

Abstract
Brain Ca2+ regulatory processes are altered during aging, disrupting neuronal, and cognitive functions. In hippocampal pyramidal neurons, the Ca2+-dependent slow afterhyperpolarization (sAHP) exhibits an increase with aging, which correlates with memory impairment. The increased sAHP results from elevated L-type Ca2+ channel activity and ryanodine receptor (RyR)-mediated Ca2+ release, but underlying molecular mechanisms are poorly understood. Previously, we found that expression of the gene encoding FK506-binding protein 12.6/1b (FKBP1b), a small immunophilin that stabilizes RyR-mediated Ca2+ release in cardiomyocytes, declines in hippocampus of aged rats and Alzheimer's disease subjects. Additionally, knockdown/disruption of hippocampal FKBP1b in young rats augments neuronal Ca2+ responses. Here, we test the hypothesis that declining FKBP1b underlies aging-related hippocampal Ca2+ dysregulation. Using microinjection of adeno-associated viral vector bearing a transgene encoding FKBP1b into the hippocampus of aged male rats, we assessed the critical prediction that overexpressing FKBP1b should reverse Ca2+-mediated manifestations of brain aging. Immunohistochemistry and qRT-PCR confirmed hippocampal FKBP1b overexpression 4–6 weeks after injection. Compared to aged vector controls, aged rats overexpressing FKBP1b showed dramatic enhancement of spatial memory, which correlated with marked reduction of sAHP magnitude. Furthermore, simultaneous electrophysiological recording and Ca2+ imaging in hippocampal neurons revealed that the sAHP reduction was associated with a decrease in parallel RyR-mediated Ca2+ transients. Thus, hippocampal FKBP1b overexpression reversed key aspects of Ca2+ dysregulation and cognitive impairment in aging rats, supporting the novel hypothesis that declining FKBP1b is a molecular mechanism underlying aging-related Ca2+ dysregulation and unhealthy brain aging and pointing to FKBP1b as a potential therapeutic target.
SIGNIFICANCE STATEMENT This paper reports critical tests of a novel hypothesis that proposes a molecular mechanism of unhealthy brain aging and possibly, Alzheimer's disease. For more than 30 years, evidence has been accumulating that brain aging is associated with dysregulation of calcium in neurons. Recently, we found that FK506-binding protein 12.6/1b (FKBP1b), a small protein that regulates calcium, declines with aging in the hippocampus, a brain region important for memory. Here we used gene therapy approaches and found that raising FKBP1b reversed calcium dysregulation and memory impairment in aging rats, allowing them to perform a memory task as well as young rats. These studies identify a potential molecular mechanism of brain aging and may also have implications for treatment of Alzheimer's disease.
Introduction
The population of developed nations is projected to age significantly over the next several decades. Because advanced age is the leading risk factor for Alzheimer's disease (AD) and other cognitive disorders, a corollary of this increase in population age will be a substantial rise in the prevalence of cognitively impaired individuals and a resulting enormous socioeconomic burden (Reitz et al., 2011; Hebert et al., 2013). Discovery of new therapeutic approaches that can delay cognitive impairment would significantly mitigate this burden, but such discovery will likely be difficult without improved understanding of the molecular mechanisms underlying unhealthy brain aging and aging-related susceptibility to Alzheimer's disease. In this regard, accumulating evidence suggests that mechanisms of deleterious brain aging can be elucidated in part by investigation of the molecular machinery regulating neuronal Ca2+ homeostasis.
Numerous studies have found that Ca2+-dependent neuronal processes are dysregulated during brain aging, generally in a cell type- and region-specific manner (Gibson and Peterson, 1987; Landfield, 1987; Khachaturian, 1989; Disterhoft et al., 1996; Foster and Norris, 1997; Lynch et al., 2006; Murchison and Griffith, 2007; Thibault et al., 2007; Toescu and Verkhratsky, 2007). In CA1 pyramidal neurons of the dorsal hippocampus, a region important for learning and memory (Moser et al., 1995; Buzsáki and Moser, 2013), one of the most consistent manifestations of aging is an increased magnitude of the Ca2+-dependent, K+-mediated slow afterhyperpolarization (sAHP; Landfield and Pitler, 1984; Moyer et al., 1992; Disterhoft and Oh, 2006; Gant et al., 2006). The sAHP is generated by hyperpolarizing Ca2+-sensitive K+ channels that are activated by action potential-induced Ca2+ influx and resulting Ca2+ release from intracellular ryanodine receptors (RyRs; Madison and Nicoll, 1984; Sah and Bekkers, 1996; Lancaster et al., 2001; Andrade et al., 2012). The aging-related increases in the hippocampal sAHP and other Ca2+-mediated processes, which act to dampen neuronal excitability, have been found to correlate with impaired learning in several species (Disterhoft et al., 1996; Thibault and Landfield, 1996; Tombaugh et al., 2005; Luebke and Amatrudo, 2012), suggesting that the mechanisms underlying Ca2+ dysregulation may be directly relevant to brain dysfunction with aging. Although aging-related augmentation of the hippocampal sAHP appears to be driven primarily by elevations in voltage-gated L-type Ca2+ channel (LTCC) activity (Moyer et al., 1992; Disterhoft et al., 1996; Thibault and Landfield, 1996; Murphy et al., 2006) and Ca2+-induced Ca2+ release (CICR) from intracellular RyRs (Kumar and Foster, 2005; Gant et al., 2006; Thibault et al., 2007), the elevated activity is not associated with upregulation of LTCC expression (Rowe et al., 2007). Thus, the molecular bases for these increases in Ca2+ sources remain unclear.
However, in microarray studies of gene expression in the rat hippocampus (Kadish et al., 2009), we found aging-related downregulation of the gene encoding FK506-binding protein 12.6/1b (FKBP1b), a small immunophilin that inhibits Ca2+ release from sarcoplasmic reticulum RyRs in cardiomyocytes (Zalk et al., 2007). Moreover, in electrophysiological and Ca2+ imaging studies, we found that knockdown/disruption of FKBP1b in rat hippocampal neurons augments Ca2+ transients generated by RyR-mediated Ca2+ release or LTCC activation (Gant et al., 2011). Those findings suggest the hypothesis that aging-related decline or dysfunction of FKBP1b is a molecular mechanism of hippocampal Ca2+ dyshomeostasis and consequent cognitive impairment. Here, using viral-mediated delivery of a transgene encoding FKBP1b, we tested a key prediction of this hypothesis, namely, that selectively counteracting the FKBP1b deficit in aged hippocampus should reduce/reverse Ca2+ dysregulation and impaired cognition. The results clearly show that overexpressing FKBP1b in hippocampal neurons is sufficient to reverse major manifestations of Ca2+-mediated brain aging.
