Abstract
Human sapoviruses (HSaV) are considered important causative agents of acute gastroenteritis in humans worldwide. However, knowledge of the genetic characteristics of the whole genome of HSaV in Brazil is limited. Here we report the complete genome sequences of six HSaVs GI.2 and two GI.3 strains obtained from children with acute gastroenteritis in the Northern region of Brazil. Next generation sequencing was used to obtain the full genome and molecular characterization of the genome was performed. Phylogenetic analysis of the genome was also performed. Only one complete HSaV GI.2 genome characterization in the country precedes that of the present study. This is the first complete genome sequence of genotype GI.3 in Brazil. The data obtained in this investigation can contribute to the augmentation of the database on the molecular diversity of HSaVs strains circulating in Brazil, and to the improvement of current typing protocols.
Key words:
Caliciviridae; sapovirus; gastroenteritis; genotypes; deep sequencing
Human sapoviruses (HSaVs) belong to the Sapovirus genus of the family Caliciviridae are considered important causative agents of acute gastroenteritis (AGE) in humans.11. Oka T, Wang Q, Katayama K, Saif LJ. Comprehensive review of human sapoviruses. Clin Microbiol Rev. 2015; 28: 32-53.,22. Grant LR, O'Brien KL, Weatherholtz RC, Reid R, Goklish N, Santosham M, et al. Norovirus and sapovirus epidemiology and strain characteristics among Navajo and Apache infants. PLoS One. 2017; 12(1): e0169491. Mortality associated with this pathogen is rare and the symptoms are generally mild.33. Zhou X, Sun Y, Shang X, Gao J, Zhao X, Shuai H, et al. Complete genome sequence of a sapovirus from a child in Zhejiang, China. Virus Genes. 2016; 52(5): 706-10. Based on the VP1 nucleotide sequence, human HSaVs are classified into genogroups GI, GII, GIV, and GV, and subdivided into 17 genotypes,11. Oka T, Wang Q, Katayama K, Saif LJ. Comprehensive review of human sapoviruses. Clin Microbiol Rev. 2015; 28: 32-53. with an additional proposed GII.8 genotype.44. Liu X, Jahuira H, Gilman RH, Alva A, Cabrera L, Okamoto M, et al. Etiological role and repeated infections of sapovirus among children aged less than 2 years in a cohort study in a Peri-urban community of Peru. J Clin Microbiol. 2016; 54(6): 1598-604. HSaV has a positive-sense, single stranded RNA genome of 7.1-7.7kb in length which contains two open reading frames (ORFs). ORF1 encodes a large polyprotein containing the nonstructural proteins followed by the major capsid protein VP1. ORF2 is predicted to encode the minor structural protein VP2.11. Oka T, Wang Q, Katayama K, Saif LJ. Comprehensive review of human sapoviruses. Clin Microbiol Rev. 2015; 28: 32-53.
Although HSaV has been accepted as one of the causes of acute gastroenteritis worldwide, little is known about the genetic characteristics of HSaV in Brazil based on whole genome analysis. In Brazil, the most common genotypes identified are GI.1, GI.2 and GII.1.55. Portal TM, Siqueira JA, Costa LC, Lima IC, Lucena MS, Bandeira RS, et al. Caliciviruses in hospitalized children, São Luís, Maranhão, 1997-1999: detection of norovirus GII.12. Braz J Microbiol. 2016; 47(3): 724-30.,66. Fioretti JM, Rocha MS, Fumian TM, Ginuino A, da Silva TP, de Assis MR, et al. Occurrence of human sapoviruses in wastewater and stool samples in Rio de Janeiro, Brazil. J Appl Microbiol. 2016; 121(3): 855-62.,77. Oliveira DMM, Souza M, Fiaccadori FS, Santos HCP, Cardoso DDP. Monitoring of Calicivirus among day-care children: evidence of asymptomatic viral excretion and first report of GI.7 Norovirus and GI.3 Sapovirus in Brazil. J Med Virol. 2014; 86(9): 1569-75.,88. Xavier MP, Oliveira SA, Ferreira MS, Victoria M, Miranda V, Silva MF, et al. Detection of caliciviruses associated with acute infantile gastroenteritis in Salvador, an urban center in Northeast Brazil. Braz J Med Biol Res. 2009; 42(5):438-44. Here we report the complete genome sequences of six HSaVs GI.2 and two GI.3 strains obtained from children with acute gastroenteritis in the Northern region of Brazil. Phylogenetic analysis was performed for comparison with other previously reported genogroups/genotypes.
