

Abstract
During neurogenesis in the embryonic cerebral cortex, the classical neurotransmitters GABA and l-glutamate stimulate ionic conductance changes in ventricular zone (VZ) neuroblasts. Lysophosphatidic acid (LPA) is a bioactive phospholipid producing myriad effects on cells including alterations in membrane conductances (for review, see Moolenaar et al., 1995). Developmental expression patterns of its first cloned receptor gene,lpA1/vzg-1 (Hecht et al., 1996; Fukushima et al., 1998) in the VZ suggested that functional LPA receptors were synthesized at these early times, and thus, LPA could be an earlier stimulus to VZ cells than the neurotransmitters GABA and l-glutamate. To address this possibility, primary cultures of electrically coupled, presumptive cortical neuroblast clusters were identified by age, morphology, electrophysiological profile, BrdU incorporation, and nestin immunostaining. Single cells from cortical neuroblast cell lines were also examined. Whole-cell variation of the patch-clamp technique was used to record from nestin-immunoreactive cells after stimulation by local administration of ligands. After initial plating at embryonic day 11 (E11), cells responded only to LPA but not to GABA or l-glutamate. Continued growth in culture for up to 12 hr produced more LPA-responsive cells, but also a growing population of GABA- orl-glutamate-responsive cells. Cultures from E12 embryos showed LPA as well as GABA and l-glutamate responses, with LPA-responsive cells still representing a majority. Overall, >50% of cells responded to LPA with depolarization mediated by either chloride or nonselective cation conductances. These data implicate LPA as the earliest reported extracellular stimulus of ionic conductance changes for cortical neuroblasts and provide evidence for LPA as a novel, physiological component in CNS development.
- lysophosphatidic acid
- LPA
- chloride currents
- cation currents
- cerebral cortex
- embryonic development
- lysophospholipids
- programmed cell death
The mammalian cerebral cortex consists of six neuronal layers that are produced over a discrete period of embryonic development (Angevine and Sidman, 1961). Neurons destined for a cortical layer are formed in a proliferative, neurogenic region overlying the lateral ventricles, thus called the “ventricular zone” (VZ; Boulder Committee, 1970). Previous studies examining the role of extracellular signaling molecules on cortical neurogenesis implicated two general families of receptor-mediated signals. Peptide growth factors acting through receptor tyrosine kinases have been shown in primary cell culture to influence the blast-precursor potential of cortical neuroblasts through responses that include both cell proliferation (through basic fibroblast growth factor) and differentiation (neurotrophin 3; NT3) (Temple and Qian, 1995). By comparison, physiological studies have revealed putative neuroblast ionotropic responses to GABA or l-glutamate, mediated by their cognate ligand-gated ion channels; these ionic changes have been suggested to influence the proliferative capacity of a stimulated blast by decreasing DNA synthesis (LoTurco et al., 1995; Owens et al., 1996). In addition to these interactions, however, it is likely that other forms of extracellular signals also exist, based in part on the normal cortical phenotype of null mutations for neurotrophins and/or their tyrosine kinase receptors, e.g., for NT3/trk receptors (Klein et al., 1993; Ernfors et al., 1994; Fariñas et al., 1994;Klein et al., 1994; Smeyne et al., 1994), or the requirement for serum and/or membrane fractions for the growth of cortical neuroblasts in culture (Kilpatrick and Bartlett, 1993; Temple and Davis, 1994; Temple and Qian, 1995).
To identify new extracellular signaling receptors expressed in the VZ, an approach was taken that combined degenerate PCR for G-protein coupled receptor (GPCR) genes with in situ hybridization; this approach allowed the identification of ventricular zone gene-1 (vzg-1), which has an expression pattern restricted to the cortical VZ during neurogenesis (Hecht et al., 1996). Functional analysis of the receptor led to the identification of a ligand;vzg-1 encodes a receptor for the bioactive lipid lysophosphatidic acid (LPA) and was thus redesignated lysophospholipid receptor A1 (lpA1), the first of a growing family of related lysophospholipid receptors (Hecht et al., 1996; Fukushima et al., 1998; Chun et al., 1999; Zhang et al., 1999). Documented cellular responses to LPA in cell lines from different lineages include cell proliferation; actin-based morphological changes involved in the formation of stress fibers in fibroblasts, neurite retraction, and rounding of cells in neuroblastoma cell lines (Moolenaar, 1995); gap–junction closure (e.g., in WB cells;Hill et al., 1994); and increases in chloride (Cl−) conductances (e.g., in Rat-1 fibroblasts; Postma et al., 1996). Operation of this signaling system would represent a stimulus distinct from those previously described in the developing cerebral cortex by being (1) mediated by an extracellular lipid molecule and (2) transduced by a GPCR. The developmental expression pattern oflpA1/vzg-1 predicted that extracellular LPA signaling could affect embryonic cortical neuroblasts. Here we report that LPA is indeed a major extracellullar stimulus for cortical neuroblasts, activating two distinct ionic conductances that cause depolarization and developmentally precede GABA and l-glutamate signaling.
MATERIALS AND METHODS
Primary cultures. All animal protocols were approved by the University of California, San Diego Animal Subjects Committee and are in accordance with National Institutes of Health guidelines and public law. Timed-pregnant Balb/c mice [day of plug, embryonic day 0 (E0)] were euthanized by cervical dislocation, and embryonic cortical regions were dissected as described previously (Chun and Jaenisch, 1996) in cold serum-free OptiMEM-I (Life Technologies, Gaithersburg, MD) supplemented with 20 mmd-glucose, 1% penicillin–streptomycin, either with or without 55 μm β-mercaptoethanol. Cortices were triturated using a sterile long-nose Pasteur pipette into small clusters (<50 cells) and plated at a density of ∼10,000–50,000 cells/12 mm coverslip previously coated with Cell-Tak (Collaborative Research, Bedford, MA). Cultures were grown at 37°C and 5% CO2. Cells in clusters appeared healthy and remained proliferative (87% incorporated BrdU at over the first 24 hr within clusters), consistent with previous reports of rodent cortical cultures (Ghosh and Greenberg, 1995). The basic morphology of cells in clusters did not change over the duration of the experiment, whereas single cells more often appeared to differentiate into neurons with large sodium currents, although some single cells appeared to remain undifferentiated. The most responsive cells appeared to be those maintained in low-density cultures that resided within the clusters, and electrophysiology was performed on cells within small clusters.