Materials and Methods
Animals.
Young mature (4–5 months old) and aged (20–22 months old) male Fischer 344 (F344) rats were obtained from the aging rodent colony maintained by the National Institute on Aging. The present study comprises experiments using four cohorts of F344 male rats. The size, composition, treatments, and measures used for each cohort are described in the corresponding Results section. All protocols and procedures were performed in accordance with institutional guidelines and were approved by the Animal Care and Use Committee.
Adeno-associated viral vector constructs and microinjection.
We induced FKBP1b overexpression in the hippocampus of male F344 rats using one-time microinjection of a recombinant adeno-associated viral (AAV) vector construct containing the transgene for rat FKBP1b under the control of the largely neuron-specific Ca2+-calmodulin kinase II (CaMKII) promoter. Because of their safety, efficacy, and long-lasting effects, AAV vector approaches are widely used for brain gene transfer studies in animal models and humans (Broekman et al., 2006; Kaplitt et al., 2007; McCown, 2010; Salegio et al., 2012; Bosch et al., 2014). Before the FKBP1b overexpression experiments, we used a reporter gene, enhanced green florescent protein (EGFP), and the FKBP1b transgene to conduct extensive pilot studies and compare transduction efficiency, tropism, and expression of three AAV serotypes (1, 8, and 9) and three promoters (CB7, CaMKII, and Synapsin) under our specific experimental conditions. These serotypes have been shown to exhibit high transduction efficiency in principal neurons of the brain (Broekman et al., 2006; Aschauer et al., 2013; Bosch et al., 2014), and in our studies all three serotypes showed good in vivo transduction of hippocampal pyramidal neurons in adult rats 4–6 weeks after microinjection into the hippocampus (see Results, below). For the FKBP1b overexpression experiments, we selected AAV serotype 9 and the CaMKII promoter, as this combination exhibited slightly higher transduction and expression in pyramidal neurons of the dorsal hippocampus with minimal spread beyond the ipsilateral hippocampus. Vectors were delivered via microsyringe under stereotaxic guidance to the apical dendrites (stratum radiatum), below the mediolateral peak of the CA1 pyramidal cell body layer (stratum pyramidale) in dorsal hippocampus. Under isoflurane anesthesia, the microsyringe tip was stereotaxically guided to the target coordinates in CA1 stratum radiatum (3.8 mm caudal, 2.8 mm lateral from bregma, and 1.7–1.8 mm ventral from the top of the cortex). For each injection, 2 μl of AAV vector were infused into the hippocampus at a rate of 0.2 μl/min using a 10 μl Hamilton syringe with a custom 30 gauge needle (Hamilton Company) and a Stoelting QSI microinjection pump. Following surgery, animals were treated with analgesics and monitored closely under the supervision of veterinary staff. The design of the AAV-FKBP1b vector was AAV2/9.CAMKII0.4.ratFKBP1b.rBG (titer, 1.99e13 genome copies per milliliter (GC/ml) or 1.99e12 GC/ml). The design of the AAV-EGFP control vector was the same except that the EGFP transgene was substituted for the FKBP1b transgene: AAV2/9.CAMKII0.4.EGFP.rBG (titer, 1.86e13 GC/ml). AAV vectors were constructed and amplified by the Penn Vector Core (Philadelphia, PA).
Morris water maze test of spatial memory.
Cognitive function was tested using the Morris water maze (MWM), with procedures similar to those described previously (Rowe et al., 2007; Kadish et al., 2009). Multiple groups have found deficient spatial learning in the MWM to be a highly sensitive and reliable index of aging-related cognitive impairment in rodents (Gallagher and Rapp, 1997; Markowska, 1999; Tombaugh et al., 2005; Kadish et al., 2009). The MWM used here consisted of a 190-cm-diameter black round tub filled with water (26°C). A 15-cm-diameter escape platform was placed in one of four pool quadrants 1 cm below the water surface and remained in this position until the memory retention probe trial, during which it was removed. The pool was surrounded by circular black curtains with three high-contrast geometric images hung on the inner side of the curtains. Tracking was achieved through contrast imaging using water maze acquisition software (Columbus Instruments). The spatial learning protocol consisted of 4 d of training (learning phase) followed by a memory retention probe trial on the fifth day and a visual acuity/motor function test (cued trial, sixth day). On each training day, subjects were given three trials in which they were placed into the pool from a different quadrant location on each trial and allowed up to 60 s to find and climb onto the escape platform. After a rat found the platform, it was left on the platform for an additional 30 s. Those animals that did not find the platform were gently guided to the platform and allowed to remain there for 30 s before being returned to their cage. Intertrial interval was 2 min. On the fifth day (retention test probe), the platform was removed, and the subjects were placed into the pool from a different location and allowed to swim for 60 s. On the sixth day, a cued trial was performed with a white contrasting marker placed 6 inches above the platform location. Performance on all trials was assessed by path length and latency to reach the platform or former location of the platform.
For the maze learning experiment, 15 aged rats (20 months old) received bilateral injections of AAV-FKBP1b, 7 rats at the original titer (1.99e13 GC/ml) and 8 rats at a 10-fold lower titer (1.99e12 GC/ml), and 10 aged control animals received bilateral injections of AAV-EGFP (1.86e13 GC/ml). Ten young control rats received bilateral injections of AAV-EGFP (1.86e13 GC/ml, n = 5) or bilateral sham surgeries (n = 5) in which the microsyringe was lowered into the hippocampus, but no injection administered. Because the two AAV-FKBP1b titer subgroups did not differ on any performance measure, they were combined into a single group, Aged-FKBP1b, for the statistical analyses. Similarly, the young control EGFP and young control sham subgroups did not differ from one another on any measure and were treated statistically as a single group, Young-Control.
Intracellular recording in hippocampal slice pyramidal neurons.
