

Abstract
Previous reports, including transplantation experiments using dominant-negative inhibition of β1-integrin signaling in oligodendrocyte progenitor cells, suggested that β1-integrin signaling is required for myelination. Here, we test this hypothesis using conditional ablation of the β1-integrin gene in oligodendroglial cells during the development of the CNS. This approach allowed us to study oligodendroglial β1-integrin signaling in the physiological environment of the CNS, circumventing the potential drawbacks of a dominant-negative approach. We found that β1-integrin signaling has a much more limited role than previously expected. Although it was involved in stage-specific oligodendrocyte cell survival, β1-integrin signaling was not required for axon ensheathment and myelination per se. We also found that, in the spinal cord, remyelination occurred normally in the absence of β1-integrin. We conclude that, although β1-integrin may still contribute to other aspects of oligodendrocyte biology, it is not essential for myelination and remyelination in the CNS.
Introduction
In the CNS, after controlled migration, proliferation, and programmed cell death, oligodendrocyte progenitors differentiate and produce myelin, a lipid-rich membrane, that forms multi-lamellar, spirally wrapped sheets around axons (Pfeiffer et al., 1993). Precise control of these processes derives, at least in part, from instructive cues originating within the extracellular environment of which proteins of the extracellular matrix (ECM) are essential components (Hynes, 1992).
Integrins are the major group of cell-surface receptors for ECM constituents (Ruppert et al., 1995; Montgomery et al., 1996). They play important instructive roles during the development of the CNS (Georges-Labouesse et al., 1998; Graus-Porta et al., 2001; Blaess et al., 2004) and are therefore attractive candidate molecules for the regulation of oligodendrocyte myelination.
In support of such a role, oligodendrocytes cultured on laminin-2, an ECM molecule expressed in the developing mammalian CNS (Hagg et al., 1997; Tian et al., 1997; Powell et al., 1998), produce enhanced myelin membranes (Buttery and ffrench-Constant, 1999), whereas mice and humans lacking the laminin α2 chain (present in laminin-2) show dysmyelination (Jones et al., 2001; Chun et al., 2003). Antibody-blocking experiments and expression of a β1-integrin dominant-negative chimeric integrin, β1-DN (Relvas et al., 2001), showed that the oligodendrocyte integrin mediating this process is probably α6β1-integrin, the putative laminin integrin receptor expressed by oligodendrocytes. This integrin also regulates oligodendrocyte survival both in vitro (Frost et al., 1999) and in vivo, as demonstrated by the increased levels of oligodendrocyte apoptosis present at embryonic day 18.5 (E18.5) in the brainstem of α6-integrin null mice (Colognato et al., 2002). Unfortunately, CNS myelination in these mice could not be analyzed because of a blistering skin defect causing perinatal death (Georges-Labouesse et al., 1996). However, transplantation experiments support a role for β1-integrin in the regulation of CNS myelination. When β1-DN-expressing oligodendrocytes were transplanted into a focal area of demyelination within adult rat spinal cord, they failed to remyelinate neighboring axons (Relvas et al., 2001).
A requirement for β1-integrin/laminin interactions during Schwann cell differentiation and myelination has been revealed by conditional ablation of β1-integrin (Feltri et al., 2002). In oligodendrocytes, however, the processes regulating differentiation and myelination are likely to be intrinsically different. Unlike Schwann cells, oligodendrocytes are not surrounded by a basal lamina to supply them with a permanent source of laminins. During oligodendrocyte differentiation, laminin molecules become available to α6β1-integrin in axons tracts just before myelination (Colognato et al., 2002), i.e., after axon contact has been made. In addition, oligodendrocytes only express α6β1 and do not express α6β4, the predominant laminin integrin receptor expressed by promyelinating Schwann cells (Previtali et al., 2001). Here, therefore, we have tested the role of β1-integrin in oligodendrocytes in the developing CNS and also in a remyelination model of the spinal cord using transgenic mice with conditionally ablated β1-integrin in cells of the oligodendrocyte lineage. Our results suggest that, although β1-integrin signaling still contributes to oligodendrocyte survival, it is not necessary for CNS myelination or remyelination per se.
Materials and Methods
Generation of conditional knock-out mice.
The generation of conditional β1-integrin mutant mice has been described previously (Brakebusch et al., 2000; Potocnik et al., 2000). Mice heterozygous for the β1 null allele (β1+/−), in a 129Sv/B6 mixed background were crossed with CNP-Cre+/− mice [in which cAMP response element (Cre) recombinase expression was under the control of 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNP) regulatory sequences] (Genoud et al., 2002; Lappe-Siefke et al., 2003; Saher et al., 2005) to obtain CNP-Cre+/− β1+/− mice. These were then crossed with mice homozygous for β1-floxed alleles (β1lox/lox), also in a 129Sv/B6 mixed background, to obtain CNP-Cre+/− β1lox/− mice (hereafter called mutant mice) and either β1lox/+ or CNP-Cre+/− β1lox/+ (hereafter called control and heterozygous control mice, respectively). Conditional heterozygous control mice were only used in experiments in which the excision of the β1-integrin gene was monitored by detection of β-galactosidase (β-gal) expression. All other experiments were conducted using CNP-Cre-negative (β1lox/+) control mice. Genotypes were determined by performing PCR on genomic DNA.
Electron microscopy.