The samples BRA/TO-07, BRA/TO-31, BRA/TO-48, BRA/TO-49, BRA/TO-65, BRA/TO-66, BRA/TO-89 and BRA/TO-90 were obtained from the Central Laboratory of Public Health of Tocantins state (LACEN/TO), located in the Northern region of Brazil (Table). All patients were experiencing acute gastroenteritis symptoms, such as diarrhea, vomiting and fever.
The protocol used to perform deep sequencing was a combination of several protocols normally applied to viral metagenomics and/or virus discovery,99. Li L, Deng X, Mee ET, Collot-Teixeira S, Anderson R, Schepelmann S, et al. Comparing viral metagenomics methods using a highly multiplexed human viral pathogens reagent. J Virol Methods. 2015; 213: 139-46. and has been partially described by da Costa et al.1010. da Costa AC, Thézé J, Komninakis SCV, Sanz-Duro RL, Felinto MRL, Moura LCC, et al. Spread of Chikungunya virus East/Central/South African genotype in northeast Brazil. Emerg Infect Dis. 2017; 23(10): 1742-4. In summary, 50 mg of each human fecal sample was diluted in 500 μL of Hanks’ buffered salt solution (HBSS), added to a 2 mL impact-resistant tube containing lysing matrix C (MP Biomedicals, USA), and homogenized in a FastPrep-24 5G Homogenizer (MP biomedicals, USA). The homogenized sample was centrifuged at 12,000×g for 10 min, and approximately 300 μL of the supernatant was then percolated through a 0.45 μm filter (Merck Millipore, Billerica, MA, USA) in order to remove eukaryotic and bacterial cell-sized particles. Approximately, 100 μL, roughly equivalent to one-fourth of the volume of the tube of cold PEG-it Virus Precipitation Solution (System Biosciences, CA, USA) was added to the obtained filtrate, and the contents of the tube were gently mixed then incubated at 4ºC for 24 h. After the incubation period, the mixture was centrifuged at 10,000×g for 30 min at 4ºC. Following centrifugation, the supernatant (~350 μL) was discarded. The pellet rich in viral particles was treated with a mixture of nuclease enzymes (14 uni TURBO Dnase and 7 uni RNase Cocktail Enzyme Mix-Thermo Fischer Scientific, CA, USA; 9 uni Baseline-ZERO DNase - Epicentre, WI, USA; 25 Benzonase - Darmstadt, Germany; and 9 RQ1 RNase- Free DNase and 0.09mg RNase A Solution - Promega, WI, USA) in order to digest unprotected nucleic acids. The resulting mixture was subsequently incubated at 37ºC for 2 h.
After incubation, viral nucleic acids were extracted using ZR & ZR-96 Viral DNA/RNA Kit (Zymo Research, CA, USA) according to the manufacturer’s protocol. The cDNA synthesis was performed with AMV Reverse transcription (Promega, WI, USA). A second strand of cDNA was synthesized using DNA Polymerase I Lar e (Klenow) Fragment (Promega, WI, USA). Subsequently, a Nextera XT Sample Preparation Kit (Illumina, CA, USA) was used to construct a DNA library, identified using dual barcodes. For size range, Pippin Prep (Sage Science, Inc.) was used to select a 300 bp insert (range 200-400 bp). The library was deep-sequenced using the HiSeq 2500 Sequencer (Illumina, CA, USA) with 126 bp ends. Bioinformatic analysis was performed according to the protocol previously described by Deng et al.1111. Deng X, Naccache SN, Ng T, Federman S, Li L, Chiu CY, et al. An ensemble strategy that significantly improves de novo assembly of microbial genomes from metagenomic next-generation sequencing data. Nucleic Acids Res. 2015; 43(7): e46. Contigs that shared percent nucleotide identities of 95% or less were assembled from the obtained sequence reads by de novo assembly. The contigs included the group A rotavirus sequences and others, such as enteric viruses (i. e., enterovirus, adenovirus, norovirus), and human, fungal, and bacterial sequences. The resulting singlets and contigs were analyzed using BLASTx to search for similarity to viral proteins in GenBank’s Virus RefSeq. The contigs were compared to the GenBank nonredundant nucleotide and protein database (BLASTn and BLASTx).