Our sharp-electrode electrophysiological methods have been described previously (Thibault et al., 2001; Gant et al., 2006, 2011). Briefly, rats were anesthetized in a CO2 chamber before rapid decapitation. Intracellular recordings were obtained from CA1 pyramidal neuron cell bodies in 350-μm-thick dorsal hippocampal transverse slices maintained in oxygenated artificial CSF [containing (in mm) 128 NaCl, 1.25 KH2PO4, 10 glucose, 26 NaHCO3, 3 KCl, 2 CaCl2, and 2 MgCl2] using sharp glass pipettes filled with HEPES (10 mm) and KMeSO4 (2 m). Electrophysiological data were acquired and analyzed using pClamp 8, a sharp-electrode amplifier in current-clamp mode (Axoclamp 2B; Molecular Devices), and a DigiData 1320 board (Molecular Devices). Voltage records were digitized at 2–20 kHz and low-pass filtered at 1 kHz. The sAHPs were triggered at 0.1 Hz by depolarizing current injection steps (200 ms duration) set at an intensity in each cell that consistently elicited a burst of four action potentials. The amplitude of the sAHP was measured at 800 ms following the end of the depolarizing step, and sAHP duration was determined as the interval from the end of the current step to the time point at which the membrane potential returned to prestimulus baseline. The integrated area under the curve (AUC; millivolts by milliseconds) of the sAHP was determined following stimulating current termination.
Ca2+ imaging.
Intracellular Ca2+ responses were recorded simultaneously with electrophysiological responses in neurons from a separate cohort of10 aged rats injected with AAV-FKBP1b in the right (ipsilateral) hippocampus and with AAV-EGFP in the contralateral hippocampus. Imaging methods used were similar to those described previously by several groups, including ours (Magee and Johnston, 1997; Thibault et al., 2001; Hemond and Jaffe, 2005; Gant et al., 2011; Oh et al., 2013). Individual hippocampal slice neurons were impaled with sharp pipettes containing the single-wavelength Ca2+ indicator Calcium Orange (5 mm). Pyramidal neurons with acceptable baseline electrophysiological characteristics (Table 1) that were also sufficiently close to the slice surface to permit simultaneous visualization of Ca2+ responses (9 AAV-FKBP1b- and 6 AAV-EGFP-treated neurons) were imaged on the stage of a Nikon E600 microscope equipped with a 40× water-immersion objective and a CCD camera (Princeton Instruments). Calcium Orange was allowed to diffuse into the cell for at least 10 min before Ca2+ fluorescence measures were obtained. Calcium Orange was excited using a wavelength switcher (Lambda DG-4; Sutter Instrument Company) and software control (Axon Imaging Workbench; Molecular Devices, version 2.2.1.54). The 575 nm wavelength was monitored through a dichroic mirror centered at 570 nm during excitation with the 550 nm wavelength. The Ca2+ response elicited during activation of the sAHP was recorded in each neuron simultaneously with electrophysiological recordings of the sAHP. In addition, in each cell we recorded the Ca2+ response elicited by 10 s of 7 Hz repetitive synaptic stimulation (RSS) delivered via a bipolar stimulating electrode on the Schaffer collaterals, which synapse on CA1 pyramidal cells. Stimulation intensity during RSS was set at action potential threshold, which at 7 Hz generates an action potential on essentially each pulse (Thibault et al., 2001). The rise time of this RSS-induced Ca2+ response is markedly accelerated with aging because of increased RYR-mediated Ca2+ release (Gant et al., 2006). Ca2+ responses were measured in the visible outline of the cell soma, excluding surrounding low-intensity diffracted light. Following background subtraction from an area devoid of cells, the percentage change in fluorescence during RSS was determined relative to baseline (%ΔF/F). The AUC (percentage of fluorescence change times seconds) of the Ca2+ response was determined for the first 5 s of the sAHP-associated Ca2+ responses and the first 5 s of the RSS-generated responses. The RSS-induced Ca2+ levels during the rise to peak Ca2+ concentration were fit with an exponential curve (confidence interval, >93%) to calculate the rising time constant (τ, time to ∼63.7% of the peak) for each cell.
Baseline electrophysiological measures for neurons from behaviorally characterized rats (means ± SEM)
Quantitative real-time polymerase chain reaction.
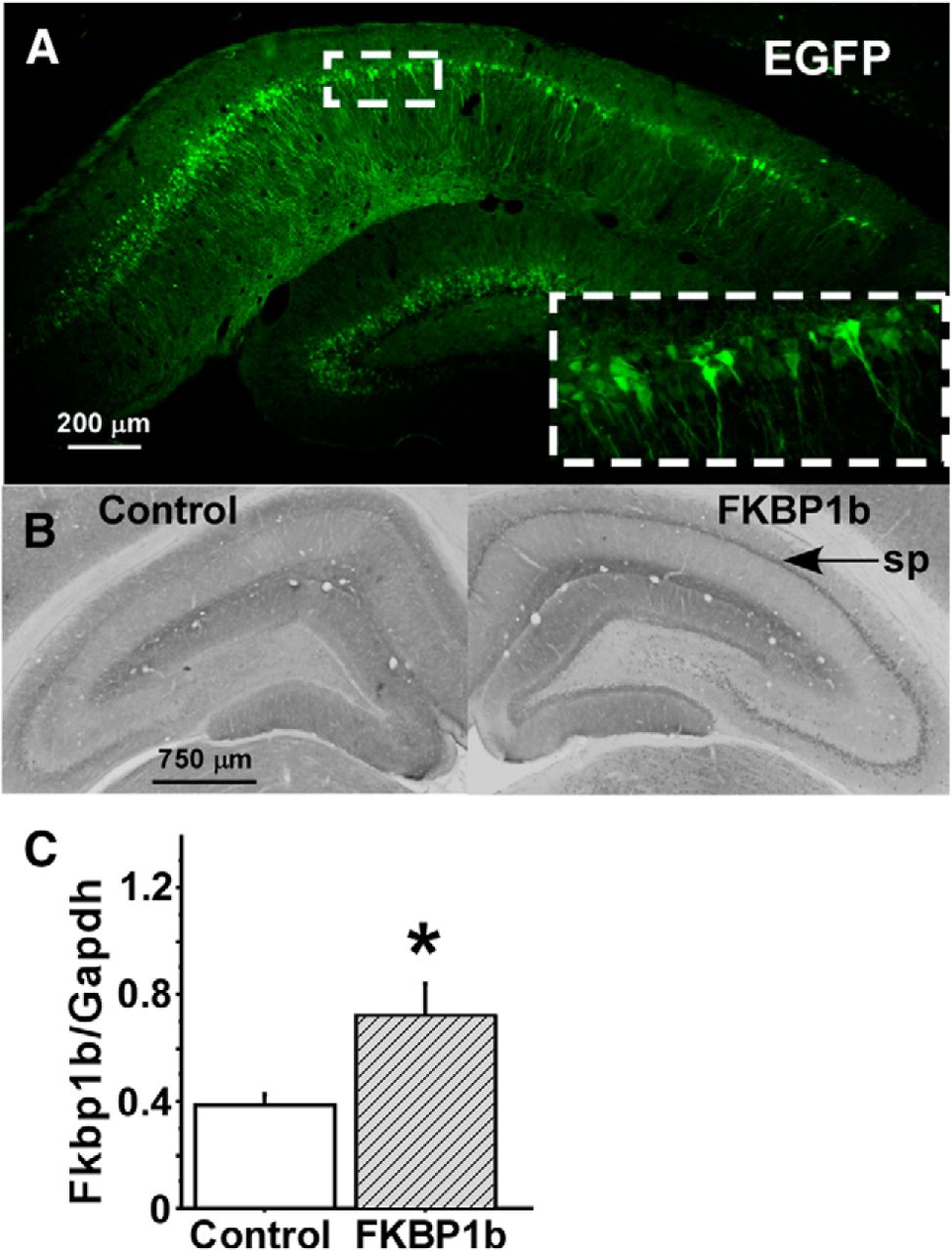