To prepare mouse tissues for electron microscopy, mice were anesthetized with a lethal dose of Pentobarbital (Nembutal; Abbott Labs, Irving, TX) and intracardially perfused with 0.1 m phosphate buffer, pH 7.4, followed by a fixative containing 3% glutaraldehyde and 4% paraformaldehyde in the same phosphate buffer. Fixed tissues were removed and postfixed overnight at 4°C. For resin embedding, tissues were postfixed in 2% osmium tetroxide overnight, dehydrated through a graded acetone series, and embedded in Spurrs resin (Electron Microscopy Sciences, Fort Washington, PA). One micrometer semithin cross sections were cut and stained with toluidine blue for analysis at the light microscope. For electron microscopic analysis, resin blocks containing the tissue samples were cut at 60 nm using an Ultracut E ultramicrotome (Leica, Nussloch, Germany) and collected on a 200 mesh grid (Agar Scientific, Stansted, UK). These sections were stained in 3% uranyl acetate and 1% lead citrate before observation in a Hitachi (Tokyo, Japan) H-600 TEM at 75 kV. The growth ratios (g-ratios) of at least 100 myelinated and remyelinated axons were measured with MCID software (Imaging Research, St. Catharines, Ontario, Canada). For remyelination experiments, mice were treated as above and a 4 mm segment of spinal cord comprising the lesion site was removed for analysis. For Bluogal electron microscopy, whole-mount optic nerves were stained for Bluogal (see below) before being processed for electron microscopy. Ultrathin sections of these optic nerves were not contrasted with uranyl acetate and lead citrate.
Lysolecithin-induced focal demyelination.
Two- to 3-month-old mutant and littermate control mice were subjected to the demyelination procedure. Mice were anesthetized by isoflurane (Forene; Abbott Labs) inhalation narcosis. Operations were performed under aseptic conditions. To prevent dehydration, a single intraperitoneal injection of 1 ml of Ringer’s lactate solution (B. Braun Medical, Melsungen, Germany) containing the antibiotic combination preparation Borgal (sulfadoxin, trimethoprim, lidocaine; 10 μl of Borgal 7.5%; Intervet, Boxmeer, The Netherlands) was administered preoperatively. For the perioperative and postoperative analgesia, buprenorphinum (Temgesic; 0.1 mg/kg bodyweight; Essex Chemie, Lucerne, Switzerland) was injected subcutaneously initially at the start of operation and thereafter every 12 h for the following 3 d. Dorsal laminectomies were performed on the midthoracic level of the spinal cord. A glass capillary, pulled to a fine tip, attached to a Hamilton syringe and mounted on a stereotactic micromanipulator, was used to inject a 1% solution of lysolecithin (l-α-lysophosphatidylcholine; Sigma, St. Louis, MO) in sterile PBS, pH 7.4. The micropipette was inserted into the dorsal funiculus, 2 μl of lysolecithin solution was injected, and the needle was slowly withdrawn. The site of injection was marked for future identification with a fine suture in the paravertebral musculature, the wound was sutured in two layers, and the animals were allowed to recover. The day of lysolecithin injection was designated as day 0. All animal experiments were approved by the veterinary office of the Canton of Zürich.
Immunofluorescence, terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling staining, 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside, and Bluogal histochemistry.
Tissue-Tek (OCT; Sakura, Tokyo, Japan) embedded frozen tissues were cut with a cryotome into 12-μm-thick sections, thaw mounted onto Superfrost slides (Mettler Scientific, Highstown, NJ), and air dried. After blocking with goat serum (10%)/PBS, tissue sections were incubated with primary antibodies diluted in PBS overnight at 4°C. The following primary antibodies were used: mouse antiserum-CC1 (1:200; Calbiochem, La Jolla, CA), rat monoclonal MBP (1:75; Serotec, Oxford, UK), and rat monoclonal-β1-integrin (1:30; BD Biosciences PharMingen, San Diego, CA). On the following day, tissue sections were washed in PBS and incubated with the appropriate secondary antibodies, including cyanine 3 (Cy3)-conjugated anti-rat (1:300; Jackson ImmunoResearch, West Grove, PA) and Alexa 488-conjugated anti-rabbit (1:250; Invitrogen, Carlsbad, CA) for 1 h at room temperature. The sections were mounted in Citifluor (Citifluor, London, UK) containing 4′,6′-diamidino-2-phenylindole (DAPI) to stain the nuclei. Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) assay was performed using indirect immunofluorescence to visualize nicked DNA according to the instructions of the manufacturer (Apoptag Red; Serologicals, Norcross, GA). For 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal) and Bluogal histochemistry, tissue sections were fixed in 2% formaldehyde and 0.2% glutaraldehyde in PBS for 7 min and stained in X-gal staining solution (5 mm K3[Fe(CN)6], 5 mm K4[Fe(CN)6], 2 mm MgCl2, and 1 mg/ml X-gal [Calbiochem]) or Bluogal (Sigma) in PBS. After staining, tissue sections were postfixed in 2% formaldehyde/PBS at 4°C. For Bluogal/CC1 double staining, tissue sections were stained with Bluogal before CC1 detection using Vectastain ABC kit and the peroxidase substrate NovaRed (Vector Laboratories, Burlingame, CA). Pictures were acquired using a Zeiss (Oberkochen, Germany) fluorescence microscope equipped with a Zeiss Axiocam CCD camera. Images were processed using Photoshop 7.0 software (Adobe Systems, San Jose, CA).
Primary cell cultures and cytochemistry.
Oligodendroglial cells were obtained from the spinal cord of E19.5 mutant or control mice by enzymatic digestion (Genoud et al., 2002). Briefly, spinal cords were incubated for 20 min at 37°C in 200 μl of DMEM (Invitrogen) containing 2 mg/ml collagenase type IV (Worthington, Freehold, NJ), 1.2 mg/ml hyaluronidase type IV-S (Sigma), and 0.3 mg/ml trypsin inhibitor (Sigma). After trituration, dissociated cells were plated onto poly-d-lysine (Sigma) coated tissue Lab-Tek chamber slides (Nalge Nunc International, Naperville, IL), maintained in DMEM (Invitrogen) supplemented with 10% fetal calf serum (Sigma) at 37°C in 5% CO2, and fixed with 4% paraformaldehyde 6–12 h later. After two washes with PBS, cells were blocked with goat serum (10%)/PBS for 1 h at room temperature. The cells were then incubated with the rabbit NG2-antiserum (1:200; Chemicon, Temecula, CA) and the rat monoclonal β1-integrin (1:30; BD Biosciences PharMingen) primary antibodies, overnight at 4°C. Cy3-conjugated anti-rat antibodies (1:300; Jackson ImmunoResearch) and Alexa 488-conjugated anti-rabbit antibodies (1:250; Invitrogen) were used as secondary antibodies. The slides were mounted in Citifluor before being visualized in a fluorescent microscope equipped with a Zeiss Axiocam CCD camera connected to a Macintosh computer (Apple Computers, Cupertino, CA). Purified oligodendrocyte precursor cell (OPC) cultures were obtained from mixed glial cultures of P0 mutant and control mice as described in by McCarthy and de Vellis (1980).
