Resumo
Simultaneous oxidation/co-precipitation of an equimolar mixture of La(III) and Co(II) nitrates and La(III) nitrate and Mn(II) chloride afforded a hydroxide gel, which was converted to LaCoO3 and LaMnO3 on calcination at 600 °C. After calcination, the obtained perovskites have been characterised by X-ray diffraction (XRD), X- ray photoelectron spectroscopy (XPS), thermogravimetric analysis (DTA - TGA) and BET specific surface determination. Specific surface areas of perovskites were 12 - 60 m²/g. XRD analysis showed that LaCoO3 and LaMnO3 are simple phase perovskite - type oxides. Traces of LaOCl, in addition to the perovskite were detected in the LaMnO3. The catalytic behavior was examined in the propane and CO oxidation. The LaCoO3 catalyst was more active to CO2 than the LaMnO3 catalyst.
perovskites; CO oxidation; propane oxidation
perovskites; CO oxidation; propane oxidation
ARTIGO
Emprego de óxidos tipo perovskita nas oxidações do propano e CO
Employment of perovskite - type oxide in the propane and CO oxidations
Paulo Roberto Nagipe da Silva
Centro de Ciência e Tecnologia, Universidade Estadual do Norte Fluminense, Av. Alberto Lamego, 2000, 28015-620 Campos dos Goytacazes - RJ
ABSTRACT
Simultaneous oxidation/co-precipitation of an equimolar mixture of La(III) and Co(II) nitrates and La(III) nitrate and Mn(II) chloride afforded a hydroxide gel, which was converted to LaCoO3 and LaMnO3 on calcination at 600 °C. After calcination, the obtained perovskites have been characterised by X-ray diffraction (XRD), X- ray photoelectron spectroscopy (XPS), thermogravimetric analysis (DTA TGA) and BET specific surface determination. Specific surface areas of perovskites were 12 60 m2/g. XRD analysis showed that LaCoO3 and LaMnO3 are simple phase perovskite type oxides. Traces of LaOCl, in addition to the perovskite were detected in the LaMnO3. The catalytic behavior was examined in the propane and CO oxidation. The LaCoO3 catalyst was more active to CO2 than the LaMnO3 catalyst.
Keywords: perovskites; CO oxidation, propane oxidation.
INTRODUÇÃO
Os óxidos tipo perovskitas têm a fórmula geral ABO3±d , sendo A um cátion grande tal como os metais alcalinos, os alcalinos terrosos e as terras raras; B um cátion menor, tal como um metal de transição. As perovskitas mais comuns são aquelas em que A é um cátion de um elemento terra rara com estado de oxidação +3 e B um metal de transição no mesmo estado de valência. Esses materiais possuem uma estrutura bem definida, sendo a perovskita ideal do tipo cúbico. Entretanto, essas estruturas aparecem freqüentemente distorcidas, principalmente para as simetrias ortorrômbicas e romboédricas1. É possível realizar substituições parciais dos cátions A e B sem alterar a estrutura fundamental da perovskita2. Muita atenção tem sido dada às perovskitas à base de lantânio (LaBO3), B = Co ou Mn, devido à sua alta atividade e estabilidade térmica em reações de oxidação de hidrocarbonetos. No caso do cobalto, as perovskitas correspondentes são denominadas de cobaltitas de lantânio (LaCoO3) e no caso do manganês, de manganitas de lantânio (LaMnO3). Essas perovskitas chegam a ter atividades comparáveis às dos metais nobres2. Entre as perovskitas de lantânio a manganita é a única rica em oxigênio3, isto é d>0. De outro lado, as manganitas podem tornar-se deficientes em oxigênio pela introdução de um outro sítio catiônico B, como é o caso do ferro4.
O efeito da substituição parcial dos cátions A, geralmente um metal terra rara, com elementos que apresentam estados de valência diferente de +3, tem sido freqüentemente estudado5,6, enquanto o efeito da substituição de cátions do tipo B, e sua aplicação na combustão catalítica tem despertado menos o interesse dos pesquisadores7.
O interesse inicial pelo estudo desses materiais foi motivado por suas propriedades ótica, elétrica e magnética. Comercialmente, as perovskitas de elementos terras raras têm sido muito aplicadas na fabricação de eletrodos8. Na catálise, vários trabalhos na área ambiental têm demonstrado a versatilidade das perovskitas contendo Co e Mn como metais de transição. Assim, as perovskitas tem sido aplicadas na redução seletiva de óxido nítrico com propeno9, na oxidação de CO e hidrocarbonetos10,11, na redução de NO com CO12 ou hidrogênio13 para produzir CO2 e N2. Outras aplicações, tais como combustão do metano14,15, acoplamento oxidativo de alcanos16, hidrogenação e hidrogenólise de hidrocarbonetos17,18 e oxidação da amônia19, evidenciam a importância dos óxidos do tipo perovskitas.