mRNA amplification was performed as described previously (Gant et al., 2011) using an ABI prism 7700 sequence detection system (Applied Biosystems) and Taqman One Step RT-PCR reagents (Applied Biosystems). All samples were run in duplicate in a final volume of 30 μl containing 50 ng of cellular RNA and a Taqman probe (5 μm) and primers (10 nm each), with an amplicon spanning the rat Fkbp1b cDNA region from nucleotides 155 to 259. Cycling parameters for all assays were as follows: 30 min at 48°C, 10 min at 95°C, followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. Primers were designed using Primer Express software (version 1.5, Applied Biosystems) and chemically synthesized by Applied Biosystems (forward primer, 5′-GCAAGCAGGAAGTCATCAAAGG-3′; reverse primer, 5′-CAGTAGCTCCATATGCCACATCA-3′; Taqman probe, 5′-AGCTCATCTGGGCAGCGCCTTCTT-3′). The RNA levels of glyceraldehyde-3-phosphate dehydrogenase (Gapdh) were used as normalization controls for Fkbp1b RNA quantification. Reported values (Fig. 1C) are log10 transforms of the Fkbp1b/Gapdh ratio.
Immunohistochemistry.
Our immunohistochemistry (IHC) methods have been described previously (Kadish et al., 2009). Animals were deeply anesthetized with sodium pentobarbital (100 mg/kg, i.p.) and perfused transcardially with cold saline. Brains were placed in 4% paraformaldehyde overnight and then transferred to 15% sucrose for cryoprotection. Before staining, 30-μm-thick sections were rinsed in TBS-T and incubated in the primary FKBP1b antibody (1:500; sc-98742; Santa Cruz Biotechnology) at room temperature for 18 h. The tissues were rinsed and incubated with appropriate secondary antibody for 2 h at room temperature, followed by incubation for 2 h in avidin-peroxidase. Staining was visualized using a metal-enhanced diaminobenzidine (DAB) solution. For tissue from animals receiving AAV-EGFP injections, 30 μm sections were mounted directly on slides and coverslipped. Imaging was performed using a Nikon Eclipse TE200 and CRi Nuance Multispectral Imaging System (Cambridge Research and Instrumentation). A wavelength of 480 nm was used to visualize EGFP. Gray scale analysis of DAB immunoreactivity was accomplished using Scion software and reported as optical density (average gray value/pixel).
Statistical analysis.
Data analyses were performed with Clampfit8 (Molecular Devices) or Sigma Plot (Systat Software), and statistical analyses performed with StatView (SAS Institute). Multigroup results were analyzed using ANOVA across all groups. Fisher's protected least significant difference test was used for post hoc group comparisons. Correlations were calculated using the Pearson product-moment correlation coefficient. P values <0.05 were considered significant. All results are expressed as mean ± SEM.
Results
Localization of overexpression
We performed extensive pilot studies with unilateral injection of AAV-EGFP or AAV-FKBP1b into the hippocampal field CA1. Fluorescence microscopy of EGFP and immunostaining of FKBP1b showed that by 4–6 weeks after injection, there was pronounced expression of EGFP (Fig. 1A) or FKBP1b (B) in stratum pyramidale neuronal cell bodies of the CA1, CA2, and, to a lesser extent, CA3 fields of the ipsilateral dorsal hippocampus. Expression was less intense in dendrites. The AAV vectors did not significantly transduce cells in the contralateral hippocampus. Granule cells in the dentate gyrus were also often transduced (Fig. 1A), likely because of AAV retrograde/anterograde spread along axonal pathways such as the mossy fibers (Cearley and Wolfe, 2006; Betley and Sternson, 2011; Castle et al., 2014). Consequently, although FKBP1b expression in the hippocampus appears to underlie the behavioral effects reported below, contributions from hippocampal cell types other than CA1 neurons cannot be excluded. In another group of six aging rats, FKBP1b overexpression 5–6 weeks following delivery of the transgene was confirmed by qRT-PCR analyses in dorsal hippocampal tissue. These analyses showed a significant increase in Fkbp1b mRNA expression in the hippocampus injected with AAV-FKBP1b compared with the contralateral control hippocampus (p < 0.05, paired t test; Fig. 1C).

Hippocampal expression of EGFP and FKBP1b following microinjection of AAV vectors harboring the encoding transgenes. A, AAV-EGFP microinjection into field CA1 of dorsal hippocampus resulted in extensive EGFP expression (green fluorescence) in CA1 pyramidal cell bodies of the stratum pyramidale. Inset, Higher magnification of CA1 pyramidal cell bodies and dendrites expressing EGFP. B, FKBP1b immunostaining in AAV-EGFP control (left) and AAV-FKBP1b-injected (right) hippocampi, showing more intense immunostaining in the FKBP1b hippocampus, particularly in CA1 pyramidal cell bodies of the stratum pyramidale (sp, arrow). C, qRT-PCR measures of Fkbp1b mRNA expression normalized to Gapdh mRNA. Shown is a comparison of right (FKBP1b) versus the left (control) hippocampus of aging rats (n = 6) 4–5 weeks following injection of AAV-FKBP1b into right hippocampus of each animal. *p < 0.05 (paired t test).