Western blot analysis.
To quantify expression of β1-integrin, optic nerve tissue was homogenized in lysis buffer (10 mm Tris-HCl, 5 mm EDTA, 150 mm NaCl buffer, and 1% Triton X-100) containing protease and phosphatase inhibitors (Sigma).
Proteins within the lysates were separated by SDS-PAGE and electroblotted onto nitrocellulose membranes (Hybond-C; Amersham Biosciences, Arlington Heights, IL) using standard protocols. Densitometry and quantification of the relative levels of protein expression were performed on scanned images of Western blots using Quantity One software (Bio-Rad, Hercules, CA).
Statistical analysis.
The data show the mean ± SD. Statistical significance was determined using Student’s t test. In addition, for the g-ratio/linear regression analysis, Welch’s two-sample t test and Wilcoxon’s rank sum test were applied. Significance was set at p < 0.05. n represents the number of animals used for each control and mutant groups, respectively. n was always 3 unless otherwise indicated.
Results
Recombination and excision of a conditional β1-integrin allele (Fassler and Meyer, 1995; Brakebusch et al., 2000; Potocnik et al., 2000) was targeted to the cells of the oligodendrocyte lineage using a “knock-in” mouse in which Cre expression was under the control of CNPase regulatory sequences (Lappe-Siefke et al., 2003) (Fig. 1A,B). CNPase is expressed in premyelinating oligodendrocytes (Pfeiffer et al., 1993), and in vivo fate mapping using the ROSA26 lacZ reporter strain (Soriano, 1999) confirmed that Cre controlled by the CNPase regulatory sequences is active in oligodendroglial cells and in some spinal motoneurons from E13 (Genoud et al., 2002) (our unpublished data). The recombination and ablation of the conditional β1-integrin gene also activates the transcription of a lacZ reporter gene under the control of the endogenous β1-integrin promoter (Fig. 1B). This feature allowed us to directly follow the fate of cells that had lost the conditional β1-integrin allele by detecting the activity of the β-gal enzyme by X-gal or Bluogal staining.

Cre-mediated recombination of the β1-integrin gene leads to loss of β1-integrin in OPCs. Schematic representation of the CNPase-Cre knock-in allele (A) and the floxed β1-integrin allele (B). In the conditional β1-integrin allele, two loxP sites (black triangles) flank exons 2–7. During CNPase-Cre-mediated recombination, this genomic region is excised, generating a null allele. The presence of a promoterless lacZ gene trailing the conditional allele and containing a splice acceptor derived from the intron upstream of exon 2 ensures that, after recombination, the oligodendroglial cells will express β-gal under the control of the β1-integrin promoter. Western blot analysis demonstrate that β1-integrin levels in mutant P14 optic nerves (C) and in freshly purified OPCs obtained from mutant P0 mice mixed glial cultures (D) are strongly reduced compared with those of controls. E, Double immunostaining for β1-integrin and the oligodendrocyte precursor marker NG2 show loss of β1-integrin immunoreactivity on E19.5-derived spinal cord NG2-positive mutant cells (arrowheads) but not in their control counterparts (arrows). NG-2 negative cells are β1-integrin positive (white arrowheads). In <10% of NG2-positive cells, some remaining β1-integrin immunoreactivity was observed (arrows in Ec–Ec″). Scale bars, 50 μm.
Targeted deletion of the β1-integrin gene results in protein depletion in oligodendroglial cells
Recombination of the conditional β1-integrin allele under the control of CNPase regulatory sequences led to the loss of β1-integrin protein in mutant OPCs. First, we showed by Western blot analysis that the levels of β1-integrin protein in optic nerves obtained from mutant mice at postnatal day 14 (P14) were significantly lower than those in corresponding control optic nerves (Fig. 1C). Second, we demonstrated that the loss of β1-integrin expression occurred specifically on mutant oligodendroglial cells. To do that, we plated cells from freshly dissociated mutant and control E19.5 spinal cords and, 6–12 h later, assessed the expression of β1-integrin in oligodendrocyte progenitor cells by double immunofluorescence using antibodies for β1-integrin and for the OPC marker NG2. Our data confirmed that β1-integrin immunoreactivity had been lost in >90% of mutant NG2-positive cells (182 of 200 counted cells) but not in their control counterparts (Fig. 1E). In contrast, NG2-negative cells derived from the mutant spinal cord were β1-integrin positive (Fig. 1Ed–Ed″), showing that the CNP-Cre transgene targeted specifically cells of the oligodendroglial lineage. Third, we also confirmed by Western blot analysis that the levels of β1-integrin protein in lysates of freshly purified mutant OPCs, established from P0 mutant mice (McCarthy and de Vellis, 1980), were considerable lower than those in lysates of control OPCs (Fig. 1D). In these cultures, the percentage of recombined, X-gal-positive OPCs was ∼65% (759 of 1144 counted cells), whereas the percentage of recombined astrocytes was <1%.