Vários trabalhos na literatura20,21 tratam do estudo da oxidação total de compostos orgânicos voláteis (COVs). Em trabalho recente, Spinicci et al.20 estudaram a oxidação de acetona, isopropanol e benzeno em presença das perovskitas LaMnO3 e LaCoO3. A atividade diminui da acetona ao benzeno na presença de ambos os catalisadores, embora o catalisador LaMnO3 seja o mais ativo. Esta maior atividade foi atribuída às espécies de oxigênio livre e com mobilidade suficiente. As cobaltitas e manganitas de lantânio têm sido igualmente aplicadas na destruição de compostos orgânicos voláteis clorados (COVCs)22.
No passado, as aplicações das perovskitas como catalisadores foram muito limitadas, devido à obtenção de áreas superficiais muito pequenas, geralmente menor que 5 m2/g. A principal razão da baixa área obtida deve-se ao fato que as perovskitas são essencialmente fases de óxidos obtidas a temperaturas elevadas e todos os métodos usados na preparação envolviam uma etapa de aquecimento pelo menos igual à essa temperatura. No caso de LaCoO3, esta temperatura pode chegar a 1073 K23. Mais recentemente, alguns autores citam a temperatura de 823 923 K, no caso de preparação a partir de solução sol-gel24. Alguns exemplos de DRX demonstram que as perovskitas altamente cristalinas somente podem ser obtidas quando submetidas a calcinações a temperaturas superiores a 1073 K. Um trabalho recente mostra que com o emprego do método de complexação de citratos, usando ZnO como aditivo solúvel, obtém-se após calcinação à 973 K, uma perovskita com boa cristalinidade23. Assim, a temperatura de calcinação é de grande importância na preparação dos óxidos do tipo perovskitas. De fato, o aumento desta temperatura resulta em um severo processo de sinterização, que provoca não somente um decréscimo da área superficial, mas também da densidade defeitos superficiais, esses provavelmente ativos como sítios catalíticos de oxidação/redução.
Na preparação das perovskitas são envolvidos métodos químicos e físicos25, mediante reações envolvendo sólido - sólido e líquido - sólido. As reações do tipo sólido - sólido são usadas, principalmente, quando as áreas superficiais resultantes não constituem um parâmetro importante, na preparação de materiais cerâmicos (método cerâmico). Os métodos químicos líquidos - sólido requerem aparelhagens mais simples e podem originar áreas superficiais maiores que 40 m2/g. Entre os métodos químicos podem-se citar cristalização, co-precipitação e complexação. Ainda sobre os métodos físicos, alguns autores constataram que a trituração de reagentes óxidos, seguida da adição de alguns aditivos, pode ser uma rota para obtenção, após calcinação a 473 K, de perovskitas de elevadas áreas superficiais26.
Alguns autores suportam as perovskitas em alguns suportes a fim de aumentar sua área específica. Assim, cobaltitas e manganitas de lantânio foram suportadas em óxido de zircônio, ZnO2, e examinadas na reação de combustão do metano e oxidação do CO10.
O método de preparação utilizado nesse trabalho foi desenvolvido por Barnard et al.27 e baseia-se na co-precipitação de sais (nitrato de cobalto e nitrato de lantânio) para obtenção de catalisadores de LaCoO3 com elevadas áreas superficicias. Neste trabalho, este método será igualmente empregado na preparação de catalisadores do tipo LaMnO3.
PARTE EXPERIMENTAL
Preparação dos catalisadores
Inicialmente, para a preparação das perovskitas à base de cobalto, foram preparadas as soluções dos sais de nitrato de cobalto II e nitrato de lantânio contendo 20 mmol de cada um desses sais em 100 ml de água destilada. A solução de nitrato de cobalto II foi preparada em laboratório pela interação do óxido de lantânio com ácido nítrico 6 M, seguido de aquecimento quase à secura e, conseqüentemente, adição de água destilada para obtenção da quantidade especificada anteriormente. Na preparação das perovskitas à base de manganês empregou-se, como sal fornecedor de cátions do tipo B, o cloreto de manganês (II). As soluções preparadas foram misturadas e colocadas em repouso por aproximadamente 1 h.
Em outro recipiente, preparou-se uma solução oxidante de hidróxido de sódio (7,2 g) em hipoclorito de sódio (3,5 M, 30 ml).
Em seguida, sob atmosfera inerte de nitrogênio, adicionou-se gota-a-gota e sob constante agitação a solução de sais na mistura oxidante. A agitação requerida foi obtida através da passagem de um fluxo elevado de nitrogênio pela solução. O emprego da atmosfera inerte é necessário para minimizar a formação de carbonato do produto final. De fato, tem sido constatado que CO2 proveniente do ar atmosférico sorve-se facilmente sobre os géis de hidróxido de metais de terras raras27, formando carbonatos. O gel obtido foi lavado cerca de três vezes com 100 ml de água deionizada de cada vez. A cada lavagem, o gel foi separado da fase líquida por filtração. Procedeu-se, então, à lavagem com acetona, aproximadamente 300 ml, sendo cada etapa de 100 ml. O material preparado foi seco à temperatura ambiente por 48 h.