Total of 110,104; 38,780; 748,755; 395,031; 28,399; 27,971; 25,835; and 21,539 paired-end reads were obtained from the BRA/TO-07, BRA/TO-31, BRA/TO-48, BRA/TO-49, BRA/TO-65, BRA/TO-66, BRA/TO-89 and BRA/TO-90 samples, respectively. Of the total reads, 6.8% (n = 7,472) from BRA/TO-07, 19.1% (n = 7,402) from BRA/TO-31, 1.1% (n = 8,032) from BRA/TO-48, 1.9% (n = 7,667) from BRA/TO-49, 26% (n = 7,337), 26.6% (n = 7,443) from BRA/TO-66, 28.9% (n = 7,471) from BRA/TO-89 and 34.7% (n = 7,476) from BRA/TO-90 showed BLASTx score (coverage 1856x, 660x, 11745x, 6491x, 485x, 473x, 435x and 363x, respectively) to HSaV. The final genome analysis was performed using Geneious software v9.1.8 (Biomatters Ltd., Auckland, New Zealand). Open reading frames were predicted with the Geneious ORF finder. Based on the bioinformatics pipeline used,1111. Deng X, Naccache SN, Ng T, Federman S, Li L, Chiu CY, et al. An ensemble strategy that significantly improves de novo assembly of microbial genomes from metagenomic next-generation sequencing data. Nucleic Acids Res. 2015; 43(7): e46. no reads related to human, fungal, or bacterial sequences were obtained.
A public accessible typing tool (http://www.rivm.nl/mpf/norovirus/typingtool) was used to assign the genogroup of the study strains.1212. Kroneman A, Vennema H, Deforche K, Van Der Avoort H, Peñaranda S, Oberste MS, et al. An automated genotyping tool for enteroviruses and noroviruses. J Clin Virol. 