Normal myelination in the optic nerve, corpus callosum, and spinal cord in the absence of β1-integrin
Previous studies on ECM function in myelination revealed important regional differences in the requirement for these molecules (Chun et al., 2003). Therefore, we analyzed myelination in the optic nerve, corpus callosum, and spinal cord. These anatomical regions contain large numbers of fibers running in parallel to each other, greatly facilitating the analysis of the relationships between axon diameter and myelin sheath thickness. We compared the relationships between axon diameter and myelin sheath thickness (g-ratio) in 2-month-old mutant and control optic nerves and corpora callosa (Fig. 2) and 3-month-old mutant and control spinal cords (Fig. 3). For the optic nerve (p = 0.37), corpus callosum (p = 0.88), and the spinal cord (p = 0.61), no significant differences were found. The average g-ratios for the control and mutant optic nerve were 0.81 ± 0.014 and 0.80 ± 0.017, respectively (Fig. 2B), for the control and mutant corpus callosum were 0.75 ± 0.005 and 0.74 ± 0.02, respectively (Fig. 2E), and for the spinal cord were 0.79 ± 0.005 and 0.77 ± 0.01, respectively (Fig. 3B). In addition, we also found no evidence of dysmyelination in mutant optic nerves, corpora callosa (Fig. 2A,D), and spinal cords (Fig. 3A). To further corroborate that the axon size threshold for myelination was the same in mutants and that there were not more unmyelinated axons, we calculated the amount of myelinated small-diameter axons (axon diameter, <0.5 μm) and the total amount of myelinated axons in relation to all axons in midsagittal sections of the corpus callosum just above the fornix (Fig. 2F). In mutants, the amount of myelinated small-diameter axons (103 ± 5% of control; p = 0.39), the total number of myelinated axons (96 ± 4.5% of control; p = 0.63), as well as the total number of axons (105 ± 2.2% of control; p = 0.51) were not significantly different.

Normal myelination in the optic nerve and corpus callosum in the absence of β1-integrin. A, D, Representative EM pictures of myelinated fibers in cross sections of the optic nerve and corpus callosum of 2-month-old control and mutant mice. The myelin sheaths within mutant optic nerve (A) and corpus callosum (D) show no signs of dysmyelination. The linear regression of the fiber measurements performed for each animal is shown for the optic nerve (B) and for the corpus callosum (E), dashed lines representing mutants. Neither in the optic nerve nor in the corpus callosum were the linear regressions from controls (co) and mutants (mu) significantly different (optic nerve, p = 0.37; corpus callosum, p = 0.88). C, Bluogal electron microscopy of mutant optic nerves showing precipitates in the soma of an oligodendrocyte (arrowhead in Ca) and in the myelin sheath inner loop of mature fibers (asterisks in Cb–Cd). Cd, Higher-magnification image of inset in Cc showing Bluogal precipitates in the inner loop (arrowheads) of the myelin sheath (arrows). The total number of axons, myelinated axons, and small myelinated axons (axon diameter <0.5 μm) were counted in 10 randomly selected nonoverlapping fields from midsagittal sections through the corpus callosum just above the fornix. The total number of axons, myelinated axons, and small myelinated axons per unit area are presented as percentages of control. No significant differences were found in all three categories. Scale bars: A, Ca–Cc, D, 1 μm; Cd, 0.5 μm.

Normal myelination in the developing spinal cord in the absence of β1-integrin. A, C, Representative EM pictures of myelinated fibers in cross sections of the spinal cord of 3-month-old and 5-d-old control and mutant mice. The linear regression of the fiber measurements performed for each animal is shown for the spinal cord of 3-month-old mice. B, Dashed lines represent mutants. No significant differences were found (p = 0.61). D, MBP immunohistochemistry performed on cross sections of thoracic spinal cord of 5-d-old control and mutant mice. No significant differences were found in MBP-positive areas in control and mutant spinal cords (p = 0.56). E, In 3-week-old mutant spinal cord, the proportion of recombined [i.e., Bluogal (Bgal)-positive] oligodendrocytes was compared with the number of all CC1-positive oligodendrocytes. Bluogal/CC1 double-positive oligodendrocytes (black arrows) and a CC1 single-positive oligodendrocyte (white arrow) are shown. Of all counted CC1-positive cells, 89% were also Bluogal positive. F, The frequency of recombined, i.e., Bluogal positive, oligodendrocytes in 3-week-old control and mutant spinal cords were analyzed, and the numbers were not significantly different (p = 0.94). Scale bars: A, 1 μm; C, 5 μm; D, 200 μm; E, 20 μm; F, 50 μm.
Despite the loss of β1-integrin, mutant oligodendrocytes were able to myelinate axons. This was shown in the optic nerve by Bluogal electron microscopy. The presence of Bluogal precipitates in oligodendrocyte somata (Fig. 2Ca), processes, outer loops, and inner loop of the myelin sheath of mature fibers (Fig. 2Cb–Cd) indicated that mutant oligodendrocytes were able to myelinate axons and that their recombination occurred before myelination.
The loss of β1-integrin did not affect the onset or the extent of myelination in the developing spinal cord. At P5, myelination in the mutant spinal cord was not impaired (Fig. 3C). MBP-positive areas, measured as a percentage of the total area of the spinal cord, in cross sections of P5 control (29 ± 1.68%) and mutant (28 ± 1.04%) spinal cords were not significantly different (p = 0.56) (Fig. 3D). In addition, no signs of dysmyelination were observed in mutant spinal cords. To demonstrate that the majority of oligodendrocytes are recombined, Bluogal/CC1-double staining was performed on 3-week-old mutant spinal cord (Fig. 3E). A total of 89% of CC1-positive cells were also Bluogal positive (1311 of 1479 counted cells). Besides a subset of motoneurons in the ventral horns, no Bluogal-positive/CC1-negative cells were observed, further substantiating the oligodendrocyte specificity of the CNP-Cre mouse line. We also measured and compared the number of oligodendrocytes per area expressing β-galactosidase in both control (312 ± 80/mm2) and mutant (308 ± 24/mm2) spinal cords at 3 weeks of age (Fig. 3F). No significant difference (p = 0.93) was observed, further demonstrating that, despite the lack of β1-integrin, mutant oligodendrocytes were capable of properly myelinating the spinal cord.