Caracterização físico química
Difração de raios X
A identificação da fase do catalisador foi conduzida em um difratômetro de raios X do tipo URD 65 - Rich Seifert & Co. As condições usadas foram radiação Cu Ka, filtro Ni, 30 kV, 40 m A, varredura 0,05 (2q)/5 s. Os difratogramas obtidos foram comparados com os padrões do JCPDS.
BET
As medidas de fisissorção foram realizadas em um equipamento do tipo Autosorb 1c da Quantachrome. A área superficial BET foi determinada através da adsorção de nitrogênio a 77 K e pressão relativa na faixa de 0,05 (p /p0 (0,20, considerando-se para a molécula de nitrogênio uma área de 0,162 nm2). Antes das medidas de adsorção as amostras foram desgaseificadas em vácuo de 10-6 torr a 573 K por 1 h.
Análise térmica ATD - ATG
As análises térmica diferencial (ATD) e térmica gravimétrica (ATG) foram realizadas em um equipamento Universal V2 6D TA Instruments. Os óxidos secos apenas à temperatura ambiente por 48 h foram tratados com fluxo de ar sintético por 15 min, sendo depois aquecidos sob temperatura controlada (10 °C/min) até 600 °C.
XPS
A espectroscopia de fotoelétrons excitados por raios X (XPS), foi realizada em um espectrômetro VG Scientific Escalab Mark II com fonte de raios X Al Ka. A pressão base na câmara de XPS foi menor que 1,0. 10-9 torr. O sinal C 1 s em 284,9 eV foi usado como referência para obter as energias de ligação.
Análise química
A valência do estado do manganês foi determinada pela dissolução da amostra em excesso conhecido de uma solução padrão de sulfato ferroso e pela titulação do Fe2+ em excesso com permanganato de potássio28.
Teste catalítico
Os catalisadores preparados e caracterizados foram testados nas reações de oxidação do propano e do CO. As medidas de atividade foram realizadas em um microrreator de leito fixo operando em pressão atmosférica. O reator, um tubo vertical de 6 mm de diâmetro interno, era feito de quartzo e colocado em um forno elétrico também vertical com altura de 250 mm. A temperatura do catalisador foi monitorada por um termopar de chromel - alumel associado a um programador linear de temperatura do tipo EDGON 3P. A alimentação do reator foi feita através de uma mistura padrão com a seguinte composição: O2 = 2,02%, CO = 0,97% e C3 H8 = 0,507%. O fluxo da mistura reacional foi de 1,8 l/h.
Os produtos foram analisados em linha por cromatografia gasosa (Finnigam 9001) com detetor de ionização de chama para análise dos compostos orgânicos e coluna do tipo Porapack Q de 3 m de comprimento e 1/8 de polegadas de diâmetro, utilizando nitrogênio como gás de arraste. O dióxido de carbono proveniente da oxidação do propano e CO foi monitorado através de um analisador FTIR (Hartmann and Braun URAS 14).
RESULTADOS E DISCUSSÃO
Na preparação dos óxidos tipo perovskitas, LaCoO3 e LaMnO3, foi empregado um método de co-precipitação - oxidação, desenvolvido anteriormente27 para preparação de catalisadores LaCoO3.
A vantagem deste método em relação a outros, em que a desidratação do gel formado é realizada apenas com aquecimento em presença de ar, é o fato de ser feita uma desidratação inicial em presença de acetona. Tal desidratação controlada de hidrogéis, usando água e um solvente miscível, já mostrou resultados positivos na obtenção de sólidos com elevadas áreas superficiais29,30. O deslocamento da água pela acetona em tal gel, permite uma diminuição da tensão superficial e origina menor força capilar, produzindo aglomerações mais fracas e mantendo a estrutura das partículas do hidróxido, sob aquecimento, mais aberta. O resultado é um menor colapso durante a calcinação30. As melhores medidas de área superficial foram encontradas após as amostras serem calcinadas inicialmente a 400 °C por 1 h e, em seguida, a 600 °C por mais 1 h. Entretanto, alterando as temperaturas de calcinação e eventualmente a lavagem com acetona, vários materiais catalíticos foram preparados.
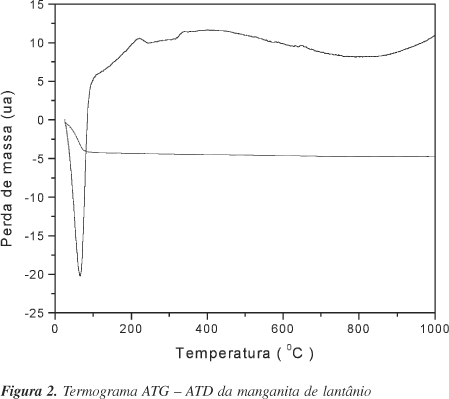
Os resultados da análise térmica sobre os catalisadores podem ser acompanhados através das Figuras 1 e 2. Os termogramas para as supostas perovskitas à base de cobalto e manganês apresentam picos endotérmicos à baixa temperatura, correspondendo à eliminação de água fisissorvida. Este resultado está de acordo com o termograma de ATG, onde a perda de massa nos dois casos corresponde a 2,7% no caso da cobaltita de lantânio e 1,16% no caso da manganita de lantânio. Em aproximadamente 550 °C, o termograma ATD relativo à cobaltita apresenta um pico exotérmico. Já no caso da manganita, um pico exotérmico de menor intensidade foi observado em aproximadamente 720 °C. Esses picos podem ser associados com a cristalização das perovskitas. Este fato pode ser comprovado pelo termograma ATG, onde se observa nessas temperaturas uma queda brusca de peso proveniente provavelmente da liberação de CO2, oriundo da decomposição de carbonato formado durante a preparação28.