Electrophysiology. The whole-cell patch-clamp technique (Hamill et al., 1981) was used to record ligand-induced currents from embryonic VZ cells maintained for 1–20 hr in serum-free media. Cells were visualized using a Nikon Diaphot 300 with DIC Nomarski optics. Cells were continuously perfused in a physiological saline (∼0.5 ml/min) unless otherwise indicated. The standard physiological saline (Tyrode’s solution) contained (in mm): 130 NaCl, 4 KCl, 5 CaCl2, 1.2 MgCl2, and 10 hemi-Na-HEPES (pH 7.37, 295–300 mOsm as measured using a Wescor 5500 vapor-pressure osmometer; Wescor, Logan, UT). In some experiments, the Ca2+ concentration of the physiological saline was decreased to 1 mm, and similar results were obtained. Recording electrodes were fabricated from borosilicate capillary tubing (BF100; Sutter Instrument Co., Novato, CA), the tips were coated with dental periphery wax (Miles Laboratories, South Bend, IN), and had resistances of 8–15 MΩ when containing intracellular saline [in mm: 100 K-gluconate, 25 KCl, 0.483 CaCl2, 3 MgCl2, 10 hemi-Na-HEPES, and 1 K4-BAPTA (100 nm free Ca2+), pH 7.4, with dextrose added to achieve 290 mOsm]. Series resistances (Rs) were 20–40 MΩ. To mark cells that had been physiologically recorded, lysine-fixable rhodamine dextran (10 kDa; Molecular Probes, Eugene, OR) was included at 500 μm in the pipette solution. To determine the TTX sensitivity of voltage-activated inward currents, a Cs+-based intracellular saline was used to block outward K+ currents (in mm): 25 CsCl, 102.5 methansulfonate, 0.483 CaCl2, 3 MgCl2, 10 hemi-Na HEPES, and 1 BAPTA free acid, pH 7.4 with CsOH. In Cl− ion substitution experiments, the cells were continuously perfused with low Cl−extracellular saline composed of (in mm): 140 Na-isethionate, 3 K-gluconate, 5 Ca2+-acetate, 1 MgCl2, and 10 hemi-Na-HEPES (305 mOsm, pH 7.37, with acetic acid). In experiments designed to shift the reversal potential (Vrev) of nonselective cation (NSC) currents, an intracellular saline containing impermeant cations (ZIS) was used (Delay et al., 1997) [in mm: 100 NMDG, 100 methanesulfonate, 5 NaCl, 1 BAPTA free acid, 0.483 CaCl2 (100 nm free Ca2+), 2 MgCl2, 10 hemi-Na-HEPES; pH 7.4, with dextrose added to achieve 290 mOsm]. Liquid junction potentials were −18 mV using standard pipette and bath solutions as determined both empirically and using the computer program JPCalc (Barry, 1994); in the low Cl− extracellular saline, voltages were corrected by +3 mV, and in experiments using ZIS and Tyrode’s, voltages were corrected by −15 mV. All voltages shown are corrected for liquid junction potential. Current and voltage signals were detected and filtered at 5 kHz with an Axopatch 200A patch-clamp amplifier (Axon Instruments, Foster City, CA), digitally recorded with a TL-1 DMA laboratory interface (Axon Instruments) and personal computer-compatible system, and stored on magnetic disk for off-line analysis. Data acquisition and analysis were performed with pClamp software. Slow changes in holding current were detected and filtered at 5 kHz and recorded with a LPF200A DC amplifier (Warner, Hamden, CT) and VR-10B digital data recorder (Instrutech, Great Neck, NY) onto video tape. The signal was later analyzed at 10 Hz using Axotape software.
The total membrane capacitance (Cm) was determined as the difference between the maximum current after a 30 mV hyperpolarizing voltage ramp from −68 mV generated at a rate of 10 mV/msec and the steady-state current at the final potential (−98 mV) (values refer to Vm when using standard saline solutions). The principles underlying this method derive from the equation describing the current IC flowing onto (and off of) the membrane, IC =Cm [dVm/dt] and are discussed in Armstrong and Gilly (1992). ICwas measured as the difference between the current at the end of the voltage ramp and the steady-state current achieved after completion of the ramp. The voltage at the end of the ramp protocol was maintained for 47 msec to ensure that the decay of the maximum current had reached steady-state. Because dV/dt is 10 V/sec in the experiments here,Cm = 0.1[IC]. TheCm calculated using this method was similar to the Cm determined using the classical method of calculating the time constant of the current response elicited by a 10 mV voltage step (data not shown).
Cell capacitance measurements provided an assessment of cell coupling (de Roos et al., 1996). The mean single cell Cmdepended on the cell morphology. Single neuroblast-like cells hadCm values of ∼2 pF similar to the lowest values obtained for cells in clusters (23 of 107 hadCm values of <3 pF). To assess the effects of cell coupling, 1-Octanol (OCT, 1 mm) was used; however, it was found to decrease the Cm from 10.3 to 4.1 pF in only one of 10 examined clusters. The Cm of these OCT-insensitive cells was 1.7, 3.3, 4.5, 4.9, 4.9, 5.7, 7.8, 8.7, and 14.4 pF. Based on the Cm values for a single cell, only one cell was not electrically coupled (1.7 pF), whereas all of the other observed cells in clusters were coupled; this offered the best biological approximation of the normal state of neuroblasts in the VZ, based on previous analyses demonstrating that VZ cells are coupled in groups of 15–90 cells (LoTurco and Kriegstein, 1991; Bittman et al., 1997).
Current densities were calculated as the peakIin or maximal Iout at a voltage step to +32 mV normalized to the cell capacitance. Voltage-activated currents were obtained by voltage steps between −88 and +112 mV from a holding potential of −98 mV. Membrane resistance (Rm) values were determined by voltage-ramp (the inverse of the slope of the ramp-induced current at negative potentials) or current-clamp protocols using small current injections and were similar (n = 9). EstimatedRs errors were no more than 1.5 mV, on average <1 mV.
Apparent reversal potentials (Vrev) of ligand-induced conductance changes were determined using a voltage-ramp protocol (Dubin and Dionne, 1994). Voltage ramps were applied every 6 sec, and the resulting whole-cell ramp-induced currents were recorded. Usually the voltage was ramped from positive to negative values to avoid the nonlinearity introduced by the voltage-activatedIin when present at high density; however, in cells with large outward currents, the protocol was also reversed to enable the detection of small LPA-induced currents. The current required to clamp the cells at −68 mV was continuously monitored. Ligand-induced conductances were determined from whole-cell currents elicited by a voltage-ramp protocol in the presence and absence of ligand. Control ramp currents were subtracted from those obtained in the presence of ligand to produce difference currents, revealing the component of the whole-cell current–voltage relationship contributed by ligand-induced currents. The voltage at which there was no net ligand-induced current was determined (Vrev). A criterion used to avoid spurious Vrev determinations was that at least three individual ramps during the response revealed a similarVrev. This was possible because the responses were quite long-lasting. The figures show the difference ramp comparing the initial ramp after onset of the response and the average of control ramps. The concentrations of the major ions in the intracellular and extracellular salines were set such that each ion had a unique equilibrium potential. The equilibrium potentials for Na+, K+, and Cl− under the normal recording conditions were +85, −85, and −40 mV, respectively. The recorded current trace was filtered off-line (at 1 Hz), and in some traces only part of the positive voltage-ramp deflection is shown (see Figs.4a,c,e, 5a,6a–c).