In hippocampal pyramidal cells, the sAHP is generated largely in the proximal dendrites (Andreasen and Lambert, 1995; Sah and Bekkers, 1996; Lancaster et al., 2001) and possibly the cell bodies (Lima and Marrion, 2007). To confirm that FKBP1b expression declines with aging and is rescued by AAV-FKBP1b transduction in the hippocampal CA1 neurons, we measured FKBP1b immunostaining optical density in the CA1 neuronal cell body layer of a separate cohort of 16 rats (five Young-Control rats, five Aged-Control rats, six Aged-FKBP1b rats). Effects of AAV-FKBP1b microinjection on FKBP1b expression in stratum pyramidale were visibly apparent (Figs. 2A–C), and semiquantitative optical density analyses revealed a main effect of age/treatment group (F(2,12) = 4.24; p < 0.05). Post hoc comparisons showed that density of FKBP1b immunostaining was significantly lower (p < 0.05) in Aged-Control (134.30 ± 4.29; n = 5) relative to Young-Control (150.90 ± 3.58; n = 5) or AAV-FKBP1b-treated aged animals (Aged-FKBP1b; 150.40 ± 4.64; n = 6).

Aging-related decline in FKBP1b expression in CA1 pyramidal cell bodies is counteracted by AAV-mediated FKBP1b overexpression. A–C, Representative examples of FKBP1b immunostaining in the CA1 pyramidal cell body layer (stratum pyramidale) of Young-Control (A), Aged-Control (B), and AAV-FKBP1b-treated (C) rats, illustrating age-related decline of FKBP1b expression and rescue in aged rats by AAV-FKBP1b microinjection. sp, Stratum pyramidale; sr, stratum radiatum.
Cognitive performance
To test whether overexpression of FKBP1b could reverse established indicators of aging-related cognitive decline, we assessed MWM spatial memory in another cohort of young and aged F344 rats (n = 35 animals total). Aged animals were divided into two groups: the Aged-Control group (n = 10) received bilateral injections of AAV-EGFP, and Aged-FKBP1b rats (n = 15) received bilateral injections of AAV-FKBP1b. Additionally, a Young-Control group (n = 10; five bilateral EGFP plus five bilateral Sham) was included to determine the normal young baseline under our conditions. Behavioral testing was initiated 5–6 weeks after bilateral AAV injections. Animals were given 4 d of training (three trials per day) to learn the fixed location of a submerged escape platform. The platform was removed on the fifth day, and animals were tested for memory (retention probe trial) of the platform's former location. On the sixth day (cued trial), sensorimotor function was evaluated in a trial during which the platform's location was visually cued. All groups exhibited a significant improvement in water maze performance over the 4 d course of training (Fig. 3A), as reflected in main effects across trials (repeated measures ANOVA across all trials) for latency and path length to platform location (F(11,352) = 16.74, p < 0.0001 and F(11,352) = 6.28, p < 0.01, respectively; for illustrative clarity, only path length values are shown in Figure 3A). No consistent group differences were seen across training, although by the last training day there was a trend for the Young-Control and Aged-FKBP1b groups to outperform the Aged-Control group (Fig. 3A).

FKBP1b overexpression in hippocampal neurons reversed spatial memory deficits in aged F344 rats. A, Path length values for the training trials (T1–T3) on each day across 4 training days. Young-Control, Aged-Control (bilateral AAV-EGFP), and Aged-FKBP1b (bilateral AAV-FKBP1b) rats all improved across training trials, although there was a trend for Aged-Control rats to perform more poorly. B, On the retention probe trial (Day 5, platform removed), Aged-FKBP1b animals exhibited enhanced memory relative to Aged-Control animals, as indicated by shorter path length (p < 0.001) and latency (p < 0.01) to reach the platform's former location; Aged-FKBP1b rats did not differ from Young-Controls on the retention test. Asterisks denote a significant difference from Aged-Control. C, No group differences in path length or latency were observed during the visually cued trial (Day 6), indicating that deficits in sensorimotor function did not account for the differences in retention performance.
The memory retention probe trial, performed 24 h after the last training session (Day 5), revealed a highly significant main effect of treatment group on both path length and latency to the platform's former location (F(2,32) = 11.15, p < 0.001 and F(2,32) = 4.92, p < 0.01 respectively). Post hoc analyses showed that the Aged-FKBP1b group and the Young-Control group both performed significantly better than the Aged-Control group on path length and latency to platform (p < 0.001 and p < 0.01, respectively; Fig. 3B), but did not differ from each other. No group differences were found in the cued test (Day 6). All groups performed at a high level (Fig. 3C), indicating that aging-related sensorimotor deficits did not account for the differences in MWM retention performance.