Após desidratação à 673K por 1 h e calcinação à 873K por igual tempo, as amostras foram submetidas à análise de DRX. O modelo de difração obtido para o catalisador LaCoO3 corresponde à estequiometria da perovskita descrita no JCPDS: 25-1060.
O modelo DRX para o catalisador à base de manganês lantânio, é mais próximo da estrutura LaMnO3 não estequiométrica, com excesso de oxigênio (JCPDF: 32-0484), a qual apresenta uma simetria ortorrômbica. Convém ressaltar que este fator por si só não comprova a existência de tal estrutura. À baixa temperatura, a estrutura romboédrica de LaMnO3 é, sem dúvida, rica em vacâncias catiônicas3, o que corresponde a um excesso de oxigênio com relação à estequiometria (d = 0,1 0,2), acompanhado pela transição espontânea de 15 30% de Mn3+ a Mn4+ para restabelecer a eletroneutralidade do sistema. De outro lado, além dos picos característicos da perovskita LaMnO3, observam-se picos característicos de LaOCl (Figura 4), aqui representados por (*), JCPDS 34-1494, indicando a presença de traços do referido óxido. A presença de LaOCl já foi constatada por outros autores22, onde o efeito do cloro foi estudado sobre os dois tipos de catalisadores.
A caracterização da superfície dos catalisadores LaCoO3 e LaMnO3 foi realizada por análise XPS com a utilização de C1s como referência. A Tabela 1 lista os valores das energias de ligação correspondentes para: O 1s, La 3d, Co 2p e Mn 2p. Vários trabalhos têm demonstrado que a maioria dos cátions está presente na forma trivalente31.
Segundo a análise dos dados para LaCoO3 e referentes ao La 3d 5/2 são registrados em 835,3 e 838,8 eV, sendo muito semelhantes aos valores apresentados na literatura22 para o óxido de lantânio puro (La2 O3) em 834,4 e 837,8 eV. Esses dados indicam que o lantânio provavelmente está presente na forma trivalente.
Os sinais em 779,9 e 795,5 eV correspondem ao Co 2p3/2 e Co 2p1/2, respectivamente, típicos da presença de Co3+. De outro lado, a ausência de picos satélites na região de 785 788 eV é prova da inexistência de Co2+. Isto comprova a teoria sobre a existência dos cátions em suas formas trivalentes.
Espectro XPS do catalisador LaCoO3 (Figura 3) apresenta a deconvolução do pico O 1s em dois picos, que correspondem a duas espécies de oxigênio. O pico de menor energia, 529,8 eV, pode ser associado com o oxigênio da rede, enquanto o de maior energia, 532,5 eV, pode ser associado a espécies de oxigênio absorvido, embora esta fração contenha também oxigênio adsorvido. A fração do oxigênio absorvido adsorvido podendo ser representado pelas espécies O- e OH-32.
Na literatura32, os valores de energia correspondentes a espécie de oxigênio O- aparecem em 528,3 e 531,9 eV. O primeiro representa o oxigênio da rede e o segundo, a parte absorvida. Já no caso da espécie OH-, esses valores passam a 529,7 e 532,2 eV. Convém ressaltar a semelhança dos resultados de energia de ligação para a espécie OH- com os apresentados nesse trabalho, indicando maior probabilidade da existência do mesmo.
No caso do catalisador LaMnO3 (Figura 4), todos os picos são deslocados de aproximadamente 6 eV em relação aos valores de energia de ligação encontrados no espectro do catalisador LaCoO3. Isto pode estar relacionado à presença de cloro, conforme indicou a presença da fase La O Cl na difração de raios X. Assim, picos referentes ao La 3d 5/2 são deslocados para região de maior valor de energia de ligação (841,10 e 845 eV). Um único pico correspondente a O 1s aparece somente em 536,60 eV. A ausência do pico na região de maior energia indica apenas o envolvimento do oxigênio da rede. Alterações na estrutura cristalina do composto e também na sua estrutura eletrônica podem ser as responsáveis pelo deslocamento citado anteriormente.
O Mn 2p apresenta três sinais correspondendo a Mn3+ (647,40 eV), Mn4+ (658,7 eV) e à presença de um pico satélite, indicando a ausência de Mn2+. A presença de cloro é constatada através de um pico bem definido em aproximadamente 274,0 eV.
No que se refere ao nível C 1s, observa-se, embora ainda pequeno, um grau maior de contaminação por carbonato da superfície de manganita em 289 eV.