2011; 51(2): 121-5. Sequences generated here and a set of cognate sequences of SaV available in GenBank were aligned using the BioEdit sequence alignment editor (version 7.0.5.2) program. Genetic analysis was performed with MEGA software version 6.0.1313. Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013; 30(12): 2725-9. The Kimura two-parameter substitution model and neighbour-joining method was selected to infer phylogenetic relationships among relevant strains. Nucleotide sequences determined in this study have been deposited in GenBank under the accession numbers MK250983-MK250990.
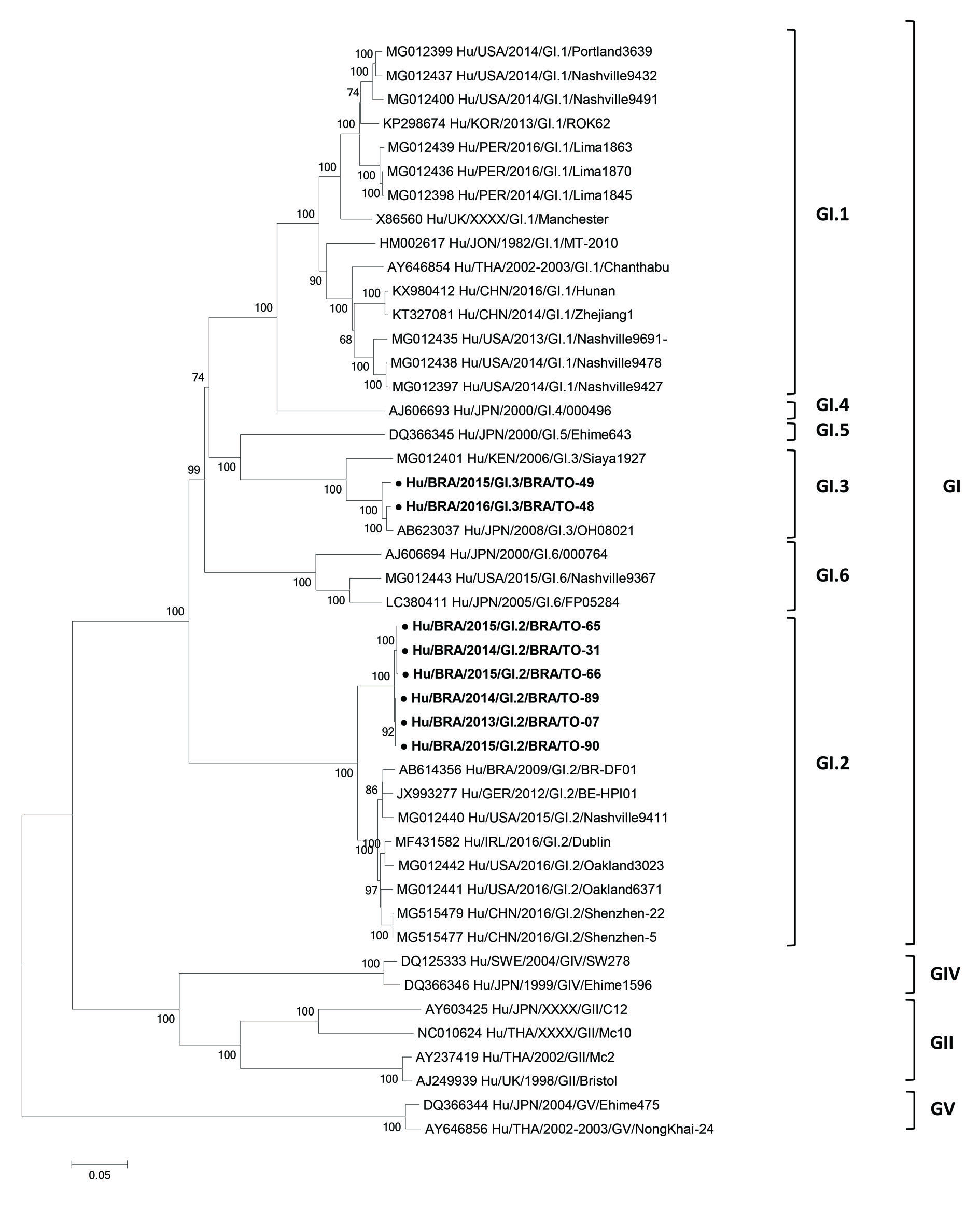
The eight HSaVs strains were classified in genogroup GI (ORF 1) based on the web tool analysis. Phylogenetic tree indicated that six HSaVs samples belong to genotype GI.2 (BRA/TO-07, BRA/TO-31, BRA/TO-65, BRA/TO-66, BRA/TO-89 and BRA/TO-90), and two HSaVs samples (BRA/TO-48 and BRA/TO-49) belong to genotype GI.3 (Table, Figure). Brazilian HSaV GI.2 sequences showed 98.4-99.9% similarity at nucleotide level (nt) (97.8-100% aa) between them, and 92.1-94.3% nt (80.0-84.3% aa) when compared to representative GI.2 strains detected in Brazil, United States, China and Ireland. The Brazilian HSaV GI.3 strains were close related to each other, since they shared 97.0% nt identity (94.6% aa). BRA/TO-48 and BRA/TO-49 GI.3 HSaVs strains exhibited high nucleotide and amino acid identity to the human strain OH08021, isolated in Japan in 2008 (90.5-93.3% nt; 86.5-91.3% aa). When compared with human GI.3 strain isolated in Kenya in 2006, strains BRA/TO-48 and BRA/TO-49 displayed lower genetic homology (87.4-88.2% nt; 73.8-75.2% aa).