The LPA-induced conductance was calculated from voltage-ramp data. The conductance was calculated from the linear portion of ramp-induced current–voltage curve at negative potentials (I = G * V, where G is the slope). The control conductance (usually the average of two or three traces before the LPA-induced response) was subtracted from the largest conductance measured in the presence of LPA. The LPA-induced conductance determined by this method was underestimated because the peak response often was not captured during the ramp protocol.
Most values are presented as the arithmetic mean ± SEM. BecauseINa values had a skewed distribution, geometric means were determined, and the reported values are the arithmetic means with the upper and lower error limits in parentheses (Hancock et al., 1988). For the latter calculation, a value of 0 pA/pF was given to cells with no detectable inward INa and averaged with the natural log of the other values.
Ligand reagents. LPA was thoroughly dissolved in Milli-Q water and used fresh or stored frozen as a 5 mm stock solution and used within 5 d. It was diluted to the indicated concentrations in extracellular saline and applied at concentrations ranging from 10 nm to 1 μm from nearby puffer pipettes. As a control for possible nonspecific lipid effects, a structurally related compound, lysophosphatidyl glycerol (LPG), was applied at concentrations between 10 nm and 10 μm. Stocks of GABA (100 mm) andl-glutamate (300 mm) were stored frozen and thawed only once and diluted in extracellular saline just before use. Ligand was applied (3–10 psi) together with 0.05% fast green dye to mark the plume using a Picospritzer (General Valve, Fairfield, NJ) for durations indicated in the figures (typically 15–25 sec).
BrdU and nestin immunofluorescence. To determine the proliferative capacity of primary cells, cultures were pulsed at the time of plating with bromodeoxyuridine (BrdU), and BrdU immunofluorescence was detected 24 hr later (cell proliferation kit; Boehringer Mannheim, Indianapolis, IN). For nestin immunofluorescence, coverslips containing cells that had been filled with lysine-fixable rhodamine-dextran during recording (see above) were fixed for 15 min with 4% paraformaldehyde in PBS. After PBS rinses, coverslips were incubated with blocking solution (2.5% BSA, 0.3% Triton X-100 in PBS) for 1 hr, followed by monoclonal anti-nestin antibody (PharMingen, La Jolla, CA) at 1:500 in blocking solution overnight. Coverslips were rinsed with PBS and incubated with FITC-conjugated anti-mouse IgG secondary antibody (Pierce, Rockford, IL) at 1:100 for 1 hr. After PBS rinses, all coverslips were counterstained with the fluorescent nuclear stain 4′6-diamidino-2-phenylindole (DAPI; Sigma, St. Louis, MO), air-dried, and mounted on glass microscope slides in GelMount (Biomeda, Foster City, CA). Coverslips were examined and photographed using a Zeiss Axioskop fluorescence microscope with rhodamine, fluorescein, or DAPI filters.
RESULTS
Cells comprising small clusters of 10–50 cells were examined for features consistent with a neuroblast identity in primary cultures derived from E11 and E12 dissociated murine cortices. Single cells that were part of clusters were studied because they offered the closest biological approximation of the actual electrical state of neuroblasts in the VZ, based on extensive previous studies demonstrating that VZ cells are normally coupled in groups of 15–90 cells (LoTurco and Kriegstein, 1991; Bittman et al., 1997). In addition, the resting membrane potentials of the examined cells in clusters were very similar to those reported previously, as were responses to GABA andl-glutamate (see below), demonstrating that this culture system supported physiological parameters of previous reports. Moreover, single cells from a cortical neuroblast cell line that were not electrically coupled showed similar results to cells in clusters. Changes in membrane conductance elicited after rapid, local exposure of these primary cells to LPA or both GABA and l-glutamate were assayed between 1 and 20 hr in vitro under serum-free conditions using the whole-cell variation of the patch-clamp technique. In addition to being tested for electrophysiological properties, recorded cells were labeled using fluorescent dextran, fixed, then double-labeled for nestin immunofluorescence. In some cases, BrdU pulses were also used to determine the percentage of cells undergoing DNA synthesis. Two distinct LPA-induced conductance changes were observed in putative neuroblasts: a Cl− conductance increase and a NSC conductance increase. LPA responsiveness always preceded that of GABA or l-glutamate during development.
LPA-responsive cells can incorporate BrdU and are nestin-immunoreactive
To determine the percentage of cells still undergoing DNA synthesis and expressing the neural precursor protein nestin (Cataneo and McKay, 1990), primary cultures at E12 were exposed to BrdU and processed for immunofluorescence. When these cultures were treated with BrdU for 24 hr to label cells undergoing DNA synthesis, 87% of cells showed BrdU immunoreactivity (data not shown). This value is consistent with the proliferative capacity of VZ cells that have been reported to be electrically coupled (LoTurco and Kriegstein, 1991; Bittman et al., 1997), as were the cells assayed here. More than 80% of the cells were also nestin-immunoreactive. To ascertain the relationship between electrophysiological properties and nestin immunoreactivity, these cells, which could be easily identified by their stereotypic clustering, were first assayed electrophysiologically, filled with lysine-fixable rhodamine dextran (Fig.1a), then immunostained for nestin (Fig. 1b). This particular cell responded to LPA (25 nm) with an increase in Cl− conductance (GCl) (Fig.1c,d).

LPA elicits conductance increases in cells that are nestin-positive. a, Recorded cell labeled with lysine-fixable rhodamine dextran (10,000 Da; arrow).b, All cells in the cluster containing the tested cell were nestin-positive, including the dextran-labeled cell itself (arrow; only some of the clustered cells are in the plane of focus). c, LPA (25 nm) elicited an inward current (the holding potential was −68 mV). The large deflections occurring throughout the current record are voltage-ramp-induced currents evoked every 6 sec to measure whole-cell conductance and the reversal potential (Vrev). The solid rectangle indicates the time and duration of LPA application.d, Difference voltage-ramp-induced currents revealed that the current activated by LPA had a reversal potential (Vrev = −39 mV) near theECl (−40 mV), consistent with an LPA-induced increase in Cl− conductance.