É também conveniente comparar as informações da estequiometria obtida pelas medidas de XPS (Tabela 1). A razão atômica La /Co na superfície é de 1,8. Isto sugere a presença de excesso de lantânio ou deficiência de Co na superfície da cobaltita LaCoO3. Alguns autores têm observado a presença de excesso de lantânio nas estruturas das perovskitas (La/Co >1). Tabata et al.33 mencionaram a razão La/Co como sendo 1,2 no caso de catalisadores LaCoO3, obtidos pela síntese com acetato. O caso mais pronunciado foi observado por Connell et al.31, que prepararam a cobaltita através da precipitação da soluções de nitrato de cobalto e lantânio, com hidróxido de tetraetilamônio. Para este caso a razão La/Co foi 4,26, evidenciando a presença de um grande excesso de lantânio na superfície. Este excesso de lantânio na superfície foi atribuído à formação de duas fases; uma correspondendo à perovskita pura e a outra formada por La/perovskita, ambas com deficiência em íons lantânio. Na primeira, as vacâncias catiônicas são compensadas pela presença das valências elevadas de cobalto e oxigênio; na segunda, pela presença de vacâncias aniônicas. Esta última sendo, cataliticamente, muito mais interessante.
De uma maneira geral, muitos autores têm considerado o sistema LaCoO3 como estequiométrico. De outro lado, o sistema é apontado como não estequiométrico como é o caso do trabalho de Wu et al.34. Nos sistemas não estequiométricos geralmente existe uma grande deficiência de lantânio na rede, que pode ser compensada pelo aumento na valência do metal do tipo B, ou do oxigênio ou, ainda, pela criação de vacâncias aniônicas.
Em nosso caso, pode-se observar que a quantidade de íons lantânio é quase o dobro dos íons cobalto. Assim, poder-se-ia supor primeiramente a formação de uma cobaltita de lantânio estequiométrica, juntamente com outros compostos à base de lantânio como, por exemplo, óxido de lantânio. Entretanto, através da difração de raios X não foi observada outra fase que as da perovskita, descartando a hipótese de formação de uma perovskita estequiométrica. Assim, já que a perovskita é não estequiométrica e não há formação de outros compostos, a razão encontrada provavelmente é devido à deficiência em cobalto.
De outro lado, a razão La/Mn é de 0,79, sugerindo que a fase da manganita de lantânio não é estequiométrica e sim, com deficiência em lantânio. Para manter o equilíbrio de cargas, a seguinte conversão pode ser considerada; Mn3+® Mn4+. Esta transição é acompanhada pela geração de vacâncias catiônicas e pela presença de excesso de oxigênio. Tagauchi et al.35 encontraram para o catalisador LaMnO3, dependendo do método de preparação, razões La/Mn que variavam de 1,5 2. Entretanto, parte do lantânio não estava incorporada à perovskita, mas se apresentava na forma de óxidos, onde parte desses óxidos estava carbonatada.
A razão oxigênio-íon metálico total (O/La + Co) no catalisador LaCoO3 é aproximadamente igual a 3,5, em perfeito acordo com o resultado encontrado por Connell et al.26. Já no catalisador LaMnO3, esta razão é de aproximadamente 2,5, ambas superiores à razão de 1,5, esperada caso a estequiometria fosse obedecida. Isto comprova que esses materiais possuem excesso de oxigênio. Este resultado comprova que o excesso de oxigênio no caso da cobaltita e da manganita caracteriza a formação de vacâncias catiônicas. Logo, estamos diante uma cobaltita com deficiência em cobalto e de uma manganita com deficiência em manganês. Entretanto, a supremacia da quantidade de oxigênio não estequiométrico da cobaltita em relação à manganita, pode indicar a presença de maior quantidade de vacâncias catiônicas, no primeiro caso. Além disso, no caso da manganita parte dessas vacâncias é minimizada pela transição Mn3+® Mn4+.
Na Tabela 1 são apresentados os resultados de XPS (energia de ligação e razão atômica) em relação aos elementos presentes na constituição dos catalisadores.
As atividades dos catalisadores preparados foram medidas através das seguintes reações:


A primeira reação, oxidação do CO, ocorre a temperaturas mais baixas e a segunda, oxidação total do propano, a temperaturas mais elevadas.
Os resultados obtidos na reação de oxidação do propano podem ser vistos nas Tabelas 2 e 3. Nessas tabelas pode-se comparar a variação da conversão com a temperatura e a área total dos catalisadores. Pode-se observar que a conversão aumenta à medida que aumenta a área superficial total e que as perovskitas LaCoO3 são mais ativas que as LaMnO3 exigindo, desta forma, menor temperatura na obtenção de um mesmo grau de conversão. Para melhor comparação, podem-se comparar catalisadores que apresentem áreas totais próximas ou semelhantes; como é o caso da cobaltita, LaCoO3-V com área de 34,0 m2/g e a manganita, LaMnO3-I, 39,0 m2/g. Nota-se que no caso da cobaltita é preciso uma temperatura de 148 °C para obtenção de uma conversão de 5,0%, enquanto no caso da manganita é preciso uma temperatura de 308 °C para obtenção do mesmo grau de conversão.