Effects of FKBP1b overexpression on the Ca2+-dependent sAHP
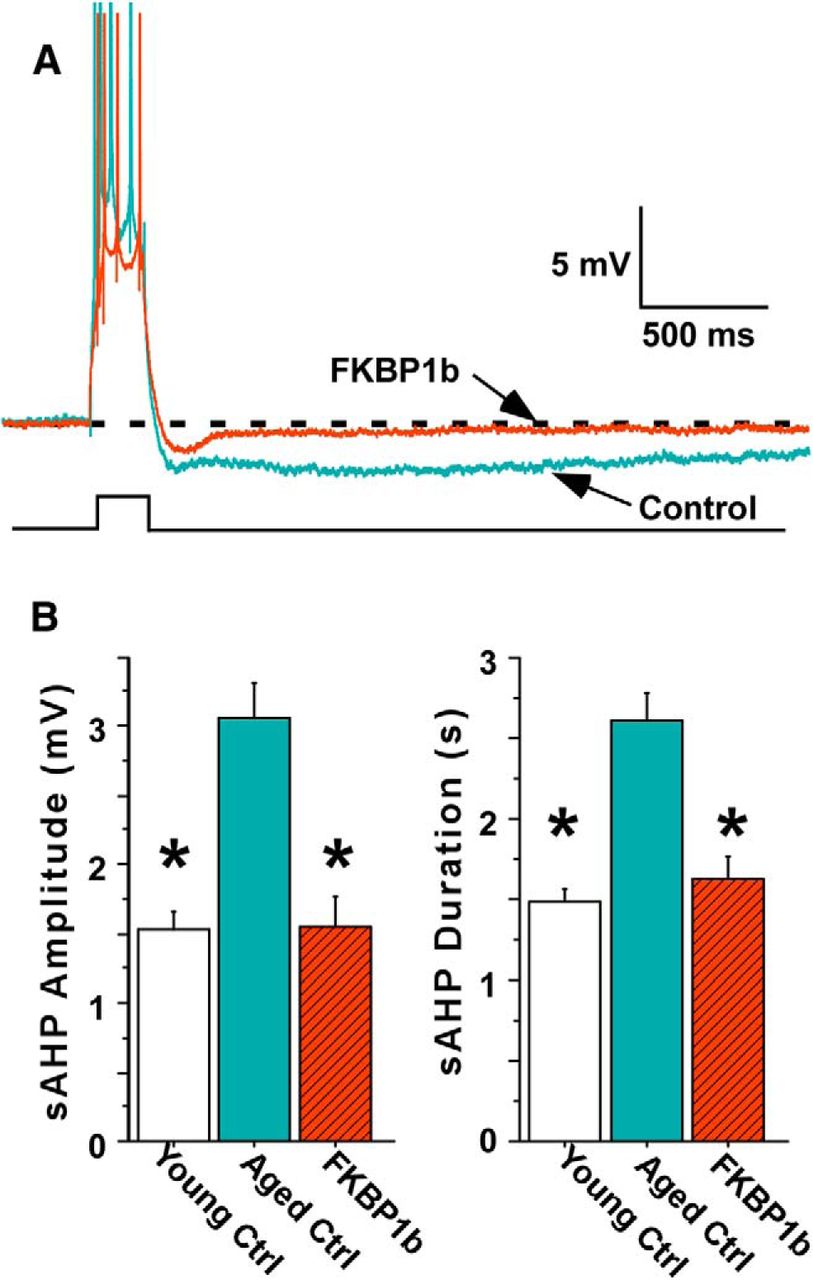
We then determined whether the AAV-FKBP1b intervention also reversed age-related changes in the sAHP in a behaviorally correlated manner. Following completion of the MWM testing, we used a subset of the same animals described above for electrophysiological studies in hippocampal slices. We obtained high-quality intracellular recordings of sAHPs in CA1 pyramidal cells (Fig. 4A) from 18 of the behaviorally characterized animals (13 neurons from five Young-Control rats; 16 neurons from six Aged-Control rats; and 17 neurons from seven Aged-FKBP1b rats). These recording studies began 7 d after behavioral testing ended and continued for ∼9 weeks, alternating animals from the different groups.

FKBP1b overexpression reversed the age-related augmentation of the sAHP in hippocampal neurons from the MWM-tested aged animals. A, Representative traces of the sAHP following a burst of four action potentials elicited by an intracellular current pulse (lower trace) in hippocampal neurons of an Aged-Control rat (blue) and an Aged-FKBP1b rat (red). B, sAHP amplitude and duration were substantially greater in neurons from Aged-Control rats than neurons from Young-Control rats (p < 0.0001) or Aged-FKBP1b (p < 0.0001) rats, but did not differ between Aged-FKBP1b and Young-Control rats. Asterisks denote a significant difference from Aged-Control.
All cells recorded met standard criteria for healthy neurons, and no group differences were seen in baseline electrophysiological parameters (Table 1). However, highly significant group differences were found for sAHP amplitude and duration, regardless of whether individual cells (F(2,43) = 16.64; p < 0.0001) or averaged values per animal (F(2,15) = 12.89; p < 0.01) were used as the statistical population. The sAHPs from Aged-FKBP1b rats were similar to those from Young-Control rats and substantially smaller than those from Aged-Control rats (Fig. 4A,B). This effect was dramatic, as there was little overlap of sAHP magnitudes between the Aged-Control and Aged-FKBP1b groups. The electrophysiological effects of FKBP1b overexpression were highly stable across the 9 week recording period, as there was no difference (p = 0.41) in sAHP amplitudes between FKBP1b-transduced cells recorded during earlier (1.66 ± 0.29 mV; n = 9) versus later (1.34 ± 0.24 mV; n = 8) phases of the recording period.
Furthermore, across individual animals, we found significant positive correlations between the degree of impairment of water maze memory performance and both sAHP amplitude (r = 0.51; p < 0.05) and sAHP duration (r = 0.67; p < 0.01; Fig. 5). Thus, the FKBP1b intervention shifted both Ca2+-dependent electrophysiology and cognitive performance in aged rats to levels similar to those in young rats, in a correlated manner. This correlation remained even when the Young-Control group was omitted from analysis to remove age as a confounding factor (e.g., without the Young-Control group, correlation of MWM path length with sAHP duration was r = 0.62; p < 0.05).

Correlation of sAHP duration with impaired spatial memory in the MWM. Pearson correlation analysis between MWM performance and averaged sAHPs for individual animals showed that enhanced spatial memory performance (shorter retention trial path length) was correlated with reduced sAHP duration and sAHP amplitude (data not shown). Note that there was very little overlap in sAHP duration between the Aged-FKBP1b group and the Aged-Control group.
Simultaneous Ca2+ imaging and electrophysiological recording
We then imaged intracellular Ca2+ responses simultaneously with electrophysiological recordings in a separate cohort of aged animals (n = 10) to test the prediction that FKBP1b overexpression should induce effects on coincident Ca2+ transients that covaried with its effects on Ca2+-dependent sAHPs. In addition, we measured Ca2+ responses elicited during trains of postsynaptic action potentials generated by 7 Hz RSS of the Schaffer collaterals. With aging, the rise time of this Ca2+ response is substantially accelerated because of increased RyR-mediated Ca2+ release from the endoplasmic reticulum (Gant et al., 2006, 2011).
In 10 aged rats, AAV-FKBP1b was injected into one hippocampus and AAV-EGFP into the contralateral hippocampus, and intracellular responses were measured in hippocampal slices over a 3 week period beginning ∼6 weeks after AAV injection. We obtained concurrent electrophysiology and optical Ca2+ imaging in nine neurons from the AAV-FKBP1b-injected hippocampi and six neurons from the AAV-EGFP control hippocampi. sAHPs were substantially smaller in the FKBP1b-treated neurons (Fig. 6A; amplitude and duration, p < 0.01; mean values not shown), essentially replicating the preceding sAHP experiment (Fig. 4). Additionally, AAV-FKBP1b significantly reduced the area under the curve of the sAHP (p < 0.01).