β1-integrin expression is not required for spinal cord remyelination
Having demonstrated that β1-integrin is not necessary for CNS myelination, we investigated whether it is required for remyelination. For this, we induced focal demyelination by injecting the membrane solubilizer lysolecithin into the dorsal funiculus of 3-month-old mutant and control spinal cords (Fig. 4A). In our previous work (Arnett et al., 2004), we have shown that, in the lysolecithin lesion paradigm, remyelination is completed 5 weeks after injury. Therefore, we chose to assess the extent of remyelination in control and mutant lesioned spinal cords 5 weeks after lesion.
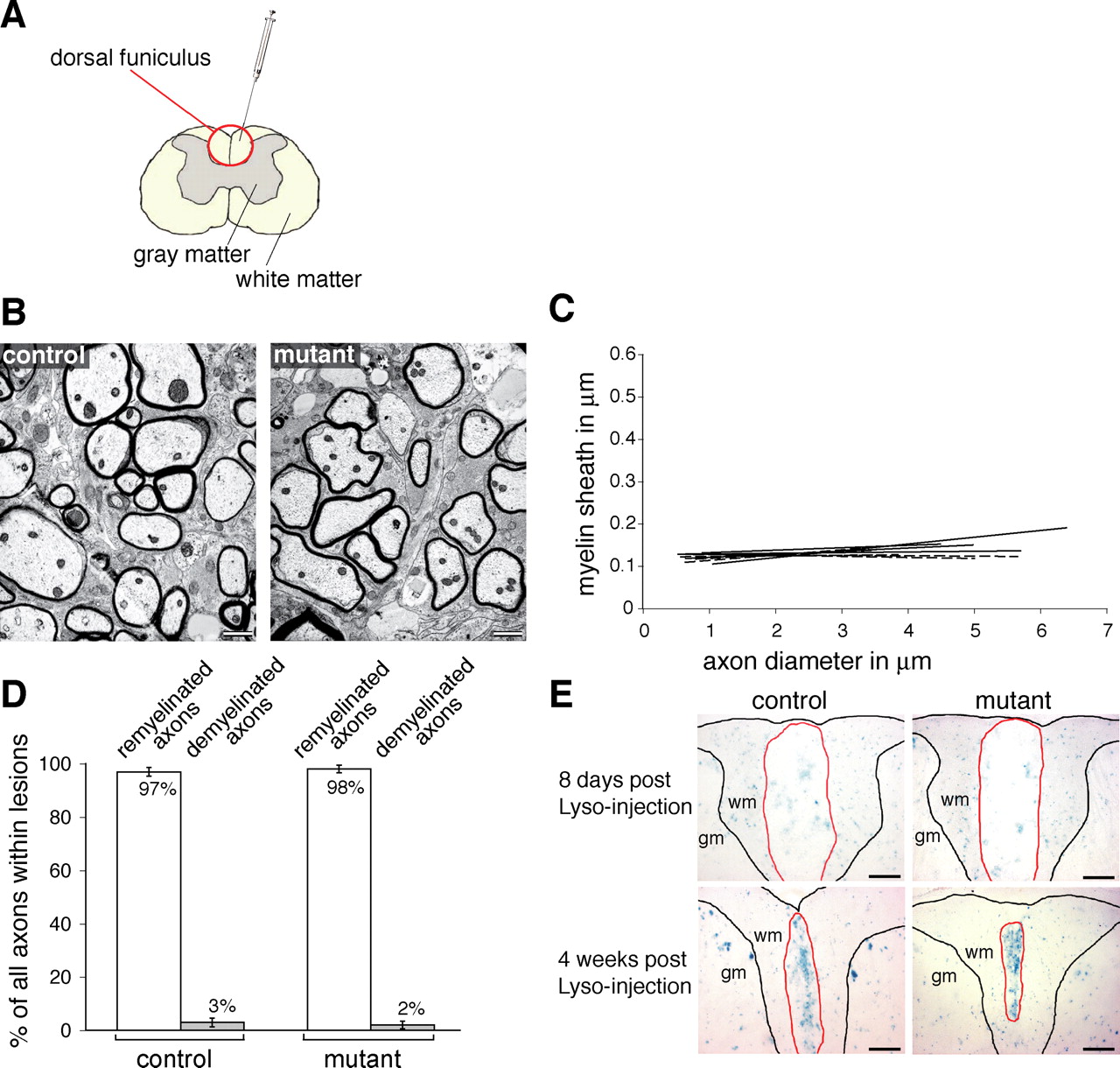
β1-integrin expression is not required for spinal cord remyelination. A, The focal demyelination was induced by the injection of the membrane solubilizer lysolecithin into the dorsal funiculus of the spinal cord. B, Representative EM pictures showing cross sections of remyelinated fibers 5 weeks after lysolecithin-induced demyelination. The myelin sheaths in the mutants show no signs of dysmyelination. C, The graphic represents the linear regressions of fiber measurements for each animal within the remyelinated lesion. The dashed lines represent mutants. The linear regressions from control and mutant animals were not significantly different (4 controls, 5 mutants; p = 0.38). D, The ratio of remyelinated and demyelinated fibers is presented as percentage of the total number of fibers. There is no significant difference (p = 0.54; n = 4) in the percentage of remyelinated fibers between control (97 ± 1.6%) and mutant (98 ± 1.4%) lesions. The extent of remyelination was assessed by counting at least 350 fibers within control and mutant lesions. E, X-gal stainings in control and mutant lesioned spinal cords 8 d and 4 weeks after lysolecithin injection. At 8 d, both control and mutant lesions display a diffuse and faint staining, probably reflecting the degraded oligodendrocytes. At 4 weeks after injection of lysolecithin, both control and mutant lesions show a very prominent staining pattern, which reflects the accumulation of recombined remyelinating cells. This indicates that a significant contribution of non-recombined cells is unlikely to explain the normal remyelination in the mutant lesion. gm, Gray matter; wm, white matter. Scale bars: B, 1 μm; E, 100 μm. Results are expressed as the mean ± SD.