De outro lado, a cobaltita atinge a conversão máxima em temperatura de 382 °C, enquanto a manganita atinge a mesma conversão a 458 °C. Convém ressaltar que esta diferença é mais pronunciada em temperaturas mais baixas.
No caso da reação de oxidação do CO, os resultados podem ser vistos nas Tabelas 4 e 5, onde as cobaltitas continuam sendo mais ativas. Os catalisadores LaCoO3 -V e o LaMnO3- I convertem 5% de CO nas temperaturas de 122 e 140 °C, respectivamente. De outro lado, eles atingem a conversão máxima em 160 e 320 °C, respectivamente. Aqui as diferenças de atividade são mais importantes nas temperaturas mais elevadas, já que a reação de oxidação de CO ocorre em temperaturas mais baixas. A excelente atividade das cobaltitas de lantânio na reação de oxidação do CO foi ressaltada por Shu et al .25.
Existem várias discussões sobre as atividades das perovskitas. Dentre as menos comuns destacam-se: estados de oxidação de íons de metais de transição; a quantidade de oxigênio não estequiométrico e a estrutura de defeitos da rede.
Aparece freqüentemente na literatura que o oxigênio não estequiométrico pode ser um fator de considerável importância na interpretação da atividade catalítica de uma série de perovskitas de metais de transição, inclusive com a participação de promotores28. Entretanto, a estrutura química da rede de uma perovskita é muito complexa, podendo haver distorções do tipo hexagonal, romboédrica e tetragonal. Esta distorção do sistema cúbico pode resultar em uma não estequiometria catiônica ou aniônica.
A oxidação de monóxido de carbono sobre óxidos do tipo perovskita tem sido muito estudada. A partir de dados cinéticos e dados da espectroscopia infravermelho, foi proposto um esquema para a oxidação do CO sobre LaCoO32:

Onde a etapa (5) seria a etapa determinante da velocidade. Oxigênio é adsorvido como espécie O2- sobre íons cobalto de baixo estado de oxidação, o qual subseqüentemente dissocia-se, originando espécies de oxigênio atômico (O-), adsorvido sobre o mesmo centro (2). CO é adsorvido sobre íons O2- da rede, produzindo espécies lábeis que interagem com oxigênio atômico adsorvido, produzindo carbonato que se decompõe produzindo CO2 e oxigênio (2). Este resultado está em perfeito acordo com o resultado de análise térmica apresentado pela amostra de LaCoO3 nesse trabalho, onde a decomposição do carbonato ocorre em aproximadamente 550 °C. Para o caso do propano, poderíamos propor um mecanismo semelhante ao anterior, diferindo apenas na etapa inicial, onde iria gerar monóxido de carbono e água.
A manganita de lantânio, apesar de ser menos ativa que o catalisador LaCoO3, possui uma atividade intrínseca considerável devido à tendência do Mn+3 oxidar-se sob aquecimento em ar, à pressão atmosférica. A transição é acompanhada pela geração de vacâncias catiônicas e pela presença de um conhecido excesso de oxigênio em relação à estequiométrica necessária. O não aparecimento do pico exotérmico nas proximidades de 550 0C pode indicar que o mecanismo de oxidação e o processo de cristalização do LaMnO3 são diferentes dos apresentados para o catalisador LaCoO3.
CONCLUSÃO
Através do método de preparação empregando a co-precipitação de sais em meio oxidante consegue-se preparar cobaltitas e manganitas de lantânio, não estequiométricas, com consideráveis áreas superficiais e com grau relativamente baixo de contaminação por carbonato. Essas perovskitas são ricas em vacâncias catiônicas, que correspondem a um excesso de oxigênio em relação à estequiometria. Este excesso foi mais pronunciado no caso das cobaltitas, o que pode ser o causador de sua maior atividade nas reações de oxidação de CO e propano.
AGRADECIMENTOS
À FAPERJ e à FINEP/CTPETRO pelo apoio financeiro. Aos técnicos M. V. Rocha e R. N. Agum, ambos bolsistas, FINEP/CTPETRO, pela ajuda na realização da caracterização das amostras.
REFERÊNCIAS
1. Tejuca, L. G.; Fierro, J. L. G. Em Properties and Applications of Perovskites Type Oxides, Dekker, M., ed.; New York, 1993.
2. Tejuca, L. G.; Fierro, J. L. G.; Tascon, J. M. D.; Adv. Catal. 1989, 36, 237.
3. Misusaki, J.; Takai, N.; Yonemura, H.; Minamiue, Y.; Tagawa, H.; Dokiya, T.; Inaba M.; Naraya, H.; Sasamoto, K.; Hashimoto, T.; Solid State Ionics 2000, 129, 163.
