Synthesis, Pergamon Press: New York, 1992, pp. 35, 36.
- 14. Patrocinio, V. L.; Correia, C. R. D.; Costa, P. R. R.; Synthesis 1994, 474.
- 15. Costa, J. S.; Dias, A. G.; Anholeto, A. L.; Patrocinio, V. L.; Costa, P. R. R.; J. Org. Chem 1997, 62, 4002.
- 16. Pinto, A. C.; Freitas, C. B. L.; Dias A.G.; Pereira, V. L. P.; Tinant, B.; Declercq, J.; Costa, P. R R.; Tetrahedron: Asymmetry 2002, 13, 1025.
- 17. For a recent review on conjugate additions of nitroalkanes to electron-poor alkenes see Balline, R.; Bosica, J.; Fiorini, D.; Palmieri, A.; Petrine, M.; Chem. Rev. 2005, 105, 933.
- 18. Bachi, M. D.; Bilokin, Y. V.; Melman, A.; Tetrahedron Lett 1998, 39, 3035.
- 19. Ceccherelli, P.; Curini, M.; Marcotullio, M. C.; Epifano, F.; Rosati, O.; Synth. Commun. 1987, 17, 195.
- 20. Pinnick, H. W. In Organic Reactions, Paquette, L. A., ed.; Wiley: New York, 1990, vol. 38, ch. 3.
- 21. For a recent review on Nef reaction see Ballini, R.; Petrini, M.; Tetrahedron 2004, 60, 1017.
- 22. Ono, N.; Miyake, H.; Kaji, A.; J. Org. Chem. 1984, 49, 4997.
- 23. Zen, S.; Koiama, M.; Koto, S.; Org. Syn. Coll 1988, 50, 797.
- 24. Öhrlein, R.; Schwab, W.; Ehrler, R.; Jäger, V.; Synthesis 1986, 535.
Publication Dates
-
Publication in this collection
29 Jan 2007 -
Date of issue
Dec 2006
History
-
Received
06 Mar 2006 -
Accepted
01 Aug 2006