Membrane electrical properties
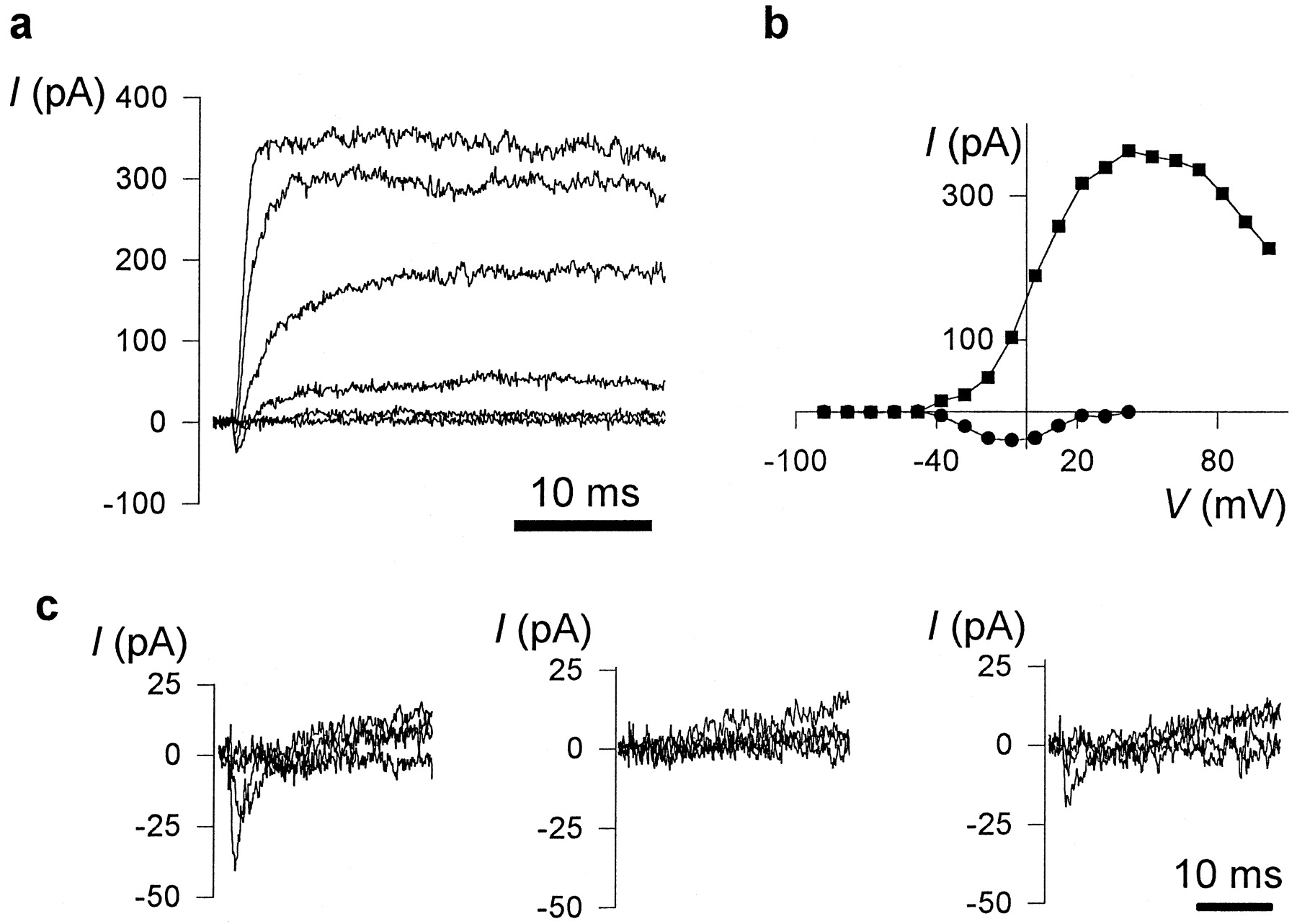
The majority of nestin-immunoreactive cell clusters produced from dissociated cortices at E11 or E12 and tested acutely, or E12 cells cultured 12–20 hr under serum-free conditions (noted as E12–E13), exhibited membrane properties that were consistent with previously reported values for rodent embryonic cortical neuroblasts (LoTurco et al., 1991) (Table 1). Many cells expressed depolarization-activated fast transient inward currents (Fig.2a,b) that remained stable over the recording period (at least 10 min). The apparent threshold voltage for Iin activation was between −40 and −50 mV, and the peak current was elicited near −10 mV (Fig.2b). Half the channels were inactivated when the membrane was held at a 500 msec prepulse potential of −62 ± 1 mV (n = 8). These inward currents were completely blocked by 2 μm tetrodotoxin under conditions in which voltage-activated K+ currents were blocked by intracellular Cs+ (to avoid outward current contamination of Iin), indicatingIin was mediated by Na+ and not Ca2+ (Fig. 2c). PeakINa current densities were often <10 pA/pF (Fig. 3a–c; 62% at E11; 42% at E12; 40% at E12–13). Except for rare cells with very large INa (>75 pA/pF) that could be maturing subplate neurons present at this embryonic stage (Chun et al., 1987), overshooting action potentials could not be elicited.
Resting membrane properties of presumptive neuroblasts from E11 (acute) and E12 cortices [acute and after 12–20 hr in vitro without serum (E12–13)]

Recorded cells from primary cultures of dissociated cortex have electrical properties of neuroblasts. Small cell clusters (∼10–50 cells) from E12 telencephala were examined between 2 and 20 hr after plating and examined as small aggregates.a, Fast transient inward currents (negative values in the figure) could be observed in LPA-responsive cells. Whole-cell currents were elicited by voltage steps (40 msec duration) between −58 and +42 mV in increments of 20 mV from a holding potential of −98 mV. The peak inward current was −15.6 pA/pF. b, The current–voltage relationship of the voltage-activated currents elicited for the neuroblast shown in Figure 2a. Peak inward (Iin; circles) and steady-state outward (Iout;squares) currents (in picoamperes) are plotted against the step voltage. c, The fast transient inward currents are TTX-sensitive. Whole-cell currents were elicited by voltage steps to −68, −28, 2, and 32 mV from a holding potential of −98 mV. The recording electrode contained Cs+ IS; bath contained Tyrode’s. Left, Fast transientIin (−12.3 pF/pA) before bath puffer application of TTX (2 μm); center, during exposure to TTX; right, partial recovery 3 min after washout of TTX.