4. Kindermann, L.; Das, D.; Nickel, H.; Hilpert, K.; Solids State Ionics 1996, 89, 215.
5. Isuprova, L. A.; Tsybulya, S. V.; Kryukoya, G. N.; Alikina, G. M.; Boldyreva, N. N.; Yakovleva, I. S.; Ivanov, V. P.; Sadykov, V. A.; Solid State Ionics 2001, 141, 417.
6. Inaba, H.; Hayasashi, H.; Suzuki, M.; Solid State Ionics 2001, 144, 99.
7. Rojas, M. L.; Fierro, J. L. G.; Tejuca, L. G.; Bell, A. T.; J. Catal. 1990, 124, 41.
8. Furusaki, A.; Konno, H.; Furuichi, R.; J. Mater. Sci. 1995, 2829.
9. Hansen, K. K.; Skou, E. M.; Chritensen, H.; Turek, T.; J. Catal. 2001, 199, 132.
10. Cimino, S.; Colonna, S.; De Rossi, S.; Faticanti, M.; Lisi, L.; Pettiti, I.; Porta, P.; J. Catal. 2002, 205, 309.
11. Song, K. S.; Klvana, D.; Kichnerova, J.; Appl. Catal. 2001, 213, 113.
12. Forni, L.; Oliva, C.; Barzetti, T.; Selli, E. A. M.; Vishiniakov, A.V.; Appl. Catal., B 1997, 13, 35.
13. Ferri, D.; Forni, L.; Dekkers, M. A. P.; Nieuwenhuys, B. E.; Appl. Catal., B 1998, 16, 339.
14. Nguyen, S. V.; Szabo, V.; Trong On, D.; Kaliaguine, S.; Microporous Mesoporous Mater. 2002, 54, 51.
15. Saracco, G.; Geobaldo, F.; Baldi, G.; Appl. Catal., B 1999, 20, 277.
16. Buyevskaya, O. V.; Rothaemel, M.; Zanthoff, H. W.; Baerns, M.; J. Catal. 1994, 71, 150.
17. Libby, W. F.; Science 1971, 171, 499.
18. Pederson, L. A.; Libby, W. F.; Science 1972, 176, 1355.
19. Yu, Z.; Gao, L.; Yuan, S.; Wu, Y.; J. Chem. Soc., Faraday Trans. 1992, 88, 3245.
20. Spinicci, R.; Faticanti, M.; Marini, P.; De Rossi, S.; Porta, P.; J. Mol. Catal. 2003, 197, 147.
21. Spinnici, R.; Tofannari, A.; Faticanti, M.; Pettiti, I.; Porta, P.; J. Mol. Catal. 2001, 176, 247.
22. Sinquin, G.; Petit, C.; Hindermann, K.; Catal. Today 2001, 70, 183.
23. Crespin, M.; Hal, K. W.; J. Catal. 1981, 69.
24. Xiong, G.; Zhi, Z. L.; Yang, X. J.; Lu, L.; Wang, X.; J. Mater. Sci. Lett. 1997, 16, 1064.
25. Shu, J.; Kaliaguine, S.; Appl. Catal., B 1998, 16, 303.
26. Kaliaguine, S.; Van Neste, A.; Szabo, V.; Gallot, J. E.; Bassir, M.; Muzychuk, R.; Appl. Catal., A 2001, 209, 345.
27. Barnard, K. R.; Foger, K.; Turney, T. W.; Williams, R. D.; J. Catal. 1990, 125, 265.
28. Lee, Y. Ng.; Lago, R. M.; Fierro, J. L. G. V.; Sapinã, F.; Martínez, E.; Appl. Catal., A 2001, 207, 17.
29. Her, R.; The Chemistry of silica, Wiley: New York, 1979.
30. Dombro, R. A.; Kirch, W.; Eur. Pat. Application 1984, 110, 78; Appl. Catal., A 2001, 207, 17
31. O Connel, M.; Norman, A.; Huttermann, C. F.; Morris, M. A.; Catal. Today 1999, 47, 123.
32. Roberts, M. W.; Chem. Soc. Rev. 1989, 18, 451.
33. Tabata, K.; Matsumoto, I.; Kohiki, S.; J. Mater Sci. 1987, 22, 1882.
34. Wu, Y.; Yu, T.; Dou, B. S.; Wang, C. X.; Xie, X. F.; Yu, Z. L.; Fan, S. R.; Wang, L. C.; J. Catal. 1989, 120, 88.
35. Taguchi, H.; Sugita, A.; Nagao, M. J.; Takeda, Y.; J. Solid State Chem. 1995, 116, 343.
Recebido em 29/11/02; aceito em 8/9/03
e-mail: nagipe@uenf.br
- 1. Tejuca, L. G.; Fierro, J. L. G. Em Properties and Applications of Perovskites Type Oxides, Dekker, M., ed.; New York, 1993.
- 2. Tejuca, L. G.; Fierro, J. L. G.; Tascon, J. M. D.; Adv. Catal 1989, 36, 237.
- 3. Misusaki, J.; Takai, N.; Yonemura, H.; Minamiue, Y.; Tagawa, H.; Dokiya, T.; Inaba M.; Naraya, H.; Sasamoto, K.; Hashimoto, T.; Solid State Ionics 2000, 129, 163.