Although the remyelinated axons had thinner myelin sheaths than those present on normal axons, as is well described after remyelination in the CNS, analysis of the relationships between axon diameter and myelin sheath thickness in the remyelinated mutant and control spinal cords did not show significant differences (four controls, five mutants; p = 0.38) (Fig. 4B,C), with the average g-ratios for the mutant spinal cord being 0.86 ± 0.008 and the control spinal cord being 0.87 ± 0.02. To investigate whether the lack of β1-integrin affected the recruitment, migration, and/or the capacity of oligodendrocyte progenitors to engage axons in the lesion, we calculated the ratio between remyelinated and non-remyelinated axons within the lesion. No significant differences (p = 0.54) were found in the number of remyelinated (98 ± 1.4%) or demyelinated (2 ± 1.4%) axons in relation to control remyelinated (97 ± 1.6%) and demyelinated (3 ± 1.6%) axons present in mutant spinal cord lesions (Fig. 4D).
To exclude a major contribution of nonrecombined cells to the remyelination of the mutant lesion, we identified the recombined cells within the lesions by X-gal staining. At 8 d after injection, the X-gal staining within the lesion was faint and diffuse (Fig. 4E), probably a consequence of the degradation of the myelin-producing cells. In contrast, at 4 weeks after lesion, the X-gal staining was prominent in both control and mutant lesions (Fig. 4E). The transcriptional activity of the lacZ gene is controlled by the endogenous β1-integrin promoter after recombination of the conditional β1-integrin allele. Thus, increased X-gal staining most likely reflected an accumulation of recombined oligodendroglial cells within the lesion. This accumulation was similar in control and mutant lesions, excluding a significant contribution of nonrecombined cells during remyelination of the mutant lesions. These results suggest that, although the β1-integrin promoter is active in oligodendrocytes during remyelination, the lack of β1-integrin does not influence the outcome of the remyelination process.
Increased apoptosis of premyelinating oligodendrocytes in the cerebellum of β1-integrin mutant mice
During development, a large excess of oligodendrocytes is produced in relation to the available axonal targets. These oligodendrocytes compete for neuron-derived survival signals, and their surplus is later eliminated by apoptosis, within a relative short time window, soon after the initial differentiation into oligodendrocytes (Raff et al., 1998). Previously, we reported that α6β1-integrin signaling participates in the regulation of this process in the brainstem by studying the α6-integrin knock-out mice (Colognato et al., 2002). To confirm the role of β1-integrin in survival and to investigate whether this was also the case in other regions of the CNS, we analyzed the cerebellum at P5 of β1-integrin mutant and control mice.
In the cerebellum, myelination proceeds from the central regions to the more peripheral regions of the presumptive white matter. At P5, oligodendrocytes in the peripheral regions are still actively engaging axons and are particularly susceptible to apoptosis (Fig. 5De,Df). Consequently, the peripheral regions of both mutant and control cerebella tend to contain higher numbers of apoptotic TUNEL-positive cells and smaller numbers of myelinating oligodendrocytes (identified as CC1-positive cells) than those found in the corresponding central regions (Fig. 5C). To investigate the effect of a loss of β1-integrin signaling in oligodendrocyte cell survival in the cerebellum, we measured both the number of apoptotic TUNEL-positive cells and CC1-positive oligodendrocytes per area in central fiber tract regions of control and mutant cerebella. We found significantly more TUNEL-positive cells in peripheral fiber tract regions of the mutant cerebellum (340 ± 105/mm2) than in the control (185 ± 61/mm2) (n = 4; p = 0.038). In contrast, the number of CC1-positive oligodendrocytes was significantly smaller (n = 4; p = 0.0017) in the same peripheral fiber tract regions of mutant (88 ± 27/mm2) compared with control (167 ± 18/mm2) cerebella. We conclude that, as in the brainstem (Colognato et al., 2002), β1-integrin signaling is involved in the control of oligodendrocyte survival in the cerebellum. No significant difference in the number of CC1-positive and TUNEL-positive oligodendrocytes was observed between the central fiber tract regions of mutant and control cerebella (Fig. 5B,C). This was expected given the later stage of development in these regions, as evidenced by reduced levels of oligodendrocyte apoptosis and greater numbers of oligodendrocytes; these oligodendrocytes may be beyond the time window during which target-dependent mechanisms regulate survival.

Increased apoptosis of premyelinating oligodendrocytes in the cerebellum of mutant mice. A, Overview of a sagittally sectioned P5 cerebellum (DAPI stained). Indicated are the analyzed central and peripheral regions. B, In the fiber tracts of the central area of the cerebellum, the number of TUNEL-positive cells in control (co) (121 ± 86/mm2) and mutant (mu) (83 ± 19/mm2) mice is not significantly different (p = 0.345; n = 4). C, The same is true (n = 4; p = 0.153) for the number of control (211 ± 95/mm2) and mutant (146 ± 29/mm2) CC1-positive cells. In contrast, in the fiber tracts of the peripheral lobar areas of the cerebellum, the number of both TUNEL- and CC1-positive cells is significantly different (B, C). In these areas, the number of TUNEL-positive cells is significantly higher (p = 0.038; n = 4) in the mutant (340 ± 105/mm2) than in control (185 ± 61/mm2) cerebella and, in the case of CC1-positive cells, significantly lower (p = 0.0017; n = 4) in the mutant (88 ± 27/mm2) than in the control (167 ± 18/mm2) mice. D, Immunostainings for MBP (Da, Db), CC1 (Dc, Dd), and TUNEL (De, Df) in the peripheral areas of control and mutant cerebella. In Da, the dashed lines delineate the different cell layers and the fiber tracts in a P5 cerebellar lobe. egl, External granular cell layer; ml, molecular layer; gl, granular cell layer; ft, fiber tracts. the continuous line indicates the pial surface. Arrowheads in De and Df point to TUNEL-positive cells within the fiber tracts. Scale bars, 10 μm. Results are expressed as the mean ± SD.