FKBP1b overexpression in aged rat neurons reduced Ca2+ transients imaged simultaneously with the sAHP. A, Representative examples of the sAHPs (top traces) and simultaneously imaged Ca2+ transients (bottom traces) in an Aged-Control neuron and an Aged-FKBP1b neuron illustrating that FKBP1b induced a decrease in both responses. Middle trace, Intracellular current pulse triggering the action potential burst. B, FKBP1b overexpression significantly reduced PeakΔCa2+ (p < 0.05) and the area under the curve (p < 0.001) of the Ca2+ response during the sAHP. Asterisks indicate a significant difference from Aged-Control. C, Across individual neurons, the FKBP1b-induced AUC Ca2+ reduction was correlated with the reduction in the AUC of the sAHP. Red triangles, Aged-FKBP1b; blue circles, Aged-Controls; AUC Ca2+, %ΔF times seconds; AUC sAHP, millivolts times milliseconds).
Optical imaging of the Ca2+ transients coinciding with the sAHPs showed that AAV-FKBP1b injection significantly reduced the peak amplitude (%ΔF/F) and AUC of the Ca2+ transients compared with AAV-EGFP-injected cells (Fig. 6B; p < 0.05 and p < 0.01, respectively). Moreover, correlation analysis showed that the AUCs of the Ca2+ responses and sAHPs covaried across individual neurons (Fig. 6C; r = 0.57; p < 0.05).
In addition, the 7 Hz RSS-induced Ca2+ response, the rise time of which is strongly accelerated by RyR-mediated Ca2+ release (Gant et al., 2006), reached peak levels more slowly in neurons from aged AAV-FKBP1b-injected hippocampi (much as in neurons from young rats) compared with aged AAV-EGFP control neurons (increased τ, p < 0.05; Fig. 7A,B). No differences were observed in peak %ΔCa2+ during RSS, although the AUC of the RSS stimulated Ca2+ response was reduced in AAV-FKBP1b-treated hippocampal neurons (p < 0.05) because of the slower rise time to peak (Fig. 7B).

FKBP1b overexpression in aged rat neurons slowed the Ca2+ rise associated with RSS. A, Examples of Ca2+ responses during the first 5 s of RSS of the Schaffer collaterals in neurons from an Aged-Control and an Aged-FKBP1b rat, illustrating the difference in the rising time constant (τ), but not the peak of the response. The bottom trace is a representative intracellular recording showing the train of action potentials elicited by the 7 Hz synaptic stimulation protocol. B, FKBP1b overexpression significantly reduced the AUC of the RSS Ca2+ response and significantly increased the rising time constant. AUC units of the Ca2+ responses, %ΔF times seconds. *p < 0.05 (significant difference from Aged-Control.
Discussion
The present study combines gene transfer approaches, behavioral assessment, intracellular electrophysiology and simultaneous Ca2+ imaging to elucidate mechanisms of brain aging-related neuronal Ca2+ dysregulation and cognitive impairment. We show, for the first time, that in aged rats in which hippocampal RyR-mediated Ca2+ transients and the Ca2+-dependent sAHP are normally increased, hippocampal overexpression of FKBP1b dramatically reduced these responses, restoring them to levels found in young rats. Furthermore, this reduction of the sAHP was significantly correlated with reversal of the spatial memory impairment characteristically present in aged animals (Fig. 5).
These data strongly support the novel hypothesis that the deficit in hippocampal CA1 FKBP1b expression/function observed previously in aging rats (Kadish et al., 2009; Gant et al., 2014) is an underlying molecular mechanism of brain aging-related Ca2+ dysregulation and cognitive dysfunction. Specifically, our present results satisfy the critical prediction of a mechanistic molecular hypothesis (Falkow, 2004) that selectively counteracting the proposed pathogenic mechanism (FKBP1b deficit) should substantially lessen or reverse pathologic manifestations. Thus, together with our prior study on the effects of hippocampal FKBP1b knockdown (Gant et al., 2011), the findings here with FKBP1b overexpression indicate that FKBP1b is a key regulator of neuronal Ca2+ homeostasis and provide strong support for an FKBP1b-deficiency mechanistic hypothesis of unhealthy brain aging.
Relationship of cognitive impairment to Ca2+ dysregulation
FKBP1b overexpression reversed both cognitive impairment and Ca2+ dysregulation, raising the question of whether the reversal of Ca2+ dysregulation played a causal role in the enhanced cognition. As noted, several prior studies have found close associations between aging-related impairment of cognition/plasticity and increased sAHP magnitude or LTCC activity in hippocampal or cortical pyramidal neurons of rabbits, rats, and monkeys (Disterhoft et al., 1996; Thibault and Landfield, 1996; Tombaugh et al., 2005; Disterhoft and Oh, 2007; Luebke and Amatrudo, 2012). In addition to those correlational data, it has been shown that L-type Ca2+ channel blockers and other agents that reduce the sAHP can improve learning (Disterhoft and Oh, 2006). Furthermore, in the present work, we show that memory performance remained inversely correlated with sAHP magnitude following a molecular intervention (FKBP1b overexpression) that significantly reduced the sAHP (Fig. 5) and parallel Ca2+ transients (Fig. 6). The persistence of this association despite strong modification of the sAHP appears to substantially increase the likelihood of a causal influence of hippocampal Ca2+ responses on cognitive function.
However, reduction of the sAHP is clearly not the only pathway through which restoration of Ca2+ regulation could have enhanced learning. Presumably, multiple Ca2+-related processes in addition to the sAHP were rescued by FKBP1b overexpression, and normalization of one or more of those might have contributed to enhanced spatial learning. For example, long-term potentiation, a lasting form of synaptic plasticity associated with learning, is a Ca2+-dependent process shown to be impaired at some pyramidal cell synapses with aging (Lynch et al., 2006). Because it depends on dendritic spine cytoskeletal reorganization mediated by the Ca2+-activated protease calpain (Lynch and Baudry, 1987; Lynch et al., 2006), excessive Ca2+ transients with aging could seemingly disrupt finely tuned spine calpain activation, resulting in impaired LTP. Accordingly, one pathway through which restoration of Ca2+ regulation by FKBP1b might have enhanced learning is via LTP rescue.