- 4. Kindermann, L.; Das, D.; Nickel, H.; Hilpert, K.; Solids State Ionics 1996, 89, 215.
- 5. Isuprova, L. A.; Tsybulya, S. V.; Kryukoya, G. N.; Alikina, G. M.; Boldyreva, N. N.; Yakovleva, I. S.; Ivanov, V. P.; Sadykov, V. A.; Solid State Ionics 2001, 141, 417.
- 6. Inaba, H.; Hayasashi, H.; Suzuki, M.; Solid State Ionics 2001, 144, 99.
- 7. Rojas, M. L.; Fierro, J. L. G.; Tejuca, L. G.; Bell, A. T.; J. Catal 1990, 124, 41.
- 8. Furusaki, A.; Konno, H.; Furuichi, R.; J. Mater. Sci. 1995, 2829.
- 9. Hansen, K. K.; Skou, E. M.; Chritensen, H.; Turek, T.; J. Catal 2001, 199, 132.
- 10. Cimino, S.; Colonna, S.; De Rossi, S.; Faticanti, M.; Lisi, L.; Pettiti, I.; Porta, P.; J. Catal 2002, 205, 309.
- 11. Song, K. S.; Klvana, D.; Kichnerova, J.; Appl. Catal 2001, 213, 113.
- 12. Forni, L.; Oliva, C.; Barzetti, T.; Selli, E. A. M.; Vishiniakov, A.V.; Appl. Catal., B 1997, 13, 35.
- 13. Ferri, D.; Forni, L.; Dekkers, M. A. P.; Nieuwenhuys, B. E.; Appl. Catal, B 1998, 16, 339.
- 14. Nguyen, S. V.; Szabo, V.; Trong On, D.; Kaliaguine, S.; Microporous Mesoporous Mater. 2002, 54, 51.
- 15. Saracco, G.; Geobaldo, F.; Baldi, G.; Appl. Catal, B 1999, 20, 277.
- 16. Buyevskaya, O. V.; Rothaemel, M.; Zanthoff, H. W.; Baerns, M.; J. Catal 1994, 71, 150.
- 17. Libby, W. F.; Science 1971, 171, 499.
- 18. Pederson, L. A.; Libby, W. F.; Science 1972, 176, 1355.
- 19. Yu, Z.; Gao, L.; Yuan, S.; Wu, Y.; J. Chem. Soc., Faraday Trans 1992, 88, 3245.
- 20. Spinicci, R.; Faticanti, M.; Marini, P.; De Rossi, S.; Porta, P.; J. Mol. Catal 2003, 197, 147.
- 21. Spinnici, R.; Tofannari, A.; Faticanti, M.; Pettiti, I.; Porta, P.; J. Mol. Catal 2001, 176, 247.
- 22. Sinquin, G.; Petit, C.; Hindermann, K.; Catal. Today 2001, 70, 183.
- 23. Crespin, M.; Hal, K. W.; J. Catal 1981, 69.
- 24. Xiong, G.; Zhi, Z. L.; Yang, X. J.; Lu, L.; Wang, X.; J. Mater. Sci. Lett 1997, 16, 1064.
- 25. Shu, J.; Kaliaguine, S.; Appl. Catal., B 1998, 16, 303.
- 26. Kaliaguine, S.; Van Neste, A.; Szabo, V.; Gallot, J. E.; Bassir, M.; Muzychuk, R.; Appl. Catal, A 2001, 209, 345.
- 27. Barnard, K. R.; Foger, K.; Turney, T. W.; Williams, R. D.; J. Catal 1990, 125, 265.
- 28. Lee, Y. Ng.; Lago, R. M.; Fierro, J. L. G. V.; Sapinã, F.; Martínez, E.; Appl. Catal, A 2001, 207, 17.
- 29. Her, R.; The Chemistry of silica, Wiley: New York, 1979.
- 30. Dombro, R. A.; Kirch, W.; Eur. Pat. Application 1984, 110, 78;
- Appl. Catal., A 2001, 207, 17
- 31. O Connel, M.; Norman, A.; Huttermann, C. F.; Morris, M. A.; Catal. Today 1999, 47, 123.
- 32. Roberts, M. W.; Chem. Soc. Rev 1989, 18, 451.
- 33. Tabata, K.; Matsumoto, I.; Kohiki, S.; J. Mater Sci 1987, 22, 1882.
- 34. Wu, Y.; Yu, T.; Dou, B. S.; Wang, C. X.; Xie, X. F.; Yu, Z. L.; Fan, S. R.; Wang, L. C.; J. Catal 1989, 120, 88.
- 35. Taguchi, H.; Sugita, A.; Nagao, M. J.; Takeda, Y.; J. Solid State Chem 1995, 116, 343.
Datas de Publicação
-
Publicação nesta coleção
09 Mar 2004 -
Data do Fascículo
Fev 2004
Histórico
-
Aceito
08 Set 2003 -
Recebido
29 Nov 2002