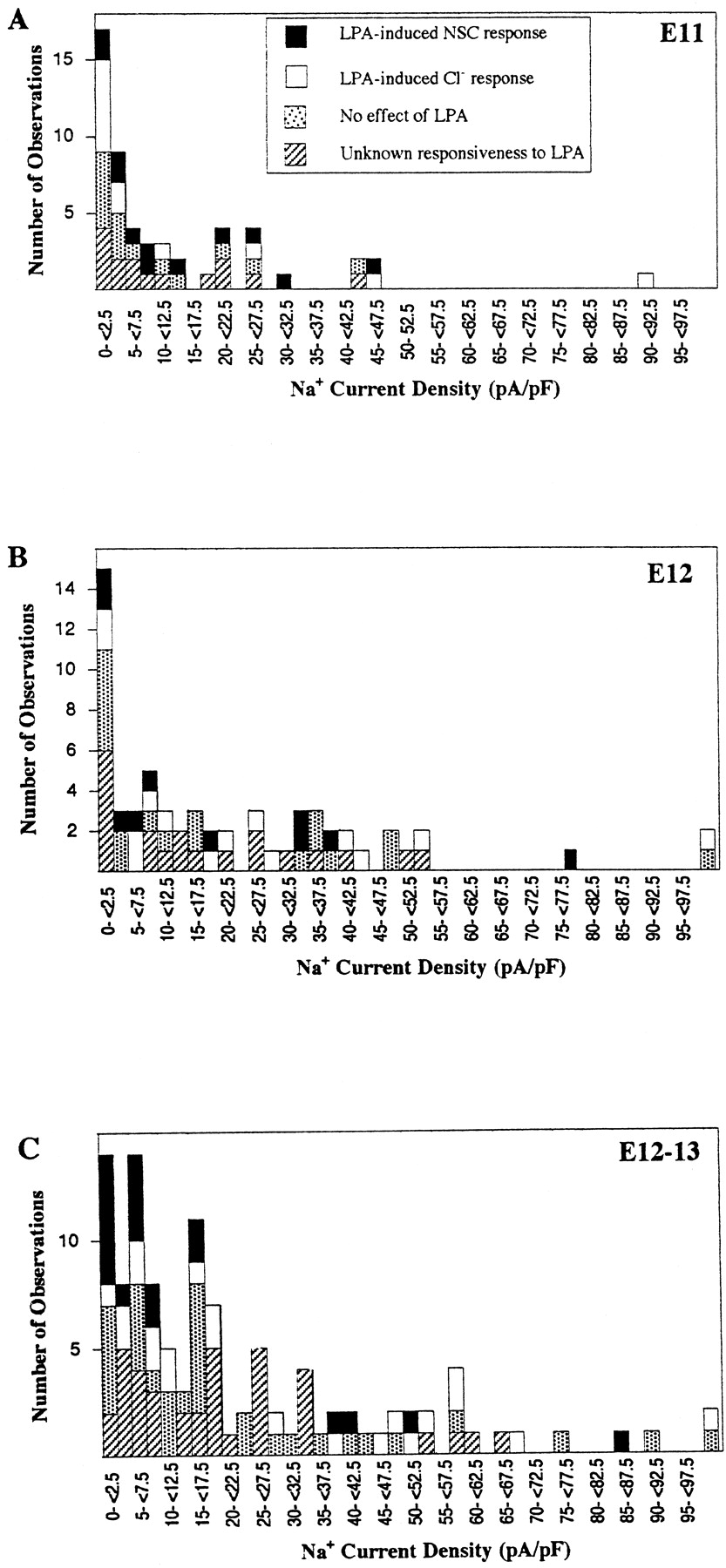
Histograms of peak Na+ current density (in picoamperes per picofarad) of cells tested. Solid bars, Cells that responded with an NSC conductance; open bars, cells that responded with an increased Cl− conductance; stippled bars, cells that revealed no detectable effect of LPA (≥10 nm);hatched bars, cells were either not tested for LPA sensitivity or possible responses were obscured by spontaneous activity (these were not included in the response data). A significant proportion of cells exhibited no voltage-activated fast transient inward currents from holding potentials of −98 mV. In E11, E12, and E12–E13 cultures, 22% (n = 53), 22% (n = 62), and 11% (n = 110) of cells, respectively, revealed no detectable inward currents, despite other cell properties indicating that they were healthy (e.g., RP,Rm). A, E11 acute;B, E12 acute; C, E12–E13.
Outward currents (Iout) were also elicited by voltage depolarizations (Fig. 2b). The maximumIout elicited by a step to +32 mV was +46.2 ± 4.9 pA/pF (n = 50), +57.0 ± 8.3 pA/pF (n = 30), and +61.6 ± 5.9 pA/pF (n = 77) at E11, E12, and E12–E13, respectively. On E11 and E12, most cells revealed a sustainedIout over a 40 msec voltage step [84% (n = 44) and 74% (n = 31) of the cells, respectively]. However, at E12–E13 most cells (57%;n = 37) revealed an inactivating component. Many cells revealed an N-shaped current–voltage relationship ofIout at very positive voltages, usually indicative of the presence of Ca2+-activated currents. Although only a quarter of the E11 cells (26%;n = 43) revealed this characteristic, 50% (n = 28) and 61% (n = 31) of E12 and E12–E13 cells did so. There were no significant differences in cell electrical properties (Cm,Rm, INa,Iout) between the populations of cells responsive to LPA (see below) and those with no detectable LPA sensitivity (data not shown).
LPA activates two distinct electrical responses in different populations of cells
LPA increased the membrane conductance without altering voltage-activated currents in 58% (n = 131) of cells from all ages tested. Two cell populations with distinct responses were identified, based on the reversal potential (Vrev) using a voltage-ramp protocol and ion substitution. In physiological solutions, 30% of cells responded to LPA with an increase in conductance that reversed near the Cl− equilibrium potential (ECl; −40 mV) (Table2, Figs. 1c,d,4a,b). The remaining group of sensitive cells (28%) responded to LPA with a conductance increase having a Vrev near 0 mV, consistent with its mediation by NSCs (Table 2, Fig.5a,b). Both types of responses displayed homologous desensitization to LPA at tested concentrations within 10 min (Fig. 5a). LPG (or other non-LPA ligands; data not shown) exposure never induced membrane conductances like those observed with LPA (Fig. 4a).