Although the lack of β1-integrin led to an increase of oligodendrocyte cell death in the mutant cerebellum, myelination itself was not impaired. We analyzed and compared the relationships between axon diameter and myelin sheath thickness (g-ratio) in peripheral lobar fiber tracts of 2-month-old mutant and control cerebella (Fig. 6A). The average g-ratios for the control and mutant cerebella were 0.75 ± 0.01 and 0.76 ± 0.02, respectively, and no significant difference was observed (Fig. 6B). The mutant cerebella also did not present any signs of dysmyelination. To confirm that the lack of a myelination phenotype was not attributable to a compensation caused by an overall increase in the number of nonrecombined (and therefore β1-expressing) oligodendrocytes, we measured and compared the number of recombined oligodendrocytes per area expressing β-galactosidase in the central and peripheral regions of both control and mutant cerebella (central, 94 ± 13/0.2 and 88 ± 27/0.2 mm2, respectively; peripheral, 98 ± 21/0.2 and 96 ± 34/0.2 mm2, respectively) (Fig. 6C,D). No significant difference (central region, p = 0.74; peripheral region, p = 0.94) was observed, confirming that the β1-integrin null oligodendrocytes were myelination competent.

Normal myelination in the cerebellum in the absence of β1-integrin. A, Representative EM pictures illustrate cross sections of myelinated fibers in the peripheral lobar area of 2-month-old control and mutant animals. The myelin sheaths in the mutant show no signs of dysmyelination. The graphic in B shows the linear regressions of the measured fibers in each animal. The dotted lines represent the mutants. The statistical analysis of the linear regressions reveals no significant difference (p = 0.83). C, D, The number of recombined, i.e., Bluogal positive, oligodendrocytes in the central and peripheral cerebellar regions of 6-week-old control and mutant animals (n = 3) were analyzed. The numbers calculated were not significantly different (central region, p = 0.74; peripheral region, p = 0.94). co, Control; mu, mutant. Scale bars: A, 1 μm; D, 50 μm. Results are expressed as the mean ± SD.
Discussion
The formation of myelin sheaths in the CNS requires a number of distinct steps after the generation of oligodendrocyte precursor cells from the neural stem cells of the CNS. Of these, previous studies led to the hypothesis that β1-integrin signaling could play an important role in precursor migration, in the target-dependent survival of newly formed oligodendrocytes, and in regulating the morphological changes associated with myelination itself. In the present work, we tested this hypothesis using tissue-specific gene ablation in transgenic mice to analyze the functional role of β1-integrin in the latter two steps: myelination and survival. Our findings do not support a requirement for β1-integrin signaling in CNS myelination during either normal development or remyelination, but they confirm a role for this integrin in oligodendrocyte cell survival.
Based on our previous in vitro findings (Buttery and ffrench-Constant, 1999; Relvas et al., 2001) and especially in our cell transplantation experiments (Relvas et al., 2001), in which β1-integrin signaling function was blocked by expression of a β1-integrin dominant-negative mutant, we suggested a possible role for β1-integrin signaling in CNS myelination. In the present work, we tested this hypothesis by targeting the ablation of this gene specifically to oligodendroglial cells during CNS myelination. In addition to providing a highly stringent test system, this approach circumvented the problems posed by the use of both the null β1-integrin mouse, which dies during embryogenesis (Fassler and Meyer, 1995), and the α6-integrin null mouse, which dies at birth (Georges-Labouesse et al., 1996). Our results show that, in the absence of β1-integrin, oligodendrocytes are still able to myelinate axons. In the developing mutant spinal cord, myelination was not impaired or delayed, and, in mutant adult mice, no detectable qualitative or quantitative differences of the myelin sheath in the optic nerve, corpus callosum, spinal cord, or cerebellum were observed.
In the light of previous studies (Jones et al., 2001; Chun et al., 2003), in which alteration of the laminin composition leads to defects of CNS myelination, our results were unanticipated. By ablating β1-integrin in oligodendrocytes, we suppressed the surface expression of α6β1-integrin, the only described laminin receptor present in oligodendrocytes, but also removed any other potential β1-integrin laminin receptors, such as α1β1, α3β1, or α7β1. These additional β1-integrins, so far, have not been detected in oligodendrocytes using conventional biochemical and molecular detection methods. Therefore, the most likely explanation is that other laminin receptors must be involved in oligodendrocyte differentiation and myelination. Oligodendrocytes express dystroglycans (H. Colognato, unpublished observations), but a function for these molecules in oligodendrocyte differentiation has still to be supported by experimental evidence.
The different conclusions reached between this study and our previous one (Relvas et al., 2001), using a dominant-negative approach, may be explained by the chosen method to suppress β1-integrin signaling. In other cell types (Calderwood et al., 2004), these dominant-negative approaches produced broader effects than those that are found when more physiological and stringent systems, such as conditional genetic loss-of-function, were used (Graus-Porta et al., 2001). One possible explanation is that the expression of β1-integrin dominant-negative constructs in oligodendroglial cells elicited a transdominant inhibitory effect impairing not only the function of β1-integrins but also the function of other oligodendrocyte integrin receptors. Apart from α6β1, these cells also express several developmentally regulated αv-integrins: αvβ1, αvβ3, αvβ5, and αvβ8 (Milner and ffrench-Constant, 1994; Shaw et al., 1996). Specific functions for some of these have been defined during oligodendrocyte differentiation in vitro. They include a role for ανβ1 in the control of OPC migration (Milner et al., 1996), a role for ανβ3, in close synergism with the platelet-derived growth factor (PDGF) receptor, in the regulation of OPC proliferation (Blaschuk et al., 2000; Baron et al., 2002), and a role for ανβ5 in the onset of oligodendrocyte precursor differentiation (Blaschuk et al., 2000). Because the dominant-negative construct was expressed using a promoter active in the precursor cells and differentiated oligodendrocytes, transdominant inhibition of any of these integrins could contribute to the phenotype observed and explain the different results of the two strategies.