Several studies demonstrate HSaV GI.2 as one of the main genotypes associated with outbreaks and sporadic cases of AGE,1414. Medici MC, Tummolo F, Albonetti V, Abelli LA, Chezzi C, Calderaro A. Molecular detection and epidemiology of astrovirus, bocavirus, and sapovirus in Italian children admitted to hospital with acute gastroenteritis, 2008-2009. J Med Virol. 2012; 84(4): 643-50.,1515. Bucardo F, Reyes Y, Svensson L, Nordgren J. Predominance of norovirus and sapovirus in Nicaragua after implementation of universal rotavirus vaccination. PLoS One. 2014; 9(5): e98201.,1616. Iritani N, Yamamoto SP, Abe N, Kubo H, Oka T, Kaida A. Epidemics of GI.2 sapovirus in gastroenteritis outbreaks during 2012-2013 in Osaka City, Japan. J Med Virol. 2016; 88(7): 1187-93. including in Brazil.66. Fioretti JM, Rocha MS, Fumian TM, Ginuino A, da Silva TP, de Assis MR, et al. Occurrence of human sapoviruses in wastewater and stool samples in Rio de Janeiro, Brazil. J Appl Microbiol. 2016; 121(3): 855-62.,1717. Reymão TK, Hernandez JD, Costa ST, Sousa MS, Oliveira DS, Silva LD, et al. Sapoviruses in children with acute gastroenteritis from Manaus, Amazon region, Brazil, 2010-2011. Rev Inst Med Trop Sao Paulo. 2016; 58: 81. The data obtained here confirmed the epidemiological role of HSaV GI.2 genotype in AGE etiology. HSaV GI.3 genotype appears to be detected more sporadically.1818. Mancini P, Ferraro GB, Iaconelli M, Suffredini E, Valdazo-González B, Della Libera S, et al. Molecular characterization of human Sapovirus in untreated sewage in Italy by amplicon-based Sanger and next-generation sequencing. J Appl Microbiol. 2019; 126(1): 324-331.,1919. Zhang HY, Qiao MK, Wang X, He M, Shi LM, Xie GX, et al. Molecular diversity of sapovirus infection in outpatients living in Nanjing, China (2011-2013). Gastroenterol Res Pract. 2016; 2016: 4210462. In Brazil, a recent study conducted in a day-care center in the Midwest region reported the detection of HSaV GI.3 genotype in asymptomatic children.77. Oliveira DMM, Souza M, Fiaccadori FS, Santos HCP, Cardoso DDP. Monitoring of Calicivirus among day-care children: evidence of asymptomatic viral excretion and first report of GI.7 Norovirus and GI.3 Sapovirus in Brazil. J Med Virol. 2014; 86(9): 1569-75. There is relatively limited sequence information about Brazilian HSaVs strains at the complete genome level. Before the present study, only one complete HSaV GI.2 genome had been characterized in the country. To the best of our knowledge these are the first complete genome sequences of genotype GI.3 in Brazil.
The data acquired in this investigation can contribute to the growing database on the molecular diversity of HSaV circulating in Brazil and also to future epidemiological studies of HSaV by providing data necessary for the development of more sensitive and specific diagnostic tools that could be used to define the worldwide distribution of the virus.

Neighbor-joining phylogenetic tree of nucleotide sequence generate with MEGA 6.0 software of the human sapoviruses (HSaV) strains detected from children with acute gastroenteritis in Brazil (highlighted in bold and ●). References of HSaVs were obtained from GenBank database. Genogroups, genotypes, accession number, isolates, countries and year of each strain are indicated. The scale indicates the number of divergent nucleotide residues. Percentage of bootstrap values is shown at the branch node.
Socio-demographic data of positive sapoviruses (SaV) samples from patients with acute gastroenteritis in Brazil
Ethics - Previous Ethics Committee approval was granted by Faculdade de Medicina da Universidade de São Paulo (CAAE: 53153916.7.0000.0065), and Centro Universitário Luterano de Palmas ― ULBRA (CAAE: 53153916.7.3007.5516). This was an anonymous unlinked study, and informed consent was not required according to resolution 466/12 concerning research involving humans (Conselho Nacional de Saúde/Ministério da Saúde, Brasília, 2012).
ACKNOWLEDGEMENTS
To Zymo Research Inc., Illumina Inc., Sage Science Inc., Promega Biotecnologia do Brasil Ltda, for the donation of reagents for this project.
REFERENCES
-
1Oka T, Wang Q, Katayama K, Saif LJ. Comprehensive review of human sapoviruses. Clin Microbiol Rev. 2015; 28: 32-53.
-
2Grant LR, O'Brien KL, Weatherholtz RC, Reid R, Goklish N, Santosham M, et al. Norovirus and sapovirus epidemiology and strain characteristics among Navajo and Apache infants. PLoS One. 2017; 12(1): e0169491.
-
3Zhou X, Sun Y, Shang X, Gao J, Zhao X, Shuai H, et al. Complete genome sequence of a sapovirus from a child in Zhejiang, China. Virus Genes. 2016; 52(5): 706-10.
-
4Liu X, Jahuira H, Gilman RH, Alva A, Cabrera L, Okamoto M, et al. Etiological role and repeated infections of sapovirus among children aged less than 2 years in a cohort study in a Peri-urban community of Peru. J Clin Microbiol. 2016; 54(6): 1598-604.