Physiological parameters of LPA responsiveness in early embryonic cortical neuroblasts

LPA exposure increases a membrane conductance mediated by chloride ions (Cl−). Two different cells from primary cortical cultures (E12–E13;a–d) and a cell from an embryonic cortical cell line (e, f) are shown. a, LPA exposure elicited an inward current and increase in conductance not produced by related compounds (shown for LPG). The large deflections occurring throughout the current record are voltage-ramp-induced currents evoked every 6 sec to measure whole-cell conductance and the reversal potential (Vrev). Rectanglesindicate time and duration of delivered ligand. Similar negative results with LPG were observed in seven of seven cells.b, Vrev for the response shown in Figure 2a is consistent with a Cl−-mediated conductance. The voltage was ramped from +82 to −118 mV at a rate of 1 mV/msec. Plotted is the difference current calculated by subtracting the control voltage-ramp-induced currents from those obtained during LPA exposure.Vrev was estimated at −28 mV, similar to the ECl (−40 mV). c, Ion substitution demonstrates that some LPA-induced responses are mediated by Cl−. Cells were recorded in low-Cl− saline, where the predictedECl is +70 mV. LPA elicited an inward current. d, Vrev for the response in c is +55 mV, consistent with a chloride conductance in low-Cl− saline. e, LPA exposure in the embryonic cortical cell line TSM stably overexpressinglpA1/vzg-1 also showed LPA-dependent inward currents (compare a).f, Vrev for the response ine is −29 mV, consistent with a Cl−conductance (compare b).

LPA exposure increases a membrane conductance mediated by NSCs. Two different cells from primary cortical cultures (a–d) are shown. a, LPA exposure elicited an inward current that showed homologous desensitization. Incomplete desensitization reflects the low concentration of LPA applied; LPA concentrations that elicited maximal responses decreased responsiveness of the remaining cells in that culture (E12–E13) to subsequent LPA challenges. b,Vrev for the response shown ina is consistent with an NSC conductance.Vrev was estimated at 0 mV.c, LPA elicited an inward current in an E12 cell. Recording electrode contained ZIS saline to shift theVrev for a NSC conductance to positive voltages. d, The Vrev of the LPA increase in conductance shown in c was +46 mV (ZIS/Tyrode’s).
LPA-induced Cl− conductances
LPA-activated Cl− conductances were observed at all ages tested (Table 2). The percentage of cells responding with an increase in GCl was similar in E11, E12, and E12–E13 cultures. Latencies were ∼30 sec (Table 2). The average peak INa density was low at E11 and increased at later times (Table 2), but it can be clearly seen in Figure 3 that many Cl− responses were observed in cells with little or no INa at each age tested (Fig. 3, open bars). The Ioutdensity was no different from that observed in the population of cells with increased NSC conductances (Table 2).
The ionic dependence of LPA responses was investigated in Cl− ion substitution experiments. Approximately 36% of examined cells bathed in low Cl− saline revealed an increased conductance with a Vrev(+39 ± 5 mV; n = 8) shifted toward the predictedECl (+70 mV) (Fig. 4c,d) and significantly more positive than that expected for a NSC conductance (p < 0.01; Fig. 5). In contrast, ∼14% of cells responded with a Vrev near 0 mV (2 ± 1 mV; n = 3) in low Cl−saline. These results demonstrate the existence of two distinct, LPA-dependent responses, one dependent on Cl−, and the other consistent with mediation by NSC channels.
GCl responses could also be elicited in a cell line derived from cortical neuroblasts of the VZ (cell line TSM; Chun and Jaenisch, 1996) stably transfected to overexpress the cloned LPA receptor gene lpA1/vzg-1 (Hecht et al., 1996) (Fig. 4e,f). In standard salines, Vrev was −31 ± 2 mV (n = 4), GLPA was 0.56 ± 0.27 nS, and the response latency was 49 ± 25 sec. These cells exhibited electrical properties similar to those expected for neuroblasts: INa was −2.2 pA/pF (−1.7, −3.0 upper and lower error limits), Rm was 3.1 ± 0.7 GΩ, and no action potentials could be elicited by depolarizing current injection. Unlike primary cells in clusters, TSM cells could be assayed as single cells. Nevertheless, these single cells showed similar electrical properties and GCl values and latencies after LPA exposure despite their lack of electrical coupling.
LPA-induced NSC conductances
LPA-activated NSC conductances were observed at all ages tested (Table 2). The percentage of cells responding with an increase inGNSC and the magnitude of the response (GLPA) was similar in E11, E12, and E12–E13 cultures (Fig. 5a,b, Table 2). Response latencies were ∼45 sec. Figure 3 shows that many NSC responses were observed in cells with little or no INa at each age tested (solid bars).
The ionic dependence of this putative NSC response was investigated by ion substitution experiments in which the intracellular milieu was perfused with impermeant cations (ZIS intracellular solution). With Tyrode’s in the bath and ZIS in the pipette, an NSC conductance is predicted to have a Vrev ≫ 0 mV andECl that is negative (−69 mV). If this class of LPA-induced response was caused by nonspecific membrane breakdown, then the Vrev should remain near 0 mV. With ZIS in the pipette, however, eight cells responded to LPA (25 nm) with an increase in conductance that had a Vrevsignificantly more positive than 0 mV (Fig. 5c,d; +32 ± 5 mV; p < 0.0005 compared with theVrev obtained under standard conditions). Other cells responded with a Vrev near −69 mV as expected for an increased GCl (data not shown).GNSC responses could be elicited in a subpopulation of cells from the TSM stable cell line (data not shown).
LPA responsiveness precedes that of GABA or l-glutamate
Previous work indicated that the onset of GABA andl-glutamate responses in rat cortex commenced at a developmental period corresponding to murine E11–E12 (LoTurco et al., 1995). To determine whether LPA effects can be observed earlier in development than GABA or l-glutamate responsiveness, E11–E12 cells were cultured and exposed to either LPA (10–25 nm) or a combined stimulus consisting of GABA (100 μm) and l-glutamate (300 μm). GABA and l-glutamate were applied simultaneously because the reversal potential of responses to these two ligands are distinct [near −40 mV (ECl) and ∼0 mV, respectively], allowing unambiguous ligand identification based on reversal potential while using a single delivery pipette. Acutely dissociated cells from E11, examined within 3 hr of plating, possessed only LPA sensitivity (n = 7; Fig.6a,d). By late E11, both LPA and neurotransmitter responses could be seen in similar proportions (Fig. 6d; an example of an E12 cell is shown in Fig.6b). E12 and E12–E13 cells were most often sensitive to a single ligand (48%), however a small population responded to both LPA and either GABA or l-glutamate (26%; Fig. 6b). It is unlikely that the lack of GABA/l-glutamate responsiveness in early E11 cells is caused by the dissociation procedure because responses were observed within 1 hr after plating E12 embryonic cortices. At E12–E13, the Vrev of responses elicited by GABA/l-glutamate application was usually near ECl (Vrev of −38 ± 2 mV; n = 8 of 9 cells), suggesting that the neurotransmitter response was mediated by GABAAreceptors. The GABA-induced conductance ranged from 0.6 ± 0.1 nS, (n = 10; p < 0.01) in the earliest examined cultures, to 1.8 ± 0.4 nS (n = 8) at later times (Fig. 6c). Rarely, responses to GABA/l-glutamate application elicited a fast increase in conductance that reversed at −3 mV (n = 1), consistent with mediation by ionotropic glutamate receptors.