FKBP1b actions on Ca2+ regulation
FKBPs are immunophilins that bind the immunosuppressant drugs FK506 and rapamycin and mediate inhibition of immune response pathways. In addition, FKBPs are characterized by peptidylprolyl isomerase activity and are present in many cell types of the body. Although FKBPs have diverse functions, including protein folding, protein chaperoning and transcriptional regulation (Kang et al., 2008), only FKBP1b and its closely related isoform, FKBP1a/FKBP12, the smallest members of the FKBP protein family, are known to importantly regulate Ca2+ in excitable cells. In myocytes, FKBP1b and FKBP1a bind to and stabilize RyRs in the closed state, thereby inhibiting Ca2+-induced Ca2+ release from the sarcoplasmic reticulum. Deletion of FKBP1b in cardiac cells (or FKBP1a in other myocytes) results in aberrant excitation–contraction coupling and “leaky” RyRs (Zalk et al., 2007; MacMillan, 2013). Although it has not been known whether the small FKBPs regulate Ca2+ release in neurons, we found previously that FKBP1b knockdown/disruption in pyramidal neurons results in an increased sAHP magnitude, faster rise time of the RSS-induced Ca2+ response, and an increase in plasma membrane LTCC activity (Gant et al., 2011). Conversely, we show here that FKBP1b overexpression had the opposite effects on these responses, reducing the sAHP and its parallel Ca2+ transient and slowing the rise of the RSS-activated Ca2+response. Since sAHP magnitude and RSS-response rise time are highly dependent on RyR-mediated Ca2+ release (i.e., are suppressed by ryanodine; Gant et al., 2006), our data show that, as in myocytes, FKBP1b in neurons inhibits CICR from RyRs. Additionally, in neurons, FKBP1b inhibits membrane LTCC activity. Thus, FKBP1b appears to exert substantial regulatory control over Ca2+-dependent signaling in neurons.
Small FKBPs also inhibit the mammalian target of rapamycin (mTOR) (Hoeffer et al., 2008; Hausch et al., 2013), an important regulator of growth and plasticity pathways (Hoeffer and Klann, 2010). In the brain, mTOR is a target of BDNF, which plays major roles in neural plasticity, neurite outgrowth, and neuroprotection (Lynch et al., 2006; Scharfman and MacLusky, 2006). Although mTOR can apparently modulate Ca2+ release from IP3 receptors in smooth muscle, it is not known to participate in RyR-mediated Ca2+ release (MacMillan and McCarron, 2009). Therefore, it appears unlikely that the FKBP–mTOR pathway contributed significantly to the FKBP1b-induced reductions in RyR-mediated Ca2+ responses seen in the present study.
Reversibility of Ca2+ dysregulation and cognitive aging
A particularly striking finding in this study is that major aging-related changes in hippocampal Ca2+ regulation and spatial memory could be fully reversed by FKBP1b overexpression at >20 months of age. In rats, detectable alterations in hippocampal Ca2+-dependent processes (Gant et al., 2006; Lynch et al., 2006) and MWM memory performance (Markowska, 1999; Kadish et al., 2009) normally emerge by midlife (∼12 months old) and are well established by 20 months of age. Reversibility of learning deficits in aging animals has also been seen previously in studies of Ca2+ channel antagonists (Disterhoft and Oh, 2006) and antioxidants (Cartford et al., 2002), among other treatments. This reversibility of cognitive deficits at late ages seems surprising considering the numerous reports of aging-related structural changes in brain synapses, axons, and dendrites, many of which have been correlated with declining cognitive functions (Peters et al., 1996; Rosenzweig and Barnes, 2003; Masliah et al., 2006; Morrison and Hof, 2007; Scheff et al., 2007). One possible explanation for reversibility despite morphological alterations may be that the aging brain compensates for some localized anatomical changes, but is unable to compensate for a widely acting and fundamental deficit such as Ca2+ dysregulation. Alternatively, morphological alterations may be more reversible than is generally recognized (Morrison and Hof, 2007). Regardless of the reasons, however, the observations on reversibility may have important translational implications.
Potential relevance to Alzheimer's disease
Advanced age is essentially the leading risk factor for AD (Reitz et al., 2011), suggesting that some processes seen in normal brain aging increase susceptibility to neurodegeneration and AD. Considerable evidence indicates that one of those processes may be neuronal Ca2+ dysregulation. Elevated intracellular [Ca2+] induces electrophysiological deterioration of hippocampal neurons (Scharfman and Schwartzkroin, 1989), and elevated Ca2+ release from RyRs is toxic to neurons (Verkhratsky, 2005; Bezprozvanny and Mattson, 2008). Importantly, significant Ca2+ dysregulation is also present in AD, as evidenced by altered calcineurin and calpain activation, modified Ca2+ release, and disturbance of Ca2+ signaling by AD-related pathology (Nixon et al., 1994; Gibson et al., 1996; Rozkalne et al., 2011; Hudry et al., 2012; Overk et al., 2014). Furthermore, mouse models of AD exhibit dysregulated Ca2+ release from RyRs and IP3 receptors, as well as altered Ca2+ channel activity (Disterhoft and Oh, 2006; Stutzmann et al., 2006; Thibault et al., 2012; Wang and Mattson, 2014). In vivo Ca2+ imaging in AD model mice also has revealed elevation of Ca2+ in cortical neuron dendrites, which is more pronounced in locations proximal to β amyloid plaques (Kuchibhotla et al., 2008).
In previous microarray studies, we found that hippocampal FKBP1b gene expression is downregulated in human subjects with incipient or advanced AD (Blalock et al., 2004) as well as in normally aging rats (Kadish et al., 2009; Gant et al., 2014). Together with the present results, those findings raise the intriguing possibility that declining expression of the gene encoding FKBP1b during normal brain aging might act synergistically or additively with other sources of pathology to exacerbate Ca2+ dysregulation and increase susceptibility to and/or progression of AD. Thus, if supported by further studies, the present findings suggest that cell-specific elevation of FKBP1b expression may be a feasible new approach for modifying the course of deleterious brain aging and, possibly, early stage Alzheimer's disease.
Footnotes
This work was supported by Grant R37-AG004542 from the National Institute on Aging.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License Creative Commons Attribution 4.0 International, which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed.
- Correspondence should be addressed to either John C. Gant or Philip W. Landfield at the above address. cgant{at}uky.edu or pwland{at}uky.edu
This article is freely available online through the J Neurosci Author Open Choice option.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}