-
5Portal TM, Siqueira JA, Costa LC, Lima IC, Lucena MS, Bandeira RS, et al. Caliciviruses in hospitalized children, São Luís, Maranhão, 1997-1999: detection of norovirus GII.12. Braz J Microbiol. 2016; 47(3): 724-30.
-
6Fioretti JM, Rocha MS, Fumian TM, Ginuino A, da Silva TP, de Assis MR, et al. Occurrence of human sapoviruses in wastewater and stool samples in Rio de Janeiro, Brazil. J Appl Microbiol. 2016; 121(3): 855-62.
-
7Oliveira DMM, Souza M, Fiaccadori FS, Santos HCP, Cardoso DDP. Monitoring of Calicivirus among day-care children: evidence of asymptomatic viral excretion and first report of GI.7 Norovirus and GI.3 Sapovirus in Brazil. J Med Virol. 2014; 86(9): 1569-75.
-
8Xavier MP, Oliveira SA, Ferreira MS, Victoria M, Miranda V, Silva MF, et al. Detection of caliciviruses associated with acute infantile gastroenteritis in Salvador, an urban center in Northeast Brazil. Braz J Med Biol Res. 2009; 42(5):438-44.
-
9Li L, Deng X, Mee ET, Collot-Teixeira S, Anderson R, Schepelmann S, et al. Comparing viral metagenomics methods using a highly multiplexed human viral pathogens reagent. J Virol Methods. 2015; 213: 139-46.
-
10da Costa AC, Thézé J, Komninakis SCV, Sanz-Duro RL, Felinto MRL, Moura LCC, et al. Spread of Chikungunya virus East/Central/South African genotype in northeast Brazil. Emerg Infect Dis. 2017; 23(10): 1742-4.
-
11Deng X, Naccache SN, Ng T, Federman S, Li L, Chiu CY, et al. An ensemble strategy that significantly improves de novo assembly of microbial genomes from metagenomic next-generation sequencing data. Nucleic Acids Res. 2015; 43(7): e46.
-
12Kroneman A, Vennema H, Deforche K, Van Der Avoort H, Peñaranda S, Oberste MS, et al. An automated genotyping tool for enteroviruses and noroviruses. J Clin Virol. 2011; 51(2): 121-5.
-
13Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013; 30(12): 2725-9.
-
14Medici MC, Tummolo F, Albonetti V, Abelli LA, Chezzi C, Calderaro A. Molecular detection and epidemiology of astrovirus, bocavirus, and sapovirus in Italian children admitted to hospital with acute gastroenteritis, 2008-2009. J Med Virol. 2012; 84(4): 643-50.
-
15Bucardo F, Reyes Y, Svensson L, Nordgren J. Predominance of norovirus and sapovirus in Nicaragua after implementation of universal rotavirus vaccination. PLoS One. 2014; 9(5): e98201.
-
16Iritani N, Yamamoto SP, Abe N, Kubo H, Oka T, Kaida A. Epidemics of GI.2 sapovirus in gastroenteritis outbreaks during 2012-2013 in Osaka City, Japan. J Med Virol. 2016; 88(7): 1187-93.
-
17Reymão TK, Hernandez JD, Costa ST, Sousa MS, Oliveira DS, Silva LD, et al. Sapoviruses in children with acute gastroenteritis from Manaus, Amazon region, Brazil, 2010-2011. Rev Inst Med Trop Sao Paulo. 2016; 58: 81.
-
18Mancini P, Ferraro GB, Iaconelli M, Suffredini E, Valdazo-González B, Della Libera S, et al. Molecular characterization of human Sapovirus in untreated sewage in Italy by amplicon-based Sanger and next-generation sequencing. J Appl Microbiol. 2019; 126(1): 324-331.
-
19Zhang HY, Qiao MK, Wang X, He M, Shi LM, Xie GX, et al. Molecular diversity of sapovirus infection in outpatients living in Nanjing, China (2011-2013). Gastroenterol Res Pract. 2016; 2016: 4210462.
-
Financial support: FAPESP (#2016/01735-2), CNPq (#400354/2016-0). ACC and AL are funded by FAPESP (#2017/00021-9 and #2015/12944-9, respectively).
Publication Dates
-
Publication in this collection
04 Apr 2019 -
Date of issue
2019
History
-
Received
06 Dec 2018 -
Accepted
18 Mar 2019