LPA responsiveness precedes that of GABA orl-glutamate in early cortical neuroblasts. E11–E12 cortices were dissociated, plated onto glass coverslips, and examined at the indicated times using whole-cell recording techniques for sensitivity to puffer application of LPA and combined GABA/l-glutamate. Solid bars indicate duration of LPA exposure, and hatched bars indicate GABA/l-glutamate exposure. a, Representative recording from an E11 neuroblast responding to LPA only. TheVrev for this response was −40 mV, consistent with an increase in Cl− conductance (1.05 nS). b, An E12 neuroblast sensitive to both LPA and GABA/l-glutamate. LPA responses,Vrev was −14 mV (NSC); increase in conductance was at least 800 pS. GABA response:Vrev was −34 mV (consistent with being mediated by Cl− and not NSC); increase in conductance was at least 1530 pS. c, An E12 neuroblast responsive to only GABA. The response to the GABA/l-glutamate application reversed at −45 mV consistent with its being mediated by GABAA receptors; the increase in conductance was at least 2 nS. d, Developmental onset of ligand sensitivity. Solid bars indicate LPA responsiveness; stippled bars indicate GABA/l-glutamate responsiveness. During the first 3 hr after plating E11 cortices, only LPA sensitivity was observed, representing ∼50% of recorded cells (the remainder being unresponsive). By E12, neuroblasts began responding to both LPA and GABA/l-glutamate. By E12–E13 (cells isolated at E12 and cultured for 12–20 hr), approximately equal percentages of cells responded to either LPA or GABA/l-glutamate.
DISCUSSION
An expectation extending from LPA receptor gene expression in the cortical VZ (Hecht et al., 1996) was the existence of receptor-mediated actions of LPA influencing the physiology of cortical neuroblasts. In this study, LPA was found to stimulate two distinct conductances in separate populations of nestin-immunoreactive, putative neuroblasts present in cell clusters. These effects commenced at the earliest stages of cortical development and preceded both GABA andl-glutamate sensitivity that appeared at subsequent ages. A majority of cells examined were LPA-responsive, consistent with a prominent role for this lysophospholipid in cortical development.
Examined cells were identified as early cortical neuroblasts based on multiple criteria. At the ages of cell isolation, E11–E12, very few neurons have become postmitotic, and few glia appear to be generated (Caviness, 1982; Blaschke et al., 1996, 1998; Weiner and Chun, 1997). The examined cells could also be easily distinguished by their spherical shape and association in clusters, contrasting with rare, flat, fibroblast-like cells that were also present in the culture. The presence of extensive electrical coupling within the examined clusters is the normal, physiological state of cells in the VZ (LoTurco and Kriegstein, 1991), and the resting membrane potential of these cells was consistent with previous measurements of cortical neuroblasts within living tissue slices (LoTurco et al., 1995). At the older ages examined, 12–20 hr after isolation at E12 (E12–E13), prominent GABA responses were also observed in the spherical cells, further consistent with previous observations on cortical neuroblasts (LoTurco and Kriegstein, 1991; LoTurco et al., 1995). Cells responding to LPA were also nestin-immunoreactive which, combined with the aforementioned criteria, is again consistent with a neuroblast identity (Cataneo and McKay, 1990). In addition, the high percentage of cells responding to LPA (>60% at E11; see Table 2) was consistent with the extent oflpA1/vzg-1 expression in the VZ observed by in situ hybridization at comparable ages, where most cells appeared to express this LPA receptor gene (Hecht et al., 1996). Moreover, the electrical properties and LPA-dependentGCl changes of the putative neuroblasts were similar to those of neuroblast cell lines derived from the embryonic cortex (Chun and Jaenisch, 1996) in which single (noncoupled) cells were examined, indicating that electrical coupling per se was not necessary for the observed physiological responses. Taken together, these data show that the examined primary cells possess developmental and phenotypic characteristics of cortical neuroblasts while responding to LPA stimulation.
LPA exposure induced responses that cannot be explained by nonspecific or known secondary effects. Nonspecific effects could hypothetically be produced by detergent properties of LPA through micelle formation, but such effects are not seen until the critical micelle concentration is reached (>1 mm) (Durieux, 1995), whereas all of our studies were performed in the nanomolar concentration range. In addition, a structurally similar ligand having potential detergent properties, LPG, did not elicit these responses (Fig. 4a). We examined the membrane for possible damage by LPA by monitoring the reversal potential after LPA exposure; these studies demonstrated an intact membrane (Fig. 5c,d). Moreover, if the conductance changes were a nonspecific perturbation, all cells should respond, and the response should be stereotyped. In fact, ∼
 of the cells did not respond at all (Table 2). Similarly, as seen in Figure 6c, application of LPA after stimulation of a cell by GABA/l-glutamate produced no conductance changes, again inconsistent with a lack of specificity. Combined, these data argue against nonspecific activation of conductance changes by LPA.
of the cells did not respond at all (Table 2). Similarly, as seen in Figure 6c, application of LPA after stimulation of a cell by GABA/l-glutamate produced no conductance changes, again inconsistent with a lack of specificity. Combined, these data argue against nonspecific activation of conductance changes by LPA.