Remyelination is a spontaneously occurring regenerative process showing some similarities to developmental myelination in that both involve oligodendrocyte progenitor proliferation, migration, engagement of axons, differentiation, and production of myelin membranes (Franklin and Hinks, 1999). However, the extracellular environment in which remyelination takes place is different from that of myelination. Experimentally induced and disease-related demyelinating lesions elicit profound and dynamic changes in the level, composition, and temporal expression of growth factors, chemokines, and proteins of the extracellular matrix within the lesions (Sobel, 1998; Franklin et al., 2001; Sobel, 2001). These changes are proposed to impact oligodendrocyte biology and may influence the outcome of the remyelination process, such as in certain types of multiple sclerosis in which demyelinated lesions fail to remyelinate and eventually predominate (Franklin, 2002). To investigate whether oligodendrocytes require β1-integrin signaling for the remyelination of the CNS, we performed lysolecithin-induced focal demyelinating lesions in the dorsal funiculus of the spinal cord of mutant and control mice. Five weeks after injury, no significant differences in the extent of remyelination were found between mutant and control spinal cords. There was no increase in the number of demyelinated axons and no alterations in the thickness of the myelin sheath, suggesting that oligodendrocytes do not require β1-integrin to remyelinate demyelinated axons. Furthermore, at 4 weeks after lesion, X-gal stainings identified similar numbers of recombined, β-galactosidase-positive cells within the lesions of control and mutant mice, indicating that mutant oligodendroglial cells were also recruited to participate in the remyelination process. Although the lysolecithin lesion paradigm does not directly assess oligodendrocyte progenitor migration, these results imply that the migration of mutant OPCs was not significantly impaired. Because OPC migration on vitronectin and fibronectin substrates is inhibited by β1-integrin function-blocking antibodies (Milner et al., 1996) and, in demyelinating lesions the levels of vitronectin and fibronectin are vastly increased (Sobel, 2001) (our unpublished data), this outcome was unexpected and warrants additional investigation.
Once contacting laminin on the axonal surface through their α6β1-integrin receptor, oligodendrocytes are believed to activate survival signaling in response to low concentrations of growth factors, such as neuregulin or PDGF. Activation of oligodendrocyte survival requires activation of MAPK (mitogen-activated protein kinase) signaling, which leads to phosphorylation and inactivation of the proapoptotic molecule BAD (Bcl-2-associated death protein) (Colognato et al., 2002). Accordingly, mice lacking the α6-integrin subunit and, as a consequence, cell-surface expression of α6β1-integrin showed increased oligodendrocyte apoptosis in the embryonic brainstem, demonstrating the need for this integrin in axon-dependent oligodendrocyte cell survival during development. In the present work, we confirm this role for integrins by showing that the β1-integrin signaling requirement for oligodendrocyte survival extends to other CNS regions, such as the cerebellum. In relation to their control counterparts, P5 mutant cerebella showed both increased apoptotic cell death and a significantly reduced number of myelinating oligodendrocytes (identified as CC1-positive cells in the cerebellum) in peripheral regions in which myelination had just started. No such effect was detected in the central regions of the cerebellum, in which active myelination was already advanced.
These observations corroborate our hypothesis (Colognato et al., 2002) that integrin signaling, in the context of oligodendrocyte cell survival, is required at a stage when cells contact axons and are starting to myelinate. At later stages, the integrin is no longer required for survival and oligodendrocyte numbers have also returned to normal; this may reflect the inherent compensatory ability of a developmental system in which oligodendrocytes are generated in considerable excess (Raff et al., 1998). By targeting the deletion of β1-integrin and, as a result, the loss of α6β1-integrin cell-surface expression to the oligodendrocyte lineage, we were also able to demonstrate that the effect of integrin loss on oligodendrocyte survival was cell autonomous. This is conceptually important because, in the α6-integrin null mice, oligodendrocyte cell death could have also been induced by alterations in cell behavior of other cell types, such as astrocytes, known to promote oligodendrocyte survival (Corley et al., 2001).
In summary, our data show that, although the absence of β1-integrin in oligodendrocytes leads to stage-specific increased oligodendrocyte cell death, CNS myelination and remyelination are not delayed or impaired. In contrast to our previous published observations, this study does not support a role for β1-integrin signaling in myelination and remyelination.
Footnotes
-
This work was supported by a Technische Hochschule grant from the Swiss Federal Institute of Technology in Zurich (Y.B., T.T., U.S., J.B.R.) and by the Swiss National Science Foundation and the National Centre for Competence in Research for “Neural Plasticity and Repair” (U.S.). H.C. was supported by a Career Transition Fellowship from the National Multiple Sclerosis Society. R.J.M.F. was supported by the United Kingdom Multiple Sclerosis Society. C.f.-C. was supported by the Wellcome Trust. We want to thank Cord Brakebusch and Reinhardt Faessler for making the floxed β1-integrin mice available to us and Y. Yonekawa and R. Frick for the use of the Neurosurgery Laboratory of the University Hospital Zurich. We thank Mike Peacock for his excellent support in the processing of EM specimens. We also thank Ned Mantei and Lukas Sommer for critically reading this manuscript.
- Correspondence should be addressed to either João B. Relvas or Ueli Suter, Institute for Cell Biology, Department of Biology, Swiss Federal Institute of Technology, CH-8093 Zurich, Switzerland. Email: joao.relvas{at}cell.biol.ethz.ch or Email: usuter{at}cell.biol.ethz.ch





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}