Another possibility is that GABA or l-glutamate could produce the conductance changes as a secondarily released factor. This explanation is inconsistent with the previous literature and our data. Based on studies by Kriegstein (LoTurco et al., 1995) in which GABA responses among embryonic VZ cells were extensively characterized, only GABAA responses were observed, i.e., the fast, ligand-gated ion channel responses, whereas GABAB responses were not observed in any of these VZ cells, and our data confirm this observation. Similarly, l-Glu responses were also mediated by ligand-gated ion channels. We observed both GABAA and ionotropic l-Glu receptor responses, both of which have a relatively brief latency (Fig. 6). This brief latency fundamentally contrasts with the prolonged latency of LPA responses, 30–45 sec, that is nearly identical to LPA latencies observed in two different cell systems (Holtsberg et al., 1997; Thoreson et al., 1997). Migrating, postmitotic cortical cells have been reported to respond through GABAB receptors (Behar et al., 1996), however GABAB-type responses were not reported in VZ cells of previous studies (LoTurco et al., 1995) and in the present study, never seen even in the presence of clearly detectable GABAAresponses (demonstrating that GABA was active); these data are compatible with GABAB receptors being expressed on at least some newly postmitotic neurons, but not being expressed on mitotic neuroblasts that are electrically coupled in the cell clusters that were recorded here. In addition, no GABA responses (fast or slow) were ever observed at E11. Still further, the increase in chloride conductance with a latency of 30 sec is not typical for a GABAB receptor (which generally reduces calcium, and increases potassium conductances) (Rang et al., 1995). The direct experimental paradigm of exposing a cell to GABA/l-Glu followed by LPA, or vice-versa (Fig. 6) again illustrates that secondary phenomena cannot account for the LPA responses. Our combined experimental data along with the previous literature show that the responses we observe cannot be explained by secondary release of the previously identified factors, GABA or l-glutamate.
LPA activated two distinct conductances in separate populations of cells. Half of the responsive cells showed increased Cl− conductance based on reversal potential and ion substitution experiments. LPA has been reported to increase Cl− conductances in non-neuronal cells, including defolliculated Xenopus oocytes (Durieux et al., 1992;Fernhout et al., 1992), wounded corneal keratocytes (Watsky, 1995), and quiescent Rat-1 fibroblasts (Fischer et al., 1995). There are differences in observed responses that include homologous desensitization, apparent ligand affinity, pertussis toxin sensitivity, and Ca2+ sensitivity. These likely reflect differences in cell type, lysophospholipid (including LPA) receptors (Hecht et al., 1996; Fukushima et al., 1998; Chun et al., 1999; Zhang et al., 1999), and/or downstream mechanisms. The mechanisms underlying activation of the LPA-induced Cl− conductance in cortical neuroblasts are under investigation. One possibility involves increases in intracellular Ca2+ and subsequent activation of Ca2+-activated Cl−channels. LPA increases intracellular Ca2+ in some neuroblasts in these primary cultures (Fluo-3 and Fura-2 imaging; A. Dubin, T. Bahnson, and J. Chun, unpublished observation). Alternatively, Cl− currents may be activated by cytoskeletal remodeling. Alterations in the actin cytoskeleton have been shown to gate Cl− channels in neocortical astrocytes (Lascola et al., 1998), embryonic human skeletal muscle myoballs (Häussler et al., 1994), and human vascular endothelial cells (Oike et al., 1994). Actin depolymerization opens CFTR Cl− channels with a time course similar to that observed after LPA stimulation (Fischer et al., 1995). Rearrangements of the actin cytoskeleton could provide such a mechanism and are consistent with LPA-dependent cell-rounding observed in neuroblast cell lines (Jalink et al., 1994; Hecht et al., 1996; Fukushima et al., 1998;Chun et al., 1999).
An NSC conductance rather than a Cl− conductance was activated by LPA in a second population of cells. Ca2+ may permeate the LPA-activated channels underlying this NSC response, and its contribution to the inward current may be considerable (Frings et al., 1995). NSC conductances that allow Ca2+ entry could participate in the extensive programmed cell death among cortical neuroblasts within the VZ (Blaschke et al., 1996, 1998; Staley et al., 1997) and could also lead to some of the myriad effects associated with Ca2+ signaling.
The conductance changes produced by LPA in neuroblasts are likely to be depolarizing. The Cl− equilibrium potential has been shown to be approximately −30 mV in rat neuroblasts using gramicidin perforated-patch techniques (Owens et al., 1996), and NSC conductances have reversal potentials near 0 mV. Thus, LPA-dependent increases in NSC as well as Cl− conductances would produce a depolarization. There were no detectable differences in resting physiological properties between cells responding to LPA with an increased Cl− or NSC conductance. The resting membrane potentials, magnitudes of INa and outward current densities, Cm, andRm were not significantly different. The biological significance of these distinct ionic groups awaits further investigation. Their existence, however, provides a potential link between LPA signaling and several outcomes known to occur within the VZ: cell proliferation, differentiation, and death. In addition to the relatively rapid effects, seconds to minutes, documented here, LPA also has longer term growth factor-like properties in other systems that are manifest over a period of hours to days (Moolenaar et al., 1992). These comparatively long-term effects complement the LPA-dependent responses observed. We note that this scenario is undoubtedly incomplete, and it will be crucial to determine which of these LPA effects take placein vivo through genetic and cell biological strategies. Regardless of the actual roles of the ionic conductance responses observed here, it is notable that they resemble neurotransmitter stimulation among neuroblasts at an early developmental stage that precedes GABA and/or l-glutamate responsiveness, thus providing an additional mechanism for producing ionic conductance changes at the earliest stages of cortical neurogenesis.
The existence of G-protein-coupled receptor-mediated LPA signaling in the embryonic cortex adds to receptor tyrosine kinase and neurotransmitter-gated ion channels as a new and potentially important ligand-receptor system influencing cerebral cortical neurogenesis. A difficult but important issue to address is the in vivocellular source of extracellular LPA. Because LPA is also produced intracellularly as an intermediate in phospholipid biosynthesis (Durieux, 1995), this source must be differentiated from the pool that acts as an extracellular signal. The presented data indicate that the stimulatory activity of LPA affects the majority of cortical neuroblasts, consistent with the spatial and temporal expression of the LPA receptor gene lpA1/vzg-1 (Jalink et al., 1994; Hecht et al., 1996; Fukushima et al., 1998; Chun et al., 1999). As the first electrophysiological evidence of extracellular lysophospholipid signaling during CNS development, these data indicate that LPA, along with other related lipid molecules and their cognate receptors, could have important influences on the development and function of the cerebral cortex as well as other parts of the nervous system.
Footnotes
This work was supported by the National Institute of Mental Health, an institutional grant from the Howard Hughes Medical Institute, the James H. Chun Memorial Fund (J.C.), and National Institutes of Health (A.E.D., T.B.). We thank Drs. V. Dionne and M. Pompeiano for reading this manuscript.
Correspondence should be addressed to Dr. Chun, The Department of Pharmacology, School of Medicine, University of California, San Diego, 9500 Gilman Drive, La Jolla, CA 92093-0636.
Dr. Dubin’s present address: RW Johnson PRI, 3535 General Atomics Court, Suite 100, San Diego, CA 92121.
Dr. Bahnson’s present address: Division of Cardiology, Duke University Medical Center, Box 2959, Durham, NC 27710.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}